Catalysis is a phenomenon by which chemicalreactions are accelerated by small quantities offoreign substances, called catalysts. A suitablecatalyst can enhance the rate of a thermodynam-ically feasible reaction but cannot change theposition of the thermodynamic equilibrium.Most catalysts are solids or liquids, but theymay also be gases.
The catalytic reaction is a cyclic process.According to a simplified model, the reactantor reactants form a complex with the catalyst,thereby opening a pathway for their transforma-tion into the product or products. Afterwards thecatalyst is released and the next cycle canproceed.
However, catalysts do not have infinite life.Products of side reactions or changes in thecatalyst structure lead to catalyst deactivation.In practice spent catalysts must be reactivated orreplaced (see Chapter Catalyst Deactivationand Regeneration).
1.1. Types of Catalysis
If the catalyst and reactants or their solutionform a commonphysical phase, then the reaction
is calledhomogeneously catalyzed.Metal salts oforganic acids, organometallic complexes, andcarbonyls of Co, Fe, and Rh are typical homoge-neous catalysts. Examples of homogeneouslycatalyzed reactions are oxidation of toluene tobenzoic acid in the presence of Co and Mnbenzoatesandhydroformylationofolefinstogivethe corresponding aldehydes. This reaction iscatalyzed by carbonyls of Co or Rh.
Heterogeneous catalysis involves systems inwhich catalyst and reactants form separatephysical phases. Typical heterogeneous cata-lysts are inorganic solids such as metals, oxides,sulfides, and metal salts, but they may also beorganic materials such as organic hydroperox-ides, ion exchangers, and enzymes.
Examples of heterogeneously catalyzed reac-tions are ammonia synthesis from the elementsover promoted iron catalysts in the gas phase andhydrogenation of edible oils on Ni – kieselguhrcatalysts in the liquid phase, which are examplesof inorganic and organic catalysis, respectively.
Electrocatalysis is a special case of hetero-geneous catalysis involving oxidation or reduc-tion by transfer of electrons. Examples arethe use of catalytically active electrodes inelectrolysis processes such as chlor-alkali elec-trolysis and in fuel cells.
2 Heterogeneous Catalysis and Solid Catalysts
In photocatalysis light is absorbed bythe catalyst or a reactant during the reaction.This can take place in a homogeneous or het-erogeneous system. One example is the utili-zation of semiconductor catalysts (titanium,zinc, and iron oxides) for photochemical deg-radation of organic substances, e.g., on self-cleaning surfaces.
In biocatalysis, enzymes or microorganismscatalyze various biochemical reactions. Thecatalysts can be immobilized onvarious carrierssuch as porous glass, SiO2, and organic poly-mers. Prominent examples of biochemical re-actions are isomerization of glucose to fructose,important in the production of soft drinks, byusing enzymes such as glucoamylase immobi-lized on SiO2, and the conversion of acryloni-trile to acrylamide by cells of corynebacteriaentrapped in a polyacrylamide gel.
The main aim of environmental catalysis isenvironmental protection. Examples are thereduction of NOx in stack gases with NH3 onV2O5 – TiO2 catalysts and the removal of NOx,CO, and hydrocarbons from automobile exhaustgases by using the so-called three-way catalystconsisting of Rh – Pt – CeO2 – Al2O3 depos-ited on ceramic honeycombs.
The term green catalytic processes has beenused frequently in recent years, implying thatchemical processes may be made environmen-tally benign by taking advantage of the possiblehigh yields and selectivities for the target pro-ducts, with little or no unwanted side productsand also often high energy efficiency.
The basic chemical principles of catalysisconsist in the coordination of reactant mole-cules to central atoms, the ligands of whichmay be molecular species (homogeneous andbiocatalysis) or neighboring atoms at the surfaceof the solid matrix (heterogeneous catalysis).Although there are differences in the details ofvarious types of catalysis (e.g., solvation effectsin the liquid phase, which do not occur insolid – gas reactions), a closer and undoubted-ly fruitful collaboration between the separatecommunities representing homogeneous, het-erogeneous, and biocatalysis should be strong-ly supported. A statement byDavid Parker (ICI)during the 21st Irvine Lectures on 24 April1998 at the University of St. Andrews shouldbe mentioned in this connection, namely, that,
“. . . at the molecular level, there is little todistinguish between homogeneous and hetero-geneous catalysis, but there are clear distinc-tions at the industrial level” [1].
1.2. Catalysis as a Scientific Discipline
Catalysis is a well-established scientificdiscipline, dealing not only with fundamentalprinciples or mechanisms of catalytic reac-tions but also with preparation, properties, andapplications of various catalysts. A number ofacademic and industrial institutes or laborato-ries focus on the study of catalysis andcatalytic processes as well as on the improve-ment of existing and development of newcatalysts.
International journals specializing in cataly-sis include Journal of Catalysis, Journal ofMolecular Catalysis (Series A: Chemical; Se-ries B: Enzymatic), Applied Catalysis (SeriesA:General; Series B: Environmental),ReactionKinetics and Catalysis Letters, Catalysis Today,Catalysis Letters, Topics in Catalysis, Advancesin Organometallic Catalysis, etc.
Publications related to catalysis can alsobe found in Journal of Physical Chemistry,Langmuir, and Physical Chemistry ChemicalPhysics.
Well-known serials devoted to catalysis areHandbuch der Katalyse [edited by G.-M.Schwab, Springer, Wien, Vol. 1 (1941) - Vol.7.2 (1943)], Catalysis [edited by P. H. Emmett,Reinhold Publ. Co., Vol. 1 (1954) - Vol. 7(1960)], Catalysis—Science and Technology[edited by J. R. Anderson and M. Boudart,Springer, Vol. 1 (1981) - Vol. 11 (1996)],Catalysis Reviews (edited by A. T. Bell and J. J.Carberry, Marcel Dekker), Advances in Cata-lysis (edited by B. C. Gates and H. Kn€ozinger,Academic Press), Catalysis (edited by J. J.Spivey, The Royal Society of Chemistry),Studies in Surface Science and Catalysis (editedby B. Delmon and J. T. Yates), etc.
Numerous aspects of catalysis were thesubject of various books. Some, published since1980, are mentioned here:
C. N. Satterfield, Heterogeneous Catalysisin Practice, McGraw Hill Book Comp., NewYork, 1980.
Heterogeneous Catalysis and Solid Catalysts 3
D. L. Trimm,Design of Industrial Catalysts,Elsevier, Amsterdam, 1980.
J. M. Thomas, R. M. Lambert (eds.), Char-acterization of Heterogeneous Catalysts, Wiley,Chichester, 1980.
R. Pearce, W. R. Patterson (eds.), Catalysisand Chemical Processes, John Wiley, NewYork, 1981.
B. L. Shapiro (ed.), Heterogeneous Cataly-sis, Texas A & M Press, College Station, 1984.
B. E. Leach (ed.), Applied Industrial Catal-ysis, Vol. 1, 2, 3, Academic Press, New York,1983 – 1984.
M. Boudart, G. Djega-Mariadassou,Kineticsof Heterogeneous Reactions, Princeton Univer-sity Press, Princeton, 1984.
F. Delannay (ed.), Characterization ofHeterogeneous Catalysts, Marcel Dekker, NewYork, 1984.
R. Hughes, Deactivation of Catalysts, Aca-demic Press, New York, 1984.
M. Graziani,M. Giongo (eds.),FundamentalResearch in Homogeneous Catalysis, Wiley,New York, 1984.
H. Heinemann, G. A. Somorjai (eds.), Ca-talysis and Surface Science, Marcel Dekker,New York, 1985.
J. R. Jennings (ed.), Selective Developmentin Catalysis, Blackwell Scientific Publishing,London, 1985.
G. Parshall, Homogeneous Catalysis, Wiley,New York, 1985.
J. R. Anderson, K. C. Pratt, Introduction toCharacterization and Testing of Catalysts, Ac-ademic Press, New York, 1985.
Y. Yermakov, V. Likholobov (eds), Homo-geneous and Heterogeneous Catalysis, VNUScience Press, Utrecht, Netherlands, 1986.
J. F. Le Page, Applied Heterogeneous Catal-ysis—Design, Manufacture, Use of Solid Cat-alysts, Technip, Paris, 1987.
G. C. Bond, Heterogeneous Catalysis, 2nded., Clarendon Press, Oxford, 1987.
P. N.Rylander,HydrogenationMethods, Ac-ademic Press, New York, 1988.
A. Mortreux, F. Petit (eds.), Industrial Ap-plication of Homogeneous Catalysis, Reidel,Dordrecht, 1988.
J. F. Liebman, A. Greenberg, MechanisticPrinciples of Enzyme Activity, VCH, New York,1988.
J. T. Richardson, Principles of Catalytic De-velopment, Plenum Publishing Corp., NewYork, 1989.
M. V. Twigg (ed.), Catalyst Handbook,Wolfe Publishing, London, 1989.
J. L. G. Fierro (ed.), Spectroscopic Charac-terization of Heterogeneous Catalysts, Elsevier,Amsterdam, 1990.
R. Ugo (ed.), Aspects of Homogeneous Ca-talysis, Vols. 1 – 7, Kluwer Academic Publish-ers, Dordrecht, 1990.
W. Gerhartz (ed.), Enzymes in Industry,VCH, Weinheim, 1990.
R. A. van Santen, TheoreticalHeterogeneousCatalysis, World Scientific, Singapore, 1991.
J. M. Thomas, K. I. Zamarev (eds.), Per-spectives in Catalysis, Blackwell ScientificPublications, Oxford, 1992.
B. C. Gates, Catalytic Chemistry, Wiley,New York, 1992.
G. W. Parshall, S. D. Ittel, HomogeneousCatalysis, 2nd ed., Wiley, New York, 1992.
J. J. Ketta (ed.), Chemical Processing Hand-book, Marcel Dekker, New York, 1993.
J. A. Moulijn, P. W. N. M. van Leeuwen,R. A. van Santen (eds.), Catalysis—An Inte-grated Approach to Homogeneous, Heteroge-neous and Industrial Catalysis, Elsevier,Amsterdam, 1993.
J. W. Niemantsverdriet, Spectroscopy in Ca-talysis, VCH, Weinheim, 1993.
J. Reedijk (ed.), Bioinorganic Catalysis, M.Dekker, New York, 1993.
G. A. Somorjai, Introduction to SurfaceChemistryandCatalysis,Wiley,NewYork,1994.
J. M. Thomas,W. J. Thomas, Principles andPractice of Heterogeneous Catalysis, VCH,Weinheim, 1996.
R. J. Wijngarden, A. Kronberg, K. R. Wes-terterp, Industrial Catalysis—Optimizing Cat-alysts and Processes, Wiley-VCH, Weinheim,1998.
G. Ertl, H. Kn€ozinger, J. Weitkamp (eds.),EnvironmentalCatalysis,Wiley-VCH,Weinheim,1999.
G. Ertl, H. Kn€ozinger, J. Weitkamp (eds.),Preparation of Solid Catalysts, Wiley-VCH,Weinheim, 1999.
B. Cornils, W. A. Herrmann, R. Schl€ogl, C.-H. Wong, Catalysis from A – Z, Wiley-VCH,Weinheim, 2000.
4 Heterogeneous Catalysis and Solid Catalysts
B. C. Gates, H. Kn€ozinger (eds.), Impact ofSurface Science on Catalysis, Academic, SanDiego, 2000.
A comprehensive survey of the principles andapplications: G. Ertl, H. Kn€ozinger, F. Sch€uth, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, 2nd ed. with 8 volumes and 3966pages, Wiley-VCH, Weinheim 2008.
The first International Congress onCatalysis(ICC) took place in 1956 in Philadelphia and hassince been held every four years in Paris (1960),Amsterdam (1964), Moscow (1968), PalmBeach (1972), London (1976), Tokyo (1980),Berlin (1984), Calgary (1988), Budapest(1992), Baltimore (1996), Granada (2000)),Paris (2004) and Seoul (2008). The 15th Con-gress will be held in Munich in 2012. Presentedpapers and posters have been published in theProceedings of the corresponding congresses.The International Congress on Catalysis Coun-cil (ICC)was renamed at the Council meeting inBaltimore 1996. The international organizationis now called International Association of Ca-talysis Societies (IACS).
In 1965 the Catalysis Society of NorthAmerica was established and holds meetings inthe USA every other year.
The European Federation of Catalysis Soci-eties (EFCATS) was established in 1990. TheEUROPACAT Conferences are organized underthe auspices of EFCATS. The first conferencetook place in Montpellier (1993) followed byMaastricht (1995), Cracow (1997), Rimini(1999), and Limerick (2001).
Furthermore, every four years (in the evenyear between two International Congresses onCatalysis) an International Symposium focusingon Scientific Basis for the Preparation of Het-erogeneous Catalysts is held in Louvain-LaNeuve (Belgium).
Other international symposia or congressesdevoted to catalysis are: International ZeoliteConferences, International Symposium of Cat-alyst Deactivation, Natural Gas ConversionSymposium, Gordon Conference on Catalysis,TOCAT (Tokyo Conference on Advanced Cata-lytic Science and Technology), InternationalSymposium of Acid-Base Catalysis, the Europe-an conference series, namely the Roermond,Sabatier- and Schwab-conference, and the Tay-lor Conference.
1.3. Industrial Importanceof Catalysis
Because most industrial chemical processes arecatalytic, the importance and economical sig-nificance of catalysis is enormous. More than80% of the present industrial processes estab-lished since 1980 in the chemical, petrochemi-cal, and biochemical industries, as well as in theproduction of polymers and in environmentalprotection, use catalysts.
More than 15 international companies havespecialized in the production of numerous cata-lystsappliedinseveralindustrialbranches.In2008the turnover in the catalysts world market wasestimatedtobeaboutUS-$ 13 � 109(seeChapterProduction of Heterogeneous Catalysts).
1.4. History of Catalysis
The phenomenon of catalysis was first recog-nized byBERZELIUS [2,3] in 1835.However, somecatalytic reactions such as the production ofalcoholic beverages by fermentation or the man-ufacture of vinegar by ethanol oxidation werepracticed long before. Production of soap by fathydrolysis and diethyl ether by dehydration ofethanol belong to the catalytic reactions thatwere performed in the 16th and 17th centuries.
Besides BERZELIUS, MITSCHERLICH [3] wasalso involved at the same time in the study ofcatalytic reactions accelerated by solids. Heintroduced the term contact catalysis. This termfor heterogeneous catalysis lasted for more than100 years.
In 1895 OSTWALD [3,4] defined catalysis asthe acceleration of chemical reactions by thepresence of foreign substances which are notconsumed. His fundamental work was recog-nized with the Nobel prize for chemistry in1909.
Between 1830 and 1900 several practicalprocesses were discovered, such as flamelesscombustion of CO on a hot platinum wire, andthe oxidation of SO2 to SO3 and of NH3 to NO,both over Pt catalysts.
In 1912 SABATIER [3,5] received the Nobelprize for his work devoted mainly to the hydro-genation of ethylene and CO over Ni and Cocatalysts.
Heterogeneous Catalysis and Solid Catalysts 5
The first major breakthrough in industrialcatalysis was the synthesis of ammonia fromthe elements, discovered by HABER [3,6,7] in1908, using osmium as catalyst. Laboratoryrecycle reactors for the testing of various am-monia catalysts which could be operated at highpressure and temperature were designed byBOSCH [3]. The ammonia synthesis was com-mercialized at BASF (1913) as the Haber –Bosch [8] process. MITTASCH [9] at BASF devel-oped and produced iron catalysts for ammoniaproduction.
In 1938 BERGIUS [3,10] converted coal toliquid fuel by high-pressure hydrogenation inthe presence of an Fe catalyst.
Other highlights of industrial catalysis werethe synthesis of methanol from CO and H2 overZnO – Cr2O3 and the cracking of heavier pe-troleum fractions to gasoline using acid-activat-ed clays, as demonstrated by HOUDRY [3,6] in1928.
The addition of isobutane to C3 – C4 olefinsin the presence of AlCl3, leading to branchedC7 – C8 hydrocarbons, components of high-quality aviation gasoline, was first reported byIPATIEFF et al. [3,7] in 1932. This invention led toa commercial process of UOP (USA).
Of eminent importance for Germany, whichpossesses no natural petroleum resources, was
the discovery by FISCHER andTROPSCH [11] of thesynthesis of hydrocarbons and oxygenated com-pounds from CO and H2 over an alkalized ironcatalyst. The first plants for the production ofhydrocarbons suitable asmotor fuel started up inGermany 1938. After World War II, Fischer-Tropsch synthesis saw its resurrection in SouthAfrica. Since 1955 Sasol Co. has operated twoplants with a capacity close to 3 � 106 t/a.
One of the highlights of German industrialcatalysis before World War II was the synthe-sis of aliphatic aldehydes by ROELEN [12] bythe addition of CO and H2 to olefins in thepresence of Co carbonyls. This homogeneous-ly catalyzed reaction was commercialized in1942 by Ruhr-Chemie and is known as OxoSynthesis.
During and after World War II (till 1970)numerous catalytic reactions were realized onan industrial scale (see also Chapter Applica-tion of Catalysis in Industrial Chemistry).Some important processes are compiled inTable 1.
Table 2 summarizes examples of catalyticprocesses representing the current status of thechemical, petrochemical and biochemical in-dustry as well as the environmental protection(see also Chapter Application of Catalysis inIndustrial Chemistry).
Table 1. Important catalytic processes commercialized during and after World War II (until 1970) [13,14]
Year ofcommercialization
Process Catalyst Products
1939 – 1945 dehydrogenation Pt – Al2O3 toluene from methylcyclohexanedehydrogenation Cr2O3 – Al2O3 butadiene from n-butanealkane isomerization AlCl3 i-C7 – C8 from n-alkanes
1946 – 1960 oxidation of aromatics V2O5 phthalic anhydride fromnaphthalene and o-xylene
hydrocracking Ni – aluminosilicate fuels from high-boiling petroleumfractions
polymerization(Ziegler – Natta)
TiCl4 – Al(C2H5)3 polyethylene from ethylene
dehydrogenation Fe2O3 – Cr2O3 – KOH styrene from ethylbenzeneoxidation (Wacker process) PdCl2 – CuCl2 acetaldehyde from ethylene
1961 – 1970 steam reforming Ni – a-Al2O3 Co, (CO2), and H2 from methaneammoxidation Bi phosphomolybdate acrylonitrile from propenefluid catalytic cracking H zeolites þ aluminosilicates fuels from high boiling fractionsreforming bimetallic catalysts (Pt, Sn, Re, Ir) gasolinelow-pressure methanolsynthesis
Cu – ZnO – Al2O3 methanol from CO, H2, CO2
isomerization enzymes immobilized on SiO2 fructose from glucose (productionof soft drinks)
distillate dewaxing ZSM-5, mordenites removal of n-alkanes from gasolinehydrorefining Ni – , CO – MoSx hydrodesulfurization, hydrodenitrification
6 Heterogeneous Catalysis and Solid Catalysts
2. Theoretical Aspects
The classical definition of a catalyst statesthat “a catalyst is a substance that changes therate but not the thermodynamics of a chemicalreaction” and was originally formulated byOSTWALD [4]. Hence, catalysis is a dynamicphenomenon.
As emphasized by BOUDART [19], the condi-tions under which catalytic processes occur onsolid materials vary drastically. The reactiontemperature can be as low as 78 K and as highas 1500 K, and pressures canvary between 10�9
and 100 MPa. The reactants can be in the gasphase or in polar or nonpolar solvents. Thereactions can occur thermally or with the assis-tance of photons, radiation, or electron transferat electrodes. Pure metals and multicomponentand multiphase inorganic compounds can act ascatalysts. Site-time yields (number of productmolecules formed per site and unit time) as lowas 10�5 s�1 (corresponding to one turnover per
day) and as high as 109 s�1 (gas kinetic collisionrate at 1 MPa) are observed.
It is plausible that it is extremely difficult, ifnot impossible, to describe the catalytic phe-nomenon by a general theory which covers theentire range of reaction conditions and observedsite-time yields (reaction rates). However, thereare several general principleswhich are consid-ered to be laws or rules of thumb that are usefulin many situations. According to BOUDART [19],the value of a principle is directly related to itsgenerality. In contrast, concepts are more spe-cialized and permit an interpretation of phenom-ena observed for special classes of catalysts orreactions under given reaction conditions.
In this chapter, important principles andconcepts of heterogeneous catalysis are dis-cussed, followed by a section on kineticsof heterogeneously catalyzed reactions. Thechapter is concluded by a section on the deter-mination of reaction mechanisms in heteroge-neous catalysis.
Table 2. Important catalytic processes commercialized after 1970 [15–18]
1. Mo, Bi oxides acrylic acid from propene2. Mo, V, PO (heteropolyacids)
oxidation (Monsanto) vanadylphosphate maleic anhydride from n-butanefluid-bed polymerization (Unipol) Ziegler – Natta type polyethylene and polypropylenehydrocarbon synthesis (Shell) 1. Co – (Zr,Ti) – SiO2 middle distillate from CO þ H2
2. Pt – SiO2
environmental control(combustion process)
Pt – Al2O3 (monoliths) deodoration
1986 – 2000 oxidation with H2O2 (Enichem) Ti silicalite hydroquinone and catecholfrom phenol
hydration enzymes acrylamide from acrylonitrileammoxidation (Montedipe) Ti silicalite cyclohexanone oxime from
cyclohexanone, NH3, and H2O2
dehydrogenation of C3, C4 alkanes(Star and Oleflex processes)
The Sabatier principle proposes the existence ofan unstable intermediate compound formed be-tween the catalyst surface and at least one of thereactants [5]. This intermediate must be stableenough to be formed in sufficient quantities andlabile enough to decompose to yield the finalproduct or products. The Sabatier principle isrelated to linear free energy relationships suchas a Brønsted relation [19]. These relations dealwith the heat of reaction q (thermodynamicquantity) and the activation barrier E (kineticquantity) of an elementary step in the exother-mic direction (q > 0). With an empirical pa-rameter a (0 < a < 1) and neglecting entropyeffects, a Brønsted relation can be written as
DE ¼ a Dq;
where DE is the decrease in activation energycorresponding to an increase Dq in the heat ofreaction. Hence, an elementary step will have ahigh rate constant in the exothermic directionwhen its heat of reaction q increases. Since theactivation barrier in the endothermic direction isequal to the sum of the activation energy E andthe heat of reaction, the rate constant will de-crease with increasing q.
The Brønsted relationship represents a bridgebetween thermodynamics and kinetics and, to-gether with the Sabatier principle, permits aninterpretation of the so-called volcano plots firstreported byBALANDIN [20].Thesevolcano curvesresult when a quantity correlated with the rate ofreaction under consideration is plotted against ameasure of the stability of the intermediatecompound. The latter quantity can be the heatof adsorption of one of the reactants or the heat offormation of a bulk compound relative to thesurface compound, or even the heat of formationof any bulk compound that can be correlatedwiththe heat of adsorption, or simply the position ofthe catalytic material (metal) along a horizontalseries in the Periodic Table [263].
As an example, Figure 1 shows the volcanoplot for the decomposition of formic acid ontransition metals [21]. The intermediate in thisreaction was shown to be a surface formate.Therefore, the heats of formationDHf of the bulk
metal formates were chosen as the measure ofthe stability of the intermediate. At lowvalues ofDHf, the reaction rate is low and corresponds tothe rate of adsorption, which increases withincreasing heat of formation of the bulk for-mates (representing the stability of the surfacecompound). At high values of DHf the reactionrate is also low and corresponds to the desorp-tion rate, which increases with decreasing DHf.As a consequence, a maximum in the rate ofreaction (decomposition of formic acid) is ob-served at intermediate DHf values which isneither a pure rate of adsorption nor a pure rateof desorption but which depends on both.
2.1.2. The Principle of Active Sites
The Sabatier principle of an unstable surfaceintermediate requires chemical bonding of re-actants to the catalyst surface, most likely be-tween atoms or functional groups of reactantand surface atoms. This leads to the principle ofactive sites. When LANGMUIR formulated hismodel of chemisorption on metal surfaces [22],
Figure 1. Volcano plot for the decomposition of formicacid. The temperature Tat which the rate of decomposition vhas a fixed value is plotted against the heat of formation DHf
of the metal formate (adopted from [31]).
8 Heterogeneous Catalysis and Solid Catalysts
he assumed an array of sites whichwere energet-ically identical and noninteracting, and whichwould adsorb just one molecule from the gasphase ina localizedmode.TheLangmuir adsorp-tion isotherm results from this model. The sitesinvolved can be considered to be active sites.
LANGMUIR was already aware that the as-sumption of identical and noninteracting siteswas an approximation which would not hold forreal surfaces, when he wrote [23]: “Most finelydivided catalysts must have structures of greatcomplexity. In order to simplify our theoreticalconsideration of reactions at surfaces, let usconfine our attention to reactions on plane sur-faces. If the principles in this case are wellunderstood, it should then be possible to extendthe theory to the case of porous bodies. Ingeneral, we should look upon the surface asconsisting of a checkerboard.” LANGMUIR thusformulated the surface science approach to het-erogeneous catalysis for the first time.
The heterogeneity of active sites on solidcatalyst surfaces and its consequences wereemphasized by TAYLOR [24], who recognizedthat “There will be all extremes between thecase in which all atoms in the surface are activeand that inwhich relatively few are so active.” Inother words, exposed faces of a solid catalystwill contain terraces, ledges, kinks, and vacan-cies with sites having different coordinationnumbers. Nanoscopic particles have edges andcorners which expose atoms with different co-ordination numbers [25]. The variation of coor-dination numbers of surface atoms will lead todifferent reactivities and activities of the corre-sponding sites. In this context, Schwab’s adli-neation theory may be mentioned [26], whichspeculated that one-dimensional defects con-sisting of atomic steps are of essential impor-tance. This view was later confirmed by surfacescience studies on stepped single-crystal metalsurfaces [27].
In addition to variable coordination numbersof surface atoms in one-component solids, thesurface composition may be different from thatof the bulk and different for each crystallogra-phic plane inmulticomponentmaterials (surfacesegregation [28]). This would lead to a hetero-geneity of the local environment of a surfaceatom and thus create nonequivalent sites.
Based on accurate kinetic measurementsand on the Taylor principle of the existence of
inequivalent active sites, BOUDART et al. [29]coined the terms structure-sensitive and struc-ture-insensitive reactions. A truly structure-in-sensitive reaction is one in which all sites seemto exhibit equal activity on several planes of asingle crystal. Surprisingly, many heteroge-neously catalyzed reactions turned out to bestructure-insensitive. Long before experimentalevidence for this phenomenonwas available andbefore a reliable interpretation was known,TAYLOR predicted it by writing [24]: “Theamount of surface which is catalytically activeis determined by the reaction catalyzed.” Inother words, the surface of a catalyst adaptsitself to the reaction conditions for a particularreaction. The driving force for this reorganiza-tion of a catalyst surface is the minimization ofthe surface free energy, which may be achievedby surface-reconstruction [30,31]. As a conse-quence, a meaningful characterization of activesites requires experiments under working (insitu) conditions of the catalytic system.
The principle of active sites is not limited tometals. Active sites include metal cations, an-ions, Lewis and Brønsted acids, acid – basepairs (acid and base acting simultaneously inchemisorption), organometallic compounds,and immobilized enzymes. Active sites mayinclude more than one species (or atom) toform multiplets [20] or ensembles [32]. Amandatory requirement for these sites to beactive is that they are accessible for chemisorp-tion from the fluid phase. Hence, they mustprovide free coordination sites. Therefore,BURWELL et al. [33,34] coined the term coordi-natively unsaturated sites in analogy with ho-mogeneous organometallic catalysts. Thus, ac-tive sites are to be considered as atoms or groupsof atoms which are embedded in the surface of amatrix in which the neighboring atoms (orgroups) act as ligands. Ensemble and ligandeffects are discussed in detail by SACHTLER [35]and quantum chemical treatments of geometricensemble and electronic ligand effects on metalalloy surfaces are discussed by HAMMER andNøRSKOV [36].
2.1.3. Surface Coordination Chemistry
The surface complexes formed by atomsor molecules are now known to usually
Heterogeneous Catalysis and Solid Catalysts 9
resemble a local structure similar to molecularcoordination complexes. The bonding in thesesurface complexes can well be described in alocalized picture [37,38]. Thus, important phe-nomena occuring at the surface of solid catalystsmay be described in the framework of surfacecoordination chemistry or surface organometal-lic chemistry [39,40].
This is at variance with the so-called bandtheory of catalysis, which attempted to corre-late catalytic performance with bulk electronicproperties [41–43]. The shortcomings of thistheory in oxide catalysis are discussed bySTONE [44].
2.1.4. Modifiers and Promoters
The performance of real industrial catalysts isoften adjusted by modifiers (additives) [45,46].A modifier is called a promoter when it in-creases the catalyst activity in terms of reactionrate per site. Modifiers may also affect a cat-alyst’s performance in an undesired manner. Inthis case the modifier acts as a catalyst poison.However, this simple distinction between pro-moters and poisons is less straightforward forreactions yielding more than one product inparallel or consecutive steps, of which only oneis the desired product. In this case not only highactivity but also high selectivity is desired. Theselectivity can be improved by adding sub-stances that poison undesirable reactions. Inexothermic reactions excessively high reactionrates may lead to a significant temperatureincrease (sometimes only locally: hot spots)which can yield undesirable products (e.g., COand CO2 in selective catalytic oxidation). Adeterioration of the catalyst due to limited cata-lyst stability may also occur. Consequently, amodifier is required which decreases the reac-tion rate so that a steady-state temperature andreaction rate can be maintained. Although themodifier acts as a poison in these cases, it is infact a promoter as far as selectivity and catalyststability are concerned.
Modifiers canchange thebindingenergyofanactive site or its structure, or disrupt an ensembleof atoms, e.g., by alloying an active with aninactive metal. A molecular approach toward anunderstanding of promotion in heterogeneouscatalysis was presented by HUTCHINGS [47].
As an example, the iron-based ammoniasynthesis catalyst is promoted by Al2O3 andK2O [48]. Alumina acts as a textural promoter,as it prevents the rapid sintering of pure ironmetal. It may also stabilize more active sites onthe iron surface (structural promoter). Potassi-um oxide appears to affect the adsorption kinet-ics and dissociation of dinitrogen and the bind-ing energy of nitrogen on adjacent iron sites(electronic promoter).
The addition of Co to MoS2-based cata-lysts supported on transitional aluminas hasa positive effect on the rate of hydrodesulfur-ization of sulfur-containing compounds at Co/(Co þ Mo) ratios belowca. 0.3 [49] (see SectionSupported Metal Catalysts). The active phaseis proposed to be the so-called CoMoS phasewhich consists of MoS2 platelets, the edges ofwhich are decorated by Co atoms. The lattermay act as structural and electronic promoterssimultaneously.
Another example concerns bifunctional cat-alysts for catalytic reforming [50], which con-sist of Pt supported on strongly acidic aluminas,the acid strength of which is enhanced by mod-ification with chloride. Since these materialslose chlorine during the catalytic process, thefeed contains CCl4 as a precursor of the surfacechloride promoter.
2.1.5. Active Phase – SupportInteractions
Several concepts have proved valuable ininterpreting phenomena which are pertinent tocertain classes of catalysts. In supported cata-lysts, the active phase (metal, oxide, sulfide)undergoes active phase-support interactions[51–53]. These are largely determined by thesurface free energies of the support and activephase materials and by the interfacial free ener-gy between the two components [51–53].Activetransition metal oxides (e.g., V2O5, MoO3,WO3) have relatively low surface free energiesas compared to typical oxidic support materialssuch as g-Al2O3, TiO2 (anatase), and SiO2.Although the interfacial free energies betweenactive phase and support are not known, theinteraction between the two components ap-pears to be favorable, with the exception ofSiO2-supported transition metal oxides. As a
10 Heterogeneous Catalysis and Solid Catalysts
consequence spreading and wetting phenomenaoccur if the thermal treatment of the oxidemixtures is carried out at temperatures suffi-ciently high to induce mobility of the activeoxide. As a rule of thumb, mobility of a solidtypically occurs above the Tammann tempera-ture, which is equal to half the melting point ofthe bulk solid. As a result, the active transitionmetal oxide tends to wet the support surface andforms a monolayer (monolayer-type catalysts).
Transition and noble metals typically havehigh surface free energies [52], and therefore,small particles or crystallites tend to agglomeratetoreduce their surfacearea.Stabilizationofnano-sizemetal particles therefore requires depositionon the surface of supports providing favorablemetal-support interactions (MSI). The smallerthe particle the more its physical properties andmorphology can be affected by these interac-tions. Therefore, the nature of the support mate-rial for agivenmetal alsocritically influences thecatalytic properties of the metal particle.
Supported metals are in a nonequilibriumstate and therefore still tend to agglomerate atsufficiently high temperatures in reducing atmo-spheres. Hence, deactivation occurs because ofthe reduced metal surface area. Regenerationcan typically be achieved by thermal treatmentin an atmosphere in which the active metal isoxidized. The surface free energies of transitionand noble metal oxides are significantly lowerthan those of the parent metals, so that theirspreading on the support surface becomesmore favorable. Subsequent reduction undersufficiently mild conditions can restore the high
degree of metal dispersion [dispersion D isdefined as the ratio of the number of metalatoms exposed at the particle surface (NS) tothe total number of metal atoms NT in theparticle (D ¼ NS/NT)].
So-called strong metal-support interactions(SMSI) may occur, e.g., for Pt – TiO2 andRh – TiO2 [51,53,54]. As shown experimen-tally, the adsorption capacity for H2 and CO isdrastically decreased when the precursorfor the catalytically active metal on the supportis reduced in H2 at temperatures aboveca. 770 K [51,54]. Simultaneously, the oxidesupport is slightly reduced. Although severalexplanations have been proposed for the SMSIeffect, the most probable explanation is encap-sulation of the metal particle by support oxidematerial. Encapsulation may occur when thesupport material becomes mobile. Althoughthe electronic properties of the metal particlemay be affected by the support oxide in theSMSI state, the decrease of the adsorptioncapacity appears to be largely due to a geomet-ric effect, namely, the resulting inaccessibilityof the metal surface.
The various possible morphologies and dis-persions of supported metals are schematicallyshown in Figure 2. The metal precursor typical-ly is well dispersed after impregnation of thesupport. Low-temperature calcination may leadto well-dispersed oxide overlayers, while directlow-temperature reduction leads to highly dis-persed metal particles. This state can also bereached by low-temperature reduction of thedispersed oxide precursor, this step being
Figure 2. Schematic representation of metal-support interactions (adopted from [50])
Heterogeneous Catalysis and Solid Catalysts 11
reversible by low-temperature reoxidation. Forthe preparation of highly dispersed Ni catalysts,it is important to remove thewater that is formedby hydrogen reduction of NiO. H2 diluted withN2 is used for this purpose. Surface compoundformation may also occur by a solid-state reac-tion between the active metal precursor and thesupport at high calcination temperatures. Re-duction at high temperatures may lead to parti-cle agglomeration when cohesive forces aredominant, and to so-called pillbox morpholo-gies when adhesive forces are dominant. In bothcases, the metal must be mobile. In contrast,when the support is mobile, sintering of thesupport can occur, and the small metal particlesare stabilized on the reduced surface area(cohesive forces). Alternatively, if adhesiveforces are dominant encapsulation (SMSIeffect) may occur.
2.1.6. Spillover Phenomena
In multiphase solid catalysts spillover may oc-cur of an active species (spillover species) ad-sorbed or formed on one phase (donor phase)onto a second phase (acceptor) which does notform the active species under the same condi-tions [55–57]. Awell-known example is hydro-gen spillover from Pt, on which dihydrogenchemisorbs dissociatively, onto WO3 with for-mation of a tungsten bronze [58]. According toSOMORJAI [59] the spillover phenomenon mustbe regarded as one of the “modern concepts insurface science and heterogeneous catalysis”.Nevertheless, the exact physical nature of spill-over processes has only rarely been verifiedexperimentally. The term is typically used toexplain nonlinear effects (synergistic effects)of the combination of chemically differentcomponents of a catalytic material on itsperformance.
Besides hydrogen spillover, oxygen spilloverhas been postulated to play an important role inoxidation reactions catalyzed by mixed oxides.For example, the addition of antimony oxide toselective oxidation catalysts enhances the cata-lytic activity at high levels of selectivity by afactor of up to five relative to the Sb-free system,although antimony oxide itself is completelyinactive.
Observations of this kind motivated DELMON
et al. [60,61] to formulate the remote-controlconcept to explain the fact that all industrialcatalysts used for the partial oxidation of hydro-carbons or in hydrotreatment are multiphasicand that particular phase compositions developsynergy effects. The remote-control concept is,however, not undisputed.
2.1.7. Phase-Cooperation and Site-Isola-tion Concepts
GRASSELLI [62] proposed the phase-cooperationconcept for partial oxidation and ammoxidationreactions. It is suggested that two phases (e.g.,a-Bi2Mo3O12 and g-Bi2MoO6) cooperate in thesense that one phase performs the actual cata-lytic function (a-phase) and the other (g-phase)the reoxidation function. The concept could beverified for many other multiphase, multicom-ponent mixed metal oxide catalysts, such asmulticomponent molybdates and multicompo-nent antimonates [62,63].
Another concept most relevant for selectiveoxidation and ammoxidation is the site-isolationconcept first formulated by CALLAHAN andGRASSELLI [64]. Site isolation refers to the sepa-ration of active sites from each other on thesurface of a heterogeneous catalyst and is con-sidered to be the prerequisite for obtaining thedesired selective partial oxidation products. Theconcept states that reactive surface lattice oxy-gen atoms must be structurally isolated fromeach other in defined groupings on a catalystsurface to achieve selectivity. The number ofoxygen atoms in a given isolated grouping de-termines the reaction channel through the stoi-chiometry requirements imposed on the reactionby the availability of oxygen at the reaction site.It was postulated that two and up to five adjacentsurface oxygen atoms would be required for theselective oxidation of propene to the desiredproduct acrolein. Lattice groupings with morethan five oxygen atomswould only produce totaloxidation products (CO and CO2), whilecompletely isolated single oxygen atoms wouldbe either inactive or could produce allyl radicals.The latter would couple in the vapor phase togive hexadiene and ultimately benzene. Thesescenarios are schematically shown in Figure 3.
12 Heterogeneous Catalysis and Solid Catalysts
2.1.8. Shape-Selectivity Concept
Zeolites and related materials have crystallinestructure and contain regular micropores, thediameters of which are determined by the struc-ture of the materials. The pore sizes are welldefined and have dimensions similar to those ofsmall organic molecules. This permits shape-selective catalysis to occur. The geometric con-straints may act on the sorption of reactants, onthe transition state of the catalyzed reaction, oron the desorption of products. Correspondingly,shape-selective effects have been classified asproviding reactant shape selectivity, restricted
transition state shape selectivity, and productshape selectivity [65,66]. These scenarios areschematically illustrated in Figure 4 for thecracking of n-heptane and 1-methylhexane (re-actant shape selectivity), for the transalkylationof m-xylene (transition state shape selectivity),and for the alkylation of toluene by methanol(product shape selectivity). In the first example,the kinetic diameter of n-heptane is smaller thanthat of 1-methylhexane. The latter is not able toenter micropores, so that shape-selective crack-ing of n-heptane takes place when both hydro-carbons are present in the feed. An examplefor shape-selective control of the transition
Figure 3. Site-isolation principle. Schematic of lattice oxygen arrangements on hypothetical surfaces. Anticipated reactionpaths of propene upon contact with these surfaces (NR ¼ no reaction; adopted from [62])
Heterogeneous Catalysis and Solid Catalysts 13
state is the transalkylation of m-xylene. Thereaction is bimolecular and the formation of1,2,4-trimethylbenzene has a less bulky transi-tion state than the formation of 1,3,5-trimethyl-benzene. The latter product can thus not beformed if the pore size and geometry is carefullyadapted to the transition state requirements.Finally, p-xylene can be selectively formed bymethylation of toluene with methanol and zeo-lites whose pore openings only allow p-xyleneto be released. The o and m isomers eitheraccumulate in zeolite cages or are isomerizedto p-xylene.
2.1.9. Principles of the Catalytic Cycle
The most fundamental principle in catalysis isthat of the catalytic cycle, whichmaybebasedona redefinition of a catalyst by BOUDART [67]: “Acatalyst is a substance that transforms reactantsinto products, through an uninterrupted andrepeated cycle of elementary steps in which thecatalyst is changed through a sequence of reac-tive intermediates, until the last step in the cycleregenerates the catalyst in its original form”.
The catalytic substance or active sites maynot be present originally, but may be formed byactivation during the start-up phase of the cata-lytic reaction. The cycle must be uninterruptedand repeated since otherwise the reaction isstoichiometric rather than catalytic. The number
of turnovers, a measure of catalyst life, must begreater than unity, since the catalyst wouldotherwise be a reagent. The total amount ofcatalyst (active sites) is typically small relativeto the amounts of reactants and products in-volved (catalytic amounts). As a consequence,the reactive intermediates can be treated by thekinetic quasi-steady-state approximation ofBODENSTEIN.
The activity of the catalyst is defined by thenumber of cycles per unit time or turnovers orturnover frequency (TOF; unit: s�1). The life ofthe catalyst is defined by the number of cyclesbefore it dies.
2.2. Kinetics of HeterogeneousCatalytic Reactions [67–76]
The catalytic cycle is the principle of catalyticaction. The mechanism of a catalyzed reactioncan be described by the sequence of elementaryreaction steps of the cycle, including adsorption,surface diffusion, chemical transformations ofadsorbed species, and desorption, and it is thebasis for deriving the kinetics of the reaction. Itis assumed that for each individual elementarystep the transition-state theory is valid. An earlytreatise of the kinetics of heterogeneously cata-lyzed reactions was published by SCHWAB [77].
The various aspects of the dynamics of sur-face reactions and catalysis have been classified
Figure 4. Classification of shape-selective effects
14 Heterogeneous Catalysis and Solid Catalysts
by ERTL [31] into five categories in terms of timeand length scales, as shown schematically inFigure 5. In the macroscopic regime, the rate ofa catalytic reaction is modeled by fitting empir-ical equations, such as power laws, to experi-mental data, so as to describe its concentrationand pressure dependence and to determine rateconstants that depend exponentially on temper-ature. This approach was very useful in chemi-cal engineering for reactor and process design.Assumptions about reaction schemes (kineticmodels) provide correlations between the sur-face coverages of intermediates and the externalvariables, an approach that led to the Temkinequation [78] modeling the kinetics of ammoniasynthesis.
Improved kinetic models could be developedwhen atomic processes on surfaces and theidentification and characterization of surfacespecies became available. The progress of acatalytic reaction is then described by a micro-kinetics approach by modeling the macroscopickinetics through correlating atomic processeswith macroscopic parameters within the frame-work of a suitable continuum model. Continu-um variables for the partial surface coveragesare, to a first approximation, correlated to ex-ternal parameters (partial pressures and temper-ature) by the Langmuir latticemodel of a surfaceconsisting of identical noninteracting adsorp-tion sites.
The formulation of rate laws for the fullsequence of elementary reactions will usuallylead to a set of nonlinear coupled (ordinary)differential equations for the concentrations(coverages) of the various surface species in-volved. The temporal behavior of the reactionsystem under constant continuous-flow condi-
tions may be nonstationary transient. In certainparameter ranges it may be oscillatory or evenchaotic. Also, there may be local variations insurface coverages which lead to coupling of thereaction with transport processes (e.g., particlediffusion, heat transfer). The formation of spa-tiotemporal concentration profiles on a meso-scopic scale is the consequence of these nonlin-ear dynamic phenomena.
Since the Langmuir lattice model is not validin reality, the continuummodel can describe thereaction kinetics only to a first approximation.Interactions between adsorbed species occur,and adsorbed particles occupy nonidenticalsites, so that complications arise in the descrip-tion of the reaction kinetics. Apart from theheterogeneity of adsorption sites, surfaces mayundergo structural transformations. Surface sci-ence investigations provide information onthese effects on an atomic scale.
As mentioned above, it is assumed that thetransition-state theory is valid for description ofthe rates of individual elementary steps. Thistheory is based on the assumption that at allstages along the reaction coordinate thermalequilibrium is established. Temperature then isthe only essential external macroscopic param-eter. This assumption can only be valid if energyexchange between all degrees of motionalfreedom of the particles interacting with thesolid acting as a heat bath is faster than theelementary stepwhich induces nuclear motions.Energy transfer processes at the quantumlevel are the basic requirements for chemicaltransformations.
Nonlinear dynamics and the phenomenaoccuring at the atomic and quantum levels werereviewed by ERTL [31].
Figure 5. Schematic classification of the various aspects of the dynamics of surface reactions (adopted from [31])
Heterogeneous Catalysis and Solid Catalysts 15
2.2.1. Concepts of Reaction Kinetics(Microkinetics)
The important concepts of (catalytic) reactionkinetics were reviewed by BOUDART [67,68,79,80], and by CORTRIGHT and DUMESIC [74].
The term microkinetics was defined to de-note reaction kinetics analyses that attempt toincorporate into the kinetic model the basicsurface chemistry involved in the catalyticreaction at a molecular level [73,74]. An im-portant prerequisite for this approach is thatreaction rates are measured in the absence ofheat- andmass-transfer limitations. The kineticmodel is based on a description of the catalyticprocess in terms of information and/or assump-tions about active sites and the nature of ele-mentary steps that make up the catalytic cycle.The ultimate goal of a kinetic analysis is thedetermination of preexponential factors andactivation energies (cf. Arrhenius equation) forall elementary steps in forward and reversedirection. Usually there is not sufficient infor-mation available to extract the values of allkinetic parameters. However, it has been es-tablished that in many cases the observed ki-netics are controlled by a limited number ofkinetic parameters [73,74]. Questions to beanswered in this situation are: (1) how manykinetic parameters are required to calculate theoverall rate from a reaction scheme? (2) Whatspecies are themost abundant intermediates onthe catalyst surface under reaction conditions?(3) Does the reaction scheme include a rate-determining step for the kinetic parameters ofinterest under the reaction conditions? Gener-ally, only a few parameters are kineticallysignificant, although it is difficult to predictwhich parameters control the overall rate of thecatalytic process. Therefore, initial estimatesrequire a larger set of parameters than areultimately necessary for the kinetic descrip-tion of the catalytic process of interest. Be-sides experimental values of kinetic para-meters for individual elementary reactions(often resulting from surface sciencestudies on single-crystal surfaces), quantumchemical calculations permit mechanistic in-vestigations and predictions of kinetic para-meters [36–38].
Assume that a kinetic model has been estab-lished which consists of n elementary steps,
each proceeding at a net rate
ri ¼ rfi�rri ði ¼ 1; 2; :::; nÞ ð1Þ
The subscripts f and r stand for “forward” and“reverse”, respectively. As mentioned above,the validity of the Bodenstein steady-state con-cept can be assumed. The kinetic steady state isthen defined by:
sir ¼ ri ð2Þ
where r is the net rate rf � rr of the overallcatalytic reaction defined by a stoichiometricequation. si is the stoichiometric number ofthe ith step, i.e., the number of times that thisstep must occur for the catalytic cycle toturnover once. If the transition-state theory isvalid for each individual elementary step, theratio of the forward rate rfi to the reverse raterri of step i is given by the De Donder rela-tion [81,82]:
rfi=rri ¼ expðAi=RTÞ ð3Þ
where Ai is the affinity of step i:
Ai ¼ ½@Gi=@ji�T ;P ð4Þ
where ji is the extent of reaction of step i.At steady state, the affinity for each step but
one may be very small as compared to theaffinity A of the overall reaction. Each step butone is then in quasi-equilibrium. The step that isnot in quasi-equilibrium (subscript d) is calledthe rate-determining step (rds) as defined byHORIUTI [83]. As a consequence of this defini-tion, the following inequalities are valid:
rfi � rfd; and rri � rrd ði 6¼ dÞ
If there is an rds, then the affinity Ai ¼ 0 forall values of i except for the rds (i 6¼ d), i.e., all(or almost all) of the affinity for the catalyticcycle is dissipated in the rds, hence
A ¼ sdAd ð5ÞIt follows that
rf=rr ¼ rfd=rrd ð6Þ
16 Heterogeneous Catalysis and Solid Catalysts
At steady state sd(rf � rr) ¼ rfd � rrd.Hence
sdrf ¼ rfd and sdrr ¼ rrd: ð7ÞThe stoichiometric equation for the overall
reaction can always be written such that sd isequal to unity. It is then clear that the rds isappropriately and uniquely named as the step forwhich the forward and reverse rates are equal tothe forward and reverse rates, respectively, ofthe overall reaction [67].
Clearly the rds (if there is one) is the onlykinetically significant step. A kinetically signif-icant step is one whose rate constants or equi-librium constant appear in the rate equation forthe overall reaction. In some cases there is no rdsin the Horiuti sense, but frequently only a few ofthe elementary steps in a catalytic cycle arekinetically significant. It is sometimes said thata rate-limiting step is the one having the smallestrate constant. However, rate constants can oftennot be compared because they have differentdimensions.
The relative importance of rate constants ofelementary steps in a catalytic cycle providesuseful guidelines for the development of activityand selectivity. This can be achieved byparametric sensitivity analysis [84], which wasfirst proposed by CAMPBELL [85] for analysis ofkinetic parameters of catalytic reactions (seealso ref. [74]). CAMPBELL [85] defined a degreeof rate control for any rate constant ki in acatalytic cycle turning over at a rate r
Xi ¼ ki=r�@r=@ki ð8Þwhere the equilibrium constant for step i and allother rate constants are held constant. The mainadvantage of this mathematical operation is itssimplicity. It turns out that HORIUTI’s rds, as theonly kinetically significant step in a catalyticcycle, has a degree of rate control Xi ¼ 1,whereas the X values for all other steps areequal to zero. Clearly, all intermediate valuesof Xi are possible, and probable in most cases.
As a catalytic cycle turns over at the quasi-steady state, the steady-state concentrations(coverages) of the reactive intermediates maybe significantly different from the values thatthey would attain if they were at equilibriumwith fluid reactants or products. The steady-state concentrations (coverages) of reactive in-termediates may be lower or higher than the
equilibrium values. The reason for this phenom-enon is kinetic coupling between elementarysteps at the steady state, where the net rate ofeach step is equal to the net rate of the overallreaction multiplied by the stoichiometric num-ber of the step. With kinetic coupling, a reactiveintermediate can accumulate as a reactant or bedepleted as a product [68,79,81].
The principle of microscopic reversibility isstrictly valid only for reactions at equilibrium.Away fromequilibrium, it remains valid providedthat transition-state theory is still applicable,which appears to be the case in heterogeneouscatalysis [19]. Hence, the principle remainsvalid for any elementary step in a heteroge-neous catalytic reaction. However, the princi-ple must be applied with caution to a catalyticcycle, as opposed to a single elementary reac-tion. If valid, the principle of microscopicreversibility allows the calculation of a rateconstant if the second rate constant andthe equilibrium constant Ki of an elementaryreaction i are known: kfi/kri ¼ Ki.
2.2.2. Application of MicrokineticAnalysis
Two of the most intensively studied systems inheterogeneous catalysis are CO oxidation overnoble metals and ammonia synthesis. In bothcases, pioneering work usingmicrokinetic anal-ysis led to a better understanding of the catalyticcycle and new fundamental insights, whichsupported design and optimization of the cata-lytic applications. In industry, CO oxidationover Pt and Pd was one of the first systems usedfor automobile emission control and is a keyintermediate step in many technical systems forhydrocarbon transformations. Ammonia syn-thesis — once the driving force for a newchemical industry — still is one of the mostimportant technical applications of heteroge-neous catalysis. These technical aspects of COoxidation and ammonia synthesis are discussedin Chapter Industrial Application and Mechan-isms of Selected Technically Relevant Reac-tions. Since CO oxidation on noble metals hasbeen the major working system in surfacescience and has led to elucidation of manyfundamental issues of reactions on catalyticsurfaces, such as oscillatory kinetics and
Heterogeneous Catalysis and Solid Catalysts 17
spatio-temporal pattern formation [86], this sys-tem will be exemplarily used for the illustrationof microkinetic analysis.
CO oxidation on noble metals (Pt, Pd, etc.)
COþ1/2O2!CO2 ð9Þis relatively well understood, based on surfacescience studies. Molecular oxygen is chemi-sorbed dissociatively, while CO binds asso-ciatively [87,88].Molecular CO then reactswithatomic oxygen in the adsorbed state:
O2þ2*!O2;ads!2Oads ð10Þ
COþ*!COads ð11Þ
COadsþOads!CO2þ2* ð12ÞHere * denotes a free surface site and thesubscript “ads” an adsorbed species. The reac-tion steps (10)–(12) suggest that COoxidation isa Langmuir – Hinshelwood process in whichboth reacting species are adsorbed on the cata-lyst surface. The reverse of reaction (10), i.e.,the recombination of two oxygen atoms is ki-netically insignificant at temperatures belowca. 600 K. Possible Eley – Rideal steps suchas (13), in which a gas-phase molecule reactswith an adsorbed species
COþOads!CO2þ* ð13Þwere found to be unlikely.
Quantitative experiments led to a schematicone-dimensional potential-energy diagram char-
acterizing the elementary steps on the Pd(111)surface (Fig. 6). Most of the energy is liberatedupon adsorption of the reactants, and the activa-tion barrier for the combination of the adsorbedintermediates is relatively small; this step is onlyweakly exothermic, and the heat of adsorption(activation energy for desorption) of CO2 is verylow.
The sequence of elementary steps (10) –(12) is quite simple. The overall kinetics, how-ever, is not. This is due to the nonuniformity ofthe surface and segregation of the reactants intosurface domains at higher coverages. As a con-sequence, the reaction between the surface spe-cies COads and Oads can only occur at theboundaries between these domains. A simpleLangmuir – Hinshelwood treatment of the ki-netics is therefore ruled out, except for thespecial case of low surface coverages by COads
and Oads, when these are randomly distributedand can be considered to a first approximation asbeing part of an ideal surface.
The Langmuir – Hinshelwood – Hougen –Watson (LHHW) approach is based on theLangmuir model describing the surface of acatalyst as an array of equivalent sites whichdo not interact either before or after chemisorp-tion. Further, for derivation of rate equations, itis assumed that both reactants and products areequilibrated with surface species that react on
Figure 6. Schematic one-dimensional potential-energy diagram characterizing the CO þ O2 reaction on Pd(111) [88]
18 Heterogeneous Catalysis and Solid Catalysts
the surface in a rate-determining step. Surfacecoverages are correlated with partial pressuresor concentrations in the fluid phase by means ofLangmuir adsorption isotherms. It was men-tioned above that the Langmuir model is unre-alistic. Moreover, it was demonstrated in Sec-tion Concepts of Reaction Kinetics (Microki-netics) that the surface coverages of adsorbedspecies are by no means identical to the equi-librium values predicted by the Langmuir ad-sorption isotherm for reaction systems in whichkinetic coupling occurs, and rate-determiningsteps do not generally exist.
Despite these weaknesses, the LHHW kinet-ics approach has proved valuable for modelingheterogeneous catalytic reactions for reactor andprocess design. The kinetic parameters whichare determined by fitting the rate equations toexperimental data, however, do not have astraightforward physical meaning. As an alter-native, simple power-law kinetics for straight-forward reactions (e.g., A ! B) can be used fortechnical application.
Often it is difficult todiscriminatebetween twoormore kinetic models within the accuracy limitsof the experimental data. Sophisticated mathe-maticalprocedureshave thereforebeendevelopedfor the discrimination of rival models [91].
As an example for a typical LHHW rateequation consider the reaction
AþBÐ C:
The form of rate equation is as follows [91]:
r ¼ krdsNTKi ðPAPB�PC=KeqÞð1þKAPAþKBPBþKCPCþ
Pj KjPjÞn
¼ rate factor� driving force
inhibition termð14Þ
The numerator is a product of the rate con-stant of the rds krds, the concentration of activesites NT, adsorption equilibrium constants Ki,and the driving force for the reaction. The latteris a measure of how far the overall reaction isfrom thermodynamic equilibrium. The overallequilibrium constantKeq, can be calculated fromthermodynamics. The denominator is an inhibi-tion term which takes into account the competi-tive adsorption of reactants and products.
A few examples of LHHW rate equations aresummarized in Table 3. A collection of usefulLHHW rate equations and kinetic data foralmost 100 industrially important catalytic re-actions is available in [92].
Table 3. General structure of Langmuir type rate equations 90
Reaction Controlling step Net rate Kineticconstant
Drivingforce
Adsorptionterm
1. AÐ P a. adsorption of A kApA(1 � SQ) � kA’QA kA pA� pPK 1 þ KApA þ Kppp
b. surface reaction single-sitemechanism
kSQp � kS’Qp kSKA pA� pPK 1 þ KApA þ Kppp
c. desorption of P kp’Qp � kppp(1 � SQ) kp’kSKA pA� pPK 1 þ KApA þ Kppp
2. AÐ PþQ surface reaction, A (ads) reactswith vacant site
Catalytic activity is expressed in terms ofreaction rates, preferably normalized to thesurface area of the active phase (e.g., metalsurface area for supported metal catalysts).These surface areas can be obtained by suitablechemisorption techniques (see Section Physi-cal Properties). As an alternative to these arealrates, specific rates are also used which arenormalized to catalyst weight. The best possi-ble measure of catalytic activity, however, isthe turnover rate or turnover frequency, since itis normalized to the number of active sites andrepresents the rate at which the catalytic cycleturns over. For comparison of rates reported bydifferent research groups, the methodology forthe determination of the number of active sitesmust be carefully reported. The hitherto unre-solved problem is that the site densities mea-sured prior to the catalytic reaction are notnecessarily identical to those available underreaction conditions.
A readily available measure of catalytic ac-tivity is space – time yield, expressed in units ofamount of product made in the reactor per unittime and unit reactor volume.
A considerable obstacle for the comparisonof catalytic activities for a given reaction thatwere obtained in different laboratories for thesame catalyst is the use of different reactors. Fora series of catalysts, reasonable comparisons ofactivities or rates are possible when relativevalues are used.
Conversion data alone, or conversion versustime plots are not sufficient as a measure ofcatalytic activity.
Selectivity can be defined as the amount ofdesired product obtained per amount of con-sumed reactant. Selectivity values are only use-ful if the conversion is also reported. A simplemeasure of selectivity is the yield (yield ¼ se-lectivity � conversion). Selectivities can alsobe used to indicate the relative rates of two ormore competing reactions; competition mayoccur when several reactants form products inparallel (type I):
when one reactant transforms into several pro-ducts in parallel (type II):
or in consecutive reactions (type III):
The selectivity is defined as the ratio of therate of formation of the desired product to therate of consumption of the startingmaterial [93].Thus, the selectivities for product X for the first-order reactions I and II is r1/(r1 þ r2), whereas itis (r1 � r2)/r1 for type III.
In the case of type I or II reactions, selectivityfor X or Y is independent of the conversion ofthe starting material. In type III reactions, theselectivity for X is 100% initially, decreasesgradually with increasing conversion, and dropsto zero at 100% conversion. At an intermediateconversion, there is a maximum yield of Xwhich depends on the ratio of the rate constantsk1 and k2 of the rates r1 and r2. The integratedrate equations are:
½A� ¼ expð�k1tÞ ð15Þ
½X� ¼ k1=ðk2�k1Þ ½expð�k1tÞ�expð�k2tÞ�ð16Þ
where [A] is the concentration of unconvertedA, [X] the concentration of A converted toproduct X, and t time. The maximum yield isreached at
t ¼ ðk1�k2Þ�1 lnðk1=k2Þ ð17Þ
2.3. Molecular Modeling inHeterogeneous Catalysis
Modeling of catalytic reactions is applied atmany levels of complexity covering severalorders of length and time scales. It ranges fromcomplete description of the dynamics of areaction through adsorbate – adsorbate interac-tions to the simple mean-field approximations
20 Heterogeneous Catalysis and Solid Catalysts
and macrokinetic models discussed in SectionLangmuir – Hinshelwood – Hougen – WatsonKinetics. The different approaches can be rep-resented in a hierarchy of models (Table 4).
In this section, frequently used models arepresented that either describe the molecular be-havior of the catalytic cycle directly or are basedon the molecular picture. Often, the output of acomputation using a more sophisticated methodserves as input for a computation using a lessdetailed model; for instance activation energiescomputedbyDFTareoften used as parameters inkinetic Monte Carlo and simulations.
2.3.1. Density Functional Theory
In real ab initio calculations, in which the time-dependent Schr€odinger equation is solved toobtain the complex N-electron wavefunctionY, the number of atoms of the system studiedis very limited, and therefore quantum mechan-ical calculations in heterogeneous catalysis arealmost exclusively based on the DFT approach.Based on the Hohenberg – Kohn theorem, theground-state energy of an atom or molecule iscompletely determined by the electron density.Eventhoughtheexact functionaldependenceof theenergy on the electron density is not known, ap-proximate functionals can be developed (Kohn –Sham formalism) that lead to the much simplercomputed electron density.
There are two major methods for DFT simu-lations of catalytic systems: In the first, thecluster algorithm, the molecules studied aremetal clusters including the adsorbed particles.The advantage of this approach is that the specialshape of catalytic clusters can be taken intoaccount, and methods developed for gas-phasechemistry can be used, so that computational
costs are relatively low. Disadvantages are thelimited number of atoms in the cluster, currently(ca. 2008) a few hundred, and the fact that metalclusters in general have different properties tothree-dimensional metals. Some prominent soft-ware tools using the cluster algorithm areGAUSSIAN [94] and TURBOMOLE [95].
The second approach, usually denoted by theterms “planar waves” or “periodic boundaries”,is much more popular in heterogeneous cataly-sis. The algorithm is based on a supercell ap-proach, i.e., structures to be calculated must beperiodic in three dimensions. This approach isespecially advantageous when considering sur-face structures, because a real solid surface isbuilt on expansion from a small metal cluster ormetal slab into three dimensions. In particular,the metallic properties are better described. The“third dimension” is a disadvantage, because thesolid cell must be periodic in this direction aswell. Aside from the problem of choosing ap-propriate functionals, the size of the cell and theconvergence criteria are significant for DFTcomputations to provide reliable information.Some prominent software tools using the planarwaves approach are CASTEP [96], DACAPO[97], and VASP [98].
Even though still very computer time con-suming, DFT can be used to calculate the stabil-ity and frequencies for all reactants, intermedi-ates, and products, as well as activation barriersof the elementary reactions [99–105]. Recently,complete reaction mechanisms including prop-erties of intermediates have been developedbased on DFT computations alone, for instance,for CO oxidation over RuO2(110) [105], epoxi-dation of ethylene over Ag [106], methanoldecomposition over Cu [107], ammonia synthe-sis over Ru [108], and decomposition of N2O onFe-ZSM-5 [109]. DFT simulation not only helps
Table 4. Hierarchy of methods of modeling catalytic reactions
Method of modeling Simplification Application
Ab initio calculation Most fundamental approach Not yet significant in heterogeneous catalysisDensity functional theory (DFT) Replacement of the N-electron wave
function by the electron densityDynamics of reactions, activation barriers, adsorbedstructures, frequencies
Kinetic Monte Carlo (kMC) Details of dynamics neglected Adsorbate – adsorbate interactions on catalytic surfacesand nanoparticles
Detailed configuration of the adsorbatestructure neglected
Microkinetic modeling of catalytic reactions in technicalsystems
Power-law kinetics All mechanistic aspects neglected Scaleup and reactor design for “black-box” systems
Heterogeneous Catalysis and Solid Catalysts 21
to understand the fine details of catalytic reac-tions, for instance, the effect of surface steps onstability of intermediates [108] and the impact ofcoverage on activation energies [110], but also toelucidate the broader picture, for example, byfinding a relationship between activation energyand chemisorption energy [111].
2.3.2. Kinetic Monte Carlo Simulation
Diffusion of adsorbates on catalytic surfaces iscrucial for catalytic reactions. Furthermore, in-teractions between adsorbates can be substantialand lead to ordered structures such as islandsand influence the energetic state of the surface,which also implies dependence of the activationbarriers for adsorption, diffusion, reaction, anddesorption on the surface coverage and theactual configuration of the adsorbates. The ad-sorbed species can be associated with a surfacesite, and thus a lattice representation of a two-dimensional surface can be constructed. In thecase of catalytic particles, a three-dimensionalstructure can be used with individual two-di-mensional facets that can differ in their catalyticactivity. In the three-dimensional case, specialcare is needed for appropriate treatment ofedges and corners. Even reconstruction of sur-faces can be taken into account. At each surfacesite the local environment (presence of adsor-bates, catalyst morphology/crystal phase) willdetermine the activation energies. If the inter-actions between the adsorbates, the surface, andthe gas phase are known, such parameters couldtheoretically be derived from DFT simulations,and the kinetics can be computed by the kineticMonte Carlo method (kMC) [105,112–118].Each molecular event, i.e., adsorption, desorp-tion, reaction, diffusion, is computed and leadsto a new configuration of adsorbed species onthe surface lattice. Aside from this very detaileddescription of the process, time averaging of thetime-dependent computed reaction rates andsurface coverage can then lead to overall rateexpressions. However, the computational effortneeded is immense, not only due to the kMCsimulation but also because of the huge numberof fundamental DFT computations needed toprovide reliable activation barriers for all possi-ble individual steps. Experimental derivation ofthis information is even more exhausting. Most
of the adsorbate – adsorbate interactions, suchas the formation of ordered structures, mayappear at low temperature and pressure, wherediffusion is slow and the rate of impingement ofgas-phase molecules is small, respectively. Un-der these conditions kMC may be the onlydescription that is accurate, while at high tem-perature and pressure, the adsorbates are ratherrandomly dispersed on the surface, and theassumptions of the mean-field approximationmay be valid.
2.3.3. Mean-Field Approximation[119–122]
In the mean-field approximation, a continuousdescription is considered instead of the detailedconfigurations of the system discussed above.Hence, the local state of the catalytic surface onthe macroscopic or mesoscopic scale can berepresented by mean values by assuming ran-domly distributed adsorbates on the surface,which is viewed as being uniform. The state ofthe catalytic surface is described by the temper-ature T and a set of surface coverages ui, that is,the fraction of the surface covered with adsor-bate i. The surface temperature and the cov-erages depend on time and spatial position in themacroscopic system (reactor), but are averagedover microscopic local fluctuations. Underthose assumptions a chemical reaction can bedefined as
XNgþNsþNb
i¼1n0ikAi!
XNgþNsþNb
i¼1n00ikAi
where Ai denote gas-phase species, surface spe-cies, and bulk species. The Ns surface speciesare those that are adsorbed on the top monoa-tomic layer of the catalytic particle, while theNb
bulk species are those found in the inner solidcatalyst.
Steric effects of adsorbed species and variousconfigurations, e.g., type of chemical bond be-tween adsorbate and solid, can be taken intoaccount by using the following concept: Thesurface structure is associatedwith a surface sitedensity G that describes the maximum numberof species that can be adsorbed on unit surfacearea. Each surface species is associated with a
22 Heterogeneous Catalysis and Solid Catalysts
coordination number si describing the numberof surface sites which are covered by this spe-cies. Under the assumptions made, a multistep(quasi-elementary) reaction mechanism can beset up. The molar net production rate is thengiven as
si ¼XKs
k¼1nikkfk
YNgþNsþNb
j¼1cn0jk
j :
where Ks is the number of surface reactions, ciare the species concentrations, which are given,e.g., in mol m�2 for the Ns adsorbed species andin, e.g., mol m�3 for the Ng and Nb gaseous andbulk species. With Qi ¼ cisiG�1, the variationsof surface coverages follow:
@Qi
@t¼ sisi
G:
Since the temperature and concentrations ofgaseous species depend on the local position inthe reactor, the set of surface coverages alsovaries with position. However, no lateral inter-action of the surface species between differentlocations on the catalytic surface is modeled inthis approach. This assumption is justified bythe fact that the computational cells in reactorsimulations are usually much larger than therange of lateral interactions of the surface pro-cesses. In each of these cells, the state of thesurface is characterized by mean values (mean-field approximation).
The binding states of adsorption of all speciesvary with the surface coverage, as discussed inSection Kinetic Monte Carlo Simulation. Thisadditional coverage dependence can be mod-eled in the expression for the rate coefficient byan additional function leading to:
kfk ¼ AkTbkexp
�Eak
RT
� �YNs
i¼1Q
miki exp
eikQi
RT
� �:
For adsorption reactions sticking coefficientsare commonly used, which can be converted toconventional rate coefficients.
2.3.4. Development of Multistep SurfaceReaction Mechanisms [122]
The development of a reliable surface reactionmechanism is a complex process. A tentative
reaction mechanism can be proposed based onexperimental surface-science studies, on analo-gy to gas-phase kinetics and organometalliccompounds, and on theoretical studies, in-cluding DFT and kMC calculations as well assemi-empirical calculations [123,124]. Thismechanism should include all possible pathsfor formation of the chemical species underconsideration in order to be “elementary-like”and thus applicable over a wide range of con-ditions. The mechanistic idea then needs to beevaluated against numerous experimentally de-rived data, which are compared with theoreticalpredictions based on the mechanism. Here,simulations of the laboratory reactors requireappropriate models for all significant processesin order to evaluate the intrinsic kinetics. Sen-sitivity analysis leads to the crucial steps in themechanism, for which refined kinetic experi-ments and data may be needed.
Since the early 1990s, many groups havedeveloped surface reaction mechanisms follow-ing these concepts. In particular, oxidation re-actions over noble metals have been modeledextensively, such as those of hydrogen [125–129], CO [130–132], methane [133–137], andethane [138–140] over Pt and formation ofsynthesis gas over Rh [141–142].More recently,mechanisms have been established for morecomplex systems such as three-way cata-lysts [143] and chemical vapor deposition(CVD) of diamond [144,145], silica [146], andnanotubes [147]. A more detailed survey onexisting microkinetic models can be foundin [121].
3. Development of Solid Catalysts
The development of a catalytic process involvesthe search for the catalyst and the appropriatereactor, and typically occurs in a sequence ofsteps at different levels. Figure 7 shows ascheme summarizing this evolutionary process.
Small-scale reactors are used for screening todetermine the optimal catalyst formulation.Since catalyst development and sequentialscreening are slow and cost-intensive processes,high-throughput experimentation (HTE) tech-niques [149–155] which permit parallel testingof small amounts of catalyst in automated sys-tems have attracted great interest (see also !
Heterogeneous Catalysis and Solid Catalysts 23
Combinatorial Methods in Catalysis and Mate-rials Science). Companies such as Symyx Tech-nologies, Santa Clara (www.symyx.com), hte,Heidelberg (www.hte-company.de), and Avan-tium Technologies, Amsterdam (www.avan-tium.nl) already offer HTE-based developmentof catalysts or other materials. The HTE-basedsearch for catalysts usually starts with a firstphase (stage I or discovery) in which largecatalyst libraries, often with several hundredmaterials, are categorized into promising andless promising candidates by use of relativelysimple and fast analysis techniques. One exam-ple is infrared thermography for detection ofexothermic reactions with spatial resolution. Todecrease the number of experiments, optimiza-tion methods based on genetic algorithms maybe used to derive subsequent catalyst genera-tions from the performance of the members ofthe preceding generation [156]. In stage II, themore interesting materials, typically less than50 candidates, are subjected to tests under muchmore realistic process conditions with moredetailed characterization. For this purpose, avariety of parallel-reactor systems has beendeveloped. A crucial point in high-throughputexperimentation is the precise and fast analyti-cal quantification of reaction starting materialsand products. Especially promising for obtain-ing fast and detailed on-line information during
catalyst testing is high-throughput multiplexinggas chromatography [155]. Instead of perform-ing time-consuming chromatographic analysesduring parallelized catalyst testing one after theother, samples are rapidly injected into theseparation by means of a special multiplexinginjector. The obtained chromatogram is a con-volution of overlapping time-shifted singlechromatograms and must therefore be mathe-matically deconvoluted. This new techniquewas successfully used for the study of palladi-um-catalyzed hydrogenation reactions [157].
High-throughput experimentation is a mod-ern and accelerated version of classical catalystdevelopment by trial and error. A famous earlyexample of this approach is the discovery of theiron-based ammonia synthesis catalyst, duringwhich 2500 catalysts were tested in 6500 ex-periments [9]. In recent years is has becomeevident that the empirical search for new orimproved catalyst formulations can be success-fully aided by knowledge-based (expert) sys-tems or molecular design [158–160]. State-of-the-art computational tools for the effectivemolecular-scale design of catalytic materialsare summarized in [161]. A striking exampleis the theoretical prediction of bimetallic am-monia synthesis catalysts [162]. As the rate-limiting step in heterogeneously catalyzed am-monia synthesis is the dissociative adsorption of
Figure 7. Scheme for catalyst development and design (from [148], modified)
24 Heterogeneous Catalysis and Solid Catalysts
N2, an optimumstrength of themetal – nitrogeninteraction is required for high ammonia syn-thesis activity. The resulting volcano-shapedrelationship shows, in agreement with experi-mental evidence, that Ru and Os, followed byFe, are the best pure metal catalysts (Figure 8).
First-principlesDFTcalculationswereusedtopredict that alloys of metals with high and lowadsorption energy should give rise to bindingenergies close to the optimum. Based on thesecalculations, a Co – Mo catalyst was developedthat hasmuchhigher ammonium synthesis activ-ity than the individual metals and is even betterthan Ru and Fe at low ammonia concentrations.
4. Classification of Solid Catalysts
Solid catalysts are extremely important in large-scale processes [163–167] for the conversion ofchemicals, fuels, and pollutants. Many solidmaterials (elements and compounds) includingmetals, metal oxides, and metal sulfides, arecatalysts. Only a few catalytic materials used inindustry are simple in composition, e.g., puremetals (e.g., Ni) or binary oxides (such as g-Al2O3, TiO2). Typical industrial catalysts, how-ever, consist of several components and phases.This complexity often makes it difficult toassess the catalytic material’s structure.
In the following a variety of families ofexisting catalysts are described, and selectedexamples are given. These families include(1) unsupported (bulk) catalysts; (2) supported
catalysts; (3) confined catalysts (ship-in-a-bot-tle catalysts); (4) hybrid catalysts; (5) polymer-ization catalysts, and several others. The select-ed examples not only include materials whichare in use in industry, but also materials whichare not yet mature for technological applicationbut which have promising potential.
4.1. Unsupported (Bulk) Catalysts
4.1.1. Metal Oxides
Oxides are compounds of oxygen inwhich theOatom is the more strongly electronegative com-ponent. Oxides of metals are usually solids.Their bulk properties largely depend on thebonding character between metal and oxygen.Metal oxides have widely varying electronicproperties and include insulators (e.g., Al2O3,SiO2), semiconductors (e.g., TiO2, NiO, ZnO),metallic conductors (typically reduced transi-tion metal oxides such as TiO, NbO, and tung-sten bronzes), superconductors (e.g., BaPb1�x-BixO3), and high-Tc superconductors (e.g.,YBa2Cu3O7�x).
Metal oxides make up a large and importantclass of catalytically active materials, their sur-face properties and chemistry being determinedby their composition and structure, the bondingcharacter, and the coordination of surface atomsand hydroxyl groups in exposed terminatingcrystallographic faces. They can develop acid-base and redox properties. Metal oxides can
Figure 8. Calculated turnover frequencies (TOF) for ammonia synthesis as a function of the adsorption energy of nitrogen forvarious transition metals and alloys (reprinted with permission from [162]).
Heterogeneous Catalysis and Solid Catalysts 25
have simple composition, like binary oxides, butmany technologically important oxide catalystsare complex multicomponent materials.
4.1.1.1. Simple Binary OxidesSimple binary oxides of basemetalsmay behaveas solid acids or bases or amphoteric materi-als [168]. These properties are closely related totheir dissolution behavior in contact with aque-ous solutions. Amphoteric oxides (e.g., Al2O3,ZnO) form cations in acidic and anions in basicmilieu. Acidic oxides (e.g., SiO2) dissolve withformation of acids or anions. Transition metaloxides in their highest oxidation state (e.g.,V2O5, CrO3) behave analogously. Basic oxides(e.g., MgO, lanthanide oxides) form hydroxidesor dissolve by forming bases or cations. Thesedissolution properties must be considered whensuch oxides are used as supports and impreg-nated from aqueous solutions of the active phaseprecursor [169,170]. The dissolution propertiesalso are closely related to the surface propertiesof the oxides in contact with a gas phase, wherethe degree of hydration/hydroxylation of thesurface is a critical parameter. Silica, aluminaand magnesia are commonly used catalysts andcatalyst supports representative for awide rangeof surface acid – base properties.
Aluminas are amphoteric oxides, whichform a variety of different phases depending onthe nature of the hydroxide or oxide hydroxideprecursor and the conditions of their thermaldecomposition. Bayerite, nordstrandite, boehm-ite, and gibbsite can be used as starting materi-als. The thermal evolution of the various poorlycrystalline transitional phases (namely h-, Q-,g-, x-, and k-Al2O3) and of the final crystalline,thermodynamically stable a-Al2O3 phase (co-rundum) is shown in Figure 9. The structures of
these oxides can be described as close-packedlayers of oxo anions with Al3þ cations distrib-uted between tetrahedral and octahedral vacan-cy positions. Stacking variations of the oxoanions result in the different crystallographicforms of alumina. The most commonly usedtransitional phases are h- and g-Al2O3, whichare often described as defect spinel struc-tures [171] that incorporate Al3þ cations inboth tetrahedral and octahedral sites. The Alsublattice is highly disordered, and irregularoccupation of the tetrahedral interstices resultsin a tetragonal distortion of the spinel structure.There is a higher occupancy of tetrahedral cat-ion positions in g-Al2O3, and a higher density ofstacking faults in the oxygen sublattice of h-Al2O3. Crystallites are preferentially terminatedby anion layers, and these layers are occupied byhydroxyl groups for energetic reasons [172].
Acidic and basic sites and acid-base pair siteshave been identified on the surfaces of alumi-nas [174]. Thermal treatment of hydroxylatedoxides leads to partial dehydroxylation withformation of coordinatively unsaturated O2�
ions (basic sites) and an adjacent anion vacancywhich exposes 3- or 5-coordinate Al3þ cations(Lewis acid sites). The remaining hydroxylgroups can be terminal or doubly or triplybridging with the participation of Al3þ in tetra-hedral and/or octahedral positions. The proper-ties of the resulting OH species range fromvery weakly Brønsted acidic to rather stronglybasic and nucleophilic [172,174]. As a resultof this complexity, alumina surfaces develop arich surface chemistry and specific catalyticproperties [175].
Besides their intrinsic catalytic propertiesand their use as catalysts in their own right(e.g., for elimination reactions, alkene isomeri-zation [175], and the Claus process [176]),
Figure 9. The dehydration sequences of the aluminum trihydroxides in air (adopted from [173])
26 Heterogeneous Catalysis and Solid Catalysts
aluminas are frequently used as catalyst sup-ports for oxides andmetals. The surface area andparticle size of aluminas can be controlled by thepreparation conditions, and their redox andthermal stability give the supported activephases high stability and ensure a long catalystlifetime.
Silicas are weakly Brønsted acidic oxideswhich occur in a variety of structures such asquartz, tridymite and cristobalite (! Sili-ca) [177,178]. The most commonly used silicain catalysis is amorphous silica. The buildingblocks of silica are linked SiO4 tetrahedra, witheach O atom bridging two Si atoms. Bondingwithin the solid is covalent. At the fully hydratedsurface, the bulk structure is terminated byhydroxyl (silanol) groups, SiOH [174,177,178].Two types of these groups are usually distin-guished: isolated groups and hydrogen-bondedvicinal groups. Fully hydrated samples, cal-cined at temperatures below 473 K, may con-tain geminal groups Si(OH)2 [174,177,178].Heating in vacuum removes the vicinal groupsby dehydroxylation, i.e., condensation to formH2OandSi – O – Si linkages(siloxanebridges).Complete removal of the hydroxyl groupsoccursat temperatureswell above 973 K invacuo and isbelieved toresult insignificantchanges insurfacemorphology.
The surface hydroxyl groups are only weaklyBrønsted acidic and thereforehardlydevelop anycatalytic activity. They are, however, amenableto hydrogen-bonding [179] and they are usuallyregarded as the most reactive native surfacespecies,which are available for functionalizationof silicas. The siloxane bridges are (at least afterheating at elevated temperatures) essentially un-reactive. For this reason and because of the lowacidity of silanol groups, silicas are not used asactive catalysts, but theyplayan important role asoxide supports and for the synthesis of functio-nalized oxide supports (see Section SupportedSulfide Catalysts).
Tailored silicas can be synthesized by con-trolling the preparation conditions [177,178].Thus, surface area, particle size andmorphology,porosity and mechanical stability can be variedby modification of the synthesis parameters.
In addition to amorphous silicas, the crystal-line microporous silica silicalite I can be ob-tained by hydrothermal synthesis [180]. This
material has MFI structure and can be consid-ered as the parent siliceous extreme of zeoliteZSM-5.
Large-pore mesoporous structures, the so-called porosils, have also been reported [180–182]. The dimensions of their linear and paral-lel pores can be varied from 2 to 10 nm in aregular fashion. These pores can thereforeaccommodate bulky molecules and functionalgroups.
The incorporation of foreign elements suchas Al3þ substituting for Si4þ induces Brønstedacidity and creates activity for acid catalysis.
Magnesium oxide is a basic solid. It hasthe simple rock salt structure, with octahedralcoordination of magnesium and oxygen. Abinitio molecular orbital calculations indicatedthat the electronic structure is highly ionic, withthe Mg2þO2� formalism being an accurate re-presentation of both bulk and surface struc-tures [183]. The lattice is commonly envisagedto terminate in (100) planes incorporating five-coordinate (5c) Mg2þ and O2� ions [184] (seeFigure 10). This model appears to be physicallyaccurate for MgO smoke, which may be re-garded as a model crystalline metal oxidesupport [185]. Although the (100) plane iselectrically neutral, hydroxyl groups are presenton the surfaces of polycrystalline MgO. Thesegroups and theO2- anions are responsible for thebasic properties, coordinatively unsaturatedMg2þ ions being only weak Lewis acid sites.The hydroxyl groups are also highlynucleophilic.
These properties dominate the surface chem-istry ofMgO. Organic Brønsted acids have beenshown to be chemisorbed dissociatively to formsurface-bound carbanions and surface hydroxylgroups [186]. Even the heterolytic dissociativeadsorption of dihydrogen on polycrystallineMgO has been reported. Mg2þO2� pairs withMg2þ and O2� in 4- or 3-coordination seem toplay a crucial role.
The presence of low-coordinate Mg2þ andO2� ions (see Figure 10) on the MgO surfaceafter activation at high temperatures has beendemonstrated [184,187,188], and the uniquereactivity of 3c centers has been discussed[189].
MgO has also been used as a host matrix fortransition metal ions (solid solutions) [190].
Heterogeneous Catalysis and Solid Catalysts 27
Thesematerials permit the properties of isolatedtransition metal ions to be studied.
Transition metal oxides [191–193] can bestructurally described as more or less densepackings of oxide anions, the interstices ofwhich are occupied by cations. The bonding,however, is never purely ionic, but rather mixedionic-covalent, sometimes also developing me-tallic character (e.g., bronzes). The surface ofthese oxides is often partially occupied by hy-droxyl groups, so they possess some acidiccharacter. However, it is the variability in oxi-dation states and the possibility of formingmixed-valence and nonstoichiometric com-pounds that are responsible for their importantredox catalytic properties. The most frequentlyused transition metal oxides are those of theearly transition metals (mostly suboxides).Fields of application are particularly selectiveoxidation and dehydrogenation reactions.
Titania TiO2 exists in two major crystallo-graphic forms: anatase and rutile. Anatase is themore frequently used modification since it de-velops a larger surface area, although it is ametastable phase and may undergo slow trans-formation into the thermodynamically stablerutile phase above ca. 900 K. Vanadium impu-rities seem to accelerate the rutilization above820 K. Other impurities such as surface sulfateand phosphate seem to stabilize the anatasephase. The anatase ! rutile phase transitionmust be sensitively controlled for supported
VOx/TiO2, which plays a significant role inselective oxidation andNOx reduction catalysis.
Titania is a semiconductor with a wide bandgap and as such is an important material forphotocatalysis [194,195].
Zirconia has attracted significant interest inthe recent past as a catalyst support and as a basematerial for the preparation of strong solid acidsby surfacemodificationwith sulfate or tungstategroups [196]. The most important crystallo-graphic phases of ZrO2 for catalytic applica-tions are tetragonal andmonoclinic. The latter isthe thermodynamically stable phase. Highersurface areas, however, are developed by themetastable tetragonal phase, which is stabilizedat low temperatures by sulfate impurities orintentional addition of sulfate or tungstate.
ZrO2 is the base material for the solid-stateelectrolyte sensor for the measurement of oxy-gen partial pressure in, e.g., car exhaust gas-es [197]. The solid electrolyte shows high bulkconductivity for O2� ions.
Other transition metal oxides are used assupported catalysts or as constituents of com-plex multicomponent catalysts.
Only a few examples are reported on theapplication of the unsupported binary oxides ascatalysts. Iron oxide Fe2O3 and chromiumoxide Cr2O3 catalyze the oxidative dehydroge-nation of butenes to butadiene. Fe2O3-basedcatalysts are used in the high-temperature wa-ter gas shift reaction [198] and in the dehydro-genation of ethylbenzene [199]. Vanadium
Figure 10. Representation of a surface plane (100) of MgO showing surface imperfections such as steps, kinks, and cornerswich provide sites for ions of low coordination (adopted from [184]).
28 Heterogeneous Catalysis and Solid Catalysts
pentoxide V2O5 is active for the selective oxi-dation of alkenes to saturated aldehydes [200].Acidic transition metal oxides such as vanadi-um pentoxide and molybdenum trioxide MoO3
can be used for the synthesis of formaldehydeby oxidative dehydrogenation of methanol,while the more basic iron oxide Fe2O3 leadsto total oxidation [201]. Zinc oxide ZnO is usedas a catalyst for the oxidation of cyclohexanolto cyclohexanone.
4.1.1.2. Complex Multicomponent OxidesComplex multicomponent oxides play a majorrole as catalytic materials.
Aluminum silicates are among the mostimportant ternary oxides. Four-valent Si atomsare isomorphously substituted by trivalent Alatoms in these materials. This substitution cre-ates a negatively charged framework of inter-connected tetrahedra. Exchangeable cations arerequired for charge compensation when protonsare incorporated as charge-compensating ca-tions, OH groups bridging Si and Al atoms arecreated which act as Brønsted acidic sites.
Amorphous silica – alumina can be pre-pared by precipitation from solution. Thismixed oxide is a constituent of hydrocarboncracking catalysts.
Zeolites Hydrothermal synthesis can beused for preparation of a large family of crystal-line aluminosilicates, known as zeolites (!Zeolites),whicharemicroporous solidswithporesizes ranging from ca. 3 to 7 A
�[180,202,203].
Characteristic properties of these structurallywell-defined solids are selective sorption of smallmolecules (molecular sieves), ion exchange, andlarge surface areas. Zeolites possess a frameworkstructure of corner-linked SiO4
4� and AlO45�
tetrahedrawith two-coordinateoxygenatoms thatbridge two tetrahedral centers (so-called Tatoms). Zeolite frameworks are open and containchannels (straight or sinusoidal) or cages ofspherical or other shapes. These cages are typi-cally interconnected by channels. The evolutionof several zeolite structures from the primarytetrahedra via secondary building blocks is dem-onstrated in Figure 11 [204]. The diameter of thechannels is determined by the number n of Tatoms surrounding the opening of the channels as
pore zeolites are constructed of 12-memberedrings (d > 7 A
�). Examples of small-pore zeolites
are sodalite and zeolite A, of medium-pore zeo-lites ZSM-type zeolites (see Figure 11), whilelarge-pore zeolites include faujasites and zeolitesX and Y (see Figure 11).
The H forms of zeolites develop strongBrønsted acidity and play a major role inlarge-scale industrial processes such as catalyticcracking, the Mobil MTG (methanol-to-gaso-line) process and several others.
Besides Si andAl as Tatoms P atoms can alsobe incorporated in zeolite structures. In addi-tion, transitionmetal atoms such as Ti, V, and Crcan substitute for Si, which leads to oxidationcatalysts of which titanium-silicalite-1 (TS1) isthe most outstanding catalyst for oxidation,hydroxylation, and ammoxidationwith aqueousH2O2 [205].
Basic properties can be created in zeolitesby ion-exchange with large alkali metal ionssuch as Csþ and additional loading withCsO [206].
Aluminum phosphates (AlPO) [207,208]are another family of materials whose structuresare similar to those of zeolites. They can beregarded as zeolites in which the T atoms are Siand Al. More recently they have been namedzeotypes, the T atoms of which are Al and P. Incontrast to aluminosilicate zeolites, AlPOs typ-ically have a Al/P atomic ratio of 1/1, so that theframework composition [AlPO4] is neutral.Therefore, these solids are nonacidic and havehardly any application as catalysts. However,acidity can be introduced by substituting Al3þ
by divalent atoms, which yields metal alumino-phosphates (MAPOs), e.g., MnAPO or CoAPO,or by partial substitution of formally pentavalentP by Si4þ to give silicoaluminophosphates(SAPO). The AlPO family contains memberswith many different topologies which span awider range of pore diameters than aluminosili-cate zeolites.
Mesoporous solid acids with well-definedpore structures can be obtained by replacing acertain amount of Si atoms inMCM-type oxidesby Al atoms.
Heterogeneous Catalysis and Solid Catalysts 29
Clays (! Clays) are aluminosilicateminerals (montmorillonite, phyllosilicates(smectites), bentonites, and others). Montmo-rillonite is an aluminohydroxysilicate and is themain constituent of most clay minerals. It is a2 : 1 clay, i.e., one octahedral AlO6 layer issandwiched between two tetrahedral SiO4
layers. Montmorillonites are reversibly swella-ble and possess ion-exchange capacity. Theycan be used as catalyst supports. The structurallayers can be linked together by introducinginorganic pillars which prevent the layers fromcollapsing at higher temperatures when theswelling agent is evaporated (pillaredclays) [209]. A bimodal micro-/mesoporouspore size distribution can thus be obtained.Pillaring can be achieved with a wide varietyof reagents including hydroxy aluminum poly-mers, zirconia hydroxy polymers, silica, andsilicate pillars. Catalytically active componentsmay be built in by the pillaring material, e.g.,transition metal oxide pillars.
Mixed metal oxides are multimetal multi-phase oxides which typically contain one ormore transition metal oxide and exhibit signifi-cant chemical and structural complexi-ty [210,211]. Their detailed characterization istherefore extremely difficult, and structure-property relationships can only be establishedin exceptional cases. Bulk mixed metal oxidecatalysts are widely applied in selective oxida-tion, oxydehydrogenation, ammoxidation andother redox reactions. Several examples ofmixed metal oxides and their application inindustrial processes are summarized in Table 5.
Vanadium phosphates (e.g., VOHPO4 �0.5 H2O) are precursors for the so-called VPOcatalysts, which catalyze ammoxidation reac-tions and the selective oxidation of n-butane tomaleic anhydride. It is proposed that the crys-talline vanadyl pyrophosphate phase (VO)2P2O7
is responsible for the catalytic properties of theVPO system. The vanadium phosphate precur-
Figure 11. Structures of four representative zeolites and their micropore systems and dimensions [204]
30 Heterogeneous Catalysis and Solid Catalysts
sor undergoes transformations in reducing andoxidizing atmospheres, as shown in the follow-ing scheme [212]:
As discussed by GRASSELLI [63] effectiveammoxidation (and oxidation) catalysts aremultifunctional and need several key properties,including active sites which are composed of atleast two vicinal oxide species of optimal met-al – oxygen bond strengths. Both species mustbe readily reducible and reoxidizable.
The individual active sites must be spatiallyisolated from each other (site-isolation concept)to achieve the desired product selectivities. Theyshould either be able to dissociate dioxygen andto incorporate the oxygen atoms into the lattice,or they must be located close to auxiliary reoxi-dation sites which contain metals having a facileredox couple. These sites are generally distinctfrom each other. They must, however, be able tocommunicate with each other electronically andspatially so that electrons, lattice oxygen, andanionvacancies can readilymove between them.The lattice must be able to tolerate a certaindensity of anion vacancies without structural
collapse [63]. It is clear that these complexrequirements can only be achieved bymulticom-ponent materials.
GRASSELLI [63,213] has listed three key cata-lytic functionalities required for effective am-moxidation/oxidation catalysts:
1. An a-H-abstracting component, which maybe Bi3þ, Sb3þ, Te4þ, or Se4þ.
2. A component that chemisorbs alkene/ammo-nia and inserts oxygen/nitrogen (Mo6þ, Sb5þ).
3. A redox couple such as Fe2þ/Fe3þ, Ce3þ/Ce4þ, or U5þ/U6þ to facilitate lattice oxygentransfer between bulk and surface of the solidcatalyst.
An empirical correlation was found betweenthe electron configurations of the various metalcations and their respective functionalities[63,213] as shown in Table 6. This correlationcan be used to design efficient catalysts.
Bismuth molybdates are among the mostimportant catalysts for selective oxidation andammoxidation of hydrocarbons [212,63]. Thephase diagram shown in Figure 12 demon-strates the structural complexity of this class ofternary oxides [214]. The catalytically mostimportant phases lie in the compositional rangeBi/Mo atomic ratio between 2/3 and 2/1 and are
Table 5. Examples of mixed metal oxide catalysts and their applications*
a-Bi2Mo3O12, b-Bi2Mo2O9, and g-Bi2MoO6.An industrially used Bi molybdate catalyst wasoptimized in several steps and has the empiricalformula (K,Cs)a(Ni,Co,Mn)9.5(Fe,Cr)2.5BiMo12Ox [63]. This material is supported on 50% SiO2
andwas subsequently optimized further to give acatalyst with the empirical formula (K,Cs)a(Ni,Mg,Mn)7.5(Fe,Cr)2.3Bi0.5Mo12Ox.
Antimonites area second important classofammoxidation catalysts [63], the most importantof which are those containing at least one of theelements U, Fe, Sn, Mn, or Ce, which all havemultiple oxidation states. Many formulations ofcatalystshavebeenproposedover theyears.Thoseof current commercial interest have extremelycomplex compositions, e.g., Na0–3(Cu,Mg,Zn,Ni)0–4(V,W)0.05–1Mo0.1–2.5Te0.2–5Fe10Sb13–20Ox
[63,215].
Scheelites Numerous multicomponentoxides adopting the scheelite (CaWO4) structurewith the general formulaABO4 are known [216].This structure tolerates cation replacements ir-respective of valency provided that A is a largercation thanB and that there is charge balance.An
additional property of the scheelite structure isthat it is often stablewith 30%ormorevacanciesin the A cation sublattice. As an example,Pb2þ1�3xBi
3þ2x &xMo6þO2�
4 , where f indicates avacancy in the Bi3þ (A cation) sublattice, pos-sesses scheelite structure. The materials areactive for selective oxidation of C3 and C4
alkenes, which involves formation of allyl spe-cies followed by extraction of O2� from thelattice. Replenishment of the created vacanciesoccurs by oxygen chemisorption at other sitesand diffusion of O2� ions within the solid. Theintroduction of A cation vacancies has a signif-icant effect on allyl formation, and the moreopen structure which prevails when cation va-cancies are present facilitates O2� transport.
Perovskite is a mineral (CaTiO3) which isthe parent solid for a whole family of multicom-ponent oxides with the general formulaABO3[191,217].Thecommonfeature,whichalsoresembles that of the scheelite-type oxides, is thesimultaneous presence of a small, often highlycharged,BcationandalargecationA,oftenhavinga low charge. The structure also tolerates a widevariety of compositions. As an example,
3�1=2x is active for methane cou-pling. Other applications of perovskite-type oxi-des in catalysis are in fuel cells, as catalysts forcombustion and for DeNOx reactions.
Hydrotalcites are another family of solidswhich tolerate rather flexible composi-tions [210,218,219]. Hydrotalcite is a clayminer-al. It is a hydroxycarbonate of Mg and Al ofgeneral formula [Mg6Al2(OH)16]CO3 � 4H2O.The compositional flexibility of the hydrotalcitelattice permits the incorporationofmanydifferentmetal cations and anions to yield solids with thegeneral formula ½M2þ
NO�3 , etc.).Hydrotalcites develop large surfaceareas and basic properties. They have conse-quently been applied as solid catalysts for base-catalyzed reactions for fine-chemicals synthe-sis, polymerization of alkene oxides, aldolcondensation, etc. Hydrotalcite-type phases(and also malachite (rosasite)- and copper zinchydroxycarbonate (aurichalcite)-type phases)can also be used as precursors for the synthesisof mixed oxides by thermal decomposition, forexample, Cu – Zn and Cu – Zn – Cr catalysts[210].
Heteropolyanions are polymeric oxo an-ions (polyoxometalates) formed by condensa-tion of more than two kinds of oxo an-ions [220,221]. The amphoteric metals ofGroups 5 (V, Nb, Ta) and 6 (Cr, Mo, W) in theþ5 and þ6 oxidation states, respectively, formweak acids which readily condense to formanions containing several molecules of the acidanhydride. Isopolyacids and their salts containonly one type of acid anhydride. Condensationcan also occur with other acids (e.g., phosphoricor silicic) to form heteropolyacids and salts.About 70 elements can act as central heteroa-toms in heteropolyanions. The structures of het-eropolyanions are classified into several familiesaccording to similarities of composition andstructure, such as Keggin type XM12O40
n�,Dawson type X2M18O62
n�, and Anderson typeXM6O24
n�, where X stands for the heteroatom.The most common structural feature is the Keg-gin anion, for which the catalytic properties havebeen studied extensively. Typically the M atomsin catalytic applications are either Mo or W.
Heteropoly compounds can be applied as het-erogeneous catalysts in their solid state. Theircatalytic performance is determined by theprimary structure (polyanion), the secondarystructure (three-dimensional arrangement ofpolyanions, counter cations, and water of crys-tallization, etc.), and the tertiary structure (parti-cle size, pore structure, etc.) [222,223]. In con-trast to conventional heterogeneous catalysts, onwhich reactions occur at the surface, the reac-tants are accommodated in the bulk of thesecondary structure of heteropoly compounds.Certain heteropolyacids are flexible, and polarmolecules are easily absorbed in interstitialpositions of the bulk solid, where they form apseudoliquid phase [222,223].
Heteropoly compounds develop acidic andoxidizing functions, so that they can be usedfor acid and redox catalysis. In addition, poly-anions are well-defined oxide clusters. Catalystdesign is therefore possible at the molecularlevel. The pseudoliquid provides a unique reac-tion environment.
Somesolidheteropolyacids havehigh thermalstability and are therefore suitable for vapor-phase reactions at elevated temperatures. Thethermal stability of several heteropolyacidsdecreases in the sequence H3PW12O40 >H4SiW12O40 > H3PMo12O40 > H4SiMo12O40
[222,223]. It can be enhanced by formation ofthe appropriate salts [224,225].
Because of their multifunctionality, hetero-polyacids catalyze a wide variety of reactionsincluding hydration and dehydration, condensa-tion, reduction, oxidation, and carbonylationchemistry with Keggin-type anions of V,Mo [223,224,226–228]. A commercially impor-tant process, the oxidation of methacrolein,is catalyzed by a Cs salt of H4PVMo11O40.Heteropoly salts with extremely complexcompositions have been proposed, e.g., for theoxydehydrogenation of ethane. A Keggin-type molybdophosphoric salt with formulaK2P1.2MO10W1Sb1Fe1Cr0.5Ce0.75On was foundto be themost efficient among the tested solids interms of activity, selectivity, and stability [229].
4.1.2. Metals and Metal Alloys
Metals and metal alloys are used as bulk, un-supported catalysts in only a few cases. Metal
Heterogeneous Catalysis and Solid Catalysts 33
gauzes or grids are used as bulk catalysts instrongly exothermic reactions which requirecatalyst beds of small height. Typical examplesare platinum – rhodiumgrids used for ammoniaoxidation in the nitric acid process [230] andsilver grids for the dehydrogenation of methaneto formaldehyde.
Skeletal (Raney-type) catalysts, particularlyskeletal nickel catalysts, are technologically im-portant materials [231] which are specificallyapplied in hydrogenation reactions. However,their application is limited to liquid-phase reac-tions. They are used in particular for the produc-tionof fine chemicals andpharmaceuticals. Skel-etal catalysts are prepared by the selective re-moval of aluminum fromNi – Al alloy particlesby leaching with aqueous sodium hydrox-ide [231]. Besides skeletal Ni, cobalt, copper,platinum, ruthenium, and palladium catalystshave been prepared, with surface areas between30 and 100 m2 g�1. One of the advantages ofskeletal metal catalysts is that they can be storedin the form of the active metal and thereforerequire no pre-reduction prior to use, unlikeconventional catalysts, the precursors of whichare oxides of the active metal supported on acarrier.
Fused catalysts are particularly used asalloy catalysts. The synthesis from a homoge-neous melt by rapid cooling may yieldmetastablematerials with compositions that canotherwise not be achieved [232]. Amorphousmetal alloys have also been prepared (metallicglasses) [232,233].
Oxide materials can also be fused for cata-lytic applications [232]. Such oxides exhibit acomplex and reactive internal interface struc-ture which may be useful either for direct cata-lytic application in oxidation reactions or inpredetermining the micromorphology of result-ing catalytic materials when the oxide is thecatalyst precursor. The prototype of such acatalyst is the multiply promoted iron oxideprecursor of catalysts used for ammoniasynthesis [48,234].
4.1.3. Carbides and Nitrides [48,235]
Monometallic carbides and nitrides of earlytransition metals often adopt simple crystalstructures inwhich themetal atoms are arranged
in cubic close-packed (ccp), hexagonal close-packed (hcp), or simple hexagonal (hex) arrays.C and N atoms occupy interstitial positionsbetween metal atoms (interstitial alloys). Thematerials have unique properties in terms ofmeltingpoint (> 3300 K), hardness (> 2000 kgmm�2), and strength (> 3 � 105 MPa). Theirphysical properties resemble those of ceramicmaterials, although their electronic andmagneticproperties are typical of metals. Carbon inthe carbides donates electrons to the d band ofthe metal, thus making the electronic character-istics of, e.g., tungsten and molybdenum resem-ble more closely those of the platinum groupmetals.
Bulk carbides and nitrides, e.g., of tungstenand molybdenum, can be prepared with surfaceareas between 100 and 400 m2 g�1 by advancedsynthetic procedures [235], so that they can beapplied as bulk catalysts. They catalyze avarietyof reactions for which noble metals are stillpreferentially used. Carbides and nitrides areexceptionally good hydrogenation catalysts,and they are active in hydrazine decomposition.Carbides of tungsten and molybdenum are alsohighly active for methane reforming, Fischer –Tropsch synthesis of hydrocarbons and alco-hols, and hydrodesulfurization, and the nitridesare active for ammonia synthesis and hydrode-nitrogenation [234]. The catalytic properties ofcarbides can be fine tuned by treatment withoxygen, which leads to the formation of oxy-carbides [236]. While clean molybdenum car-bide is an excellent catalyst for C – N bondcleavage (cracking of hydrocarbons), molybde-num oxide carbide is selective for skeletalisomerization [236].
In conclusion, carbides and nitrides, espe-cially those of tungsten and molybdenum, maywell be considered as future substitutes forplatinum and other metals of Groups 8 – 10 ascatalysts.
4.1.4. Carbons [237]
Although carbons are frequently used as cata-lyst supports, they may also be used as catalystsin their own right [238,239]. Carbons exist in avariety of thermodynamic phases (allotropesof carbon) and metastable structures, which areoften ill defined (see also ! Carbon,
34 Heterogeneous Catalysis and Solid Catalysts
1. General). The surface chemistry of carbons israther complex [174,237]. Carbon surfaces maycontain a variety of functional groups, particu-larly those containing oxygen, depending on theprovenience and pretreatment of the carbon. Ata single adsorption site several chemically in-equivalent types of heteroatombondsmay form.Strong interactions between surface functionalgroups further complicate the variety of surfacechemical structures derived for the most impor-tant carbon – oxygen system. Two functions ofthe carbon surface act simultaneously during acatalytic reaction. Firstly, the reactants are che-misorbed selectively on the carbon surface byion exchange via oxygen functional groups ordirectly by dispersion forces involving thegraphite valence-electron system. The secondfunction is the production of atomic oxygenoccurring on the graphene basal faces of all sp2
carbon materials [237].Carbon can already be catalytically active
under ambient conditions and in aqueousmedia.Therefore efforts have been made to applycarbons as catalysts in condensed phases. Itsapplication in the gas phase under oxidizingconditions is severely limited by its tendencyto irreversible oxidation.
Catalytic applications of carbons include theoxidation of sulfurous to sulfuric acid, the se-lective oxidation of hydrogen sulfide to sulfurwith oxygen in the gas phase at ca. 400 K, thereaction between phosgene and formaldehyde,and the selective oxidation of creatinine by air inphysiological environments.
A potential technological application of car-bon catalysts involves the catalytic removal ofNO by carbon [237].
More recently, carbon nanotubes (CNT) andnanofibers (CNF) have found significant inter-est as catalysts and catalyst supports [237,240].These materials, especially nanotubes, exhibitinteresting electronic, mechanical and thermalproperties that are clearly different from those ofactivated carbons. High mechanical strengthand resistance to abrasion in combination withhigh accessibility of active sites are advantagesof CNT-based catalysts which make them veryattractive for liquid-phase reactions, where themicroporosity of activated carbons often limitsthe catalytic performance. Due to their highelectrical conductivity and oxidation stability,CNTs are also highly interesting carrier materi-
als for proton-exchange membrane fuel cell(PEMFC) and directmethanol fuel cell (DMFC)catalysts [241].
4.1.5. Ion-Exchange Resins and Ionomers
Ion-exchange resins (! Ion Exchangers) arestrongly acidic organic polymers which areproduced by suspension copolymerization ofstyrene with divinylbenzene and subsequentsulfonation of the cross-linked polymer ma-trix [242]. This matrix is insoluble in water andorganic solvents. Suspension polymerizationyields spherical beads which have differentdiameters in the range 0.3 – 1.25 mm. TheGaussian size distribution of the beads can beinfluenced by the polymerization parameters.
A network of micropores is produced duringthe copolymerization reaction. The pore size isinversely proportional to the amount of cross-linking agent. In the presence of inert solventssuch as isoalkanes during the polymerization,which dissolve the reactive monomers and pre-cipitate the resulting polymers, beads with anopen spongelike structure and freely accessibleinner surface are obtained. The matrix is then aconglomerate of microspheres which are inter-connected by cavities or macropores. Macro-porous resins are characterized by microporesof 0.5 – 2 nm and macropores of 20 – 60 nm,depending on the degree of cross-linking.
Strongly acidic polymeric resins are thermal-ly stable at temperatures below 390 – 400 K.Above 400 K, sulfonic acid groups are split offand a decrease in catalytic activity results.
Industrially, acidic resins are used in theproduction of methyl tert-butyl ether [243].
The ionomer Nafion is a perfluorinated poly-mer containing pendant sulfonic acid groupswhich is considered to develop superacidicproperties. It can be used as a solid acid catalystfor reactions such as alkylation, isomerization,and acylation [244].
4.1.6. Molecularly Imprinted Catalysts[245]
Molecular imprinting permits heterogeneoussupramolecular catalysis to be performed onsurfaces of organic or inorganic materials with
Heterogeneous Catalysis and Solid Catalysts 35
substrate recognition. Heterogeneous catalystswith substrate specificity based on molecularrecognition require a material having a shape-and size-selective footprint on the surface or inthe bulk. The stabilization of transition states byimprinting their features into cavities or adsorp-tion sites by using stable transition-state analo-gues as templates is of particular interest.
Imprinted materials can be prepared on thebasis of Al3þ-doped silica gel [246] and ofcross-linked polymers [247,248]. Chiral molec-ular footprint cavities have also been designedand imprinted on the surface of Al3þ-dopedsilica gel by using chiral template molecules.
When transition-state or reaction-intermedi-ate analogues are used as templates for molecu-lar imprinting, specific adsorption sites are cre-ated. Such molecular footprints on silica gelconsist of a Lewis site and structures comple-mentary to the template molecules. These struc-tures can stabilize a reacting species in thetransition state and lower the activation energyof the reaction, thus mimicking active sites ofnatural enzymes and catalytic antibodies.
Although this approach seems to have a highpotential for heterogeneous catalysis, the realapplication of imprinted materials as catalystsstill remains to be demonstrated.
4.1.7. Metal – Organic Frameworks[249,250]
Metal – organic frameworks (MOFs) are highlyporous, crystalline solids consisting of a three-dimensional network of metal ions attached tomultidentate organic molecules. Similar to zeo-lites, the spatial organization of the structuralunits gives rise to a system of channels andcavities on the nanometer length scale. A mile-stone for the development of MOFs was thesynthesis ofMOF-5 in 1999 [251]. Thismaterialconsists of tetrahedral Zn4O
6þ clusters linkedby terephthalate groups and has a specific sur-face area of 2900 m2 g�1. MOF-177 has aneven larger specific surface area of 4500 m2
g�1 [252]. By selection of the linker length, thesize of the resulting pores can be tailored.
Due to their extremely high surface areas andtheir tunable pore structure with respect to size,shape, and function, MOFs are highly interest-ingmaterials for various applications. Examples
are the adsorption of gases such as hydrogen ormethane targeted at the replacement of com-pressed-gas storage, removal of impurities innatural gas, pressure-swing separation of noblegases (krypton, xenon), and use as catalysts[253]. Despite their higher metal contentcompared to zeolites, the use of MOFs inheterogeneous catalysis is restricted due totheir relatively low stability at elevated temper-ature and in the presence of water vapor orchemical reagents. In addition, the metal ionsin MOFs are often blocked by the organiclinker molecules and are therefore not accessi-ble for catalytic reactions. However, successfulapplications of especially stable Pd MOFsin alcohol oxidation, Suzuki C–C coupling andolefin hydrogenation have been reported[254]. It can be expected that the number ofsuccessful catalytic studies using MOFs willgrow considerably.
4.1.8. Metal Salts
Although salts can be environmentally harmful,they are still used as catalysts in some techno-logically important processes. FeCl3 – CuCl2 isa catalyst for chlorobenzene production, andAlCl3 is still used for ethylbenzene synthesisand n-butane isomerization.
4.2. Supported Catalysts
Supported catalysts play a significant role inmany industrial processes. The support provideshigh surface area and stabilizes the dispersion ofthe active component (e.g., metals supported onoxides). Active phase – support interactions,which are dictated by the surface chemistry ofthe support for a given active phase, are responsi-ble for thedispersionand thechemical stateof thelatter. Although supports are often considered tobe inert, this is not generally the case. Supportsmay actively interfere with the catalytic process.Typicalexamplesfortheactive interplaybetweensupport and active phase are bifunctional cata-lysts such as highly dispersed noble metals sup-ported on the surface of an acidic carrier.
To achieve the high surface areas and stabi-lize the highly disperse active phase, supportsare typically porous materials having high
36 Heterogeneous Catalysis and Solid Catalysts
thermostability. For application in industrialprocesses they must also be stable towards thefeed and they must have a sufficient mechanicalstrength.
4.2.1. Supports
Many of the bulk materials described in SectionUnsupported (Bulk) Catalysts may also func-tion as supports. The most frequently usedsupports are binary oxides including transitionalaluminas, a-Al2O3, SiO2, MCM-41, TiO2 (ana-tase), ZrO2 (tetragonal), MgO etc., and ternaryoxides including amorphous SiO2 – Al2O3 andzeolites. Additional potential catalyst supportsare aluminophosphates, mullite, kieselguhr,bauxite, and calcium aluminate. Carbons invarious forms (charcoal, activated carbon) canbe applied as supports unless oxygen is requiredin the feed at high temperatures. Table 7 sum-marizes important properties of typical oxideand carbon supports.
Silicon carbide, SiC, can also be used as acatalyst support with high thermal stability andmechanical strength [255]. SiC can be preparedwith porous structure and high surface area bybiotemplating [256]. This procedure yields ce-ramic composite materials with biomorphicmicrostructures. Biological carbon preforms arederived from different wood structures by high-temperature pyrolysis (1100 – 2100 K) andused as templates for infiltration with gaseousor liquid Si to form SiC and SiSiC ceramics.During high-temperature processing the micro-structural details of the bioorganic preforms areretained, and cellular ceramic composites withunidirectional porous morphologies and aniso-tropic mechanical properties can be obtained.These materials show low density, high specificstrength, and excellent high-temperature stabil-
ity. Although they have not yet found applica-tion in catalysis, the low-weight materials maywell be advantageous supports for high-temper-ature catalysis processes.
Monolithic supports with unidirectionalmacrochannels are used in automotive emissioncontrol catalysts (! Automobile Exhaust Con-trol) where the pressure drop has to be mini-mized [257]. The channelwalls are nonporous ormay contain macropores. For the above applica-tion the monoliths must have high mechanicalstrength and low thermal expansion coefficientsto give sufficient thermal shock resistance.Therefore, the preferred materials of monolithstructures are ceramics (cordierite) or high-quality corrosion-resistant steel. Cordierite is anatural aluminosilicate (2 MgO � 2 Al2O3 � 5SiO2). The accessible surface area of these ma-terials corresponds closely to the geometric sur-face area of the channels. High surface area iscreated by depositing a layer of amixture of up to20 different inorganic oxides, which includetransitional aluminas as a common constituent.This so-called washcoat develops internal sur-face areas of 50 to 300 m2/g [258,259].
Silica, MCM-41, and polymers can be func-tionalized for application as supports for thepreparation of immobilized or hybrid cata-lysts [174,177,260–266]. The functional groupsmay serve as anchoring sites (surface boundligands) for complexes and organometalliccompounds. Chiral groups can be introducedfor the preparation of enantioselective catalysts(see Section Hybrid Catalysts).
4.2.2. Supported Metal Oxide Catalysts
Supported metal oxide catalysts consist of atleast one active metal oxide componentdispersed on the surface of an oxide support
Table 7. Properties of typical catalyst supports
Support Crystallographic phases Properties/applications
Al2O3 mostly a and g SA up to 400; thermally stable three-way cat., steam reforming and many other cats.SiO2 amorphous SA up to 1000; thermally stable; hydrogenation and other cats.Carbon amorphous SA up to 1000; unstable in oxid. environm., hydrogenation cats.TiO2 anatase, rutile SA up to 150; limited thermal stability; SCR cats.MgO fcc SA up to 200; rehydration may be problematic; steam reforming cat.Zeolites various (faujasites, ZSM-5) Highly defined pore system; shape selective; bifunctional cats.Silica/alumina amorphous SA up to 800; medium strong acid sites; dehydrogenation cats.; bifunctional catalysts.
SA ¼ surface area in m2/g
Heterogeneous Catalysis and Solid Catalysts 37
[52,267,268]. The active oxides are often transi-tion metal oxides, while the support oxidestypically include transitional aluminas (prefer-entially g-Al2O3), SiO2, TiO2 (anatase), ZrO2
(tetragonal), and carbons.Supported vanadia catalysts are extremely
versatile oxidation catalysts. V2O5/TiO2 is usedfor the selective oxidation of o-xylene to phtha-lic anhydride [269,270] and for the ammoxida-tion of alkyl aromatics to aromatic nitriles [270].The latter reaction is also catalyzed by V2O5/Al2O3 [270]. The selective catalytic reduction(SCR) of NOx emissions with NH3 in tail gasfrom stationary power plants is a major appli-cation of V2O5 – MoO3 – TiO2 and V2O5 –WO3 – TiO2 [271,272]. MoO3 – Al2O3 andWO3 – Al2O3 (promoted by oxides of cobaltor nickel) are the oxide precursors for sulfidedcatalysts (see Section Supported Sulfide Cata-lysts) for hydrotreating of petroleum (hydrode-sulfurization, hydrodenitrogenation, hydro-cracking) [49,273,274]. WO3 – ZrO2 developsacidic and redox properties [275,276]. Whenpromoted with Fe2O3 and Pt it can be applied asa highly selective catalyst for the low-tempera-ture isomerization of n-alkanes to isoalk-anes [277]. Re2O7 – Al2O3 is an efficient me-tathesis catalyst [278]. Cr2O3 – Al2O3 andCr2O3 – ZrO2 are catalysts for alkane dehydro-genation and for dehydrocyclization of, e.g., n-heptane to toluene [279].
The above-mentioned transition metal oxi-des have lower surface free energies than thetypical support materials [52,280]. Therefore,they tend to spread out on the support surfacesand form highly dispersed active oxide over-layers. These supported oxide catalysts are thusfrequently calledmonolayer catalysts, althoughthe support surface is usually not completelycovered, even at loadings equal to or exceedingthe theoretical monolayer coverage. This isbecause most of the active transition metaloxides (particularly those of V, Mo, and W)form three-dimensional islands on the supportsurface which have structures analogous to mo-lecular polyoxo compounds [52,267].
4.2.3. Surface-Modified Oxides
The surface properties, that is acidity and basic-ity, of oxides can be significantly altered by
deposition of modifiers. The acid strength ofaluminas is strongly enhanced by incorporationof Cl� into or on the surface. This may occurduring impregnation with solutions containingchloride precursors of an active compo-nent [169] or by deposition of AlCl3. Chlorinat-ed aluminas are also obtained by surface reac-tion with CCl4 [174]. The presence of chlorineplays an important role in catalytic reformingwith Pt – Al2O3 catalysts [50].
Strongly basic materials are obtained bysupporting alkali metal compounds on the sur-face of alumina [281]. Possible catalysts includeKNO3, KHCO3, K2CO3, and the hydroxides ofthe alkali metals supported on alumina.
Sulfation of several oxides, particularly te-tragonal ZrO2, yields strong solid acids, whichwere originally considered to develop superaci-dic properties [282,284], because, like tung-stated ZrO2 (see Section SupportedMetal OxideCatalysts), they also catalyze the isomerizationof n-alkanes to isoalkanes at low temperature.
4.2.4. Supported Metal Catalysts
Metals typically have high surface free ener-gies [280] and therefore a pronounced tendencyto reduce their surface areas by particle growth.Therefore, for applications as catalysts they aregenerally dispersed on high surface area sup-ports, preferentially oxides such as transitionalaluminas, with the aim of stabilizing small,nanosized particles under reaction condi-tions [169, 285]. This can be achieved by somekind of interaction between ametal nanoparticleand the support surface (metal – support inter-action:), which may influence the electronicproperties of the particles relative to the bulkmetal. This becomes particularly significant forraftlike particles of monatomic thickness, forwhich all atoms are surface atoms. Furthermore,the small particles expose increasing numbersof low-coordinate surface metal atoms. Bothelectronic and geometric effects may influencethe catalytic performance of a supported metalcatalyst (particle-size effect). Aggregation ofthe nanoparticles leads to deactivation.
Model supported metal catalysts having uni-form particle size and structure can be preparedby anchoring molecular carbonyl clusters onsupport surfaces, followed by decarbonyla-
38 Heterogeneous Catalysis and Solid Catalysts
tion [286,287]. Examples are Ir4 and Ir6 clusterson MgO and in zeolite cages.
Bimetallic supported catalysts contain twodifferent metals, which may either be miscibleor immiscible as macroscopic bulk alloys. Thecombination of an active and an inactive metal[e.g., Ni and Cu (miscible) or Os and Cu (im-miscible)] dilutes the activemetal on the particlesurface. Therefore, the catalytic performance ofreactions requiring ensembles of several activemetal atoms rather than single isolated atoms isinfluenced [288,289]. Selectivities of catalyticprocesses can thus be optimized. Typically, thesurface composition of binary alloys differs fromthat of the bulk. The component having the lowersurface free energy is enriched in the surfacelayer. For example, Cu is largely enriched at thesurface of Cu – Ni alloys, even at the lowestconcentration. Also, surface compositions ofbinary alloys may be altered by the reactionatmosphere.
In industrial application, supported metalcatalysts are generally used as macroscopicspheres or cylindrical extrudates. By specialimpregnation procedures, metal concentrationprofiles within the pellet can be created in acontrolled way. Examples are schematicallyshown in Figure 13 [169]. The choice of theappropriate concentration profilemay be crucialfor the selectivity of a process because of theinterplay between transport and reaction inthe porous mass of the pellet. For example forthe selective hydrogenation of ethyne impuritiesto ethene in a feed of ethene, eggshell profilesare preferred.
Applications of supported metal catalysts,such as noble (Pt, Pd, Rh) or non-noble (Ru,Ni, Fe, Co) metals supported on Al2O3, SiO2, oractive carbon include hydrogenation and dehy-drogenation reactions. Ag on Al2O3 is used forethene epoxidation. Supported Au catalysts areactive for low-temperature CO oxidation.
Multimetal catalysts Pt – Rh – Pd on Al2O3
modified byCeO2 as oxygen storage componentare used on a large scale in three-way carexhaust catalysts [259]. Pt supported on chlori-nated Al2O3 is the bifunctional catalyst used forcatalytic reforming, isomerization of petroleumfractions, etc.
Modification of supported Pt catalysts bycinchona additives yields catalysts for the en-antioselective hydrogenation of a-ketoesters[290].
4.2.5. Supported Sulfide Catalysts
Sulfided catalysts ofMo andW supported on g-Al2O3 or active carbons are obtained by sulfi-dation of oxide precursors (supported MoO3 orWO3; see Section SupportedMetal Oxide Cat-alysts) in a stream of H2/H2S. They are typi-cally promoted with Co or Ni and serve (inlarge tonnage) for hydrotreating processes ofcrude oil, including hydrodesulfurization(HDS) [49,273,274], hydrodenitrogenationHDN [274], and hydrodemetalation HDM[291]. Currently, the CoMoS and NiMoS mod-els are most accepted for describing the activephase. These phases consist of a single MoS2layer or stacks of MoS2 layers in which thepromoter atoms are coordinated toedges [49,274], as shown in Figure 14. Thisfigure also indicates that Co may simulta-neously be present as Co9S8 and as a solidsolution in the Al2O3 support matrix. It isinferred that the catalytic activity of the MoS2layers is related to the creation of sulfur vacan-cies at the edges of MoS2 platelets. Thesevacancies have recently been visualized onMoS2 crystallites by scanning tunneling mi-croscopy (STM) [292].
Figure 13. The four main categories of macroscopic distri-bution of a metal within a support
Figure 14. Three forms of Co present in sulfided Co – Mo/Al2O3 catalysts: as sites on the MoS2 edges (the so-calledCo – Mo – S phase), as segregated Co9S8, and as Co
2þ ionsin the support lattice.
Heterogeneous Catalysis and Solid Catalysts 39
4.2.6. Hybrid Catalysts[260,262,263,265,266]
Hybrid catalysts combine homogeneous andheterogeneous catalytic transformations. Thegoal of the approach is to combine the positiveaspects of homogeneous catalysts or enzymes interms of activity, selectivity, and variability ofsteric and electronic properties by, e.g., theappropriate choice of ligands (including chiralligands [293]) with the advantages of heteroge-neous catalysts such as ease of separation andrecovery of the catalyst. This can be achieved byimmobilization (heterogenization) of activemetal complexes, organometallic compounds,or enzymes on a solid support.
There are several routes for the synthesis ofimmobilized homogeneous catalysts:
1. Anchoring the catalytically active speciesvia covalent bonds on the surface of suitableinorganic or organic supports such as SiO2,mesoporous MCM-41, zeolites, polystyr-enes, and styrene – divinylbenzene copoly-mers [260–262,266]. The polymerization orcopolymerization of suitably functionalizedmonomeric metal complexes is also known.
2. Chemical fixation by ionic bonding using ionexchange.
3. Deposition of active species on surfaces ofporous materials by chemi- or physisorption,or chemical vapor deposition (CVD). The“ship-in-bottle” principle belongs to thissynthetic route, but is treated separately inSection Ship-in-a-Bottle Catalysts.
4. Molecularly defined surface organometallicchemistrymay also yield immobilized activeorganometallic species.
The reagents for covalent bonding on sili-ceous materials (SiO2, MCM-41) are often alk-oxy- or chlorosilanes which are anchored to thesurface by condensation reactions with surfacehydroxyl groups [260–262,266]. Functionalgroups thus created on the surface can includephosphines, amines etc., which serve as an-chored ligands for active species that undergoligand-exchange reactions. Careful control ofthe density of functional groups leads to spatialseparation of active complexes (site isolation)and thus helps to avoid undesired sidereactions [294].
Immobilized enzymes (! ImmobilizedBio-catalysts) are frequently used in biocatalysis andin organic synthesis. The synthesis and catalyticperformance of this class of heterogenized ma-terials is discussed in several review articles[266,295].
Dendrimers [296] which are functionalizedat the ends of the dendritic arms can be used forimmobilization of metal complexes. A catalyticeffect is thus generated at the periphery of thedendrimer. Dendrimers with core functionali-ties have also been synthesized. The resem-blance of the produced structures to prostheticgroups in enzymes led to the introduction of theword dendrizymes [297]. Dendrimers havefound application, e.g., in membrane reactors.
Immobilized homogeneous catalysts areused for selective oxidation reactions, for hy-drogenation, and for C – C coupling reactions.They have proved very efficient in asymmetricsynthesis [262,265,298].
Special processes with immobilized cata-lysts are supported (solid) liquid-phase cataly-sis (SLPC) [299] and supported (solid) aque-ous-phase catalysis (SAPC) [300]. In SLPC asolution of the homogeneous catalyst in a high-boiling solvent is introduced into the pore vol-ume of a porous support by capillary forces, andthe reactants pass the catalyst in the gas phase.For example, the active phase—a mixture ofvanadium pentoxide with alkali metal sulfatesor pyrosulfates— is present as a melt in thepores of the SiO2 support under the workingconditions of the oxidation of SO2 [301]. InSAPC hydrophobic organic reactants are con-verted in the liquid phase. The catalyst consistsof a thin film of water on the surface of a support(e.g., porous SiO2) and contains an active hy-drophilic organometallic complex [300]. Thereaction takes place at the interface betweenthe water film and an organic liquid phasecontaining the hydrophobic reactant. The natureof these catalyst systems is schematically shownin Figure 15.
A new and improved version of SLPC usesionic liquids for immobilizationofhomogeneouscatalysts in supported ionic liquid phase (SILP)systems. The advantage of ionic liquids over thepreviously used solvents is their extremely lowvapor pressures that allow for long-term immo-bilization of homogeneous catalysts. A varietyof reactions have already been successfully
40 Heterogeneous Catalysis and Solid Catalysts
studied [302,303]. A related novel concept usessolid catalysts with ionic liquid layer (SCILL) asa method to improve the selectivity of heteroge-neous catalysts. The sequential hydrogenation ofcyclooctadiene to cyclooctene and cyclooctaneon a commercial Ni catalyst coated with an ionicliquid was tested as model system. Compared tothe original catalyst, the coating of the internalsurface with the ionic liquid strongly enhancedthe maximum intrinsic yield of the intermediateproduct [304].
4.2.7. Ship-in-a-Bottle Catalysts [305]
Metal complexes which are physically en-trapped in the confined spaces of zeolite cages(confined catalysts) are known as ship-in-a-bottle catalysts or tea-bag catalysts. The en-trapped complexes are assumed to retain manyof their solution properties. The catalytic per-
formance can be modified in a synergistic man-ner by shape selectivity, the electrostatic envi-ronment, and the acid-base properties of thezeolite host. Ligands for metal centers in thezeolite cages include ethylenediamine, di-methylglyoxime, various Schiff bases, phthalo-cyanines, and porphyrins [305,306]. The en-trapped complexes can be obtained via threeprincipal routers [305]:
1. Reaction of the preformed flexible ligandwith transition metal previously introducedinto the zeolite cages (flexible ligand meth-od). The synthesis of a zeolite entrappedmetal salen complex is schematically shownin Figure 16.
2. Assembling the ligand from smaller speciesinside the zeolite cavities (ship-in-a-bottletechnique). For example, the synthesis of azeolite-entrapped metal phthalocyanine isschematically shown in Figure 17.
Figure 15. Schematic representation of SAP catalysis
Heterogeneous Catalysis and Solid Catalysts 41
3. Synthesis of the zeolite structure around thepreformed transition metal complex (zeolitesynthesis technique).
The success of ship-in-a-bottle catalysts incatalytic processes has still to be demonstrated.
Zeolite-encapsulated complexes have alsobeen suggested as model compounds for mim-icking enzymes. These zeolite-based enzymemimics are called zeozymes to describe a cata-lytic system, in which the zeolite replaces theproteinmantle of the enzyme, and the entrappedmetal complex mimics the active site of theenzyme (e.g., an iron porphyrin) [307].
Host – guest supramolecular compoundsmay also be mentioned in this context[245,308].
4.2.8. Polymerization Catalysts
Ziegler – Natta (! Polyolefins – Ziegler Cat-alysts) catalysts are mixtures of solid and liquid
compounds containing a transition metal suchas Ti or V [309,310]. TiCl4 combined with Al(C2H5)3 or other alkyl aluminum compoundswere found to be active for olefin polymeriza-tion. More active catalysts were produced com-mercially by supporting the TiCl4 on solidMgCl2, SiO2 or Al2O3 to increase the amountof active titanium. Currently, Ziegler – Nattacatalysts are produced by ball milling MgCl2with about 5% of TiCl4, and the cocatalyst is Al(C2H5)3.
The Phillips catalyst (! Polyolefins –Phillips Catalysts) consists of hexavalent sur-face chromate on high surface area silicatesupports. Cr6þ is reduced by ethylene or otherhydrocarbons, probably to Cr2þ and Cr3þ, thecatalytically active species [309,310].
More recently, so-called single-site catalystsusing metallocenes as active species were devel-oped (! Metallocenes, ! Polyolefins)[310,311]. The activity of these materials isdramatically enhancedbyactivationwithmethy-laluminoxane (MAO), obtained by incomplete
Figure 16. Synthesis of zeolite-entrapped metal salen complexes by the flexible ligand method
Figure 17. Ship-in-a-bottle synthesis of zeolite-encaged metal phthalocyanines
42 Heterogeneous Catalysis and Solid Catalysts
hydrolysis of Al(CH3)3, the catalytic perfor-mance of which is significantly more versatilethan that of the classical Ziegler – Natta orPhillips catalysts. Activities and the nature ofthe polymeric product can be tailored by thechoice of the metal and ligands.
4.3. Coated Catalysts
In addition to bulk and supported catalysts,coated catalysts can be considered as a thirdclass of catalysts. In contrast to traditional cata-lyst geometries such as powders, tablets,spheres, and rings, coated catalysts are catalyti-cally active layers applied on inert structuredsurfaces. These active layers consist of bulk orsupported catalysts. The use of coated catalystshas recently become increasingly popular.Examples for such systems are:
* Egg-shell catalysts deposited on an inertcarrier
* Monolithic honeycombs for environmentalapplications or for multiphase reactions[312,313]
* Structured packings [314]* Foams and sponges [315]* Fibers and cloths [316]* Catalytic-wall reactors* Catalytic filters for flue gas treatment and
diesel exhaust after-treatment [317]* Membrane – electrode assemblies for fuel
cells [318]* Microstructured reactors with coated chan-
nels [319]
Advantages of coated catalysts are optimalusage of the active mass, high selectivity at lowdiffusion lengths, highly efficient mass transferfrom fluid phases to the solid catalyst layer, andlow pressure drop.
5. Production of HeterogeneousCatalysts
The development of heterogeneously catalyzedreactions for the production of chemicals initi-ated the preparation of the required catalysts ona technical scale. Up to the end ofWorldWar II,solid catalysts were produced predominantly in
process companies such as IG Farben and BASFin Germany, and Standard Oil Company andUOP in the USA [320,321]. About ten yearslater some independent catalyst producing com-panies were founded in the USA, WesternEurope, and Japan [320]. At present more than15 international companies [320,322] are pro-ducing solid catalysts on multitonne scale; forexample:
* Synetix (ICI Catalysts and ICI Catalco)* Davison Chemicals and Grace* S€UD-Chemie Catalyst Group (incl. UCI,
Houdry, Prototec in the USA, NGC, CCIFEin Japan, UCIL in India and AFCAT, SYN-CAT in South Africa)
* UOP and Katalystiks* BASF (incl. Engelhard Corp., Calsicat)* Monsanto* Shell and Criterion Catalysts* Akzo Chemicals* Johnson Matthey* Haldor Topsøe* Evonik Degussa* Nippon Shokubai* Nikki Chemical
In 1991 the catalyst world market achieved aturnover of about $ 6 � 109 [320,321,323],grew to $ (8 – 9) � 109 in 1996, and reached$ 13 � 109 in 2008.
Approximately 24 – 28% of produced cat-alysts were sold to the chemical industry and38 – 42% to petrochemical companies includ-ing refineries. 28 – 32% of solid catalysts wereused in environmental protection, and 3 – 5%in the production of pharmaceuticals [320,323].
The catalytic properties of solid catalysts arestrongly affected by the preparation method,production conditions, and quality of sourcematerials. Therefore, it is necessary to controleach production step and the physical or me-chanical properties of all intermediates. To at-tain a better reproducibility of catalyst produc-tion, batch procedures were mainly replaced bycontinuous operations, such as precipitation,filtration, drying, calcination, and forming.
Automation of various operations and com-puter control of different equipments were in-stalled in catalyst production lines [320]. Re-cently, SPC (statistic process control) and QA(quality assurance) were integrated into the
Heterogeneous Catalysis and Solid Catalysts 43
catalyst production process. Some companies,especially in Western Europe and in the USA,produce solid catalysts according to ISOStandard which guarantees a standard catalystquality to the customer [320,324].
Catalysts applied in several industrialprocesses can be subdivided into the followingcategories:
Unsupported catalysts represent a large categoryandare applied in numerous industrial processes.Various preparationmethodswere adopted in thepast decades in the commercial production ofunsupported catalyst, such as mechanical treat-ment or fusion of catalyst components, precipi-tation, coprecipitation, flame hydrolysis, andhydrothermal synthesis[232,322,325–329].
Mechanical treatment, for example, mix-ing, milling, or kneading of catalytic activematerials or their precursors with promoters,structure stabilizers, or pore-forming agents, isone of the simplest preparation meth-ods [322,325–327]. In some cases, however, therequired intimate contact of catalyst compo-nents could not be achieved and therefore theactivity, selectivity, or thermal stability of cat-alysts prepared in this way was lower than ofthose prepared by other methods. However,recent improvements in the efficiency of variousaggregates for the mechanical treatment of so-lids resulted in activity enhancement. An im-portant advantage of these methods is that for-mation of wastewater is avoided.
Industrial catalysts produced by mechanicaltreatment are summarized in Table 8.
Fusion of components or precursors isused for the production a small group of unsup-ported catalysts. The fusion process [232] per-mits the synthesis of alloys consisting of ele-ments which do not mix in solution or in thesolid state. However, preparation of unsupport-ed catalysts by fusion is an energy-consumingand quite expensive process.
The most important application of this meth-od is the production of ammonia synthesis
catalysts based on the fusion of magnetite(Fe3O4) with promoters such as oxides of K,Al, Ca, and Mg [232]. Another example is thepreparation of SO2 oxidation catalysts by fusionof V2O5 with K pyrosulfate (K2S2O7) [232].Some producers incorporate Cs oxide as anactivity promoter in this catalyst.
Quite recently, amorphous alloys composed,e.g., of Pd and Zr, so-called metallic glasseswere found to be active in catalyticoxidations [232,233].
Industrial catalysts produced by the fusionprocess are listed in Table 8.
Precipitation and coprecipitation are themost frequently applied methods for the prepa-ration of unsupported catalysts or catalyst sup-ports [322,325–329]. However, both methodshave the major disadvantage of forming largevolumes of salt-containing solutions in the pre-cipitation stage and in washing the precipitate.
Source materials are mainly metal salts, suchas sulfates, chlorides, and nitrates. Acetates,formates, or oxalates are used in some cases.In industrial practice nitrates or sulfates arepreferred. Basic precipitation agents on an in-dustrial scale are hydroxides, carbonates, andhydroxocarbonates of sodium, potassium, orammonium.
Alkali metal nitrates or sulfates formed asprecipitation byproducts must be washed out ofthe precipitate. Thermally decomposable an-ions, such as carbonates and carboxylates andcations like NH4
þ are especially favored incatalyst production.
Table 8. Unsupported catalysts prepared by mechanical treatment(MT) or by fusion (F)
Coprecipitation of two or more metal cationsis a suitable operation for the homogeneousdispersion of the corresponding oxides, espe-cially if the catalyst precursors have a definedcrystalline structure, for example, Cu(OH)NH4CrO4 or Ni6Al2(OH)16CO3. After thermaltreatment, binary oxides such as CuO – Cr2O3
and NiO – Al2O3 are formed [322,325–329].Precipitation and coprecipitation can be car-
ried out in batch or continuous operations.If the metal salt solution is placed in the
precipitation vessel and the precipitating agentis added, then the pH changes continuouslyduring the precipitation. Coprecipitation shouldbe carried out in the reverse manner (addition ofthemetal salt solution to the precipitation agent)to avoid sequential precipitation of two or threemetal species.
If the metal salt solution and the precipitatingagent are simultaneously introduced into theprecipitation vessel, then it is possible to keepthe pH constant. However, the residence time oftheprecipitate inthevesselchangescontinuously.
Finally, if the metal salt solution and the pre-cipitatingagentarecontinuouslyintroducedin theprecipitationvessel, and the reaction products areremoved continuously, then pH and residencetime can be kept constant [322,325–329].
Besides pH and residence time, other pre-cipitation parameters, such as temperature, agi-tation, and concentration of starting solutions,affect the properties of the precipitate. Thechoice of anions, the purity of raw material,and the use of various additives also play animportant role [322,325–329].
In general, highly concentrated solutions,low temperatures and short ageing times resultin finely crystalline or amorphous materialswhich are difficult to wash and filter. Lower
concentrations of the solutions, higher tempera-tures, and extended ageing provide coarse crys-talline precipitates which are easier to purifyand separate [322,325–329].
The industrial production of precipitatedcatalysts usually involves the following steps:
* Preparation of metal salt solution and ofprecipitating agent (dissolution, filtration)
* Precipitation* Ageing of the precipitate* Washing of the precipitate by decantation* Filtration* Washing of the filter cake (spray drying)* Drying* Calcining* Shaping* Activation
Operations such as filtration, drying, calci-nation etc. are discussed in Section Unit Opera-tions in Catalyst Production.
Typical unsupported industrial catalysts pro-duced by precipitation or coprecipitation arecompiled in Table 9.
The sol – gel process [330] involves theformation of a sol, followed by the creation of agel. A sol (liquid suspension of solid particlessmaller than 1 mm) is obtained by the hydrolysisand partial condensation of an inorganic salt or ametal alkoxide. Further condensation of solparticles into a three-dimensional network re-sults in the formation of a gel. The porosity andthe strength of the gel are strongly affected byconditions of its formation. For example, slowcoagulation, elevated temperature, or hydro-thermal posttreatment increase the crystallinefraction of the gel.
Table 9. Catalysts (their precursors) or supports prepared by precipitation or coprecipitation
Catalysts (precursors) or supports Source materials Application
Alumina Na aluminate, HNO3 support, dehydration, Claus processSilica Na silicate (water glass), H2SO4 supportFe2O3 Fe(NO3)3, NH4OH ethylbenzene dehydrogenation (styrene production)TiO2 Fe titanate, titanyl sulfate, NaOH support, Claus process, NOx reductionCuO – ZnO – (Al2O3) Cu, Zn, (Al) nitrates, Na2CO3 LTS, methanol synthesisFe molybdate Fe(NO3)3, (NH4)2MoO4, NH4OH methanol oxidation to formaldehydeVanadyl phosphate vanadyl sulfate, NaHPO4 butane oxidation to maleic acid anhydrideNiO – Al2O3 Ni, Al nitrates, Na2CO3 hydrogenation of aromaticsNiO – SiO2 Ni nitrate, Na silicate, Na2CO3 hydrogenation of aromatics
Heterogeneous Catalysis and Solid Catalysts 45
Alumina and silica can be produced fromsodium aluminate or sodium silicate by treat-ment with nitric, hydrochloric, or sulfuric acid.In this process, first sols and then gels areformed. Washing the sodium from the gels isessential [327,328,330].
Spherical silica or silica-alumina gels areproduced directly by injecting drops of a gellingmixture into oil at a proper rate to allow settingof the gel. To avoid bursting during drying, thegel beads are washed to reduce the salt content(NaNO3, NaCl, or Na2SO4). Finally, the beadsare dried and calcined [327,328].
Based on the sol – gel process, high-puritymaterials such as alumina, TiO2, ZrO2 are pro-duced on an industrial scale. Raw materials arethe corresponding metal alkoxides, e.g., Al(C12 – C18 alkoxide)3 and Ti(n-C4H9O)4 [330].
Flame Hydrolysis In flame hydroly-sis [331] a mixture of the catalyst or supportprecursor, hydrogen, and air is fed into the flameof a continuously operating reactor. Precursors(mainly chlorides such as AlCl3, SiCl4, TiCl4 orSnCl4) are hydrolyzed by steam (formed by H2
oxidation). The products of flame hydro1ysisare oxides. More than 100 000 t/a of so-calledfumed silica, alumina, or titania are produced byDegussa, Wacker (both Germany), and Cabot(USA).
Thermal decomposition of metal – inor-ganic or metal – organic catalyst precursors issometimes used in industrial catalyst produc-tion. For example, mixtures of Cu- and Zn(NH3)4(HCO3)2 decompose at 370 K to formbinary Cu – Zn carbonates, which are trans-formed during calcination into the correspond-ing binary oxides, used as low temperaturewater gas shift catalysts [327].
Industrial production of Cu – Cr oxides(copper chromites), used in the hydrogenationof carbonyl compounds, is based on the thermaldecomposition of a basic copper ammoniumchromate [CuNH4(OH)CrO4] at 620 – 670 K[327,328].
Highly active Ni catalysts for the hydrogena-tion of fats and oils are obtained by the thermaldecomposition of Ni formate [(HCOO)2Ni] at390 – 420 K. The decomposition is usuallycarried out in hard fat, which protects Ni againstoxidation [327].
Catalysts or supports produced by flamehydrolysis or thermal decomposition of inor-ganic complexes are summarized in Table 10.
Hydrothermal synthesis [180,216] is avery important preparation method for zeolitesand other molecular sieves.
Currently, the importance of zeolites in in-dustrial catalysis is still increasing. They areused as catalysts or supports not only in petro-chemical operations but also in the productionof fine chemicals.
In hydrothermal synthesis (see also! Zeo-lites–Zeolite Synthesis: Routes and RawMaterials) a mixture of silicon and aluminumcompounds containing alkali metal cations, wa-ter, and in some cases organic compounds (so-called templates) is converted into microporous,crystalline aluminosilicates [328,333].
Common sources of silicon are colloidalsilica, water glass, fumed silica, and siliconalkoxides. Aluminum can be introduced as alu-minum hydroxide,metahydroxide, or aluminatesalts. Common templates are tetrapropyl- ortetraethylammonium bromides or hydroxides[328,333].
Hydrothermal synthesis is a complex processconsisting of three basic steps: achievement
Table 10. Catalysts (their precursors) or supports prepared by sol – gel process (SG), flame hydrolysis (FH), or thermal decomposition (TD) ofinorganic metal complexes
Catalyst (precursor) or support Source material Preparation method Application
Alumina (high-purity) Al alkoxides hydrolysis, SG support for noble metalsAlumina (acidic, low bulk density) AlCl3 FH support or additiveSilica (low bulk density) SiCl4 FH support or additiveTiO2 Ti(n-C4H9O)4 hydrolysis, SG supportTiO2 (low bulk density) TiCl4 FH support or additiveCuO – ZnO Cu, Zn (NH3)4HCO3 TD low-temperature shift, methanolCuO – Cr2O3 Cu(NH4)OHCrO4 TD hydrogenation of carbonyl compoundsNi (kieselguhr) Ni formate TD hydrogenation of fats and oils
46 Heterogeneous Catalysis and Solid Catalysts
of supersaturation, nucleation, and crystalgrowth. It is affected by the hydrogel molarcomposition, a1kalinity, temperature, and time[328,333].
In general, synthesis is carried out at 360 –450 K under atmospheric or autogenous waterpressure (0.5 – 1 MPa) with residence times of1 – 6 d. After synthesis, the crystalline productis separated by filtration or centrifugation,washed, dried, and calcined. Sodium-containingzeolites, which are the products of the hydro-thermal synthesis, are converted into acidicforms by exchange of sodium ions with theammonium, followed by thermal treatment[328,333].
Zeolites used in the industrial catalysis areabove all Y zeolite, mordenite, ZSM-5, ZSM-11, and zeolite b [180,215].
To improve the thermal stability of zeolites,especially of Y zeolite, Al ions are extractedfrom the lattice by steaming or acid treatment.For example, fluid-cracking catalysts (amor-phous aluminosilicates) contain 10 – 50% ofultrastable Y zeolites [328,333].
Related to zeolites are other molecular sievessuch as aluminum phosphates (AlPO) and sili-co-aluminum phosphates (SAPO), the impor-tance of which in industrial catalysis is growing.SAPO-11 was applied recently in the isomeri-zation of cyclohexanone oxime to e-caprolac-tam, instead of sulfuric acid in a demonstrationunit [180].
Other preparation methods includecondensation of more than two kinds of oxoanions, such as MoO4
2�;WO42�; HPO4
2�, etc.to give heteropolyacids such as H3PW12O40 orH3PMo12O40 [222].
In industrial practice, the sourcematerials areNa2HPO4, Na2WO4, or Na2MoO4 solutions.Hydrolysis and subsequent condensation arecarried out with HCl. The heteropolyacids areextractedwith organic solvents.Heteropolyacidsare very strong acids with Hammett acidityfunction Ho < � 8. They have found industrialapplication in acid-catalyzed reactions con-ducted in the liquid phase, such as hydration,esterification, and alkylation. Their activity isevidently higher than that of inorganic acids[222]. Their K or Cs salts are used as catalystsin the selective vapor phase oxidation of propeneto acrolein or isobutene to methacrolein [222].
Another preparation method is based on thetreatment of alumina or aluminosilicate withgases such as HF, HCl, BF3, AlCl3 to createhighly acidic centers. Such catalysts are activein the skeletal isomerization of hydrocarbons, e.g., n-C4 or n-C5 [328].
Skeletal catalysts, also called porous me-tals, consist of the metal skeleton remaining afterthe less noble component of an alloywas removedby leaching with alkali, preferentially NaOH.Skeletal catalysts were discovered in 1925 andintroduced into chemistrybyRaney, and thereforesome bear his name, e.g., Raney Ni or Co.
The group of skeletal catalysts [231] includesNi, Co, Cu, Fe, Pt, Ru, and Pd. The second alloycomponent can be Al, Si, Zn, or Mg, with Albeing used preferentially.
Small amounts of a third metal such as Cr orMo have been added to the binary alloy asactivity promoter.
Ni – Al, Cu – Al, and Co – Al alloys withdifferent grain sizes are commercially availableand can be leached out before use. Davison-Grace and Degussa provide finished extremelypyrophoric Ni or Co skeletal catalysts protectedby water in commercial quantities. Skeletal Nifound technical application in the hydrogena-tion of aliphatic or aromatic nitro compoundsand nitriles.
5.2. Supported Catalysts
The main feature of supported catalysts isthat the active material forms only a minorpart and is deposited on the surface of thesupport [326,328,332].
In some cases, the support is more or lessinert, e.g., a-alumina, kieselguhr, porous glass,ceramics. In other cases the support takes part inthe catalytic reaction, as in the case of bifunc-tional catalytic systems, e.g., alumina, alumino-silicate, zeolites, etc. [326,328,332].
Additionally, some supports can alter thecatalytic properties of the active phase. Thisso-called strong metal – support interaction(SMSI) can decrease, for example, the chemi-sorption capacity of supported metals (Pt –TiO2) or can hinder the reduction of supportedmetal oxides (Ni silicate, Ni and Cu aluminates,etc.) [326,328].
Heterogeneous Catalysis and Solid Catalysts 47
5.2.1. Supports
Currently, various industrial supports are avail-able in multitonne quantities possessing a widerange of surface areas, porosities, shapes, andsizes.
Widely used supports include alumina, sili-ca, kieselguhr, porous glass, aluminosilicates,molecular sieves, activated carbon, titania, zincoxide, silicates such as cordierite (2 MgO �Al2O3 � 5 SiO2) and mullite (3 Al2O3 � 2SiO2), and Zn and Mg aluminates [326,328].The supports are produced by specialized
producers or directly by catalyst producers.Well known support manufacturers are:
In the past, mostly natural supports, e.g.,bauxite, pumice and kieselguhr, were used incatalyst production. At present (with the excep-tion of kieselguhr) mainly synthetic supportswith “tailored” physical properties are preferredin industrial catalysis.
Because the majority of supports also havecatalytic properties (e.g., alumina, aluminosili-cates, zeolites) their production methods aredescribed in Section Unsupported Catalysts.
5.2.2. Preparation of SupportedCatalysts
The broad application of supported catalysts inindustrial catalysis led to the development ofnumerous preparation methods applicable on atechnical scale. Some of these methods areidentical with those used in the production ofunsupported catalysts, e.g., mechanical treat-ment, precipitation, thermal decomposition ofmetal – inorganic or metal – organic com-plexes and therefore they will be discussed herevery briefly.
Mechanical treatment, e.g., kneading ofa catalyst precursor with a support is applied,
e.g., in the production of kieselguhr-supportedNi (precursor NiCO3) [325,327,328]. Also,MoO3 supported on Al2O3 is sometimes pro-duced by this process. However, the distributionof the active phase on the support is in somecases not sufficient [325].
Better results are obtained by the combina-tion of mechanical and thermal treatmentwhich, results in spreading [52] of, e.g., MoO3
on Al2O3 or of V2O5 on Al2O3 or TiO2.
Impregnation by pore filling of a carrierwith an active phase is a frequently used pro-duction method for supported cata-lysts [169,325]. The object of this method is tofill pores of the support with a solution of thecatalyst precursor, e.g., a metal salt of sufficientconcentration to achieve the desired loading. Ifhigher loadings with active phases are required,it is mostly necessary to repeat the impregnationafter drying or calcination of the intermediate.Examples of catalysts prepared by the porefilling method are Ni or Co on Al2O3 – MoO3,MoO3 on aluminosilicates including zeolites, Nior Ag on a-alumina, noble metals on activecarbon, etc. [322,325–328].
Adsorption is a very good method toachieve uniform deposition of small amountsof active component on a support. Powders orparticles exposed to metal salt solutions adsorbequilibrium quantities of salt ions, in accor-dance with adsorption isotherms. Adsorptionmay be either cationic or anionic, dependingon the properties of the carrier surface. Forexample, alumina (depending on the adsorptionconditions, mainly on the pH of the solution)adsorbs both cations and anions. Silica weaklyadsorbs cations, while magnesia strongly ad-sorbs anions [322].
Adsorption of PdCl2 from aqueous solutionon different aluminas is very fast, and a highequilibrium concentration (ca. 2 wt%) can beobtained. The Pd deposition takes place mainlyin an outer shell (egg shell profile) of shapedparticles [322,328].
With H2[PtCl4] only 1 wt% Pt loading onalumina is possible owing to from the flatadsorption isotherm [322,328].
The addition of oxalic, tartaric, and citricacid to the metal salt solution changes theprofiles of active component on the carrier. In
48 Heterogeneous Catalysis and Solid Catalysts
general, with increasing acid strength the metalions are forced deeper into the supportparticles [322].
Ion exchange [169,322] is very similar toionic adsorption but involves exchange of ionsother than protons. Lower valence ions, such asNaþ or NHþ4 can be exchanged with highervalence ions, for example, Ni2þ or Pt4þ. Thismethod is used mainly in the preparation ofmetal-containing zeolites, e.g., Ni- or Pd-con-taining Y zeolites or mordenites used in petro-leum-refining processes.
Thermal decomposition of inorganic ororganic complexes in the presence of a support.This method is identical with that used in thepreparation of unsupported catalysts by thermaldecomposition of precursors. Supports can beused either as powder or preshaped. For exam-ple, Ni or Co deposited on kieselguhr or silica isproduced by this method [327].
Precipitation onto the support is carriedout in a similarway as in the case of unsupportedcatalysts [322,325,332]. Supports, mainly aspowders, are slurried in the salt solution, andalkali is added. Rapid mixing is essential toavoid precipitation in the bulk.
Uniform precipitation can be achieved byusing urea rather than conventional alka-lis [332]. An appropriate amount of urea isadded to the metal salt – support slurry and themixture is heated while stirring. At 360 K ureadecomposes slowly to NH3 and CO2, and pre-cipitation takes place homogeneously over thesurface and in pores of the support. This methodis called deposition – precipitation [332] and isused especially in the production of highlyactive Ni – SiO2 or Ni – Al2O3 catalysts.
Reductive deposition is a preparationmethod in which especially precious metals aredeposited on the carrier surface by reduction ofaqueous metal salts, mainly chlorides or ni-trates, with agents such as H2, Na formate,formaldehyde, and hydrazine. Examples ofcommercial catalysts produced by this methodare precious metals on active carbon, SiO2 or a-Al2O3. Reductive deposition is preferred espe-cially in the case of bimetallic supportedcatalysts such as Pt – Re or Pd – Rh [334].
Heterogenization of homogeneous cata-lysts is based on the binding of metal
complexes to the surface or entrapment in poresof the inorganic or organic support [266]. Suchcatalysts are used mainly in stereospecific hy-drogenations in the production of fine chemicalsor pharmaceuticals.
Enzymes [266] can also be heterogenized.They found industrial application in biochemi-cal processes. A prominent example is theisomerization of glucose to fructose in the pro-duction of soft drinks.
5.3. Unit Operations in CatalystProduction
As in other branches of the chemical industry,unit operations have also been established incatalyst production in the past few decades, forexample, in operations such as filtration, drying,calcination, reduction and catalyst form-ing [322,324,328]. Continuous operations arefavored because of larger throughputs, loweroperating costs, and better quality control. Ad-ditionally, factors such as environmental pollu-tion and hazards to human health can be mini-mized more easily.
Filtration, Washing The main purpose ofthese operations is to separate precipitates andto remove byproducts and possible impurities.In batch operations mainly plate-and-frame fil-ter presses are used [322,328]. The washing ofthe filter cake proceeds in countercurrent to thedirection of the filtration.
Continuous vacuum rotary filters are widelyused. By changing the speed of the filter drum(covered with filter cloth), the quantity of slurryfiltered and the thickness of the filter cake can bevaried over a broad range. As the drum rotates,there is a washing phase in which water issprayed against the moving filter cake. Finally,the filter cake is scraped or blown to remove itfrom the drum [328].
Another filtration equipment is the centri-fuge. However, its application is possible onlywhen the filtered material is grainy or crystal-line, e.g., zeolites.Washing can be carried out byintroducing washing water into the centrifuge.Centrifuges can operate either in a discontinu-ous or continuous manner [328].
Heterogeneous Catalysis and Solid Catalysts 49
Drying Because drying conditions such asrate, temperature, duration, or gas flow rate canchange the physical properties of the resultingmaterial, it is important to measure and keepthese parameters constant. For example theporosity of precipitated catalysts depends onthe drying procedure. The drying of impregnat-ed supports can change the distribution of activecomponents. Their uniform distribution can beobtained only if all the liquid is evaporatedspontaneously [322,325,328]. Usually, dryingproceeds up to 400 K.
For the drying of filter cake, various tools areused, e.g., box furnaces with trays, drum dryers,rotary kilns, and spray dryers [322,325,328].
The main problems with drums and rotarydryers are the feeding of the wet filter cake andremoval of adhering material from the walls.Because lumps are usually formed in the dryingprocess, the resulting material must pass a gran-ulator equipped with a sieve [328].
Spray dryers provide microspherical materi-als with a narrow particle-size range. Spraydryers are equipped with a nozz1e or a rotatingdisk to disperse the watery slurry of the filtercake in a stream of hot air [322,328,335].
All the above drying equipment operates incontinuous mode.
Small batches of catalyst precursors are driedin box furnaces with trays [325].
For the drying of extrudates, continuouslyoperating belt dryers have found technicalapplication [328].
Calcination The main object of calcina-tion (thermal treatment in oxidizing atmo-sphere) is to stabilize physical and chemicalproperties of the catalyst or its precursor. Duringcalcination, thermally unstable compounds(carbonates, hydroxides, or organic com-pounds) decompose and are usually convertedto oxides. During calcination new compoundsmay be formed, especially if the thermal treat-ment is carried out at higher temperatures [268].For example, in the thermal decomposition ofCu or Ni nitrate deposited on alumina, not onlyCuO or NiO but also Cu or Ni aluminate isadditionally formed [268].
Furthermore amorphous material can be-come crystalline. Various crystalline modifica-tions can undergo reversible or irreversiblechanges.
Physical and mechanical properties and porestructures can also change. The calcinationtemperature is usually slightly higher than thatof the catalyst operating temperature[322,325,328,335].
For the calcination of powder or granulate,rotary kilns are preferably used [325,328].Smaller batches of powdered catalysts are cal-cined in box or muffle furnaces with trays, as inthe case of drying. The gases that are mainlyused for heating are in direct contact with thematerial being calcined [322,325,328].
Pellets or extrudates are calcined in belt ortunnel furnaces. The tunnel-type calciner canoperate at substantially higher temperatures(close to 1270 – 1470 K) than the belt type(870 – 1070 K). The tunnel calciner is especial-ly suitable for firing ceramic carriers. The ma-terial being calcined is taken in boxes or cartswhich are recycled to the entrance via a contin-uous chain or belt [328].
Reduction, Activation, and ProtectionReduction, activation, or passivation, is in sev-eral cases the last step in catalyst production.These operations are performed by the catalystproducer or in the plant of the client.
For example, in the production of Ni cata-lysts for the liquid-phase hydrogenation of fatsand oils, the reduction of NiO deposited onkieselguhr is carried out exclusively by thecatalyst producer. The reduction of powders(50 – 500 mm particle size) is performed onan industrial scale in fluid-bed reactors. Thereduced material is pyrophoric and must beprotected with a hard fat such as tallow, tomake its handling easy and safe. The finishedcatalyst is supplied in the form of flakes orpastilles [322,328,336].
The reduction of metal oxides such as NiO,CuO, CoO, or Fe2O3 is carried out with H2 atelevated temperature (> 470 K) and has twosteps. In the first step metal nuclei are formed.In the second, nuclei accumulate to form metalcrystallites. The rates of both processes de-pend on temperature and on the nature of thesubstrate [322]. Reduction at lower tempera-tures (< 570 K) provides a narrow distribu-tion of small metal crystallites. Reduction athigher temperatures (> 670 K) gives a broad-er distribution and larger metal crystallites[322].
50 Heterogeneous Catalysis and Solid Catalysts
Reduction of some oxides, such as those ofCu and Fe, is exothermic and needs to be carriedout carefully with H2 diluted with N2.
Water, the reduction product, has negativeeffects on the rate and on the extent of reduc-tion [322]. In industrial practice, where H2 isrecycled, the removal of water by freezing out(below 270 K) and by adsorption on molecularsieve is essential.
To achieve optimal activity, partial reductionof oxidic catalysts is common [322,337]. Forexample, Ni catalysts for fat and oil hydrogena-tion contain about 50 – 60 wt% of metallic Ni,45 – 35 wt% NiO, and about 5 wt% Nisilicate.
When the reduction of shaped oxidic cata-lysts is conducted by the catalyst producer, thenthe active material is protected either with ahigh-boiling liquid such as higher aliphaticalcohols or C14 – C18 paraffins [337] or it ispassivated. In this procedure, chemisorbed hy-drogen is removed in a gas stream composed ofN2 and about 0.1 – 1.0 vol% of O2 at ambienttemperature. After this treatment catalysts canbe handled in air without any precautions [336].The activity is restored in the client’s plant bytreatment with H2 [322].
The activation of hydrotreating catalystscomposed of Ni- or Co-promoted MoO3 –Al2O3 is carried out with H2 containing10 vol% of H2S [268]. In the past, this activa-tion was performed exclusively in the hydro-desulfurization plants. However, presulfiding atcatalyst producers is becoming more common[322].
Mainly electrical or gas-heated shaft reactorsare used for the reduction of extrudates, spheres,or pellets in plants of catalyst producers.
Catalyst Forming The size and shape ofcatalyst particles depend on the nature of thereaction and on the type of applied reactor.
Reactions in the liquid phase require smallparticles or even powders (50 – 200 mm). Suchmaterials are made by grinding of a dried orcalcined precursor, e.g., filter cake, using gran-ulators equipped with sieves to give uniformparticle size [322,328,335,337].
Catalysts for fluidized bed reactors (0.05 –0.25 mm) are usually made by spray drying orby cooling molten material droplets (V2O5) inan air stream [322,328,335].
Spheres consisting of Al2O3, SiO2, or alu-minosilicate with 3 – 9 mm diameter are usedpreferentially as a support for catalysts inmoving-bed or ebullating-bed reactors. Theyare produced by the so-called oil-drop method(see Section Unsupported Catalysts). Spheresprepared in this way possess sufficient abrasionresistance [322,328,335].
Another method for producing spheres isbased on agglomeration of powder bymoisteningon a rotating disk (spherudizer) [322,328,335].Asthe spheres reach the desired diameter they areremoved automatically and transported to thedryer and calciner. Such spheres are suitable forfixed bed reactors.
Other methods for forming spherical parti-cles include tumbling short, freshly extrudedcylinders in a rotating drum [328].
In the briquetting technique, the plastic mix-ture of catalyst powder with a binder is fedbetween two rotating rolls provided with hol-lowed-out hemispheres [328].
Extrusion of pastes containing catalyst pow-der, binders and lubricants is a frequently usedindustrial shaping method [322,328,335].Depending on the properties of the paste, pressor screw extruders are applied. Press extrudersare principally suitable for viscous pastes.Screw extruders are preferred for thixotropicmasses. In both cases, pastes are forced througha die, and the extruded material is cut with aspecial device to a desired length and falls onto amoving belt that transports it through a drier orcalciner [322,328,335]. Poly(vinyl alcohol),powdered stearine, and Al stearate are used aslubricants. If the mass being extruded containsalumina, then peptizing agents such as nitricacid are added mainly to improve the mechani-cal strength [322,327,328].
Another type of binder is calcium aluminatecement, which sets up by treatment withsteam [322,328].
The extruded material can have differentshapes, such as cylinders (noodles), hollowcylinders (macaroni), or ribbed cylinders. Thesizes depend on the shape and are in the range of1.5 – 15 mm diameter [328,337].
Added organic lubricants and pore-formingagents can be removed by calcination in astream of air.
Special extrusion techniques and equipmentare necessary to produce honeycombs.
Heterogeneous Catalysis and Solid Catalysts 51
Extruding is less expensive than pelletizing,but extrudates have less resistance to abrasionthan pellets. Extrudates are suitable for differenttypes of fixed-bed reactors operating in the gasor trickle phase.
Pelletizing is a very common method forcatalyst forming. It is based on compression ofa certain volume of powder in a die between twomoving punchers, one of which also serves toeject the formed pellet [322,328,335]. Depend-ing on the size and the shape of the preparedpellets, the material being pelletized must becrushed and forced through a correspondingsieve [328]. Furthermore, lubricants such asgraphite, Al stearate, pol(vinyl alcohol), kaolin,and bentonite are added before the materialenters the tabletting machine. The fluidity ofthe material is required to assure homogeneousfilling of the die [322,328,335]. As in the case ofextrudates, organic lubricants can be removedby calcination of the pellets.
Industrial pelleting machines are equippedwith around thirty dies and produce about 10 li-ter of pellets per hour or more, depending ontheir shape and size. Pressures in the range 10 –100 MPa in the pelleting machine arecommon [322,328,335].
Commercially, cylindrical pellets with sizessuch as 3 � 3, 4.5 � 4.5, 5 � 5, or 6 � 6 mmare offered [322,328,335]. Production of 3 � 3mm pellets is more expensive than that of largersizes. Besides cylindrical pellets, various com-panies provide rings, cogwheels, spokedwheels, multihole pellets, etc. [336,337].
Pellets of different shapes and sizes are suit-able for various types of fixed-bed reactors.
Coating of inert supports [338]with a thinlayer of catalytically active material is requiredfor manufacture of coated catalysts. Avariety ofmethods for coating with catalysts are available.One can distinguish between material-depen-dent methods for the preparation of thin catalyt-ically active layers on supports and material-independent coating methods [319]. Material-dependent methods are anodic oxidation ofaluminum or aluminum alloys, which gives riseto a layer with a one-dimensional and unidirec-tional pore system with adjustable proper-ties [339], and formation of porous layers onFeCrAl alloys by heat treatment. Material-in-dependent coating technologies can be grouped
according to the state of aggregation of thecatalyst precursor [338].
Gaseous catalyst precursors can be trans-formed into coatings by chemical vapor depo-sition (CVD) or physical vapor deposition(PVD).Coatingmethods based on a liquid phasecomprise sol – gel methods, deposition of cat-alyst suspensions, and combinations of bothtechniques. Depending on the adjusted viscosityof the sols or suspension, the liquid precursorsmay be applied on surfaces by dip coating,spraying, printing, or rolling. Solid catalystpowders can be applied, e.g., by flame spraydeposition or powder plasma spraying.
A coating procedure that has been intensive-ly studied is the manufacture of monoliths, e.g.,as catalysts for pollution control [340]. Oxidessuch as Al2O3, CeO2, and ZrO2 (washcoat) aredeposited on monoliths with a honeycomblikestructure by dipping into an aqueous slurrycontaining primary particles (about 20 nm indiameter) of these materials [259]. The excessslurry is blown out, and after drying and calci-nation a thin catalyst layer is obtained, thethickness of which can be tailored by adjustingthe slurry properties and repetition of the dip-coating step.
6. Characterization of SolidCatalysts
Catalytic activity and selectivity critically de-pend on the morphology and texture, surfacechemical composition, phase composition, andstructure of solid catalysts. Therefore, manyphysical and chemical methods are used in ca-talysis research to characterize solid catalystsand to search for correlations between structureand performance of catalysts. These methodsinclude classical procedures [341] as well astechniquesdevelopedmore recently for the studyof the chemistry and physics of surfaces [341].
6.1. Physical Properties
6.1.1. Surface Area and Porosity[342,343]
The specific surface area of a catalyst or support(in m2/g) is determined by measuring the vol-
52 Heterogeneous Catalysis and Solid Catalysts
ume of gas, usually N2, needed to provide amonomolecular layer according to the Bru-nauer – Emmett – Teller (BET) method.
In this approach, the determination of themonolayer capacity is based on the physisorp-tion of the test gas. The volume adsorbed at agiven equilibrium pressure can be measured bystatic methods, namely, volumetric or gravimet-ric measurements. Flow or dynamic techniquesare also applied.
The total surface area of a porous material isgiven by the sum of the internal and externalsurface areas. Pores are classified asmicropores(pore width < 2 nm), mesopores (pore width2 – 50 nm), and macropores (pore width > 50nm) according to IUPAC definitions [344].
The specific pore volume, pore widths, andpore-size distributions formicro- andmesoporesare determined by gas adsorption. For meso-pores, the method is based on the dependenceof the pressure of capillary condensation on theradius of a pore in which condensation takesplace, which is given by the Kelvin equation:
lnðp=poÞ ¼ V
RT
2s cosu
rk
� �ð18Þ
where p/po ¼ pressure/saturation pressure, V isthe molar volume, s the surface tension of theliquid adsorbate, Q the contact angle betweenadsorbate and adsorption layer on pore walls(hence, Q ¼ 0 is a good approximation), andrK isKelvin radius of a pore assuming cylindricalshape. Since an adsorption layer is typicallyformed before capillary condensation occurs,the geometric radius rp of a pore is given by thesum of the Kelvin radius rK and the thickness ofthe adsorption layer t: rp ¼ rK þ t. Mesoporesize distributions can be calculatedwhen adsorp-tion and desorption isotherms are available in thefull pressure range up to p/po ¼ 0.95. Themesopore volume Vp is assumed to be complete-ly filled at this relative pressure, which corre-sponds to rp � 20 nm.
In themicropore range (pore width< 2 nm),where the pore dimensions are comparable tomolecular dimensions, pore filling occurs ratherthan condensation [343]. The Dubinin – Ra-dushkevich and the Dubinin – Stoeckli theoriesthen permit the estimation of pore dimensionsfrom physisorption data. In addition, severalempirical methods exist, such as the t-meth-
od [345] and the as-method [346]. In the origi-nal t-method the amount of nitrogen adsorbed at77 K was plotted against t, the correspondingmultilayer thickness calculated from a universalN2 isotherm, while in the as method the multi-layer thickness t is replaced by the reducedadsorption as. Here, as is defined as the dimen-sionless adsorption na=nax such that as ¼ 1 at p/po ¼ 0.4, and na is the adsorbed amount inmoles of the adsorbate (e.g., N2) at a givenrelative pressure and nax is the amount adsorbed(in moles) at a relative pressure of 0.4.
For meso- and macroporous materials (porewidth > 2 nm), the pore size distribution isdetermined by measuring the volume of mercu-ry (or another nonwetting liquid) forced into thepores under pressure [322]. The measurement,carried outwith amercury pressure porosimeter,depends on the following relation:
P ¼ 2ps cosa
rpð19Þ
where P is pressure, s is surface tension ofmercury, and a is the contact angle of mercurywith solid. At pressures of 0.1 – 200 MPa, poresize distributions in the range of 3.75 –7500 nm can be measured. Because the actualshape of the pores is not exactly cylindrical asassumed in the derivation of the above equation,the calculated pore sizes and distributions candeviate appreciably from the actual valuesshown by electron microscopy.
For pore systems with narrow pore size dis-tributions, the average pore radius can be ap-proximated by using
rp ¼ 2 Vp=Sp ð20Þwhere Vp is pore volume, and Sp is the surfacearea. The pore volume of a catalyst or support isgiven by
Vp ¼ 1=rp�1=r ð21Þwhere rp and r are the particle and true densi-ties, respectively. The former is determined by apycnometer using a nonpenetrating liquid, suchas mercury, whereas the true density is obtainedby measuring the volume of the solid part of aweighed sample by helium displacement.
In certain instances, pore dimensions can bedetermined by high-resolution electron micros-copy (HREM) [347].
Heterogeneous Catalysis and Solid Catalysts 53
6.1.2. Particle Size and Dispersion [348]
The surface area of active metals dispersed on asupport deserves particular consideration sincethe metal surface area and particle size (whichare interrelated quantities) determine the cata-lytic properties of supported metal catalysts.The metal dispersion D is given by D ¼ NS/NT,where NS is the number of metal atoms exposedat the surface andNT is the total number ofmetalatoms in a given amount of catalyst. The fractionof surface atoms D can be determined if NS isexperimentally available. It can be determinedby chemisorption measurements with adsorp-tives that strongly bind to the metal but whichinteract negligiblywith the support at the chosentemperatures and pressures. H2, CO, NO, andN2O have been used for this purpose at or aboveroom temperature [348], and static, dynamic,and desorption methods have been applied.Saturation values of the chemisorbed amountspermit NS to be calculated if the chemisorptionstoichiometries are known.
Dispersion is directly related to particle sizeand particle size distribution. Assuming reason-able model shapes for the metal particles, aver-age particle sizes can be calculated from thechemisorption data.
Average crystallite size distributions can bedetermined independently from X-ray diffrac-tion line broadening [348,350], and small-angleX-ray scattering (SAXS) permits the determi-nation of particle sizes and particle size distri-butions, but also of the specific surface area ofthe metal and of the support [348,350].
Electron microscopy offers the uniqueopportunity to observe catalyst morphologiesover the entire range of relevant particlesizes [347,348,351–353]. Particle shapes andsizes of the support or active phase and theirsize distributions can be extracted from micro-graphs, but structural information can be alsoobtained by electron-diffraction and lattice-imaging techniques [347].
6.1.3. Structure and Morphology
X-ray powder diffraction (see also !Structure Analysis by Diffraction – Diffractionby Polycrystalline Specimens) (XRD) is aroutine technique for the identification of
phases present in a catalyst [349,354]. It is basedon the comparison of the observed set of reflec-tions of the catalyst sample with those of purereference phases, or with a database (PowderDiffraction File (PDF) distributed by ICDD, theInternational Centre for Diffraction Data). XRDstudies can now be carried out in situ on theworking catalyst [354], and the use of synchro-tron radiation permits dynamic experiments inreal time [355]. Time-resolved studies on atimescale of seconds are now becoming possi-ble. Quantification of phase compositions canalso be performed.
More sophisticated analysis of the diffractionpatterns of crystalline materials provides de-tailed information on their atomic structure. TheRietveld method is used for structure refine-ments. Perhaps more importantly for catalyticmaterials, the local atomic arrangement ofamorphous catalysts is based on the Debyeequation, which gives the intensity scattered bya collection of randomly distributed atoms. TheFourier transform of the Debye equation givesthe radial distribution function (RDF) of elec-trons, from which the number of atoms (elec-trons) located in the volume between twospheres of radius r and r þ dr around a centralatom, i.e., the radial density of atoms, can beobtained [354]. This approach has been appliedfor the structural analysis of amorphous orpoorly crystalline catalyst materials and ofsmall metal particles.
X-ray Absorption Spectroscopy (XAS)[356,358], is the method of choice where theapplicability of XRD for structure analysesceases to be possible. Because of their highphoton flux, synchrotron facilities are the pre-ferredsources forXASexperiments.Thephysicalprinciple of XAS is the ejection of a photoelec-tron from a core level of an atomby absorption ofan X-ray photon. The position of the absorptionedge gives the binding energy of the electron inthe particular core level and is thus characteristicof the respective element and its chemical state(see also ! Surface and Thin-Film Analysis)and the shape of the absorption edge providesinformation on the distribution of the local den-sity of states (LDOS).
The ejected photoelectron wave is backscat-tered at neighboring atoms, and the scatteredwave interfereswith the outgoing primarywave.This interference results in a modulation of the
54 Heterogeneous Catalysis and Solid Catalysts
absorption coefficient at energies between 50and 1000 eV beyond the absorption edge(extended X-ray absorption fine structure,EXFAS).Analysis of these oscillations providesinformation on the chemical nature of atoms atwell-defined distances from the central (ion-ized) atom and gives coordination numbers.Qualitative information on coordination of thecentral atom may also be obtained from theobservation of pre-edge peaks. Information ondynamic and static disorder can also beextracted from the EXAFS. Hence, a detailedmicroscopic picture of the structure of a catalystcan be derived.XAS is particularly attractive forstudies of catalysts under working conditions,although there are limitations regarding temper-ature [356]. The combined application of XASandXRD on the same sample using synchrotronradiation for in situ studies is an ideal tool incatalysis research [359].
Electron Microscopy and Diffraction[347,351–353], (see also ! Microscopy–Electron Microscopy). When electrons pene-trate through matter in an electron microscope,contrast is formed by differential absorption(amplitude contrast) or by diffraction phenom-ena (phase contrast). Electron micrographs ofcatalyst materials can provide for identificationof phases, images of surfaces and theirmorphol-ogies, and elemental compositions and distri-butions. Image interpretations are often notstraightforward and need expert analysis. Sev-eral variants of electron microscopy use differ-ent electron optics and working principles andtherefore have to be chosen according to theproblem to be solved.
Conventional transmission electron micros-copy (CTEM) operates in the 100 – 200 kVrange of electron energies, and imaging is basedon amplitude contrast in the bright-field mode.Point resolutions of 0.2 – 0.3 nm can beachieved in favorable cases. A typical applica-tion of CTEM in catalysis research is theexamination of metal particle sizes and theirdistributions in supported catalysts.
Dark-field images are produced when thedirectly transmitted electron beam is excludedby the objective aperture, and only diffractedelectrons are used for imaging. This mode ofoperation selectively detects crystallites withcrystallographic spacingswithin a narrow range.
High-resolutionelectronmicroscopy (HREM)can be performed in CTEM instruments bymod-ifying the mode of imaging, or in dedicatedinstruments operating at electron energies of0.5 – 1.0 MeV. HREM images can be directlyrelated to the atomic structure of the materi-al [360]. Lattice fringes can be resolved, and thedetermination of the spacings of atomic planes isenabled. Support particles can thus be identified,and the crystal structure of heavy metal particleshaving sizes in the range down to 1 nm can beinvestigated.
In dedicated scanning transmission electronmicroscopes (STEM) an annular detector pro-vides the image formed from diffracted beams,while the central transmitted beam can be fur-ther analyzed by using an electron spectrometerto simultaneously provide elemental analysis.The intensity distribution of electrons scatteredat high angles (40 – 150 mrad) depends on thesquare of the atomic number Z according to theRutherford scattering cross section. The STEMimages are therefore also called Z-contrastimages, and they are particularly useful for thestudy of catalysts containing small metalparticles [347].
In scanning electron microscopy (SEM) theimage is produced by scanning a finely focusedprobe beam in a raster pattern across the speci-men surface. Emitted signals such as backscat-tered and secondary electrons are detected andused for image formation. Secondary electronsare most commonly used. The best resolutionsthat can be achieved with current generationSEM instruments are approximately 1 nm.SEM is most useful for studying sample topo-graphies, and it can be applied with a significantbackground pressure of a reactive gas while thesample is observed (environmental SEM orESEM).
Selected area electron diffraction (SAED)provides information on phase compositionsand structures at a microscopic level. The com-bination ofmicrodiffraction patterns and bright-field images enables the determination ofshapes and exposed facets in dispersed phasesin solid catalysts.
Analytical electron microscopy (AEM) per-mits the determination of the elemental compo-sition of a solid catalyst at the microscopic levelby energy dispersive detection of the electron-induced X-ray emission. Energy dispersive
Heterogeneous Catalysis and Solid Catalysts 55
spectroscopy (EDS) is sensitive for elementswith atomic numbers Z > 11. For lighter ele-ments (Z < 11), electron energy loss spectros-copy EELS is applied.
Controlled atmosphere electron microscopy(CAEM) [347,361] is arousing considerableinterest as it will permit the observation ofchanges in the catalyst structure and morpho-logy under reaction conditions.
Vibrational Spectroscopy [362] (! In-frared and Raman Spectroscopy). Vibrationalspectroscopy is one of the most promising andmost widely used methods for catalyst charac-terization, since it provides detailed structuralinformation on the solid catalyst material and onsurface groups and adsorbates. Several vibra-tional spectroscopic methods can be applied insitu, and they can be successfully used for studieson ill-defined high surface area porousmaterials.In situations where X-ray diffraction techniquesare not applicable, vibrational spectroscopy canoften provide information on phase transitionsand changes in compositions of bulk catalystmaterials, on their crystallinity, and on the natureof surface functional groups. Most vibrationalspectroscopic methods are not surface-sensitive,but they become surface-sensitive when vibra-tional spectra are recorded for groups oradsorbates that are present exclusively at thematerial’s surface. Representative examples forthe structural characterization of solid catalystsby vibrational spectroscopy are bulk oxides (in-cluding simple binary oxides, multicomponentmaterials such as oxidation catalysts, and zeo-lites andmolecular sieves), and supported oxides(e.g., monolayer-type catalysts), and sulfides.The vibrational analysis of surface groups, par-ticularly of hydroxyl groups, can also be ad-dressed. In many cases surface hydroxyl groups(e.g., on oxides) are simply formed by dissocia-tive chemisorption of water molecules, whichreduces the surface free energy.Hydroxyl groupscan also be constituents of the solid-state struc-ture, for example as in zeolites.
There are several methods and techniques ofvibrational spectroscopy which are particularlysuitable in catalysis research. Infrared transmis-sion – absorption spectroscopy is the mostcommonly used technique. The KBr disk tech-nique is routine for transmission spectroscopyof powder samples. However, for in situ inves-
tigations pressed self-supporting wafers have tobe used. Samples which exhibit only weak bulkabsorption, and the average particle size d ofwhich is smaller than the wavelength of theinfrared radiation (d < l) are optimally suitedfor the transmission mode. Transmission – ab-sorption infrared spectroscopy has been partic-ularly successful in elucidating the structure ofhydroxyl groups [174,362]. More strongly ab-sorbing materials, and particularly those havingaverage particle sizes greater than the wave-length of the infrared radiation, which thereforecause significant scattering losses in transmis-sion, may preferentially be studied by diffusereflectance spectroscopy (DRS, DRIFT). Apowerful technique for structural studies oncatalytic materials under extreme temperatureand pressure conditions is laser Raman spec-troscopy (LRS), although laser heating andlaser-induced fluorescence may cause seriousproblems. One way, among others [362], toavoid fluorescence is to use of UV light insteadof visible radiation for spectral excitation [363].LRS has been successfully applied for the struc-tural characterization of complex oxides, zeo-lites, and supported oxides and sulfides [362].Surface-enhanced Raman spectroscopy (SERS)has found some application in studies of finelydividedmetal catalysts, particularly silver [364].Second harmonic generation (SHG) and sumfrequency generation (SFG) [365,366] are non-linear optical techniques with high surface sen-sitivity which will probably find increasingapplication in studies relevant to catalysis.
Neutron techniques [367] include neu-tron diffraction and inelastic neutron scattering(INS). Both techniques are particularly sensitiveto light elements (such as H or D) and providecomplimentary structural information to XRD.
6.1.4. Local Environment of Elements
Nuclear spectroscopic methods provide infor-mation on the local environment of severalselected elements.
M€ossbauer spectroscopy and time differen-tial perturbed angular correlation
(TDPAC) belong to the class of techniqueswhich detect solid-state properties mediated by
56 Heterogeneous Catalysis and Solid Catalysts
hyperfine interactions via nuclear spectrosco-py [368]. Both techniques are g spectroscopies;they are bulk techniques and can be appliedunder in situ conditions, although M€ossbauerspectroscopy requires low temperatures.
M€ossbauer spectroscopy (M€ossbauer Spec-troscopy) [368,369] provides information onoxidation states, phases, lattice symmetry, andlattice vibrations. Its application is limited toelements which exhibit the M€ossbauer effect,such as iron, cobalt, tin, iridium, ruthenium,antimony, and platinum. Particularly valuableinformation on catalyst structures has been ob-tained for iron catalysts for Fischer-Tropsch andammonia synthesis, and for cobalt-molybde-num hydrodesulfurization catalysts.
The time differential observation of the per-turbed angular correlation ofg rays emitted fromradioactive nuclei (TDPAC) [368,370,371] is ag-spectroscopic techniquewhich also allows thedetermination of hyperfine interactions such asnuclear electric quadrupole interactions (NQI).The NQI parameters enable local structural in-formation around the g emitter to be extracted.The technique has been successfully applied instudies on molybdenum-containing catalysts,and its application to tungsten seems promising.
Solid State NuclearMagnetic Resonance[372–374] (see also ! Nuclear MagneticResonance and Electron Spin Resonance Spec-troscopy – NMR of solids and HeterogeneousSystems). NMR spectroscopy in heterogeneouscatalysis principally allows the characterizationof the chemical and structural environment ofatoms in the catalysts (or in species adsorbed oncatalyst surfaces). NMR studies on catalysts canbe carried out over a wide range of temperaturesand pressures, as well as in the presence of gasesand liquids. Information can therefore be derivedabout the structures of catalysts and theirthermal or chemical transformations. In addi-tion, specific adsorbent – adsorbate interac-tions, the nature of chemically bonded surfacespecies, and chemical reactions occurring at thecatalyst surface can be studied.Most elements ofinterest in catalysis have isotopes that can bestudied with modern NMR spectrometers. Iso-tope enrichments may be desirable or even nec-essary for certain elements, for example, 17O.
NMR spectra of solids are often complexsince structure-dependent interactions such as
dipolar interactions, chemical shift interactions,quadrupolar interactions (for nuclei with spinI > 1/2) contribute strongly to the shape andposition of NMR lines. Because of their struc-ture-dependence these interactions are the mainsource of information on the structural environ-ment of the nucleus in question. The selectivedetermination of the related interaction para-meters of structurally inequivalent nuclei is themajor goal of an NMR experiment. In well-crystallized samples, the interaction parametersadopt unique values, while in poorly crystal-lized or amorphous powders they must bedescribed by distribution functions.
The anisotropy of the above-mentioned inter-actions results in line broadening, and the spectraof polycrystalline samples consist of a broadsuperposition of signals arising from differentorientations of the crystallites relative to thedirection of the external magnetic field Bo,weighted by the statistical probability withwhich each orientation occurs (powder patterns).Special techniques have been developed whichremove or at least reduce substantially these line-broadening effects and permit highly resolvedNMR spectra of powders with individual linesfor inequivalent nuclei to be recorded. The mostimportant of these techniques are dipolar decou-pling, magic-angle spinning (MAS), and doubleoriented rotation (DOR). Cross-polarization(CP) improves the sensitivity for nuclei withlow natural abundance and allows the spatialproximity of nuclei to be monitored.
Typical examples for structural characteriza-tions by solid-state NMR [372] are studies onzeolites using 27Al and 29Si NMR. Informationon the distribution ofAl in the environment of Siatoms and on the possible presence of nonfra-mework Al species has been obtained. Thelocation of exchangeable alkali metal ions hasbeen studied by 23Na and 133Cs NMR. Vanadi-um- and molybdenum-based catalysts have suc-cessfully been characterized by 51V and 95MoNMR.
6.2. Chemical Properties
6.2.1. Surface Chemical Composition
The atomic composition of a catalyst surfaceplays a decisive role for the catalytic properties.
Heterogeneous Catalysis and Solid Catalysts 57
Electron and ion spectroscopies [375] are sur-face-sensitive analytical tools which provideinformation on the atomic composition withinthe topmost atomic layers. The informationdepth, i.e., the number of atomic layers contrib-uting to the measured signal, depends on themethod. Concentration profiles can be obtainedby sputter etching of the surface by ion bom-bardment. The application of these particlespectroscopies requires ultrahigh-vacuum(UHV) conditions.
The basis for the identification of atoms onsurfaces of solid materials by electron spectro-scopies, such as Auger electron spectroscopy(AES) and X-ray photoelectron spectroscopy(XPS) are the electronic binding energies. Withion spectroscopies, such as low-energy ion scat-tering (LEIS) and Rutherford backscattering(RDS), surface atoms are identified by theirnuclear masses. Ion bombardment of surfacesis accompanied by sputtering processes (surfaceetching) which lead to the removal of secondaryionic and neutral particles. These are analyzedby mass spectroscopic techniques, such as sec-ondary ion mass spectroscopy (SIMS), andsecondary neutral mass spectroscopy (SNMS).Less frequently used is laser microprobe massanalysis (LAMMA). Relevant information onthe properties of the various surface analyticaltechniques is summarized in Table 11.
The physical principles of the various tech-niques have been discussed in several articlesand monographs [375,376].
Electron Spectroscopy (AES, XPS) (seealso ! Surface and Thin-film Analysis –Auger Electron Spectroscopy (AES)). Thesetechniques use electrons as information carriers.The electrons can be produced by the absorptionof photons resulting in photoemission. In XPS,X-ray photons are used to ionize core levels, and
the kinetic energy Ek of the emitted photoelec-trons is measured. The energy balance is givenby:
Ek ¼ hn�Eb�F ð22ÞThis equation permits the electron binding
energy Eb (relative to the Fermi level) to bemeasured when the photon energy hn and thework functionF of the spectrometer are known.The binding energies are characteristic for aparticular element.
As a result of the photoionization a singlyionized atom is formed, which can also beproduced by electron impact. The core hole(e.g., in the K shell) can be filled by an electronfrom a higher shell (e.g., the L1 shell) and theenergy of this de-excitation process can bereleased by emission of an X-ray photon (X-rayfluorescence, XRF) or can be transferred toanother electron (e.g., in the L2 shell) which isthen emitted with a well-defined kinetic energy(Auger process). This kinetic energy is deter-mined by the orbital energiesEK,EL1, andEL2 ofthe three orbitals involved. The Auger energyEKLL is then given by:
EKLL ¼ EK�EL1�EL3�dE�F ð23Þwhere dE is a relaxation energy, and F thespectrometer work function. Clearly, EKLL ischaracteristic for an element and independent ofthe initial ionization process. Thus, both tech-niques permit the elemental constituents of asurface to be identified.
The information depth of both electron spec-troscopies is determined by the mean free pathof the emitted electrons, which depends on thekinetic energy of the electron in the solidmatrix.This dependence is known [375–377]. The elec-tron mean free path is typically larger in oxidesthan in metals at equal energy, and it is particu-larly large for zeolites because of their low
Table 11. Characteristics of surface analytical techniques in standard applications (adapted from ref. [375])
*Increasing number of positive signs indicates better capabilities; parentheses indication of restriction to special conditions.
58 Heterogeneous Catalysis and Solid Catalysts
density. Together with reported ionization crosssections and, in the case of AES, Auger decayprobabilities, quantitative surface analysis ispossible. The ratios of integral peak areas areproportional to concentration ratios. These canbe analyzed as a function of preparation andtreatment conditions of a given catalyst system(e.g., supported metal, oxide, or sulfide cata-lysts) and compared with model calcula-tions [376]. Information on the elementaldistributions and on dispersions of active com-ponents thus becomes available.
Ion-scattering Spectroscopies [375] (seealso ! Surface and Thin-Film Analysis). Inion-scattering spectroscopies solid surfaces arebombarded with monoenergetic ions, which arescattered on the top atomic layer (ion energies ofabout 0.5 – 5 keV, low-energy ion scattering(LEIS) [378,379]) or within near-surface re-gions (ion energies of about 0.1 – 23 MeV,Rutherford backscattering (RBS) [380,381]). Inboth cases the collision kinematics can bedescribed as simple binary collisions, so thatthe kinetic energy of the backscattered ion isdirectly dependent on the ratio of the masses ofthe projectile and the scattering target atom andon the scattering angle. The mass of the projec-tile is known and the scattering angle is fixedand determined by the geometry of the spec-trometer. Thus, the mass, and hence the identity,of the scattering target atoms can be determinedunequivocally.
The LEIS technique provides information onthe nature of the atomic constituents of thetopmost atomic layers. Quantitative analysis,however, is difficult since neutralization proba-bility, which makes the technique surface sen-sitive, is not easily available. Only a few percentof the primary ions are backscattered as ions inthe case of noble gas ion (e.g., Heþ). Thetechnique can be applied for the characteriza-tion of real catalyst surfaces, although surfaceroughness reduces the signal intensity.
In contrast, in the energy regime of RBS thescattering cross sections can be calculated ex-actly. As a consequence, quantitative analysis ispossible by RBS, but the surface sensitivity islower than for LEIS. In optimal cases an infor-mation depth of 1 – 5 nm can be achieved. Acombined application of LEIS, RBS, andperhaps XPS is often most informative [375].
Secondary Particles Ion bombardment ofa surface leads to ion etching with the release ofatoms and molecular fragments with varyingcharges (anions, cations, and neutrals) and exci-tation states.Themass analysis of secondary ionsbymass spectrometry [secondary ionmass spec-troscopy (SIMS)] has been developed as a highlysensitive andpowerful surface analyticalmethod(! Surface and Thin-Film Analysis – Second-ary ionMass Spectrometry) [382,383].Althoughdestructive because of the need for sputtering,the sputtering rate can be kept low in theso-called static mode (low primary-ion currentdensity) so that the surface remains essentiallyunchanged. Since the sputtered particles arepreferentially released from the first two atomiclayers, the SIMS technique is surface-sensitive.In contrast to the ion-scattering techniques, notonly atomic constituents of a surface can bedetected but information on the local environ-ment of an atom in the surface can be obtained byanalysis of molecular fragments. The detectionof light elements, particularly hydrogen, is alsopossible. Quantification of the method is diffi-cult, although not entirely impossible.
A high percentage of the sputtered secondaryparticles are neutral and must be postionized formass spectroscopic analysis [secondary neutralmass spectroscopy (SNMS), ! Surface andThin-Film Analysis – Secondary Natural MassSpectrometry (SNMS)] [384,173]. Post-ioniza-tion can be achieved by electron impact in aplasma or by an electron beam. Alternatively,resonant and nonresonant laser ionization canbe applied. Applications of SNMS for catalystcharacterization have still not been reported.
6.2.2. Valence States and RedoxProperties
Electron Spectroscopies X-ray photoelec-tron spectroscopy (XPS) and Auger electronspectroscopy (AES), in addition to elementalanalysis, permit information to be obtained onthe valence and bonding states of a given ele-ment. This is due to the fact that the core-levelbinding energies and Auger kinetic energies aredependent on the chemical state, which leads tocharacteristic chemical shifts. In solids, theMadelung potential also plays an importantrole [376,385,386]. In addition, the ionization
Heterogeneous Catalysis and Solid Catalysts 59
of an atom leads to relaxation phenomenawhichprovide a relaxation energy that is carried on bythe emitted photoelectron. The binding energyof a core electron in level C of an atom is givenby:
EbðCÞ ¼ const:þXj
ðqj=RjÞþkq�Rea ð24Þ
where qj are the charges of all other atoms and Rj
their distances from the core-ionized atom. Theconstant is the Hartree – Fock energy of the coreelectron in the atomic level C for the free atom, kis the change of the core potential resulting fromthe removal of an electron, andRea represents theextra-atomic polarization energy.
For an Auger transition involving the atomiclevels C, C0, and C00, the kinetic energy Ek(C, C
0,C00) of the Auger electron is related to thephotoelectron binding energy Eb(C) in goodapproximation by:
a0 ¼ Eb ðCÞþEk ðC;C0 :C00Þ ¼ const:þRea
ð25Þwhere a0 is the so-called Auger parameter.
Chemical shifts and the Auger parameterprovide detailed information on the chemicalstate of an element as regards its oxidation stateand its local environment. The latter is reflectedin the Auger parameter, which is dependent onthe extra-atomic polarization energy and hence,on the structural and bonding characteristics ofthe atom under consideration. So-calledWagnerplots [386,387] in which the XP binding energy,the Auger energy, and the Auger parameter arecorrelated for families of compounds often per-mit the analysis of a compound of unknownstructural and bonding characteristics.
Optical Spectroscopy and Electron Para-magnetic Resonance Optical excitations
in the UV, VIS, and NIR regions and electronparamagnetic resonance (EPR) are classicaltechniques which provide information on theelectron configuration (oxidation state) of ametal center and on the symmetry of the ligandsphere [388–391].While optical spectroscopy isapplicable to practically all systems, EPR islimited to paramagnetic species, i.e. thosewhichcontain one or more unpaired electrons.
UV – VIS – NIR spectroscopy covers a widerange of energies (typically 0.5 – 6 eVor 4000
to 50 000 cm�1 or wavelengths (2500 to200 nm) as shown in Figure 18. Several typesof transitions occur in this range, namely,charge-transfer (CT) and d – d transitions, asalso indicated in Figure 18. The first class ofexcitations involves two adjacent atoms, one ofwhich is typically a metal center and the other aligand or another metal atom. Electromagneticradiation can promote charge transfer from theligand (L) to the metal (M), from the metal (M)to the ligand (L) or from one metal center toanother. These transitions are therefore calledligand-to-metal CT (LMCT), metal-to-ligandCT (MLCT), and metal-to-metal CT (MMCT),respectively. Such transitions occur in molecu-lar complexes and in nonmolecular solids, suchas metal oxides. The energy of CT transitionsdepends on the symmetry and oxidation state ofthe metal center and on the nature of the ligandor of the second metal atom [392]. Hence,information on these properties can be extractedfrom CT spectra. These spectra are relativelyintense since they are dipole-allowed. In con-trast, metal-centered or intra-atomic transitionsin transition metal atoms or ions (ligand-field ord – d transitions) are of moderate or weak in-tensity because they are forbidden by the La-porte (orbital) selection rule (and also by thespin selection rule) unless the selection rules arerelaxed by vibronic or spin – orbit coupling.The d – d transitions also provide informationon the electron configuration and on the sym-metry of a complex (or local environment in a
Figure 18. Energy ranges of different types of electronictransitions (adopted from [388])
60 Heterogeneous Catalysis and Solid Catalysts
solid). Typical systems that have been studiedby optical spectroscopy are transition metal andbase metal oxides, transition metal ions or com-plexes grafted on support surfaces, and transi-tion metal ion-exchanged zeolites.
While spectra of liquid samples can be re-corded in the transmission mode, catalyst pow-ders must be studied by the diffuse reflectancetechnique (DRS) [389,390,393]. The measureddiffuse reflectance can be converted into the so-called Schuster – Kubelka – Munk (SKM)function, which is directly proportional to theabsorption coefficient. The wavelength depen-dence of the SKM function is thus equivalent toan absorption spectrum, provided the scatteringcoefficient is independent of the wavelength.This condition is often fulfilled in the UV andVIS spectral regions. Simple quartz cells for insitu treatments can be designed, to which anEPR tube can also be connected for simulta-neous optical spectroscopy and EPR on thesame sample [388].
Luminescence spectroscopy [394,395] hasproved to be a valuable addition to the spectros-copy techniques for characterization of solidcatalysts under well-defined conditions.
As mentioned above, electron paramagneticresonance (EPR) is used to study paramagneticspecies in catalytic materials. Besides the sim-ple qualitative (and quantitative) detection ofthe presence of paramagnetic sites, the spindensity distribution at the paramagnetic centerand on the neighboring atoms can be deducedfrom the spectra. Simulations of EPR spectra areoften useful for full interpretation. The extreme-ly high sensitivity of the EPR technique canbe an advantage but also a drawback because theimportance of minority radical species may beoveremphasized. EPR is not surface-sensitive.However, radical species in the surface caneasily be identified by exposing the sample toparamagnetic O2, which leads to significantbroadening or disappearance of signals ofsurface species because of dipole – dipoleinteractions.
Several reviews on the use of EPR incatalyst characterization have been published[388,396,397]. Typical applications of EPR arethe detection of paramagnetic states of transi-tion metal ions and analysis of the symmetry oftheir ligand sphere and/or their coordination,redox properties of catalytic materials and their
surfaces, and surface anion or cation radicalsdeliberately produced by organic molecules asprobes for the redox properties of the solidcatalyst. Radical species (e.g., in connectionwith coke formation) formed during catalyticreactions have also been detected.
ThermalAnalysis Thermoanalytical tech-niques such as differential thermal analysis(DTA), thermogravimetry (TG), and differentialscanning calorimetry (DSC) are well-estab-lished methods (! Thermal Analysis andCalorimetry) in solid-state chemistry [398,399]which have successfully been applied to inves-tigating the genesis of solid catalytic materials.They can also be used to follow reduction andoxidation processes by measuring either ther-mal effects and/or weight changes. When com-bined with an on-line mass spectrometer,changes in the gas-phase composition occurringduring chemical transformations of the solidsample can be monitored simultaneously.
In temperature-programmed reduction(TPR), as first described by ROBERTSON etal. [400], a streamof inert gas (N2 orAr) contain-ing ca. 5 vol% H2 is passed through the catalystbed of a flow reactor containing a reducible solidcatalyst [401]. By monitoring continuously theH2 concentration in the gas stream and its even-tual consumption with a thermal conductivitydetector while heating the sample with a lineartemperature ramp of ca. 10 K/min, the rates ofreduction are obtained as a function of time (ortemperature). The total amount of H2 consumeddetermines the reduction equivalents present inthe catalyst, and detailed analysis of the experi-ment permits the kinetic parameters of the re-duction process to be determined and providesinformation on reduction mechanisms. Charac-teristic numbers which depend on the experi-mental parameters (amount of reducible speciespresent, H2 concentration, flow rate, and tem-perature ramp) have been defined [328,329].These numbers must be kept in certain rangesfor optimal performance of the experiment.
TPR experiments have been used to investi-gate the reduction behavior of bulk andsupported reducible species, solid solutions,promoted metal catalysts, metals in zeolites,and of supported sulfides and of nitrides [401].
Temperature-programmed oxidation (TPO)is an equally valuable technique for investigat-
Heterogeneous Catalysis and Solid Catalysts 61
ing the oxidation kinetics and mechanisms ofreduced materials [401]. Cyclic application ofTPR and TPO provides information on theredox behavior of catalytic materials, e.g., ofcatalysts for selective catalytic oxidations.
6.2.3. Acidity and Basicity
Acid-catalyzed reactions are among the indus-trially most important hydrocarbon conver-sions. Acid sites can be classified as Lewisacidic sites, such as coordinatively unsaturatedcations (e.g., Al3þ on the surface of partiallydehydroxylated alumina, and Brønsted acidicsites, which are typically surface OH groups as,e.g., in H forms of zeolites. Carbenium andcarbonium ions are thought to be formed byprotonation of hydrocarbons on these groups.Surface oxygen ions may function as Lewisbasic centers, and if strong enough they mayabstract protons from hydrocarbonmolecules toform carbanion intermediates. A typical solidbase is MgO.
For characterization of acid and base prop-erties, the nature (Lewis or Brønsted) of thesites, the acid or base strength, and the numberof sites per unit surface area of a solid catalystmust be determined. Brønsted acidity is almostcertainly required for all acid-catalyzed reac-tions. However, the mechanistic details on sur-faces are significantly different from the well-known carbenium and carbonium ion chemistryin solution, because of the lack of the stabilizingeffect of solvation in heterogeneously catalyzedgas-phase reactions. As shown by KAZANS-
KY [404], the electronic ground state of surfaceacidic OH groups of oxides and in H forms ofzeolites is essentially covalent. The main differ-ences in their acid strength are thought to be dueto the energetic positions of their electronicallyexcited heterolytic terms. Similarly, the interac-tion of acid groups with alkenes does not resultin the formation of adsorbed carbenium ions butrather in the formation of more stable covalentalkoxides. Basic sites (surface O2� ions) in thevicinity of OH groups could be involved in thisprocess. Carbenium ions (and even more socarbonium ions) are therefore not consideredto be reaction intermediates in solid acid catal-ysis, but rather excited unstable ion pairs ortransition states resulting from electronic
excitation of covalent surface alkoxy species.Because of the proposed bifunctional nature ofactive acid sites in heterogeneous acid catalysis,it is necessary to characterize both the acidic andbasic properties of solid catalysts. Many differ-ent methods have been developed for the char-acterization of acidity, but only little is knownabout the basic character, particularly of mate-rials that are typically considered to be acidcatalysts.
Chemical Characterization [405]. Titra-tion methods in aqueous medium are not veryinformative, becauseH2O tends to stronglymod-ify surface properties by molecular or dissocia-tive chemisorption. Therefore, nonaqueousmethods have been proposed, in which the sol-vent (e.g., benzene or isooctane) does not or onlyweakly interact with the catalyst surface. Ham-mett indicators were used to determine the acidstrength in terms of the Hammett – Deyrup H0
function:
H0 ¼ �log aHþðfB=fBHþÞ ð26Þwhere aþH is the proton activity and fB and fBH
þ
are the activity coefficients of the basic probeand its protonated form, respectively. A series ofHammett indicators covers the range of �18 <H0 < 4, where H0 ¼ �12 corresponds to 100%H2SO4.
Site densities and acid strength distributionswere determined by the n-butylamine titrationmethod [406]. As these titration and indicatormethods can yield erroneous results [407], theyare not frequently used today.
Isosteric heats of adsorption of strong bases(e.g., pyridine) may be considered as measuresof acid strength. However, a discriminationbetween Lewis and Brønsted sites is notpossible.
Temperature-programmed desorption [alsocalled thermal desorption spectroscopy (TDS)]of basic probe molecules has been developed asa powerful tool for the characterization of solidacids [408,409]. In this method, a strong base isisothermally pre-adsorbed on an acidic catalystand then exposed to a stream of inert gas (e.g.,He). Heating by a temperature ramp (ca. 10 K/min) leads to desorption of the base. The inte-gral area of the desorption peak gives the totalacid site density, and the position of the peakmaximum provides the activation energy of
62 Heterogeneous Catalysis and Solid Catalysts
desorption (which may be close or identical tothe heat of adsorption), which can be consideredto be a measure of the acid strength. Thisapproach has been applied for investigations ofH forms of zeolites using ammonia as theprobe [410,411]. However, discriminationbetween Lewis and Brønsted acid sites is againonly possible with the assistance of, e.g.,vibrational spectroscopy.
Microcalorimetry [408].Differential heatsof adsorption of probe molecules can be mea-sured with high accuracy by heat-flow calorim-etry and differential scanning calorimetry. Thesedata provide information on the acid (or base)strength distribution. Ammonia and otheramines have been used as probes for acid siteson oxides [412] and in H forms of zeo-lites [413,414], and carbon dioxide and sulfurdioxide were adsorbed as acidic probes on sev-eral oxides [412].
Vibrational spectroscopy [415,420].Transmission infrared spectroscopy is the mostfrequently applied technique for investigationsinto acidic and basic properties of solid cata-lysts. Surface hydroxyl groups can easily bedetected since they function as dipolar oscilla-tors. However, the stretching frequency of un-perturbed OH groups can not be taken as ameasure of the acid strength. Lewis acidic andbasic centers can only be detected by vibrationalfrequencies with the adsorption of suitableprobe molecules. Criteria for the selection ofoptimal probe molecules have been defined byKNOEZINGER et al. [415,420].
The use of basic probe molecules permits adiscrimination between Brønsted and Lewisacid sites. When a base B is adsorbed on anacidic OH group, hydrogen bonding followedeventually by protonation of the base may occur(Eq. 20):
OHþBÐ OH���BÐ O����HþB ð27ÞThe strength of the hydrogen bond and the
ability of the OH group to protonate the base isdetermined by the acid strength of the surfaceOH group and by the base strength (or protonaffinity) of B. When hydrogen bonding occurs,the induced frequency shift of the O – Hstretching mode Dn‘OH is a measure of thestrength of the hydrogen bond DHB
(Eq. 28) [421] and hence, of the acidity of theOH group.
jDnOHj1=2 � DHB ð28ÞSimultaneously, internal molecular modes of
the base B are modified, particularly whenprotonation occurs. These changes can also beused for the interpretation of the bonding type ofthe probe. Strong bases such as the traditionalprobe molecules ammonia and pyridine areprotonated by even very weak Brønsted siteswhichmay not be at all relevant in acid catalysis.Weaker bases such as nitriles, carbonmonoxide,and even dinitrogen and dihydrogen only un-dergo hydrogen bonding (hydrogen bondingmethod [416]), but due to their weak interac-tions they are very specific and can provide verydetailed information on the properties of acidicsurfaces.
The same bases can be used for the detectionof Lewis acid sites L, with which they formsurface coordination compounds:
LþBÐ L B ð29ÞThe frequency shifts of the internal B modes
are a measure of the nature and strength of thecoordinative bond. For example, carbon mon-oxide when coordinated to L sites undergoesvery typical shifts of the C – O stretching fre-quencywhich provide information on the natureof the element and of the coordinative bond, onthe oxidation state, and on the coordination ofthe L site [415,420].
The investigation of basic sites O2� by acidicprobe molecules AH with analysis of the vibra-tional spectra is much less advanced than that ofacid sites. The hydrogen-bond method can inprinciple be applied:
O2�þH�AÐ O2����H�A ð30ÞHere the shift of theH–A stretchingmode is a
measure of the hydrogen-bond strength andhence, the basic character (proton affinity) ofthe surface O2� site. Recently, CH compoundssuch as trichloromethane [416], acetylene andsubstituted acetylenes [420,422] and evenmeth-ane [423] were proposed and successfully testedas acidic probe molecules. Pyrrole [419] andseveral Lewis acids [415] have also been used.
Surface chemical transformations of, e.g.,CO2, alcohols, ketones, acetonitrile, and
Heterogeneous Catalysis and Solid Catalysts 63
pyridine gave detailed information on the bi-functional acid – base pair character of severaloxides, particularly of alumina [415,424].
Nuclear Magnetic Resonance [425,427].Solid-state 1H magic-angle spinning (MAS)NMR spectroscopy measures proton chemicalshifts, which were thought to reflect the depro-tonation energies of surface OH groups. How-ever, proton chemical shifts are also very sensi-tive to hydrogen bonding. Therefore, changes inproton chemical shifts induced by hydrogenbonding of probe molecules can also be usedfor the characterization of protic acidity.
Dissociative chemisorption of CH3I was pro-posed for characterization of surface basicity by13C NMR spectroscopy [428].
6.3. Mechanical Properties [429]
Catalyst particles are exposed to diverse me-chanical strains during transportation, chargingto the reactor, and operation. In fixed bed re-actors catalyst particles must withstand pressurecaused by the mass of the catalyst charge anderosion by high-velocity gas streams. In fluid-ized- and moving-bed reactors, the particlesmust resist attrition from rubbing against eachother and from colliding with the walls of thereactor system. The technical performance ofcatalysts depends on their mechanical strengthto maintain integrity for a reasonable time inspite of these strains.
There are three types of methods for deter-mining the strength of catalysts used under staticconditions [430]. For pellets and rings with noareas of distortion (preferably sized 1 cm orlarger), the crushed (or crush) strength is deter-mined by exerting pressure on the specimenplaced between two horizontal plates of a hy-draulic press. The upper plate moves down untilthe specimen is crushed, at which point thepressure is recorded. The test is repeated forseveral particles, and the values are averaged. Inthe knife-edge hardness test, the upper plate ofthe press is replaced by a knife with a 0.3 mmedge. A mass of 1 kg is applied to the knife andthe percentage of broken samples is recorded.The mass is then raised in increments of 1 kg,and the test is repeated until 100% of theparticles are broken or until a mass of 10 kg is
reached. Catalyst particles of irregular form aretested in a cylinder provided with a ram. After adefinite pressure is applied, the sample is dis-charged, and the weight percent of fines formedduring the test is determined by screening.
Tests for impact strength and resistanceagainst abrasion or attrition are carried outunder dynamic conditions. For impact testingof very strong catalysts (e.g., ammonia synthe-sis catalysts), amass of 500 – 1000 g is droppedon the particle from a standard height and thepercentage of unbroken, split, and broken sam-ples out of 20 ormore is recorded.Abrasion testson tableted and extruded catalysts are carriedout in a rotating horizontal steel cylinder pro-vided with one baffle. The percent of fines(based on themass of the catalyst tested) formedafter 1 h is reported as attrition loss. The attritionloss of fluid catalysts is measured by exposingthe catalyst particles to a high-velocity airstream in a glass pipe. The fines formed duringthe test (prevented from escaping by a filter) arereported as attrition loss expressed as the per-centage of the sample charged [431].
6.4. Characterization of SolidCatalysts under Working Conditions
The best description of a catalytic mechanism isthe corresponding catalytic cycle. As a first step,a detailed product analysis is required to differ-entiate between a single clean reaction andsystems undergoing parallel and/or consecutivereactions. The microkinetic approach, as out-lined in Section Kinetics of HeterogeneousCatalytic Reactions, for the prediction of theoverall rate of a catalytic reaction taking intoaccount the surface chemistry of the catalyst andthe elementary reactions involved, is the mostpromising procedure to predict a mechanism.However, the results of microkinetic analysesmay not always be unequivocal, and discrimi-nation between different kineticmodelsmay notbe straightforward. Therefore, additional infor-mation is necessary to prove or disprove thesequence of elementary steps (catalytic cycle)that represents the mechanism of a catalyzedreaction at the molecular level.
Quantum chemical calculations at variouslevels of sophistication and computer modelingprocedures now permit structures of adsorbed
64 Heterogeneous Catalysis and Solid Catalysts
intermediates and transition states to beelucidated and reaction energy diagrams to becomputed (see Section Molecular Modeling inHeterogeneous Catalysis).
6.4.1. Temporal Analysis of Products(TAP Reactor)
Transient kinetics measurements can also pro-vide quantitative values of kinetic parametersand elucidate individual reaction steps[432,433]. Pulse reactors are one type of tran-sient reactors. A valuable laboratory pulsereactor (transient operation) is the TAP reactor(TAP ¼ temporal analysis of products). Pulsescontaining small amounts of reactants (1013 –1017 molecules per pulse) are injected into theevacuated reactor containing the catalyst bed.The reactant/product molecules leaving thereactor (response signal) are analyzed by massspectroscopy with a time resolution of less than100 ms. This approach permits surface pro-cesses on solid catalysts such as adsorption,reaction, and desorption to be studied, andreaction mechanisms and kinetic models to beestablished [434,436].
In another kind of transient experiment, stepchanges in concentrations are effected, and theresponse of product concentration is measuredas a function of time. The analysis of thisresponse provides details of the course ofreaction and permits kinetic parameters to bedetermined.
6.4.2. Use of Isotopes
A powerful technique for the kinetic and mech-anistic study of heterogeneous catalytic reac-tions is steady-state isotopic-transient kineticanalysis (SSITKA) [433,437]. The technique isbased on the detection of isotopic labels in thereactor effluent species versus time following astep change in the isotopic labeling of one of thereactants in the reactor feed. Reactant andproduct concentrations and flow rates remainundisturbed during the step change and — inthe absence of isotopic mass effects — steady-state conditions are maintained under isotopic-transient operation. In contrast to othertransient experiments, the steady-state kinetic
behavior of the catalyst surface can be studied.Steady-state kinetic and mechanistic informa-tion which can be obtained from SSITKAincludes concentrations of different types ofadsorbed reaction intermediates, coverages,surface lifetimes, site heterogeneity, activitydistributions, and identification of possiblemechanisms [433].
The use of isotopes can greatly aids theelucidation of catalytic mechanisms [438]. Themost frequently used isotopes are 2H, 13C, 14C,and 18O. Deuterium-exchange reactions withorganic reactants yield isotopic distribution pat-terns which are often specific enough to elimi-nate a number of conceivable mechanisms.When carried out in conjunction with structurevariations, isotopic distribution patterns may beeffective in narrowing the range of possiblemechanisms, even though such studies cannotgive “the mechanism” [34]. Deuterium labelingis also used to determine which carbon atomsend up where or whether a reaction is inter- orintramolecular [34]. 13C labeling can be used forthe same purpose. Although nonradioactivelabels are preferred, radioactive tracers such as14C have also been used [439]. 18O labeling hasbeen applied to elucidate the relative rates of COand CO2 in methanol synthesis [440].
Kinetic isotope effects [441,442] are due tothe different masses of a given element and itscorresponding isotope. The resulting differencein zero-point energy may lead to an increase inactivation energy of the labeled molecule andtherefore a reduction of the rate. Whether akinetic isotope effect occurs or not when anatom in a certain position or group is isotopi-cally labeled (i.e., an X�H bond is replaced byX�D) for a catalytic reaction of interestprovides information on whether weakening orrupture of the X�H bond is involved in akinetically significant elementary step.
6.4.3. Use of Substituents, SelectiveFeeding, and Poisoning
Modification of organic molecules with suit-able substituent groups may provide valuableinformation on reaction mechanisms from thestereochemistry of the reaction of inter-est [34,443,13]. Substituents generally alsohave electronic effects on the reactivity of a
Heterogeneous Catalysis and Solid Catalysts 65
parent reactant (substituent effects). Resultinglinear free-energy relationships for a series ofsubstituents also assist the determination ofkinetically significant reaction steps of a con-ceivable reaction mechanism [444,445,14,18],since the substituents directly affect the rela-tive energy of the transition state and hence theactivation barrier of a kinetically significantstep.
Modification of molecules by substituentsmay also cause intra- or intermolecular stericeffects [34], and steric interactions betweenadsorbate and catalyst surface can be studied.The latter studies provide almost the onlyway todirectly probe the steric nature of active catalyt-ic sites without confusion with adsorption sitesthat are not catalytic sites [34].
Selective feeding and scavenging have beenproposed for the characterization of reactionintermediates [34]. Suppose Q is a suspectedintermediate for a particular reaction. Thishypothesis can be tested by adding (feeding)a compound to the reaction feed which is sup-posedly adsorbed to form the suspected inter-mediate Q, and by testing whether the addedcompound is indeed converted to the expectedproduct. In scavenging, a compound is addedwhich should react with the intermediate Q toform another compoundwhich is not normally aproduct.
The nature of catalytic sites can be tested andtheir density estimated by selective poisoning[446].
6.4.4. Spatially Resolved Analysis of theFluid Phase over a Catalyst
Analysis of the temperature and concentrationprofiles in the fluid phase over aworking catalystoften provides valuable information on the func-tional behavior of the catalyst. Since, at hightemperatures, conversion of reactants can alsoproceed via homogeneous reactions in the fluidphase aside from catalytic conversion, interac-tion of homogeneous and heterogeneous chemi-cal reactions and mass and heat transfer in thecatalyst-containing reactor becomes importantto fully understand the function of the catalyst. Inparticular in these cases, temporally and spatial-ly resolved profiles provide a more stringent testfor model development and evaluation.
Useful data arise from the experimental reso-lution of local velocity profiles by laserDoppler anemometry/velocimetry (LDA, LDV)[447,449] and of spatial and temporal speciesprofiles by in situ, noninvasive methods such asRaman and laser-induced fluorescence (LIF)spectroscopy. For instance, an optically accessi-ble catalytic channel reactor can be used toevaluate models for heterogeneous and homo-geneous chemistry as well as transport by thesimultaneous detection of stable species by Ra-man measurements and OH radicals by planarlaser-induced fluorescence (PLIF) [450,451].For example, the onset of homogeneous ignitionof methane oxidation in a platinum-coated cata-lytic channel can be monitored by means of thedistribution of OH radicals. While catalytic oxi-dation of methane along the channel walls re-leases some OH radicals, at a certain point in thereactor a transition to homogeneous oxidationoccurs accompanied by high concentrations ofOH radicals in the flame region. Since transientphenomena such as ignition, extinction, andoscillations of reactions are very sensitive totransport and kinetics, they can serve as mea-sures for critical evaluation of theoretical mod-els. For instance, the reliability of differentheterogeneous and homogeneous reactionschemes proposed in the literature was investi-gated by comparison of the experimentallyderived ignition distances with numericalelliptic two-dimensional simulations of the flowfield by using combinations of a variety ofschemes [452,453].
6.4.5. Spectroscopic Techniques
Any spectroscopic technique which is surface-sensitive, has sufficiently high sensitivity, andcan be applied under catalytic working condi-tions can provide valuable information on thenature of sufficiently long lived intermediates.However, spectroscopy often detects spectatorspecies rather than reaction intermediates.Therefore, it is mandatory to demonstrate thattrue intermediates are in fact seen. This can bedone by varying critical reaction parameters andmonitoring the response of spectroscopic signalintensities as a function of time. Sufficientlyhigh temporal resolution of the applied spectro-scopic technique is therefore required.
66 Heterogeneous Catalysis and Solid Catalysts
7. Design and Technical Operationof Solid Catalysts
7.1. Design Criteria for SolidCatalysts [454]
Solid catalysts are used in a surprising varietyof shapes including powders and irregularlyshaped particles, regular particles such asspheres and cylinders, and more complex ge-ometries like monolithic honeycombs, gauzes,and fibers. The most suitable geometry must becarefully selected and adjusted according to theproperties of the catalytically active materialand specific requirements of the chemical reac-tion and catalytic reactor. Only rarely is thecatalytic reaction so fast that the outer geometricsurface area of a nonporous catalytic body issufficiently large. Hence, porous catalysts aremainly used in which the catalytically activesurface area inside the structure often exceedsthe geometric surface area by several orders ofmagnitude. In these cases, the pore structuremust be accessible to the reactants while pro-ducts are allowed to leave. Design criteria forsolid catalysts comprise the choice of appropri-ate geometries with respect to highest possiblecatalyst utilization and product selectivity in agiven reactor. These goals should be achieved atthe lowest possible pressure drop over thereactor.
actions take place on the external and internalsurfaces (pores) of the catalyst. External andinternal concentration and temperature gradi-ents can build up in the fluid (gas or liquid)boundary layer around the catalyst particles andinside the pores if mass and heat transfer be-tween the bulk of the fluid and the active sur-faces are not sufficiently fast. Such gradientstend to exist (1) in fixed-bed reactors chargedwith large, porous catalysts; (2) during opera-tion at low mass flow velocities (mass flow rateper unit cross-sectional area); or (3) in the caseof highly exothermic reactions. On the otherhand, gradient effects usually are absent whensmall catalyst particles are used in fluidized-bedreactors.
Because such internal and external gradientscan substantially reduce the activity and selec-
tivity of the catalyst, conditions have beendelineated under which their adverse effectscan be minimized [89,455,456]. By carefullymatching operating conditions, catalyst, andreactor, optimum catalyst performance can beensured.
Mass and heat transfer in heterogeneouscatalytic reactions occur in two ways. Externaltransport to the external surface involves diffu-sion through the more or less stationary hydro-dynamic boundary layer that surrounds the cat-alyst particle. The thickness of this layer de-pends on the characteristics of the fluid and itsflow rate past the particle, and affects the rate ofmass and heat transfer. Internal transport to thestationary fluid in the pores of catalyst particle iscontrolled by diffusion alone.
Depending on the relative rates of the trans-port processes and the catalytic reaction, threeor four types of regimes can be distinguished. Inthe kinetic regime the rates of external andinternal mass transport are much higher thanthe rate of the chemical reaction. Therefore,concentration and temperature gradientsbetween the fluid and the center of a catalystparticle are negligible, and the catalyst is fullyutilized.
In the internal diffusion regime, mass trans-port in the catalyst pores is about as fast as, orslower than, the chemical reaction. In this casethere are considerable concentration gradientsalong the length of the pores, the effectivenessof the catalyst is impaired, and the apparentenergy of activation is lower than that observedin the kinetic regime.
At an even lower ratio of rates of transportand conversion, there is an intermediate regimein which the reaction takes place only on theexternal surface of the catalyst particles whilethe internal surface area in the pores is inactive.Because of the limited heat transfer in thisregime, exothermic reactions can overheat thecatalyst, and this results in a higher activity thanthat corresponding to the temperature of thefluid.
Finally, on further decreasing the ratio of therates of transport and conversion (e.g., by rais-ing the temperature of the fluid), the externalmass transfer regime is reached in which thereaction rate is controlled by mass transfer, andthe concentration of the reactants at the surfaceof the catalyst particles drops. Raising the
Heterogeneous Catalysis and Solid Catalysts 67
reactor temperature in this regime has littleeffect on the reaction rate, and the apparentactivation energy drops.
Effectiveness Factor [89,455,456] Theeffectiveness factor h is the ratio of the actualreaction rate observed on a porous catalystparticle to the rate that would be obtained if theinside of the particle were exposed to the tem-perature and reactant concentrations of the flu-id.Mathematical analysis [89,457–466] ofmasstransfer in porous particles of different shapeshas shown that the effectiveness factor is afunction of a dimensionless quantity, called theThiele modulus w [457]: for a sphere of radius Rand for a plate sealed on one side and on theedges the thickness of which isL,w is defined bythe following equations:
sphere : ws ¼ Rkvc
m�1s
Deff
� �1/2
ð31Þ
plate : wL ¼ Lkvc
m�1s
Deff
� �1/2
where kv is the reaction rate constant per unit ofgross catalyst volume, cs is the concentration onthe surface, m is the reaction order, and Deff theeffective diffusion coefficient, given by
Deff ¼ DQt
ð32Þ
where D is the diffusion coefficient for a pair offluids taking into account binary and Knudsendiffusion, Q the void fraction of the porousmass, and t a factor allowing for tortuosity andvarying cross sections of the pores.
Equation (31) for the plate can also be usedfor arbitrary catalyst geometry if L is interpretedas characteristic diffusion length, i.e. the ratio ofcatalyst particle volume and its external surfacearea.
For first-order reactions (m ¼ 1), the effec-tiveness factors are as follows (tan is hyperbolictangent):
Sphere : h ¼ 3
ws
1
tanh ws� 1
ws
� �ð33Þ
Plate : h ¼ tanh wL
wL
Correlation between the effectiveness factor andthe Thielemodulus for nonexothermic reactionsis shown in Figure 19 [462]. The effectivenessfactor is about unity for w < 1 and inverselyproportional to w for w > 3.
If the intrinsic velocity rate constant kv(Eq. 32) cannot be determined directly, anotherdimensionless modulus Q has been de-rived [460,462]. For first-order reactions occur-ring in a sphere, it is defined by
Q w2h ¼ R2
DeffVcCs
� �dn
dt
� �ð34Þ
where dn/dt is the conversion rate in moles persecond of the reactant in the catalyst volume Vc.The effectiveness factor as a function of Q isshown in Figure 20 for a moderate energy ofactivation (E ¼ 10 RT; first-order reaction in aspherical particle) and variable enthalpy changeDH (l is the thermal conductivity of the cata-lyst). For exothermic reactions (b > 0), theeffectiveness factor goes through a maximumvalue exceeding unity because of the interactionof two opposing effects. Poor mass transferlowers the efficiency of the catalyst, whereasinsufficient heat transfer raises catalyst temper-ature and reaction rate.
Effects on Selectivity [89,455,456] Theeffect of mass- and heat-transport processes onthe selectivity of reactions yielding more thanone product depends on the selectivity type. In atype I reaction at low effectiveness factors, theobserved selectivity factor changes from k1/k2 to
Figure 19. Effectiveness factorw as a function of the Thielemodulus ws or sL
Flat plate sealed on one side and on edges, first-orderreaction; D Same, second-order reaction;þ Spherical parti-cle, first-order reaction* Reproduced with permission [462]
68 Heterogeneous Catalysis and Solid Catalysts
ðk1DAeffk�12 D�1BeffÞ1/2, provided the order of the
two reactions (A! X and B! Y) is the same.If, as usual, the ratio of the effective diffusivitiesis smaller than k1/k2, the selectivity will drop atlow effectiveness factors. However, if the dif-fusivity ratio is high, the selectivity can increasein a porous catalyst. This is the situation in theso-called shape-selective zeolites [89]. If thereaction orders are different, the reaction withthe lower order is favored in the porous catalystat low h values.
In reaction type II, the effectiveness factorhas no influence on the selectivity if the ordersof the two reactions are equal. Otherwise, theeffect of different orders is the same as in type IIreactions [456,467].
For an isothermal first-order reaction of typeIII occurring at low effectiveness factor on aporous plate, the observed selectivity is approx-imately the square root of the intrinsic rateconstant ratio k1/k2 [468]. Figure 21 showscomparative conversions of A to X for type IIIat effectiveness factorsh¼ 1 andh< 0.3, for k1/k2 ¼ 4; the maximum yield and selectivity bothdrop by ca. 50 % at the low effectivenessfactor [468].
Temperature gradients caused by exothermicreactions favor reactions with higher apparentenergies of activation. Because such undesiredside reactions as decomposition and oxidativedegradation generally have high energies ofactivation, large catalysts having narrow porestructure can have an unfavorable effect on theselectivity and product yield.
In the regime of external mass transfer, re-sistance of the boundary layer to diffusion hassimilar effects on the selectivity of parallel andconsecutive reactions as does diffusion in pores.
Catalyst Geometries and Transfer Proce-sses Industrial processes that occur in the
kinetic regime include reactions conducted influidized-bed reactors using catalysts 0.05 –0.25 mm in size [89,456]. Because diffusioncoefficients in liquids are smaller by severalorders of magnitude than those in the gaseousphase, liquid-phase operation in the kineticregime requires a finely powdered catalyst.
Because the resistance to flow of catalystsincreases steeply with decreasing size, use ofcatalysts smaller than 2 – 3 mm in fixed-bedreactors is restricted to radial-flow reactors withsmall bed length. Ring-shaped and tablet cata-lysts show relatively favorable pressure dropand diffusion characteristics.
Invarious partial oxidation processes (e.g., o-xylene to phthalic anhydride), good results havebeen obtained with so-called eggshell catalystsin which the active catalyst mass is applied in athin layer of a few tenths of a millimeter to theexternal surface of an inert, nonporous support.
If the reaction rate is higher and the regime ofinternal diffusion cannot be avoided, it is oftenadvantageous to use catalysts with a bimodalpore system in which micropores (< 2 nm) are
Figure 20. Effectiveness factor as a function of modulus(Eq. 34)--- Unstable region* Reproduced with permission [467]
Figure 21. Effect of the effectiveness factor on catalystselectivity* Reproduced with permission [468]
Heterogeneous Catalysis and Solid Catalysts 69
connected by macropores (> 50 nm) to theexternal surface area of the catalyst particle.The micropores provide the needed high activesurface area, whereas the macropores facilitatemass transport to and from themicropores. Suchcatalysts are especially suitable for operation atprocess pressures below 3 MPa. At these pres-sures mass transport into pores smaller than100 – 1000 nm occurs increasingly throughKnudsen diffusion involving molecular colli-sions with the pore wall rather than betweenmolecules [89,456].
In fast processes occurring in the regime ofexternal mass transfer, the overall conversionrate is independent of the porosity of the catalystand is essentially proportional to its externalsurface area [463]. Examples of such reactionsare oxidation of ammonia to nitric acid andammoxidation of methane to hydrogen cyanidecarried out at 1070 – 1270 K on stacks of fine-mesh Pt – Rh gauze.
7.2. Catalytic Reactors [469–473]
7.2.1. Classification of Reactors[167,469,471]
Catalytic reactors can be classified by theirmode of operation under steady-state or tran-sient conditions or on their mode of contacting/mixing of reactants and solid catalyst.
Typical steady-state reactors are packed-bedtubular reactors under continuous flow condi-tions, either plug flow or mixed flow. In an idealtubular reactor ideal mixing takes place in theradial direction, but there is no mixing in theaxial direction. Plug flow is attained in this case.Plug flow reactors can be operated either inintegral or differential mode. In the latter case,single-pass experiments in small-scale reactorsprovide the data for differential conditions re-quired for analysis of the reaction kinetics. Asan alternative, the effluent from the differential-ly operating reactor can also be recycled exter-nally or internally, thus approaching a well-mixed reactor system, the continuous-flow stir-red tank reactor (CSTR). Without inlet andoutlet feed a continuous recycle flow results,characteristic of a batch reactor in which thefeed composition changes with time (transientconditions as opposed to steady-state condi-
tions). The catalyst must not necessarily be keptin a packed bed but can be suspended in theliquid or gaseous fluid reactant mixture. In thefluidized-bed mode, the solid catalyst consist-ing of fine powder (particle diameter 10 –200 mm) is kept in motion by an upward gasflow (fluidized-bed reactor, see Section Indus-trial Reactors). If the fluid is a liquid the catalystcan be suspended easily in a CSTR by efficientstirring (slurry reactor, see Section IndustrialReactors). In so-called riser reactors, catalystmaterial is continuously introduced into andremoved from the reactor with the reactant andproduct feeds.
Batch and semi-batch reactors operate undertransient conditions. Discontinuous step orpulse operation necessarily results in transientconditions.
Several catalytic reactor types are schemati-cally shown in Figure 22. Tubular fixed bedreactors (A) have an inlet flow n0 and an outletflow n of the reactant-product mixture.The adiabatic fixed-bed reactor is shown inFigure 22 B. Multitube fixed-bed reactors (C)are used for highly exothermic reactions such asthe oxidation of o-xylene to phthalic anhydride.The principle of aCSTR is demonstrated asD.Afluidized-bed reactorwith catalyst recirculationis sketched in E. A slurry CSTR reactor isschematically shown in F. An alternative forthree-phase reactions (gas – liquid – solid) isthe packed bubble column or slurry reactor (G).Discontinuous batch reactors with internal andexternal recycling operating under transientconditions are depicted as H and I, respectively.
7.2.2. Laboratory Reactors [469,470]
A compilation of laboratory reactors and theiroperation mode is given in Figure 23.
Laboratory reactors are small-scale reactors.Steady-state fixed-bed tubular flow reactors aremost frequently used for catalyst testing anddetermination of the reaction kinetics on thelaboratory scale. These reactors are favoredbecause of the small amounts of catalyst re-quired, the ease of operation, and the low cost.Stirred tank reactors (e.g., with fixed or rotatingbasket) and batch suspension reactors are lessfrequently used. Provided that heat- and mass-transfer limitations can be neglected under the
70 Heterogeneous Catalysis and Solid Catalysts
operating conditions in a tubular flow reactor,the catalyst bed is isothermal, and the pressuredrop across the catalyst bed is negligible, thereaction rate can be determined from the massbalance for a component i. This is usually veri-fied in microreactors operating under differen-tial conditions. The reaction rate perunit mass rW (mol s�1 kg�1) is then given by
Equation (35):
dxi
d WF0i
� � ¼ �nirw; ð35Þ
where Xi is the conversion of component i, ni itsstoichiometric coefficient, W the catalyst mass(kg), andFi
o themolar rate of component i at the
Figure 22. Schematic representation of several types of catalytic reactorsA) Tubular fixed bed reactor; B) Adiabatic fixed-bed reactor; C) Multitube fixed-bed reactor; D) CSTR; E ) Fluidized-bedreactor with catalyst recirculation; F) Slurry CSTR reactor; G) Packed bubble column or slurry reactor; H), I) Discontinuousbatch reactors
Heterogeneous Catalysis and Solid Catalysts 71
reactor inlet (mol s�1). The ratio W/Fio is the
space – time.Ancillary techniques in laboratory units for
catalyst testing such as generation of feedstreams and product sampling are discussedin [474].
The TAP reactor, a laboratory pulse reactor,is described in Section Temporal Analysis ofProducts (TAP Reactor).
7.2.3. Industrial Reactors [167,471]
Various types of industrial reactors and theiroperation mode are listed in Figure 24.
Catalytic Fixed-Bed Reactors [475,477]In the chemical industry fixed-bed reactors arethe standard type of reactors for heterogeneous-ly catalyzed gas-phase reactions (two-phasereactors). Fixed catalyst beds can be realized
in various ways. Randomly packed beds (deepbeds) require catalyst particles having differentshapes such as spheres, cylinders, rings, flat diskpellets, or crushed material of a defined sievefraction. The geometries and dimensions of thecatalyst particles are dictated by pressure dropand heat- and mass-transfer considerations. Theuse ofmonolithic catalysts significantly reducesthe pressure drop across the catalyst bed. Thistype of catalyst is employed for example forautomobile exhaust gas purification and for theremoval of nitrogen oxides from tail gases ofpower stations.
Fixed-bed reactors can be operated underadiabatic or nonadiabatic conditions. Adiabaticreactors can be applied for reactions with lowheats of reaction such as gas purification. Theyconsist of a cylindrical tube inwhich the catalystis packed on a screen and is traversed in axialdirection (Fig. 22 B). This design is particularlysuitable when short residence times and high
Figure 23. Classification of laboratory reactors
72 Heterogeneous Catalysis and Solid Catalysts
temperatures are required. In this case a fixedbed of large diameter and small height (5 –20 mm) is used (shallow bed). As an example,for ammonia oxidation in nitric acid plants thefixed bed consists of several layers of platinumwire gauze with bed diameters up to severalmeters. This type of reactor is limited to smallcatalyst volumes. The radial flow concept ispreferred when large amounts of catalyst arerequired [475]. In this reactor type, the catalystis charged in the annular space around an axiallylocated tube. The reactants are traversing radi-ally, either from the inside or from the outside ofperforated plate rings. Because of the low pres-sure drop, smaller catalyst pellets (4 � 4 or3 � 3 mm) can be used in this reactor type.
Only limited conversions can be achieved byadiabatic reactors because of the necessary con-trol of the adiabatic temperature change. Multi-stage reactors consisting of several sequentialadiabatic stages which are separated by inter-stage heat exchangers have therefore beenintroduced.
Nonadiabatic operation can be achievedwithfixed-bed reactors which are cooled or heatedthrough the reactor walls. Efficient heat ex-change results in so-called isothermal reactors.A typical example is the multitubular reactorschematically shown in Fig. 22 C, which isused for highly exothermic and temperaturesensitive reactions (e.g., o-xylene oxidation)with, e.g., salt melts as heat-transfer media. Ina multitube reactor, the pressure drop must bethe same in each tube so that the gas flow isdistributed uniformly over the tubes. Smallchanges in the packing density in the tubes cancause uneven heat transfer and, in the case ofhighly exothermic reactions, hot spots and se-lectivity loss. For these reasons multitube re-actors are filled by special equipment thatcharges each tube with the same amount ofcatalyst at a definite rate. After filling, the tubesare checked for pressure drop, and if necessary,the charge is adjusted.
Autothermal reactors can favorably be ap-plied for exothermic and temperature sensitive
Figure 24. Classification of industrial reactors
Heterogeneous Catalysis and Solid Catalysts 73
reaction systems. The conventional reactor de-sign consists of an adiabatic packed-bed reactorcoupled with a countercurrent heat exchanger inwhich the cold reactant feed is brought to reac-tion temperature.
Multifunctional reactors are being devel-oped [478] with the goal of improving operationconditions which are not necessarily optimallydetermined in standard fixed-bed reactorconfigurations.
The following industrial processes areperformed in various types of fixed-bedreactors:
i. “deep-bed” adiabatic system:* Isomerization of C4 – C6 alkanes or of
light gasoline at 620 – 770 K, 20 –40 bar on Pt-alumina (HCI activated) oron Pt-H-mordenite.
* Catalytic reforming of heavy gasolineusing a cascade of single bed reactors at700 – 820 K, 20 – 25 bar on K-promot-ed Cr2O3-Al2O3 catalyst.
* Hydrocracking of vacuum gas oil at670 – 770 K, 20 – 40 bar, using singleor two stage processes on Ni-MoO3-Y-zeolite-alumina and Pt-mordenite-alu-mina, respectively.
ii. “multibed” adiabatic system:* Ammonia synthesis at 670 – 770 K,
200 – 300 bar on K-, Mg-, Al-promotediron catalysts.
* Oxidation of SO2 in the sulfuric acidproduction at 720 – 770 K, atmosphericpressure on K2SO4-V2O5 catalysts. Re-actor with externally located heat ex-changers are in operation.
iii. “radial flow” system:* Water gas shift (HTS) at 620 – 670 K,
25 – 50 bar on Cr2O3-Fe2O3 catalysts.
iv. “shallow-bed” system:* Methanol dehydrogenation to formalde-
hyde at 870 K, atmospheric pressure onmetallic Ag (granulate).
* Ammonia oxidation to NOx at 1170 Katmospheric pressure on Pt/Rh-grids.
v. “quench” system:* Methanol synthesis at490 – 520 K,50 –
100 bar on Cu-ZnO-Al2O3 catalysts.
vi. “multitube” system:* Methanol synthesis at 490 – 520 K,
50 – 100 bar on Cu-ZnO-Al2O3 cata-lysts.
* Oxidation of ethylene to ethylene oxideat 470 – 520 K, atmospheric pressure onAg-a-alumina.
* Oxidation of o-xylene to phthalic anhy-dride at 640 – 680 K, at atmosphericpressure on V2O5-TiO2 catalysts.
* Hydrogenation of benzene to cyclohex-ane at 470 – 520 K, 35 bar on Ni-SiO2
catalysts.* Dehydrogenation of ethylbenzene to sty-
rene at 770 – 870 K, atmospheric or re-duced pressure on promoted (K, Ce, Mo)Fe-oxide.
Fluidized Bed Reactors [479–482] Fluid-ized-bed reactors are preferred over fixed-bedreactors if rapid catalyst deactivation occurs oroperation in the explosive regime is required.In this type of reactor, an initially stationarybed of catalyst is brought to a fluidized-state byan upward stream of gas or liquid when thevolume flow rate of the fluid exceeds a certainlimiting value, the minimum fluidization vol-ume flow rate. The catalyst particles are heldsuspended in the fluid stream at this or higherflow rates. The pressure drop of fluid passingthrough the fluidized bed is equal to the differ-ence between the weight of the solid catalystparticles and the buoyancy divided by thecross-sectional area of the bed. Major advan-tages of fluidized-bed reactors are excellentgas – solid contact, good gas – solid heat andmass transfer, and high bed-wall heat transfercoefficients.
The gas distribution in the fluidized bed isperformed industrially by, for example, perfo-rated plates, nozzles, or bubble caps which aremounted at the reactor bottom.
Depending on the volume flow rate of thefluid different types of fluidized beds form.Fluidizationwith a liquid feed leads to a uniformexpansion of the bed. In contrast, solid-freebubbles form when fluidization is carried outin a gas stream. These bubbles move upwardsand tend to coalesce to larger bubbles as theyreach increasing heights in the bed. At high gasvolume flow rates, solid particles are carried outof the bed. Tomaintain steady-state operation of
74 Heterogeneous Catalysis and Solid Catalysts
such a turbulent fluidized bed, the solid catalystparticles entrained in the fluidizing gas must becollected and transported back to the reactorbed. This can be achieved most easily with anintegrated cyclone, as schematically shown inFigure 25 A. A circulating fluidized bed is fi-nally formed at still higher gas volume rates. Anefficient external recycle system, as shown inFigure 25 B, is required for such operatingconditions because of the high solidsentrainment.
Catalytic cracking is carried out in fluidized-bed reactors because the solid acid catalysts arerapidly deactivated by coke deposition. Thecatalyst must therefore continuously be dis-charged from the reactor and regenerated in anair-fluidized regenerator bed where the coke isburned off. The regenerated catalyst is thenreturned to the fluidized-bed reactor. The heatof combustion of the coke can be used forpreheating of the reactant feed.
The main advantages of fluidized-bed reac-tors are:
* Uniform temperature distribution (due tointensive solid mixing)
* Large solid-gas exchange area* High heat-transfer coefficient between bed
and immersed heating or cooling surfaces.
However, these reactors have also some dis-advantages, e.g.:
* Expensive catalyst separation and purifica-tion of reaction products (installation ofcyclones and filters)
* Undesired bypass of reactants due to bubbledevelopment
* Catalyst attrition* Erosion of internals resulting from high
solids velocities
Fluidized-bed reactors found very broad ap-plication in the petroleum refining and produc-tion of chemicals, for example:
* Catalytic cracking of vacuum gas oil togasoline at 720 – 820 K on aluminosili-cates containing ultrastable Y-zeolites.
* Fischer – Tropsch synthesis (Syntholprocess) from CO und H2 at 620 – 670 Kand 15 – 30 bar on promoted Fe-oxidecatalysts.
* Ammoxidation of propylene to acrylonitrile(SOHIO-process) at 670 – 770 K, 1 –2 bar on promoted Bi-MoOx catalysts.
* Oxidation of naphthalene or o-xylene tophthalic anhydride at 620 – 650 K, at atmo-spheric pressure on V2O5–SiO2 catalysts.
A special type of the fluidized-bed reactor isthe so-called riser reactor. This reactor consistsof a vertical tube in which the reaction takesplace in the presence of the entrained catalyst.Catalyst coming from the riser tube is collectedin the vessel, before passing through the stripperto the regenerator (fluidized-bed type). The riserreactor is mainly used in the catalytic crackingof heavy oils on highly active zeolitic catalysts.
The moving-bed reactor [479] operates withspherical catalyst particles larger (2 – 6 mm)than those used in the fluid-bed system. In thestandard arrangement, catalyst particles aremoving slowly through the agitated bed. Cata-lyst reaching the reactor top is transported intothe regenerator. Using mechanical or pneumaticconveyer the regenerated catalyst is returning tothe bottom of the reactor.
The main advantage of the moving-bed re-actor is lower catalyst attrition than in thefluidized-bed system. The disadvantage is apoor heat transfer, and therefore this reactor isnot suitable for exothermic reactions.
The moving-bed reactor found applicationmainly in petroleum cracking.
Slurry Reactors [483] The aim of slurryreactors is intimate contact between a gas
Figure 25. Schematic representation of two forms of gas –solid fluidized beds under turbulent flow conditions
Heterogeneous Catalysis and Solid Catalysts 75
phase component (which is to be dissolved in aliquid-phase component) and a finely dis-persed solid catalyst (three-phase reactors).The particle size of the solid catalyst is keptsufficiently small (< 200 mm) that it remainssuspended by the turbulence of the liquid in theslurry reactor. This is in contrast to three-phasefluidized-bed reactors, in which an upwardliquid flow is required to suspend the largercatalyst particles.
On the basis of the contacting pattern of thephases and the mechanical devices that achievecontact and the mass transfer, nine types ofslurry reactors can be distinguished [483]:
Slurry reactors are industrially applied for amultitude of processes [484], many of which areheterogeneously catalyzed processes for hydro-genation of edible oils. A new development isthe continuous Fischer – Tropsch slurry synthe-sis process of SASOL in South Africa [485].
Three-phase reactors are classified as fixed-bed or suspension reactors depending on thecatalyst arrangement and shape:
Fixed-bed reactors operate either in the trick-le-bed or in the bubble-flow mode [483]. In thefirst case, liquid reactants or reactants dissolvedin a solvent are flowing downward through thecatalyst bed and the gaseous reactants are con-ducted in the countercurrent or concurrentdirection.
In the bubble-flow reactors, liquid and gas-eous reactants are fed into the bottom of thecolumn and are flowing upward through thecatalyst fixed-bed.
The trickle-bed arrangement has some ad-vantages such as:
* Fast diffusion of gases through the liquidfilm to the catalyst surface
* Lower back-mixing* No problems with catalyst separation
* Selective removal of catalytic poison in theentrance zone of the bed
* Simple catalyst regeneration
Drawbacks of trickle-bed reactors are:
Poor heat transferPartial utilization of the catalyst in case of the
incomplete wettingPossibility of “brooks” formation
The successful performance of the trickle-phase reactors depends on the suitable diameter/length ratio, catalyst shape and size, and theliquid flow distribution through the catalyst bed.The catalyst particle size is limited by theallowed pressure drop. Larger sizes (6 – 10 mmdiameter) are therefore preferred, which,however, can bring diffusion problems.
The “bubble-flow” version is favored in par-ticular for reactions with a low space velocity.A good heat transfer and no problems withan incomplete catalyst wetting are the mainadvantages of the “bubble-flow” reactors.
Both types of the three-phase reactorshave found numerous industrial applications,e.g.:
* Hydrotreating of petroleum fractions at570 – 620 K, 30 – 60 bar on Ni–MoO3–Al2O3 catalysts.
* Hydrocracking of high boiling distillates at570 – 670 K, 200 – 220 bar on Ni–MoO3–Y–zeolite–alumina catalysts.
* Selective hydogenation of C4-fractions (re-moval of dienes and alkines) at 300 –325 K, 5 – 20 bar on Pd-Al2O3 catalysts.
* Hydrogenation of aliphatic carbonyl com-pounds to alcohols at 370 – 420 K, 30 baron CuO–Cr2O3 catalysts.
Suspension reactors [483,484] are operatedsuccessfully in the chemical industry because oftheir good heat transfer, temperature control,catalyst utilization, and simple design. Becauseof the small catalyst particle size, there are noproblems with internal diffusion. Suspensionreactors operate either in the discontinuous orin the continuous mode. One serious disadvan-tage is the difficult catalyst separation, especial-ly if fine particles have to be removed from theviscous liquid.
76 Heterogeneous Catalysis and Solid Catalysts
Currently two types of suspension reactorsare in use: stirred vessels and three-phase bubblecolumns.
In the case of stirred vessels the catalystparticles (mainly smaller than 200 mm) aresuspended in the liquid reactant or solutions ofreactants, whereas gaseous reactants are intro-duced at the bottom of the vessel through per-forated tubes, plates or nozzles. The vessels areequipped with different types of stirrers orturbines keeping the catalyst suspended. Cool-ing and heating coils as well as the gas recyclesystem belong to the standard equipment. Stir-red vessels operate mostly discontinuously.However, if continuous operation is favored,then stirred vessels are arranged in a cascadeto complete the required conversion.
Bubble columns are mainly continuouslyoperating three-phase reactors. The gas is intro-duced at the bottom of the column throughnozzles, perforated plates or tubes. In the stan-dard arrangement the liquid reactant flows in thecocurrent direction with the gas. In some casesstirrers are installed to keep powdered catalystsin the suspension.
The gaseous reactants can be recycled by anexternal loop or by an internal system, such as aVenturi jet tube. This equipment is driven by arecycle of the slurry using a simple pump. TheVenturi tube is sucking thegas fromthe freeboardabove the reactor back into the slurry. Heat ex-changerscanbe installed in the loop inbothcases.
The main advantages of bubble columns aresimple and low-priced construction, good heattransfer, and good temperature control.
Suspension reactors are used predominant-ly for fat and oil hydrogenation 420 – 470 K, at5 – 15 bar using various Ni–kieselguhr catalysts.
Also, hydrogenolysis of fatty acid methylesters to fatty alcohols is performed in suspen-sion reactors at 450 – 490 K, 200 – 300 bar onpromoted copper chromites (CuO–Cr2O3).
Further applications are: syntheses of acetal-dehyde and of vinyl acetate according to theWacker process.
7.2.4. Special Reactor Typesand Processes
Microstructured Reactors [486–491] Amicrostructured reactor can be defined as a
series of interconnecting channels having dia-meters between 10 and 1000 mm that areformed in a planar surface in which smallquantities of reagents aremanipulated (see also! Microreactors –Modeling and Simulation).Among the advantages of microstructured re-actors over conventional catalytic reactors arehigh heat-transfer coefficients, increased sur-face-to-volume ratios of up to 50 000 m2 m�3,as opposed to 1000 m2 m�3 for conventionalcatalytic laboratory reactors, shorter mixingtimes, and localized control of concentrationgradients. The small scales reduce exposure totoxic or hazardous materials, and the enclosednature of the microstructured reactors permitsgreater ease of containment in the event of arunaway reaction. Furthermore, the highly ef-ficient heat transfer as well as high surfaceareas available for adsorption of radicals allowreactions to be carried out beyond the explo-sion limit [488,492,493].
Despite their small size, microstructured re-actors can be used for synthetic chemistry [494],since as few as 1000 microchannels operatingcontinuously could produce 1 kg of product perday. While several types of laboratory-scalemicrostructured reactors are already commer-cially available, further developments are stillrequired to make microstructured reactors ma-ture for industrial application. One of the mostadvanced designs targeted at large-scale appli-cations for exothermic gas-phase reactions isthe DEMiS project of Degussa and Uhde [495].The state of development of microstructuredreactors for heterogeneously catalyzed gas-phase and liquid-phase reactions has been sum-marized [319]. Coating of wall materials withcatalysts, strategies for replacement of spentcatalysts, and the reduction of overall apparatussize appear to be the most challenging obstaclesto be overcome. It is also evident that in manycases more active catalysts are required for fullutilization of microstructured reactors.
Unsteady-State Reactor Operation [496]Forced unsteady-state reactor operation hasbeen applied to continuous catalytic processesin fixed and f1uidized-bed reactors. This oper-ating mode can lead to improved reactorperformance. Nonlinearity of chemical reac-tion rates and complexity of reaction systemsare responsible for conversion or selectivity
Heterogeneous Catalysis and Solid Catalysts 77
improvements under forced unsteady-stateconditions [497].
An unsteady-state in a fixed-bed reactor canbe created by oscillations in the inlet composi-tion or temperature (control function) such asschematically shown in Figure 26. The mostwidely applied technique in a fixed-bed reactoris periodic flow reversal. Examples of this oper-ation mode in industrial applications are SO2
oxidation, NOx reduction by NH3, and oxidationof volatile organic compounds (VOC). In fluid-ized-beds for exothermic reactions, favorableunsteady-state conditions of the catalyst can beachieved by catalyst circulation inside the reac-tor. The unsteady-state operation in the fluid-ized-bed is applied for example for the partialoxidation of n-butane to maleic anhydride onvanadyl pyrophosphate catalysts developed byDuPont. In the first step, n-butane dilutedwith aninert gas is contacted in the riser reactor (resi-dence time10 – 30 s)with spherical catalystpar-ticles (100 mm).Thepartial oxidationofn-butaneproceeds on account of the lattice oxygen in thesurface layer of vanadyl pyrophosphate. In thesecond step, the partially reduced catalyst istransported into the fluidized-bed reactorwhere the catalyst reoxidation takes place.The obtained selectivity was about 10% higherthan that in the multitubular fixed-bed reactor,operating under steady-state conditions. Afurther group of forced unsteady-state processesuses the combination of a chemical reactionwith the separation of products (chromato-graphic reactor). Systems applied till todayoperating on the principle of chromato-graphic columns are filled with a catalystpossessing suitable adsorption properties, suchas Pt - Al2O3. Pulses of reactant are periodical-ly injected into this reactor which is purged bycarrier gas during periods between the pulses.This operation can provide a higher conversion
for reversible reactions if one of the reactionproducts is adsorbed on the catalyst morestrongly than the other one. The feasibility ofthis principlewas tested in the dehydrogenationof cyclohexane to benzene on pilot scale.
Membrane Reactors [498–500] In mem-brane reactors (! Membrane Reactors) cata-lytic conversion is coupled with a separationeffect provided by the integrated membrane.High conversions can be achieved for equilibri-um-restricted reactions when one of the reactionproducts can be removed from the reactionmixture by diffusion through a permselectivemembrane. Hydrocarbon dehydrogenation re-actions are the best example for this application.In addition, catalytic membrane reactors havebeen proposed for improved control of theselectivity of some catalytic reactions. For ex-ample, controlled introduction of a reactant intothe reactor by selective or preferential perme-ation may limit possible secondary reactions ofthe target product.
Two types of membranes can be distin-guished: dense membranes and porous mem-branes. Dense metallic membranes consist ofthin metal foils. Palladium and palladium al-loys (PdAg, PdRu) are specific for hydrogenpermeation. Dense oxide membranes are usu-ally solid electrolytes such as ZrO2 and CeO2,which are permeable for O2 [500]. Porousmembranes are typically made of oxides, al-though carbon membranes have also beenused [501]. Ceramic membranes consist ofseveral layers of material with progressivelydecreasing pore size. The top layer with thesmallest pore size controls the separation.Mostof these membranes are produced by sol – geltechniques. Intrinsic catalytic properties can beintroduced into thesemembranes, which can beproduced as cylindrical tubes forming the basisfor tubular reactors. Zeolite membranes canalso be prepared, and the structure of the zeoliteshould permit high selectivity in separationprocesses.
Despite the potential of membrane reactors,their development is still not mature for indus-trial application.
Reactive Distillation [502–504] In reac-tive distillation (! Reactive Distillation) frac-tional distillation and chemical reaction are
Figure 26. Stepwise variation of inlet parameters to createunsteady state conditions
78 Heterogeneous Catalysis and Solid Catalysts
performed simultaneously, e.g., for a reaction ofthe type
AþB!CþD
in which at least one of the products has avolatility which is different form those of theother compounds. The most attractive featuresof reactive distillation are:
1. The separation of at least one of the productsby distillation drives reactions to completionwhich are otherwise equilibrium-limited.
2. Reactions in which high concentrations of aproduct or one of the reactants lead to unde-sired side reaction, can be carried out.
Despite these advantages, several chemicaland physical limitations exist in practice for itsuse in chemical processes, so that reactive dis-tillation has found application for only a fewimportant reactions. Industrially important pro-cesses are various etherification, esterification,alkylation, and isomerization reactions.
IFP,Mobil, Neste Oy, Snamprogetti, Texaco,and UOP are providing licenses for plants toproduce tert-butyl and tert-amyl ethers from thecorresponding olefins and methanol or ethanolusing strong acidic resins as solid catalysts.
Reactions under Supercritical Conditions[505–508] The major advantage of super-critical fluids as solvents in catalytic reactionsis the fact that carbon dioxide and water can beused as environmentally benign solvents.When in their supercritical state, these non-toxic compounds are good solvents for manyorganic compounds. A multicomponent sys-tem under supercritical conditions may behavelike a single gaslike phase with advantageousphysical properties. Under reaction conditionsthis leads to [505]:
limitations in multiphase reaction systems3. Easier separations and downstream pro-
cessing4. In situ extraction of coke precursors5. Strongly pressure and temperature depen-
dent solvent properties near the critical point6. Higher diffusivities than in liquid solvents
7. Better heat transfer than in gases8. Use of clustering to alter selectivities
Hence, supercritical fluids offer strategiesfor more economical and environmentally be-nign process design, mainly because of en-hanced reaction rates, prolongation of catalystlifetime, and simplification of downstreamprocessing.
7.2.5. SimulationofCatalyticReactors [122]
Catalytic reactors are generally characterized bya complex interaction of various physical andchemical processes. Therefore, the challenge incatalysis is not only the development of newcatalysts to synthesize a desired product, butalso the understanding of the interaction of thecatalystwith the surrounding reactive flow field.Sometimes, the exploitation of these interac-tions can lead to the desired product selectivityand yield. This challenge calls for the develop-ment of reliable simulation tools that integratedetailed models of reaction chemistry andcomputational fluid dynamics (CFD) modelingof macroscale flow structures. The consider-ation of detailed models for chemical reactions,in particular for heterogeneous reactions, how-ever, is still very challenging due to the largenumber of species mass-conservation equa-tions, their highly nonlinear coupling, and thewide range of timescales introduced by thecomplex reaction networks.
Currently available multipurpose commer-cial CFD codes can simulate very complexflow configurations including turbulence andmulticomponent species transport. However,CFD codes still have difficulties in implement-ing complex models for chemical processes, inwhich an area of recent development is theimplementation of detailed models for hetero-geneous reactions. Several software packageshave been developed for modeling complex re-action kinetics in CFD such asCHEMKIN [509],CANTERA[510],DETCHEM[511],which alsooffer CFD codes for special reactor configura-tions such as channel flows and monolithic re-actors. These kinetic packages and also a varietyof userwritten subroutines formodeling complexreaction kinetics have meanwhile been coupledto several commercial CFD codes. Aside from
Heterogeneous Catalysis and Solid Catalysts 79
the commercially widespreadmultipurpose CFDsoftware packages such as FLUENT [512],STAR-CD[513],FIRE[514],CFD-ACEþ [515],and CFX [516], a variety of multipurpose andspecialized CFD codes have been developedin academia and at research facilities such asMP-SALSA [517].
From a reaction-engineering perspective,computational fluid dynamics simulations havematured to a powerful tool for understandingmass and heat transport in catalytic reactors.Initially, CFD calculations focused on a betterunderstanding of mixing, mass transfer to en-hance reaction rates, diffusion in porous media,and heat transfer. Later, the flow field and heattransportmodels were also coupled withmodelsfor heterogeneous chemical reactions. So far,most of these models are based on the mean-field approximation as discussed in SectionMean-Field Approximation, in which the localstate of the surface is described by its coveragewith adsorbed species averaged on amicroscop-ic scale.
Detailed CFD simulations of catalytic reac-tors, often including multistep reaction mec-hanisms, have been carried out for catalyticchannel reactors with laminar [518] and tur-bulent [519] flow fields, monolithic reac-tors [520–524], fixed-bed reactors [525,526],wire-gauze reactors [527,528], reactors withmultiphase flow [512], and others. CFD simula-tions are becoming a powerful tool for under-standing the behavior of catalytic reactors andin supporting the design and optimization ofreactors and processes.
7.3. Catalyst Deactivationand Regeneration
7.3.1. Different Types of Deactivation
As has been observed in the laboratory and inindustrial application, heterogeneous catalystsare deactivated during time on stream. Forexample, in fluid-bed catalytic cracking andpropene ammoxidation the catalyst life is limit-ed to a few seconds or minutes, while in otherreactions, such as NH3 and CO oxidation thecatalyst remains active for several years. Not
only loss of activity but also a decrease inselectivity is usually caused by catalyst deacti-vation [529–531].
The activity loss can be compensated withincertain limits by increasing the reaction temper-ature. However, if such compensation is notefficient enough, the catalyst must be regener-ated or replaced.
The main reasons for catalyst deactivationare:
1. Poisoning2. Fouling3. Thermal degradation4. Volatilization of active components
Some types of catalyst deactivation are re-versible, e.g., catalyst fouling and some specialtypes of poisoning. Other types of deactivationare mostly irreversible [529,530,532].
Catalyst Poisoning The blocking of activesites by certain elements or compounds accom-panied by chemisorption or formation of surfacecomplexes are the main causes of catalyst de-activation [529,530,532]. If the chemisorptionis weak, reactivation may occur; if it is strong,deactivation results. Chemical species oftenconsidered as poisons can be divided in fiveclasses:
1. Group 15 and 16 elements such as As, P, S,Se, and Te
2. Metals and ions, e.g., Pb, Hg, Sb, Cd3. Molecules with free electron pairs that are
or aromatic amines, pyridine, and quinoline5. Various compounds which can react with dif-
ferent active sites, e.g., NO, SO2, SO3, CO2.
Elements of class 1 and their compounds, forexample, H2S, mercaptans, PH3, and AsH3, arevery strong poisons for metallic catalysts, espe-cially for those containing Ni, Co, Cu, Fe, andnoble metals [529,530,532].
Elements of the class 2 can form alloys withactive metals and deactivate various systems inthis way [529,533].
CO is chemisorbed strongly on Ni or Co andblocks active sites. Below 450 K and at elevated
80 Heterogeneous Catalysis and Solid Catalysts
pressure the formation of volatile metal carbo-nyls is possible [529,530], and catalyst activityis strongly reduced.
Ammonia, amines, alcohols, and water arewell known poisons for acidic catalysts, espe-cially for those based on zeolites [529,534].
Catalysts or carriers containing alkali metalsare sensitive to CO2, SO2, and SO3.
Catalyst Fouling Because most catalystsand supports are porous, blockage of pores,especially of micropores, by polymeric com-pounds is a frequent cause of catalyst deactiva-tion. At elevated temperatures (> 770 K) suchpolymers are transformed to black carbona-ceousmaterials generally called coke [529,535].Catalysts possessing acidic or hydrogenating –dehydrogenating functions are especially sensi-tive to coking.
There are different types of coke, such as Ca,Cb, carbidic or graphitic coke, and whiskercarbon [529]. Ca is atomic carbon formed asa result of hydrocarbon cracking on nickelsurfaces above 870 K. Ca carbon can be trans-formed at higher temperatures to polymericcarbon (Cb) which has a strongly deactivatingeffect. Ca carbon can also dissolve inmetals andforms metal carbides, and it may precipitate atgrain boundaries. Metal-dissolved carbon mayalso initiate the growth of carbon whiskers,which can bear metal particles at their tops.
Coke formation can be minimized, for ex-ample, in methane steam reforming by suffi-ciently high steam/methane ratio or/and by thealkalization of the carrier.
Thermal Degradation One type of ther-mal degradation is the agglomeration of smallmetal crystallites below the melting point,called sintering [529]. The rate of sinteringincreases with increasing temperature. Thepresence of steam in the feed can accelerate thesintering of metal crystallites.
Another type of thermal degradation are sol-id-solid reactions occurring especially at highertemperatures (above 970 K). Examples are re-actions between metals, such as Cu, Ni, Co, andalumina carriers which result in the formation ofinactive metal aluminates [529,536].
Also, phase changes belong to the categoryof thermal degradations. A prominent example
is the reduction of the surface area of aluminafrom 250 m2 g�1 (g phase) to 1 – 2 m2 g�1
(a phase) by thermal treatment between 870and 1270 K.
Alternating oxidation and reduction of thesystem, as well as temperature fluctuations, areoften accompanied by activity losses and cancause mechanical strain in catalyst pellets.Therefore, mechanical strength of catalyst par-ticles has major industrial importance.
Volatization of Active Components Somecatalytic systems containingP2O5,MoO3,Bi2O3,etc. lose their activity on heating close to thesublimation point. Cu, Ni, Fe and noble metalscan escape from catalysts after conversion tovolatile chlorides if traces of chlorine are presentin the feed.
7.3.2. Catalyst Regeneration
The regeneration of metallic catalysts poisonedby Group 16 elements is generally rather diffi-cult. For example, oxidation of sulfur-poisonedmetallic catalysts converts metal sulfides toSO3, which desorbs from metals. However, ifthe catalyst or carrier contains Al2O3, ZnO,MgO then SO3 forms the corresponding sul-fates.When the catalyst is subsequently broughton-line under reducing conditions, then H2S isformed from sulfates and the catalyst will berepoisoned [529,537].
Therefore it is necessary to remove poisonsfrom the feed as completely as possible. Furtherprevention is the installation of a guard-bedcontaining effective poison adsorbents in frontof the reactor.
Ni catalysts poisoned with CO or HCN canbe regenerated by H2 treatment at temperaturesthat allow formation of methane and NH3,respectively.
The original activity of acidic catalysts poi-soned partially by H2O, alcohols, NH3, andamines can be restored by thermal treatment atsufficiently high temperatures.
From the industrial point of view, the regen-eration of coked catalysts is very important. Theremoval of coke depends on its structure and onthe catalyst composition. Alkali metals, espe-cially potassium, accelerate coke gasification.
Heterogeneous Catalysis and Solid Catalysts 81
Oxidation is the fastest gasification reaction, butit is highly exothermic [529,538]. To maintainthe temperature within allowed limits, mixturesof O2, steam, and N2 are mainly used to removethe coke.
Catalysts deactivated by thermal degradationare very difficult to regenerate. Certain Pt –Al2O3 catalysts, deactivated as a result of ther-mal Pt sintering, can be partly regenerated bychlorine treatment at elevated temperatures,which makes Pt redistribution possible.
7.3.3. Catalyst Reworking and Disposal
The leaching of precious metals from spentcatalysts is widely practiced. Producers of noblemetal catalysts, such as Engelhard Corp. BASF,Johnson Matthey, and Umicore have plants forthis purpose. Yields of recovered precious me-tals are 90 – 98% depending on the originalmetal content and on the nature of the supportused.
Besides precious metals, Ni from spent Ni –kieselguhr catalysts used in fat and oil hydro-genations are reworked to a large extent. BeforeNi leaching, fats must be removed.
Hydrotreating catalysts containing Mo, Nior Co are also reworked. Before leaching ofthe metals, coke and sulfur are removed byroasting [529,530].
In various cases, however, problems arisewhen the price of recovered components doesnot cover the costs of catalyst reworking. Thefinal alternative is the disposal of the spentcatalyst. In general, catalysts containing Al, Si,Fe can be disposed of without any special pre-cautions or can be used in construction materi-als. However, if such catalysts contain Ni or Vaccumulated during their use, then removalof these elements below legislative limits isnecessary [529].
Spent Cu- and Cr-containing catalysts aresometimes accepted by metallurgical plants.
Before disposal, spent catalysts containingvarious contaminants need to be encapsulatedto avoid their release into water. Materials usedfor encapsulation are, e.g., bitumen, cement,wax, and polyethylene [529]. Nevertheless,the disposal of encapsulated catalysts is notonly expensive but is becoming increasinglydifficult.
8. Industrial Application andMechanisms of Selected TechnicallyRelevant Reactions
8.1. Synthesis Gas and Hydrogen[539,540]
All fossil fuels, i.e., coal, petroleum, heavy oil,tar sands, shale oil, and natural gas, but also so-called renewable sources such as biomass, canbeused for the productionof synthesis gas (syngas),a mixture of hydrogen and carbon monoxide invarious ratios. Syngas is an important raw mate-rial for many catalytic syntheses in the chemicalindustry as described in consecutive sections.
By conversion of CO to CO2 in the water-gasshift reaction (WGS), and CO2 separation, hy-drogen can be produced.
H2OþCO!H2þCO2
Direct hydrogen production can also be basedon water electrolysis using electricity fromnuclear power, solar, wind, hydro, geothermal,and oceanic sources as well as combustion(power plants) of any fuel. Due to currentchanges in energy resources, a variety of newroutes are being discussed or are on the way tocommercialization (see also! Gas Productionand! Hydrogen), in many of which heteroge-neous catalytic reactions play a significant role.
Syngas is manufactured from coal by coalgasification (see also! Hydrogen–Productionfrom Coal and Hydrocarbons), and fromgaseous or liquid hydrocarbons by endothermicsteam reforming (SR),
CnHmþnH2O!n COþðnþm/2ÞH2
exothermic partial oxidation (POX)
CnHmþðn/2ÞO2!n COþðm/2ÞH2
and a combination of both, called autothermalreforming (ATR) [541]. Today, natural gas is thedominant feedstock for syngas production.
TheH2/CO ratio can be adjusted by reformingand shift reactions according to the application,e.g., 1/1 for oxo synthesis, 2/1 for production ofmethanol andDME aswell as Fischer – Tropsch(FT) synthesis, 3/1 for methanation. Syngas forammonia synthesis (H2/N2 ¼ 3) ismanufacturedby nitrogen addition in a second reformingstep [542].
82 Heterogeneous Catalysis and Solid Catalysts
Asurveyonsteamreforming isgiven in [542].Nickel is the preferred catalyst but other group8 – 10 metals are active as well, in particular Coand Fe [543]. The expensive metals Pt, Ru, andRh show even higher activities [544]. A varietyof industrial catalysts are available, most ofwhich are based on Ni/alumina with alkali metalpromoters, produced in the shape of pellets withlarge external diameter and high void fraction(rings, cylinders with holes, or ceramic foams),and used in tubular reformers [542]. Presentdevelopments focus on compact reformers, effi-cient coupling with heat exchangers, and heatrecovery from the reformed gas.
An alternative technology for syngas gener-ation is exothermic catalytic partial oxidation(CPOX) of hydrocarbons to syngas over metalcatalysts, in particular Rh. The fuel (natural gas,vaporized liquid hydrocarbons, alcohols), pre-mixedwith oxygen at an atomic C/O ratio of 1 isfed into a catalytic bed ormonolith, in which thefuel is almost completely converted to synthesisgas within few milliseconds at ca. 1000 �C(high-temperature catalysis) [545-547]. Thelargest hurdle for widespread application ofCPOX seems to be safety issues; therefore,membrane [548] and microreactor [549] con-cepts are under development.
Autothermal reforming is a combination ofSR and POX, inwhich the heat for the reformingreaction is supplied by internal combustion ofthe fuel with oxygen. Actually, CPOX can alsobe considered to be a two-stage process, inwhich the oxygen is first used to burn some ofthe fuel and consecutive steam reforming of themajor part of the fuel produces the desiredsyngas [524]. ATR has already been widelyused in chemical industry, and now is alsoconsidered for syngas production in new GTL(gas-to-liquid) plants [550]. ATR runs either onNi- or Rh-based catalysts, usually with aluminaor magnesium alumina supports to improvethermal stability and strength at the high oper-ating temperatures. Current developments in-clude air-blown ATR, in which eliminating theneed for an oxygen plant is traded off by thecompression costs, in particular inGTLwith FT.ATR can also be operated without catalysts.
There are a variety of proposed methods forsyngas production from alternative feeds [551]such as ethanol or any other biomass-derivedfuel. In particular, CPOX and ATR techniques
using noble metal catalysts have been shown toprovide high syngas yields when operated athigh temperatures and millisecond contacttimes [547,552–554].
WGS is the most important step in the indus-trial production of hydrogen, ammonia, andother bulk chemicals utilizing syngas in respectto the adjustment of the CO/H2 ratio [540,555].Depending on the required CO conversion thereaction is carried out in several stages. The firsthigh-temperature shift (HTS) is carried out overFeCr catalysts at temperatures between 280 and350 �C. Since the equilibrium at high tempera-ture is unfavorable for complete conversion, asecond stage, the low-temperature shift (LTS,180 – 260 �C), is added, in which CuZn- orCuZnAl-based catalysts are used to give a COcontent of 0.05 – 0.5 vol %. The produced CO2
can be removed by scrubbing. Complete COremoval, e.g., as needed for ammonia synthesis,can be achieved by subsequent methanationover Ni catalysts. Since sulfur-containing com-pounds such as H2S and carbonyl sulfide (COS)are not removed, separate catalytic treatment isusually necessary, e.g., hydrolysis of COS toH2S or oxidation with SO2 (Claus COS conver-sion) and sour gas shift over Mo catalysts [556].
The production of syngas and subsequenthydrogen is currently also of interest in the areaof energy-related catalysis such as fuel cells.While for stationary applications natural gas isthe major fuel option, logistic fuels are consid-ered as potential feed inmobile applications, forinstance, to provide electricity by an auxiliarypower unit in a vehicle. Logistic fuels can, forinstance, efficiently be converted to syngas incompact on-board CPOX reactors; the syngas isthen fed to a SOFC or a PEMFC; in the lattercase an additional fuel-processing system isneeded for CO removal.
8.2. Ammonia Synthesis[48,557,534,558]
In the Haber – Bosch process, ammonia is syn-thesized over a promoted iron metal catalystsfrom its constituents, nitrogen and hydrogen, atapproximately 400 �C and 15 MPa (for moredetails see also! Ammonia).
N2þ3 H2!2 NH3
Heterogeneous Catalysis and Solid Catalysts 83
Reactors with capacities up to 1000 t/d are used.The reaction toward the target product NH3 istherefore thermodynamically favored at lowtemperature and high pressure. This equilibriumlimitation at practical conditions requires loopoperation with recovery of the easily condens-able product gas. The feed gases are preparedfrom air (nitrogen) and hydrogenvia syngas (seealso Section Synthesis Gas and Hydrogen).
Besides Fe, only Ru has been found to be apractically useful catalyst, although thousand ofsystems have been tested over the years [558].
The most important single application ofammonia is the production of artificial fertilizer.Ammonia is also required in the production ofexplosives, dyestuffs, plastics, and life-scienceproducts. In environmental catalysis, ammoniais applied as reducing agent for nitrogen oxidesemitted in power plants and more recently alsoin automotive vehicles (see also Section Envi-ronmental Catalysis).
The mechanism of ammonia synthesis is oneof the best known in heterogeneous catalysis,besides CO oxidation on Pt (see Chap. Theoreti-cal Aspects), the reaction served as prototype forthe understanding of heterogeneous catalysis byelucidation of the molecular behavior on thecatalytic surface, and represents one of the fewsuccessful examples of bridging the materialsand pressure gap between surface science andindustrial heterogeneous catalysis. Three Nobelprizes in chemistry are closely related to ammo-nia synthesis (HABER 1921, BOSCH 1931, ERTL
2007). Therefore, the mechanism of ammoniasynthesis is discussed in more detail here.
The adsorption of N2 on iron is slow and ischaracterized by a very low sticking coefficient(ca. 10�6) and high activation energy [557].Single-crystal surfaces of iron are reconstructedupon adsorption of nitrogen. Dinitrogen is dis-sociated above 630 K [559] and forms complexsurface structures. These have been inferred tobe surface nitrides with depths of several atomiclayers [557]. Their composition is roughly Fe4N.The corresponding bulk compound is thermody-namically unstable under conditions for whichthe surface structure is stable. The rate of disso-ciative adsorption of dinitrogen is structure-sen-sitive, the Fe(111) face being by far the mostactive, since the activation energy is the smallestand the rate of adsorption the highest [560]. Thesame crystal face is also the catalytically most
active. These observations are consistent withthe earlier suggestion [561] that dinitrogen ad-sorption is an activated process and that it is therate-determining step in the catalytic cycle.
In contrast, the adsorption of dihydrogen oniron is fast and characterized by a high stickingcoefficient (ca. 10�1) and an extremely smallactivation barrier. This chemisorption is disso-ciative and yields covalently bonded H atomswhich have high mobility on the iron surface.
Atomic nitrogen was shown to be the moststable and predominant chemisorbed species onFe(111) after evacuation [87,88], and it wasinferred to be an intermediate in the catalyticreaction. Adsorbed dinitrogen could be excludedas a reactive intermediate. The involvement ofadsorbedNatoms in the rate-determining stepwasalso demonstrated by kinetics experiments [562].Other less stable and less abundant surface inter-mediates include NH and NH2 species.
Based on these results the following se-quence of elementary steps was formulated(* denotes a surface site):
N2þ2*!2 Nads ð36Þ
H2þ2*!2 Hads ð37Þ
NadsþHads!NHadsþ* ð38Þ
NHadsþHads!NH2adsþ* ð39Þ
NH2adsþHads!NH3þ2* ð40ÞA schematic potential-energy diagram for the
catalytic cycle is shown in Figure 27. Decompo-sition of N2 is exothermic, whereas the stepsinvolved in successive hydrogenation yieldingNHx species are endothermic. The addition ofthe first H atom is the most difficult step.
The promotion of the iron catalyst withpotassium lowers the activation energy for dis-sociative N2 chemisorption [563].
8.3. Methanol and Fischer – TropschSynthesis
8.3.1. Methanol Synthesis [148]
Methanol is one of the most important organicchemicals (see also ! Methanol). It is mainly
84 Heterogeneous Catalysis and Solid Catalysts
used as an intermediate for production of form-aldehyde, methyl tert-butyl ether (MTBE),acetic acid, amines, and others. Methanol isproduced from synthesis gas according to thefollowing stoichiometry.
COþ2 H2!CH3OH
It is now generally accepted that this reactionproceeds by conversion of CO via the water-gasshift reaction followed by hydrogenation ofcarbon dioxide [564].
COþH2O!H2þCO2
CO2þ3 H2!CH3OH
All these reactions are exothermic and equilib-rium-limited. The achievable methanol yield isfavored by high pressure and low temperature.
The first process for methanol synthesis,operating at about 30 MPa and 300 – 400 �Cover a Zn/Cr2O3 catalyst, was developed byBASF in Germany in 1923. A substantial im-provement was made by ICI in the 1960sthrough introduction of the more active Cu/ZnO/Al2O3 catalyst, which allowed for synthe-sis under much milder reaction conditions of50 – 100 bar and 200 – 300 �C. Today, the vast
majority of methanol plants use this more ad-vanced low-pressure synthesis.
Althoughmethanol synthesis on copper-basecatalysts has been intensively studied for severaldecades, no general agreement about the natureof the active sites and the reaction mechanismcould be achieved. Regarding the active site, itappears that metallic copper in close contact toZnO is a requirement for an active and selectivecatalyst. This synergy has been explained byvarious mechanisms including hydrogen spill-over fromZnO [565], stabilization of intermedi-ates on ZnO or the interface between Cu andZnO [566], and spreading of Cu on the ZnOsurface [567]. Themost important intermediatesappear to be formate, methoxy, and formylspecies. A possible reaction mechanism in-volves dissociative adsorption of hydrogen, hy-drogenation of adsorbed CO to CO2, conversionof atomic hydrogen to formate, further additionof hydrogen to give H2COO, hydrogenation ofthis species to a methoxy species, and finallyhydrogenation of this group to methanol. Simu-lations suggested that the rate-determining stepin this sequence is hydrogenation of the H2COOspecies to the methoxy group [568].
Several empirical and mechanistically basedrate equations for methanol synthesis have been
Figure 27. Potential-energy diagram for the sequence of elementary steps of the ammonia synthesis reaction (energies inkJ mol�1) [234]
Heterogeneous Catalysis and Solid Catalysts 85
proposed. An example using only statisticallysignificant and physically meaningful para-meters is given in [569]. The usual catalystgeometry comprises pellets of typically fivemillimeters in size. Under the commercial reac-tion conditions, pore diffusion resistances mayoccur [570].
In industrial practice, a variety of differentreactor types for low-pressure methanol synthe-sis are used, but generally fixed beds of catalystoperated in the gas phase are employed. Onepossibility for temperature control during theexothermic reaction is a staged catalyst bedwithinterstage cooling through heat exchangers orinjection of cold synthesis gas. The other fre-quently used possibility for exothermic reac-tions is a cooled multitubular reactor with fixedbed of catalyst in each tube. While the multi-tubular reactor allows the best temperature con-trol and thus the longest catalyst life, the capitalcosts for adiabatic reactors are lower. Two-phase fluidized-bed reactors and three-phasereactors with an additional liquid product phasehave also been extensively tested. However, thelack of mechanical stability and/or low catalysteffectiveness has until now prevented commer-cial implementation of these reactor designs.
Much effort has also been devoted to over-coming the equilibrium-limited methanol con-version in a single-pass reactor by removal ofmethanol from the reaction mixture. A particu-larly interesting system removes methanol byselective adsorption on a porous adsorbent trick-ling through a fixed-bed of catalyst [571]. How-ever, this elegant multifunctional reactor suffersfrom severe practical mechanical problems.Another approach concentrates on operationclose to the dew point of the product to allowremoval of liquid methanol between beds ofcatalyst. In this case, highly active catalysts foroperation at very low temperature and highpressure are required.
A possible future trend is the further proces-sing of methanol to synthetic fuels via metha-nol-to-olefins as proposed by Lurgi.
8.3.2. Fischer – Tropsch Synthesis [148]
Fischer – Tropsch synthesis (FTS) is the directproduction of hydrocarbon chains from synthe-sis gas. Details of the process can also be found
in ! Coal Liquefaction. The most importantreaction, which is exothermic with a reactionenthalpy of about �150 kJ/mol, can be des-cribed by the following equation.
n COþð2nþ1ÞH2!CnH2nþ2þnH2O
In side reactions, olefins and oxygenates areformed, and undesirable CO2 and additionalCH4 may be produced via the water-gas shiftreaction and CO methanation.
FTS has a long history starting with FISCHERand TROPSCH reporting the synthesis of liquidhydrocarbons from synthesis gas under moder-ate conditions in 1923 [572]. Within a shortperiod, this new process was commercializedand provided, together with coal liquefaction,synthetic fuel on a large scale in Germanyduring World War II. After the era of cheap oilbegan in the 1950s it became evident that FTSwas uneconomical at that time. Only SouthAfrica continued production of fuels by FTSbased on coal-derived synthesis gas for politicalreasons. The 1973 oil crisis stimulated newinterest in FTS, and Shell started developmentof its middle distillate process. In 1993, the firstplant based on natural gas came into operation inMalaysia. This gas-to-liquids (GTL) process iscurrently being realized on industrial scale atseveral sites. Commissioning of a plant with acapacity of 70 000 barrels per day built by Sasoland Qatar Petroleum took place in 2006 inQatar. Further large-scale industrial plants inEscravos, Nigeria (Sasol Chevron) and Qatar(Shell, Qatar Petroleum) are under construction.It appears that FTS will play a major role for thefuture production of synfuels based on alterna-tive feedstocks (natural gas, coal, biomass).
FTS has been considered as an ideal poly-merization reaction [573]. According to thisapproach the distribution of mole fractions xnof products can be described as a function of thenumber of carbon atoms n in the chain.
xn ¼ ð1�aÞan�1
The ideal product composition depends onlyon the chain-growth probability a, which isdetermined by the catalyst used. In reality sig-nificant deviations from ideal polymerizationbehavior are observed. Usually the methanemole fraction is higher, while the ethene/ethanemole fractions are lower than calculated. Many
86 Heterogeneous Catalysis and Solid Catalysts
mechanistic studies on FTS support the carbenemechanism, which starts with the decomposi-tion of CO and involves the insertion of methy-lene (CH2) species into the growing alkylchain [574].
As FTS catalysts metals like iron, cobalt, andruthenium can be used [575]. Due to the highprice of ruthenium only iron and cobalt haveindustrial relevance. A disadvantage of ironcatalysts is kinetic inhibition by the co-productwater, whereas an advantage is the activity forthe water-gas shift reaction that allows the useof carbon-dioxide-containing or hydrogen-depleted synthesis gas mixtures [576]. Com-pared to iron, cobalt catalysts are already activeat lower reaction temperatures and have a dura-bility of up to five years on stream comparedto about six months in the case of iron [577]. Onthe other hand cobalt is more expensive thaniron. In addition to the active component differ-ent promoters (Pt, Pd, Ru, Re, K) can be em-ployed [578]. As carrier materials alumina,silica, and titania can be utilized. Typical chain-growth probabilities are 0.5 – 0.7 for iron and0.7 – 0.8 for cobalt [579]. Currently the deve-lopment of cobalt catalysts is aimed at maxi-mizing the chain-growth probability to values ofup to 0.95 [580]. Since the product mixturesobtainedwith these catalyst cannot directly usedand must be further processed to achieve thedesired fractions (diesel and gasoline fuels), ithas been suggested to couple Fischer – Tropschcatalysts with hydrocracking catalysts in onereactor [581,582].
As liquid products often fill the pore systemof working catalysts, resistances caused by porediffusion may occur even with small catalystparticles. Catalyst efficiency is significantlyreduced at characteristic catalyst dimensionsabove 100 mm [583]. Furthermore, the higherdiffusion coefficient of hydrogen compared tocarbon monoxide increases the H2/CO ratioinside the porous catalyst. This leads to anincrease of the chain-termination probabilityand thus to a decrease in chain length of theproducts [584].
The development of catalysts with very highchain-growth probabilities resulted in the devel-opment of the more advanced low-temperatureFTS, in which synthesis gas and liquid productsare present under reaction conditions. Industrialreactors are operated at typical conditions of
2 – 4 MPa and 220 – 240 �C.Two reactor typesare presently applied in low-temperature FTS: acooled fixed-bed reactor mainly used by Shelland a slurry bubble column developed by Sasol.Disadvantages of fixed-bed reactors are devel-opment of a hot spot, low catalyst utilization dueto pore diffusion, and, especially in case of gasrecycle for improved heat removal, high pres-sure drop. On the other hand, mechanical stresson catalyst particles, the need for separation ofsolid catalyst and liquid products and highlydemanding scaleup are drawbacks of slurrybubble column reactors. New trends in FTSreactors are the use of monolith reactors forimproved gas – liquid mass transfer [585] andisothermal microstructured reactors.
8.4. Hydrocarbon Transformations
8.4.1. Selective Hydrocarbon OxidationReactions
Selective hydrocarbon oxidation reactionsinclude several important classes of heteroge-neously catalyzed reactions, which find large-scale industrial application for the synthesis ofbulk chemicals. Reviews on the mechanisms ofselective hydrocarbon oxidation [200], oxida-tive dehydrogenation of alkanes [586], ammox-idation of alkenes, aromatics and alkanes [63],and epoxidation of alkenes [587] are available.Here, some mechanistic aspects of the epoxida-tion of alkenes and of the ammoxidation ofhydrocarbons are discussed.
8.4.1.1. Epoxidation of Ethylene and Pro-pene [587,588]The epoxidation of ethylene by dioxygen iscatalyzed by silver metal and yields ethyleneoxide (! Ethylene Oxide), an important inter-mediate for the synthesis of glycols and polyols.
Total oxidation of the reactant and thetarget product limit the selectivity of the
Scheme 1.
Heterogeneous Catalysis and Solid Catalysts 87
process. Scheme 1 shows the three competingreactions.
The catalyst therefore must be tuned suchthat the optimal selectivity for ethylene oxide isachieved. The active phase consists of large Agparticles supported on low surface area a-Al2O3
promoted by alkali metal salts. A beneficialeffect is also obtained by adding chlorine-con-taining compounds such as vinyl chloride to thereaction feed. Under reaction conditions thisadditive is readily combusted on silver, andchlorine is adsorbed on the metal surface.
Oxygen can be adsorbed on transition metalsin general and on silver in particular in threedifferent states: (1) molecular dioxygen, (2)adsorbed atomic oxygen, and (3) subsurfaceatomic oxygen [588]. Molecular oxygen is sta-ble on anAg(111) surface at temperatures belowca. 220 K. It dissociates at higher temperatures.Oxygen dissociation occurs at high-coordina-tion sites, since at least two neighboring metalatoms must be available. It has been shown thatensembles with a minimum of five silver atomsare required [589,590]. Oxygen atoms adsorbedoriginally on the external silver metal surfacemay move to subsurface lattice positions. Sub-surface oxygen atoms have been proved to formon transition metals including Rh, Pd, andAg [591]. The maximum oxygen coverage onsilver surfaces is one oxygen atom per silveratom, corresponding to the composition ofAgO [588].
The presence of subsurface oxygen atomsreduces the electron density on adjacent silveratoms. Hence, oxygen atoms adsorbed on theexternal surface which share bonds to silversurface atoms with subsurface oxygen atomsbecome highly polarizable. When exposed toethylene, the interaction of the surface oxygenatoms with thep electrons of ethylene leads to aflowof electron density from the surface oxygenatom to the positively charged surface silveratom [592]. The surface oxygen atoms behavechemically as electrophilic oxygen atoms,which preferentially react with the part of thereactant molecule having the highest electrondensity. This situation is most likely at highoxygen coverages, consistent with the experi-mental observation that the epoxidation selec-tivity is dramatically enhanced by increasingoxygen coverage [593]. Scheme 2 illustratesthis scenario [587]. At low oxygen coverages
the density of subsurface oxygen atoms is alsoreduced so that the polarizability of oxygenatoms adsorbed on the external surface is re-duced. Consequently, these oxygen atoms be-have as nucleophilic oxygen atoms and tendto interact preferentially with hydrogen atomsof the ethylene molecule, thus leading to totaloxidation. This situation is schematically shownin Scheme 3 [587]. Therefore, epoxidationselectivity must decrease with decreasing oxy-gen coverage. The fact that vacant silver sitesexist in the vicinity of an adsorbed oxygenatom at low coverage (see Scheme 3), is alsodetrimental.
The influence of alkali metal and chlorinemodifiers is complex. The effect of chlorine istwofold: (1) it suppresses vacant silver sites, and(2) it enhances the electron deficiency of silver.The latter effect is due to the ability of chlorineto also occupy subsurface positions [594] andthus to adopt the role of subsurface oxygen asillustrated in Scheme 4 [587]. These effectsimprove the initial selectivity r1/r2 (ri denotesa reaction rate, see Scheme 1). The overallselectivity is also reduced by subsequent com-bustion of the epoxide (r3 in Scheme 1), partic-ularly at high conversions. The combustionof the epoxide is induced by the residualacidity of the a-Al2O3 support. The presence
Scheme 2.
Scheme 3.
88 Heterogeneous Catalysis and Solid Catalysts
of alkali metal reduces the density of acid sitesand thus has a beneficial effect on selectivity byblocking reaction step r3.
The rate-limiting step of the epoxidationreaction is the dissociative chemisorption ofdioxygen. Alkali metal compounds enhance thedissociation rate of dioxygen by reducing theactivation barrier, and consequently the alkalimetal modifier enhances the epoxidation rateas its coverage increases [587]. Interestingly,when a chlorine-modified catalyst is promotedby alkali metal compounds, the reaction ratedecreases, and this is suggestive of an enhance-ment of the steady-state concentration of ad-sorbed chlorine, which leads to site blocking.Therefore, there is a very subtle interplay be-tween the two additives which must be carefullycontrolled to optimize conversion and selectivityof the ethylene epoxidation reaction.
Details on the catalytic and engineering as-pects of ethylene epoxidation can be foundin [148,595]. The reaction is carried out inmultitubular reactors in the gas phase, eitherwith air or with pure oxygen, at residence timesof about 1 s, temperatures between 230 and290 �C and pressures between 1 and 3 MPa. Inthe earlier air-based process, a series of two orthree reactors was employed. The ethylene con-version in the first reactor is kept relatively low(ca. 40%) to maintain high ethylene oxide se-lectivity,while the following reactors are used toincrease the overall ethylene conversion. Mod-ern ethylene oxide processes use pure oxygen ina single-stage reactor in recycling mode. Incontrast to air-based processes, ethylene con-centrations are relatively high (25 – 30 vol %)in order to stay above the upper flammabilitylimit of the reaction mixture. The oxygen pro-cess gives rise to higher ethylene oxide yield,
smaller equipment size, and smaller amount ofvent gas and is therefore nowadays preferredover the air-based process.
The epoxidation of propenewith dioxygen isunfavorable because of the enhanced reactivityof the methyl group for nucleophilic attack.Activation of the methyl group leads to theallyl or combustion of the propylene epoxide.Alternative oxidants are hydrogen peroxide orhydroperoxide (! Propylene Oxide–IndirectOxidation Routes). The reaction of propenewith hydrogen peroxide yields the target prod-uct propylene epoxide and water (Eq. 41).
(41)
The preferred catalyst for this reaction istitanium silicalite-1 (TS-1) (see Section MetalOxides), in which four coordinate Ti4þ plays thedecisive role [596]. Although the exact nature ofthe reaction intermediate is not known yet,hydrogen peroxide may coordinate nondisso-ciatively to a Lewis acidic tetrahedral Ti4þ siteas shown in Scheme 5. This induces electrondeficiency on the oxygen atoms of the peroxi-des, which is favorable for epoxidation. Ananalogous reaction path has been proposed forthe homogeneous epoxidation of propene byperoxides [587].
The development of technical processesbased on hydrogen peroxide and TS-1 catalystshas recently been reviewed [597]. These HPPO
Scheme 4.
Scheme 5.
Heterogeneous Catalysis and Solid Catalysts 89
processes have been developed further by De-gussa and Uhde, as well as by BASF and Dow,and startup of first production plants is sched-uled for 2008 [598]. Degussa and Uhde havealso investigated the gas-phase epoxidation ofpropene. Due to the safety risks associated withmixtures of gaseous propene and hydrogenperoxide as well as the danger of hydrogenperoxide decomposition during evaporation,new technical concepts based on microstruc-tured devices had to be developed [495]. Highproductivities of more than 1 kg of propyleneoxide per kilogram of catalyst and hour could beobtained at high propane-to-propene oxide se-lectivity of more than 90 %. If the decomposi-tion of hydrogen peroxide can be significantlyreduced, the gas-phase process could becomeinteresting alternative to the commercial liquid-phase processes.
8.4.1.2. Ammoxidation of Hydrocarbons[63,599,600]In ammoxidation, ammonia reacts with a reduc-ible organic molecule, most frequently an al-kene, alkane, or aromatic, in the presence ofdioxygen to yield nitriles (e.g., Eq. 42).
2 CH2 ¼ CRCH3þ 2 NH3þ 3 O2!2 CH2
¼ CRCNþ 6 H2O
The ammoxidation of an alkene is a six-electron oxidation that produces an unsaturatednitrile and water. The reaction is related to thefour-electron oxidation of alkenes (Eq. 42) [75]producing unsaturated aldehydes andwater, andto the two-electron oxydehydrogenation of al-kenes to dienes and water (Eq. 43) [75].
CH2 ¼ CRCH3þO2!CH2 ¼ CRCHOþH2O
2 CH2 ¼ CHCH2CH2RþO2!2 CH2
¼ CHCH ¼ CHRþ 2 H2O
Catalysts for these reactions are complexmixedmetal oxides containing variable-valenceelements (see Section Metal Oxides), the am-moxidation catalysts typically being the mostcomplex. These materials possess redox prop-erties, i.e., they can readily be reduced byammonia and reoxidized by dioxygen presentin the gas phase. It is the lattice oxygen of thecatalyst which reacts with ammonia and the
hydrocarbon, and the reduced solid is reoxi-dized by gas-phase oxygen (Mars – van Kreve-len mechanism [601], see also [200]).
The most important alkene ammoxidation isthat of propene to acrylonitrile (Sohio Acryloni-trile Process, Eq. 44; see also! Acrylonitrile–Quality Specifications and Chemical Analysis)[586]
2 CH2 ¼ CH�CH3þ 2 NH3þ 3 O2!2 CH2
¼ CHCNþ6 H2O
Molybdates and antimonates can be used ascatalysts for this reaction. The active sites arethought tohave bifunctional nature [63,602,603].Ageneralized catalytic cycle for alkene ammoxi-dation is shown in Figure 28 [63]. Ammonia isproposed to interact first with the bifunctionalactive site generating an ammoxidation site. Thealkene coordinates to this site to form an allylicintermediate. After several rearrangements andoxidation steps, the surface intermediate is trans-formed into the nitrile, which subsequently des-orbs. A reduced surface site is thus formed,which is restored to its original fully oxidizedstate by lattice oxygenO2�, which is provided byadjacent reoxidation sites. These sites then dis-sociate dioxygen to lattice oxygen. The newlyformed lattice oxygen then diffuses to the oxy-gen-deficient reduced surface site, from wherevacancies simultaneously penetrate through thelattice of the solid to the reoxidation sites. Clear-ly, these sites must communicatewith each othervia a common solid-state latticewhich is capableof facile transport of electron, anion vacancies,and lattice oxygen [63]. As an example, theproposed bifunctional active site of Bi2MoO6
(see Section Metal Oxides) is schematically illu-strated in Figure 29 [213]. The various function-alities were assigned to specific elements and tospecific lattice oxygen positions. Bridging oxy-gen atoms Bi–O–Mo are considered to be re-sponsible for a-hydrogen abstraction from thealkene, while oxygen atoms associated with Moare responsible for oxygen (Mo¼O) andnitrogen(Mo¼NH) insertion into an allylic intermediate.The oxygen dissociation and its reduction tolattice oxygen is assumed to occur in theregion of high electron density generated by thetwo lone pair electron orbitals of Bi–O–Bisites. More easily reducible elements than Biare Fe, Ce, U, and Cu, which are components of
(42)
(43)
(44)
90 Heterogeneous Catalysis and Solid Catalysts
more complex, multicomponent catalysts (seeSection Metal Oxides) [63]. As an illustration ofthe mechanisms of ammoxidation and selectiveoxidation of propene, Figure 30 shows the pro-posed catalytic cycles for the two reactions [604].
More recently, selective catalytic oxidationand ammoxidation of alkanes as lower costalternatives to alkenes has attracted consider-able interest [600,605]. Multicomponent metaloxide catalysts have been intensively studied.Promising results have been obtained especiallywith the MoV – TeNbO system, both for oxida-
tive dehydrogenation of ethane to ethylene andfor ammoxidation of propane to acrylonitrile.
(see also ! Oil Refining–Environmental Pro-tection in Oil Refining)
Hydroprocessing treatment, including hy-drodesulfurization (HDS), hydrodenitrogena-tion (HDN), hydrodeoxygenation (HDO),hydrometalation (HDM), hydrogenation, andhydrocracking, are among the largest industrialprocesses in terms of catalyst consumption.Crude petroleum contains particularly organo-sulfur and organonitrogen compounds, whichare most abundant in heavy petroleum fractions.These contaminants must be removed for envi-ronmental reasons. The reactions take place inthe presence of H2 at high temperatures (ca.600 – 700 K) and pressures of 500 kPa to1 MPa. Because of the lower reactivity of or-ganonitrogen compounds as compared to orga-nosulfur compounds, the reaction conditions aremore severe for HDN than for HDS.
Catalysts for hydroprocessing are highly dis-persed metal sulfides (mainly MoS2, but also
Figure 28. Generalized mechanistic cycle for alkene ammoxidation
Figure 29. Schematic representation of the active site ofBi2MoO6 [446]O0 ¼ Oxygen responsible for a-H abstraction; O00 ¼ Oxy-gen associated with Mo; responsible for oxygen insertioninto the allylic intermediate; f ¼ Proposed center for O2
reduction and dissociative chemisorption.
Heterogeneous Catalysis and Solid Catalysts 91
WS2) supported on g-Al2O3. The materials arepromoted by cobalt or nickel, depending onapplication.
Although the detailed mechanisms have notyet been elucidated, significant progress in un-derstanding the chemistry of the various hydro-processing reactions at a molecular level hasbeen made [49,273,274,607,608]. In the follow-ing, however, the focus is on reaction networksof several hydroprocessing reactions with pseu-do-first-order rate constants for individual reac-tion steps.
The organosulfur compounds in petroleuminclude sulfides, disulfides, and aromatics (in-cluding thiophene, benzothiophene, dibenzo-thiophene, and related compounds). Benzo- anddibenzothiophene are predominant in heavyfuels. The reaction network for hydrodesulfur-ization of dibenzothiophene, a representativemember of organosulfur contaminants in fuel,is shown in Figure 31 [609]. Hydrogenation andhydrogenolysis take place in parallel. The latterreaction is essentially irreversible and leads tothe formation of H2S and biphenyl. At low H2Sconcentrations in the feed, the sulfide catalystsare highly selective for hydrogenolysis. The
selectivity, however, drops sharply as the H2Sconcentration in the feed increases.
Hydroprocessing reactions accompanyinghydrogenation and hydrodesulfurization includehydrodenitrogenation, whereby organonitrogencompounds in the feed react with H2 to give NH3
and hydrocarbons. As an example, a reactionnetwork for the hydroprocessing of quinoline isshown in Figure 32 [606]. The supported metalsulfide catalysts are much less selective fornitrogen removal than for sulfur removal.
Hydroprocessing reactions are carried out indifferent reactor types [610]. Themost common-ly used is a fixed-bed reactor operated in thetrickle-flow regime with cocurrent up- or down-flow of gas and liquid. Alternative reactor de-signs aremoving-bed and ebullated-bed reactorswith greater flexibility, e.g., owing to easy re-placement of spent catalyst during operation.Recently, structured packed columns of mono-lithic catalysts operated in countercurrent modeare gaining importance in hydroprocessing re-search, because higher conversions can be ob-tained. A future trend is the development ofprocesses for the treatment of increasingly heavyoils and of various residues [611].
Figure 30. Mechanism of selective ammoxidation and oxidation of propene over bismuth molybdate catalysts [604]
92 Heterogeneous Catalysis and Solid Catalysts
Figure 31. Reaction network for hydrodesulfurization and hydrogenation of dibenzothiophene catalyzed by sulfided Co –Mo/Al2O3 at 570 K and 10 MPa [609]Numbers next to the arrows represent the pseudo-first-order rate constants in units of L/(g of catalyst � s) when the H2Sconcentration is very small. Addition of H2S markedly decreases the selectivity for hydrodesulfurization.
Figure 32. Reaction network for hydrogenation and the hydrogenolysis of quinoline catalyzed by sulfided Ni – Mo/g-Al2O3
at 620 K and 3.5 MPa [606]Numbers next to the arrows represent the pseudo-first-order rate constants in units of L/(g of catalyst � s) when the H2Sconcentration is small but sufficient to maintain the catalysts in the sulfided forms.
Heterogeneous Catalysis and Solid Catalysts 93
8.5. Environmental Catalysis
8.5.1. Catalytic Reduction of NitrogenOxides from Stationary Sources [313]
Fossil fuels such as coal, oil, gas, and others areburnt or gasified for energy conversion. InWestern European countries and Japan mea-sures have been implemented since 1980 forreducing emissions, especially of NOx frompower plants. The preferred method to removeNOx from exhaust gases of power plants, indus-trial boilers, and gas turbines is based on the so-called selective catalytic reduction (SCR) withammonia in the presence of oxygen. The stoi-chiometry of the main desired reactions can bedescribed as follows:
The standard SCR reaction is most importantif NOx originates from high-temperature com-bustion processes, where very little NO2 ispresent.However, in exhaust streams containinghigher amounts of nitrogen dioxide, the fastSCR reaction, which proceeds at least ten timesfaster than the standard SCR reaction, maybecome the predominant reaction [612].
At higher temperatures above ca. 450 �C, thereducing agent ammonia reacts with oxygen inundesirable parallel reaction to give the pro-ducts N2, N2O, or NO. On the other hand, attemperatures below 200 �C, ammonia and NOx
may form solid deposits of ammonium nitrateand nitrite.
The SCR of nitrogen oxides was first carriedout with Pt catalysts [613]. Due to the highnitrous oxide selectivity of this catalyst, basemetal catalysts have been developed for NOx
reduction. Vanadia supported on titania (in theanatase form) and promoted with tungsten ormolybdenum oxide exhibits the best catalyticproperties.While BASFwas the first to describevanadia as active component for SCR [614],TiO2-supported vanadia for treatment of ex-haust gases was developed in Japan [615]. Ana-tase is the preferred support for SCR catalysts
for two main reasons. Firstly, it is only moder-ately sulfated under real exhaust gas conditions,and catalytic activity even increases after sulfa-tion [616]. Secondly, vanadia is able to spread inthin layers on the anatase support to give highlyactive structures with large surface area. How-ever, the amount of vanadia in technical cata-lysts is limited to only a few weight percent,because it is also catalytically active for SO2
oxidation.The mechanism of the standard SCR reac-
tion over vanadia-based catalysts is generallyassumed to proceed via an Eley – Ridealmechanism involving adsorbed ammonia andgas-phase NO. Based on this mechanism, thefollowing rate equation can be derived thathas been successfully used to model the SCRreaction.
rNO ¼ k � KNH3cNH3cNOð1þKNH3cNH3Þ
Water vapor has an additional inhibitingeffect on the rate of NO removal. In recentstudies, it was also possible to model and simu-late the transient behavior of SCR catalystsexposed to changes in reactant concentrationand temperature [313].
As the rate of the SCR reaction under indus-trially relevant conditions is quite high, externaland intraparticle diffusion resistances play animportant role, especially for the frequentlyused honeycomb monolith or plate-type cata-lysts operating in laminar flow regime. Thesegeometries must be used to minimize the pres-sure drop over the catalyst bed. Monolithicelements usually have channel sized of 3 –7 mm, cross sections of 15 � 15 cm, andlengths of 70 – 100 cm. Monoliths or packagesof plate catalysts are assembled into standardmodules, which are then placed in the SCRreactors as layers. These modules can be easilyreplaced to introduce fresh or regeneratedcatalysts.
SCR reactors can be used in different con-figurations, depending on fuel type, flue gascomposition, NOx threshold, and other factors.The first possibility is the location directly afterthe boiler (high-dust arrangement) where theflue gas usually has the optimal temperature forthe catalytic reaction. On the other hand, dustdeposition and erosion as well as catalyst deac-tivation are more pronounced than in other
94 Heterogeneous Catalysis and Solid Catalysts
configurations. A second possibility, which iscommon in Japan, uses the SCR reactor after ahigh-temperature electrostatic precipitator fordust removal (low-dust arrangement). In thatcase catalyst damage by dust can be prevented.On the other hand, ammonium sulfate deposi-tion,which in the high-dust configurationmainlytakes place on the particulate matter in the gasstream, may become more critical. For thisreason, especially low limits for ammonia slipmust be met. Finally, the SCR reactor may belocated in the cold part after the flue gas desul-furization unit in the so-called tail-end arrange-ment. To achieve the required reaction tempera-ture, the exhaust gases must be reheated bymeans of a regenerative heat exchanger and anadditional burner. On the other hand, catalystswith very high activity can be used, since poisonsare absent and SO2 oxidation need not to beconsidered.
New promising catalysts for the removalof nitrogen oxides are iron-exchanged zeolites(e.g., MFI, BEA). Although field tests in fluegases of power plants have shown quite strongdeactivation, notably by mercury [617], thesecatalysts appear to be especially suited for“clean” exhaust gases such as in nitric acidplants. Advantages of iron zeolite catalysts area broader temperature window for operationand the ability to reduce N2O emissions aswell. Uhde has recently developed the EnviNOx
process for simultaneous reduction of NOx andN2O, which uses iron zeolite catalysts providedby S€ud-Chemie [618].
8.5.2. Automotive Exhaust Catalysis [619–622]
This section focuses on the catalysis-relateditems of automobile emission control. A moredetailed discussion considering all aspects canbe found in ! Automobile Exhaust Control.
Internal combustion engines in automobilesrepresent a major source for the emission ofNOx, CO, and unburnt hydrocarbons (HC),while diesel engines contribute to the emissionof soot as well. The most appropriate way tominimize these air pollutants is themodificationof the combustion process. Aside from theseprimary measures, current legislative emissionstandards can only be met by additional, sec-
ondary measures for exhaust purification byapplication of catalysts. The importance of en-vironmental catalysis will further increase infuture due to the tightening of emission limitsand an increasing number of automobiles. Al-ready today (ca. 2008), environmental applica-tions exhibit a worldwide market share of 35 %among all catalytic processes, and more than70 � 106 automotive catalyst devices are pro-duced per year.
The catalytic system used for aftertreatmentof the exhaust gas primarily depends on the fuel(gasoline, diesel, biofuels) and the operatingconditions. In principle, a distinction is madebetween stoichiometrically operated gasolineengines, lean-operated gasoline engines, anddiesel engines producing different major pollu-tants, namely, CO/NOx/HC,NOx, andNOx/soot,respectively.
Three-Way Catalyst The most frequenttype of catalytic converter in automobiles is thethree-way catalyst (TWC) for stoichiometri-cally operated gasoline engines with an annualproduction of over 60 � 106 units. TWC sys-tems have been applied in gasoline enginessince the 1980s and contain Pt/Rh or Pd/Rh inthe mass ratio of approximately 5/1 with a totalloading of precious metals of ca. 1.7 g/L. TheTWC simultaneously converts NOx, CO, andHC to N2, CO2, and H2O [619–623]. Thecatalytic components are supported by a cor-dierite honeycomb monolith coated with highsurface area g-Al2O3. This washcoat layer ad-ditionally contains thermal stabilizers, for in-stance, La2O3, as well as the oxygen-storagecomponent CeO2. Ceria is able to release oxy-gen under rich conditions and thus maintainHC and CO abatement and avoid the emissionof H2S. The TWC process exclusively occurswithin a narrow range of O2 content that isclose to stoichiometric combustion conditions,i.e., when the air coefficient l ranges from 0.99to 1.01. To realize these conditions an oxygensensor is used which measures the air coeffi-cient of the exhaust stream and forces theengine management system to regulate theair/fuel ratio. The TWC process involves acomplex network of numerous elementary re-actions [624], whereby the effectiveness ofthe catalyst is closely related to the specificactivity of the precious metals and their surface
Heterogeneous Catalysis and Solid Catalysts 95
coverage. The transfer of TWC technology tolean-burn gasoline and diesel motors is prob-lematic because of the insufficient NOx abate-ment. This is associated with the lower rawemissions of reducing agents aswell as the highcontent of O2, which enhances oxidation of HCand CO and thus suppresses NOx reduction.Therefore, alternative concepts are required forthe reduction of NOx under lean-burn condi-tions. For this purpose selective catalytic re-duction by NH3 and NOx storage and reductioncatalysts are being considered in the automo-tive industry.
Selective Catalytic Reduction (SCR) of NOx
by Ammonia. The SCR procedure is theonly technique that selectively converts NOx toN2, even under strongly oxidizing conditions.Thus, SCR has been considered as the technol-ogy of choice for NOx removal in lean-burnengines. Indeed, the SCR process covers therelevant temperature range of diesel engines andprovides effective NOx abatement. Thus, SCRhas advanced to a state-of-the-art technology forheavy-duty vehicles. However, in mobile appli-cations the storage of NH3 is a problem. There-fore, an aqueous solution of urea (32.5 wt %)called AdBlue is currently used. The urea solu-tion is sprayed into the tailpipe, where ammoniais produced after thermolysis and hydrolysis ofthe vaporizing urea – water droplets. Currentresearch focuses on optimization of the dosingsystem and the development of vanadia-freecatalysts, for instance, by substitution withFe-ZSM5 zeolites [622]. Alternative reducingagents such as hydrocarbons and hydrogen hasbeen discussed as well.
NOx Storage Reduction Catalysts NOx
storage reduction catalysts (NSC) were origi-nally developed for lean spark-ignition en-gines and are currently being transferred todiesel passenger cars. The NSC procedure isbased on periodic adsorption and reduction ofNOx [625]. The catalysts consist of Pt, Pd, andRh in the mass ratio of approximately 10/5/1with a total precious metal load of ca. 4 g L�1.NSCs contain basic adsorbents like Al2O3
(160 g L�1), CeO2 (98 g L�1) and BaCO3
(29 g L�1, as BaO equivalent) [626]. In thelean phase of the engine (general operationmode), NOx of the exhaust is adsorbed on the
basic components of the NSC, mainly onbarium carbonate to form the nitrate. Whenthe storage capacity is reached, the engineis operated under rich conditions for a fewseconds to give an exhaust containing CO,HC, and H2 as reducing agents for catalystregeneration (back-transformation of the ni-trate to the carbonate).The effect of the Bacomponent is to adsorb NOx at temperaturesabove 250 �C, whereas substantial storage isalso provided by Al2O3 and CeO2 at lowertemperatures [626].
Catalytic CO oxidation Catalytic COoxidation is an essential reaction of TWC andNSC and has also applied in diesel enginessince the 1990s in the so-called direct oxidationcatalyst (DOC). Furthermore, the catalyticabatement of CO is also a state-of-the-art tech-nology for gas turbine engines fed by naturalgas. DOCs usually contain Pt as an activecomponent showing outstanding performance.The expensive platinum can be substituted bythe less active but cheaper palladium. Theprecious metal load of a DOC is ca. 3 g L�1.DOCs also oxidize gaseous HC and HC ad-sorbed on soot particles.
Removal of Soot The diesel particulatefilter (DPF) is used for the removal of soot fromdiesel exhaust. DPFs mechanically separatethe particles by forcing the exhaust gas todiffuse through porous walls thus leading tohigh filtration efficiency [627]. The DPF ap-plication requires regeneration, i.e., oxidationof the stored soot particles. Soot deposits canproduce a substantial backpressure leading toincreased fuel consumption and decreased en-gine efficiency. The preferred method for DPFregeneration is the CRT (continuously regen-erating trap) technology involving the initia-tion of soot oxidation by NO2 produced byoxidation of NO on Pt catalysts, as in NSCand SCR. The Pt catalyst can be applied in formof a precatalyst, and coating on the DPF. Fur-thermore, so-called fuel-borne catalysts(FBC), which are organometallic compoundsbased on Ce or Fe, e.g., ferrocene, can be addedto the fuel [628]. FBCs also decrease sootemissions from the engine by direct oxidationof soot in the engine. Additionally, they areembedded in the soot particles.
96 Heterogeneous Catalysis and Solid Catalysts
References
1 Appl. Catal. A, General 173 (1998) N3.2 J. J. Berzelius, Ann. Chim. Phys. 61 (1836) 146.3 B. H. Davis, “Development of the Science ofCatalysis” in G. Ertl, H. Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim, 1997, p. 13.
4 W. Ostwald, Nature 65 (1902) 522.5 P. Sabatier, J. B. Senderens, C. R. Acad. Sci. 134(1902) 514.
6 H. Heinemann, “Development of IndustrialCatalysis” in G. Ertl, H. Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 35.
7 S. A. Topham in J. R. Anderson, M. Boudart(eds.): Catalysis: Science and Technology,Vol. 7, Springer, Berlin 1987, p. 1.
8 F.Aftalion:AHistory of the InternationalChem-ical Industry, University of Pennsylvania Press,Philadelphia 1991.
9 A. Mittasch, Adv. Catal. 2 (1950) 81.10 DE 301 231, 1919(F. Bergius, J. Billwiller).11 F. Fischer, H. Tropsch, Brennstoff-Chem. 4
(1923) 276.12 DE 103 362, 1943(O. Roelen).13 J. K. A. Clarke, J. J. Rooney, Adv. Catal. 25
(1976) 125.14 A. J. Gellman,Curr. Opin. Solid StateMater. Sci.
5 (2001) 85.15 J. N. Armor, Appl. Catal. 78 (1991) 141.16 A. Chauvel, B. Delmon, W. F. Hoelderich, Appl.
Catal. A 115 (1994) 173.17 M. Misono, N. Nojiri, Appl. Catal. 64 (1990) 1.18 A. J. Gellman, Acc. Chem. Res. 33 (2000) 19.19 M. Boudart in G. Ertl, H. Kn€ozinger, J. Weit-
kamp (eds.): Handbook of Heterogeneous Ca-talysis, Wiley-VCH, Weinheim 1997, p. 1.
20 A. Balandin, Adv. Catal. Rel. Subj. 19 (1969) 1.21 W. J. M. Rootsaert, W. M. H. Sachtler, Z. Phys.
Chem. 26 (1960) 16.22 I. Langmuir, J. Am. Chem. Soc. 37 (1915) 1139.23 I. Langmuir, Trans. Faraday Soc. 17 (1922)
607.24 H. S. Taylor, Proc. Roy. Soc. (London) A 108
(1925) 105.25 M. Che, C. O. Bennett,Adv. Catal. 36 (1989) 55.26 G.-M. Schwab, E. Pietsch, Z. Phys. Chem. 131
(1929) 385.27 S. M. Davis, G. A. Somorjai in D. A. King, D. P.
Woodruff (eds.): The Chemical Physics of SolidSurfaces and Heterogeneous Catalysis, Vol. 4,Elsevier, New York 1982, p. 217.
28 P. G. Menon, T. S. R. Prasada Rao, Catal. Rev.-Sci. Eng. 20 (1979) 97.
29 M. Boudart, A. Aldag, J. E. Benson, N. A.Dougharty, C. G. Harkins, J. Catal. 6 (1966)92;M. Boudart, A. Aldag, L. D. Ptak, J. E.Benson, J. Catal. 11 (1968) 35.
30 G. A. Somorjai, Ann. Rev. Phys. Chem. 45(1994) 721.
31 G. Ertl, Adv. Catal. 45 (2000) 1.32 V. Ponec, W. M. H. Sachtler in G. C. Bond, P. B.
Wells, F. C. Tompkins (eds.): Proc. 5th Intern.Congr. Catal., Vol. 1, The Chemical Society,London 1976, p. 645.
33 R. L. Burwell, G. L. Haller, K. C. Taylor, J. F.Read, Adv. Catal. 20 (1969) 1.
34 R. L. Burwell in J. R. Anderson, M. Boudart(eds.): Catalysis: Science and Technology,Vol. 9, Springer, Heidelberg 1991, p. 1.
35 W. M. H. Sachtler in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 3, Wiley-VCH, Weinheim 1997,p. 1077.
36 B.Hammer, J. K.Nørskov,Adv. Catal. 45 (2000)71.
37 R. A. van Santen: Theoretical HeterogeneousCatalysis, World Scientific, Singapore 1991.
38 R. A. van Santen, M. Neurock in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 3, Wiley-VCH,Weinheim 1997, p. 991.
39 J. M. Basset, J. P. Candy, A. Choplin, B. Didil-lon, F.Quignard,A. Theolier in J.M.Thomas,K.I. Zamaraev (eds.): Perspectives in Catalysis,Blackwell Scientific Publications, London1992, p. 125.
40 J. M. Basset, B. C. Gates, J. P. Candy, A.Choplin, M. Leconte, F. Quignard, C. Santini(eds.): Surface Organometallic Chemistry: Mo-lecular Approaches to Surface Catalysis,Kluwer Academic Publ., Dordrecht 1988.
41 K. Hauffe, H.-J. Engell, Z. Elektrochem. 56(1952) 366.
47 G. J. Hutchings, Catal. Lett. 75 (2001) 1.48 R. Schl€ogl in G. Ertl, H. Kn€ozinger, J.Weitkamp
(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 1697.
Heterogeneous Catalysis and Solid Catalysts 97
49 H. Topsøe, B. S. Clausen, F. E. Massoth in J. R.Anderson, M. Boudart (eds.): Catalysis: Sci-ence and Technology, Vol. 11, Springer, Berlin1996.
50 J.H.Sinfelt inG.Ertl,H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 1939.
51 S. A. Stevenson, J. A. Dumesic, R. T. K. Baker,E. Ruckenstein: Metal-Support Interactions inCatalysis, Sintering and Redispersion, Van Nos-trand Reinhold, New York 1987.
52 H. Kn€ozinger, E. Taglauer in G. Ertl, H. Kn€ozin-ger, J. Weitkamp (eds.): Handbook of Heteroge-neous Catalysis, Vol. 1, Wiley-VCH,Weinheim1997, p. 216.
53 G. C.Bond inG. Ertl, H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 752.
54 S. J. Tauster, Acc. Chem. Res. 20 (1987) 389.55 G. M. Pajonk in G. Ertl, H. Kn€ozinger, J. Weit-
kamp (eds.): Handbook of Heterogeneous Ca-talysis, Vol. 3, Wiley-VCH, Weinheim 1997,p. 1064.
56 W. C. Conner, G. M. Pajonk, S. J. Teichner, Adv.Catal. 34 (1986) 1.
57 W. C. Conner, Jr., J. L. Falconer, Chem. Rev. 95(1995) 759.
58 E. Keren, A. Soffer, J. Catal. 50 (1977) 43.59 G. A. Somorjai, J. Phys. Chem. 94 (1990) 1013.60 B. Delmon, Stud. Surf. Sci. Catal. 77 (1993) 1.61 L. T. Weng, P. Ruiz, B. Delmon, Stud. Surf. Sci.
Catal. 72 (1982) 399.62 R. K. Grasselli, Top. Catal. 15 (2001) 93.63 R. K. Grasselli in G. Ertl, H. Kn€ozinger, J.
64 J. L. Callahan, R. K.Grasselli,AIChE J. 9 (1963)755.
65 P. B. Weisz, V. J. Frilette, R. W. Maatman, E. B.Mower, J. Catal. 1 (1962) 307.
66 S. M. Csicsery, J. Catal. 19 (1970) 394.67 M. Boudart in J. M. Thomas, K. I. Zamaraev
(eds.): Perspectives in Catalysis, Blackwell Sci-entific Publications, London 1992, p. 183.
68 M.Boudart inG.Ertl,H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 958.
69 M. Boudart, Catal. Rev.-Sci. Eng. 23 (1981) 1.70 G. F. Froment, L. Hosten in J. R. Anderson, M.
Boudart (eds.): Catalysis: Science and Technol-ogy, Vol. 2, Springer, Heidelberg 1980, p. 97.
71 M. Boudart, G. Dj�ega-Mariadassou: Kinetics ofHeterogeneous Catalytic Reactions, PrincetonUniversity Press, Princeton 1984.
72 O. A. Hougen, K.M.Watson:Chemical ProcessPrinciples. Part Three: Kinetics and Catalysis,Wiley, New York, 1947.
73 J. A. Dumesic, D. F. Rudd, L. M. Aparicio, J. E.Rekoske, A. A. Trevino: The Microkinetics ofHeterogeneous Catalysis, American ChemicalSociety, Washington 1993.
74 R. D. Cortright, J. A. Dumesic, Adv. Catal. 46(2001) 161.
75 R.A. van Santen, J.W.Niemantsverdriet:Chem-ical Kinetics and Catalysis, Plenum Press, NewYork 1995.
76 Catalysis Today, 1999, Vol. 3, No. 2.77 G. M. Schwab: Katalyse vom Standpunkt der
chemischen Kinetik, Springer, Berlin, 1931.78 M. I. Temkin, V. Pyzhev, Acta Physicochim.
URSS 12 (1940) 217.79 M. Boudart in Catalysis, Vol. 14, The Royal
Society of Chemistry, Cambdrigep. 93.80 M. Boudart, Topics Catal. 14 (2001) 181.81 M. Boudart, I &ECFundamentals 25 (1986) 70.82 R. D. Cortright, J. A. Dumesic, R. J. Madon,
Topics Catal. 4 (1997) 15.83 J. Horiuti, J. Res. Inst. Catal. (Hokkaido Univ.) 5
(1957) 1.84 A. Varma, M. Morbidelli, H. Wu: Parametric
Sensitivity in Chemical Systems, CambridgeUniv. Press, Cambridge 1999.
85 C. T. Campbell, Topics Catal. 1 (1994) 364.86 G. Ertl in G. Ertl, H. Kn€ozinger, F. Sch€uth, J.
Weitkamp (eds.), Handbook of HeterogeneousCatalysis, 2nd ed., Vol. 3, Wiley-VCH, Wein-heim 2008, pp. 1492.
87 T. Engel, G. Ertl, Adv. Catal. 28 (1979) 1.88 T. Engel, G. Ertl in D. P. Woodruff (ed.): The
Chemical Physics of Solid Surfaces and Hetero-geneous Catalysis, Vol. 4, Elsevier, Amsterdam1982, p. 73.
89 G. Emig, R. Dittmeyer in G. Ertl, H. Kn€ozinger,J.Weitkamp (eds.):Handbook ofHeterogeneousCatalysis, Vol. 3, Wiley-VCH, Weinheim 1997,p. 1209.
90 W. J. Thomas in J. M. Thomas, K. I. Zamaraev(eds.): Perspectives in Catalysis, Blackwell Sci-entific Publications, London 1992, p. 251.
91 F. Kapteijn, J. A. Moulijn in G. Ertl, H. Kn€ozin-ger, J. Weitkamp (eds.): Handbook of Heteroge-neous Catalysis, Vol.Wiley-VCH, Weinheim1997, p. 1189.
92 R. Mezaki, H. Inoue: Rate Equations of SolidCatalyzed Reactions, University of Tokyo Press,Tokyo 1991.
93 A. Wheeler, Adv. Catal. 2 (1951) 250.94 http://www.gaussian.com/95 http://www.turbomole.com/
98 Heterogeneous Catalysis and Solid Catalysts
96 http://www.tcm.phy.cam.ac.uk/castep/ and http://accelrys.com/
97 http://www.camd.dtu.dk/98 http://cms.mpi.univie.ac.at/vasp/99 M.Neurock, Stud. Surf. Sci. Catal. 109 (1997), 3.100 L. D. Kieken, M. Neurock, D. H. Mei, J. Phys.
Chem. B 109 (2005), 2234.101 E. W. Hansen, M. Neurock, Chem. Eng. Sci. 54
(1999) 3411102 A. A. Gokhale, S. Kandoi, J. P. Greely, M.
Mavrikakis, J. A. Dumesic, Chem. Eng. Sci.59 (2004) 4679.
103 R. A. van Santen, M. Neurock, Chem. Rev. Sci.Eng. 37 (1995) 557.
104 H. Toulhoat, P. Raybaud, J. Catal. 216 (2003) 63.105 K. Reuter, D. Frenkel, M. Scheffler, Phys. Rev.
Lett. 93 (2004) 116105.106 S. Linic, M. A. Barteau, J. Catal. 214 (2003)
200.107 J. Greeley, M. Mavrikakis, J. Catal. 208 (2002)
291.108 A. Logadottir, J. K. Nørskov, J. Catal. 220
(2003) 273.109 A. Heyden, A. T. Bell, F. J. Keil, J. Catal. 233
(2005) 26.110 O. R. Inderwildi, D. Lebiedz, O. Deutschmann,
J. Warnatz, J. Chem. Phys. 122 (2005) 034710.111 A. Logadottir, T. H. Rod, J. K. Nørskov, B.
Hammer, S. Dahl, C. J. H Jacobsen, J. Catal.,197 (2001) 229.
112 P. Stoltze, Prog. Surf. Sci. , 65 (2000) 65.113 H. C. Kang, W. H. Weinberg, Surf. Sci., 299/300
(1994) 755.114 V. P. Zhdanov, Surf. Sci. Rep., 45 (2002) 231.115 M.W. Deem,W. H.Weinberg, H. C. Kang, Surf.
Sci. 276 (1992) 99.116 V. P. Zhdanov, B. Kasemo, Surf. Sci. Rep. 20
(1994) 111.117 D. J. Dooling, L. J. Broadbelt, Ind. Eng. Chem.
Res. 40 (2001) 522.118 S. Ovesson, B. I. Lundqvist, W. F. Schneider, A.
Bogicevic, Phys. Rev. B 71 (2005) 115406.119 M. E. Coltrin, R. J. Kee, F. M. Rupley,
SURFACE CHEMKIN (Version 4.0): A FortranPackage for AnalyzingHeterogeneous ChemicalKinetics at a Solid-Surface – Gas-Phase Inter-face, SAND91-8003B, Sandia National Labora-tories, 1991.
120 R. J. Kee,M. E. Coltrin, P. Glarborg,ChemicallyReacting Flow, John Wiley & Sons, Hoboken,New Jersey, 2003, pp. 929
121 P. Stolze, J. K. Norskov in G. Ertl, H. Kn€ozinger,F. Sch€uth, J. Weitkamp (eds.), Handbook ofHeterogeneous Catalysis, 2nd ed., Vol. 3, Wi-ley-VCH, Weinheim 2008, pp. 1479.
122 O. Deutschmann in G. Ertl, H. Kn€ozinger, F.Sch€uth, J. Weitkamp (eds.), Handbook of Het-erogeneous Catalysis, 2nd ed., Vol. 3, Wiley-VCH, Weinheim 2008, p. 1815.
123 E. Shustorovich, Surface Science 176 (1986)L863 – L872
124 E. Shustorovich, H. Sellers, Surf. Sci. Rep. 31(1998) 1 – 119.
125 W. R. Williams, C. M. Marks, L. D. Schmidt, J.Phys. Chem. 96 (1992) 5922.
126 B. Hellsing, B. Kasemo, V. P. Zhdanov, J. Catal.132 (1991) 210.
127 M. Rinnemo, O. Deutschmann, F. Behrendt, B.Kasemo, Combust. Flame 111 (1997) 312.
128 G. Veser, Chem. Eng. Sci. 56 (2001) 1265.129 P.-A. Bui, D. G. Vlachos, P. R. Westmoreland,
130 J. Mai, W. von Niessen, A. Blumen, J. Chem.Phys. 93 (1990) 3685.
131 V. P. Zhdanov, B. Kasemo, Appl. Surf. Sci. 74(1994) 147.
132 P. Aghalayam, Y. K. Park, D. G. Vlachos, Proc.Combust. Inst. 28 (2000) 1331.
133 O.Deutschmann, F. Behrendt, J.Warnatz,Catal.Today 21 (1994) 461 – 470.
134 G. Veser, J. Frauhammer, L. D. Schmidt, G.Eigenberger, in “Dynamics of Surfaces andReaction Kinetics in Heterogeneous Catalysis”,G. F. Froment, K. C. Waugh (eds.):Studies inSurface Science and Catalysis 109, Elsevier,Amsterdam 1997, p. 273.
135 P.-A. Bui, D. G. Vlachos, P. R. Westmoreland,Surf. Sci. 386 (1997) L1029.
136 P. Aghalayam, Y. K. Park, N. Fernandes, V.Papavassiliou, A. B. Mhadeshwar, D. G. Vla-chos, J. Catal. 213 (2003) 23.
137 D. A. Hickman, L. D. Schmidt, Am. Inst. Chem.Eng. J. 39 (1993) 1164.
138 D. K. Zerkle, M. D. Allendorf, M. Wolf, O.Deutschmann, J. Catal. 196 (2000) 18.
139 F. Donsi, K. A. Williams, L. D. Schmidt, Ind.Eng. Chem. Res. 44 (2005) 3453.
140 M. C. Huff, I. P. Androulakis, J. H. Sinfelt, S. C.Reyes, J. Catal. 191 (2000) 46.
141 D. A. Hickman, L. D. Schmidt, Am. Inst. Chem.Eng. J. 39 (1993) 1164.
142 R. Schwiedernoch, S. Tischer, C. Correa, O.Deutschmann, Chem. Eng. Sci. 58 (2003) 633.
143 D. Chatterjee, O. Deutschmann, J. Warnatz,Faraday Discuss. 119 (2001) 371.
144 B. Ruf, F. Behrendt, O. Deutschmann, J. War-natz, Surf. Sci. 352 (1996) 602.
145 S. J. Harris, D. G. Goodwin, J. Phys. Chem. 97(1993) 23.
Heterogeneous Catalysis and Solid Catalysts 99
146 S. Romet, M. F. Couturier, T. K. Whidden, J.Electrochem. Soc. 148 (2001) G82.
147 C. D. Scott, A. Povitsky, C. Dateo, T. Gokcen, P.A. Willis, R. E. Smalley, J. Nanosci. Nanotech-nol. 3 (2003) 63.
148 C. H. Bartholomew, R. J. Farrauto: Fundamen-tals of Industrial Catalytic Processes, 2nd ed.,Wiley, Hobboken 2006.
159 D. Wolf, O. V. Buyevskaya, M. Baerns, Appl.Catal. A: General 200 (2000) 63.
160 K. Kochloefl, Chem. Eng. Technol. 24 (2001) 3.161 J. Greeley, J. K. Nørskov, M. Mavrikakis, Annu.
Rev. Phys. Chem. 53 (2002) 319.162 C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S.
Bahn, A. Logadottir, J. K. Nørskov, J. Am.Chem. Soc. 123 (2001) 8404.
163 B. C. Gates: Catalytic Chemistry, Wiley, NewYork 1992.
164 C. N. Satterfield: Heterogeneous Catalysis inIndustrial Practice, McGraw-Hill, New York1991.
165 R. J. Farrauto, C. H. Bartholomew: Fundamen-tals of Industrial Catalytic Processes, BlackieAcademic and Professional, London 1997.
166 G. Ertl, H.Kn€ozinger, J.Weitkamp (eds.):Hand-book of Heterogeneous Catalysis,Vols. 1 – 5,Wiley-VCH, Weinheim 1997.
167 J. Hagen: Industrial Catalysis, A Practical Ap-proach, Wiley-VCH, Weinheim 1999.
168 K. Tanabe: Solid Acids and Bases, Kodansha,Tokyo, Academic Press, New York, London1970.
169 M. Che, O. Clause, Ch. Marcilly in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 1, Wiley-VCH,Weinheim 1997, p. 191.
170 J. P. Brunelle,Pure Appl. Chem. 50 (1978) 1211.171 B. G. Linsen (ed.): Physical and Chemical As-
pects of Adsorbents and Catalysts, AcademicPress, New York 1970.
172 H. Kn€ozinger, P. Ratnasamy, Catal. Rev.-Sci.Eng. 17 (1978) 31.
173 H. Oechsner, Scanning Microsc. 2 (1988) 9.174 H. P.Boehm,H.Kn€ozinger in J. R.Anderson,M.
Boudart (eds.): Catalysis: Science and Technol-ogy, Vol. 4, Springer, Berlin 1983, p. 39.
175 C. S. John, M. S. Scurrell: Catalysis, The Chem-ical Society, London, 1 (1977) 136.
176 J. Wagner, W. Nehb in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 4, Wiley-VCH, Weinheim 1997,p. 1761.
177 E. F. Vansant, P. Van Der Voort, K. C. Vrancken(eds.): Stud. Surf. Sci. Catal. 93 (1995).
178 A. E. Legrand (ed.): The Surface Properties ofSilicas, Wiley, Chichester, New York, Wein-heim, Brisbane, Singapore, Toronto 1998.
179 H. Kn€ozinger in P. Schuster, G. Zundel, C.Sandorfy (eds.): The Hydrogen Bond, Vol. 3,North Holland, Amsterdam 1976, p. 1263.
180 J. M. Thomas, R. G. Bell, C. R. A. Catlow in G.Ertl, H. Kn€ozinger, J. Weitkamp (eds.): Hand-book of Heterogeneous Catalysis, Vol. 1, Wiley-VCH, Weinheim 1997, p. 286.
181 C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C.Vartuli, J. S. Beck, Nature 359 (1992) 710.
182 J. S. Beck, J. C. Vartuli, W. J. Roth, M. E.Leonowicz, C. T. Kresge, K. D. Schmidt, C.T. W. Chu, D. H. Olson, E. W. Sheppard, S. B.McCullan, J. B. Higgins, J. L. Schlenker, J. Am.Chem. Soc. 114 (1992) 10834.
183 E. A. Colbourn, W. C. Mackrodt, Surf. Sci. 143(1984) 391.
184 S. Coluccia, A. J. Tench: Proc. 7th Intern. Congr.Catal., Tokyo, 1980, Kodansha, Tokyo, Elsevier,Amsterdam 1981, p. 1154.
185 A. Zecchina, D. Scarano, S. Bordiga, G. Spoto,C. Lamberti, Adv. Catal. 46 (2001) 265.
186 R.N.Spitz,J.E.Barton,M.A.Barteau,R.H.Staley,A. W. Sleight, J. Phys. Chem. 90 (1986) 4067.
187 A. Zecchina, M. G. Lofthouse, F. S. Stone, J.Chem. Soc. Faraday Trans. 1 71 (1975) 1476.
188 S. Coluccia, A. M. Deane, A. J. Tench, J. Chem.Soc. Faraday Trans. 1 74 (1978) 2913.
189 H. Kn€ozinger, Science 287 (2000) 1407.190 A. Cimino, F. S. Stone in G. Ertl, H. Kn€ozinger,
191 C. N. R. Rao, B. Raveau: Transition MetalOxides, VCH, Weinheim 1995.
192 P. A. Cox: Transition Metal Oxides, ClarendonPress, Oxford 1995.
193 H. H. Kung: Transition Metal Oxides: SurfaceChemistry and Catalysis, Elsevier, Amsterdam1995.
194 P. Pichat in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 2111.
195 A. L. Linsebigler, G. Lu, J. T. Yates, Jr., Chem.Rev. 95 (1995) 735.
196 K. Arata, Adv. Catal. 37 (1990) 165.197 W. G€opel, K. D. Schierbaum in G. Ertl, H.
Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 3, Wiley-VCH,Weinheim 1997, p. 1284.
198 K. Kochloefl in G. Ertl, H. Kn€ozinger, J. Weit-kamp (eds.): Handbook of Heterogeneous Ca-talysis, Vol. 4, Wiley-VCH, Weinheim 1997,p. 1831.
199 K. Kochloefl in G. Ertl, H. Kn€ozinger, J. Weit-kamp (eds.): Handbook of Heterogeneous Ca-talysis, Vol. 5, Wiley-VCH, Weinheim 1997,p. 2151.
200 J. Haber in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 5, Wiley-VCH, Weinheim 1997, p. 2253.
201 M.Muhler inG. Ertl, H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 5, Wiley-VCH, Weinheim 1997, p. 2274.
202 R. M. Barrer: Hydrothermal Chemistry of Zeo-lites, Academic Press, London 1982.
203 W.M.Meier, D. H. Olson, Ch. Baerlocher: Atlasof Zeolite Structure Types, 4th Ed., Butterworth-Heinemann, London 1996.
204 J. Weitkamp, Solid State Ionics 131 (2000) 175.205 G. Bellussi, V. Fattore in P. A. Jacobs, N. Jaeger,
L. Kubelkova, B. Wichterlova (eds.): ZeoliteChemistry and Catalysis, Elsevier, Amsterdam1991, p. 79.
206 D. Barthomeuf, Catal. Rev.-Sci. Eng. 38 (1996)521.
207 J. M. Thomas, R. G. Bell, C. R. A. Catlow, G.Ertl, H. Kn€ozinger, J. Weitkamp (eds.): Hand-book of Heterogeneous Catalysis, Vol. 1, Wiley-VCH, Weinheim 1997, p. 286.
208 T. Ishihara, H. Takita, Catalysis 12 (1996) 21.209 J. J. Fripiat inG. Ertl, H.Kn€ozinger, J.Weitkamp
(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 387.
210 P.G.Menon,B.Delmon inG.Ertl, H.Kn€ozinger,J. Weitkamp (eds.):Handbook of Heterogeneous
223 M.Misono,Catal. Rev.-Sci. Eng. 30 (1988) 339.224 J. B. Moffat: Metal-Oxygen Clusters, Kluwer
Academic/Plenum Publishers, New York 2001.225 J. A. Gamelas, F. A. S. Couto, M. C. N. Trovao,
A. M. V. Cavaleiro, J. A. S. Cavaleiro, M.Guelton, Thermochim. Acta 326 (1999) 165.
226 T. Okuhara, M. Yamashita, K. Na, M. Misono,Chem. Lett.(1994) 1450.
227 Y. Izumi, M. Ogawa, W. Nohara, K. Krabe,Chem. Lett.(1992) 1987.
228 G. Centi, J. L. Nieto, C. Iapalucci, K. Br€uckman,E. M. Serwicka, Appl. Catal. 46 (1989) 197.
229 F. Cavani, M. Koutyrev, F. Trifiró, Catal. Today28 (1996) 319.
230 E. Wagner, T. Fetzer in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 4, Wiley-VCH, Weinheim 1997,p. 1748.
231 M. S. Wainright in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 1, Wiley-VCH, Weinheim, Ger-many 1997, p. 64.
232 R. Schl€ogl in G. Ertl, H. Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 54.
Heterogeneous Catalysis and Solid Catalysts 101
233 A. Baiker in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 803.
234 J. R. Jennings (ed.): Catalytic Ammonia Synthe-sis: Fundamentals and Practice, Plenum Press,New York 1991.
235 S. T. Oyama in G. Ertl, H. Kn€ozinger, J. Weit-kamp (eds.): Handbook of Heterogeneous Ca-talysis, Vol. 1, Wiley-VCH, Weinheim 1997,p. 132. in ref. [4], p. 132.
236 C. Bouchy, C. Pham-Huu, B. Heinrich, C. Chau-mot, M. J. Ledoux, J. Catal. 190 (2000) 92.
237 R. Schl€ogl in G. Ertl, H. Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 138.
238 H. J€untgen, H. K€uhl, Chem. Phys. Carbon 22(1990) 145.
239 L. R. Radovic, F. Rodriguez-Reinoso, Chem.Phys. Carbon 35 (1997) 243.
240 P. Serp, M. Corria, P. Kalck, Appl. Catal. A:General 253 (2003) 337.
241 R. P. Raffaelle, B. J. Landi, J. D. Harris, S. G.Bailey,A. F.Hepp,Mater. Sci. Eng. B 116 (2005)233.
242 F. de Dardel, T. V. Arden: Ullmann’s Encyclo-pedia of Industrial Chemistry, 5th ed., Vol.A 14, VCH Verlagsgesellschaft, Weinheim1987, p. 393.
243 A. St€uwe, C.-P. H€alsig, H. Tschorn in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 4, Wiley-VCH,Weinheim 1997, p. 1986.
244 G. A. Olah, A.Molnar:Hydrocarbon Chemistry,Wiley, New York 1995.
245 J.-P. Vigneron in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 2, Wiley-VCH, Weinheim1997, p. 888.
246 K. Morihara, S. Doi, M. Takiguchi, T. Shimada,Bull. Chem. Soc. Jpn. 66 (1993) 2977.
247 G. Wulff in C. G. Gebelein (ed.): BiomimeticPolymers, Plenum Press, New York, 1990, p. 1.
248 K. Morihara, M. Takaguchi, T. Shimada, Bull.Chem. Soc. Jpn. 67 (1994) 1078.
249 M. Eddaoudi, D. B. Moler, H. L. Li, B. L. Chen,T. M. Reineke, M. O’Keefe, O. M. Yaghi, Acc.Chem. Res. 34 (2001) 319.
250 S. Kaskel in F. Sch€uth, K. S. W. Sing, J. Weit-kamp (eds.):Handbook of Porous Solids, Vol. 2,Wiley-VCH, Weinheim, 2002, p. 1190.
251 H. Li, M. Eddaoudi, M. O’Keefe, O. M. Yaghi,Nature 402 (1999) 276.
252 H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go,M. Eddaoudi, A. J. Matzger, M. O’Keefe, O. M.Yaghi, Nature 427 (2004) 523.
253 U. Mueller, M. Schubert, F. Teich, H. P€utter, K.Schierle-Arnd, J. Pastre, J. Mater. Chem. 16(2006) 626.
254 F. X. L. I. Xamena, A. Abad, A. Corma, H.Garcia, J. Catal. 250 (2007) 294.
255 M. J. Ledoux, C. Pham-Huu, CATTECH 5(2001) 226.
256 H. Sieber, C. Hoffmann, A. Kaindl, P. Greil,Adv.Eng. Mater. 2 (2000) 105.
257 J. L. Williams, Catal. Today 69 (2001) 3.258 M. Valentini, G. Groppi, C. Cristiani,M. Levi, E.
Tronconi, P. Forzatti,Catal. Today69 (2001) 307.259 E. S. J. Lox, B. H. Engler in G. Ertl, H. Kn€ozin-
ger, J. Weitkamp (eds.): Handbook of Heteroge-neous Catalysis, Vol. 4, Wiley-VCH,Weinheim1997, p. 1569.
260 J. M. Thomas, Angew. Chem. 111 (1999) 380;Angew. Chem. Int. Ed. 38 (1999) 3588.
261 A. Stein, B. J. Melde, R. C. Schroden, Adv.Mater. 12 (2000) 1403.
262 D. E. DeVos, B. F. Sels, P. A. Jacobs, Adv. Catal.46 (2001) 2.
263 D. C. Sherrington, A. P. Kybett (eds.): SupportedCatalysts and their Applications, The RoyalSociety of Chemistry, Cambridge 2001.
264 B. K. Hodnett, A. Kybett, J. H. Clark, K. Smith:Supported Reagents and Catalysts in Chemistry,The Royal Society of Chemistry, Cambridge1998.
265 B.Clapham, T. . Reger, K.D. Janda,Tetrahedron57 (2001) 4637.
266 W. Keim, B. Driessen-H€olscher in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 1, Wiley-VCH,Weinheim 1997, p. 231.
267 I. E. Wachs: Catalysis, Vol. 13, The RoyalSociety of Chemistry, Cambridge 1997, p. 37.
268 B.Delmon inG.Ertl, H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 264.
269 G. C. Bond, J. C. Vedrine, Catal. Today 20(1994) 1.
270 G.Centi,Appl.Catal. A:General147 (1996) 267.271 F. J. Janssen inG.Ertl,H.Kn€ozinger, J.Weitkamp
(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 1633.
272 J. F. Armor, Chem. Mat. 6 (1994) 730.273 R. Prins in G. Ertl, H. Kn€ozinger, J. Weitkamp
(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 1908.
274 R. Prins, Adv. Catal. 46 (2001) 399.275 M.Hino, K. Arata:Chem. Commun. 1988, 1259.276 S. Kuba, P. Concepcion, R. K. Grasselli, B. C.
Gates, M. Che, H. Kn€ozinger, Phys. Chem.Chem. Phys. 3 (2001) 146.
102 Heterogeneous Catalysis and Solid Catalysts
277 S. Kuba, B. C. Gates, P. Vijayanand, R. K. Grass-elli, H. Kn€ozinger: Chem. Commun. 2001, 57.
278 J. C. Mol in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 5, Wiley-VCH, Weinheim 1997, p. 2387.
279 F. Buonomo, D. Sanfilippo, F. Trifiro in G. Ertl,H. Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 5, Wiley-VCH,Weinheim 1997, p. 2140.
280 S. H. Overbury, P. A. Bertrand, G. A. Somorja,Chem. Rev. 75 (1975) 547.
281 Y. Ono, T. Baba: Catalysis, Vol. 15, The RoyalSociety of Chemistry, Cambridge 2000, p. 1.
282 K. Tanabe, H. Hattori in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 1, Wiley-VCH, Weinheim 1997,p. 404.
283 X. Song, A. Sayari, Catal. Rev.-Sci. Eng. 38(1996) 329.
284 Topics in Catalysis 6 (1998).285 K. Foger in J. R. Anderson, M. Boudart (eds.):
Catalysis: Science and Technology, Vol. 6,Springer, Berlin 1984, p. 228.
286 B.C.Gates inG. Ertl,H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 793.
287 B. C. Gates, Chem. Rev. 95 (1995) 511.288 J. H. Sinfelt: Bimetallic Catalysts, Wiley, New
York 1983.289 C. T. Campbell in G. Ertl, H. Kn€ozinger, J.
Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 2, Wiley-VCH, Weinheim1997, p. 814.
290 A. Baiker, Curr. Opin. Sol. State Mater. Sci. 3(1998) 86.
291 J. Wei in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 1928.
292 S. Helveg, J. V. Lauritsen, E. Lægsgaard, I. Stens-gaard, J. K. Nørskov, B. S. Clausen, H. Topsøe, F.Besenbacher, Phys. Rev. Lett. 84 (2000) 951.
293 D. E. De Vos, I. F. J. Vankelecom, P. A. Jacobs:Chiral Catalyst Immobilization and Recycling,Wiley-VCH, Weinheim 2000.
294 D. E. De Vos, S. de Wildman, B. F. Sels, P. J.Grobet, P. A. Jacobs, Angew. Chem. 111 (1999)1033;Angew. Chem. Int. Ed. 38 (1999) 980.
295 K. Dranz, H. Waldmann: Enzyme Catalysis inOrganic Synthesis, VCH Verlagsgesellschaft,Weinheim 1994.
296 A. W. Bosman, H. M. Janssen, E. W. Meijer,Chem. Rev. 99 (1999) 1665.
297 H. Brunner, J. Organomet. Chem. 500 (1995) 39.298 A. Kirschning, H. Monenschein, R. Wittenberg,
Angew. Chem. Int. Ed. 40 (2001) 650.
299 B. Cornils, W. A. Herrmann: Applied Homoge-neous Catalysis with Organometallic Com-pounds, Wiley-VCH, Weinheim 1996, p. 619.
300 J. P. Arhancet, M. E. Davis, J. S. Merola, B. E.Hanson,Nature 339 (1989) 454;K.T.Wan,M.E.Davis, Nature 370 (1994) 449.
301 J. Adlkofer inG. Ertl, H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 1774.
302 A. Riisager, P. Wasserscheid, R. van Hal, R.Fehrmann, J. Catal. 219 (2003) 452.
303 A. Riisager, R. Fehrmann, M. Haumann, P.Wasserscheid, Top. Catal. 40 (2006) 91.
304 U. Kernchen, B. Etzold, W. Korth, A. Jess,Chem. Eng. Technol. 30 (2007) 985.
305 G. Schulz-Ekloff, S. Ernst in G. Ertl, H. Kn€ozin-ger, J. Weitkamp (eds.): Handbook of Heteroge-neous Catalysis, Vol. 1, Wiley-VCH,Weinheim1997, p. 374.
306 D. E.DeVos, P. P.Knops-Gerrits, R. F. Parton,B.M. Weckhuysen, P. A. Jacobs, R. A. Schoon-heydt, J. Incl. Phnom. 21 (1995) 185.
307 R. Parton, D. E. De Vos, P. A. Jacobs in E. G.Derouane, F. Lemos, C. Naccache, F. RamoaRibeiro (eds.): Zeolite Microporous Solids: Syn-thesis, Structure and Reactivity, Kluwer Aca-demic Publ., Dordrecht 1995, p. 555.
309 M. P. McDaniel in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 5, Wiley-VCH, Weinheim 1997,p. 2400.
310 W. Kaminsky, Adv. Catal. 46 (2001) 89.311 W. Kaminsky, Macromol. Chem. Phys. 197
(1996) 3907.312 S. Roy, T. Bauer, M. Al-Dahhan, P. Lehner, T.
Turek, AIChE J. 50 (2004) 2918.313 I. Nova, A. Beretta, G. Groppi, L. Lietti, E.
Tronconi, P. Forzatti in A. Cybulski, J. A. Mou-lijn (eds.)Structured Catalysts and Reactors,2nd ed., Taylor & Francis, Boca Raton 2006.
314 K. Pangarkar, T. J. Schildhauer, J. R. van Om-men, J. Nihenhuis, F. Kapteijn, J. A. Moulijn,Ind. Eng. Chem. Res. 47 (2008) 3720.
315 M. V. Twigg, J. T. Richardson, Ind. Eng. Chem.Res. 46 (2007) 4166.
316 Y. Melatov-Meytal, M. Sheintuch, Appl. Catal.A: General 231 (2002) 1.
317 B. A. A. L. van Setten, M.Makee, J. A. Moulijn,Catal. Rev. Sci. Eng. 43 (2001) 489.
318 V. Mehta, J. S. Cooper, J. Power Sources 114(2003) 32.
319 E.Klemm,H.D€oring,A.Geisselmann, S. Schirr-meister, Chem. Eng. Technol. 30 (2007) 1615.
Heterogeneous Catalysis and Solid Catalysts 103
320 K. Kochloefl, Quo vadis heterogene Katalyse,Dechema Tagung, XXVI Jahrestreffendeutscher Katalytiker, Schloß Reinhardsbrunn,Germany, 1993.
321 H. Heinemann: “Development of Industrial Cat-alysis” in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 35.
322 J. T. Richardson in M. V. Twigg, M. S. Spencer(eds.): Principle of Catalyst Development, Ple-num Press, New York 1989, p. 95.
323 J. A. Cusumano in J.M. Thomas, K. I. Zamaraev(eds.): Perspectives in Catalysis, BlackwellScient. Publ., Oxford 1991, p. 1.
324 K. Fouhy, G. Samdani, S. Moore, Chem. Eng.,October (1992) 47.
325 J. M. Fulton, Chem. Eng. 7 (1986) 59.326 P. Courty, C. Marcilly in G. Poncelet, P. Grange,
P. Jacobs (eds.): Preparation of Catalysts III,Elsevier, Amsterdam 1983, p. 485.
327 M. Sitting: Handbook of Catalyst Manufacture,Noyes Data Corp., Park Ridge 1971.
328 B. Stiles, T. AKoch,Catalyst Manufacture, 2nd.ed., M. Dekker, NewYork 1995.
329 F. Sch€uth, K. Unger in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 1, Wiley-VCH, Weinheim 1997,p. 72.
330 E. I. Ko in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 86.
331 H. Jacobsen, P. Kleinschmit in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 1, Wiley-VCH,Weinheim 1997, p. 94.
332 J. W. Geus, J. van Dillen: “Preparation of Sup-ported Catalysts by Deposition – Precipitation”in G. Ertl, H. Kn€ozinger, J. Weitkamp (eds.):Handbook of Heterogeneous Catalysis, Vol. 1,Wiley-VCH, Weinheim 1997, p. 240.
333 E. J. P. Feijen, J. A. Martens, P. A. Jacobs in G.Ertl, H. Kn€ozinger, J. Weitkamp (eds.): Hand-book of Heterogeneous Catalysis, Vol. 1, Wiley-VCH, Weinheim 1997, p. 311.
334 J. Barbier in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 1, Wiley-VCH, Weinheim 1997, p. 257.
335 J. F. Le Page in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 1, Wiley-VCH, Weinheim1997, p. 412.
336 J. W. Fulton, Chem Eng.,May 12 (1986) 97.337 General Catalogue, S€ud-Chemie AG, Catalyst
339 D. H€onicke, E. Dietzsch in F. Sch€uth, K. S. W.Sing, J. Weitkamp (eds.): Handbook of PorousSolids, Vol. 3, Wiley-VCH, Weinheim 2002,p. 1395.
340 T. A. Nijhuis, A. E. W. Beers, T. Vergunst, I.Hoek, F. Kapteijn, J. A.Moulijn,Catal. Rev. Sci.Eng. 43 (2001) 345.
341 G. Ertl, H.Kn€ozinger, J.Weitkamp (eds.):Hand-book of Heterogeneous Catalysis, Vol. 2, Wiley-VCH, Weinheim 1997.
342 K. S. W. Sing, D. H. Everett, R. A. W. Haul, L.Moscou, R. A. Pierotti, J. Rouquerol, T. Siemie-niewska, Pure Appl. Chem. 57 (1985) 603.
343 K. S. W. Sing, J. Rouquerol in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 1, Wiley-VCH,Weinheim 1997, p. 427.
344 J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H.Everett, J. M. Haynes, N. Pernicone, J. D. F.Ramsay, K. S. W. Sing, K. K. Unger, Pure Appl.Chem. 66 (1994) 1739.
345 B. C. Lippens, J. H. de Boer, J. Catal. 4 (1965)319.
346 K. S. W. Sing, D. H. Everett, R. H. Ottewill(eds.): Surface Area Determination, Butter-worths, London 1970, p. 25.
347 K. Datye in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 493.
348 G. Bergeret, P. Gallezot in G. Ertl, H. Kn€ozinger,J.Weitkamp (eds.):Handbook ofHeterogeneousCatalysis, Vol. 2, Wiley-VCH, Weinheim 1997,p. 439.
349 P. Gallezot in J. R. Anderson, M. Boudart (eds.):Catalysis: Science and Technology, Vol. 5,Springer, Berlin 1984, p. 221.
350 R. J. Matyi, L. R. Schwartz, J. B. Butt, Catal.Rev.-Sci. Eng. 29 (1987) 41.
351 A.K.Datye,D. J. Smith,Catal. Rev.-Sci. Eng. 34(1992) 129.
352 H. Poppa, Catal. Rev.-Sci. Eng. 35 (1993) 359.353 M. J.Yacaman,G.Diaz,A.Gomez,Catal. Today
(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 464.
355 B. S. Clausen, G. Steffensen, B. Fabius, J.Villadsen, L. R. Feidenhaus, H. Topsøe, J. Catal.132 (1991) 524.
356 M. Vaarkamp, D. C. Konigsberger in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 2, Wiley-VCH,Weinheim 1997, p. 475.
357 J. H. Sinfelt, G. D.Meitzner, Acc. Chem. Res. 26(1993) 1.
104 Heterogeneous Catalysis and Solid Catalysts
358 J. C. Conesa, P. Esteban, H. Dexpert, D. Bazin,Stud. Surf. Sci. Catal. 57 (1990) 225.
359 J. M. Thomas, Chem. Eur. J. 3 (1997) 1557.360 D. J. Smith, M. R. McCartney, J. K. Weiss,
Ultramicroscopy 52 (1993) 591.361 R. T. K. Baker, Catal. Rev.-Sci. Eng. 19 (1979)
161;R. T. K. Baker, N. M. Rodriguez, Energyand Fuels 8 (1994) 330.
362 G.Mestl, H. Kn€ozinger in G. Ertl, H. Kn€ozinger,J.Weitkamp (eds.):Handbook ofHeterogeneousCatalysis, Vol. 2, Wiley-VCH, Weinheim 1997,p. 539.
363 C. Li, P. C. Stair, Catal. Lett. 36 (1995) 119.364 H. Kn€ozinger, Catal. Today 32 (1996) 71.365 Y. R. Shen, Surface Sci. 299/300 (1994) 551.366 K. B. Eisenthal, Chem. Rev. 96 (1996) 1343.367 H. Jobic in G. Ertl, H. Kn€ozinger, J. Weitkamp
(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 574.
368 J. W. Niemantsverdriet, T. Butz in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 2, Wiley-VCH,Weinheim 1997, p. 512.
369 A. M. van der Kraan, J. W. Niemantsverdriet inG. J. Lang, J. G. Stevens (eds.): IndustrialApplications of the M€ossbauer Effect, PlenumPress, New York 1985, p. 609.
370 A. Lerf, T. Butz, Angew. Chem. 99 (1987) 113.371 P. Mottner, T. Butz, A. Lerf, G. Ledezma, H.
Kn€ozinger, J. Phys. Chem. 99 (1995) 8260.372 G. Engelhardt in G. Ertl, H. Kn€ozinger, J.
Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 2, Wiley-VCH, Weinheim1997, p. 525.
373 G. Engelhardt, D. Michel: High-Resolution Sol-id-State NMR of Silicates and Zeolites, Wiley,Chichester 1987.
374 A. T. Bell, A. Pines (eds.): NMR Techniques inCatalysis, M. Dekker, New York 1994.
375 E.Taglauer inG.Ertl,H.Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 614.
376 J. W. Niemandsverdriet: Spectroscopy in Catal-ysis, VCH Verlagsgesellschaft, Weinheim 1995.
377 D. Briggs, M. P. Seah: Practical Surface Analy-sis by Auger and X-ray Photoelectron Spectros-copy, Wiley, New York 1985.
378 E. Taglauer in A.W. Czanderna, D. M. Hercules(eds.): Ion Spectroscopies for Surface Analysis,Plenum Press, New York 1991, p. 363.
379 H. Niehus, W. Heiland, E. Taglauer, Surf. Sci.Rep. 17 (1993) 217.
380 W. K. Chu, J. W. Mayes, M. A. Nicolet: Back-scattering Spectrometry, Academic Press, NewYork 1978.
381 L. C. Feldman in A. W. Czanderna, D. M.Hercules (eds.): Ion Spectroscopies for SurfaceAnalysis, Plenum Press, NewYork 1991, p. 311.
382 A. Benninghoven, F. G. R€udenauer, H. W. Wer-ner: Secondary Ion Mass Spectrometry, Wiley,New York 1987.
383 H. J. Borg, J.W.Niemandsverdriet in J. J. Spivey,S. Agarwal (eds.): Catalysis, Vol. 11, The RoyalSociety of Chemistry, Cambridge 1994, p. 1.
384 J. C. Vickerman, A. Swift in J. C. Vickerman(ed.): Surface Analysis—The Principal Techni-ques, Wiley, Chichester 1997,
385 J. C. Vickerman (ed.): Surface Analysis—ThePrincipal Techniques, Wiley, Chichester 1997,p. 135.
386 G.Moretti in G. Ertl, H. Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 632.
387 C. D.Wagner, A. Joshi, J. Electron Spectrosc. 47(1988) 283.
388 M. Che, F. Bozon-Verduraz in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 2, Wiley-VCH,Weinheim 1997, p. 641.
389 W. N. Delgass, G. L. Haller, R. Kellerman, J. H.Lunsford: Spectroscopy in Heterogeneous Ca-talysis, Academic Press, New York 1979.
390 R. A. Schonheydt in F. Delannay (ed.): Charac-terization of Heterogeneous Catalysts, Dekker,New York 1984, p. 125.
391 F. Stone in J. P. Bonella, B. Delmon, E. G.Deronane (eds.): Surface Properties and Cataly-sis by Non-Metals, Reidel, Boston 1983, p. 237.
392 M. Gerlach, E. C. Constable: Transition MetalChemistry, VCH Verlagsgesellschaft, Wein-heim, Germany 1994.
393 G. Kort€um: Reflexionsspektroskopie, Springer,Berlin 1969.
394 M. Anpo in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 664.
395 M. Anpo, M. Che, Adc. Catal. 44 (1999) 119.396 J. H. Lunsford in J. R. Anderson, M. Bondart
(eds.): Catalysis—Science and Technology,Vol. 8, 1997, p. 227.
397 K. Dyrek, M. Che, Chem. Rev. 97 (1997) 305.398 P. D. Garn: Thermoanalytical Methods of Inves-
tigation, Academic Press, New York 1965.399 R. C.Mackenzie:Differential Thermal Analysis,
Academic Press, London, New York 1972.400 S. D. Robertson, B. D.McNicol, J. H. de Bass, S.
C. Kloet, J. W. Jenkins, J. Catal. 37 (1975) 424.401 H. Kn€ozinger in G. Ertl, H. Kn€ozinger, J. Weit-
kamp (eds.): Handbook of Heterogeneous Catal-ysis, Vol. 2,Wiley-VCH,Weinheim 1997, p. 676.
Heterogeneous Catalysis and Solid Catalysts 105
402 D. A. M. Monti, A. Baiker, J. Catal. 83 (1983)323.
403 P. Malet, A. Caballero, J. Chem. Soc. FaradayTrans. I 84 (1988) 2369.
404 V. B. Kazansky in G. Ertl, H. Kn€ozinger, J. Weit-kamp (eds.): Handbook of Heterogeneous Catal-ysis, Vol. 2,Wiley-VCH,Weinheim 1997, p. 740.
405 W.K. Hall in G. Ertl, H. Kn€ozinger, J.Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 692.
406 H. A. Benesi, J. Phys. Chem. 61 (1957) 970.407 M. Deeba, W. K. Hall, J. Catal. 60 (1979) 417.408 B. E. Spiewak, R. D. Cartright, J. A. Dumesic in
G. Ertl, H.Kn€ozinger, J.Weitkamp (eds.):Hand-book of Heterogeneous Catalysis, Vol. 2, Wiley-VCH, Weinheim 1997, p. 698.
409 J. L. Falconer, J. A. Schwarz, Catal. Rev.-Sci.Eng. 25 (1983) 414.
410 H. Karge, V. Dondur, J. Phys. Chem. 94 (1990)765.
411 S. Chatterjee, H. L. Greene, Y. J. Park, J. Catal.138 (1992) 179.
412 A. Auroux, A. Gervasini, J. Phys. Chem. 94(1990) 6371.
413 D. T. Chen, L. Zhang, C. Yi, J. A. Dumesic, J.Catal. 146 (1994) 257.
414 W. E. Farneth, R. J. Gorte,Chem. Rev. 95 (1995)615.
415 H. Kn€ozinger in G. Ertl, H. Kn€ozinger, J. Weit-kamp (eds.): Handbook of Heterogeneous Catal-ysis, Vol. 2,Wiley-VCH,Weinheim 1997, p. 707.
416 E. A. Paukshtis, E. N. Yurchenko, Russ. Chem.Rev. 52 (1983) 42.
417 J. C. Lavalley, Trends Phys. Chem. 2 (1991) 305.418 J. A. Lercher, C. Gr€undling, G. Eder-Mirth,
Catal. Today 27 (1996) 353.419 J. C. Lavalley, Catal. Today 27 (1996) 377.420 H. Kn€ozinger, S. Huber, J. Chem. Soc. Faraday
Trans. 94 (1998) 2047.421 G. C. Pimentel, A. L. McClellan: The Hydrogen
Bond, Freeman, San Francisco, London 1960.422 S. Huber, H. Kn€ozinger, J. Mol. Catal. 141
(1999) 117.423 A. M. Ferrari, S. Huber, H. Kn€ozinger, K. M.
Neyman, N. R€osch, J. Phys. Chem. B 102 (1998)4548.
424 H. Kn€ozinger, H. Krietenbrink, H. D.M€uller,W.Schulz: Proceedings of the 6th InternationalCongress on Catalysis, London, 1976, TheChemical Society, London 1977, p. 183.
425 H. Pfeifer in G. Ertl, H. Kn€ozinger, J. Weitkamp(eds.): Handbook of Heterogeneous Catalysis,Vol. 2, Wiley-VCH, Weinheim 1997, p. 732.
426 V. M. Mastikhin, I. L. Mundrakovsky, A. V.Nosov, Progress NMR Spectrosc. 23 (1991) 259.
427 M. Hunger, Solid State Nucl. Magn. Res. 6(1996) 1.
428 V. Bos�acek, J. Phys. Chem. 97 (1993) 10732;andZ. Phys. Chem. 189 (1995) 241.
429 J. F. Le Page: Applied Heterogeneous Catalysis�Design, Manufacture, Use of Solid Catalysts,Editions Technip, Paris 1987.
430 J. C. Dart,Chem. Eng. Prog. 71 (1975) 46; andE.R. Beaver, Chem. Eng. Prog. 71 (1975) 44.
431 W. L. Forsythe, W. R. Hertwig, Ind. Eng. Chem.41 (1949) 1200.
432 C. O. Bennett, Adv. Catal. 44 (1999) 329.433 K. Tamaru in J. R. Anderson, M. Boudart (eds.):
Catalysis: Science and Technology, Vol. 9,Springer, Berlin, 1991, p. 87.
434 J. T. Gleaves, J. R. Ebner, T. C. Kuechler, Catal.Rev.-Sci. Eng. 30 (1988) 49.
435 O. V. Buyevskaya, M. Rothaemel, H. W. Zanth-off, M. Baerns, J. Catal. 150 (1994) 71.
436 G. Creten, D. S. Lafyatis, G. F. Froment, J.Catal. 154 (1995) 151.
437 S. L. Shannon, J. G. Goodwin, Jr.,Chem. Rev. 95(1995) 677.
438 A. Ozaki: Isotopic Studies of HeterogeneousCatalysis, Kodansha, Tokyo and AcademicPress, New York, 1977.
439 G. F. Berndt in Catalysis, Vol. 6, The RoyalSociety of Chemistry, London, 1983, p. 144.
440 G. Liu, D. Willcox, M. Garland, H. H. Kung, J.Catal. 96 (1985) 251.
441 R. P. Bell, Chem. Soc. Rev. 3 (1974) 513.442 L.Melander,W.H. Sauder, Jr.,ReactionRates of
Isotopic Molecules, Wiley, New York 1980.443 S. Siegel, Adv. Catal. 16 (1966) 124.444 M. Kraus, Adv. Catal. 29 (1980) 151.445 M. Kraus in G. Ertl, H. Kn€ozinger, J. Weitkamp
(eds.): Handbook of Heterogeneous Catalysis,Vol. 3, Wiley-VCH, Weinheim 1997, p. 1051.
446 H. Kn€ozinger, Adv. Catal. 25 (1976) 184.447 C. Appel, J. Mantzaras, R. Schaeren, R. Bom-
bach, A. Inauen, Combust. Flame, 140 (2005)70.
448 T. Horstmann, H. Leuckel, B. Maurer, U. Maas,Proc. Safety Progr. 20 (2001) 215.
449 H. P. A. Calis, J. Nijenhuis, B. C. Paikert, F. M.Dautzenberg, C. M. van den Bleek, Chem. Eng.Sci. 56 (2001) 1713.
450 C. Appel, J. Mantzaras, R. Schaeren, R. Bom-bach, B. Kaeppeli, A. Inauen, Proc. Combust.Inst. 29 (2003) 1031.
451 M. Reinke, J. Mantzaras, R. Schaeren, R. Bom-bach, W. Kreutner, A. Inauen, Proc. Combust.Inst. 29 (2002) 1021.
452 U. Dogwiler, P. Benz, J. Mantzaras, Combust.Flame 116 (1999) 243.
106 Heterogeneous Catalysis and Solid Catalysts
453 M. Reinke, J. Mantzaras, R. Schaeren, R. Bom-bach, A. Inauen, S. Schenker, Combust. Flame136 (2004) 217.
454 U. Kunz, U. Peuker, T. Turek, M. Estenfelder inU. Br€ockel,W.Meier, G.Wagner (eds.):ProductDesign and Engineering, Wiley-VCH, Wein-heim 2007.
455 C. N. Satterfield, T. K. Sherwood: Role of Dif-fusion in Catalysis, Addison-Wesley, Reading,Mass. 1963, p. 56.
456 C. N. Satterfield: Mass Transfer in Heteroge-neous Catalysis, MIT Press, Cambridge, Mass.,1970, p. 129.
457 E. W. Thiele, Ind. Eng. Chem. 31 (1939) 916.458 G. Damk€ohler, Chem. Ing. 3 (1939) 430.459 Y. B. Zeldowitch, Acta Physicochim. USSR 10
(1939) 582.460 P. B. Weisz, Adv. Catal. 13 (1962) 137.461 C. D. Prater, Chem. Eng. Sci. 8 (1958) 284.462 P.B.Weisz,C.D.Prater,Adv.Catal.6 (1954) 143.463 E. Wicke, Angew. Chem. 19 (1947) 57.464 E. Wicke, Z. Elektrochem. 60 (1956) 774.465 R. Aris: The Mathematical Theory of Diffusion
and Reaction in Permeable Catalysts,Vols. 1and 2, Clarendon Press, Oxford 1975.
466 J. J. Carberry in J. R. Anderson, M. Boudart(eds.): Catalysis—Science and Technology,Vol. 8, Springer, Berlin 1987, p. 131.
467 P. B. Weisz, J. S. Hicks, Chem. Eng. Sci. 17(1962) 265.
484 L.K.Doraiswamy,M.M.Sharma:HeterogeneousReactions, Vol. 2, Wiley, New York 1984, p. 9.
485 B. Jager, R. Espinoza,Catal. Today 23 (1995) 17.486 W. Ehrfeld, V. Hessel, H. L€owe:Microreactors,
Wiley-VCH, Weinheim 2000.487 K.-F. Jensen, Chem. Eng. Sci. 56 (2001) 293.488 S. J. Haswell, R. J. Middleton, B. O’Sullivan, V.
Skelton, P. Watts, P. Styring, J. Chem. Soc.Chem. Commun. 2001, 391.
489 T. Wirth (ed.): Microreactors in Organic Syn-thesis and Catalysis, Wiley-VCH, Weinheim2008.
490 K. J€ahnisch, V. Hessel, H. L€owe, M. Baerns,Angew. Chem. Int. Ed. 43 (2004) 406.
491 L. Kiwi-Minsker, A. Renken, Catal. Today 110(2005) 2.
492 G. Veser, Chem. Eng. Sci. 56 (2001) 1265.493 T. Inoue, M. A. Schmidt, K. F. Jensen, Ind. Eng.
Chem. Res. 46 (2007) 1153.494 P. D. I. Fletcher, S. J. Haswell, Chem. Br. 35
(1999) 38.495 E. Klemm, E. Dietzsch, T. Schwarz, T. Kruppa,
A. L. de Oliveira, F. Becker, G. Markowz, S.Schirrmeister, R. Sch€utte, K. J. Caspary, F.Sch€uth, D. H€onicke, Ind. Eng. Chem. Res. 47(2008) 2086.
496 Y. Sh. Matros, G. A. Bunimovich in G. Ertl, H.Kn€ozinger, J. Weitkamp (eds.): Handbook ofHeterogeneous Catalysis, Vol. 3, Wiley-VCH,Weinheim 1997, p. 1464.
497 A. Renken, Int. Chem. Eng. 33 (1993) 61.498 J.-A. Dalmon in G. Ertl, H. Kn€ozinger, J. Weit-
kamp (eds.):Handbook of Heterogeneous Cataly-sis, Vol. 3,Wiley-VCH,Weinheim1997, p. 1387.
499 H. P. Hsieh, Catal. Rev.-Sci. Eng. 33 (1991) 1.500 G. Sarraco, V. Specchia,Catal. Rev.-Sci. Eng. 36
(1994) 305.501 R. Soria, Catal. Today 25 (1995) 285.502 G. Donati, N. Habashi, I. Miracca, D. Sanfilippo
in G. Ertl, H. Kn€ozinger, J. Weitkamp (eds.):
Heterogeneous Catalysis and Solid Catalysts 107
Handbook of Heterogeneous Catalysis, Vol. 3,Wiley-VCH, Weinheim 1997, p. 1479.
503 D. B. Keyes, Ind. Eng. Chem. 24 (1932) 1096.504 D. F. Othmer, Ind. Eng. Chem. 33 (1941) 1106.505 P. E. Sauvage in G. Ertl, H. Kn€ozinger, J. Weit-
kamp (eds.): Handbook of Heterogeneous Ca-talysis, Vol. 3, Wiley-VCH, Weinheim 1997,p. 1339.
506 A. Baiker, Chem. Rev. 99 (1999) 453.507 P. G. Jessop, W. Leitner (eds.): Chemical Syn-
thesis Using Supercritical Fluids, Wiley-VCH,Weinheim 1999.
508 R. Wandeler, A. Baiker, Cattech 4 (2000) 128.509 R. J. Kee, F. M. Rupley, J. A. Miller, M. E.
Coltrin, J. F.Grcar, E.Meeks,H.K.Moffat, A. E.Lutz, G. Dixon-Lewis, M. D. Smooke, J. War-natz, G. H. Evans, R. S. Larson, R. E. Mitchell,L. R. Petzold, W. C. Reynolds, M. Caracotsios,W. E. Stewart, P. Glarborg, C.Wang, O. Adigun,CHEMKIN, 3.6 ed., Reaction Design, Inc.,www.chemkin.com, San Diego, 2000.
510 D. G. Goodwin,CANTERA. An open-source,extensible software suite for CVD process sim-ulation, www.cantera.org, 2003.
511 O. Deutschmann, S. Tischer, C. Correa, D.Chatterjee, S. Kleditzsch, V. M. Janardhanan,DETCHEM software package, 2.0 ed., www.detchem.com, Karlsruhe, 2004.
513 CD-adapco, London Office, 200 ShepherdsBushRoad, London,W67NY,UnitedKingdom,www.cd-adapco.com.
514 FIRE, AVL LIST GmbH, www.avl.com, Graz,Austria, 2005.
515 CFD-ACþ, CFD Research Corporation, www.cfdrc.com, Huntsville, AL, 2005.
516 CFX, www-waterloo.ansys.com, 2005.517 J. Shadid, S. Hutchinson, G. Hennigan, H. Mof-
fat, K. Devine, A. G. Salinger, Parallel Comput.23 (1997) 1307.
518 L. L. Raja, R. J. Kee, O. Deutschmann, J. War-natz, L. D. Schmidt, Catal. Today 59 (2000) 47.
519 J. Mantzaras, C. Appel, P. Benz, U. Dogwiler,Catal. Today 59 (2000) 3.
520 R. Jahn, D. Snita, M. Kubicek, M.Marek,Catal.Today 38 (1997) 39.
521 G. C. Koltsakis, P. A. Konstantinidis, A. M.Stamatelos, Appl. Catal. B 12 (1997) 161.
522 S. Tischer, C. Correa, O. Deutschmann, Catal.Today 69 (2001) 57.
523 S. Tischer, O. Deutschmann, Catal. Today 105(2005) 407.
524 R. Schwiedernoch, S. Tischer, C. Correa, O.Deutschmann, Chem. Eng. Sci. 58 (2003) 633.
525 M. Nijemeisland, A. G. Dixon, Am. Inst. Chem.Eng. J. 2004, 50 () 906.
526 T. Zeiser, P. Lammers, E. Klemm, Y. W. Li, J.Bernsdorf, G. Brenner, Chem. Eng. Sci. 56(2001) 1697.
527 J. Neumann, H. Golitzer, A. Heywood, I. Ticu,Revista Chim. 53 (2002) 721.
528 R. Quiceno, J. P�erez-Ram�ırez, J. Warnatz, O.Deutschmann, Appl. Catal. A 303 (2006) 166.
529 D. L. Trimm in G. Ertl, H. Kn€ozinger, J. Weit-kamp (eds.): Handbook of Heterogeneous Ca-talysis, Vol. 3, Wiley-VCH, Weinheim 1997,p. 1263.
530 J. R. Rostrup-Nielsen in C. H. Bartholomew, J.B. Butt (eds.): Catalyst Deactivation, ElsevierScience, Amsterdam 1991.
531 J. Barbier in J. Oudar, H. Wise (eds.): Deactiva-tion andPoisoning ofCatalysts,M.Dekker,NewYork 1985.
532 C. H. Bartholomew, P. K. Agrawal, J. R. Katzer,Adv. Catal. 31 (1982) 135.
533 P. Dufresne, A. Quesada, S. Miguarel in D. L.Trimm, S. Akasheh, M. Absi-Halabi, A. Bishara(eds.): Catalysis in Petroleum Refining, ElsevierScience, Amsterdam 1990.
534 K. Tanabe, M. Misono, Y. Ono, H. Hattori: NewSolid Acids and Bases, Elsevier, Amsterdam1989.
535 D.L. Trimm,Chem.Eng.Process 18 (1984) 137.536 D. C. McCulloch in B. Leach (ed.): Applied
Industrial Catalysis, Vol. 1, Academic Press,New York 1983, p. 103.
537 Y. Huang, N.W. Cant, J. Guerbios, D. L. Trimm,A. Walpole: Proc. Third Intern. Congress onCatal. and Automotive Pollution Control, Brus-sels, Elsevier, Amsterdam 1995, p. 56.
538 C. A. Bernardo, D. L. Trimm,Carbon 17 (1979)115.
539 H. Hiller et al.: “Gas Production”, in Ullmann’sEncyclopedia of Industrial Chemistry, Vol.A12,Wiley-VCH, Weinheim 2006.
540 P. H€aussinger, R. Lohm€uller, A. M. Watson:“Hydrogen”, in Ullmann’s Encyclopedia of In-dustrial Chemistry, Vol. A13, Wiley-VCH,Weinheim 2002, p. 189.
541 S. Ernst,W. Petzny in R. Dittmeyer,W.Keim, G.Kreysa, A. Oberholz (eds.):Winnacker-K€uchler,5. ed, Vol. 4, Wiley-VCH, Weinheim 2005,p. 523.
542 J. R. Rostrup-Nielsen inG. Ertl, H.Kn€ozinger, F.Sch€uth, J. Weitkamp (eds.): Handbook of Het-erogeneous Catalysis, 2nd ed., Vol. 6, Wiley-VCH, Weinheim 2008, p. 2882.
543 J. R. Rostrup-Nielsen, J. Sehested, J. K. Nors-kov.Adv. Catal. 47 (2002) 65.
108 Heterogeneous Catalysis and Solid Catalysts
544 J. Wei, E. Iglesia, J. Phys. Chem. B 108 (2004)4094.
545 D. A. Hickman, L. D. Schmidt, J. Catal. 138(1992) 267.
546 J. J. Krummenacher, K. N. West, L. D. Schmidt,J. Catal. 215 (2003) 332.
547 J. R. Salge, G. A. Deluga, L. D. Schmidt, J.Catal. 235 (2005) 69.
548 J. N. Armor, J. Membrane Sci. 147 (1998) 217.549 I. Aartun, B. Silberova, H. Venvik, P. Pfeifer, O.
Gorke, K. Schubert, A. Holmen, Catal. Today105 (2005) 469.
550 T. J. Remans, G. Jenzer, A. Hoek in G. Ertl, H.Kn€ozinger, F. Sch€uth, J.Weitkamp (eds.):Hand-book of Heterogeneous Catalysis, 2nd ed., Vol.6, Wiley-VCH, Weinheim 2008, p. 2994.
551 G. A Olah, A. Goeppert, G. K. S. Prakash:Beyond Oil and Gas: The Methanol Economy,Wiley-VCH, Weinheim 2006.
552 D. C. Rennard, P. J. Dauenhauer, S. A. Tupy, L.D. Schmidt, Energy Fuels 22 (2008) 1318.
553 P. J. Dauenhauer, B. J. Dreyer, N. J. Degenstein,L. D. Schmidt, Angew. Chem. Int. Ed. 46 (2007)5864.
554 E. C. Wanat, B. Suman, L. D. Schmidt, J. Catal.235 (2005) 18.
555 K.-O. Hinrichsen, K. Kochloefl, M.Muhler in G.Ertl,H.Kn€ozinger, F. Sch€uth, J.Weitkamp (eds.):Handbook of Heterogeneous Catalysis, 2nd ed.,Vol. 6, Wiley-VCH, Weinheim 2008, p. 2905.
556 D. S. Newsome, Catal. Rev. Sci. Eng. 21 (1980)275.
557 G. Ertl in J. R. Jennings (ed.): Catalytic Ammo-nia Synthesis: Fundamentals and Practice, Fun-damental and Applied Catalysis, Plenum Press,New York 1991, p. 109.
558 R. Schl€ogel in G. Ertl, H. Kn€ozinger, F. Sch€uth,J.Weitkamp (eds.):Handbook ofHeterogeneousCatalysis, 2nd ed., Vol. 5, Wiley-VCH, Wein-heim 2008, p. 2501.
559 K. Aika, A. Ozaki, J. Catal. 16 (1970) 97.560 F. Bozso, G. Ertl, M. Weiss, J. Catal. 50 (1977)
519.561 P. H. Emmett, S. Brunauer, J. Am. Chem. Soc. 59
(1937) 310.562 M. Boudart, Catal. Rev.-Sci. Eng. 23 (1981) 1.563 G. Ertl, M. Weiss, S. B. Lee, Chem. Phys. Lett.
60 (1979) 391.564 M. Muhler, E. T€ornqvist, L. P. Nielsen, B. S.
Clausen, H. Topsøe, Catal. Lett. 25 (1994) 1.565 R. Burch, R. J. Chappell, S. E. Golunski, J.
Chem. Soc. Faraday Trans. 1 85 (1989) 3569.566 J. Nakamura, I. Nakamura, T. Uchima, T. Wa-
tanabe, T. Fujitani, Stud. Surf. Sci. Catal. 101(1996) 1389.
567 H. Topsøe, N. Topsøe, Top. Catal. 8 (1999) 267.568 T. S. Askgaard, J. K. Norskov, C. V. Ovesen, P.
Stoltze, J. Catal. 156 (1995) 229.569 K. M. Vanden Bussche, G. F. Froment, J. Catal.
161 (1996) 1.570 B. Lommerts, G. Graff, A. Beenackers, Chem.
Eng. Sci. 55 (2000) 5589.571 R. G. Herman in L. Guczi (ed.): New Trends in
CO Activation, Elsevier, New York 1991.572 F. Fischer, H. Tropsch, Brennst. Chem. 4 (1923)
276.573 R. B. Anderson in P. H. Emmett (ed.): Catalysis,
Vol. 4, VanNostrand-Rheinhold, NewYork 1956.574 A. T. Bell, Catal. Rev. Sci. Eng. 23 (1981) 203.575 G. P. van der Laan, A. A. C. M. Beenackers,
(TH), Karlsruhe2002.577 B. H. Davis, Top. Catal. 32 (2005) 143.578 R. Oukaci, A. H. Singleton, J. G. Goodwin, Jr.,
Appl. Catal. A 186 (1999) 129.579 M. E. Dry, J. Mol. Catal. 17 (1982) 133.580 S. T. Sie, Rev. Chem. Eng. 14 (1988) 109.581 F. G. Botes, W. B€ohringer, Appl. Catal A: Gen-
eral 267 (2004) 217.582 A.M. Subiranas, G. Schaub, Int. J. Chem. React.
Eng. 5 (2007) A78.583 M. F. M. Post, A. C. van’t Hoog, J. K. Mind-
erhoug, S. T. Sie, AIChE J. 35 (1989) 1107.584 Y.-N.Wang, Y.-Y. Xu, H.-W.Xiang, Y.-W. Li, B.
J. Zhang, Ind. Eng. Chem. Res. 40 (2001) 4324.585 A.-M. Hilmen, E. Bergene, O. A. Lindvag, D.
Schanke, S. Eri, A. Holmen, Catal. Today 69(2001) 227.
586 R. K. Grasselli in G. Ertl, H. Kn€ozinger, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, Vol. 5, Wiley-VCH, Weinheim 1997,p. 2302.
587 R. A. van Santen G. Ertl, H. Kn€ozinger, J. Weit-kamp (eds.):Handbook of HeterogeneousCatal-ysis, Vol. 5, Wiley-VCH, Weinheim 1997,p. 2244.
588 R. A. van Santen, H. C. P. E. Kuipers,Adv. Catal.35 (1988) 265.
589 M. C. Zonnevylle, J. J. C. Geerlings, R. A. vanSanten, J. Catal. 148 (1994) 417.
590 M. Neurock, R. A. van Santen,W. Biemolt, A. P.J. JansenJ. Am. Chem. Soc. 116 (1994) 6860.
591 J.W.He,U.Memmert,K.Griffiths, P. R.Norton,J. Chem. Phys. 90 (1989) 5082.
592 P. J. van den Hoek, E. J. Baerends, R. A. vanSanten, J. Phys. Chem. 93 (1989) 6469.
593 L. M. Akella, H. N. Lee, J. Catal. 86 (1984)465.
594 M.Bowker, R. C.Waugh, Surf. Sci. 155 (1984) 1.
Heterogeneous Catalysis and Solid Catalysts 109
595 P. Arpentinier, F. Cavani, F. Trifiro: The Tech-nology of Catalytic Oxidations, Vol. 1, EditionsTechnip, Paris 2001, p. 245.
596 G. Bellussi, M. S. Rigutto, Stud. Surf. Sci. Catal.85 (1991) 177.
597 M. G. Clerici, Oil Gas Eur. Mag. 32 (2006) 77.598 B. Ford, Chem. Market. Rep. 269 (2006) 24.599 P. N. Rylander in J. R. Anderson, M. Boudart
(eds.): Catalysis: Science and Technology,Vol. 4, Springer, Berlin 1983, p. 1.
600 J. F. Brazdil, Top. Catal. 38 (2006) 289.601 P. Mars, D. W. van Krevelen, Chem. Eng. Sci.
Special Suppl. 3 (1954) 41.602 R. K. Grasselli, J. D. Burrington, I & EC Res. &
Dev. 23 (1984) 393.603 T. P. Snyder, C. G. Hill, Catal. Rev.-Sci. Eng. 31
(1989) 43.604 J. D. Burrington, C. T. Kartisek, R. K. Grasselli,
J. Catal. 87 (1984) 363.605 J. M. Lopez Nieto, Top. Catal. 41 (2006) 3.606 B. C. Gates: Catalytic Chemistry, Wiley, New
York 1992, p. 406.607 R. Prius in G. Ertl, H. Kn€ozinger, J. Weitkamp
(eds.): Handbook of Heterogeneous Catalysis,Vol. 4, Wiley-VCH, Weinheim 1997, p. 1908.
608 D. D. Whitehurst, T. Isoda, I. Mochida, Adv.Catal. 42 (1998) 345.
609 M. Houalla, N. K. Nag, A. V. Sapre, D. H.Broderick,B.C.Gates,AIChE J. 24 (1978) 1015.
610 A. Kundu, K. D. P. Nigam, A. M. Duquenne, H.Delmas, Rev. Chem. Eng. 6 (2003) 531.
611 M. S. Rana, V. S�amano, J. Ancheyta, J. A. I.Diaz, Fuel 86 (2007) 1216.
612 M. Koebel, M. Elsener, G. Madia, Ind. Eng.Chem. Res. 40 (2001) 52.
613 US 2 975 025, 1961(G. Cohn, D. Steele, H.Andersen).
614 US3 279 884, 1966(K.Kartte,H.Nonnenmaker).
615 US 4 085 193, 1978(F. Nakajima, M. Takeuchi,S. Matsuda, S. Uno, T. Mori, Y. Watanabe, M.Inamuri).
616 C. Orsenigo, L. Lietti, E. Tronconi, P. Forzatti, F.Bregani, Ind. Eng. Chem. Res. 37 (1998) 2350.
617 G. S. Qi, R. T. Yang, R. Chang, S. Cardoso, R. A.Smith, Appl. Catal. A: General 275 (2005) 207.
618 M. Groves, M. Schwefer, R. Siefert, TCE 778(2006) 30.
619 M. Votsmeier, T. Kreuzer, G. Lepperhoff:“Automobile Exhaust Control” in Ullmann’sEncyclopedia of Industrial Chemistry, Vol.A3, Wiley-VCH, Weinheim 2005, p. 189
620 S. J. Lox in G. Ertl, H. Kn€ozinger, F. Sch€uth, J.Weitkamp (eds.): Handbook of HeterogeneousCatalysis, 2nd ed., Vol. 5 Wiley-VCH, Wein-heim 2008, p. 2274.
621 R.M.Heck,R. J. Farrauto, S. T.Gulati,CatalyticAir Pollution Control, Wiley-InterSci., NewYork 2002.
622 S. Kureti,Habilitation thesis, Fakult€at f€ur Che-mie und Biowissenschaften, Universit€at Karls-ruhe (TH), 2008.
623 M. V. Twigg, Appl. Catal. B 70 (2007) 2.624 D. Chatterjee, O. Deutschmann, J. Warnatz,
Faraday Discuss. 119 (2001) 371.625 N. Fekete, R. Kemmler, D. Voigtl€ander, B.
Krutzsch, E. Room, G. Wenninger, W. Streh-lau, J. A. A. van den Tillaart, J. Leyrer, E. S.Lox, W. M€uller, SAE Technical Paper 960133(1996)
626 F. Rohr, S. D. Peter, E. Lox, M. K€ogel, A. Sassi,L. Juste, C. Rigaudeau, G. Belot, P. Gelin, M.Primet, Appl. Catal. B 56 (2005) 201.
627 P. Zelenka, W. Cartellieri, P. Duke, Appl. Catal.B 7 (1996) 3.
628 O. Salvat, P. Marez, G. Belot, SAE TechnicalPaper 2000-01-0473(2000).












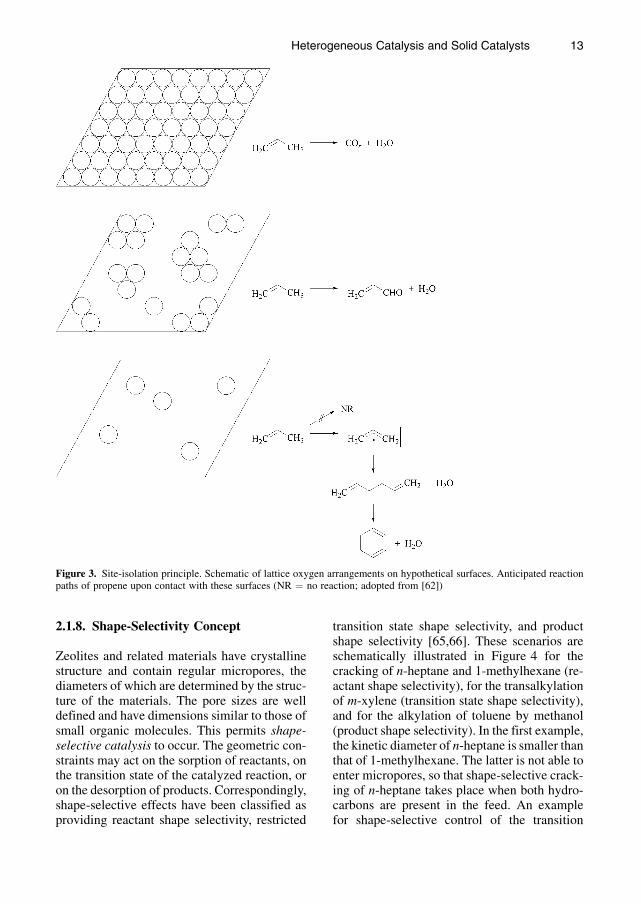













































































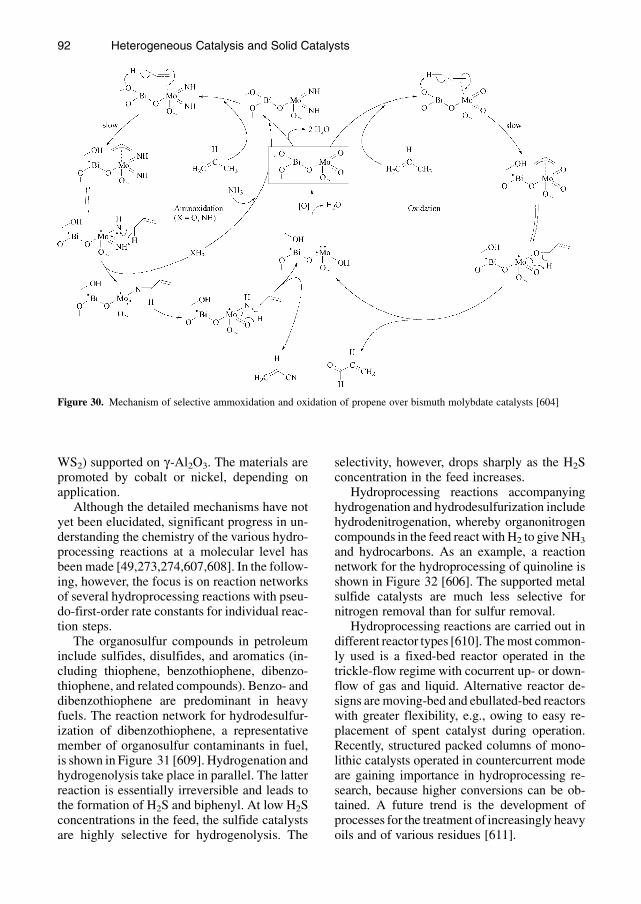






























![[Charles N. Satterfield] Heterogeneous Catalysis](https://static.documents.pub/doc/80x56/577cc1a51a28aba71193945e/charles-n-satterfield-heterogeneous-catalysis.jpg)




