Naturally occurring aminoacyl-tRNA synthetases editing-domain mutations that cause mistranslation in Mycoplasma parasites Li Li a , Michal T. Boniecki b , Jacob D. Jaffe c , Brian S. Imai d , Peter M. Yau d , Zaida A. Luthey-Schulten a,e , and Susan A. Martinis a,b,e,1 a Center for Biophysics and Computational Biology, b Department of Biochemistry, e Department of Chemistry, d Roy J. Carver Biotechnology Center, University of Illinois, Urbana, IL 61801; and c Proteomics Platform, The Broad Institute, 7 Cambridge Center, Cambridge, MA 02142 Edited by Paul Schimmel, The Skaggs Institute for Chemical Biology, La Jolla, CA, and approved April 20, 2011 (received for review November 8, 2010) Mycoplasma parasites escape host immune responses via me- chanisms that depend on remarkable phenotypic plasticity. Identi- fication of these mechanisms is of great current interest. The aminoacyl-tRNA synthetases (AARSs) attach amino acids to their cognate tRNAs, but occasionally make errors that substitute closely similar amino acids. AARS editing pathways clear errors to avoid mistranslation during protein synthesis. We show here that AARSs in Mycoplasma parasites have point mutations and deletions in their respective editing domains. The deleterious effect on editing was confirmed with a specific example studied in vitro. In vivo mistranslation was determined by mass spectrometric analysis of proteins produced in the parasite. These mistranslations are uni- form cases where the predicted closely similar amino acid replaced the correct one. Thus, natural AARS editing-domain mutations in Mycoplasma parasites cause mistranslation. We raise the possibi- lity that these mutations evolved as a mechanism for antigen diversity to escape host defense systems. amino acid editing ∣ fidelity ∣ quality control ∣ statistical proteins ∣ host-pathogen interactions M ycoplasma are characterized by their lack of a cell wall and dependence on a vertebrate host (1). Their relationship with the host can be parasitic or they can coexist as an obligate commensal. The persistent survival of Mycoplasma within their host has been attributed to a phenotypic plasticity that allows these pathogens to facilely alter their antigenic properties (2). Paradoxi- cally, this phenotypic plasticity is generated in spite of the Myco- plasma’s extremely small genomes, which have lost many of the components that typically comprise signaling pathways to adapt to changing environments (2). Remarkably, these wall-less bacteria have a highly dynamic surface architecture comprised of mem- brane proteins that confer antigenic and functional versatility (2). As with other pathogenic and nonpathogenic organisms, Mycoplasma contain a complete set of AARSs (aminoacyl-tRNA synthetases), which are essential to translate the genetic code into functional proteins (3). Each AARS has evolved for specificity to a single standard amino acid to maintain the fidelity of the genetic code. The AARS enzyme family activates and transfers amino acids to their cognate tRNA isoacceptor. Once the tRNA is “charged” with an amino acid, it is shuttled to the ribosome for incorporation into nascent polypeptides. About half of the AARSs are prone to mistakes by activating structurally similar amino acids and mischarging them to tRNA. To minimize the potential for creating statistical proteins that contain mistranslated amino acids, these AARSs have developed a second “sieve” (4); they have adapted to include hydrolytic editing domains that are distinct from their canonical aminoacy- lation core domains. In some cases, aminoacylation accuracy is also enhanced by independent editing domains that function as tRNA-specific deacylases (5, 6). We have identified multiple AARSs with editing domains in Mycoplasma and closely related species that appeared to be degenerate based on substitutions at key sites in the hydrolytic active site. This was surprising because functional defects in AARS editing that decrease the fidelity of tRNA aminoacylation have been clearly shown to result in amino acid toxicities, cell death, as well as neurological disease in mammals (7–9). As such, in order to achieve the threshold levels of translational fidelity that are required for cell viability, these amino acid editing func- tions have been broadly conserved across all three domains of life. In contrast, we determined that Mycoplasma mobile exhibits AARS-dependent translational infidelities. Editing-defective AARSs mischarge tRNA, which subsequently results in mistran- slation in vivo. It is possible that this AARS-dependent mechan- ism could provide a unique pathway to introduce heterogeneity into the cell’s proteome that could confer phenotypic plasticity in Mycoplasma pathogens. Results Mycoplasma Have Evolved AARSs with Inactivated Editing Domains. Using bioinformatic approaches to broadly scrutinize genomes across the three domains of life, we identified AARSs with unu- sual amino acid editing domains. In Mycoplasma and closely related species, we discovered AARSs with deletions and substi- tutions that we hypothesized would abolish their editing activities (Fig. 1). These Mycoplasma AARSs have substitutions at key residues in the hydrolytic active sites of the editing domains. In one extreme case, the editing domain was completely deleted from the M. mobile leucyl-tRNA synthetase (LeuRS). In six different Mycoplasma and also two closely related species, threonyl-tRNA synthetase (ThrRS) contained editing domains in which the editing site had amino acid substitutions at critical residues (Fig. 1A). In each of these cases, critical histidine, aspartic acid, and cysteine residues (Fig. S1) within two distal peptides that folded to form the editing active site were replaced (10–12). The editing domains of the editing-defective ThrRSs have a lower average sequence identity (24.4%) compared to the canonical cases (44.6%), while retaining similar levels of conser- vation for their aminoacylation domains (54.7% versus 56.1%). This suggested that the editing domain in these Mycoplasma spe- cies was preferentially prone to retaining substitutions (Table S1). Author contributions: L.L., M.T.B., and S.A.M. designed research; L.L., J.D.J., B.S.I., and P.M.Y. performed research; L.L. contributed new reagents/analytic tools; L.L., M.T.B., J.D.J., B.S.I., P.M.Y., Z.A.L.-S., and S.A.M. analyzed data; and L.L., Z.A.L.-S., and S.A.M. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1 To whom correspondence should be addressed at: Department of Biochemistry, 419 Roger Adams Laboratory, Box B4, 600 South Mathews Avenue, University of Illinois, Urbana, IL 61801. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/lookup/suppl/ doi:10.1073/pnas.1016460108/-/DCSupplemental. 9378–9383 ∣ PNAS ∣ June 7, 2011 ∣ vol. 108 ∣ no. 23 www.pnas.org/cgi/doi/10.1073/pnas.1016460108
Transcript
Naturally occurring aminoacyl-tRNA synthetasesediting-domain mutations that cause mistranslationin Mycoplasma parasitesLi Lia, Michal T. Bonieckib, Jacob D. Jaffec, Brian S. Imaid, Peter M. Yaud,Zaida A. Luthey-Schultena,e, and Susan A. Martinisa,b,e,1
aCenter for Biophysics and Computational Biology, bDepartment of Biochemistry, eDepartment of Chemistry, dRoy J. Carver Biotechnology Center,University of Illinois, Urbana, IL 61801; and cProteomics Platform, The Broad Institute, 7 Cambridge Center, Cambridge, MA 02142
Edited by Paul Schimmel, The Skaggs Institute for Chemical Biology, La Jolla, CA, and approved April 20, 2011 (received for review November 8, 2010)
Mycoplasma parasites escape host immune responses via me-chanisms that depend on remarkable phenotypic plasticity. Identi-fication of these mechanisms is of great current interest. Theaminoacyl-tRNA synthetases (AARSs) attach amino acids to theircognate tRNAs, but occasionally make errors that substitute closelysimilar amino acids. AARS editing pathways clear errors to avoidmistranslation during protein synthesis. We show here that AARSsin Mycoplasma parasites have point mutations and deletions intheir respective editing domains. The deleterious effect on editingwas confirmed with a specific example studied in vitro. In vivomistranslation was determined by mass spectrometric analysis ofproteins produced in the parasite. These mistranslations are uni-form cases where the predicted closely similar amino acid replacedthe correct one. Thus, natural AARS editing-domain mutations inMycoplasma parasites cause mistranslation. We raise the possibi-lity that these mutations evolved as a mechanism for antigendiversity to escape host defense systems.
Mycoplasma are characterized by their lack of a cell walland dependence on a vertebrate host (1). Their relationship
with the host can be parasitic or they can coexist as an obligatecommensal. The persistent survival of Mycoplasma within theirhost has been attributed to a phenotypic plasticity that allows thesepathogens to facilely alter their antigenic properties (2). Paradoxi-cally, this phenotypic plasticity is generated in spite of the Myco-plasma’s extremely small genomes, which have lost many of thecomponents that typically comprise signaling pathways to adaptto changing environments (2). Remarkably, thesewall-less bacteriahave a highly dynamic surface architecture comprised of mem-brane proteins that confer antigenic and functional versatility (2).
As with other pathogenic and nonpathogenic organisms,Mycoplasma contain a complete set of AARSs (aminoacyl-tRNAsynthetases), which are essential to translate the genetic code intofunctional proteins (3). Each AARS has evolved for specificity toa single standard amino acid to maintain the fidelity of the geneticcode. The AARS enzyme family activates and transfers aminoacids to their cognate tRNA isoacceptor. Once the tRNA is“charged” with an amino acid, it is shuttled to the ribosome forincorporation into nascent polypeptides.
About half of the AARSs are prone to mistakes by activatingstructurally similar amino acids and mischarging them to tRNA.To minimize the potential for creating statistical proteins thatcontain mistranslated amino acids, these AARSs have developeda second “sieve” (4); they have adapted to include hydrolyticediting domains that are distinct from their canonical aminoacy-lation core domains. In some cases, aminoacylation accuracy isalso enhanced by independent editing domains that function astRNA-specific deacylases (5, 6).
We have identified multiple AARSs with editing domainsin Mycoplasma and closely related species that appeared to be
degenerate based on substitutions at key sites in the hydrolyticactive site. This was surprising because functional defects inAARS editing that decrease the fidelity of tRNA aminoacylationhave been clearly shown to result in amino acid toxicities, celldeath, as well as neurological disease in mammals (7–9). As such,in order to achieve the threshold levels of translational fidelitythat are required for cell viability, these amino acid editing func-tions have been broadly conserved across all three domainsof life.
In contrast, we determined that Mycoplasma mobile exhibitsAARS-dependent translational infidelities. Editing-defectiveAARSs mischarge tRNA, which subsequently results in mistran-slation in vivo. It is possible that this AARS-dependent mechan-ism could provide a unique pathway to introduce heterogeneityinto the cell’s proteome that could confer phenotypic plasticity inMycoplasma pathogens.
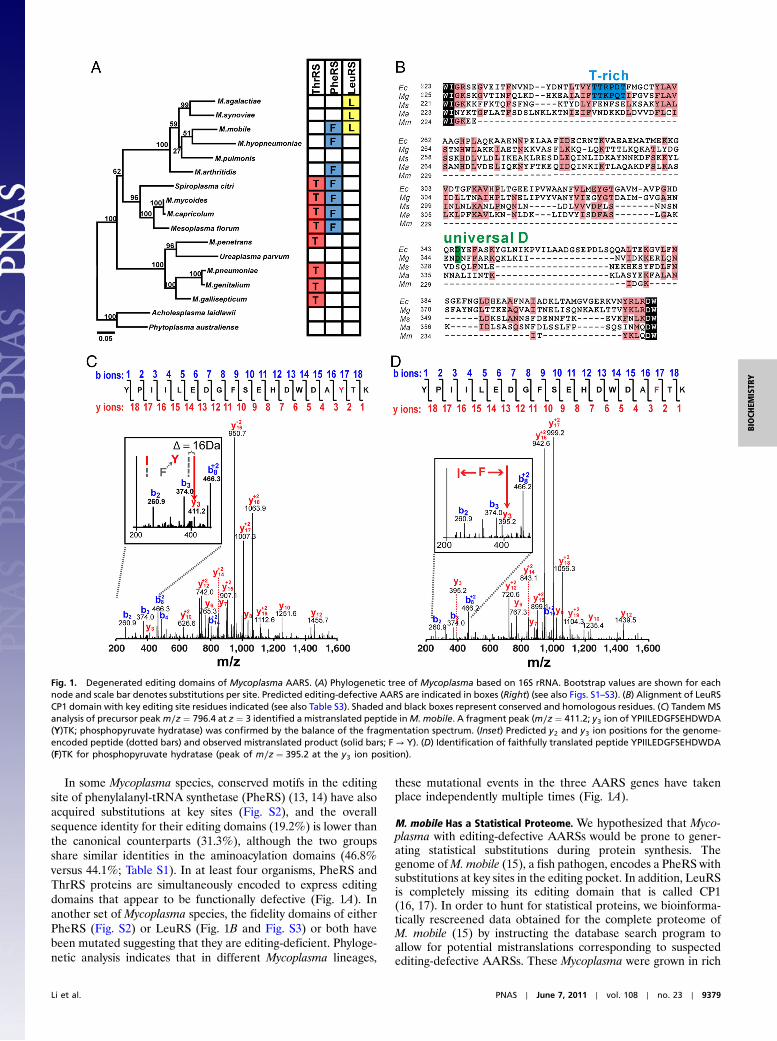
ResultsMycoplasma Have Evolved AARSs with Inactivated Editing Domains.Using bioinformatic approaches to broadly scrutinize genomesacross the three domains of life, we identified AARSs with unu-sual amino acid editing domains. In Mycoplasma and closelyrelated species, we discovered AARSs with deletions and substi-tutions that we hypothesized would abolish their editing activities(Fig. 1). These Mycoplasma AARSs have substitutions at keyresidues in the hydrolytic active sites of the editing domains. Inone extreme case, the editing domain was completely deletedfrom the M. mobile leucyl-tRNA synthetase (LeuRS).
In six different Mycoplasma and also two closely relatedspecies, threonyl-tRNA synthetase (ThrRS) contained editingdomains in which the editing site had amino acid substitutions atcritical residues (Fig. 1A). In each of these cases, critical histidine,aspartic acid, and cysteine residues (Fig. S1) within two distalpeptides that folded to form the editing active site were replaced(10–12). The editing domains of the editing-defective ThrRSshave a lower average sequence identity (24.4%) compared to thecanonical cases (44.6%), while retaining similar levels of conser-vation for their aminoacylation domains (54.7% versus 56.1%).This suggested that the editing domain in these Mycoplasma spe-cies was preferentially prone to retaining substitutions (Table S1).
Author contributions: L.L., M.T.B., and S.A.M. designed research; L.L., J.D.J., B.S.I., andP.M.Y. performed research; L.L. contributed new reagents/analytic tools; L.L., M.T.B.,J.D.J., B.S.I., P.M.Y., Z.A.L.-S., and S.A.M. analyzed data; and L.L., Z.A.L.-S., and S.A.M. wrotethe paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should be addressed at: Department of Biochemistry, 419 RogerAdams Laboratory, Box B4, 600 South Mathews Avenue, University of Illinois, Urbana,IL 61801. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1016460108/-/DCSupplemental.
In some Mycoplasma species, conserved motifs in the editingsite of phenylalanyl-tRNA synthetase (PheRS) (13, 14) have alsoacquired substitutions at key sites (Fig. S2), and the overallsequence identity for their editing domains (19.2%) is lower thanthe canonical counterparts (31.3%), although the two groupsshare similar identities in the aminoacylation domains (46.8%versus 44.1%; Table S1). In at least four organisms, PheRS andThrRS proteins are simultaneously encoded to express editingdomains that appear to be functionally defective (Fig. 1A). Inanother set of Mycoplasma species, the fidelity domains of eitherPheRS (Fig. S2) or LeuRS (Fig. 1B and Fig. S3) or both havebeen mutated suggesting that they are editing-deficient. Phyloge-netic analysis indicates that in different Mycoplasma lineages,
these mutational events in the three AARS genes have takenplace independently multiple times (Fig. 1A).
M. mobile Has a Statistical Proteome. We hypothesized that Myco-plasma with editing-defective AARSs would be prone to gener-ating statistical substitutions during protein synthesis. Thegenome ofM. mobile (15), a fish pathogen, encodes a PheRS withsubstitutions at key sites in the editing pocket. In addition, LeuRSis completely missing its editing domain that is called CP1(16, 17). In order to hunt for statistical proteins, we bioinforma-tically rescreened data obtained for the complete proteome ofM. mobile (15) by instructing the database search program toallow for potential mistranslations corresponding to suspectedediting-defective AARSs. These Mycoplasma were grown in rich
Fig. 1. Degenerated editing domains of Mycoplasma AARS. (A) Phylogenetic tree of Mycoplasma based on 16S rRNA. Bootstrap values are shown for eachnode and scale bar denotes substitutions per site. Predicted editing-defective AARS are indicated in boxes (Right) (see also Figs. S1–S3). (B) Alignment of LeuRSCP1 domain with key editing site residues indicated (see also Table S3). Shaded and black boxes represent conserved and homologous residues. (C) TandemMSanalysis of precursor peakm∕z ¼ 796.4 at z ¼ 3 identified a mistranslated peptide inM. mobile. A fragment peak (m∕z ¼ 411.2; y3 ion of YPIILEDGFSEHDWDA(Y)TK; phosphopyruvate hydratase) was confirmed by the balance of the fragmentation spectrum. (Inset) Predicted y2 and y3 ion positions for the genome-encoded peptide (dotted bars) and observed mistranslated product (solid bars; F → Y). (D) Identification of faithfully translated peptide YPIILEDGFSEHDWDA(F)TK for phosphopyruvate hydratase (peak of m∕z ¼ 395.2 at the y3 ion position).
Li et al. PNAS ∣ June 7, 2011 ∣ vol. 108 ∣ no. 23 ∣ 9379
media under conditions that would not be expected to bias abacteria to incorporate mutations (15). However, analysis ofthe proteome identified examples of statistical mutations includ-ing an F322Y substitution in phosphopyruvate hydratase (Fig. 1C)that would be indicative of an editing-defective PheRS, whichgenerates mischarged Tyr-tRNAPhe (13). In parallel, a faithfullytranslated peptide from phosphopyruvate hydratase was alsodetected that did not contain the F322Y mutation via mass spec-trometry analysis of the peptide pool (Fig. 1D), which supportedthe statistical nature of these mistakes.
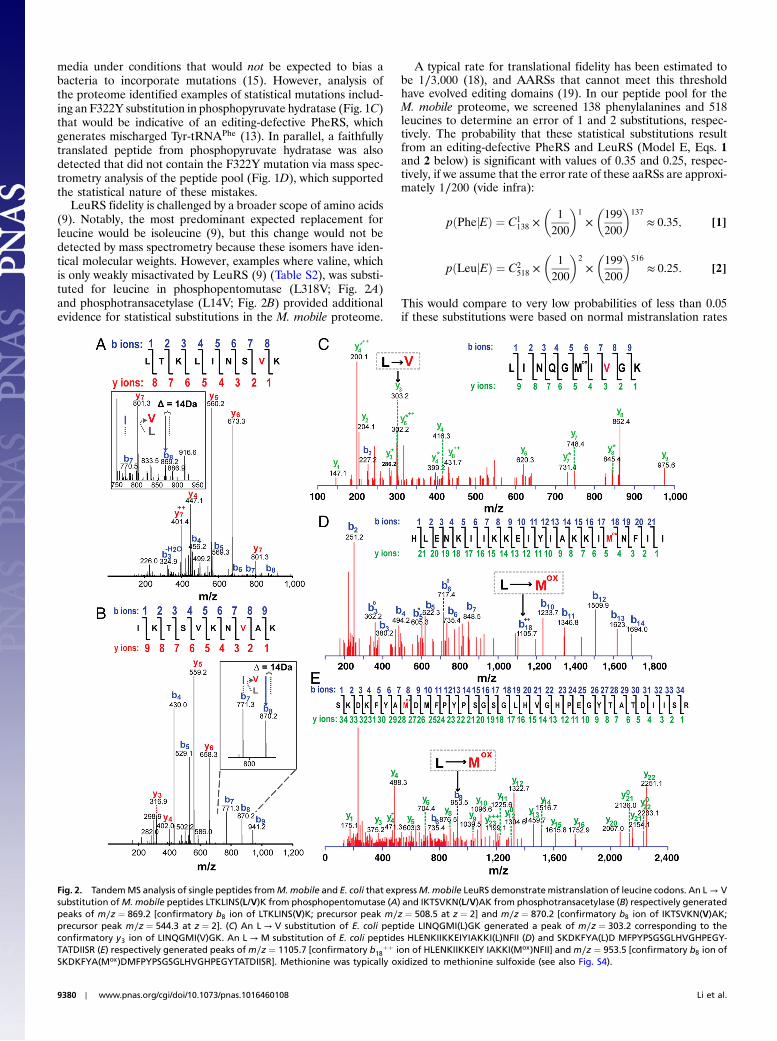
LeuRS fidelity is challenged by a broader scope of amino acids(9). Notably, the most predominant expected replacement forleucine would be isoleucine (9), but this change would not bedetected by mass spectrometry because these isomers have iden-tical molecular weights. However, examples where valine, whichis only weakly misactivated by LeuRS (9) (Table S2), was substi-tuted for leucine in phosphopentomutase (L318V; Fig. 2A)and phosphotransacetylase (L14V; Fig. 2B) provided additionalevidence for statistical substitutions in the M. mobile proteome.
A typical rate for translational fidelity has been estimated tobe 1∕3;000 (18), and AARSs that cannot meet this thresholdhave evolved editing domains (19). In our peptide pool for theM. mobile proteome, we screened 138 phenylalanines and 518leucines to determine an error of 1 and 2 substitutions, respec-tively. The probability that these statistical substitutions resultfrom an editing-defective PheRS and LeuRS (Model E, Eqs. 1and 2 below) is significant with values of 0.35 and 0.25, respec-tively, if we assume that the error rate of these aaRSs are approxi-mately 1∕200 (vide infra):
pðPhejEÞ ¼ C1138 ×
�1
200
�1
�199
200
�137
≈ 0.35; [1]
pðLeujEÞ ¼ C2518 ×
�1
200
�2
�199
200
�516
≈ 0.25: [2]
This would compare to very low probabilities of less than 0.05if these substitutions were based on normal mistranslation rates
Fig. 2. TandemMS analysis of single peptides fromM.mobile and E. coli that expressM.mobile LeuRS demonstratemistranslation of leucine codons. An L → Vsubstitution ofM. mobile peptides LTKLINS(L/V)K from phosphopentomutase (A) and IKTSVKN(L/V)AK from phosphotransacetylase (B) respectively generatedpeaks of m∕z ¼ 869.2 [confirmatory b8 ion of LTKLINS(V)K; precursor peak m∕z ¼ 508.5 at z ¼ 2] and m∕z ¼ 870.2 [confirmatory b8 ion of IKTSVKN(V)AK;precursor peak m∕z ¼ 544.3 at z ¼ 2]. (C) An L → V substitution of E. coli peptide LINQGMI(L)GK generated a peak of m∕z ¼ 303.2 corresponding to theconfirmatory y3 ion of LINQGMI(V)GK. An L → M substitution of E. coli peptides HLENKIIKKEIYIAKKI(L)NFII (D) and SKDKFYA(L)D MFPYPSGSGLHVGHPEGY-TATDIISR (E) respectively generated peaks ofm∕z ¼ 1105.7 [confirmatory b18
þþ ion of HLENKIIKKEIY IAKKI(Mox)NFII] and m∕z ¼ 953.5 [confirmatory b8 ion ofSKDKFYA(Mox)DMFPYPSGSGLHVGHPEGYTATDIISR]. Methionine was typically oxidized to methionine sulfoxide (see also Fig. S4).
9380 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1016460108 Li et al.
of 1∕3;000. Thus, we propose that this high level of mistranslationis statistical for M. mobile and is caused by editing-defectivePheRS and LeuRS that cannot clear their own mischarged tRNAproducts.
We also tested for M. mobile LeuRS-dependent mistransla-tions in Escherichia coli. Previously, we showed that LeuRSmisactivates valine, isoleucine, and methionine (9). Becauseisoleucine cannot be distinguished from leucine via mass spectro-metry, we focused on valine and methionine substitutions. Toincrease sensitivity, we induced expression of M. mobile LeuRSin E. coli BL21 cells (17 nmol∕g cells) in the presence of increas-ing valine or methionine concentrations of up to 10 mM. Tandemmass spectrometry of purified M. mobile LeuRS that was trypsin-digested identified statistical substitutions for multiple sets ofpeptides, where valine or methionine was substituted for leucine(Fig. 2) as well as correlating examples where these specific siteshad been faithfully translated (Fig. S4). In comparison, there wasno mistranslation detected in BL21 cells that expressed E. coliLeuRS (22 nmol∕g cells) under the same condition, even at highconcentrations of noncognate amino acids. In addition, E. coliBL21 cells that expressed M. mobile LeuRS had a lengthy lagphase that slowed cell growth (Fig. S5).
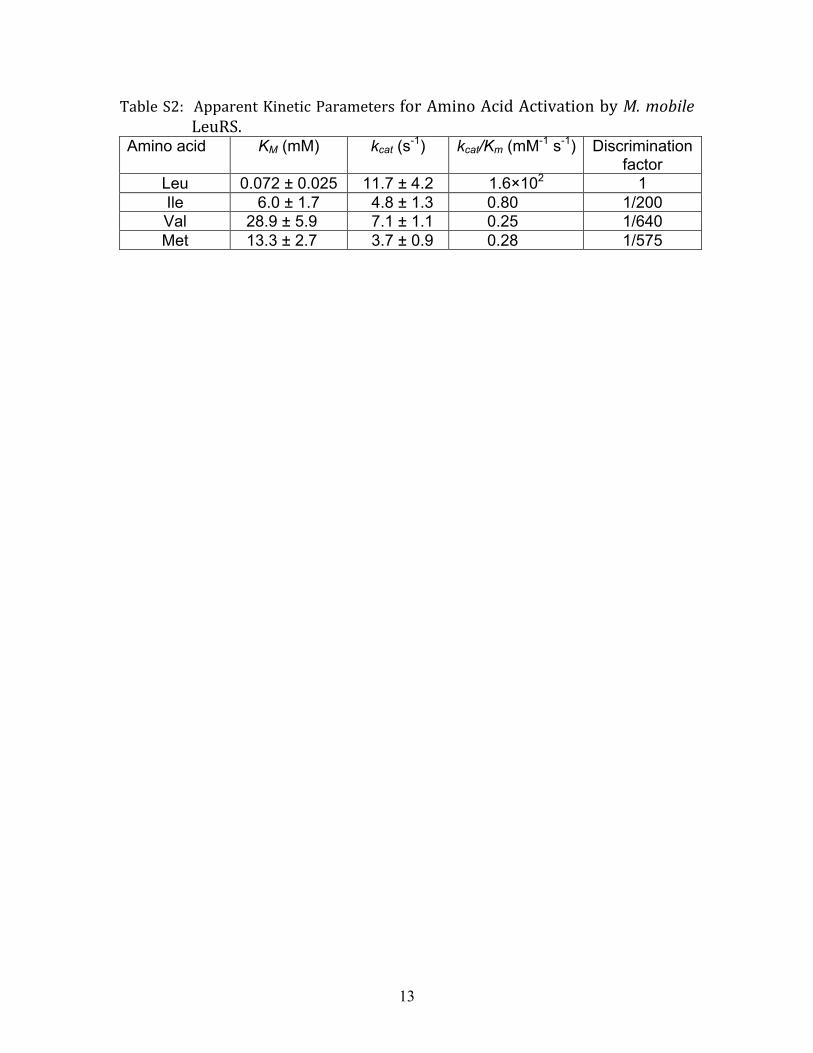
Statistical Proteome Substitutions Correlate to AARS Amino AcidEditing Defect. Preparation of pure ðαβÞ2 PheRS and dimericThrRS for in vitro analysis could be complicated by heterogeneityof their respective quaternary structures. Thus, we focused on themonomeric M. mobile LeuRS for enzymatic characterization ofits putative editing defect to understand the molecular mechan-ism underlying error-prone translation in M. mobile. The genewas synthesized in order to convert TGA triplets (which are usedto encode tryptophan in M. mobile) to TGG and also to optimizecodon usage frequencies for expression in E. coli. The monomericLeuRS was purified by affinity chromatography via an N-terminalsix-histidine tag and the enzyme robustly aminoacylated in vitrotranscribed M. mobile tRNALeu (Fig. S6) as well as E. colitRNALeu (Fig. S7). Based on the kcat∕KM for amino acid activa-tion by M. mobile LeuRS (Table S2), the discrimination factorsfor noncognate amino acids versus leucine were well below theaccepted threshold of 3,000. Indeed, the discrimination factorof 200 for isoleucine was consistent with our threshold calcula-tions for misaminoacylation in Mycoplasma (vide supra). It alsosuggests that M. mobile LeuRS is even more prone to isoleucinemisactivation than a typical LeuRS (Table 1).
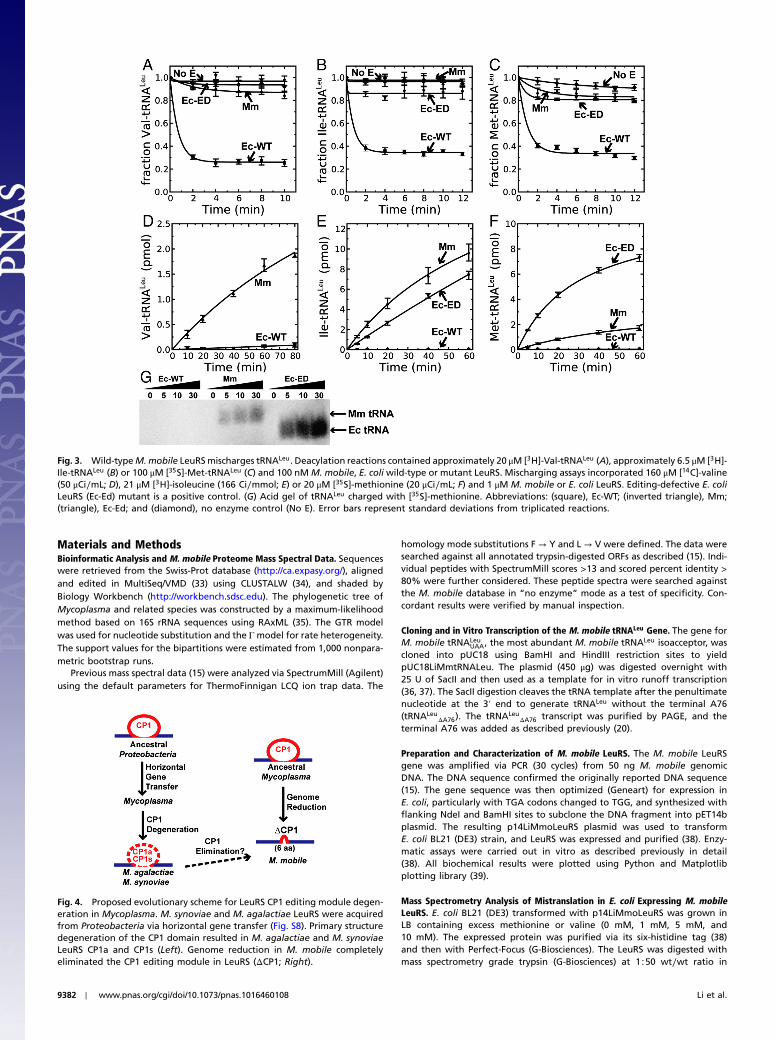
As would be expected, M. mobile LeuRS failed to dea-cylate mischarged Val-tRNALeu, Ile-tRNALeu, and Met-tRNALeu
(Fig. 3), because it does not have a CP1 editing domain. Thesenoncognate amino acids can be charged to M. mobile tRNA(Fig. 3). Consistent with the introduction of leucine to methio-nine or valine statistical substitutions in the E. coli proteome(Fig. 2), M. mobile LeuRS also mischarged E. coli tRNALeu withmethionine or valine (Fig. S7).
The propensity of M. mobile LeuRS to produce stably mis-charged tRNALeu contrasts sharply with other examples ofLeuRSs that were determined to rely on additional mechanismsto ensure fidelity. E. coli or yeast mitochondrial LeuRS main-tained fidelity in the absence of their CP1 modules by activating
an alternate pretransfer editing pathway (20). Also, M. mobileLeuRS has not evolved the higher threshold of amino acid dis-crimination that has been measured for human mitochondrialLeuRS, which lacks editing activity (21).
DiscussionIn all three domains of life, LeuRS contains the CP1 editingdomain as does isoleucyl (IleRS)- and valyl (ValRS)-tRNAsynthetases (22). These homologous proteins have differentiatedto accommodate different amino acid specificities for both ami-noacylation and editing. They also can rely selectively or com-binatorially on pre- and posttransfer editing mechanisms torespectively target the aminoacyl-adenylate intermediate or mis-charged tRNA product to clear misactivated amino acids (23–25).To our knowledge, M. mobile LeuRS is the only known examplefor LeuRS, IleRS, or ValRS that is completely missing its CP1editing module (Fig. 1B). Previously, we had shown that thefidelity of E. coli and yeast mitochondrial LeuRS were protectedin the absence of its CP1 module (LeuRS-ΔCP1) by a latentpretransfer editing activity that cleared misactivated aminoa-cyl-adenylate intermediate (20). In this case, M. mobile LeuRSwith its naturally missing CP1 editing module failed to recapturean alternate editing mechanism to posttransfer editing that wassufficient to completely protect the proteome.
Statistical proteins have been proposed to provide an advan-tage in primitive cells during the evolution of the modern proteinsynthesis machinery (26, 27). They have also been suggested toincrease fitness in contemporary bacteria by providing a proteinreservoir that is phenotypically diverse (28). In a transcription-dependent error-prone experimental model for Bacillus subtilis,proteome diversity caused by frame-shift or nonsense mutationsenabled the organism to adapt to fluctuations in the environment,such as changes in temperature (28). In yeast, CUG codon ambi-guity originating in Candida albacans was experimentally intro-duced to confer an adaptive advantage under stress conditionsvia induction of a unique set of stress proteins (29). Under spe-cific stress conditions in mammalian systems, codon ambiguityincreases significantly via activation of MetRS misacylation ofnoncognate tRNA isoacceptors (30). An artificial mutation in theValRS editing domain also allowed E. coli to adapt for subsis-tence on the nonstandard amino acid α-aminobutyrate, ratherthan valine (31).
We have identified natural examples in Mycoplasma speciesand related species in which the gene encoding AARSs containsmutations that disrupt the amino acid editing function. The shed-ding of the editing domain of LeuRS in M. mobile was likelyfacilitated by existing mechanisms in Mycoplasma that haveresulted in the dramatic evolutionary reduction of its genomes.Accordingly, the erosion of AARS-dependent translational fide-lity may be a stepwise response based on the LeuRS CP1 do-main’s massive sequence degeneration in Mycoplasma agalactiaeand Mycoplasma synoviae, and then complete elimination of theentire module in M. mobile (Fig. 4).
Editing-defective AARSs in M. mobile introduce errors atthe translational level of protein synthesis. Because of the strongselection pressure throughout the three domains of life to main-tain translational fidelity, we hypothesize that the loss of AARSediting in Mycoplasma is likely due to idiosyncratic demands onthe organism, which are benefited by the introduction of statis-tical proteomes (19). It is possible that this mechanism to intro-duce statistical proteins could confer antigenic diversity andphenotypic plasticity (2). This unique AARS-dependent mechan-ism at the translational level would be akin to the replication-dependent high rates of retroviral mutations that confer viruspopulation heterogeneity and resistance (32) to escape hostdefense mechanisms.
Table 1. Apparent kinetic parameters for amino acid activationby LeuRS
Materials and MethodsBioinformatic Analysis and M. mobile Proteome Mass Spectral Data. Sequenceswere retrieved from the Swiss-Prot database (http://ca.expasy.org/), alignedand edited in MultiSeq/VMD (33) using CLUSTALW (34), and shaded byBiology Workbench (http://workbench.sdsc.edu). The phylogenetic tree ofMycoplasma and related species was constructed by a maximum-likelihoodmethod based on 16S rRNA sequences using RAxML (35). The GTR modelwas used for nucleotide substitution and the Γmodel for rate heterogeneity.The support values for the bipartitions were estimated from 1,000 nonpara-metric bootstrap runs.
Previous mass spectral data (15) were analyzed via SpectrumMill (Agilent)using the default parameters for ThermoFinnigan LCQ ion trap data. The
homology mode substitutions F → Y and L → V were defined. The data weresearched against all annotated trypsin-digested ORFs as described (15). Indi-vidual peptides with SpectrumMill scores >13 and scored percent identity >80% were further considered. These peptide spectra were searched againstthe M. mobile database in “no enzyme” mode as a test of specificity. Con-cordant results were verified by manual inspection.
Cloning and in Vitro Transcription of the M. mobile tRNALeu Gene. The gene forM. mobile tRNALeu
UAA, the most abundant M. mobile tRNALeu isoacceptor, wascloned into pUC18 using BamHI and HindIII restriction sites to yieldpUC18LiMmtRNALeu. The plasmid (450 μg) was digested overnight with25 U of SacII and then used as a template for in vitro runoff transcription(36, 37). The SacII digestion cleaves the tRNA template after the penultimatenucleotide at the 3′ end to generate tRNALeu without the terminal A76(tRNALeu
ΔA76). The tRNALeuΔA76 transcript was purified by PAGE, and the
terminal A76 was added as described previously (20).
Preparation and Characterization of M. mobile LeuRS. The M. mobile LeuRSgene was amplified via PCR (30 cycles) from 50 ng M. mobile genomicDNA. The DNA sequence confirmed the originally reported DNA sequence(15). The gene sequence was then optimized (Geneart) for expression inE. coli, particularly with TGA codons changed to TGG, and synthesized withflanking NdeI and BamHI sites to subclone the DNA fragment into pET14bplasmid. The resulting p14LiMmoLeuRS plasmid was used to transformE. coli BL21 (DE3) strain, and LeuRS was expressed and purified (38). Enzy-matic assays were carried out in vitro as described previously in detail(38). All biochemical results were plotted using Python and Matplotlibplotting library (39).
Mass Spectrometry Analysis of Mistranslation in E. coli Expressing M. mobileLeuRS. E. coli BL21 (DE3) transformed with p14LiMmoLeuRS was grown inLB containing excess methionine or valine (0 mM, 1 mM, 5 mM, and10 mM). The expressed protein was purified via its six-histidine tag (38)and then with Perfect-Focus (G-Biosciences). The LeuRS was digested withmass spectrometry grade trypsin (G-Biosciences) at 1∶50 wt∕wt ratio in
Fig. 3. Wild-typeM.mobile LeuRSmischarges tRNALeu. Deacylation reactions contained approximately 20 μM [3H]-Val-tRNALeu (A), approximately 6.5 μM [3H]-Ile-tRNALeu (B) or 100 μM [35S]-Met-tRNALeu (C) and 100 nM M. mobile, E. coli wild-type or mutant LeuRS. Mischarging assays incorporated 160 μM [14C]-valine(50 μCi∕mL; D), 21 μM [3H]-isoleucine (166 Ci∕mmol; E) or 20 μM [35S]-methionine (20 μCi∕mL; F) and 1 μM M. mobile or E. coli LeuRS. Editing-defective E. coliLeuRS (Ec-Ed) mutant is a positive control. (G) Acid gel of tRNALeu charged with [35S]-methionine. Abbreviations: (square), Ec-WT; (inverted triangle), Mm;(triangle), Ec-Ed; and (diamond), no enzyme control (No E). Error bars represent standard deviations from triplicated reactions.
Fig. 4. Proposed evolutionary scheme for LeuRS CP1 editing module degen-eration in Mycoplasma. M. synoviae and M. agalactiae LeuRS were acquiredfrom Proteobacteria via horizontal gene transfer (Fig. S8). Primary structuredegeneration of the CP1 domain resulted in M. agalactiae and M. synoviaeLeuRS CP1a and CP1s (Left). Genome reduction in M. mobile completelyeliminated the CP1 editing module in LeuRS (ΔCP1; Right).
9382 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1016460108 Li et al.
25 mM ammonium bicarbonate. Digestion was performed using a CEMDiscover Microwave Digestor (CEM Corporation) for 15 min at 75 W and55 °C. The digested peptides were lyophilized to dryness and reconstitutedin 5% acetonitrile with 0.1% formic acid at 200 μg∕mL. A 10-μL aliquotwas used for liquid chromatography/mass spectrometry (LC/MS) analysis.
LC/MS spectrometry was performed using a Waters Q-ToF connectedto a nanoAcquity UPLC (Waters Corporation) and an Atlantis dC18 nanoAc-quity UPLC column (75 μm × 150 mm; 3-μm particle size) with a flowrate of 250 nL∕min. The 60-min gradient was from 100% A to 60% B(A ¼ waterþ 0.1% formic acid; B ¼ acetonitrile þ 0.1% formic acid). Datacollection was performed with MassLynx 4.1 using Data Directed Analysis.The top four intensive precursor ions from each survey scan were subjectedto MS/MS by collision-induced dissociation. The raw mass spectrometricdata were processed using ProteinLynx Global Server 2.2.5 (Waters) for data
filtering, smoothing, and deisotoping. The refined peak lists were analyzedwith Mascot 2.2 (Matrix Science) using a tolerance of �0.4 Da for both theprecursor ions and fragment ions. The searches were conducted againstthe National Center for Biotechnology Information nonredundant proteindatabase. Specific modifications such as L → V or L → M (or its oxidized state)substitutions were analyzed as variable modifications in conjunction withthe error-tolerant mode in Mascot.
ACKNOWLEDGMENTS. We thank Ke Chen (University of Illinois, Urbana, IL)for providing aligned 16S rRNA sequences and Dr. Makoto Miyata (OsakaCity University, Osaka, Japan) for his gift of M. mobile genomic DNA. Thiswork was supported by grants from the National Science Foundation(MCB-0843611 and MCB-0844670) and the National Institutes of Health(GM063789 and P41RR05964).
1. Maniloff J (2002) Molecular Biology and Pathogenicity of Mycoplasmas, eds S Razinand R Herrmann (Kluwer Academic, New York), pp 31–44.
2. Rottem S (2003) Interaction of Mycoplasmas with host cells. Physiol Rev 83:417–432.3. Ibba M, Söll D (1999) Quality control mechanisms during translation. Science
286:1893–1897.4. Fersht AR (1998) Sieves in sequence. Science 280:541.5. An S, Musier-Forsyth K (2004) Trans-editing of Cys-tRNAPro by Haemophilus influenzae
YbaK protein. J Biol Chem 279:42359–42362.6. Ruan B, Söll D (2005) The bacterial YbaK protein is a Cys-tRNAPro and Cys-tRNACys
deacylase. J Biol Chem 280:25887–25891.7. Nangle LA, Motta CM, Schimmel P (2006) Global effects of mistranslation from an
editing defect in mammalian cells. Chem Biol 13:1091–1100.8. Lee JW, et al. (2006) Editing-defective tRNA synthetase causes protein misfolding
and neurodegeneration. Nature 443:50–55.9. Karkhanis VA, Mascarenhas AP, Martinis SA (2007) Amino acid toxicities of Escherichia
coli that are prevented by leucyl-tRNA synthetase amino acid editing. J Bacteriol189:8765–8768.
10. Beebe K, Ribas De Pouplana L, Schimmel P (2003) Elucidation of tRNA-dependent edit-ing by a class II tRNA synthetase and significance for cell viability. EMBO J 22:668–675.
11. Dock-Bregeon AC, et al. (2004) Achieving error-free translation; the mechanism ofproofreading of threonyl-tRNA synthetase at atomic resolution. Mol Cell 16:375–386.
12. Beebe K, Merriman E, Ribas De Pouplana L, Schimmel P (2004) A domain for editing byan archaebacterial tRNA synthetase. Proc Natl Acad Sci USA 101:5958–5963.
13. Ling J, Roy H, Ibba M (2007) Mechanism of tRNA-dependent editing in translationalquality control. Proc Natl Acad Sci USA 104:72–77.
14. Roy H, Ling J, Irnov M, Ibba M (2004) Post-transfer editing in vitro and in vivo by theβ-subunit of phenylalanyl-tRNA synthetase. EMBO J 23:4639–4648.
15. Jaffe JD, et al. (2004) The complete genome and proteome of Mycoplasma mobile.Genome Res 14:1447–1461.
16. Starzyk RM, Webster TA, Schimmel P (1987) Evidence for dispensable sequencesinserted into a nucleotide fold. Science 237:1614–1618.
17. Cusack S, Yaremchuk A, Tukalo M (2000) The 2 Å crystal structure of leucyl-tRNAsynthetase and its complex with a leucyl-adenylate analogue. EMBO J 19:2351–2361.
18. Loftfield RB (1963) The frequency of errors in protein biosynthesis. Biochem J89:82–92.
19. Reynolds NM, et al. (2010) Cell-specific differences in the requirements for translationquality control. Proc Natl Acad Sci USA 107:4063–4068.
20. Boniecki MT, Vu MT, Betha AK, Martinis SA (2008) CP1-dependent partitioning ofpretransfer and posttransfer editing in leucyl-tRNA synthetase. Proc Natl Acad SciUSA 105:19223–19228.
21. Lue SW, Kelley SO (2005) An aminoacyl-tRNA synthetase with a defunct editing site.Biochemistry 44:3010–3016.
22. Lin L, Hale SP, Schimmel P (1996) Aminoacylation error correction. Nature 384:33–34.23. Martinis SA, Boniecki MT (2010) The balance between pre- and post-transfer editing in
tRNA synthetases. FEBS Lett 584:455–459.24. Schimmel P, Schmidt E (1995) Making connections: RNA-dependent amino acid recog-
nition. Trends Biochem Sci 20:1–2.25. Dulic M, Cvetesic N, Perona JJ, Gruic-Sovulj I (2010) Partitioning of tRNA-dependent
editing between pre- and post-transfer pathways in class I aminoacyl-tRNA synthe-tases. J Biol Chem 285:23799–23809.
26. Woese CR (1965) On the evolution of the genetic code. Proc Natl Acad Sci USA54:1546–1552.
27. Woese CR (2002) On the evolution of cells. Proc Natl Acad Sci USA 99:8742–8747.28. Meyerovich M, Mamou G, Ben-Yehuda S (2010) Visualizing high error levels during
gene expression in living bacterial cells. Proc Natl Acad Sci USA 107:11543–11548.29. Santos MA, Cheesman C, Costa V, Moradas-Ferreira P, Tuite MF (1999) Selective
advantages created by codon ambiguity allowed for the evolution of an alternativegenetic code in Candida spp. Mol Microbiol 31:937–947.
30. Netzer N, et al. (2009) Innate immune and chemically triggered oxidative stressmodifies translational fidelity. Nature 462:522–6.
31. Doring V, et al. (2001) Enlarging the amino acid set of Escherichia coli by infiltration ofthe valine coding pathway. Science 292:501–504.
33. Roberts E, Eargle J, Wright D, Luthey-Schulten Z (2006) MultiSeq: Unifying sequenceand structure data for evolutionary analysis. BMC Bioinformatics 7:382.
34. Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: Improving the sensitivityof progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680.
35. Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyseswith thousands of taxa and mixed models. Bioinformatics 22:2688–2690.
36. Sampson JR, Uhlenbeck OC (1988) Biochemical and physical characterization of anunmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc Natl AcadSci USA 85:1033–1037.
37. Zhai Y, Martinis SA (2005) Two conserved threonines collaborate in the Escherichia colileucyl-tRNA synthetase amino acid editing mechanism. Biochemistry 44:15437–15443.
38. Karkhanis VA, Boniecki MT, Poruri K, Martinis SA (2006) A viable amino acid editingactivity in the leucyl-tRNA synthetase CP1-splicing domain is not required in the yeastmitochondria. J Biol Chem 281:33217–33225.
39. Hunter JD (2007) Matplotlib: A 2D graphics environment. Comput Sci Eng 9:90–95.
Li et al. PNAS ∣ June 7, 2011 ∣ vol. 108 ∣ no. 23 ∣ 9383
BIOCH
EMISTR
Y
1
Supporting Information
Naturally Occurring Aminoacyl-tRNA Synthetases Editing-Domain Mutations that Cause Mistranslation in Mycoplasma Parasites
Li Li, Michal T. Boniecki, Jacob D. Jaffe, Brian S. Imai, Peter M. Yau, Zaida Luthey-
Schulten, and Susan A. Martinis
2
Fig. S1
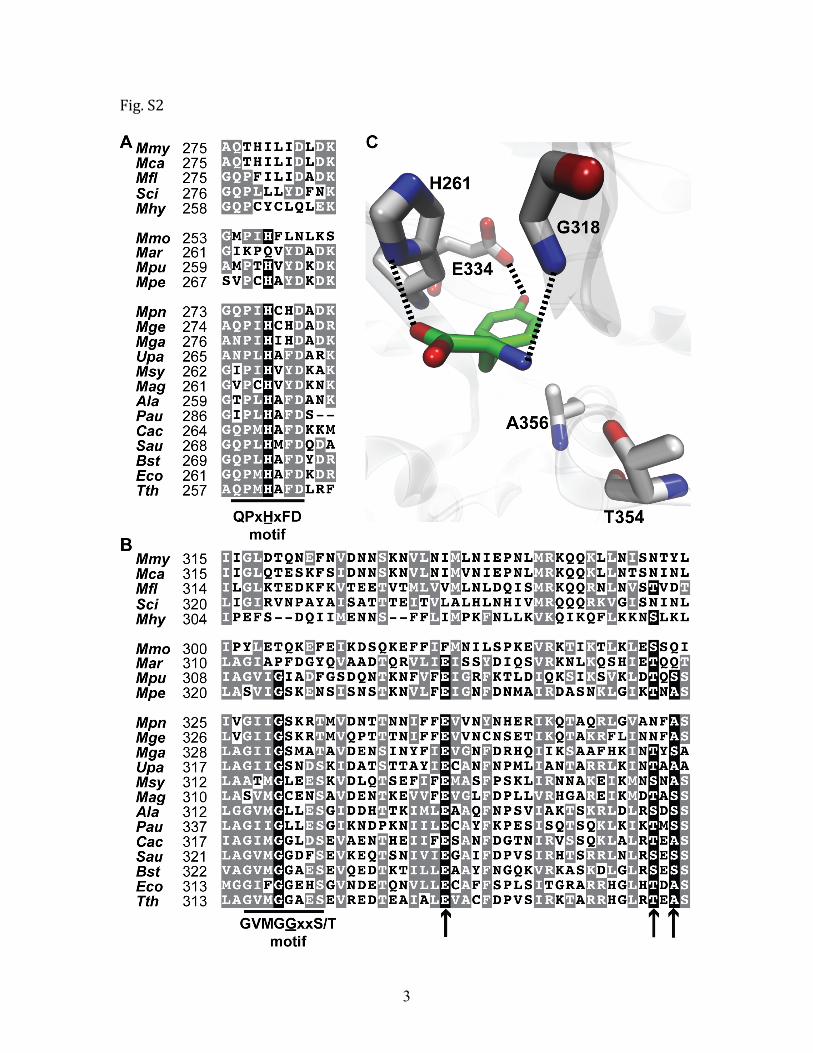
Fig. S1, related to Figure 1. Natural substitutions of key editing residues in Mycoplasma ThrRS. Sequence alignment of the editing domain from Mycoplasma and its closely‐related species ThrRSs (A) are shown. Key residues (Table S3) for editing are highlighted in black and indicated by arrows. B. These key residues are mapped to the crystal structure of E. coli ThrRS (PDB code 1TKY). Interactions between these residues and the post‐transfer editing analog (green) are indicated by dashed lines. The numbering is based on the E. coli ThrRS sequence. Organism abbreviations are as follows: Mycoplasma mycoides subsp. mycoides (Mmy), Mycoplasma capricolum (Mca), Mesoplasma florum (Mfl), Spiroplasma citri (Sci), Mycoplasma pneumoniae (Mpn), Mycoplasma genitalium (Mge), Mycoplasma gallisepticum (Mga), Mycoplasma penetrans (Mpe), Ureaplasma parvum (Upa), Mycoplasma synoviae (Msy), Mycoplasma agalactiae (Mag), Mycoplasma pulmonis (Mpu), Mycoplasma mobile (Mmo), Mycoplasma arthritidis (Mar), Mycoplasma hyopneumoniae (Mhy), Acholeplasma laidlawii (Ala), Phytoplasma australiense (Pau), Clostridium acetobutylicum (Cac), Bacillus subtilis (Bst), Staphylococcus aureus (Sau), Escherichia coli (Eco) and Thermus thermophilus (Tth).
3
Fig. S2
4
Fig. S2, related to Figure 1. Natural substitutions of key editing residues in Mycoplasma PheRSs. Sequence alignment of the PheRS editing domain from Mycoplasma and its closely‐related species PheRSs (A, B) are shown. Key residues for editing (Table S3) are highlighted in black and indicated by arrows. C. These key residues are mapped to the crystal structure of T. thermophilus PheRS (PDB code 2AMC). Interactions between these residues and the tyrosine (green) are indicated by dashed lines. The numbering is based on the T. thermophilus PheRS sequence. Organism abbreviations are defined in Fig. S1.
5
Fig. S3
6
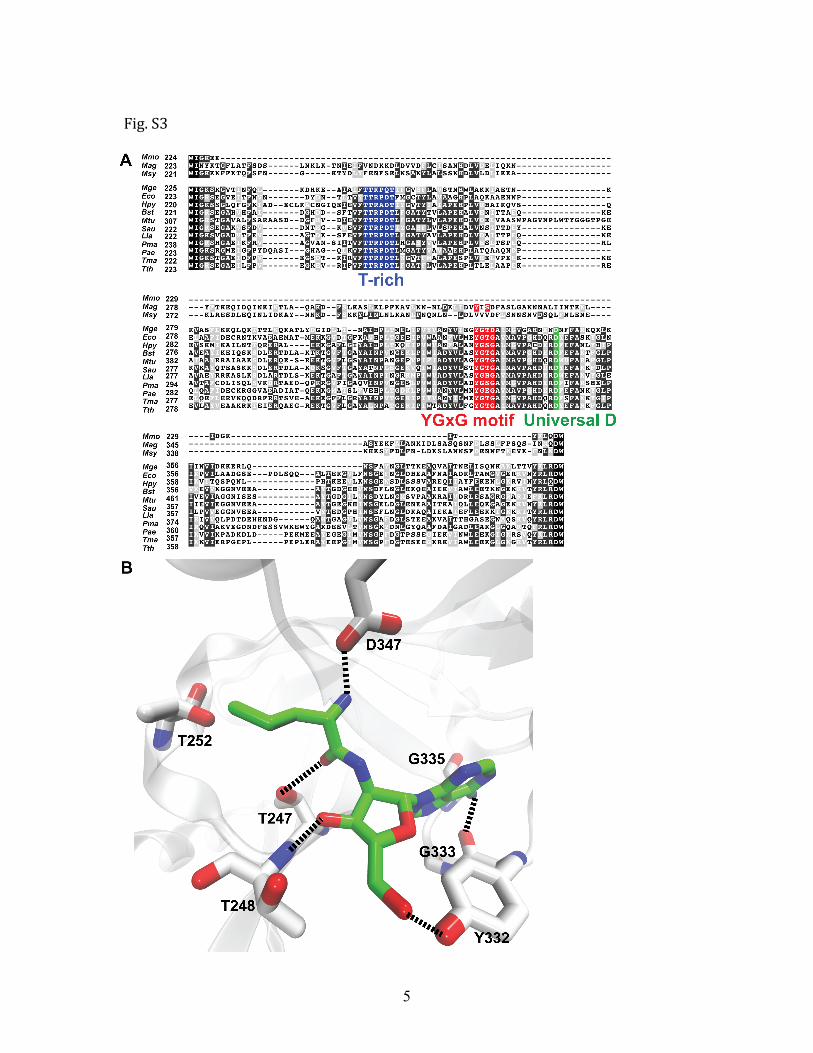
Fig. S3, related to Figure 1. Natural substitutions of key editing residues in Mycoplasma LeuRSs. A. Sequence alignment of the CP1 editing domain from bacterial LeuRSs as well as Mycoplasma LeuRSs with predicted editing deficiency. Key residues for editing (Table S3) are highlighted in blue (threonine‐rich region), red (YGxG adenine‐binding motif) and green (universally conserved aspartic acid). B. These key residues are mapped to the crystal structure of T. thermophilus LeuRS (PDB 1OBC). Interactions between these residues and the post‐transfer editing analog (green) are indicated by dashed lines. The numbering is based on T. thermophilus LeuRS sequence. Organism abbreviations are as follows: Mycoplasma mobile (Mmo), Mycoplasma synoviae (Msy), Mycoplasma agalactiae (Mag), Mycoplasma genitalium (Mge), Escherichia coli (Eco), Helicobacter pylori (Hpy), Bacillus subtilis (Bst), Mycobacterium tuberculosis (Mtu), Staphylococcus aureus (Sau), Lactococcus lactis (Lla), Prochlorococcus marinus (Pma), Pseudomonas aeruginosa (Pae), Thermotoga maritima (Tma) and Thermus thermophilus (Tth).
7
Fig. S4
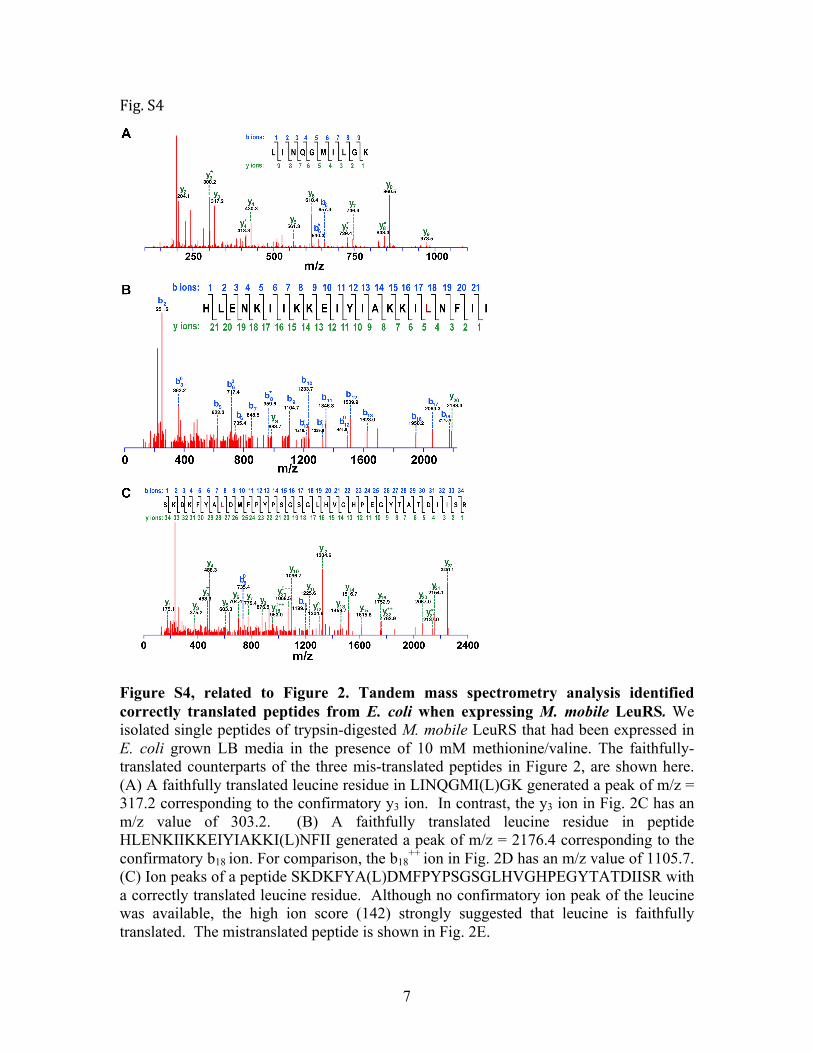
Figure S4, related to Figure 2. Tandem mass spectrometry analysis identified correctly translated peptides from E. coli when expressing M. mobile LeuRS. We isolated single peptides of trypsin-digested M. mobile LeuRS that had been expressed in E. coli grown LB media in the presence of 10 mM methionine/valine. The faithfully-translated counterparts of the three mis-translated peptides in Figure 2, are shown here. (A) A faithfully translated leucine residue in LINQGMI(L)GK generated a peak of m/z = 317.2 corresponding to the confirmatory y3 ion. In contrast, the y3 ion in Fig. 2C has an m/z value of 303.2. (B) A faithfully translated leucine residue in peptide HLENKIIKKEIYIAKKI(L)NFII generated a peak of m/z = 2176.4 corresponding to the confirmatory b18 ion. For comparison, the b18
++ ion in Fig. 2D has an m/z value of 1105.7. (C) Ion peaks of a peptide SKDKFYA(L)DMFPYPSGSGLHVGHPEGYTATDIISR with a correctly translated leucine residue. Although no confirmatory ion peak of the leucine was available, the high ion score (142) strongly suggested that leucine is faithfully translated. The mistranslated peptide is shown in Fig. 2E.
8
Fig. S5
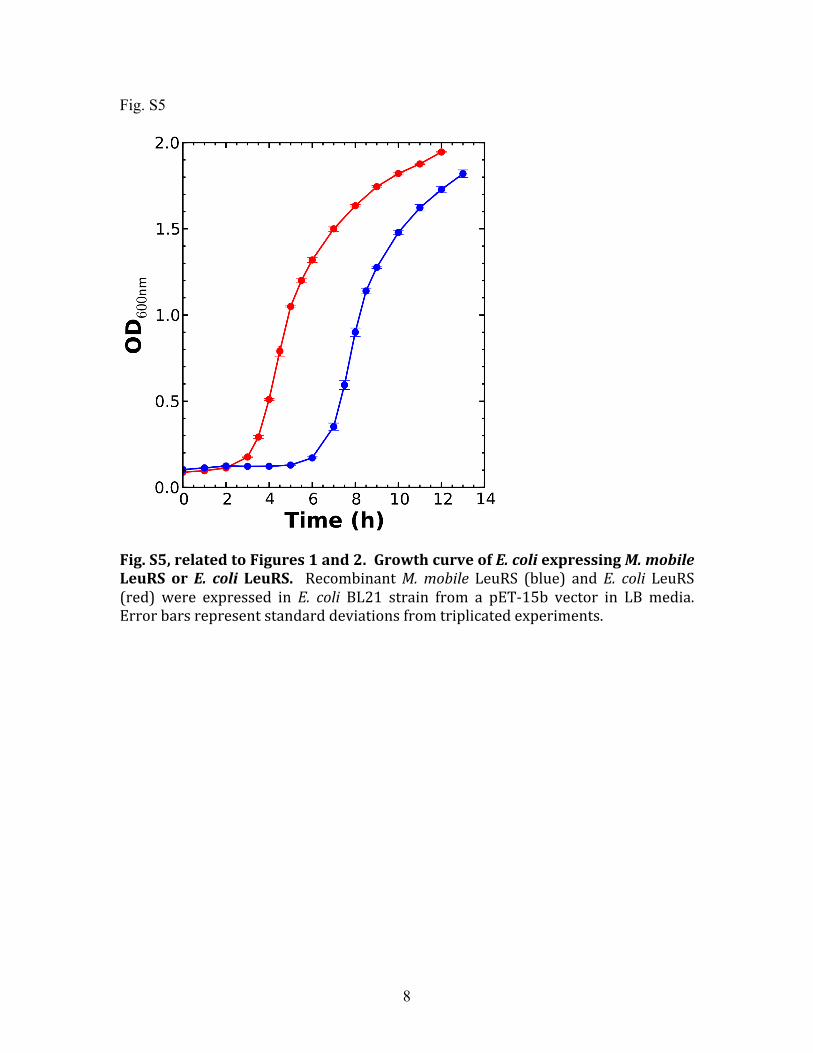
Fig. S5, related to Figures 1 and 2. Growth curve of E. coli expressing M. mobile LeuRS or E. coli LeuRS. Recombinant M. mobile LeuRS (blue) and E. coli LeuRS (red) were expressed in E. coli BL21 strain from a pET‐15b vector in LB media. Error bars represent standard deviations from triplicated experiments.
9
Fig. S6
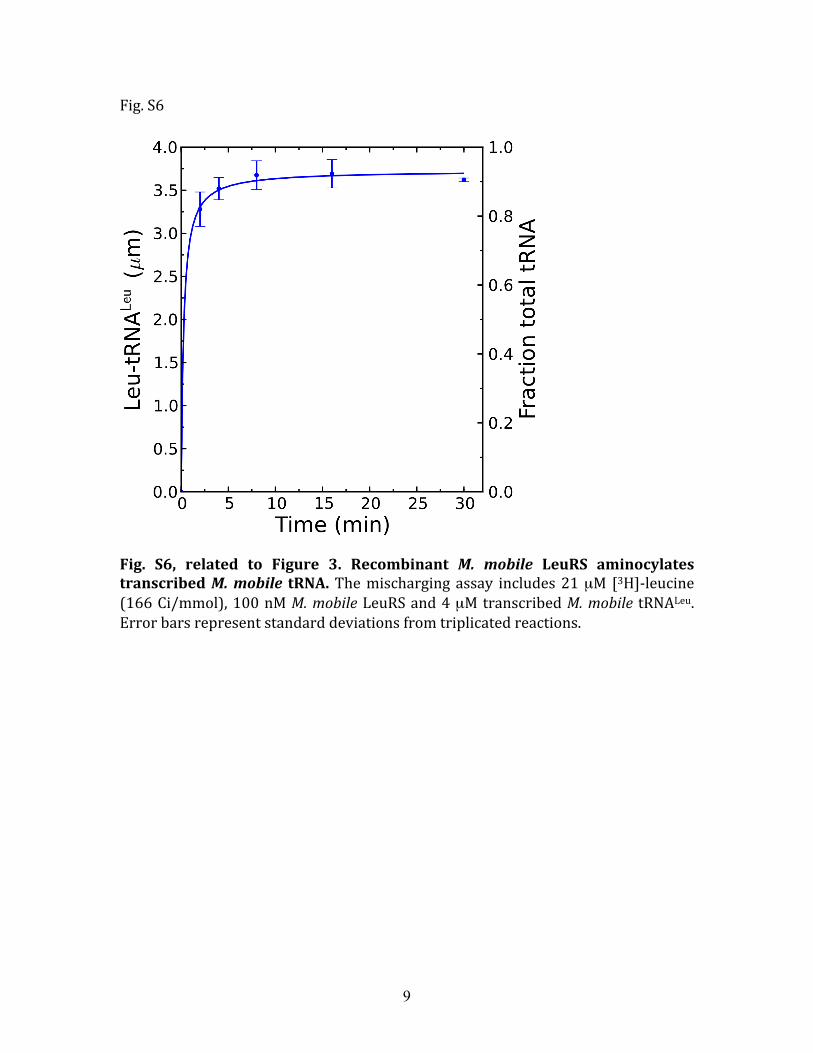
Fig. S6, related to Figure 3. Recombinant M. mobile LeuRS aminocylates transcribed M. mobile tRNA. The mischarging assay includes 21 µM [3H]‐leucine (166 Ci/mmol), 100 nM M. mobile LeuRS and 4 µM transcribed M. mobile tRNALeu. Error bars represent standard deviations from triplicated reactions.
10
Fig. S7
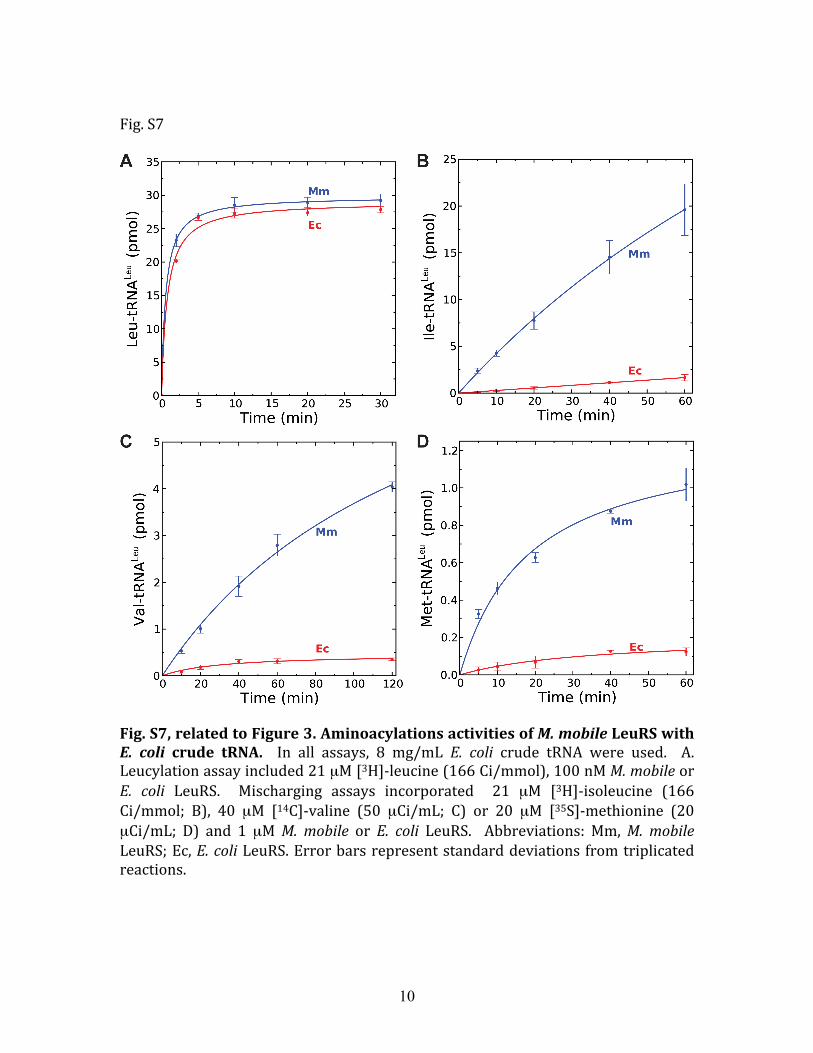
Fig. S7, related to Figure 3. Aminoacylations activities of M. mobile LeuRS with E. coli crude tRNA. In all assays, 8 mg/mL E. coli crude tRNA were used. A. Leucylation assay included 21 µM [3H]‐leucine (166 Ci/mmol), 100 nM M. mobile or E. coli LeuRS. Mischarging assays incorporated 21 µM [3H]‐isoleucine (166 Ci/mmol; B), 40 µM [14C]‐valine (50 µCi/mL; C) or 20 µM [35S]‐methionine (20 µCi/mL; D) and 1 µM M. mobile or E. coli LeuRS. Abbreviations: Mm, M. mobile LeuRS; Ec, E. coli LeuRS. Error bars represent standard deviations from triplicated reactions.
11
Fig. S8
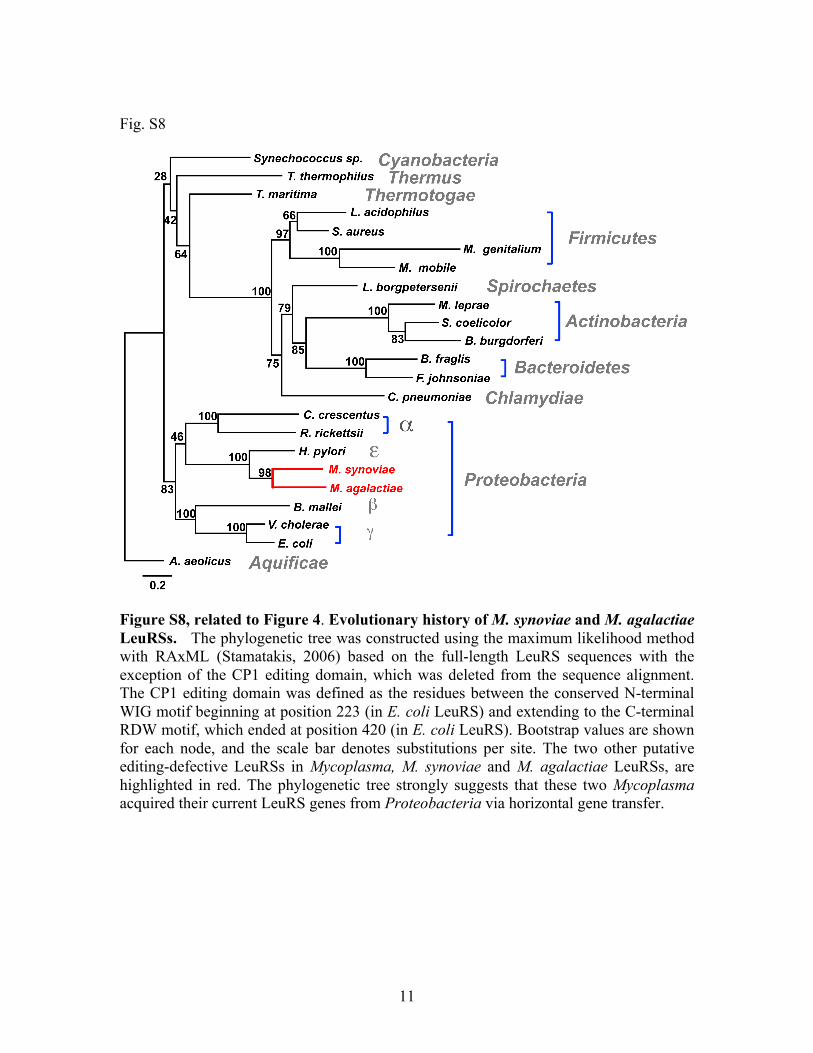
Figure S8, related to Figure 4. Evolutionary history of M. synoviae and M. agalactiae LeuRSs. The phylogenetic tree was constructed using the maximum likelihood method with RAxML (Stamatakis, 2006) based on the full-length LeuRS sequences with the exception of the CP1 editing domain, which was deleted from the sequence alignment. The CP1 editing domain was defined as the residues between the conserved N-terminal WIG motif beginning at position 223 (in E. coli LeuRS) and extending to the C-terminal RDW motif, which ended at position 420 (in E. coli LeuRS). Bootstrap values are shown for each node, and the scale bar denotes substitutions per site. The two other putative editing-defective LeuRSs in Mycoplasma, M. synoviae and M. agalactiae LeuRSs, are highlighted in red. The phylogenetic tree strongly suggests that these two Mycoplasma acquired their current LeuRS genes from Proteobacteria via horizontal gene transfer.
12
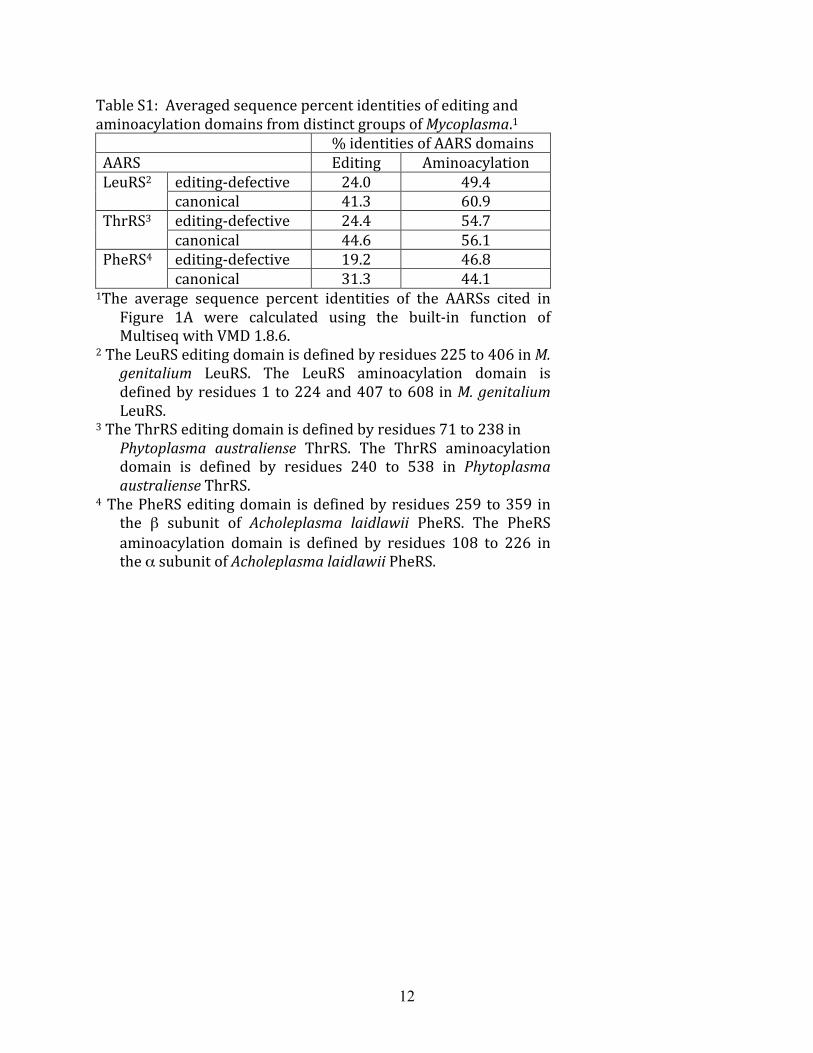
Table S1: Averaged sequence percent identities of editing and aminoacylation domains from distinct groups of Mycoplasma.1 % identities of AARS domains AARS Editing Aminoacylation
1The average sequence percent identities of the AARSs cited in Figure 1A were calculated using the built‐in function of Multiseq with VMD 1.8.6.
2 The LeuRS editing domain is defined by residues 225 to 406 in M. genitalium LeuRS. The LeuRS aminoacylation domain is defined by residues 1 to 224 and 407 to 608 in M. genitalium LeuRS.
3 The ThrRS editing domain is defined by residues 71 to 238 in Phytoplasma australiense ThrRS. The ThrRS aminoacylation domain is defined by residues 240 to 538 in Phytoplasma australiense ThrRS.
4 The PheRS editing domain is defined by residues 259 to 359 in the β subunit of Acholeplasma laidlawii PheRS. The PheRS aminoacylation domain is defined by residues 108 to 226 in the α subunit of Acholeplasma laidlawii PheRS.
13
Table S2: Apparent Kinetic Parameters for Amino Acid Activation by M. mobile LeuRS.
Leu 0.072 ± 0.025 11.7 ± 4.2 1.6×102 1 Ile 6.0 ± 1.7 4.8 ± 1.3 0.80 1/200 Val 28.9 ± 5.9 7.1 ± 1.1 0.25 1/640
Met 13.3 ± 2.7 3.7 ± 0.9 0.28 1/575
14
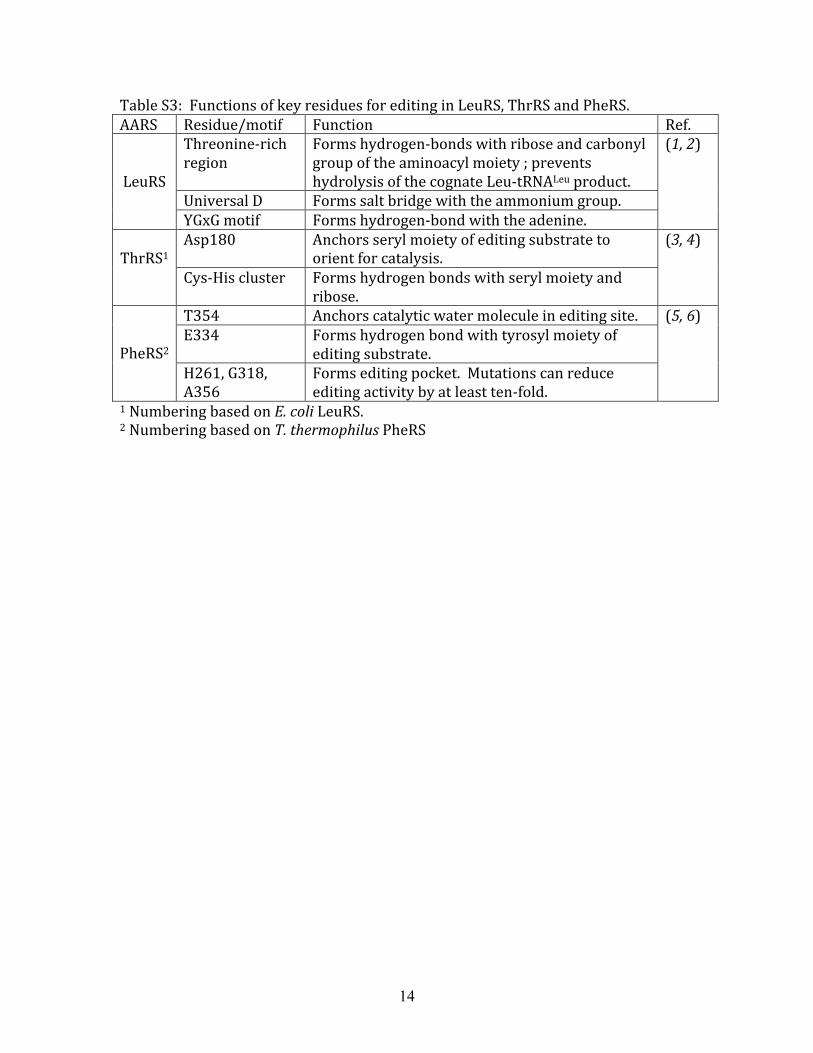
Table S3: Functions of key residues for editing in LeuRS, ThrRS and PheRS. AARS Residue/motif Function Ref.
Threonine‐rich region
Forms hydrogen‐bonds with ribose and carbonyl group of the aminoacyl moiety ; prevents hydrolysis of the cognate Leu‐tRNALeu product.
Universal D Forms salt bridge with the ammonium group.
LeuRS
YGxG motif Forms hydrogen‐bond with the adenine.
(1, 2)
Asp180 Anchors seryl moiety of editing substrate to orient for catalysis.
ThrRS1
Cys‐His cluster Forms hydrogen bonds with seryl moiety and ribose.
(3, 4)
T354 Anchors catalytic water molecule in editing site. E334 Forms hydrogen bond with tyrosyl moiety of
editing substrate.
PheRS2 H261, G318, A356
Forms editing pocket. Mutations can reduce editing activity by at least ten‐fold.
(5, 6)
1 Numbering based on E. coli LeuRS. 2 Numbering based on T. thermophilus PheRS
15
SI text Expression level of LeuRS The expression level of M. mobile LeuRS is ~17 nmol per gram of E. coli cells. The expression level of E. coli LeuRS is ~22 nmol per gram of E. coli cells.
References 1. R. S. Mursinna, K. W. Lee, J. M. Briggs, S. A. Martinis, Molecular Dissection of a
Critical Specificity Determinant within the Amino Acid Editing Domain of Leucyl‐tRNA Synthetase. Biochemistry 43, 155 (2003).
2. T. L. Lincecum Jr, M. Tukalo, A. Yaremchuk, R. S. Mursinna, A. M. Williams, B. S. Sproat, W. Van Den Eynde, A. Link, S. Van Calenbergh, M. Grotli, S. A. Martinis, S. Cusack, Structural and Mechanistic Basis of Pre‐ and Posttransfer Editing by Leucyl‐tRNA Synthetase. Mol. Cell 11, 951 (2003).
3. A.‐C. Dock‐Bregeon, R. Sankaranarayanan, P. Romby, J. Caillet, M. Springer, B. Rees, C. S. Francklyn, C. Ehresmann, D. Moras, Transfer RNA‐Mediated Editing in Threonyl‐tRNA Synthetase: The Class II Solution to the Double Discrimination Problem. Cell 103, 877 (2000).
4. A.‐C. Dock‐Bregeon, B. Rees, A. Torres‐Larios, G. Bey, J. Caillet, D. Moras, Achieving Error‐Free Translation: The Mechanism of Proofreading of Threonyl‐tRNA Synthetase at Atomic Resolution. Mol. Cell 16, 375 (2004).
5. H. Roy, J. Ling, M. Irnov, M. Ibba, Post‐transfer Editing in vitro and in vivo by the β Subunit of Phenylalanyl‐tRNA Synthetase. EMBO J. 23, 4639 (2004).
6. J. Ling, H. Roy, M. Ibba, Mechanism of tRNA‐Dependent Editing in Translational Quality Control. Proc. Natl. Acad. Sci. 104, 72 (2007).