High sensitivity EndoV mutation scanning through real-time ligase proofreading Hanna Pincas, Maneesh R. Pingle, Jianmin Huang, Kaiqin Lao 1 , Philip B. Paty 2 , Alan M. Friedman 3 and Francis Barany* Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York, NY 10021, USA, 1 Applied Biosystems, Foster City, CA 94404, USA, 2 Colorectal Service, Department of Surgery, Memorial Sloan Kettering Cancer Center, New York, NY 10021, USA and 3 Department of Biological Sciences, Purdue University, West Lafayette, IN 47907, USA Received September 9, 2004; Revised and Accepted October 11, 2004 ABSTRACT The ability to associate mutations in cancer genes with the disease and its subtypes is critical for under- standing oncogenesis and identifying biomarkers for clinical diagnosis. A two-step mutation scanning method that sequentially used endonuclease V (EndoV) to nick at mismatches and DNA ligase to reseal incorrectly or nonspecifically nicked sites was previously developed in our laboratory. Herein we report an optimized single-step assay that enables ligase to proofread EndoV cleavage in real-time under a compromise between buffer conditions. Real-time proofreading results in a dramatic reduction of background cleavage. A universal PCR strategy that employs both unlabeled gene-specific primers and labeled universal primers, allows for multiplexed gene amplification and precludes amplification of primer dimers. Internally labeled PCR primers elimi- nate EndoV cleavage at the 5 0 terminus, enabling high-throughput capillary electrophoresis readout. Furthermore, signal intensity is increased and artifacts are reduced by generating heteroduplexes containing only one of the two possible mismatches (e.g. either A/C or G/T). The single-step assay improves sensitivity to 1:50 and 1:100 (mutant:wild type) for unknown mutations in the p53 and K-ras genes, respectively, opening prospects as an early detection tool. INTRODUCTION Multiple somatic alterations lie at the root of cancer develop- ment and tumor heterogeneity. Of cancer genes reported in the current databases, 90% have somatic mutations, 20% have germline mutations and 10% have both (1). A major challenge in cancer biology is to unravel the molecular anatomy of individual cancers and tumor subtypes, which would allow a better understanding of the role of genetic alterations in tumor progression, and provide diagnostic and prognostic markers. The molecular basis for the development and progression of colorectal cancers is well developed in the scientific literature. Colorectal tumors have been classified as either demonstrating chromosomal instability (CIN) or microsatellite instability (MIN). The former is associated with sequential sporadic mutations in the APC (70%), K-ras (40%) and p53 (50%) genes, while the latter contains additional sporadic mutations in the TGFBRII (90%) and BAX (50%) genes (2–6). Muta- tional patterns may reflect alternative routes to activating/ inactivating signaling, growth and apoptosis pathways. APC mutations disrupt the association of APC and b-catenin, result- ing in excessive amounts of b-catenin and overactivation of the Wingless/Wnt signaling pathway. Oncogenic mutations in b-catenin were also observed in some colorectal cancers that lacked APC mutations (3). Likewise, colorectal tumors lack- ing K-ras mutations may have their ras pathway activated through B-raf mutations (7). In addition to these shared mutations, a recent study showed a high frequency of mutations of the PIK3CA gene (a subunit of the phosphatidylinositol 3-kinase; PI3K) in colon cancers, making this gene a potential biomarker for early detection and/ or prognosis of the disease (8). Similarly, the discovery of mutations in various tyrosine kinase and tyrosine phosphatase genes in colorectal cancers suggests that these could become targets for personalized therapy (9,10). As the number of genes linked to cancer grows, there is an increasing demand for the development of new methods for high-throughput detection of unknown mutations. Different mutation detection technologies have been developed to identify known mutations: these include DNA microarrays (11–13), the polymerase chain reaction/ligase detection reaction (PCR/LDR) (14), now used in combination with the universal DNA microarray (15–17) and primer extension assays (18). Different technologies are used for detection of unknown mutations: hybridization analysis using high-density oligonucleotide arrays (12), denaturing high-performance liquid chromatography (DHPLC) (19), capillary electrophoresis-based single strand conformation polymorphism (CE-SSCP) (20), denaturing gradient gel elec- trophoresis (DGGE) (21) and heteroduplex analysis (HA) (22) *To whom correspondence should be addressed. Tel: +1 212 746 6509; Fax: +1 212 746 7983; Email: [email protected]Nucleic Acids Research, Vol. 32 No. 19 ª Oxford University Press 2004; all rights reserved Nucleic Acids Research, 2004, Vol. 32, No. 19 e148 doi:10.1093/nar/gnh150 Published online October 28, 2004 Downloaded from https://academic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
Transcript
High sensitivity EndoV mutation scanning throughreal-time ligase proofreadingHanna Pincas, Maneesh R. Pingle, Jianmin Huang, Kaiqin Lao1, Philip B. Paty2,
Alan M. Friedman3 and Francis Barany*
Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York, NY 10021, USA,1Applied Biosystems, Foster City, CA 94404, USA, 2Colorectal Service, Department of Surgery, Memorial SloanKettering Cancer Center, New York, NY 10021, USA and 3Department of Biological Sciences, Purdue University,West Lafayette, IN 47907, USA
Received September 9, 2004; Revised and Accepted October 11, 2004
ABSTRACT
The ability to associate mutations in cancer geneswith the disease and its subtypes is critical for under-standing oncogenesis and identifying biomarkersfor clinical diagnosis. A two-step mutation scanningmethod that sequentially used endonuclease V(EndoV) to nick at mismatches and DNA ligase toreseal incorrectly or nonspecifically nicked siteswas previously developed in our laboratory. Hereinwe report an optimized single-step assay that enablesligase to proofread EndoV cleavage in real-time undera compromise between buffer conditions. Real-timeproofreading results in a dramatic reduction ofbackground cleavage. A universal PCR strategy thatemploys both unlabeled gene-specific primers andlabeled universal primers, allows for multiplexedgene amplification and precludes amplification ofprimer dimers. Internally labeled PCR primers elimi-nate EndoV cleavage at the 50 terminus, enablinghigh-throughput capillary electrophoresis readout.Furthermore, signal intensity is increased andartifacts are reduced by generating heteroduplexescontaining only one of the two possible mismatches(e.g. either A/C or G/T). The single-step assayimproves sensitivity to 1:50 and 1:100 (mutant:wildtype) for unknown mutations in the p53 and K-rasgenes, respectively, opening prospects as an earlydetection tool.
INTRODUCTION
Multiple somatic alterations lie at the root of cancer develop-ment and tumor heterogeneity. Of cancer genes reported in thecurrent databases, �90% have somatic mutations, 20% havegermline mutations and 10% have both (1). A major challengein cancer biology is to unravel the molecular anatomy ofindividual cancers and tumor subtypes, which would allowa better understanding of the role of genetic alterations in
tumor progression, and provide diagnostic and prognosticmarkers.
The molecular basis for the development and progression ofcolorectal cancers is well developed in the scientific literature.Colorectal tumors have been classified as either demonstratingchromosomal instability (CIN) or microsatellite instability(MIN). The former is associated with sequential sporadicmutations in the APC (70%), K-ras (40%) and p53 (50%)genes, while the latter contains additional sporadic mutationsin the TGFBRII (90%) and BAX (50%) genes (2–6). Muta-tional patterns may reflect alternative routes to activating/inactivating signaling, growth and apoptosis pathways. APCmutations disrupt the association of APC and b-catenin, result-ing in excessive amounts of b-catenin and overactivation ofthe Wingless/Wnt signaling pathway. Oncogenic mutations inb-catenin were also observed in some colorectal cancers thatlacked APC mutations (3). Likewise, colorectal tumors lack-ing K-ras mutations may have their ras pathway activatedthrough B-raf mutations (7).
In addition to these shared mutations, a recent study showeda high frequency of mutations of the PIK3CA gene (a subunitof the phosphatidylinositol 3-kinase; PI3K) in colon cancers,making this gene a potential biomarker for early detection and/or prognosis of the disease (8). Similarly, the discovery ofmutations in various tyrosine kinase and tyrosine phosphatasegenes in colorectal cancers suggests that these could becometargets for personalized therapy (9,10). As the number of geneslinked to cancer grows, there is an increasing demand for thedevelopment of new methods for high-throughput detection ofunknown mutations.
Different mutation detection technologies have beendeveloped to identify known mutations: these include DNAmicroarrays (11–13), the polymerase chain reaction/ligasedetection reaction (PCR/LDR) (14), now used in combinationwith the universal DNA microarray (15–17) and primerextension assays (18). Different technologies are used fordetection of unknown mutations: hybridization analysisusing high-density oligonucleotide arrays (12), denaturinghigh-performance liquid chromatography (DHPLC) (19),capillary electrophoresis-based single strand conformationpolymorphism (CE-SSCP) (20), denaturing gradient gel elec-trophoresis (DGGE) (21) and heteroduplex analysis (HA) (22)
*To whom correspondence should be addressed. Tel: +1 212 746 6509; Fax: +1 212 746 7983; Email: [email protected]
Nucleic Acids Research, Vol. 32 No. 19 ª Oxford University Press 2004; all rights reserved
/nar/article/32/19/e148/1244789 by guest on 30 January 2022
[for review, see (13,23,24)]. However, these techniqueseither lack high sensitivity and are unable to determine theapproximate position of the polymorphism, or suffer fromlow throughput (19,22,25–28). Finally, dideoxy-sequencinghas difficulty in detecting heterozygous mutations, and is oflimited utility in the analysis of solid tumors where mutantDNA may represent as little as 15% of the total. Other methodsinclude in vitro transcription/translation-based approaches(29–33), chemical and enzymatic mismatch cleavagedetection (e.g. Cleavase, RNase, T4 endonuclease VII,MutS enzymes and CEL I). In vitro mismatch cleavagemethods encounter variability in signal intensity comparedwith background bands (23,34–43).
We recently developed a two-step mutation scanning assaybased on enzymatic mismatch cleavage by thermostable TmaEndonuclease V, followed by a proofreading step with ther-mostable DNA ligase to suppress noise arising from the back-ground of matched cleavage (44). Briefly, 50 fluorescentlylabeled mutant and wild-type PCR amplicons are hetero-duplexed, EndoV recognizes and cleaves DNA one base to the30 side of the mismatch, in addition to nicking matched DNAat low levels. Ligase reseals these background nicks (see steps3 and 4 in Figure 1). Fluorescent products are separated on aDNA sequencing gel, which reveals the approximate positionof the mutation. Subsequent DNA sequencing of the specificregion identifies the exact sequence variation. This method hasbeen successfully applied to detect mutations and small inser-tions/deletions in p53, VHL, K-ras, APC, BRCA1 and BRCA2(17,44), except for certain GC-rich sequences that are refrac-tory to cleavage present at a frequency of only 2% in humanDNA. However, background cleavage by EndoV of someexons makes it difficult to distinguish the correct mutationcleavage signal, especially with pooled samples (17,44).
Here, we describe a series of improvements resulting insubstantial reduction in background and thus improved muta-tion scanning. These improvements include a universal ampli-fication strategy with nuclease resistant labeling, a single-stepEndoV/real-time ligase proofreading reaction, and separationon a capillary electrophoresis array. Optimizing the single-step reaction conditions represents a critical advance giventhat these two enzymes have opposing buffer requirements.EndoV possesses a variety of endonuclease activity towarddeaminated DNA bases deoxyinosine, deoxyxanthine, de-oxyoxanosine, deoxyuridine and other aberrant sites suchas apurinic and urea sites (45–51), and in general, non-physiological reaction conditions are required to elicit cleav-age at mismatched sites (50,52). The challenge in developingreal-time proofreading was to find conditions that allowedEndoV to nick at mismatched sites, while simultaneouslypermitting the ligase to proofread by predominantly resealingnicks at matched sites. The sensitivity of the present methodallows detection of 1 mutant in 100 wild-type sequences,making it amenable to sample pooling and early detectionstrategies.
MATERIALS AND METHODS
Materials
All routine chemical reagents were purchased from SigmaChemicals (St. Louis, MO, USA) or Fisher Scientific (Fair
Lawn, NJ, USA). GeneScanTM –500 (LIZTM) Size Standard,Hi-Di formamide, polymer POP-7, AmpliTaq and AmpliTaqGold DNA polymerases, and deoxyribonucleoside tripho-sphates (dNTPs) were purchased from Applied Biosystems(Foster City, CA). BSA and ATP were purchased fromBoehringer-Mannheim (Indianapolis, IN, USA). Proteinase Kwas purchased from QIAGEN (Valencia, CA, USA). Lambdaexonuclease and T4 polynucleotide kinase were purchasedfrom New England Biolabs (Beverly, MA). Unlabeled deoxy-oligonucleotides were purchased from Integrated DNATechnologies Inc. (Coralville, IA, USA), while HPLC-purified VIC- and NED-labeled deoxyoligonucleotideswere obtained from Applied Biosystems. Thermotoga mar-itima Endonuclease V and Thermus species AK16D DNAligase were purified as previously described (50,53). Tumorand normal tissue were obtained from surgical resection ofcolon cancers at Memorial Sloan Kettering Cancer Center,which were snap-frozen in liquid nitrogen within 15 min oftumor removal. DNA from these tissues was extracted andpurified using a proteinase-K/lithium chloride/ethanol proto-col as described (QIAGEN). Tumor samples known to con-tain Q192Ter (C!T) or Y205F (A!T) mutations in exon 6of p53 were used, as well as their matched normal colonicmucosa. Genomic DNA from cell lines was extracted usingDNeasy tissue kit from QIAGEN. HT-29 cell line containsthe wild-type K-ras gene, while SW480 and SW620 containpure exon 1 G12V (G!T) mutation. LoVo cell line containswild-type p53 gene, while HT-29, SW480 and SW620 celllines contain exon 8 R273H (G!A) mutation.
PCR amplification and 50 phosphorylation ofdeoxyoligonucleotides
DNA sequences of PCR primers used in this study are listed inTable 1. Fifty-microliter PCR reactions with gene-specificprimers alone, contained 20 mM Tricine, pH 8.7, 16 mM(NH4)2SO4, 2.5 mM MgCl2, 0.2 mM dNTP, 0.2 mM ofeach primer and 100 ng genomic DNA. The reaction mixturewas incubated at 95�C for 2 min (hot start), followed by theaddition of 5 U of AmpliTaq DNA polymerase. PCR ampli-fication conditions were as follows: 35 cycles of 94�C for 20 s,68�C for 30 s, 72�C for 1 min, followed by a final extensionstep at 72�C for 7 min.
Universal PCR reactions for amplification of specific genesfollowed by universal primer amplification (50 ml) contained20 mM Tricine, pH 8.7, 16 mM (NH4)2SO4, 2.5 mM MgCl2,0.2 mM of dNTP, 0.2 mM of each universal primer, 0.02 mM ofeach gene-specific primer, 5 U of AmpliTaq Gold DNA poly-merase and 150 ng of genomic DNA. Thermo-cycling condi-tions were as follows: 95�C for 10 min to activate AmpliTaqGold polymerase, followed by 20 cycles of 94�C for 30 s, 65�Cfor 1 min, 72�C for 1 min (for gene-specific amplification) andthen 30 cycles of 94�C for 30 s, 55�C for 1 min, 72�C for 1 min(for universal amplification), followed by a final extension stepat 72�C for 7 min. In the universal PCR reaction, the universalprimer pair consisted of a forward VIC-labeled primer anda reverse NED-labeled primer for the standard denaturation/renaturation’ procedure. In the ‘split label, denaturation,renaturation’ procedure, PCR amplification used one VIC-(or NED-) labeled universal primer and one unlabeled univer-sal primer. In the ‘split label, lambda exonuclease’ procedure,
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
Figure 1. Universal PCR to prepare heteroduplexed DNA substrates for EndoV/DNA ligase mutation scanning: split label, denaturation, renaturation or lambdaexonuclease treatment. Mutant and normal DNA are PCR amplified separately using Taq DNA polymerase (solid diamond) and gene-specific and universal primers,as detailed in the scheme. Substrates for EndoV/ligase are prepared from the PCR products by either of the following methods. (A) ‘Split label, denaturation,renaturation’ procedure. (B) ‘Split label, lambda exonuclease’ procedure: lambda exonuclease (Pacman icon) degrades 50 phosphorylated DNA, allowing the newlygenerated single-stranded DNA to anneal and generate labeled heteroduplexed fragments. EndoV preferentially nicks DNA one base 30 to the mismatch (largetriangle), but also generates nonspecific nicks with minor activity (small triangle). DNA ligase (solid circle) is used either subsequently or concurrently with EndoV toreseal these background nicks. Separation of products by capillary electrophoresis under various assay conditions is illustrated: first with a standard 50 fluorescentlabel (50 Fl-Oligo) or 20-O-methyl cytosine linkage added to the label [50 Fl-(20OMeC)2-Oligo]. In both these cases, EndoV cleaves the label off the PCR product,which results in false signal migrating at about 100 nt (cleaved label). Attachment of the fluorescent label internally in the primer sequence via the C6 position of acytosine (50 CCGC(C-Fl)-Oligo) renders labeled products resistant to EndoV cleavage; intensity of the mutation signal is stronger when using the lambda exonucleasemethod than in the denaturation, renaturation method.
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
PCR amplification used one VIC- (or NED-) labeled universalprimer and one 50 phosphorylated universal primer.
Prior to PCR amplification, primers were phosphorylatedat the 50 end by incubating 200 pmol of each unlabeled primerwith 10 U of T4 polynucleotide kinase for 1 h at 37�C in a25 ml-reaction volume containing 1· T4 polynucleotide kinasebuffer (70 mM Tris–HCl, pH 7.6, 10 mM MgCl2, and 5 mMDTT) and 1 mM ATP. The enzyme was then heat inactivatedfor 20 min at 65�C, and aliquots of the phosphorylationreaction were stored at �20�C.
Preparation of heteroduplexed DNA substrates
In the standard denaturation/renaturation procedure, approxi-mately equal ratios of VIC/NED-labeled wild-type PCRamplicons were mixed with VIC/NED-labeled mutant PCRamplicons in a 12 ml final volume (�1500 ng total DNA).The wild-type control consisted of VIC/NED-labeled wild-type DNA PCR products alone in a 12 ml final volume(�1500 ng total DNA). Taq DNA polymerase was inactivatedby adding 1 ml of proteinase K (20 mg/ml) to each mixture, andincubating at 65�C for 30 min, followed by a 10 min incuba-tion at 80�C to inactivate proteinase K. For denaturation/rena-turation, PCR mixtures were then heated at 95�C for 2 min,and gradually cooled down to room temperature as follows:95�C for 2 min, 95�C for 15 s, followed by a 0.2�C decrease intemperature every 15 s down to 45�C, and finally a 10 minincubation at 25�C.
For the ‘split label, denaturation, renaturation’ procedure,two distinct PCR mixtures were prepared as follows: approxi-mately equal yields of VIC-labeled wild-type PCR productswere mixed with NED-labeled mutant PCR products in a 12 mlfinal volume (�1500 ng total DNA; PCR mixture 1); likewise,VIC-labeled mutant PCR products were mixed with NED-labeled wild-type PCR products in another 12 ml reaction
(PCR mixture 2). The wild-type control consisted of a mixtureof VIC-labeled wild-type PCR products with NED-labeledwild-type PCR products in a 12 ml final volume (�1500 ngtotal DNA). As described above, Taq DNA polymerase wasinactivated by adding proteinase K and PCR mixtures weresubjected to denaturation/renaturation.
In the ‘split label, lambda exonuclease’ procedure, two dis-tinct PCR mixtures were prepared as follows: approximatelyequal yields of VIC/Phosphate-labeled wild-type PCR productswere mixed with Phosphate/NED-labeled mutant PCR productsin a 12 ml final volume (�1500 ng total DNA; PCR mixture 1);likewise, VIC/Phosphate-labeled mutant PCR products weremixed with Phosphate/NED-labeled wild-type PCR productsin another 12ml reaction (PCR mixture 2). The wild-type controlconsisted of a mixture of VIC/Phosphate-labeled wild-typePCR products with Phosphate/NED-labeled wild-type PCRproducts in a 12 ml final volume (�1500 ng total DNA). TaqDNA polymerase was inactivated by adding proteinase K asdescribed above. PCR mixtures were then incubated at 37�Cfor 1 h with 1 U of lambda exonuclease, which degrades50 phosphorylated DNA. Lambda exonuclease was then heatinactivated by incubating the reaction at 75�C for 10 min.
EndoV/ligase mutation scanning assay
The various buffer conditions tested for optimization of theEndoV/ligase assay are summarized in Table 2. StandardHEPES–Tris buffer conditions as well as Tricine buffer con-ditions I, II, III, IV, V and VII, all consisted of two-stepprocedures. In step 1, �6.5 ml of each heteroduplex PCRmixture, including the homoduplex wild-type control, wereincubated at 65�C (for 40 or 60 min, as indicated) in a20 ml first step reaction mixture containing the appropriatereagents. Then, in step 2, 15 ml of each step 1 reaction mixturewas subjected to the second step reaction in a 20 ml final
Table 1. PCR primers used for analysis on the ABI-3730 DNA analyzer
F, forward primer; R, reverse primer. Bases highlighted in bold correspond to the universal sequence.aUnderlined bases (CCGCC) have a 30 to 50 phosphate (inverted) linkage. The fluorescent dye is coupled to the 30 end of C instead of the 50 end.
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
volume at 65�C (for 30 or 60 min, as indicated), by adding theappropriate reagents. Conditions VI and VIII were single-stepreactions, in which �6.5 ml of each heteroduplex PCR mix-ture, including the homoduplex wild-type control, were incu-bated for 120 min at 65�C in a 20 ml reaction containing theindicated reagents. For determination of the sensitivity of thesingle-step EndoV/ligase assay, reactions were performedunder condition VIII, except that 1 mM EndoV and 12 nMligase were used instead of 500 nM and 6 nM, respectively.
Finally, all reactions were terminated by adding EDTA toa final concentration of 10 mM. This inhibited any furtherEndoV cleavage activity during capillary electrophoresis.One microliter aliquots of the reaction mixtures were dena-tured at 95�C for 2 min in 9 ml Hi-Di formamide along with0.4 ml GeneScanTM –500 (LIZTM) Size Standard, and run onthe ABI 3730 fluorescence-based capillary electrophoresisinstrument (Applied Biosystems, Foster City, CA). Electro-phoresis was carried out for 1200 s at 15 kV in POP-7 polymerat 60�C. On completion of the electrophoretic run, a capillaryarray image was displayed with ABI collection software v1.0.Data analysis was achieved using Gene Mapper fragmentanalysis software v3.0 (Applied Biosystems, Foster City,CA). Ultimately, capillary array images of the fragment ana-lysis data were displayed with Gel Render, an unpublishedprogram developed in our laboratory to correct for differencesin capillary length and fragment mobility in individualcapillaries by using internal size standards, and presented asan electrophoretogram in the form of the familiar gel picture.
Signal intensity and signal-to-noise ratio measurement
Fluorescence intensity values of mutation and backgroundcleavage bands obtained from Gene Mapper fragment analysissoftware v3.0 were normalized to the intensity of the 200
base fragment of the (LIZTM) internal Size Standard for eachcapillary lane. Signal-to-noise ratios were calculated usingthe normalized intensity for the brightest VIC- and NED-labeled background bands, which were 144 and 115 bases,respectively.
RESULTS
Reduction of EndoV cleavage of the 50 fluorescentlabel off the PCR products and validation of auniversal labeling strategy
To achieve high-throughput detection of somatic mutations intumors, we sought to transfer the EndoV/Ligase scanningassay to an automated capillary electrophoresis format. Pre-liminary EndoV cleavage data (using standard 50-end labeledPCR fragments as substrates) revealed the presence of twounanticipated bands migrating at the same mobility as frag-ments of 102 (VIC) and 94 (NED) bases, respectively. Theseanomalously migrating bands were removable by filtrationwith a 10 kDa cutoff. Thus, they most likely resulted fromEndoV cleavage near the label, most likely one base 30 to it,containing the 50 fluorescent label, a phosphate group (to pro-vide charge), and an additional base. This activity may corre-spond to the 50 exonuclease activity reported for Escherichiacoli EndoV (54). These bands created two problems: (i) theywould obscure authentic signal migrating at the same position,and (ii), this cleavage would reduce the overall intensity of thedesired mutation signal.
Three sets of p53 exon 8 PCR primers with differentlymodified 50 fluorescent labels (listed in Table 1) were testedfor their ability to resist EndoV cleavage one base 30 to thelabel. These primers all harbored on their 50 end a CGCCGCsequence, known to be refractory to EndoV cleavage when this
Table 2. Summary of the various buffer conditions tested for optimization of the EndoV/ligase assay
Standard conditions40 min 1. 1· EndoV buffer = 20 mM HEPES, pH 7.5, 1 mM DTT, with 1 mM EndoV30 min 2. 1· Ligase buffer = 20 mM Tris, pH 8.5, 50 mM KCl, 10 mM DTT, with 3 nM Ligase + 1 mM NAD
Condition I60 min 1. 1· EndoV buffer = 20 mM Tricine, pH 8, 1 mM DTT, with 1 mM EndoV60 min 2. 1· Ligase buffer = 40 mM Tricine, pH 8, 10 mM DTT, with 6 nM Ligase + 5 mM NAD
Condition II60 min 1. 1· EndoV buffer = 20 mM Tricine, pH 8, 5 mM DTT, with 1 mM EndoV + 6 nM Ligase + 5 mM NAD60 min 2. 1· Ligase buffer = 40 mM Tricine, pH 8, 6.25 mM DTT
Condition III60 min 1. 1· EndoV buffer = 40 mM Tricine, pH 8, 1 mM DTT, with 1 mM EndoV60 min 2. 1· Ligase buffer = 40 mM Tricine, pH 8, 10 mM DTT, with 6 nM Ligase + 5 mM NAD
Condition IV60 min 1. 1· EndoV buffer = 40 mM Tricine, pH 8, 5 mM DTT, with 1 mM EndoV + 6 nM Ligase + 5 mM NAD60 min 2. 1· Ligase buffer = 40 mM Tricine, pH 8, 6.25 mM DTT
Condition V60 min 1. 1· EndoV buffer = 40 mM Tricine, pH 8, 5 mM DTT, with 500 nM EndoV + 6 nM Ligase + 5 mM NAD60 min 2. 1· Ligase buffer = 40 mM Tricine, pH 8, 6.25 mM DTT
Condition VI120 min 1· EndoV/ligase buffer = 80 mM Tricine, pH 8, 5 mM DTT, with 500 nM EndoV + 6 nM Ligase + 5 mM NAD
Condition VII60 min 1. 1· EndoV buffer = 40 mM Tricine, pH 8, 5 mM DTT, with 500 nM EndoV + 6 nM Ligase + 1 mM NAD60 min 2. 1· Ligase buffer = 40 mM Tricine, pH 8, 6.25 mM DTT
Condition VIII120 min 1· EndoV/ligase buffer = 80 mM Tricine, pH 8, 5 mM DTT, with 500 nM EndoV + 6 nM Ligase + 1 mM NAD
All 1· EndoV buffers and 1· EndoV/ligase buffers contain 5 mM MgCl2, 5% DMSO, 1.5 M Betain, and 2% glycerol. All 1· Ligase buffers contain 1.25 mM MgCl2and 20 mg/ml BSA.
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
sequence is present in the middle of a fragment (44). The firstprimer set was 50-end labeled, the second set had two 20-O-methylated C residues added to the 50 end label, and the thirdcarried an internal label, i.e. a C5-dye labeled cytosine insertedwithin the EndoV resistant sequence. As shown on the capil-lary array image (Supplementary Material, Figure S1), whenusing either the 50-end labeled primers (panel A) or the 20-O-methylated C primers (panel B), both heteroduplexed (Mut)and homoduplexed (WT) PCR fragments treated with EndoV(both top panels, lanes 2 and 5) showed a dramatic decrease influorescence intensity as compared to the untreated ones(lanes 1 and 4). Note that the labeled PCR amplicons arenot precisely aligned, due to differences in capillary arraylengths. Quenching the EndoV reaction with EDTA postincubation (lanes 3 and 6), resulted in a less dramatic decreasein fluorescence intensity in both cases, suggesting thatthermostable EndoV retained residual activity even in highformamide during the capillary run. In contrast, internallylabeled PCR fragments treated with EndoV showed only asmall decrease in fluorescence intensity (top panel C, lanes2 and 5) as compared to the untreated ones (lanes 1 and 4).Protection from cleavage was independent of post-incubationEDTA quenching (lanes 3 and 6). These results demonstratethat internally labeled primers eliminate false bands migratingat �100 bases, and should increase the intensity of authenticsignal.
Since synthesis of new internally labeled primers for eachgene is both costly and time consuming, we developed auniversal labeling strategy. This strategy would also beamenable to multiplexed amplification of dozens of genes,addressing the practical reality that tumor DNA is often limit-ing. This strategy employs two sets of primer pairs: unlabeledgene-specific primers that harbor universal tails on their 50
ends (see Table 1 and Figure 1), and fluorescently labeled orphosphorylated universal primers that can be used for anygene/exon amplification (Figure 1). During the first 20 PCRcycles, an annealing temperature of 65�C allows gene-specific primers at low concentration to anneal selectivelyto their target DNA template, while universal primers do nothybridize to the gene-specific primer universal tails. By shift-ing the annealing temperature to 55�C for an additional 30PCR cycles, the dye-labeled universal primers predominate inamplifying labeled product. The universal primers are ident-ical except for the last two bases on the 30 end, assuring thateach primer amplifies and labels only the intended strand.Since the wrong universal primer may hybridize but notextend, the efficiency of this type of universal amplificationis <2-fold per cycle, requiring the additional 30 cycles.Primer dimers and inappropriately short amplicons form hair-pins (or panhandles) and are thus unable to amplify. In thecourse of optimizing reaction conditions, we also improvedPCR yield by switching from a standard 10 mM Tris–HCl,pH 8.3 PCR buffer to the 20 mM Tricine, pH 8.7, 16 mM(NH4)2SO4 buffer previously developed in our lab (55).
When testing the internally labeled primers for universalprimer PCR amplification of p53 exon 6, we detected two highmolecular weight artifacts of �600 bases (SupplementaryMaterial, Figure S2A). Thus, we also explored an invertedlinkage labeled primer pair (i.e. fluorescent dye-30-CCGCC-50-50-universal sequence-30; see also Table 1), whichdid not yield the artifacts, but the fluorescent label was
somewhat susceptible to cleavage by EndoV (SupplementaryMaterial, Figure S2B). Despite these distinct features, signalintensity of the EndoV mutation cleavage products isstrong and clearly detectable in both cases even in the absenceof the ligase proofreading step (Supplementary Material,Figure S2A and S2B, lanes 1 and 3 are wild type, lanes 2and 4 are heteroduplexes of wild type to mutant). Thus,two different chemistries for attaching fluorescent groupsmay be used, each one overcoming minor shortcomings inthe other.
Reduction of noise resulting from artifact andbackground bands
Artifactual and background bands could be grouped into thefollowing categories: (i) inappropriate bands after PCR ampli-fication and prior to EndoV treatment (SupplementaryMaterial, Figure S3A, lanes 1 and 3), (ii) multiple EndoV gen-erated bands at near full length, and at slightly larger thanprimer lengths (Supplementary Material, Figure S3A, lanes 6,8 and 10), and (iii) specific EndoV generated bands that are notat the position of the mutation (Supplementary Material,Figure S3A, lanes 6, 8 and 10). Detection of the R273H(G!A) mutation in p53 exon 8 was used as a challengingtest system for the experiments below. In the original assay,this mutation gives weak signal, and its 350 base substrategenerates higher levels of background cleavage (44).
Category (i) artifactual bands are a consequence of theparticular dyes used in the PCR amplification, and could beeliminated by labeling each strand in separate amplificationreactions as described in Figure 1. This may be achieved byeither modifying the standard method (PCR amplificationusing two labeled primers in one reaction) to eliminate theunlabeled strands using lambda exonuclease (SupplementaryMaterial, Figure S3A, lanes 5, 7 and 9) or by the ‘split label,denaturation, renaturation’ method (Supplementary Material,Figure S3A, lanes 11, 13 and 15). Use of lambda exonucleaseprovides the added advantage of generating 100% hetero-duplexed substrate (i.e. containing only the G/T or A/Cmismatch), and consequently doubles the mutation signalintensity (Supplementary Material, Figure S3A, comparelanes 8 and 10 to lanes 14 and 16).
Category (ii) artifactual bands resulted from sequential non-specific EndoV cleavage on labeled and unlabeled strands nearthe dynamically fraying ends of the DNA. The problem isameliorated by the concurrent presence of ligase that resealsone strand before the opposite can be cleaved (see nextsection). In pursuing the source of this background we alsodetermined that shorter PCR amplicons (150–250 bp) givegreater background (not shown), while longer PCR amplicons(591 bp) give less background (Supplementary Material,Compare Figure S3A, lane 16 to Figure S3C, lane 10).Longer strands presumably have a reduction in the frequencyof unpaired conformations, leading to a slower rate ofnon-specific EndoV cleavage. To completely eliminateinterference of category (ii) artifactual bands with authenticsignal, all PCR primers were redesigned to reside at least 60bases upstream and downstream of the scanning region.
Category (iii) artifactual bands are sequence-specific bandsthat reflect the ability of that particular sequence to becometransiently unpaired under the particular buffer conditions
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
required to achieve cleavage of authentic mismatched bases.Consistent with this hypothesis is the observation that theintensity of such bands varied with changes in buffer pH(see below), as well as with choice of organic co-solvent(5% DMSO + 1.5 M betaine versus 5% tetramethylenesulfone alone, data not shown).
Several other potential causes for these bands were consid-ered and ultimately eliminated following experimentation.One possibility was that PCR errors introduced during ampli-fication were subsequently cleaved after heteroduplex forma-tion. This hypothesis was rejected when no change in thesebands was observed following amplification with two differentcommercially available proofreading polymerases (TaqPlusPrecision PCR System, Stratagene, La Jolla, CA or PlatinumTaq DNA Polymerase High Fidelity, Gibco BRL LifeTechnologies, Inc., Gaithersburg, MD; data not shown). Asecond possible explanation for category (iii) artifactualbands is the formation of multi-stranded structures with unre-solved Holliday junctions or Flap structures. These structuresare known substrates for EndoV (54). This possibility was alsoeliminated when there was no reduction in bands following theaddition of mesophilic or thermophilic DNA binding proteins(e.g., RecA, T4 gene 32 protein, Aquifex ssb, RuvA andRuvB) to the reaction. Even though these proteins minimizeformation of potential tertiary or secondary structures bylambda exonuclease-generated single strands during theheteroduplex formation step, the artifactual bands wereunaffected. A third hypothesis to account for the category(iii) bands was that single-stranded fragments form transientsecondary Y structures that become substrates for EndoV (54).Although EndoV does cleave single-stranded DNA atnon-random positions, the bands differ from those generatedby cleavage of heteroduplexed DNA (Supplementary Mate-rial, Figure S3B, lanes 2, 4, 6, and 8). Category (iii) artifactualbands are also rectified by combining EndoV cleavage withresealing by ligase in a single step assay (see below).
Real-time Ligase proofreading in a homogeneoussingle-step EndoV/ligase assay
Optimization of the original EndoV/ligase mutation scanningassay entailed finding artificial buffer conditions for EndoVthat minimized cleavage of matched sites while allowing themismatched site to be cleaved (44,50). This was followedby shifting the reaction to conditions that maximized ligaseresealing of nicks at matched sites, while allowing mismatchedsites to remain unligated (44). The task is exacerbated byEndoV cleavage one base 30 to the mismatch. Such substrateswith penultimate mismatches are more difficult to discriminateby ligases than substrates with mismatches at the 30 end (53).If ligase could be made to work during the EndoV reaction, itcould continuously proofread and reseal nicks at perfectmatches. Signal would be optimized when the desiredEndoV cleavage at mismatches minus their unwanted re-ligation exceeds the unwanted EndoV cleavage at matchesminus their desired ligation. We have previously demonstratedthat EndoV shows optimal mismatch cleavage activity in highpH and no salt, minimal ionic strength buffer conditions (50),whereas the ligase enzyme exhibits higher fidelity at lower pHand 50–100 mM KCl (53). The inherent challenge for a one-step reaction was to find conditions that allow EndoV to cleave
at the mismatch (in 5% DMSO, 1.5 M betaine), while allowingligase to exhibit suitable fidelity and reactivity under the sameconditions.
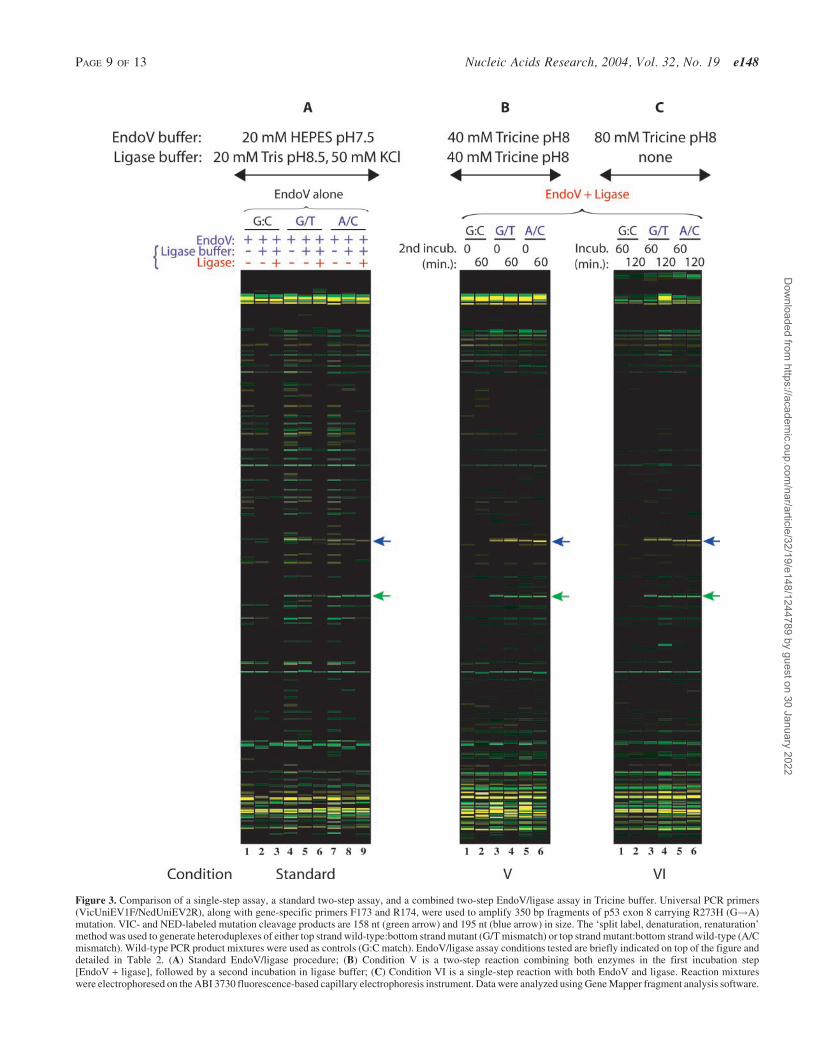
We tested a variety of buffer and incubation conditions (seeTable 2) in over 500 capillary electrophoresis runs for theability to detect the challenging p53 R273H G!A mutation.When EndoV and ligase are used together, a dramatic reduc-tion in bands from background cleavage results (see Figure 2,lanes 16–24, and Figure 3B and 3C). Compared to thesequential use of EndoV and ligase in a two-step reaction(see Figure 2, lanes 3, 5, 8, 10, 13 and 15 and Figure 3A,lanes 3, 6 and 9), fewer background bands are created. Anadditional incubation with higher concentrations of Tricinebuffer to enhance ligation fidelity (see Figure 2, lanes 20,21, 23 and 24 and Figure 3B, lanes 2, 4 and 6) does not providean enhancement in mutation signal intensity nor any furtherreduction of background cleavage bands compared to a singleincubation with EndoV and ligase (see Figure 2, lanes 16, 19and 22 and Figure 3C, lanes 1–6). While increasing the pHfrom 7.5 to 8.0 initially creates more EndoV cleavage events(compare Figure 3A, lanes 4 and 7 to Figure 2, lanes 6 and 11),the higher ionic strength in the buffer also allows ligase tocontinuously proofread and reseal these undesired nicks(Figure 2, lanes 17, 18, 20, 21, 23 and 24). Increasing the pHfrom our original conditions to a pH of 8 provides an effectivecompromise between optimal conditions for the two enzymes,at the cost of some increase in background EndoV cleavage(readily religated by ligase). There is also some loss of ligationfidelity at the compromise pH, resulting in the disappearanceof some correctly-cleaved bands after ligase treatment (Sup-plementary Material, Figure S4C, compare mutation signalintensity in Conditions I and III to Conditions II and IV).Nevertheless, reduction in category (iii) background bandsby simultaneous enzyme incubation is readily apparent bycomparing lanes 1–15 with lanes 16–24 in Figure 2.
The intensity of each authentic mutation signal was quanti-fied, and the brightest background band for each fluorescentdye was chosen for estimating a conservative, worst casesignal-to-noise ratio for various buffer and incubation condi-tions (SupplementaryMaterial,FiguresS4andS5). Ineachcase,incubation of proofreading ligase during the EndoV cleavagestep provided a 5- to 13-fold increase in signal intensity(Supplementary Material, Figure S4, panels C and D, and S5,panels C and D), with significant improvement of signal-to-noise compared to the original conditions. Dilution experimentswith known mutations were performed to test for the abilityto detect tumor mutations in the presence of an excess ofwild-type DNA (see Figure 4). In the case of K-ras exon 1,the G12V G!T mutation could be detected in a 100-fold excessof wild-type DNA on three out of four strands of the two hetero-duplexes and in a 20-fold excess for the fourth strand. Even forthe more difficult p53 R273H G!A mutation, the combinedsingle-step assay can detect the mutation in a 50-fold excess ofwild-type DNA in 3 out of 4 strands. These results represent a 5-to 10-fold improvement over the original two-step assay (44).
DISCUSSION
Mutation and polymorphism detection is of increasing impor-tance in the field of molecular genetics, resulting in a profusion
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
Figure 2. Improvement of signal-to-noise ratio in the EndoV/ligase assay when combining both enzymes in a single incubation step. Universal PCR primers(VicUniEV1F/NedUniEV2R), along with gene-specific primers F173 and R174, were used to amplify 350 bp fragments of p53 exon 8 carrying R273H (G!A)mutation. VIC- and NED-labeled mutation cleavage products are 158 nt (green arrow) and 195 nt (blue arrow) in size. The ‘split label, denaturation, renaturation’method was used to generate heteroduplexes of either top strand wild-type:bottom strand mutant (G/T mismatch) or top strand mutant:bottom strand wild-type (A/Cmismatch). Wild-type PCR product mixtures were used as controls (G:C match). Tested EndoV/ligase assay conditions are briefly indicated on top and bottom of thefigure and detailed in Table 2. Condition III is a standard two-step reaction, while condition IV combines both enzymes in the first incubation step [EndoV + ligase],followed by a second incubation in ligase buffer. Reaction mixtures were electrophoresed on the ABI 3730 fluorescence-based capillary electrophoresis instrument.Data were analyzed using Gene Mapper fragment analysis software.
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
Figure 3. Comparison of a single-step assay, a standard two-step assay, and a combined two-step EndoV/ligase assay in Tricine buffer. Universal PCR primers(VicUniEV1F/NedUniEV2R), along with gene-specific primers F173 and R174, were used to amplify 350 bp fragments of p53 exon 8 carrying R273H (G!A)mutation. VIC- and NED-labeled mutation cleavage products are 158 nt (green arrow) and 195 nt (blue arrow) in size. The ‘split label, denaturation, renaturation’method was used to generate heteroduplexes of either top strand wild-type:bottom strand mutant (G/T mismatch) or top strand mutant:bottom strand wild-type (A/Cmismatch). Wild-type PCR product mixtures were used as controls (G:C match). EndoV/ligase assay conditions tested are briefly indicated on top of the figure anddetailed in Table 2. (A) Standard EndoV/ligase procedure; (B) Condition V is a two-step reaction combining both enzymes in the first incubation step[EndoV + ligase], followed by a second incubation in ligase buffer; (C) Condition VI is a single-step reaction with both EndoV and ligase. Reaction mixtureswere electrophoresed on the ABI 3730 fluorescence-based capillary electrophoresis instrument. Data were analyzed using Gene Mapper fragment analysis software.
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
of enzymatic, chemical and biophysical based methods formutation detection (13,23). Few of them can detect previouslyunknown mutations and at the same time position themaccurately. Moreover, current methods often lack high speci-ficity or sensitivity, or are too labor-intensive to be amenableto high-throughput mutation screening (23). This work sys-tematically evaluated and developed the following improve-ments in all aspects of our original EndoV/ligase mutation
scanning assay (44): (i) shifting the assay to automatedcapillary electrophoresis readout, (ii) development of fluores-cent labeling chemistries (50 internal and inverted linkagelabeled) that resist removal by EndoV from the PCR amplicon,(iii) development of universal PCR primers for more costeffective and robust labeling, and (iv) development ofreal-time ligase proofreading conditions. Often, these improve-ments worked synergistically or revealed unanticipated
Figure 4. Sensitivity of the EndoV/ligase single-step assay. EndoV/ligase assay conditions are detailed in Materials and Methods. (A) Relative fluorescenceintensities of cleavage products for K-ras exon 1 G12V mutation with their respective mutant-to-wild type dilution ratios. Universal PCR primers (VicUniEV1F/NedUniEV2R), along with gene-specific primers F161 and R162, were used to amplify 300 bp PCR fragments of K-ras exon 1 carrying mutation G12V (G!T). The‘split label, denaturation, renaturation’ method was used to generate heteroduplexes with either a G/A mismatch or a T/C mismatch for various mutant:wild-typeratios. Wild-type PCR product mixtures (wt) were used as controls. Relative fluorescence intensity is defined as the area under a signal’s peak as determined by GeneMapper fragment analysis software. (B) Relative fluorescence intensities of cleavage products of p53 exon 8 R273H mutation with their respective mutant-to-wildtype dilution ratios. Universal PCR primers (VicUniEV1F/NedUniEV2R), along with gene-specific primers F173 and R174, were used to amplify p53 exon 8fragments carrying R273H mutation (G!A). The ‘split label, denaturation, renaturation’ method was used to generate heteroduplexes with either a G/T mismatchor a A/C mismatch for various mutant:wild-type ratios. Wild-type PCR mixtures (wt) were used as controls.
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
benefits. For example, suppressing fluorescent dye removal byEndoV was a necessary precondition in achieving theenhanced signal that resulted from continuous EndoV cleav-age and ligase proofreading. The universal primer design wasconceived to reduce primer synthesis cost and allow for simul-taneous amplification of multiple target regions from valuableand limited tumor DNA samples. This design change alsoenabled significant reduction in PCR generated backgroundbands, both through panhandle formation of primer dimers andshorter fragments, and through the two split-label protocols(Steps 2A and 2B in Figure 1). The latter allowed us to seam-lessly double signal intensity by using lambda exonucleaseto form 100% mismatched heteroduplexes. Complementarysingle-strand specific nuclease-based mismatch detectionassays (CEL I and S1) demonstrate nibbling of end-labeledduplexes and nuclease-resistant background (56), and mayalso benefit from the approaches outlined in this report.
Dilution experiments with K-ras and p53 gene mutationsshow that our EndoV with real-time ligase proofreading scan-ning assay can detect mutations at a sensitivity of 1:100 and1:50 (mutant:wild-type DNA ratio), respectively, in three outof four strands of the two heteroduplexes. While anotherapproach based on ligation-mediated PCR and the use ofglycosylases TDG and MutY has similar sensitivity (57), itrequires multiple steps and cannot detect A!T or G!C trans-version mutations, or single base deletion/insertions (asEndoV/ligase can). When the position of the potential muta-tion is known, even greater sensitivities may be achieved byPCR/LDR/Universal array (15–17), or by suppression of wild-type DNA amplification using PNA (32,58), although the latteris limited to a single hot-spot region per assay. However, ourconcern here is with detecting unknown, potentially rare muta-tions in clinical samples. The current single-step real-timeEndoV/ligase assay, represents a 5- to 10-fold improvementover our original two-step conditions (44). EndoV detects 98%of mutations including all insertions and deletions even whenthe position is unknown. This new higher level of sensitivityallows for additional pooling of the same amplicon from multi-ple tumor samples within the same EndoV/ligase reaction,beyond what we have previously demonstrated (17). This iscritical, since tumor samples that have not been microdis-sected typically contain 10–50% of stromal infiltration (i.e.normal cells), and the ability to detect mutations in pools of5 or even 10 samples represents a great advantage over auto-mated fluorescent sequencing. Real-time EndoV/ligase proof-reading represents a compromise of sub-optimal bufferconditions for both enzymes, and sensitivities may yet beimproved an additional order of magnitude by use of opti-mized variant or engineered EndoV and/or DNA ligase.
Developing cancer screening tools for clinical samples(biopsies, stool or bodily fluids) present special problems.They are often limited in either the total amount of recoveredDNA, or the percentage of authentic neoplastic DNA(33,59–64). Future diagnostic tests will require screeningnumerous candidate genes. For example, the current list ofbiologically important genes for colon cancer include APC,b-catenin (3,5), K-ras (2), B-raf (7,65), p53 (66), PI3KCA (8),hCDC4 (67), Tyr kinases (9), phosphatases (10), BAX,TGFBRII and TCF4 (1,6,5). Clinically useful diagnosticswill likely incorporate detection of the appearance of somaticmutations in these genes (60). The universal primer labeling
strategy in this report is compatible with multiplexed pre-amplification of candidate exons in a single reaction, followedby uniplexed amplification/labeling of individual fragmentsfor mutation scanning. Since DNA is usually limiting, clinicalsamples are generally not amenable to very narrow assays thatselectively amplify or detect mutations in a few codons at atime. Likewise, oligonucleotide array-based assays sufferfrom low inherent sensitivity due to only minor differencesin hybridization kinetics between wild-type and mutant probes(11). They have thus failed to provide comprehensive cover-age of all mutations in a gene, e.g. scoring 81% (68), 84% (69)and 92% (70) for p53. Furthermore, the number of potentialinactivating frameshift mutations is in vast excess of thenumber of inactivating missense mutations; hybridizationchips do not contain addresses for such frameshift mutations,and hence completely fail to detect insertions and deletions. Incontrast, in vitro transcription–translation, followed by eithermulticolor size-based or MALDI-TOF detection, allows iden-tification of truncating mutations (29–31). The sensitivity of invitro transcription–translation approaches has been extendedto 2.5% for identifying APC gene mutations in fecal DNAsamples from colorectal cancer patients (31). This sensitivityis obtained by diluting sample to �10 molecules of DNAtemplate in each of dozens to hundreds of individual PCRamplifications. We could combine this ‘digital’ approach inour assay, or most likely just use our EndoV/real-time ligaseapproach to directly detect mutations that are present in a verysmall portion of the total cells, especially for pre-symptomaticdiagnosis.
The importance of a sensitive method capable of detectingmultiple unknown mutations in a cancer gene is emphasizedby a recent study showing that polyclonal resistance to Glee-vec (a small molecule inhibitor of BCR-ABL kinase) inchronic myeloid leukemia patients is conferred by BCR-ABL point mutations, where two or more distinct mutationsare sometimes harbored in a single patient (71,72). In this case,the authors sub-cloned the PCR products and sequenced 10independent clones per patient, a time-consuming approachthat was required when sequencing missed critical mutations(71). Once again, a robust and sensitive assay such as theEndoV/real-time ligase reaction could be used in this contextto identify mutations in a minority of cells. As newer directedtherapies that overcome most Gleevec resistance mutationsbecome available (73), accurate profiling of the mutationalstatus becomes critical for patient care. This emerging needfor unknown mutation detection is recapitulated in the treat-ment of non-small cell lung cancers (NSCLC), where somaticmutations in the EGF receptor were found only in tumorsamples from patients who responded to Iressa therapy, butnot in Iressa insensitive tumors (74,75). Here too, detection ofmutations directly from sputum or non-microdissected lungbiopsies, either by our EndoV/real-time ligase mutation scan-ning assay, or harmonized with PCR/LDR/Universal arrayidentification of known mutations (76), can help predict ther-apeutic response.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
ic.oup.com/nar/article/32/19/e148/1244789 by guest on 30 January 2022
ACKNOWLEDGEMENTS
We thank Donald Bergstrom, Daniel Notterman, Weiguo Cao,and Yu-Wei Cheng for helpful discussion, and Reyna Favisand Daniel Turner for critical reading of the manuscript. Wethank Michael O’Donnell for gifts of Aquifex ssb, RuvA andRuvB. We thank Seiki Kuramitsu for gift of thermostableRecA, and Hideo Shinagawa for gifts of thermostable RuvAand RuvB. Work in the Barany laboratory is sponsored in partby a sponsored research grant from Applied Biosystems Inc.,for which Francis Barany also serves as a consultant. This workwas supported by grants from the National Cancer Institute(P01-CA65930 and RO1-CA81467).
REFERENCES
1. Futreal,P.A., Coin,L., Marshall,M., Down,T., Hubbard,T., Wooster,R.,Rahman,N. and Stratton,M.R. (2004) A census of human cancergenes. Nat. Rev. Cancer, 4, 177–183.
2. Adjei,A.A. (2001) Blocking oncogenic Ras signaling for cancer therapy.J. Natl Cancer Inst., 93, 1062–1074.
3. Grady,W.M. and Markowitz,S.D. (2002) Genetic and epigeneticalterations in colon cancer. Annu. Rev. Genomics Hum. Genet., 3,101–128.
4. Lindblom,A. (2001) Different mechanisms in the tumorigenesis ofproximal and distal colon cancers. Curr. Opin. Oncol., 13, 63–69.
5. Narayan,S. and Roy,D. (2003) Role of APC and DNA mismatch repairgenes in the development of colorectal cancers. Mol. Cancer, 2, 41.
6. Duval,A. and Hamelin,R. (2002) Mutations at coding repeat sequences inmismatch repair-deficient human cancers: toward a new concept oftarget genes for instability. Cancer Res., 62, 2447–2454.
8. Samuels,Y., Wang,Z., Bardelli,A., Silliman,N., Ptak,J., Szabo,S.,Yan,H., Gazdar,A., Powell,S.M., Riggins,G.J. et al. (2004) Highfrequency of mutations of the PIK3CA gene in human cancers. Science,304, 554.
9. Bardelli,A., Parsons,D.W., Silliman,N., Ptak,J., Szabo,S., Saha,S.,Markowitz,S., Willson,J.K., Parmigiani,G., Kinzler,K.W. et al. (2003)Mutational analysis of the tyrosine kinome in colorectal cancers.Science, 300, 949.
10. Wang,Z., Shen,D., Parsons,D.W., Bardelli,A., Sager,J., Szabo,S., Ptak,J.,Silliman,N., Peters,B.A., van der Heijden,M.S. et al. (2004) Mutationalanalysis of the tyrosine phosphatome in colorectal cancers. Science,304, 1164–1166.
11. Hacia,J.G., Brody,L.C., Chee,M.S., Fodor,S.P. and Collins,F.S. (1996)Detection of heterozygous mutations in BRCA1 using high densityoligonucleotide arrays and two-colour fluorescence analysis. NatureGenet., 14, 441–447.
15. Gerry,N.P., Witowski,N.E., Day,J., Hammer,R.P., Barany,G. andBarany,F. (1999) Universal DNA microarray method for multiplexdetection of low abundance point mutations. J. Mol. Biol., 292, 251–262.
16. Favis,R., Day,J.P., Gerry,N.P., Phelan,C., Narod,S. and Barany,F. (2000)Universal DNA array detection of small insertions and deletions inBRCA1 and BRCA2. Nature Biotechnol., 18, 561–564.
25. Cooper,C.S. (2001) Applications of microarray technology in breastcancer research. Breast Cancer Res., 3, 158–175.
26. Gross,E., Arnold,N., Goette,J., Schwarz-Boeger,U. and Kiechle,M.(1999) A comparison of BRCA1 mutation analysis by direct sequencing,SSCP and DHPLC. Hum. Genet., 105, 72–78.
27. Footz,T., Somerville,M.J., Tomaszewski,R., Sprysak,K.A. andBackhouse,C.J. (2003) Heteroduplex-based genotyping with microchipelectrophoresis and dHPLC. Genet. Test, 7, 283–293.
28. Andersen,P.S., Jespersgaard,C., Vuust,J., Christiansen,M. andLarsen,L.A. (2003) Capillary electrophoresis-based single strand DNAconformation analysis in high-throughput mutation screening.Hum. Mutat., 21, 455–465.
29. Garvin,A.M., Parker,K.C. and Haff,L. (2000) MALDI-TOF basedmutation detection using tagged in vitro synthesized peptides.Nat. Biotechnol., 18, 95–97.
30. Gite,S., Lim,M., Carlson,R., Olejnik,J., Zehnbauer,B. and Rothschild,K.(2003) A high-throughput nonisotopic protein truncation test.Nat. Biotechnol., 21, 194–197.
31. Traverso,G., Diehl,F., Hurst,R., Shuber,A., Whitney,D., Johnson,C.,Levin,B., Kinzler,K.W. and Vogelstein,B. (2003) Multicolor in vitrotranslation. Nat. Biotechnol., 21, 1093–1097.
32. Sun,X., Hung,K., Wu,L., Sidransky,D. and Guo,B. (2002) Detection oftumor mutations in the presence of excess amounts of normal DNA.Nat. Biotechnol., 20, 186–189.
33. Traverso,G., Shuber,A., Levin,B., Johnson,C., Olsson,L.,Schoetz,D.J.,Jr, Hamilton,S.R., Boynton,K., Kinzler,K.W. andVogelstein,B. (2002) Detection of APC mutations in fecal DNAfrom patients with colorectal tumors. N. Engl. J. Med., 346,311–320.
34. Cotton,R.G., Rodrigues,N.R. and Campbell,R.D. (1988) Reactivity ofcytosine and thymine in single-base-pair mismatches withhydroxylamine and osmium tetroxide and its application to the study ofmutations. Proc. Natl Acad. Sci. USA, 85, 4397–4401.
35. Mashal,R.D., Koontz,J. and Sklar,J. (1995) Detection of mutations bycleavage of DNA heteroduplexes with bacteriophage resolvases. NatureGenet., 9, 177–183.
36. Youil,R., Kemper,B.W. and Cotton,R.G. (1995) Screening for mutationsby enzyme mismatch cleavage with T4 endonuclease VII. Proc. NatlAcad. Sci. USA, 92, 87–91.
37. Marshall,D.J., Heisler,L.M., Lyamichev,V., Murvine,C., Olive,D.M.,Ehrlich,G.D., Neri,B.P. and de Arruda,M. (1997) Determination ofhepatitis C virus genotypes in the United States by cleavasefragment length polymorphism analysis. J. Clin. Microbiol., 35,3156–3162.
38. Goldrick,M.M., Kimball,G.R., Liu,Q., Martin,L.A., Sommer,S.S. andTseng,J.Y. (1996) NIRCA: a rapid robust method for screening forunknown point mutations. Biotechniques, 21, 106–112.
39. Goldrick,M.M. (2001) RNase cleavage-based methods for mutation/SNPdetection, past and present. Hum. Mutat., 18, 190–204.
40. Babon,J.J., McKenzie,M. and Cotton,R.G. (1999) Mutation detectionusing fluorescent enzyme mismatch cleavage with T4 endonuclease VII.Electrophoresis, 20, 1162–1170.
41. Smith,J. and Modrich,P. (1996) Mutation detection with MutH, MutL,and MutS mismatch repair proteins. Proc. Natl Acad. Sci. USA, 93,4374–4379.
42. Beaulieu,M., Larson,G.P., Geller,L., Flanagan,S.D. and Krontiris,T.G.(2001) PCR candidate region mismatch scanning: adaptation to
43. Oleykowski,C.A., Bronson Mullins,C.R., Godwin,A.K. and Yeung,A.T.(1998) Mutation detection using a novel plant endonuclease. NucleicAcids Res., 26, 4597–4602.
44. Huang,J., Kirk,B., Favis,R., Soussi,T., Paty,P., Cao,W. and Barany,F.(2002) An endonuclease/ligase based mutation scanning methodespecially suited for analysis of neoplastic tissue. Oncogene, 21,1909–1921.
45. Yao,M., Hatahet,Z., Melamede,R.J. and Kow,Y.W. (1994) Purificationand characterization of a novel deoxyinosine-specific enzyme,deoxyinosine 30 endonuclease, from Escherichia coli. J. Biol. Chem.,269, 16260–16268.
46. Yao,M., Hatahet,Z., Melamede,R.J. and Kow,Y.W. (1994) Deoxyinosine30 endonuclease, a novel deoxyinosine-specific endonuclease fromEscherichia coli. Ann. N. Y. Acad. Sci., 726, 315–316.
47. Yao,M. and Kow,Y.W. (1997) Further characterization of Escherichiacoli endonuclease V. Mechanism of recognition for deoxyinosine,deoxyuridine, and base mismatches in DNA. J. Biol. Chem., 272,30774–30779.
48. He,B., Qing,H. and Kow,Y.W. (2000) Deoxyxanthosine in DNA isrepaired by Escherichia coli endonuclease V. Mutat. Res., 459,109–114.
49. Moe,A., Ringvoll,J., Nordstrand,L.M., Eide,L., Bjoras,M., Seeberg,E.,Rognes,T. and Klungland,A. (2003) Incision at hypoxanthine residues inDNA by a mammalian homologue of the Escherichia coli antimutatorenzyme endonuclease. V Nucleic Acids Res., 31, 3893–3900.
50. Huang,J., Lu,J., Barany,F. and Cao,W. (2001) Multiple cleavageactivities of endonuclease V from Thermotoga maritima: recognition andstrand nicking mechanism. Biochemistry, 40, 8738–8748.
51. Hitchcock,T.M., Gao,H. and Cao,W. (2004) Cleavage ofdeoxyoxanosine-containing oligodeoxyribonucleotides by bacterialendonuclease V. Nucleic Acids Res., 32, 4071–4080.
52. Yao,M. and Kow,Y.W. (1994) Strand-specific cleavage of mismatch-containing DNA by deoxyinosine 30-endonuclease from Escherichia coli.J. Biol. Chem., 269, 31390–31396.
53. Tong,J., Cao,W. and Barany,F. (1999) Biochemical properties of a highfidelity DNA ligase from Thermus species AK16D. Nucleic Acids Res.,27, 788–794.
54. Yao,M. and Kow,Y.W. (1996) Cleavage of insertion/deletionmismatches, flap and pseudo-Y DNA structures by deoxyinosine 30-endonuclease from Escherichia coli. J. Biol. Chem., 271, 30672–30676.
55. Day,J.P., Hammer,R.P., Bergstrom,D. and Barany,F. (1999) Nucleotideanalogs and new buffers improve a generalized method to enrich forlow abundance mutations. Nucleic Acids Res., 27, 1819–1827.
56. Till,B.J., Burtner,C., Comai,L. and Henikoff,S. (2004) Mismatchcleavage by single-strand specific nucleases. Nucleic Acids Res., 32,2632–2641.
57. Zhang,Y., Kaur,M., Price,B.D., Tetradis,S. and Makrigiorgos,G.M.(2002) An amplification and ligation-based method to scan for unknownmutations in DNA. Hum. Mutat., 20, 139–147.
58. Prix,L., Uciechowski,P., Bockmann,B., Giesing,M. and Schuetz,A.J.(2002) Diagnostic biochip array for fast and sensitive detection of K-rasmutations in stool. Clin. Chem., 48, 428–435.
59. Traverso,G., Shuber,A., Olsson,L., Levin,B., Johnson,C., Hamilton,S.R.,Boynton,K., Kinzler,K.W. and Vogelstein,B. (2002) Detection ofproximal colorectal cancers through analysis of faecal DNA. Lancet, 359,403–404.
60. Dong,S.M., Traverso,G., Johnson,C., Geng,L., Favis,R., Boynton,K.,Hibi,K., Goodman,S.N., D0Allessio,M., Paty,P. et al. (2001) Detectingcolorectal cancer in stool with the use of multiple genetic targets.J. Natl Cancer Inst., 93, 858–865.
61. Su,Y.H., Wang,M., Brenner,D.E., Ng,A., Melkonyan,H., Umansky,S.,Syngal,S. and Block,T.M. (2004) Human urine contains small,150 to 250 nucleotide-sized, soluble DNA derived from the circulationand may be useful in the detection of colorectal cancer. J. Mol. Diagn.,6, 101–107.
62. Hoque,M.O., Lee,J., Begum,S., Yamashita,K., Engles,J.M.,Schoenberg,M., Westra,W.H. and Sidransky,D. (2003) High-throughputmolecular analysis of urine sediment for the detection of bladder cancerby high-density single-nucleotide polymorphism array. Cancer Res., 63,5723–5726.
63. Taback,B., Chan,A.D., Kuo,C.T., Bostick,P.J., Wang,H.J., Giuliano,A.E.and Hoon,D.S. (2001) Detection of occult metastatic breast cancer cellsin blood by a multimolecular marker assay: correlation with clinicalstage of disease. Cancer Res., 61, 8845–8850.
64. Mike Makrigiorgos,G. (2004) PCR-based detection of minority pointmutations. Hum. Mutat., 23, 406–412.
65. Davies,H., Bignell,G.R., Cox,C., Stephens,P., Edkins,S., Clegg,S.,Teague,J., Woffendin,H., Garnett,M.J., Bottomley,W. et al. (2002)Mutations of the BRAF gene in human cancer. Nature, 417, 949–954.
66. Beroud,C. and Soussi,T. (2003) The UMD-p53 database: new mutationsand analysis tools. Hum. Mutat., 21, 176–181.
67. Rajagopalan,H., Jallepalli,P.V., Rago,C., Velculescu,V.E.,Kinzler,K.W., Vogelstein,B. and Lengauer,C. (2004) Inactivation ofhCDC4 can cause chromosomal instability. Nature, 428, 77–81.
68. Ahrendt,S.A., Halachmi,S., Chow,J.T., Wu,L., Halachmi,N., Yang,S.C.,Wehage,S., Jen,J. and Sidransky,D. (1999) Rapid p53 sequence analysisin primary lung cancer using an oligonucleotide probe array. Proc. NatlAcad. Sci. USA, 96, 7382–7387.
69. Wikman,F.P., Lu,M.L., Thykjaer,T., Olesen,S.H., Andersen,L.D.,Cordon-Cardo,C. and Orntoft,T.F. (2000) Evaluation of the performanceof a p53 sequencing microarray chip using 140 previously sequencedbladder tumor samples. Clin. Chem., 46, 1555–1561.
70. Wen,W.H., Bernstein,L., Lescallett,J., Beazer-Barclay,Y.,Sullivan-Halley,J., White,M. and Press,M.F. (2000) Comparison of TP53mutations identified by oligonucleotide microarray and conventionalDNA sequence analysis. Cancer Res., 60, 2716–2722.
71. Shah,N.P., Nicoll,J.M., Nagar,B., Gorre,M.E., Paquette,R.L., Kuriyan,J.and Sawyers,C.L. (2002) Multiple BCR-ABL kinase domain mutationsconfer polyclonal resistance to the tyrosine kinase inhibitor imatinib(STI571) in chronic phase and blast crisis chronic myeloid leukemia.Cancer Cell, 2, 117–125.
72. Shah,N.P. and Sawyers,C.L. (2003) Mechanisms of resistance to STI571in Philadelphia chromosome-associated leukemias. Oncogene, 22,7389–7395.
73. Shah,N.P., Tran,C., Lee,F.Y., Chen,P., Norris,D. and Sawyers,C.L.(2004) Overriding imatinib resistance with a novel ABL kinase inhibitor.Science, 305, 399–401.
74. Lynch,T.J., Bell,D.W., Sordella,R., Gurubhagavatula,S., Okimoto,R.A.,Brannigan,B.W., Harris,P.L., Haserlat,S.M., Supko,J.G., Haluska,F.G.et al. (2004) Activating mutations in the epidermal growth factor receptorunderlying responsiveness of non-small-cell lung cancer to gefitinib.N. Engl. J. Med., 350, 2129–2139.
75. Paez,J.G., Janne,P.A., Lee,J.C., Tracy,S., Greulich,H., Gabriel,S.,Herman,P., Kaye,F.J., Lindeman,N., Boggon,T.J. et al. (2004) EGFRmutations in lung cancer: correlation with clinical response to gefitinibtherapy. Science, 304, 1497–1500.
76. Fouquet,C., Antoine,M., Tisserand,P., Favis,R., Wislez,M., Commo,F.,Rabbe,N., Carette,M.F., Milleron,B., Barany,F. et al. (2004)Rapid and sensitive p53 alteration analysis in biopsies from lungcancer patients using a functional assay and a universaloligonucleotide array: a prospective study. Clin. Cancer. Res., 10,3479–3489.