Page 1
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8061
ISSN: 0975-766X
CODEN: IJPTFI
Available Online through Research Article
www.ijptonline.com NEW PYRIMIDINE DERIVATIVES: SYNTHESIS, ANTITUMOR AND
ANTIOXIDANT EVALUATION Eman R. Kotb
1, Eman M.H. Morsy
1, Manal M. Anwar
2*, Eman M. Mohi El-Deen
2, Hanem , M. Awad
3
1Photochemistry Department, National Research Centre, Dokki, Giza, Cairo, Egypt.
2Therapeutical Chemistry Department, National Research Centre, Dokki, Giza, Cairo, Egypt.
3Department of Tanning Materials and Leather Technology, National Research Centre, Dokki, Cairo, Egypt.
Email: [email protected]
Received on 15-06-2015 Accepted on 07-07-2015
Abstract:
This study represents the synthesis of new derivatives of pyrimidine nucleus fused or conjugated with various
heteroaryl ring systems. Some of the new compounds were evaluated as antitumor and antioxidant agents. The results
indicated that the derivatives 9, 5b produced approximate equipotent antitumor activity to that of doxorubicin (%
growth inhibition; 75-56% respectively) against human liver (HepG2) and colorectal (HCT-116) carcinoma cell lines.
On the other hand, the antioxidant evaluation demonstrated strong scavenging activity of the tested derivatives
against the DPPH radicals. They exhibited more potency than rutin and ascorbic acid of IC50; 13.9-33.4 µM.
Keywords: thiopyrimidine, thiazolo[3,2-a]pyrimidine, antitumor, antioxidant activity.
Introduction
Pyrimidine heterocyclic core has a great value in medicinal chemistry since it comprises the base for thiamine, uracil
and cytosine nitrogen bases which are the building blocks of the nucleic acids [1,2]. Furthermore, pyrimidine
derivatives have registered their importance in the development of various pharmaceuticals of broad spectra of
therapeutical activities such as: anti-microbial [3], anti-viral, anti- HIV [4,5], anti-tubercular [6,7], anti-malarial [8,9],
analgesic, anti-inflammatory [10,11], diuretic [12], cardiovascular [13,14], hypnotic for the nervous system [15,16]
and antioxidant [17,18].
Other pyrimidine derivatives are considered as antagonists of calcium-sensing receptor [19] and the human A2A
adenosine receptor [20]. In addition, various drugs containing pyrimidine nucleus were synthesized and used as
anticancer agents like 5-Fluorouracil (5-FU), Tegafur and Thioguanine [21] (Fig. 1).
Page 2
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8062
Fig. (1): Pyrimidine derivatives as anticancer agents.
Moreover, it was documented that many fused pyrimidine analogues were reported to be inhibitors of tyrosine kinase
and cyclin-dependent kinases, which are involved in mediating the transmission of mitogenic signals and numerous
other cellular events, including, cell proliferation, migration, differentiation, metabolism, and immune responses
[22,23].
Cancer is one of the most dreadful diseases in the world. Despite the great development of the basic and clinical
research, which increased the cure rates for a number of malignancies, cancer remains the second leading cause of
human mortality in developing and developed countries. Although chemotherapy is the mainstay of cancer therapy,
the use of available chemotherapeutics is often limited mainly due to the undesirable side effects. Also, drug
resistance constitutes lack of response to many chemically and mechanically unrelated anticancer agents by cancer
cells. It is one of the main reasons for failure of chemotherapy and recurrence of the disease or even death may occur.
Therefore, the development of new drugs with more effective treatment strategies for cancer with well-defined
pharmacokinetic properties always remains an important and challenging goal in medicinal chemistry [24-26].
At the same time, reactive oxygen species (ROS), such as superoxide radicals, hydroxyl (OH) radicals and peroxyl
radicals, are natural byproducts of the normal metabolism of oxygen in living organisms with important roles in cell
signaling. However, excessive amounts of ROS may be a primary cause of biomolecular oxidation and may result in
significant damage to cell structure, DNA, protein, and lipid contributing to various diseases, such as cancer, stroke,
diabetes and degenerative processes associated with ageing. Minimizing oxidative damage may be an important
approach to the primary prevention or treatment of these diseases. Antioxidants are important inhibitors of lipid
peroxidation not only for food protection but also as a defense mechanism of living cells against oxidative damage
since they may stop the free-radical formation, or interrupt an oxidizing chain reaction [27-29].
Page 3
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8063
In view of the above facts and in continuation of our previous efforts in the field of synthesis of novel anticancer
chemotherapeutics [30-33], the target in this study was to synthesize new hybrid compounds having the pyrimidine
moiety fused or conjugated with different aromatic/heterocylic/side chains of documented cytotoxic potency against
several cancer cell lines such as: naphthalene, benzenesulfonamide, thiazolidine, isoxzole, pyrazoline, isoindoline,
pyrrole, chloroethoxy ethyl thio and methyl mannich side chains [26,30,34-40]. Some of the new compounds were
selected as representative examples to evaluate their antitumor activity against human liver (HepG-2) and colorectal
(HCT116) carcinoma cell lines using doxorubicin as a reference drug. Moreover, the free-radical scavenging
properties were also screened for the same derivatives using rutin and vitamin C as reference standards.
Materials and Methods
Chemistry
Regents were purchased from Acros (Geel, Belgium) and Aldrich (St. Louis, MO, USA) and were used without
purification. Melting points were determined by open capillary tubes on an electrical melting point apparatus and are
uncorrected. Nuclear Magnetic Resonance (1H NMR and
13C NMR) spectra were taken on Varian Mercury (500
MHz) spectrometer (Varian, UK) at National Research Centre (NRC), Cairo, Egypt. Chemical shifts (δ) were
expressed in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard. The splitting pattern
abbreviations are as follows: s, singlet; br, broad singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet. The
exchangeable protons (OH and NH) were verified by D2O exchange. Mass spectra were recorded at 70 eV on EI Ms-
QP 1000 EX (Shimadzu, Japan), at the Faculty of Science, Cairo University, Egypt, presented as m/z. IR spectra were
recorded on Perkin-Elmer 1650 spectrophotometer, at NRC by means of KBr pellets of the compounds. Elemental
analyses were carried out using Vario El-Mentar apparatus (Shimadzu, Japan) at NRC. The values found were within
± 0.4% of the theoretical values. The steps of the chemical reactions and purity of the synthesized products were
confirmed on precoated thin layer chromatographic plates (silica gel 60 F254, Merck) using a mixture of chloroform
and methanol (5 μ 1, v/v) as an eluent. The chromatograms were visualized under iodine vapor/UV light at β54 nm
General procedure for the synthesis of compounds 1a,b:
A mixture of urea and/or thiourea (0.01 mol), 1-naphthalenaldehyde (1.5 g; 0.01 mol) and acetylacetone (1.0 g; 0.01
mol) in ethanolic KOH solution (2 g KOH in 100 mL ethanol) was refluxed for 8h. The reaction solution was allowed
to cool and the resultant precipitate was collected by filtration, washed with water, dried and recrystallized from
acetic acid to give the corresponding derivatives 1a,b.
Page 4
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8064
1-(6-Methyl-4-(naphthalen-1-yl)-2-oxo-1,2,3,4-tetrahydropyrimidin-5-yl)ethanone (1a)
Yield: 80%, mp. 210-202 °C, IR (KBr) υ (cm-1): 3250, 3165 (2NH), 1730, 1703 (2C=O).
1H NMR (DMSO-d6, 500
MHz, δ ppm)μ 1.7γ (s, γH, CH3), 2.35 (s, 3H, -COCH3), 5.15 (s, 1H, pyrimidine- H4), 6.61, 6.65 (2s, 2H, 2NH,
exchangeable with D2O), 7.20-7.85 (m, 7H, Ar-H). 13
C NMR (DMSO-d6, 500 MHz) δ (ppm)μ 17.4 (CH3), 22.5 (-CO-
CH3), 51.1 (pyrimidine-C4), 117.0, 123.9, 125.2, 125.4, 126.0, 126.2, 126.5, 128.3, 132.5, 133.4, 134.0, 150.0 (Ar-C,
pyrimidine-C), 156.0, 178.2, (2C=O). MS (EI) m/z: 280 (M+, 70%). Anal. Calcd. for C17H16N2O2 (280.32): C, 72.84;
H, 5.75; N, 9.99. Found: C, 72.41; H, 5.42; N, 10.21.
1-(6-Methyl-4-(naphthalen-1-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl)ethanone (1b)
Yield: 85%, mp. 210-β1β °C, IR (KBr) υ (cm-1): 3200, 3115 (2NH), 1703 (C=O), 1224 (C=S).
1H NMR (DMSO-d6,
500 MHz, δ ppm)μ 1.70 (s, γH, CH3), 2.32 (s, 3H, -COCH3), 2.25, 2.50 (2s, 2H, 2NH, exchangeable with D2O), 5.10
(s, 1H, pyrimidine-H4), 7.10-7.69 (m, 7H, Ar-H). 13
C NMR (DMSO-d6, 500 MHz) δ (ppm)μ 18.0 (CH3), 22.5 (-CO-
CH3), 51.5 (pyrimidine-C4), 123.9, 125.2, 125.4, 126.0, 126.2, 126.5, 128.3, 132.5, 133.4, 134.0, 150.0 (Ar-C,
pyrimidine-C), 156.0, 196.1 (C=O, C=S). MS (EI) m/z: 296 (M+, 70%). Anal. Calcd. for C17H16N2OS (296.39): C,
68.89; H, 5.44; N, 9.45; S, 10.82. Found: C, 68.41; H, 5.22; N, 10.56; S, 11.13.
General procedure for the synthesis of 6-Acetyl-7-methyl-5-(naphthalen-1-yl)-2H-thiazolo[3,2-a]pyrimidin-
3(5H)-one (2)
The pyrimidine-2-thioxo derivative 1b (2.8 g; 0.01 mol) and bromoacetic acid (1.3 g; 0.01 mol) were dissolved in a
mixture solution of acetic acid (30 mL)/acetic anhydride (15 mL) containing anhydrous sodium acetate (2 g). The
reaction mixture was refluxed for 7h. After the reaction completion, the solution mixture was cooled, poured onto
cold water. The formed product was washed several time with water, filtered, dried and recrystallized from acetic
acid to give the target compound 2.
Yield: 60%, mp. 240-β4β °C, IR (KBr) υ (cm-1): 1730, 1723 (2C=O), 1638 (C=N).
1H NMR (DMSO-d6, 500 MHz, δ
ppm): 1.75 (s, 3H, CH3), 2.33 (s, 3H, -COCH3), 3.78 (s, 2H, CH2, thiazolidine ring), 5.15 (s, 1H, pyrimidine-H4),
7.10-7.77 (m, 7H, Ar-H). 13
C NMR (DMSO-d6, 500 MHz, δ ppm)μ 18.7 (CH3), 22.6 (-CO-CH3), 33.6 (-CH2,
thiazolidine ring), 51.8 (pyrimidine-C4), 117.0, 123.9, 125.5, 125.7, 126.0, 126.7, 127.5, 128.9, 133.5, 133.9, 135.0,
145.3, 150.0 (Ar-C, pyrimidine-C), 157.0, 176.2 (2C=O). MS (EI) m/z: 336 (M+, 70%). Anal. Calcd. for
C19H16N2O2S (336.41): C, 67.84; H, 4.79; N, 8.33; S, 9.53. Found: C, 68.20; H, 4.54; N, 8.72; S, 9.12.
Page 5
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8065
General procedure for the synthesis of (Z)-6-acetyl-2-(4-chlorobenzylidene)-5-methyl-7-(naphthalen-1-yl)-2,7-
dihydro-1H-thiazolo[3,2-a]pyrimidin-1-one (3)
A mixture of the pyrimidine-2-thioxo derivative 1b (2.8 g; 0.01 mol), bromoacetic acid (1.3 g; 0.01 mol), p-
chlorobenzaldehyde (1.4 g; 0.01 mol), anhydrous sodium acetate (1 g) in a mixture of acetic acid (30 mL)/acetic
anhydride (15 mL) was heated under reflux for 12h. The reaction mixture was cooled to room temperature and
poured onto cold water with vigorous stirring. The precipitated solid was filtered under suction, washed with cold
water and recrystallized from acetic acid to obtain the required product 3.
Yield: 70%, mp. 257-β5λ °C, IR (KBr) υ (cm-1): 1745, 1705 (2C=O), 1638 (C=N).
1H NMR (DMSO-d6, 500 MHz,
δ ppm)μ 1.75 (s, γH, CH3), 2.35 (s, 3H, -COCH3), 5.52 (s, 1H, pyrimidine- H4), 7.20 (s, 1H, exocyclic vinylic-H),
7.35-7.79 (m, 11H, Ar-H). MS (EI) m/z: 458 (M+, 70%); 460 (M
+ +2, 23%). Anal. Calcd. for C26H19ClN2O2S
(458.96): C, 68.04; H, 4.17; N, 6.10; S, 6.99. Found: C, 68.31; H, 4.47; N, 5.85; S, 7.35.
General procedure for the synthesis of 6-Acetyl-2,7-dimethyl-5-(naphthalen-1-yl)-2H-thiazolo[3,2-a]pyrimidin-
3(5H)-one (4)
The derivative 1b (2.8 g; 0.01 mol) and 2-bromopropionic acid (1.5 g; 0.01 mol) were dissolved in a mixture solution
of acetic acid (30 mL)/acetic anhydride (15 mL) containing anhydrous sodium acetate. (2 g). The reaction mixture
was refluxed for 7h. Upon the reaction completion, the solution mixture was cooled, poured onto cold water. The
formed precipitate was washed several times with water, filtered, dried and recrystallized from acetic acid to give the
title compound 4.
Yield: 65%, mp. 248-β50 °C, IR (KBr) υ (cm-1): 1740, 1700 (2C=O), 1640 (C=N).
1H NMR (DMSO-d6, 500 MHz, δ
ppm): 1.48, (d, 3H, J= 7.6 Hz, CH3), 1.73 (s, 3H, CH3), 2.32 (s, 3H, -COCH3), 3.70-3.74 (q, 1H, -CH, thiazolidine
ring), 5.60 (s, 1H, pyrimidine-H4), 7.12-7.77 (m, 7H, Ar-H). 13
C NMR (DMSO-d6, 500 MHz, δ ppm)μ 16.5, 17.5,
22.0 (3CH3), 42.6 (-CH, thiazolidine ring), 51.1 (pyrimidine-C4), 123.6, 125.1, 125.7, 126.3, 127.5, 128.5, 132.5,
133.9, 135.4, 143.3, 149.5.0 (Ar-C, pyrimidine-C), 168.0, 171.01 (2C=O). MS (EI) m/z: 350 (M+, 20%). Anal. Calcd.
for C20H18N2O2S (350.43): C, 68.55; H, 5.18; N, 7.99; S, 9.15. Found: C, 68.21; H, 4.88; N, 8.31; S, 9.00.
General procedure for the synthesis of compounds 5a-c:
A mixture of compound 2 (3.31 g; 0.01 mol) and the appropriate monosaccharides namely: D-arabinose, D-glucose
and D-mannose in absolute ethanol in the presence of few drops of glacial acetic acid was refluxed for 6 h. The
Page 6
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8066
reaction mixture was cooled and the formed precipitate was filtered, dried and recrystallized from ethanol to obtain
the target compounds 5a-c, respectively.
(Z)-6-Acetyl-5-methyl-7-(naphthalen-2-yl)-2-((2R,3S,4R)-2,3,4,5-tetrahydroxy pentylidene)-2,7-dihydro-1H-
thiazolo[3,2-a]pyrimidin-1-one (5a)
Yield: 68%; m.p.: 232~234°C. IR (KBr) υ (cm-1): 3529-3442 (br. OH), 1733, 1720 (2C=O), 1640 (C=N).
1H NMR
(DMSO-d6, 500 MHz, δ ppm)μ 1.7γ (s, γH, CH3), 2.30 (s, 3H, -COCH3), 3.30-3.42 (m, 3H, pentylidene side chain),
3.70-3.78 (m, 2H, CH2OH), 4.22-5.00 (m, 4H, 4OH, D2O exchangeable), 5.50 (s, 1H, pyrimidine-H4), 5.82 (m, 1H,
exocyclic vinylic-H), 7.12-7.67 (m, 7H, Ar-H). MS (EI) m/z: 468 (M+, 24%). Anal. Calcd. for C24H24N2O6S (468.52):
C, 61.52; H, 5.16; N, 5.98; S, 6.84. Found: C, 61.42; H, 4.85; N, 5.63; S, 6.40.
(E)-6-acetyl-5-methyl-7-(naphthalen-2-yl)-2-((2S,3R,4R,5R)2,3,4,5,6- pentahydroxy hexylidene)-2,7-dihydro-
1H-thiazolo[3,2-a]pyrimidin-1-one (5b)
Yieldμ 65%; m.p.μ β54~β56°C. IR (KBr) υ (cm-1): 3545-3450 (br. OH), 1729, 1725 (2C=O), 1630 (C=N).
1H NMR
(DMSO-d6, 500 MHz, δ ppm)μ 1.75 (s, 3H, CH3), 2.30 (s, 3H, -COCH3), 3.30-3.42 (m, 4H, hexylidene side chain),
3.81-3.89 (m, 2H, CH2OH), 4.50-5.00 (m, 5H, 5OH, D2O exchangeable), 5.52 (s, 1H, pyrimidine-H4), 5.62 (m, 1H,
exocyclic vinylic-H), 7.35 -7.74 (m, 7H, Ar-H). MS (EI) m/z: 498 (M+, 30%). Anal. Calcd. for C25H26N2O7S
(498.55): C, 60.23; H, 5.26; N, 5.62; S, 6.43. Found: C, 60.62; H, 5.51; N, 5.31; S, 6.10.
(E)-6-acetyl-5-methyl-7-(naphthalen-2-yl)-2-((2S,3S,4S,5S)-2,3,4,5,6-pentahydroxy hexylidene)-2,7-dihydro-
1H-thiazolo[3,2-a]pyrimidin-1-one (5c)
Yieldμ 67%; m.p.μ β5β~β54°C. IR (KBr) υ (cm-1): 3550-3453 (br. OH), 1729, 1723 (2C=O), 1637 (C=N).
1H NMR
(DMSO-d6, 500 MHz, δ ppm)μ 1.75 (s, γH, CH3), 2.25 (s, 3H, -COCH3), 3.27-3.38 (m, 4H, hexylidene side chain),
3.80-3.90 (m, 2H, CH2OH), 4.55-5.07 (m, 5H, 5OH, D2O exchangeable), 5.61 (s, 1H, pyrimidine H-4), 5.60 (m, 1H,
exocyclic vinylic-H), 7.12-7.81 (m, 7H, Ar-H). MS (EI) m/z: 498 (M+, 35%). Anal. Calcd. for C25H26N2O7S (498.55):
C, 60.23; H, 5.26; N, 5.62; S, 6.43. Found: C, 60.62; H, 5.51; N, 5.31; S, 6.10.
General procedure for the synthesis of compounds 6,7:
A mixture of the compound 3 (4.5 g; 0.01 mol) and hydrazine hydrate 98% (0.02 mol) and/or phenyl hydrazine (0.01
mol) in ethanol (30 mL) was refluxed for 7h. The solution was cooled and the formed precipitate was filtered, dried
and recrystallized from dioxane to give the desired products 6,7 respectively.
Page 7
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8067
1-(3-(4-Chlorophenyl)-6-methyl-8-(naphthalen-1-yl)-2,3,3a,8-tetrahydropyrazole [3',4':4,5] thiazolo [3,2-
a]pyrimidin-7-yl)ethanone (6)
Yield: 70%, mp. 240-β4β °C, IR (KBr) υ (cm-1): 3346 (NH), 1705 (C=O), 1640 (C=N).
1H NMR (DMSO-d6, 500
MHz, δ ppm)μ 1.68 (s, γH, CH3), 2.12 (s, 3H, -COCH3), 3.30, 4.42 (2d, J= 6.9 Hz, 2H, pyrazoline-H3, H4), 5.60 (s,
1H, pyrimidine-H4), 7.12-7.72 (m, 11H, Ar-H), 7.72 (s, 1H, NH, exchangeable with D2O). MS (EI) m/z: 472 (M+,
30%), 474 (M+ +2, 9%). Anal. Calcd. for C26H21ClN4OS (472.99): C, 66.02; H, 4.48; N, 11.85; S, 6.78. Found: C,
66.21; H, 4.88; N, 11.51; S, 6.50.
1-(3-(4-chlorophenyl)-6-methyl-8-(naphthalen-1-yl)-2-phenyl-2,8-dihydropyrazolo [3',4':4,5] thiazolo [3,2-a]
pyrimidin-7-yl) ethanone (7)
Yield: 60%, mp. 248-β50 °C, IR (KBr) υ (cm-1): 1703 (C=O), 1638 (C=N).
1H NMR (DMSO-d6, 500 MHz) δ (ppm)μ
1.75 (s, 3H, CH3), 2.41 (s, 3H, -COCH3), 3.30, 4.42 (2d, J= 6.9 Hz, 2H, pyrazoline-H3, H4), 5.23 (s, 1H, pyrimidine-
H4), 7.12-8.00 (m, 16H, Ar-H). MS (EI) m/z: 549 (M+, 35%), 551 (M
+ +2), 12%). Anal. Calcd. for C32H25ClN4OS
(549.09): C, 70.0; H, 4.59; N, 10.20; S, 5.84. Found: C, 70.45; H, 4.78; N, 10.51; S, 6.10.
General procedure for the synthesis of 1-(3-(4-chlorophenyl)-6-methyl-8-(naphthalen-1-yl)-8H-isoxazolo
[3',4':4,5] thiazolo [3,2-a]pyrimidin-7-yl)ethanone (8)
A mixture of compound 3 (4.5 g; 0.01 mol) and hydroxylamine hydrochloride (0.7 g, 0.01 mol) in ethanolic sodium
hydroxide (5%, 20 mL) was refluxed for 10h. The reaction mixture was cooled, poured onto ice/cold water and
acidified by diluted hydrochloric acid. The formed precipitate was filtered, dried and recrystallized from dioxane to
give the target compound 8.
Yield: 75%, mp. 250-β5β °C, IR (KBr) υ (cm-1): 1710 (C=O), 1640 (C=N).
1H NMR (DMSO-d6, 500 MHz, δ ppm)μ
1.81 (s, 3H, CH3), 2.36 (s, 3H, -COCH3), 5.23 (s, 1H, pyrimidine-H4), 7.12-8.00 (m, 11H, Ar-H). MS (EI) m/z: 471
(M +
, 33%), 473 (M +
+2, 10%). Anal. Calcd. for C26H18ClN3O2S (471.96): C, 66.17; H, 3.84; N, 8.90; S, 6.79. Found:
C, 66.51; H, 4.16; N, 9.32; S, 6.47.
General procedure for the synthesis of 2-((5-Acetyl-6-methyl-4-(naphthalen-1-yl)-1,2,3,4-tetrahydropyrimidin-
2-yl)thio)acetic acid (9)
A solution mixture of compound 1b (2.8 g; 0.01 mol) and chloroacetic acid (1.0 g, 0.01 mol) in ethanolic sodium
hydroxide (5%, 20 mL) was refluxed for 4h. After the reaction was completed, the reaction solution was poured onto
Page 8
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8068
cold water and the formed precipitate was filtered, dried and recrystallized from the ethanol to get the title compound
9.
Yield: 60%, mp. 192-1λ4 °C, IR (KBr) υ (cm-1): 3426 (OH), 3230 (NH), 1730, 1710 (2C=O).
1H NMR (DMSO-d6,
500 MHz) δ (ppm)μ 1.7γ (s, γH, CH3), 2.20 (s, 1H, NH, exchangeable with D2O), 2.36 (s, 3H, -COCH3), 3.45 (s, 2H,
CH2), 5.15 (s, 1H, pyrimidine-H4), 7.12-7.77 (m, 7H, Ar-H), 11.40 (s, 1H, OH, exchangeable with D2O). 13
C NMR
(DMSO-d6, 500 MHz, δ ppm)μ 18.5, ββ.7 (βCH3), 31.5 (CH2), 50.5 (pyrimidine-C4), 123.4, 125.8, 126.0, 126.3,
127.5, 128.1, 132.0, 133.7, 135.4, 143.3, 150.1 (Ar-C), 176.0, 195.5 (2C=O). MS (EI) m/z: 354 (M +
, 50%). Anal.
Calcd. for C19H18N2O3S (354.42): C, 64.39; H, 5.12; N, 7.90; S, 9.05. Found: C, 64.12; H, 4.83; N, 7.54; S, 9.31.
General procedure for the synthesis of 1-(2-((2-(2-Chloroethoxy) ethyl) thio)-4-methyl-6-(naphthalen-1-yl)-1,6-
dihydropyrimidin -5-yl) ethanone (10)
The compound 1b (2.8 g; 0.01 mol) was dissolved in ethyl alcohol (20 mL) containing potassium hydroxide (0.56 g;
0.01 mol) and the solution mixture was heated with continuous stirring at 60 °C for 3h. Then, bis(chloroethyl) ether
(1.43 g; 0.01 mol) was added and the reaction mixture was continued heated under reflux for further 3h.
Upon reaction completion, the excess solvent was evaporated under reduced pressure and the obtained residue was
washed with cold water several times, filtered, dried and recrystallized from ethanol to give the desired product 10.
Yield: 62%, mp. 126-1β8 °C, IR (KBr) υ (cm-1): 3206 (NH), 1730 (C=O).
1H NMR (DMSO-d6, 500 MHz, δ ppm)μ
1.65 (s, 3H, CH3), 2.21 (s, 1H, NH, exchangeable with D2O), 2.34 (s, 3H, -COCH3), 2.71 ( t, J= 2.6 Hz, 2H, S-CH2-),
3.50, 3.91 (2t, J= 2.9 Hz, 2.8 Hz, 4H, O(CH2)2), 3.60 (t, J= 2.7 Hz, 2H, Cl-CH2-), 5.15 (s, 1H, pyrimidine-H4), 7.12-
7.97 (m, 7H, Ar-H). MS (EI) m/z: 402 (M +
, 100%), 404 (M +
+2, 30%). Anal. Calcd. for C21H23ClN2O2S (402.94): C,
62.60; H, 5.75; N, 6.95; S, 7.96. Found: C, 62.94; H, 5.83; N, 6.57; S, 7.61.
General procedure for the synthesis of 1-(4-Methyl-6-(naphthalen-1-yl)-2-(piperidin-1-yl)-1,6-
dihydropyrimidin-5-yl)ethanone (11)
A mixture solution of compound 1b (2.8 g; 0.01 mol) and piperidine (0.1 g; 0.01 mol) in methyl alcohol (20 mL) was
refluxed for 5h. The resultant solid was collected by filtration, dried and recrystallized from ethanol to give the target
product 11.
Yield: 60%, mp. 170-17β °C, IR (KBr) υ (cm-1): 3226 (NH), 1703 (C=O), 3059 (CH-Ar), 2941 (CH, alicylic).
1H
NMR (DMSO-d6, 500 MHz, δ ppm)μ 1.55-1.65 (m, 6H, , 3CH2 of piperidine ring), 1.73 (s, 3H, CH3), 2.21 (s, 1H,
NH, exchangeable with D2O), 2.35 (s, 3H, -COCH3), γ.1β (m, 4H, α βCH2 of piperidine ring), 5.15 (s, 1H,
Page 9
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8069
pyrimidine-H4), 7.12-7.75 (m, 7H, Ar-H). 13
C NMR (DMSO-d6, 500 MHz, δ ppm)μ 18.0 (CH3), 22.5 (-COCH3),
23.5, 25.4, 25.9 (3CH2, piperidine ring), 44.4 (-N(CH2)2, piperidine ring), 50.1 (pyrimidine-C4), 111.5, 123.5, 125.6,
126.5, 127.1, 127.5, 128.5, 131.0, 133.2, 134.4, 145.3, 150.6 (Ar-C, pyrimidine-C), 175.0 (C=O). MS (EI) m/z: 347
(M +
, 43%). Anal. Calcd. for C22H25N3O (347.45): C, 76.05; H, 7.25; N, 12.09. Found: C, 76.47; H, 7.51; N, 12.42.
General procedure for the synthesis of 1-(2-Hydrazinyl-4-methyl-6-(naphthalen-1-yl)-1,6-dihydropyrimidin-5-
yl)ethanone (12)
A mixture of compound 1b (2.8 g; 0.01 mol) and hydrazine hydrate 98% (0.10 mL, 0.02 mol) in absolute ethanol (10
mL) was refluxed for 4h. The formed precipitate was filtered, dried and recrystallized from ethanol to give the title
compound 12.
Yield: 60%, mp. 238-β40 °C, IR (KBr) υ (cm-1): 3422, 3245, 3150 (NH2, NH), 1703 (C=O).
1H NMR (DMSO-d6,
500 MHz) δ (ppm)μ 1.75 (s, γH, CH3), 2.25, 2.29 (2s, 2H, 2NH, exchangeable with D2O), 2.32 (s, 3H, -COCH3), 4.51
(s, 2H, NH2), 5.10 (s, 1H, pyrimidine-H4), 7.10-7.80 (m, 7H, Ar-H). 13
C NMR (DMSO-d6, 500 MHz) δ (ppm)μ 17.β
(CH3), 22.9 (-OCH3), 50.6 (C-6, pyrimidine ring), 115.5, 122.5, 124.6, 126.4, 126.9, 127.5, 128.1, 131.7, 132.5,
134.4, 142.5, 151.7 (Ar-C), 175.6 (C=O). MS (EI) m/z: 294 (M +
, 100%). Anal. Calcd. for C17H18N4O (294.35): C,
69.37; H, 6.16; N, 19.03. Found: C, 69.04; H, 5.86; N, 19.43.
General procedure for the synthesis of compounds 13a,b:
To a solution of the hydrazinyl compound 12 (2.9 g; 0.01 mol) in glacial acetic acid (30 mL), phthalic anhydride
and/or dichloromaleic anhydride (0.01 mol) was added. The reaction solution was refluxed for 8h. Upon reaction
completion, the solution was poured onto ice/water and the formed precipitate was filtered, washed with water, dried
and recrystallized from dioxane to obtain the corresponding compounds 13a,b.
2-((5-Acetyl-4-methyl-6-(naphthalen-1-yl)-1,6-dihydropyrimidin-2-yl)amino) isoindoline-1,3-dione (13a)
Yield: 60%, mp. > γ00 °C, IR (KBr) υ (cm-1): 3256, 3216 (2NH), 1730, 1698 (3C=O).
1H NMR (DMSO-d6, 500
MHz, δ ppm)μ 1.70 (s, γH, CH3), 2.25, 3.20 (2s, 2H, 2NH, exchangeable with D2O), 2.32 (s, 3H, -COCH3), 5.10 (s,
1H, pyrimidine-H4), 7.20-8.20 (m, 11H, Ar-H). MS (EI) m/z: 424 (M +
, 28%), 445 (M + +2, 9%). Anal. Calcd. for
C25H20N4O3 (424.45): C, 70.74; H, 4.75; N, 13.20. Found: C, 70.41; H, 4.32; N, 13.17.
1-((5-Acetyl-4-methyl-6-(naphthalen-1-yl)-1,6-dihydropyrimidin-2-yl)amino)-3,4-dichloro-1H-pyrrole-2,5-
dione (13b)
Page 10
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8070
Yield: 69%, mp. 287-β8λ °C, IR (KBr) υ (cm-1): 3220, 3190 (2NH), 1737, 1703 (3C=O).
1H NMR (DMSO-d6, 500
MHz, δ ppm)μ 1.68 (s, γH, CH3), 2.00, 3.20 (2s, 2H, 2NH, exchangeable with D2O), 2.36 (s, 3H, -COCH3), 5.52 (s,
1H, pyrimidine–H4), 7.10-7.80 (m, 7H, Ar-H). MS (EI) m/z: 443 (M +
, 27%). Anal. Calcd. for C21H16Cl2N4O3
(443.28): C, 56.90; H, 3.64; N, 12.64. Found: C, 56.48; H, 3.30; N, 12.32.
General procedure for the synthesis of 1-(2-Chloro-4-methyl-6-(naphthalen-1-yl)-1,6-dihydropyrimidin-5-
yl)ethanone (14)
A suspension of compound 1a (2.8 g, 0.01 mol) and PCl5 (0.5 g) in POCl3 (8 mL) was heated under reflux for 5h on a
water bath. After cooling, the reaction mixture was poured slowly on crushed ice (30 g) then neutralized with NaOH
solution. The solid formed was filtered, washed with cold water and dried to give the chloro derivative 14.
Yield: 75%, mp. 160-16β °C, IR (KBr) υ (cm-1): 3340 (NH), 1705 (C=O).
1H NMR (DMSO-d6, 500 MHz, δ ppm)μ
1.75 (s, 3H, CH3), 2.36 (s, 3H, -COCH3), 2.78 (s, 1H, NH, exchangeable with D2O), 5.22 (s, 1H, pyrimidine-H4),
7.10-7.72 (m, 7H, Ar-H). MS (EI) m/z: 298 (M +
, 100%), 300 (M +
+2, 30%). Anal. Calcd. for C17H15ClN2O (298.77):
C, 68.34; H, 5.06; N, 9.38. Found: C, 68.62; H, 5.37; N, 9.59.
General procedure for the synthesis of compounds 15a,b:
To a solution of the chloro derivative 14 (2.9 g; 0.01 mol) in absolute ethanol (30 mL) containing few drops of
triethylamine, the appropriate sulfonamide derivatives namely: sulfapyridine and/or sulfadiazine (0.01 mol) was
added. The reaction mixture was refluxed for 6h. Upon cooling, the formed solid was filtered, dried and recrystallized
from DMF/ethanol to get the title derivatives 15a,b, respectively.
4-((5-Acetyl-4-methyl-6-(naphthalen-1-yl)-1,6-dihydropyrimidin-2-yl)amino)-N-(pyridin-2-
yl)benzenesulfonamide (15a)
Yield: 65%, mp. 258-β60 °C, IR (KBr) υ (cm-1): 3345, 3339, 3330 (3NH), 1706 (C=O), 1396, 1176 (SO2).
1H NMR
(DMSO-d6, 500 MHz, δ ppm)μ 1.7γ (s, γH, CH3), 2.32 (s, 3H, -COCH3), 2.78, 3.20, 4.50 (3s, 3H, 3NH,
exchangeable with D2O), 5.03 (s, 1H, pyrimidine-H4), 6.80-8.00 (m, 15H, Ar-H). MS (EI) m/z: 511 (M +
, 28%). Anal.
Calcd. for C28H25N5O3S (511.59): C, 65.74; H, 4.93; N, 13.69; S, 6.27. Found: C, 65.38; H, 4.50; N, 13.31; S, 6.01.
4-((5-Acetyl-4-methyl-6-(naphthalen-1-yl)-1,6-dihydropyrimidin-2-yl)amino)-N-(pyrimidin-2-
yl)benzenesulfonamide (15b)
Yield: 65%, mp. 240-β4β °C, IR (KBr) υ (cm-1): 3360, 3345, 3332 (3NH), 1705 (C=O), 1338, 1195 (SO2).
1H NMR
(DMSO-d6, 500 MHz, δ ppm)μ 1.70 (s, γH, CH3), 2.30 (s, 3H, -COCH3), 2.61, 3.32, 4.27 (3s, 3H, 3NH,
Page 11
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8071
exchangeable with D2O), 5.51 (s, 1H, pyrimidine-H4), 6.80-8.50 (m, 14H, Ar-H). MS (EI) m/z: 512 (M +
, 28%). Anal.
Calcd. for C27H24N6O3S (512.58): C, 63.27; H, 4.72; N, 16.40; S, 6.26. Found: C, 62.81; H, 4.43; N, 16.12; S, 6.51.
General procedure for the synthesis of compounds 16a-c:
A solution of paraformaldehyde (0.90 g, 10 mmol) and the appropriate amines namely: N,N-dimethyl amine,
morpholine and/ p-toluidine (0.015 mol) was refluxed in absolute ethanol (20 mL) for 30 min till complete solubility
of para-formaldehyde. Then, a solution of the compound 1b (2.8 g; 0.01 mol) in absolute ethanol (10 mL) was added
to the previous mixture and refluxed for 8h. The product obtained upon cooling the reaction solution was filtered,
dried and recrystallized from ethanol to give the corresponding Mannich bases 16a-c.
1-(1,3-Bis((dimethylamino)methyl)-6-methyl-4-(naphthalen-1-yl)-2-thioxo-1,2,3,4- tetrahydropyrimidin-5-
yl)ethanone (16a)
Yield: 73%, mp. 247-249 °C, IR (KBr) υ (cm-1): 1700 (C=O), 1139 (C=S).
1H NMR (CDCl3-d6, 500 MHz, δ ppm)μ
1.73 (s, 3H, CH3), 2.34 (s, 3H, -COCH3), 2.74, 2.76 (2s, 6H, 6H, -2N(CH3)2), 4.51 (s, 4H, 2CH2), 5.21 (s, 1H,
pyrimidine-H4), 7.10-7.77 (m, 7H, Ar-H). MS (EI) m/z: 410 (M +
, 43%). Anal. Calcd. for C23H30N4OS (410.58): C,
67.28; H, 7.36; N, 13.65; S, 7.81. Found: C, 67.61; H, 7.05; N, 13.81; S, 7.50.
1-(6-Methyl-1,3-bis(morpholinomethyl)-4-(naphthalen-1-yl)-2-thioxo-1,2,3,4-tetra hydropyrimidin-5-
yl)ethanone (16b)
Yield: 70%, mp. 245-247 °C, IR (KBr) υ (cm-1): 1709 (C=O), 1120 (C=S).
1H NMR (CDCl3-d6, 500 MHz, δ ppm)μ
1.75 (s, 3H, CH3), 2.24 (s, 3H, -COCH3), 2.62 (m, 4H, 4H, -2N(CH2)2, morpholine ring), 3.42 (m, 4H, 4H, -
2O(CH2)2, morpholine ring ), 4.54 (s, 4H, 2CH2), 5.25 (s, 1H, pyrimidine-H4), 7.10-7.79 (m, 7H, Ar-H). MS (EI) m/z:
494 (M+, 50%). Anal. Calcd. for C27H34N4O3S (494.65): C, 65.56; H, 6.93; N, 11.33; S, 6.48. Found: C, 65.21; H,
6.63; N, 11.81; S, 6.13.
1-(6-methyl-4-(naphthalen-1-yl)-2-thioxo-1,3-bis((p-tolylamino)methyl)-1,2,3,4-tetrahydro pyrimidin-5-
yl)ethanone (16c)
Yield: 73%, mp. 255-β57 °C, IR (KBr) υ (cm-1): 3365 (2NH), 1705 (C=O), 1135 (C=S).
1H NMR (CDCl3-d6, 500
MHz, δ ppm)μ 1.71 (s, γH, CH3), 2.26 (s, 3H, -COCH3), 2.29, 3.10 (2s, 2H, 2NH, exchangeable with D2O), 2.62 (s,
3H, 3H, -2CH3, p-toulidine ring), 4.50 (s, 4H, 2CH2), 5.53 (s, 1H, pyrimidine-H4), 7.23-7.83 (m, 15H, Ar-H). MS
(EI) m/z: 534 (M+, 46%). Anal. Calcd. for C27H34N4O3S (534.71): C, 74.12; H, 6.41; N, 10.48; S, 6.00. Found: C,
73.81; H, 6.32; N, 10.26; S, 6.47.
Page 12
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8072
Biological Methods
In-vitro antitumor evaluation
HepG-2 (Human liver carcinoma) and HCT116 (human colorectal carcinoma) cell lines were kindly supplied by
Applied Research Sector, VACSERA-Egypt and maintained in RPMI-1640 medium which was supplemented with
10% heat-inactivated FBS, 100U/ml penicillin and 100U/ml streptomycin. The cells were grown at 37°C in a
humidified atmosphere of 5% CO2. All experiments were conducted thrice in triplicate (n = 3). All the values were
represented as means ± SD.
MTT antitumor assay
The cytotoxic
ity activity against HepG2 and HCT-116 cancer cell lines was estimated using the 3-[4,5-dimethyl-2-thiazolyl)-2,5-
diphenyl-2H-tetrazolium bromide (MTT) assay, which is based on the cleavage of the tetrazolium salt by
mitochondrial dehydrogenases in viable cells [41,42]. Cells were dispensed in a 96 well sterile microplate (5 x 104
cells/well), and incubated at 37oC with 100 µM/ml of each tested compound or Doxorubicin (positive control) for 48
h in a serum free medium prior to the MTT assay. After incubation, media were carefully removed, 40 µL of MTT (5
mg/mL) were added to each well and then incubated for an additional 4 h. The purple formazan dye crystals were
solubilized by the addition of 200 µL of acidified isopropanol. The absorbance was measured at 570 nm using a
microplate ELISA reader (Biorad, USA). The relative cell viability was expressed as the mean percentage of viable
cells compared to the untreated control cells.
Statistical analysis
All experiments were conducted in triplicate (n = 3). All the values were represented as mean ± SD. Significant
differences between the means of parameters as well as IC50s were determined by probit analysis using SPSS
software program (SPSS Inc., Chicago, IL).
In-vitro antioxidant activity
1,1-Diphenyl-2-picryl hydrazyl (DPPH) and Roswell Park Memorial Institute (RPMI) 1640 medium were purchased
from Sigma Chem. Co. (St. Louis, MO, USA). Fetal bovine serum (FBS) and fetal calf serum (FCS) were purchased
from Gibco, UK. Dimethyl sulfoxide (DMSO) and methanol were of HPLC grade and all other reagents and
chemicals were of analytical reagent grade. Antioxidant activity of each compound and standards (ascorbic acid and
Page 13
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8073
rutin) was assessed based on the radical scavenging effect of stable DPPH free radical [βλ]. 10 l of each tested
compound or standard (from 0.0 to 100 M) was added to λ0 l of a 100 M methanolic solution of DPPH in a λ6-
well microtitre plate (Sigma-Aldrich Co., St. Louis, MO, US). After incubation in dark at 37°C for 30 min, the
decrease in absorbance of each solution was measured at 520 nm using an ELISA micro plate reader (Model 550,
Bio-Rad Laboratories Inc., California, USA). Absorbance of blank sample containing the same amount of DMSO and
DPPH solution was also prepared and measured. All experiments were carried out in triplicate. The scavenging
potential was compared with a solvent control (0% radical scavenging) and the standard compounds. Radical
scavenging activity was calculated by the following formula:
% Reduction of absorbance = [(AB - AA) / AB] x 100, where: AB – absorbance of blank sample and AA –
absorbance of tested compound (t = 30 min). The concentration of each compound required to scavenge 50% of
DPPH (IC50) was determined as well [43,44].
Results and discussion
Chemistry
The synthetic strategies adopted for constructing the target molecules are illustrated in Schemes 1-3. The structures of
the isolated products were evidenced by their spectral data together with elemental analyses. The starting materials,
6-methyl-4-(naphthalen-1-yl)-2-oxo/thioxo-1,2,3,4-tetrahydropyrimidine derivatives 1a,b were prepared in
quantitative yields by condensation of urea/thiourea with 1-naphthaldehyde and acetyl acetone in refluxing ethanolic
KOH solution based on the previously reported similar methods [45-48]. IR spectra of the products displayed in each
case the absorption bands at the region of 3250-3115 cm-1
due to the (NH) groups and at 1730-1703 cm_1
due to
(2C=O) in case of 1a, while 1b exhibited two bands at 1703 and 1224 cm_1
due to C=O and C=S groups,
respectively. 1H NMR spectra of the two products showed two singlet signals at the region δ 1.7γ-2.35 ppm due to
6H of CH3 and -COCH3 groups and another singlet at δ 5.15 ppm assignable to H-4 of pyrimidine ring, in addition to
the other signals of the molecules that appeared at their expected regions. At the same time, 13
C NMR of 1a,b showed
two signals at δ 17.4, ββ.5 ppm corresponding to CH3 and COCH3, respectively, at δ 156.0, 178.β, 1λ6.1 ppm due to
C=O and C=S groups, besides the other expected signals of the other carbons of the two molecules. Condensation
reaction of the pyrimidine-2-thioxo derivative 1b with bromoacetic acid in refluxing acetic acid/acetic anhydride
solution containing sodium acetate anhydrous led to the formation of the thiazolo[3,2-a]pyrimidine derivative 2. IR
spectrum of compound 2 displayed two absorption bands at 1730, 1723 cm_1
due to the presence of the acetyl and the
Page 14
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8074
lactamic C=O groups. While its 1H NMR spectrum revealed four singlets at δ 1.75, β.γγ, γ.78, 5.15 ppm assigned to
the corresponding methyl, acetyl and CH2, H-5 of thiazolo[3,2-a]pyrimidine residue, in addition to multiplet signals
at δ 7.10-7.77 ppm due to Ar-H. Furthermore, the arylidene analogue 3 has been synthesized in a one pot multi-
component reaction [49,50] involving compound 1b, bromoacetic acid and p-chlorobenzaldehyde in the presence of
anhydrous sodium acetate in a mixture solution of acetic acid/acetic anhydride (Scheme 1). IR spectrum of
compound-3 revealed the presence of two absorption bands at 1745, 1705 cm_1
assignable to 2C=O. Its 1H NMR, in
addition to the expected signals due to the aromatic protons, exhibited four singlet signals at δ 1.75, β.γ5, 5.5β ppm
referred to the protons of methyl, acetyl and pyrimidine H-4, respectively. The Z configuration of the exocyclic C=C
bond was assigned on the basis of 1H NMR spectroscopy according to literature data for analogous 4-thiazolidinones
[51,52]. The methine proton, deshielded by the adjacent C=O, was detected at 7.20 ppm. Similarly, compound 1b
was allowed to react with bromopropionic acid, under the same conditions, in a refluxing solution of acetic
acid/acetic anhydride containing sodium acetate anhydrous to form 2,7-dimethyl thiazolo[3,2-a]pyrimidine analogue
4. 1
H NMR spectrum of compound 4 showed in addition to the expected two methyl and aromatic signals of the
parent thiopyrimidine core, new douplet and quartet signals appeared at δ 1.48 and γ.70 ppm assigned to the methine
and methyl protons of the thiazolidine ring. 13
C NMR spectrum of the same derivative displayed eighteen carbon
signals, the most important signals appeared at δ 16.5, 17.5, ββ.0, 4β.6 representing the carbons of βCH3 of
thiopyrimidine ring and the carbons of CH3 and CH of the thiazolidine ring, respectively. The thiazolo[3,2-
a]pyrimidine derivative 2 was allowed to react with different monosaccharides namely: D-arabinose, D-glucose and
D-mannose in absolute ethanol acidified with acetic acid to give the corresponding derivatives 5a-c. Mass spectra of
the latter derivatives represented the molecular ion peaks which were in agreement with their molecular formulae.
The derivative 3 was a versatile precursor for the synthesis of pyrazoline and isoxazole derivatives 6-8. Thus, upon its
condensation with hydrazine hydrate 98% and phenyl hydrazine in ethanol led to the formation of 2,3,3a,8-tetrahydro
and/or (2-phenyl-2,8-dihydro) pyrazolo [3',4':4,5] thiazolo [3,2-a]pyrimidine derivatives 6, 7, respectively. While,
heating of compound 3 with hydroxylamine hydrochloride in ethanolic sodium hydroxide solution furnished the
corresponding 8H-isoxazolo [3',4':4,5] thiazolo[3,2-a]pyrimidine analogue 8 (scheme 1). IR spectra of the novel
pyrazoline and isoxazole derivatives revealed the presence of an absorption band at 1703-1710 cm_1
due to the acetyl
C=O group and the disappearance of C=O band of the thiazolidine ring, instead a stretching absorption band appeared
at 3346 cm_1
referring to NH group in case of the pyrazoline derivative 6. 1H NMR spectra of compound 6,7 showed
Page 15
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8075
two doublets at the range δ γ.γ0-4.42 corresponding to the two methine protons of the pyrazoline ring besides the
other expected protons that were presented at their exact ranges. Mass spectra of the 6,7,8 exhibited their molecular
ion peaks that confirmed their molecular formulae.
The reaction of 1b with different chloro derivatives such as chloroacetic acid and bis(chloroethyl) ether in ethanolic
potassium hydroxide solution furnished the corresponding compounds 9, 10, respectively. While, condensation
reaction of the thioxo derivative 1b with a secondary nitrogen nucleophile such as piperidine yielded the
corresponding piperidin-1,6-dihydropyrimidine derivative 11. IR spectrum of the thioacetic acid 9 displayed four
absorption bands at 3426, 3230, 1730, 1710 cm_1
due to the presence of OH, NH and 2C=O groups, respectively. At
the same time, 1H NMR spectrum of 9 revealed a new singlet at δ γ.45 ppm contributed to the methylene protons of
the thioacetic acid side chain, besides the two singlet signals of the methyl and acetyl protons of the parent core. 13
C
NMR spectrum of the same compound showed five characteristic signals at δ 18.5, ββ.7, γ1.5, 176.0, 1λ5.5 ppm due
to the carbons of CH3, COCH3, CH2 and 2C=O groups, in addition to the other expected signals that appeared at their
correct places. With regard to compound 10, its 1H NMR spectrum represented four triplets at δ β.71, γ.50, γ.60, γ.λ1
ppm that are referring to the 4CH2 of bis(chloroethyl) ether side chain, in addition to the other expected signals of the
molecule. IR spectrum of compound 11 showed absorption bands at 3226 and 1703 due to NH and C=O cm_1
,
Page 16
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8076
respectively and the disappearance of C=S absorption band. Also, its 1H NMR spectrum exhibited, besides the
expected methyl and aromatic signals, two new multiplet signals at δ 1.55-1.65 ppm due to , CH2 and at δ γ.1β
contributing to α βCH2 groups of piperidine ring.
Hydrazinolysis of 1b was performed by its heating with excess hydrazine hydrate in ethyl alcohol to achieve the
hydrazinyl derivative 12, which was used as a precursor for condensation with different acid anhydride derivatives
such as phthalic anhydride and/or dichloromaleic anhydride in acetic acid to obtain isoindoline 1,3-dione and
dichloro-1H-pyrrole-2,5-dione derivatives 13a,b, respectively (scheme 2). IR spectra of the obtained compounds
13a,b displayed characteristic peaks at the regions 3220-3190 and 1737-1698 cm_1
contributing to NH, C=O groups,
respectively. Mass spectra of the compounds 12, 13a,b revealed their molecular ion peaks at m/z: 294, 424 and 443
respectively which confirm their molecular formulae.
Page 17
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8077
Since the chloropyrimidine moiety is a versatile precursor for the preparation of various nitrogen bridge heterocyclic
compounds, the 2-oxopyrimidine derivative 1a was heated with PCl5 /POCl3 for 5h on a water bath for its conversion
to 2-chloropyrimidine derivative 14. Nucleophilic displacement of chlorine atom was carried out by the treatment of
the chloro derivative 14 with different sulfa drugs namely: sulfapyridine and sulfadiazine to get the corresponding
sulfonamide derivatives 15a,b. IR spectra of 15a,b revealed different absorption bands at the ranges 3360-3330
(NH), 1706 (C=O) and 1396-1338, 1195-1176 (SO2). Mass spectra of the same compounds represented their
molecular ion peaks at m/z: 511 and 512, respectively. Moreover, the compound 1b was allowed to react with
paraformaldehyde and different secondary amines such as N,N-dimethylamine, morpholine and p-toluidine to obtain
the corresponding Mannich bases 16a-c (scheme 3). IR spectra of the mannich bases 16a,b revealed the
disappearance of NH bands of the parent molecules, while the absorption peaks of the acetyl C=O groups appeared at
their expected ranges. Also, 1H NMR spectra were in agreement with the proposed structures of the gained products.
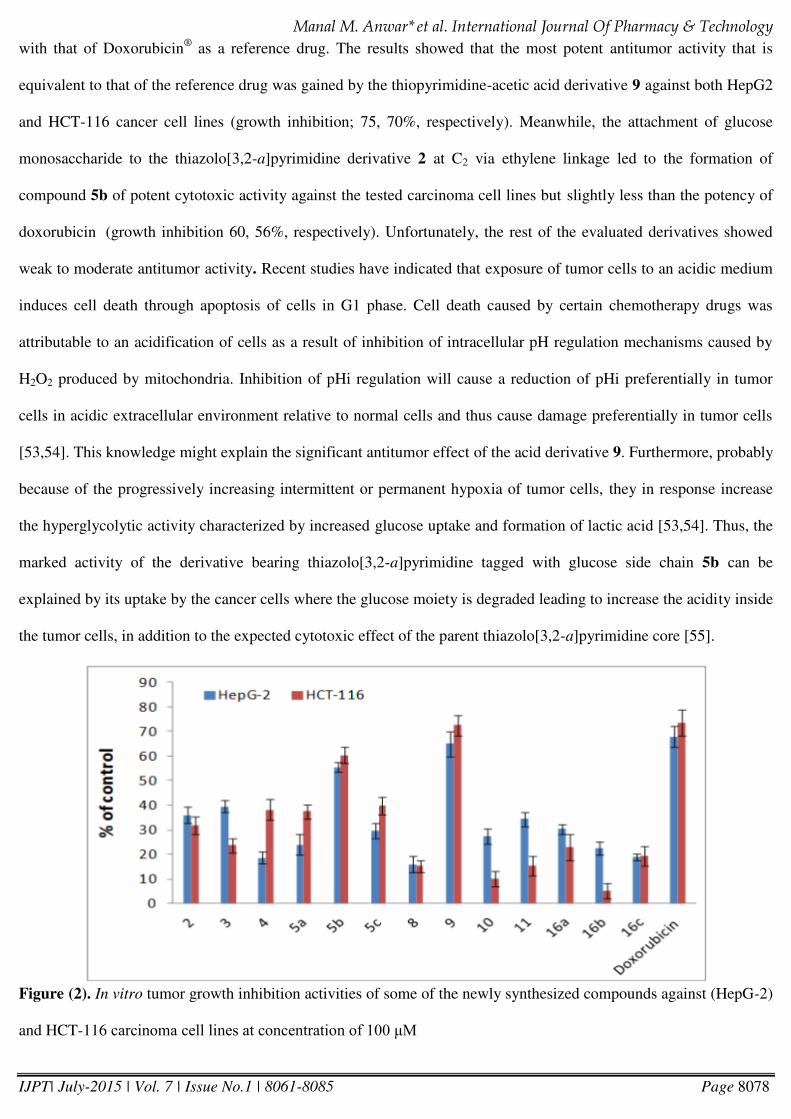
In vitro antitumor Activity
Thirteen of the newly synthesized compounds were selected as representative examples to examine their in vitro
antitumor activities against human liver (HepG2) and colorectal (HCT-116) cancer cell lines using MTT assay. The
examined derivatives were 2, 3, 4, 5a-c, 8, 9, 10, 11, 16a-c. The percentage of the intact cells was measured and
compared to the control (Figure 2). The activities of the derivatives against the tested carcinoma cells were compared
Page 18
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8078
with that of Doxorubicin®
as a reference drug. The results showed that the most potent antitumor activity that is
equivalent to that of the reference drug was gained by the thiopyrimidine-acetic acid derivative 9 against both HepG2
and HCT-116 cancer cell lines (growth inhibition; 75, 70%, respectively). Meanwhile, the attachment of glucose
monosaccharide to the thiazolo[3,2-a]pyrimidine derivative 2 at C2 via ethylene linkage led to the formation of
compound 5b of potent cytotoxic activity against the tested carcinoma cell lines but slightly less than the potency of
doxorubicin (growth inhibition 60, 56%, respectively). Unfortunately, the rest of the evaluated derivatives showed
weak to moderate antitumor activity. Recent studies have indicated that exposure of tumor cells to an acidic medium
induces cell death through apoptosis of cells in G1 phase. Cell death caused by certain chemotherapy drugs was
attributable to an acidification of cells as a result of inhibition of intracellular pH regulation mechanisms caused by
H2O2 produced by mitochondria. Inhibition of pHi regulation will cause a reduction of pHi preferentially in tumor
cells in acidic extracellular environment relative to normal cells and thus cause damage preferentially in tumor cells
[53,54]. This knowledge might explain the significant antitumor effect of the acid derivative 9. Furthermore, probably
because of the progressively increasing intermittent or permanent hypoxia of tumor cells, they in response increase
the hyperglycolytic activity characterized by increased glucose uptake and formation of lactic acid [53,54]. Thus, the
marked activity of the derivative bearing thiazolo[3,2-a]pyrimidine tagged with glucose side chain 5b can be
explained by its uptake by the cancer cells where the glucose moiety is degraded leading to increase the acidity inside
the tumor cells, in addition to the expected cytotoxic effect of the parent thiazolo[3,2-a]pyrimidine core [55].
Figure (2). In vitro tumor growth inhibition activities of some of the newly synthesized compounds against (HepG-2)
and HCT-116 carcinoma cell lines at concentration of 100 M
Page 19
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8079
Furthermore, IC50 of compounds 5b and 9 were measured against both human liver (HepG2) and colorectal (HCT-
116) cancer cell lines. Table (2) showed IC50 of 88.9 and 85.4 µM corresponding to the compounds 5b and 9 against
HepG2 cancer cells. While, both compounds showed IC50 of 79.5 and 89.3µM against the colorectal cancer cell,
respectively.
Table (1). IC50 (µM) of compounds 5b, 9 against (HepG-2) and HCT-116 carcinoma cell lines.
In vitro Antioxidant Activity
Also, this study deals with the investigation of in vitro antioxidant activities against DPPH of the same thirteen
compounds, 2, 3, 4, 5a-c, 8, 9, 10, 11, 16a-c (Figure 3). The results obtained revealed that all of the tested
compounds showed dose dependent DPPH inhibition activities, which were reflected by their IC50 values as
summarized in table (2). The activities of the compounds appeared in the following order: 16a > 3 > 10 > rutin > 4 >
5b > 5c > 2> 16b > 16c > 5a > 8 > 11 > 9 > vit C.
Comparing the activity of the thirteen compounds to the standard antioxidants and well known potent DPPH
inhibitors (rutin and vit C); It is obvious that all the newly synthesized compounds showed better or comparable
scavenging properties to that of rutin and vitamin C.
Figure (3). Antioxidant activity of the tested derivatives using DPPH.
Compound HepG-2 HCT-116
IC50
5b 88.9 79.5
9 85.4 89.3
Page 20
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8080
Table-2: The Antioxidant activities (IC50; µM) of the tested compounds.
Compound IC50
2 24.6
3 17.3
4 23.1
5a 30. 7
5b 24.3
5c 24.5
8 32.6
9 33.4
10 20.5
11 32.9
16a 13.9
16b 25.1
16c 27.9
Rutin 21.7
Vit C 39.3
Conclusion
This study reveals successful synthesis of new derivatives of pyrimidine either fused or conjugated with different
heteroaryl ring systems. Anticancer evaluation of some of the synthesized derivatives exhibited that compounds 9, 5b
appeared to be the most active cytotoxic compounds against human liver (HepG2) and colorectal (HCT-116) cancer
cell lines. They produced growth inhibition of the tested cancer cells equal to or slightly less than that of the standard
drug doxorubicin. It is noteworthy that the antioxidant examination of these derivatives exhibited that all of them are
potent antioxidants when comparing with rutin and vitamin C as reference drugs. Thus, further biological studies
could be carried out for these compounds as they can be considered as templates for antioxidant supplements.
Page 21
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8081
Acknowledgements
The authors are grateful to National Research Centre/ Cairo/ Egypt for providing infrastructures and other facilities
for carrying out this study.
Conflict of interests
The authors confirm that this article content has no conflict of interest.
References
1. Hussain MMM, Bhat KI, Revanasiddappab BC, Bharathi DR. Synthesis and Biological Evaluation of Some
Novel 2-Mercapto Pyrimidines. Int J Pharm Pharm Sci 2013; 5 (2): 471-473.
2. Mohamed MS, Awad SM, Zohny YM, Mohamed ZM. New Thiopyrimidine Derivatives of Expected Anti-
inflammatory Activity. Pharmacophore 2012; 3 (1): 62-75.
3. Nag S, Pathak R, Kumar M, Shukla PK, Batra S. Synthesis and antibacterial evaluation of ureides of Baylis–
Hillman derivatives. Bioorg & Med Chem. 2006; 16: 3824–3828.
4. Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. Broad-spectrum antiviral that interferes with de novo
pyrimidine biosynthesis. Proc Natl Acad Sci U S A 2011; 108 (14):5777–5782.
5. Clercq ED, Holý Somnath Naga, A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. NAT REV
DRUG DISCOV 2005; 4:928-940.
6. Trivedi AR, Dodiya DK, Ravat NR, Shah VH. Synthesis and biological evaluation of some new pyrimidines via
a novel chalcone series. ARKIVOC 2008; (xi): 131-141.
7. Trivedi AR, Siddiqui AB, Shah VH. Design, synthesis, characterization and antitubercular activity of some 2-
heterocycle-substituted phenothiazines. ARKIVOC 2008; (ii): 210-217.
8. Agarwal A, Srivastava K, Puri SK, Chauhan PMS. Synthesis of 2,4,6-trisubstituted pyrimidines as antimalarial
agents. Bioorg Med Chem 2005; 13 (15), 4645–4650.
9. Agarwal A, Srivastava K, Puri SK, Sinha S, Chauhan PMS. A small library of trisubstituted pyrimidines as
antimalarial and antitubercular agents. Bioorg. Med. Chem. Lett. 2005; 15 (23): 5218–5221.
10. Sondhi SM, Dinodia M, Rani R, Shukla R, Raghubir R. Synthesis, anti-inflammatory and analgesic activity
evaluation of some pyrimidine derivatives. Indian J. Chem 2009; 49b: 273-281.
11. EL-gazzar ABA, Hussein HAR, Hafez HN. Synthesis and biological evaluation of thieno[2,3-d]pyrimidine
derivatives for anti-inflammatory, analgesic and ulcerogenic activity. Acta Pharm. 2007; 57:395–411.
Page 22
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8082
12. Majeed J, Shaharyar M. Synthesis and in vivo diuretic activity of some novel pyrimidine derivatives. J. Enzyme
Inhib. Med. Chem 2011; 26 (6): 819-826.
13. Morris GW, Iams TA, Slepchenko KG, McKee EE. Origin of pyrimidine deoxyribonucleotide pools in perfused
rat heartμ implications for γ′-azido-γ′-deoxythymidine-dependent cardiotoxicity. Biochem. J. 2009; 422: 513–
520.
14. Reading SA, Earley S, Waldron BJ, Welsh DG, Brayden JE. TRPC3 mediates pyrimidine receptor-induced
depolarization of cerebral arteries. Am. J. Physiol. Heart Circ. Physiol 2005; 288 (5): H2055-H2061.
15. Jain KS, Chitre TS, Miniyar PB, Kathiravan MK, Bendre VS, Veer VS, Shahane SR, Shishoo .J. Biological and
medicinal significance of pyrimidines. Curr. Sci 2006; 90 (6):793-803.
16. Mishra R, Tomar I. Pyrimidine: The Molecule of Diverse Biological and Medicinal Importance. IJPSR 2011;
2(4):758-771.
17. Abu-Hashem AA, El-Shehry
MF, Badria FA. Design and synthesis of novel thiophene- carbohydrazide,
thienopyrazole and thienopyrimidine derivatives as antioxidant and antitumor agents. Acta Pharm 2010; 60:
311–323.
18. Padmaja A, Payani T, Reddy GD. Synthesis, antimicrobial and antioxidant activities of substituted pyrazoles,
isoxazoles, pyrimidine and thioxopyrimidine derivatives. Eur J Med Chem 2009; 44 (11): 4557–4566.
19. Yan W, Ruan Z, Wang Y, Van Kirk K, Zhengping M, Brian JA. Discovery and Structure−Activity
Relationships of Trisubstituted Pyrimidines/Pyridines as Novel Calcium-Sensing Receptor Antagonists. J Med
Chem 2009; 52 (4): 1204–1208.
20. Gillespie RJ, Bamford S J, Botting R, Comer M, Denny S, Gaur S. Antagonists of the Human A2A Adenosine
Receptor. 4. Design, Synthesis, and Preclinical Evaluation of 7-Aryltriazolo[4,5-d]pyrimidines, J Me. Chem
2009; 52 (1): 33–47.
21. Ahmed SA, Naji TS, Mohammad FI. Synthesis, Characterization and Cytotoxic Activity of Some Pyrimidine
Derivatives. J N U S 2013; 16 (2): 84-92.
22. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009; 9:
28–39.
Page 23
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8083
23. Fathalla, O.A.M.; Ismail, M.A.H.; Anwar M.M.; Abouzid, K.A.M.; Ramadan, A. A.K. Novel 2-thiopyrimidine
derivatives as CDK2 inhibitors: molecular modeling, synthesis, and anti-tumor activity evaluation. Med Chem
Res. 2013; 22: 659–673.
24. Nasr T, Bondock S, Youns M. Anticancer activity of new coumarin substituted hydrazide-hydrazone derivatives.
Eur J Med Chem 2014; 76: 539-548.
25. Sashidhara V.K, Kumar A, Kumar M, Sarkar J, Sinha S. Synthesis and in vitro evaluation of novel coumarin–
chalcone hybrids as potential anticancer agents. Bioorg Med Chem Lett 2010; 20:7205–7211.
26. Hassan GS. Synthesis and antitumor activity of certain new thiazolo [2,3-b]quinazoline and thiazolo[3,2-
a]pyrimidine analogs Med Chem Res 2014; 23: 388-401.
27. De Oliveira, A.M.F.; Pinheiro, L.S.; Pereira, C.K.S; Matias, W.N.; Gomes, R.A.; Chaves, O.S. Total Phenolic
Content and Antioxidant Activity of Some Malvaceae Family Species. Antioxidants 2012; 1:33-43.
28. Abdalla MY. Glutathione as Potential Target for Cancer Therapy; More or Less is Good?. Jordan j. biol. sci.
2011; 4 (3):119 – 124.
29. Hamdy NA, Anwar MM, Abu-zied KM, Awad HM. Synthesis, Tumor Inhibitory, and Antioxidant Activity of
New Polyfunctionally 2-Substituted 5,6,7,8-Tetrahydronaphthalene Derivatives Containing Pyridine,
Thioxopyridine and Pyrazolopyridine Moieties. Acta Poloniae Pharmaceutica n Drug Research 2013; 70 (6),
987-1001.
30. Kotb ER, Anwar MA, Soliman MS, Salama MA. Synthesis and Reactions of Some Novel Quinoxalines for
Anticancer Evaluation. Phosphorus, Sulfur, and Silicon and the Related Elements 2007; 182:1119–1130.
31. Amin KM, Anwar MM, Syam YM, Khedr M, Kamel MM, Kassem EMM. A Novel Class of Substituted
Spiro[Quinazoline-2,1’-Cyclohexane] Derivatives as Effective PARP-1 Inhibitors: Molecular Modeling,
Synthesis, Cytotoxic and Enzyme Assay Evaluation. Acta Poloniae Pharmaceutica-Drug Research 2013; 70
(4):687-708.
32. Mohi El-Deen EM, Anwar MM, Kotb ER. Synthesis and Cytotoxic Evaluation of New 6,7,8,9-
Tetrahydropyrido[3',2':4,5]thieno[3,2-b] quinoline Derivatives. RJPBCS 2014; 5(6):1535-1549.
33. Amin KM, Anwar MM, Kamel M, Kassem EMM, Syam YM, EL-seginy SA. Synthesis, Cytotoxic Evaluation
and Molecular Docking Study of Novel Quinazoline Derivatives as PARP-1 Inhibitors. Acta Poloniae
Pharmaceutica-Drug Research 2013; 70 (5):833-849.
34. Ghorab MM, Bashandy MS, Alsaid MS. Novel thiophene derivatives with sulfonamide, isoxazole,
benzothiazole, quinoline and anthracene moieties as potential anticancer agents. Acta Pharm 2014; 64:419–431.
35. Youssef MM, Amin MA. Microwave Assisted Synthesis of Some New Thiazolopyrimidine,
Thiazolodipyrimidine and Thiazolopyrimidothiazolopyrimidine Derivatives with Potential Antioxidant and
Antimicrobial Activity. Molecules 2012; 17: 9652-9667.
Page 24
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8084
36. Ghorab MM, El-Gazzar MG, Alsaid MS. Synthesis, Characterization and Anti-Breast Cancer Activity of New 4-
Aminoantipyrine-Based Heterocycles. Int J Mol Sci 2014; 15: 7539-7553.
37. Nofal ZM, Soliman EA, Abd El-Karim SS, El-Zahar MI, Srour AM, Sethumadhavan S, Maher TJ. Synthesis of
Some New Benzimidazole–Thiazole Derivatives as Anticancer Agents. J. Heterocyclic Chem 2014, 51:1797.
38. Sabastiyan A, Suvaikin MY. Synthesis, characterization and antimicrobial activity of 2-
(dimethylaminomethyl)isoindoline-1,3-dione and its cobalt(II) and nickel(II) complexes. Adv App. Sci Res 2012;
3 (1):45-50.
39. Jin YZ, Fu DX, Ma N, Li ZC, Liu QH, Xiao L, Zhang RH. Synthesis and Biological Evaluation of 3-Substituted-
indolin-2-one Derivatives Containing Chloropyrrole Moieties. Molecules 2011; 16: 9368-9385.
40. Bala S, Sharma N, Kajal A, Kamboj S, Saini V. Mannich Bases: An Important Pharmacophore in Present
Scenario. Int J Med Chem 2014; 2014:1-15.
41. Awad HM, Abd-Alla HI, Mahmoud KH, El-Toumy SA. In vitro anti-nitrosative, antioxidant, and cytotoxicity
activities of plant flavonoids: a comparative study. Med Chem Res 2014; 23 (7):3298-3307.
42. Almajhdi FN, Fouad H, Khali KA, Awad HM, Mohamed SHS.; Elsarnagawy T, Albarrag AM, Al-Jassi, FF,
Abdo HS. In-vitro anticancer and antimicrobial activities of PLGA/silver nanofibers composites prepared by
electrospinning. J Mater Sci Mater Med 2014; 25:1–9.
43. Michael HN, Awad HM, El-Sayed NH, Pare PW. Chemical and antioxidant investigations: Norfolk pine needles
(Araucaria excelsa). Pharm Biol 2010; 48 (5):534-538.
44. Soliman, H.A.; Yousif, M.N.M.; Said, M.M.; Hassan, N.A.; Ali, M.M.; Awad, H.M.; Abdel-
Megeid, F.M.E. Synthesis of novel 1,6-naphthyridines, pyrano[3,2-c]pyridines and pyrido[4,3-d]pyrimidines
derived from 2,2,6,6-tetramethylpiperidin-4-one for in vitro anticancer and antioxidant evaluation. Der Pharma
Chemica, 2014; 6(3):394-410.
45. Narsaiah AV, Basak AK, Nagaiah K. Cadmium chloride: an efficient catalyst for one-pot synthesis of 3,4-
dihydropyrimidine-2(1H)-ones. Synthesis 2004; 8:1253-1256.
46. Srinivas KVNS. Iodine catalyzed one-pot synthesis of 3, 4-dihydropyrimidine-2(1H)-ones and thiones: a simple
and efficient procedure for the Biginelli reaction. Synthesis 2004; 13:2091-2093.
47. De SK, Gibbs RA. Ruthenium (III) chloride-catalyzed one-pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones
under solvent-free conditions. Synthesis 2005; 11:1748-1750.
48. Peng, J.; Deng, Y. Ionic liquids catalyzed Biginelli reaction under solvent-free conditions. Tetrahedron Lett
2001; 42:5917-5919.
49. Solanki MJ, Chavda MM. Synthesis and Antimicrobial Activity of Some Novel Derivatives of Thiazolo[2,3-
b]dihydropyrimidine Containing 4-Pyrazole Moiety. Rasayan J Chem 2011; 4 (3):605-608.
50. Yossef MM, Mohamed SF, Kotb ER, Salama MA. A novel Synthesis of Some New Pyrimidine,
Thiazolopyrimidine and Pyrazole Derivatives Using Diarylepoxypropanones as Precursors. World J Chem 2009;
4(2):149-156.
51. Vicini P, Geronikaki A, Anastasia K, Incerti M, Zani F. Synthesis and antimicrobial activity of novel 2-
thiazolylimino-5-arylidene-thiazolidinones. Bioorg Med Chem 2006; 14:3859-3864.
Page 25
Manal M. Anwar* et al. International Journal Of Pharmacy & Technology
IJPT| July-2015 | Vol. 7 | Issue No.1 | 8061-8085 Page 8085
52. Ottana R, Maccari R, Barreca ML, Bruno G, Rotondo A, Rossi A, Chiricosta G, Di-Paola R., Sautebin L,
Cuzzocrea S, Vigorita MG. 5-Arylidene-2-imino-4-thiazolidinones: Design and synthesis of novel anti-
inflammatory agents. Bioorg Med Chem 2005; 13: 4243-4252.
53. Hirpara JL, Clements MV, Pervaiz A. Intracellular acidification triggered by mitochondrial-derived hydrogen
peroxide is an effector mechanism for drug-induced apoptosis in tumor cells. J Biol Chem 2001; 276: 514–521.
54. Ahmad KS, Iskandar KB, Hirpara JL, Clement MV, Pervaiz S. Hydrogen peroxide-mediated cytosolic
acidification is a signal for mitochondrial translocation of Bax during drug-induced apoptosis of tumor cells.
Cancer Res 2004; 64:7867–7878.
55. Abdo NYM. Synthesis and Antitumor Evaluation of Novel Dihydropyrimidine, Thiazolo[3,2-a]pyrimidine and
Pyrano[2,3-d]pyrimidine Derivatives. Acta Chim. Slov 2015; 62:1-13.
* Corresponding author:
Manal M. Anwar*,
Therapeutical Chemistry Department, National Research Center, Dokki, Giza, Cairo, Egypt.
Tel: 0123956970; Fax: + (202) 337-0931
E-mail: [email protected]
E-mail: [email protected]