INDIAN DRUGS 50(05) MA Y 2013 5 REVIEW ARTICLE PULSATILE DRUG DELIVERY “A PROGRAMMED POLYMERIC DEVICE” Rawat S. * , Bisht S. and Kothiyal P. (Received 15 February 2013) (Accepted 26 April 2013) ABSTRACT Pulsatile Drug Delivery Systems are gaining a lot of interest as they deliver the drug at the right place, at the right time and in the right amount, thus providing spatial, temporal and smart delivery and increasing patient compliance. The use of pulsatile release of the drugs is desirable where constant drug release is not desired. These systems are designed according to the circadian rhythm of the body. According to Latin literature circameans about and Diem means day. This could be advantageous for many drugs or therapies including asthma, peptic ulcer & arthritis etc. To correlate with our biological needs, “precisely timed drug delivery,” which could be accomplished with “programmable dosage forms,” is desirable. Precisely timed drug delivery may maximize therapeutic efcacy, minimize dose frequency, and may reduce toxicity. This paper outlines the concepts that have been proposed to release drugs in a pulsed manner from pharmaceutical device. *For correspondence Shri Guru Ram Rai institute of Technology & Sciences Dehradun - 248001, Uttarakhand E-mail: [email protected]m Keywords: Lag time, Pulsatile drug release, Circadian rhythm, Single unit, Multiple units, Pulsatile release pulsincap. INTRODUCTION Over the last 30 years the pharmaceutical market has focussed increasing preferably for controlled and targeted drug delivery system. Such systems have been focused on constant, variable; sustain drug release and/or targeting the therapeutic agent to a specic site/tissue/ organ. However, recently there are certain conditions for which such release pattern is not suitable. Such conditions lead to the requirements of a time programmed therapeutic system, capable of releasing drug after predetermined time delay and maintain constant drug levels throughout the day 1, 2 . For bioactive agents such as hormones, many have suggested that pulsed release may offer advantages over continuous release as hormones are gen erally secreted by the body in a pulsed manner 3, 4 . Also, a pulsatile drug release pattern could be advantageous for drugs with an extensive rst-pass metaboli sm, for drugs that develop biological tolerance are constantly needed to be present at their target site, and for drugs that require dosing at night. “Pulsed drug release” is dened as the rapid and transient release of a drug after a predetermined off-release period 5, 6 . One way to classify “pulsed drug delivery systems” is based on the physicochemical and biological principles that trigger the release. These devices are classied into “programmed” and “triggered” drug delivery systems. In programmed delivery systems, the release is completely governed by the inner mechanism of the device 7 (i.e., the lag time prior to the drug release is controlled primarily by the delivery system). In triggered delivery systems, the release is governed by changes in the physiologic environment of the device (biologica lly triggered systems) or by external stimuli (externally triggered systems). Some examples of biologically triggered pulsed delivery systems include the delivery of insulin in response to glucose levels 8, 9 and the delivery of anti-inammatory drugs in response to increased concentrations in hydroxyl radicals and hyaluronidase as may occur at inammatory sites 10 . In externally triggered systems, external stimuli such as magnetism, ultrasound, temperature changes, electrical effects and irradiation activate the drug
PULSATILE DRUG DELIVERY “A PROGRAMMED POLYMERIC DEVICE”
Rawat S.*
, Bisht S.
and Kothiyal P.
(Received 15 February 2013) (Accepted 26 April 2013)
ABSTRACT
Pulsatile Drug Delivery Systems are gaining a lot of interest as they deliver the drug at the right place, atthe right time and in the right amount, thus providing spatial, temporal and smart delivery and increasingpatient compliance. The use of pulsatile release of the drugs is desirable where constant drug releaseis not desired. These systems are designed according to the circadian rhythm of the body. According toLatin literature circa means about and Diem means day. This could be advantageous for many drugs ortherapies including asthma, peptic ulcer & arthritis etc. To correlate with our biological needs, “preciselytimed drug delivery,” which could be accomplished with “programmable dosage forms,” is desirable.Precisely timed drug delivery may maximize therapeutic efcacy, minimize dose frequency, and may
reduce toxicity. This paper outlines the concepts that have been proposed to release drugs in a pulsed
manner from pharmaceutical device.
*For correspondence
Shri Guru Ram Rai institute of Technology & Sciences
Keywords:Lag time, Pulsatile drug release, Circadianrhythm, Single unit, Multiple units, Pulsatile releasepulsincap.
INTRODUCTION
Over the last 30 years the pharmaceutical market
has focussed increasing preferably for controlled andtargeted drug delivery system. Such systems havebeen focused on constant, variable; sustain drugrelease and/or targeting the therapeutic agent to aspecic site/tissue/ organ. However, recently there are
certain conditions for which such release pattern isnot suitable. Such conditions lead to the requirementsof a time programmed therapeutic system, capableof releasing drug after predetermined time delay andmaintain constant drug levels throughout the day1, 2.
For bioactive agents such as hormones, many havesuggested that pulsed release may offer advantagesover continuous release as hormones are generallysecreted by the body in a pulsed manner3, 4. Also, apulsatile drug release pattern could be advantageous
for drugs with an extensive rst-pass metabolism, for
drugs that develop biological tolerance are constantlyneeded to be present at their target site, and for drugsthat require dosing at night. “Pulsed drug release” isdened as the rapid and transient release of a drug
after a predetermined off-release period5, 6. One way
to classify “pulsed drug delivery systems” is basedon the physicochemical and biological principles thattrigger the release. These devices are classied into
“programmed” and “triggered” drug delivery systems.In programmed delivery systems, the release iscompletely governed by the inner mechanism of thedevice7 (i.e., the lag time prior to the drug releaseis controlled primarily by the delivery system). Intriggered delivery systems, the release is governed bychanges in the physiologic environment of the device(biologically triggered systems) or by external stimuli
(externally triggered systems). Some examples ofbiologically triggered pulsed delivery systems includethe delivery of insulin in response to glucose levels 8, 9
and the delivery of anti-inammatory drugs in response
to increased concentrations in hydroxyl radicals and
hyaluronidase as may occur at inammatory sites 10 .In externally triggered systems, external stimuli such
as magnetism, ultrasound, temperature changes,electrical effects and irradiation activate the drug
release. Such systems are known as pulsatile drugdelivery systems (PDDS), time-controlled systems,or sigmoidal release systems (Fig. 1).
There are three types of biological rhythms in ourbody. They are:-13
1. Ultradian Rhythms: Oscillations of shorterduration are termed Ultradian Rhythms (more
than one cycle per 24 h). E.g. 90 minutes sleepcycle.
2. Infradian Rhythms: Oscillations that are longerthan 24 hours are termed as Infradian Rhythms(less than one cycle per 24 hours). E.g., monthlymenstruation.
3. Circadian Rhythms: Circadian rhythms areself-sustaining, endogenous oscillations thatoccur with a periodicity of about 24 Hours. Theterm circadian is derived from the Latin circa
which means “about” and diem which can bedened as “a day”. Normally, circadian rhythms
are synchronized according to internal biologicalclocks related to the sleep wake cycle.
Table I: Diseases and Chronotherapeutics
Chronological
behavior
Drugs used Diseases
Acid secretion ishigh in the afternoon
and at night
H2 blockers Pepticulcer
Precipitation of at-tacks during night orat early morning
β2 agonist,Antihistamines
Asthma
BP is at its lowestduring the sleep cy-cle and rises steeplyduring the earlymorning
Cholesterol syn-thesis is generallyhigher during nightthan day time.
HMG CoAreductaseinhibitors
Hyper-choleste-rolemia
Fig. 1: Schematic representation of different drugdelivery systems where:
(A) = sigmoidal release after lag time,
(B) = delayed release after lag time,(C) = sustained release after lag time,(D) = extended release without lag time.
PDDS have been developed in close connectionwith emerging chronotherapeutic views. In this respect,it is well established that the symptoms of manypathologies (Fig. 2), as well as the pharmacokineticand pharmacodynamic proles of most drugs, are
subject to circadian variation patterns. Circadianrhythm regulates many body functions in humans,viz., metabolism, physiology, behavior, sleep patterns,
hormone production, and other11, 12 .
To introduce the concept of chronotherapeutics,it is important to dene the following concepts.
“Chronopharmaceutics” consists of two wordschronobiology and pharmaceutics.
Chronobiology is a science concerned with thebiological mechanism of the diseases according to atime structure shown in table I. ‘chrono’ pertains to
time and ‘biology’ pertains to the study, or scienceof life.
Pharmaceutics is the discipline of pharmacythat deals with the process of turning a new chemicalentity (NCE) into a medication to be used safely andeffectively by patients. It is also called the science ofdosage form design and deals with the formulation ofa pure drug substance into a dosage form.
Of all the diseases studied, asthma may be themost prominently circadian. The role of circadianrhythms in the pathogenesis and treatment ofasthma indicates that airway resistance increasesprogressively at night in asthmatic patients. Normallung function undergoes circadian changes and
reaches a low point in early morning hours. Therefore,vast majority of bronchospastic attacks occur in theearly morning 2 a.m. and 6 a.m. each day. Thusdosage forms should be designed to deliver theactive agent at the time of maximum possibility
of bronchospastic attack. In one study, use oftimed-release formulation of theophylline achievedtherapeutic drug concentrations during the night andavoided toxic levels during the day when the dose
was ingested at 3 p.m.
Cardiovascular diseases16, 17
Several functions such as Blood pressure, heartrate, stroke volume, cardiac output, blood ow of
the cardiovascular system are subject to circadianrhythms. For instance, platelet aggregability isincreased and brinolytic activity is decreased
in the morning, leading to a state of relativehypercoagulability of the blood capillary resistance
and vascular reactivity are higher in the morning anddecrease later in the day. It is the widely acceptedfact that majority of heart attacks occur in the morninghours. The reason behind it is that the BP is at its
lowest during the sleeping period and rises steeplyduring the early morning period.
Arthritis18, 19
Arthritic diseases such as rheumatoid arthritis,osteoarthritis, ankylosing spondylitis and gout exhibit
profound circadian rhythms in the manifestation.The symptoms of rheumatoid arthritis are alwaysworse in the morning. Ratings of the severity of joint pain swelling and stiffness were about 3times higher between 08:00 and 11:00 am. People
with osteoarthritis tend to have less pain in themorning and more at night. Ankylosing spondylitisis prominent during 6 a.m. and 9 a.m. Taking nonsteroidal anti-inammator like urbiprofen, ibuprofen
and ketoprofen and indomethacin once-a-day formsoptimizes their therapeutic effect and minimizestheir side effects.
Diabetes 20, 21
In type I diabetes the circadian rhythms of insulin
level varies throughout the day & Increase in theblood sugar level after meal. The most widespreadapplication of chronotherapy is insulin pump, whichis used to administer insulin for the treatment ofdiabetes mellitus. With the insulin pump, patientscan customize insulin delivery to meet their particularrequirements.
Hypercholesterolemia22, 23
Diverse directions of circadian changes in
lipid fractions in patients and normal subjects maycontribute to alteration in the rhythmicity of othermetabolisms. A circadian rhythm occurs duringhepatic cholesterol synthesis which is generallyhigher during the night. The maximal production
occurs early in the morning, i.e. 12 h after the lastmeal. Studies with HMG CoA reductase inhibitorshave suggested that evening dosing was moreeffective.
Many of the functions of the gastrointestinal tractare subject to circadian rhythms: gastric acid secretionis highest at night, while gastric and small bowel
motility and gastric emptying are all slower at night.Suppression of nocturnal acid is an important factor induodenal ulcer healing. Therefore, for active duodenalulcer, once daily at bedtime is the recommendeddosage regimen for an H2 antagonist.
Necessities of Pulsatile Drug Delivery
System25
1. First pass metabolism: Some drugs, suchas beta blockers and salicylamide, undergoextensive rst pass metabolism and require fast
drug input to saturate metabolizing enzymes.Thus, a constant/sustained oral method of deliverywould result in reduced oral bioavailability.
2. Biological tolerance: Drug plasma proles
are often accompanied by a decline in thepharmacotherapeutic effect of the drug, e.g.,biological tolerance of transdermal nitroglycerin,salbutamol sulphate.
3. Special chronopharmacological needs: Circadian rhythms in certain physiologicalfunctions are well established. It has beenrecognized that many symptoms and onset ofdisease occur during specic time periods of the
24 hour day, e.g., asthma and angina pectorisattacks are most frequently in the morninghours.
4. Local therapeutic need: For the treatmentof local disorders such as inammatory bowel
disease, the delivery of compounds to the siteof inammation with no loss due to absorption in
the small intestine is highly desirable to achievethe therapeutic effect.
5. Gastric irritation or drug instability in gastric
uid: Protection from gastric environment isessential for the drugs that undergo degradationin gastric acidic medium (e.g., peptide drugs),irritate the gastric mucosa (NSAIDS) or inducenausea and vomiting.
Advantages of Pulsatile Drug Delivery System25
1. Extended daytime or night time activity
2. Reduced side effects
3. Reduced dosage frequency
4. Reduction in dose size
5. Improved patient compliance
6. Lower daily cost to patient due to fewer dosageunits are required by the patient in therapy.
7. Drug adapts to suit circadian rhythms of bodyfunctions or diseases.
8. Drug targeting to specic site like colon.
9. Protection of mucosa from irritating drugs.
10. Drug loss by extensive rst pass metabolism isprevented.
11. Patient comfort and compliance: Oral drug deliveryis the most common.
Limitations of Pulsatile Drug Delivery System25, 26
1. Multiple manufacturing steps in multiparticulatepulsatile drug delivery system.
2. Low drug load.
3. Incomplete release.
4. In-vivo variability in single unit pulsatile drugdelivery system.
Types of pulsatile drug delivery systems27
Pulsatile drug delivery system can be broadlyclassied into three classes;
3. Pulsatile system with erodible or solublebarrier coatings.
a. The chronotropic system
b. ‘TIME CLOCK’ System.
c. Compressed tablets
d. Multilayered Tablets
4. Pulsatile system with rupturable coating
B. Multiparticulate / Multiple unit systems
1. Pulsatile system with rupturable coatingE.g. Time –controlled explosion system
(TCES)2. Osmotic based rupturable coating system
E.g. Permeability controlled system
3. Pulsatile delivery by change in membranepermeabilityE.g. Sigmoidal release system.
II. Stimuli induced pulsatile drug delivery
1. Temperature-induced pulsatile release:
2. Chemical stimuli-induced pulsatile release:
a) Glucose-responsive insulin releasedevices
b) Inammation-induced pulsatile release
c) Drug release from intelligent gels respondingto antibody concentration.
d) Electric stimuli-responsive pulsatile release
III. Externally regulated pulsatile drug delivery
1. Magnetically induced release
2. Ultrasound induced release3. Electric eld induced release
4. Light induced release
I. Time controlled pulsatile release system
In time controlled drug delivery systempulsatile release is obtained after a specifictime interval in order to mimic the circardianrhythm.
A. Single unit pulsatile systems
Single-unit systems are mostly developed incapsule form. The lag time is controlled by a plug,which gets pushed away by swelling or erosion,
and the drug is released as a “Pulse” from theinsoluble capsule body.
1. Capsule based systems28, 29
Pulsincap ® system is one of the most used pulsatilesystems based on capsule illustrated in Fig. 3.It was developed by R.P Scherer InternationalCorporation, Michigan, USA. The Pulsincap ® system is an example of such a system that
is made up of a water-insoluble capsule bodylled with drug formulation. The body is closed
at the open end with a swellable hydrogel plug.Upon contact with dissolution medium or gastro-intestinal uids, the plug swells, pushing itself out
of the capsule after a time lag. This is followed bya spontaneous release of the drug. The lag timecan be controlled by manipulating the dimensionand the position of the plug.
The PORT system was developed by Thera-peutic System Research laboratory Ann Arbor,Michigan, USA, and consists of a capsulecoated with a semi permeable membrane.Inside the capsule an insoluble was plugconsisting of osmotically active agent and thedrug formulation. When capsule shell comes
in contact with the G.I uid, the semi perme-able membrane allows the entry of gastricuid. As a consequence the plug swells andcreate osmotic pressure. When this pressureexceeds the tensile strength of the membrane
it bursts out and time taken to rapture themembrane is known as lag time. After lagtime the plug is expelled to release the drug
as shown in g. 4. Methylphenidate used in
the treatment of attention decit hyperactivity
disorder (ADHD) is presented as the pulsatile
port system. This system avoided secondtime dosing, which was benecial for school
children during daytime30.
b) System based on expandable orice
In order to deliver drug in liquid form, an osmoti-cally driven capsular system was developed.In this system, liquid drug is absorbed intohighly porous particles, which release thedrug through an orice of a semi permeable
capsule supported by an expanding osmotic
layer after the barrier layer is dissolved 31.
Fig. 3: Design of Pulsincap system
Fig. 4: Drug release from PORT system Fig.5: System based on expandable orice
media but are released in the intestinal envi-ronment (Fig. 8). The major drawbacks of thetechnique are that relatively large amounts ofcoating materials are needed and it is difcult
to position the cores correctly.
d) Multi layered tablet38, 42
With the three layered tablet release patternwith two pulses was obtained, two drug layersare separated by a drug free gellable poly-meric barrier layer. This three-layered tabletwas coated on three sides with in imperme-able ethyl cellulose, and the top portion wasleft uncoated. Upon contact with dissolution
medium, the initial dose incorporated intothe top layer was released rapidly from the
noncoated surface as shown in g. 9. Thesecond pulse was obtained from the bottomlayer after the gelling barrier layer of HPMCwas eroded and dissolved . The rate of gellingand/or dissolution of the barrier layer controlthe appearance of the second pulse.
4. Pulsatile system with rupturable coating39, 40
These systems are dependent on the disintegrationof the coating for the release of drug. The pressurenecessary for the rupture of the coating can
be achieved by the swelling, disintegrants,effervescent excipients, or osmotic pressure
as illustrated in g.10. Water permeation and
mechanical resistance of the outer membraneare major factors affecting the lag time.
B. Multiparticulate / Multiple unit systems
Recent trends indicate that multiparticulatedrug delivery systems are especially suitable
for achieving controlled or delayed release oralformulations with low risk of dose dumping,exibility of blending to attain different release
patterns as well as reproducible and short
gastric residence time (Fig. 11). The release ofdrug from microparticles depends on a varietyof factors including the carrier used to form themultiparticles and the amount of drug containedin them. Advantages over single-unit systemsis of their small size, less inter and intra-subjectvariability in gastrointestinal transit time, reducedadverse effects and improved tolerability andnally Improve stability However, there are some
draw backs in this system, which include lackof manufacturing reproducibility, high cost ofproduction, multiple formulation steps and alsothe need of advanced technologies.
1. Pulsatile System Based on RupturableCoating41
This is a multiparticulate system in which drug iscoated on non-pareil sugar seeds followed by a
swellable layer and an insoluble top layer. Uponingress of water, the swellable layer expands,
resulting in rupture of lm with subsequent rapid
drug release. The lag time can be varied by
varying coating thickness or adding high amountsof lipophilic plasticizer in the outermost layer.
E.g. Time–controlled explosion system
(TCES)
This system is based on a combination of osmoticand swelling effects. The core contains the drug,a low bulk density solid and/or liquid lipid materialand a disintegrant. The core is further coated withcellulose acetate. Upon immersion in aqueousmedium, water penetrates the core displacing the
lipid material. After the depletion of lipid material,internal pressure increases until a critical stressis reached, which results in rupture of the coatingmaterial g. 1239.
2. Osmotic based rupturable coating system
This system is based on a combination of osmoticand swelling effects. The core containing the
Fig. 11: Hypothetical design and plasma drug prole of a multiparticulate pulsatile system. (A) Design of a pellet withmultiple coatings, and (B) Predicted bi-modal plasma concentration prole
drug, a low bulk density solid and/or liquid lipidmaterial and a disintegrant was prepared. Thiscore was then coated with cellulose acetate.Upon immersion in aqueous medium, water
penetrates the core displacing lipid material. Afterthe depletion of lipid material, internal pressureincreases until a critical stress is reached, whichresults in rupture of coating43.
3. Pulsatile delivery by change in membranepermeability
Sigmoid type of release pattern was obtained frompellet core having drug and succinic acid coatedwith amino methylacrylate copolymers. The waterin the medium dissolve succinic acid .The druginside and acid solution increase the permeabilityof the polymer lm. Actually permeability and
water uptake of acrylic polymers with quaternaryammonium groups can be influenced bythe presence of different counter-ions in themedium44. Eudragit RS 30D is reported tobe a polymer of choice for this purpose. Ittypically contains positively polarized quaternaryammonium group in the polymer side chain, whichis always accompanied by negative hydrochloridecounter-ions. The ammonium group being
hydrophilic it facilitates the interaction of polymerwith water, thereby changing its permeability andallowing water to permeate the active core in acontrolled.
Eg. Sigmoidal release system
II. Stimuli induced pulsatile drug delivery
In these systems the polymer undergoes swellingor deswelling phase in response of temperature,chemical reaction with membrane, alteration ofpH and Inammation induce, g. 13 releases drug
from polymer by swelling the polymer is compiled intable II.
1. Temperature-induced pulsatile release
This deviation sometimes can act as a stimulusthat triggers the release of therapeutic agentsfrom several temperature responsive drugdelivery systems for diseases accompanyingfever.
a) Thermoresponsive hydrogel systems
Thermo-responsive hydrogel systemsemploy hydrogels which undergo reversiblevolume changes in response to changes intemperature. These gels shrink at a transitiontemperature that is referred to the lowercritical solution temperature of the linearpolymer. Thermo-sensitive hydrosensitive
hydrogels have a certain chemical attractionfor water, and therefore they absorb waterand swell at temperatures below thetransition temperature whereas they shrink ordeswell at temperatures above the transitiontemperature by expelling water.
2. Chemical stimuli-induced pulsatile release
a) Glucose-responsive insulin releasedevices
In case of diabetes mellitus there is rhythmicincrease in the levels of glucose in the body,requiring injection of the insulin at propertime. Several systems have already beendeveloped which are able to respond toglucose concentration changes. One suchsystem includes pH sensitive hydrogelcontaining glucose oxidase immobilized in
the hydrogel. When glucose concentration
Fig.13: Effect of different chemical stimuli on drugrelease.
converts glucose into gluconic acid whichchanges the pH of the system. This pHchange induces swelling of the polymerwhich results in insulin release. Insulin byvirtue of its action reduces blood glucoselevel and consequently gluconic acid levelalso gets decreased and system turns to
the deswelling mode thereby decreasing theinsulin release. Examples of the pH sensitive
polymers include N, N-dimethylaminoethylmethacrylate, chitosan, etc46.
b) Inammation-induced pulsatile release
When human beings receive physical orchemical stress, such as injury, broken bones,etc., inammation reactions take place at
the injured sites. At the inammatory sites,
inammation- responsive phagocytic cells,
such as macrophages and polymorphonuclearcells play a role in the healing process of theinjury. During inammation, hydroxyl radicals
are produced from these inammation-
responsive cells. Degradation via hydroxyl
radicals however, is usually dominant andrapid when hyaluronic acid gel is injectedat inammatory sites. Thus, it is possible to
treat patients with inammatory diseases like
rheumatoid arthritis; using anti-inammatory
drug incorporated HA gels as new implantabledrug delivery systems 47.
c) Drug release from intelligent gels respondingto antibody concentration
Miyata et al. focused on the development ofstimuli responsive cross- linking structures
into hydrogels. Special care was given toantigen-antibody complex formation as the
cross-linking units in the gel, since specic
antigen recognition of an antibody can providethe foundation for a new device fabrication.Using the difference in association constantsbetween polymerized antibodies andnaturally derived antibodies towards specic
antigens, reversible gel swelling/deswellingand drug permeation changes could occur.Thus, biological stimuli- responsive hydrogels
were created 48.d) Electric stimuli-responsive pulsatile release
Kishi et al. developed an electric stimuli induceddrug release system using the electricallystimulated swelling /deswelling characteristicsof polyelectrolyte hydrogels. They utilized achemomechanical system, which containeda drug model within the polyelectrolyte gel
Table II: Effect of different chemical stimuli on the release of drug from hydrogels 45
Stimulus Hydrogel Type release mechanism
pH Acidic or basic hydrogel Change in pH—swelling—release of drug
Ionic strength Ionic hydrogel Change in ionic strength—change in concentration of ionsChemicalspecies
Hydrogel containingelectron-accepting groups
Electron-donating compounds—formation of charge-transfercomplexes—change in swelling—release of drug
Enzymesubstrate
Hydrogel containingimmobilized enzymes
Substrate present—enzymatic conversion—product changesswelling of gel—release of drug
Magnetic Magnetic particles dispersedin microspheres
Applied magnetic eld—change in pores in gel—change in
swelling—release of drug
Thermal Thermo-responsive hydrogel Thermal Thermo-responsive hydrogel Change intemperature—change in polymer–polymer and water–polymer interactions—change in swelling—release of drug
Electrical Polyelectrolyte hydrogel Applied electric eld—membrane charging—electrophoresisof charged drug—change in swelling—release of drug
swelling / shrinking behavior in response toon–off switching of an electric stimulus. Thus,drug molecules within the polyelectrolyte
gels might be squeezed out from the electricstimuli-induced gel contraction along withthe solvent ow. To realize this mechanism,
poly (sodium acrylate) microparticulate gelscontaining pilocarpine as a model drug wereprepared 49.
III. Externally regulated pulsatile drug delivery
1. Magnetically induced release
Magnetic carriers receive their magnetic re-sponse to a magnetic eld from incorporated
materials such as magnetite, iron, nickel,cobalt etc. Magnetic-sensitive behavior ofintelligent ferrogels for controlled releaseof drug was studied by Tingyu Liu, et al50.An intelligent magnetic hydrogel (ferrogel)was fabricated by mixing poly (vinyl alcohol)
hydrogels and Fe3O
4 magnetic particles
through freezing-thawing Cycles. Althoughthe external direct current magnetic eld
was applied to the ferrogel, the drug gotaccumulated around the ferrogel, but the
accumulated drug spurt to the environmentinstantly when the magnetic elds instantly
switched “off”. Furthermore, rapid slow drugrelease could be tunable while the magneticeld was switched from “off” to “on” mode.
2. Ultrasound induced release
Ultrasound is mostly used as an enhancer forthe improvement of drug permeation throughbiological barriers, such as skin. The interac-tions of ultrasound with biological tissues are
divided into two broad categories: thermaland nonthermal effects. Thermal effects areassociated with the absorption of acousticenergy by the uids or tissues. Non-thermal
bio-effects are generally associated withoscillating or cavitating bubbles, but alsoinclude noncavitation effects such as radia-tion pressure, radiation torque, and acousticstreaming51.
3. Electric eld induced release
Electrically responsive delivery systems areprepared by polyelectrolytes and are thuspH- responsive as well as electro-responsive.
Under the inuence of electric eld, electro-responsive hydrogels generally bend, de-pending on the shape of the gel which liesparallel to the electrodes whereas deswellingoccurs when the hydrogel lies perpendicularto the electrodes. An electroresponsive drugdelivery system was developed using poly(acrylamide-grafted-xanthan gum) hydrogel
for transdermal delivery of ketoprofen 52.
4. Light induced release
Light-sensitive hydrogels have potential appli-cations in developing optical switches, displayunits, and opthalmic drug delivery devices.The interaction between light and materialcan be used to modulate drug delivery. Whenhydrogel absorb the light and convert it to heat,raising the temperature of composite hydrogelabove its LCST41, hydrogel collapses andresult in an increased rate of release of solubledrug held within the matrix53.
Marketed Technologies of Pulsatile Drug Delivery
In examples o f cur rent ly marketed
chronopharmaceutical dosage forms, themanufacturing techniques applied, drug releasemechanisms, and the timing of drug administrationare collected and compiled in Table III.
1. PulsincapTM technology57
Pulsincap was developed by R.R. SchererInternational Corporation (Michigan). This device
consists of a non-disintegrating half capsule bodysealed at the open end with a hydrogel plug thatis covered by a water-soluble cap. The wholeunit is coated with an enteric polymer to avoidthe problem of variable gastric emptying. Whenthis capsule comes in contact with the dissolutionuid, it swells, and after a lag time, the plug push
itself outside the capsule and rapidly releasesthe drug. Study was to explore the time and pH-
dependent controlled drug delivery of diclofenacsodium using the pulsincap system illustrated inFig. 3 31-32.
2. ORBEXA ® Technology
Orbexa ® technology is a multiparticulatesystem that enables high drug loading andprovides a formulation choice for products thatrequire granulation. This technology producesbeads that are of controlled size and density
and suitable for formulation as controlled
release multiparticulate using granulation,spheronization and extrusion techniques asshown in Fig. 14. The resultant beads can becoated with functional polymer membranes foradditional release rate control and may be lled
into capsules or provided in sachet form.
3. DIFFUCAPS ® Technology56
Diffucaps is a multiparticulate bead systemcomprised of multiple layers of drug, excipients,
and release-controlling polymers is shownin Fig. 15. The beads contain a layer of organicacid or alkaline buffer to control the solubility of adrug by creating an optimal pH microenvironmentfor drugs that exhibit poor solubility in intestinal
pH, in environments with pH greater than 8.0.Alternatively, the beads can contain a solid-solution of drug and crystallization inhibitor toenhance bioavailability by maintaining the drug
in its amorphous state. Diffucaps technology isespecially suitable for drugs that traditionallyrequire multiple daily doses or drugs needingcustomized release formulations.
4. DIFFUTAB ®
Diffutab ® technology enables customized releaseproles and region-specic delivery. The Diffutab ® technology incorporates a blend of waxes and
hydrophilic polymers that control drug release
through diffusion and erosion of a matrix tabletas shown in g. 16. Diffutabs ® are particularlyuseful for high-dose products and drugs thatrequire sustained release and/or once-a-daydosing. Eurand applied this technology to bothsoluble and insoluble products 60.
5. SODAS ®Technology
SODAS ® (Spheroidal Oral Drug Absorption System)is Élan’s multiparticulate drug delivery system.Based on the production of controlled release beads,
this technology is characterized by its inherent
exibility, enabling the production of customized
dosage forms that respond directly to individualdrug candidate needs. This technology is basedon the production of uniform spherical beads of1-2 mm in diameter containing drug plus excipients
and coated with product specific controlledrelease polymers as proposed in Fig.17. Detailsare described in Patent No: US6066339.
Table III: Marketed Technologies of Pulsatile Drug Delivery54-59
Programmable Oral Drug Absorption System(PRODAS ® ) is a multiparticulate technology,which is unique in that it combines the benets
of tabletting technology within a capsule. ThePRODAS ® delivery system is presented as anumber of minitablets combined in a hard gelatincapsule and can be used to pre-program therelease rate of a drug. It is possible to incorporatemany different minitablets, each one formulated
individually and programmed to release drug atdifferent sites within the gastro-intestinal tract.Details are described in Patent No: US650045.
7. CHRONOTOPIC ® Technology
It is also described in system with erodible,soluble or rupturable membrane system. It isbasically drug-containing core coated with anouter release-controlling layer. Both single and
multiple-unit dosage forms such as tablets andcapsules or minitablets and pellets have beenemployed as the inner drug formulation.
8. Three-dimensional printing ®55
Three-dimensional printing (3DP) is a novelsolid freeform fabrication technology that hasbeen applied to the fabrication of complex
pharmaceutical drug devices. Prototypinginvolves constructing specic layers that use
powder processing and liquid binding materials.
3DP technology is exible in that it can be usedin applications linked to linear drug deliverysystems, colon-targeted delivery systems, oralfast disintegrating delivery systems, oating
delivery systems, time-controlled and pulserelease delivery systems as well as dosageforms with multiphase release properties andimplantable DDS. In addition, 3DP can alsoprovide solutions for resolving difculties relating
to the delivery of poorly water-soluble drugs,peptides and proteins, highly toxic and potent
drugs and controlled release of multidrugs in asingle dosage forms.
Two types of zero-order tablets were inventedand fabricated by 3DP. These contained drugconcentration gradients designed to complement thevolumetric non-uniformity of eroding shells. Threeformulations showed constant release of diclofenacsodium over 1–7 h (9.6 mg/hr), 1–15 h (6.8 mg/hr)and 1–36 h (2.5 mg/ hr)61.
9. CODAS ® (chronotherapeutic oral drug
absorption system)
E l an D r ug T echno l ogy deve l o ped
CODAS ® technology to achieve this prolongedinterval. Advantages of the CODAS ® technologyinclude a delivery prole designed to compliment
circadian pattern, controlled onset, an extended
release delivery system, rate of release essentiallyindependent of pH, posture and food, “sprinkle”dosing by opening the capsule and sprinkling thecontents on food, reduction in effective daily dose
and drug exposure, gastrointestinal tract targetingfor local effect and reduced systemic exposure
to achieve a target prole62. Verelan ® PM usesthe CODASTM technology, which is designed forbedtime dosing, incorporating a 4 to 5 h delayin drug delivery results in a maximum plasma
concentration of verapamil in the morning hours.
10. OROS ® technology54, 58
OROS delivery systems were adopted for poorlywater soluble drugs. The push-pull system is
comprised of a bilayer or trilayer tablet coreconsisting of one push layer and one or more druglayers. The drug layer contains the poorly solubledrugs, osmotic agents and a suspending agent.
Fig. 18: OROS ® Technology
The push layer contains amongother things, an osmotic agentand water swellable polymers.A semipermeable membranesurrounds the tablet core. Avariety of OROS ® systems (ALZACorp.) have been developed:P r o c a r d i a X L ® , D i t r opanXL ® and Concerta ® are notableexamples. The recently developed
L-OROS ® SOFTCAPTM deliverysystem combines the featuresof a control led-release andbioavailability-enhanced deliverysystem to enhance compliance andtherapeutic effect.
Controlled Release of Non Aqueous
Liquid Formulation is shownin Fig.18:
L-OROS Hard cap
L-OROS Soft cap
11. IPDAS ®
The intestinal protective drugabsorption system is a new oral
drug delivery approach that is applicable togastrointestinal irritant drugs, including thenonsteroidal anti-inflammatory drug class.This technology is composed of numerous
high-density, controlled-release beads, whichare compressed into a tablet form. Once anIPDAS ® tablet is ingested, it disintegrates anddisperses beads containing a drug in the stomach,which subsequently passes into the duodenumand along the gastrointestinal tract in a controlledmanner.
12. Geoclock ®
Geoclock ® tablets have an active drug insidean outer tablet layer consisting of a mixture of
hydrophobic wax and brittle material in orderto obtain a pH-independent lag time prior tocore drug delivery at a predetermined releaserate. This dry coating approach is designed toallow the timed release of both slow release andimmediate release active cores by releasing theinner tablet rst, after which time, the surrounding
outer shell gradually disintegrates. In additionto controlled release, the Geoclock ® technologyalso has applications for the improved release ofcolonic drug delivery as well as for multiple pulse
drug delivery to deliver doses of a drug at specictimes throughout the day. Using SkyePharma’sproprietary GeoclockTM technology, took the form ofa specially formulated tablet, which, once ingested,did not release the active ingredient, prednisone,until approximately four hours later 63.
13. Covera-HS
Covera-HS is the rst once-daily formulation
of an antihypertensive/anti-anginal agent thatuses an advanced tablet coating and a novel
drug delivery system to mimic the body’s typical24 h circadian variations in blood pressure andheart rate. This unique delivery technology,called COER-24TM (Controlled-Onset-Extended-
Release), was developed in conjunction with AlzaCorp. Covera-HS is the only controlled-releaseverapamil formulation that is currently approvedwith an indication for the management of bothhypertension and angina pectoris. Covera-HS
is designed for oral dosing at bedtime. Peakconcentration of Covera-HS is delivered in theearly waking hours, when blood pressure andheart rate are rise at their highest rate.
14. GeomatrixTM
Geomatrix ® Technology consists of a hydrophilicmatrix core, containing the active ingredient and
one or two impermeable or semi-permeablepolymeric coatings applied on one or bothbases of the core. This technology is applied toachieve customized levels of controlled releaseof specic drugs and can achieve simultaneous
release of two different drugs and different ratesfrom a single tablet. SkyePharma has developed
LodotraTM
, containing a rheumathoid arthritis drugthat delivers the active pharmaceutical ingredientat the most suitable time of day to treat thedisease.
CONCLUSION
Nowadays, pulsatile drug delivery is gainingtremendous popularity. The prime advantage in thisdrug delivery is that the drug is released when thenecessity arises. As a result the chance of developmentof drug resistance, which is seen in conventional and
sustained release formulations, can be reduced. Nowmany FDA-approved pulsaitle drugs are available inthe market. This therapy is mainly applicable wheresustained action is not required and the drugs aretoxic. Key point of development of this formulation
is to nd out the circadian rhythm, that is, a suitable
indicator that will trigger the release of the drug fromthe device. Pulsatile drug delivery is one such systemthat, by delivering a drug at right time, right place, andin right amounts, holds good promises of benet to the
patients suffering from chronic problems like arthritis,asthma, hypertension, etc. Since the drug deliverywould be in accordance with the actual concentration judged as per the chronological need, pulsatile drugrelease systems hold a promising future.
REFERENCES1. Saigal N., Baboota S., Ahuja A. and Ali J.. Site Specic
Chronotherapeutic Drug Delivery Systems: Recent Patentson Drug Delivery & Formulation 2009; 3: 64-70.
2. Jha N, Bapat S. Chronobiology and chronotherapeutics,Kathmandu University Medical Journal 2004; 2(8): 384-388.
3. Heller J. Modulated release from drug delivery devices.Crit. Rev. Ther. Drug Carr. Syst. 1993; 10:253–305.
4. Mandal A. S., Biswas N., Kazi M.K., Guha A., ChatterjeeS.. "Drug Delivery System Based On Chronobiology,AReview." Journal of Controlled Release, 2010: 314-325.
5. Bussemer T, Peppas NA, Bodmeier R. Evaluation of theswelling, hydration and rupturing properties of the swellinglayer of a rupturable pulsatile drug delivery system. Eur
J Pharm. Biopharm 2003; 56:261-70.
6. Alessandra Maroni, Lucia Zema, Matteo Cerea, "OralPulsatile Drug Delivery Systems." Exp Opin Drug Del,2005: 855-871.
7. Soppimath K. S., Aminabhavi T. M., Dave A. M., KumbarS. G., and Rudzinski W. E.. Stimulus-responsive “smart”
hydrogels as novel drug delivery systems. Drug Dev.Ind. Pharm. 2002; 28:957–974.
8. Tanna S., Sahota T., Clark J., and Taylor M. J. A covalentlystabilised glucose responsive gel formulation with aCarbopol carrier. J. Drug Target. 2002; 10:411–418.
9. K. Moriyama, T. Ooya, and N. Yui. Hyaluronic acid graftedwith poly(ethylene glycol) as a novel peptide formulation.J. Control. Rel. 1999; 59:77–86.
10. Yui N., Okano T., and Sakurai Y.. Inammation responsive
deg- radation of crosslinked hyaluronic acid gels. J.
Control. Rel. 1992; 22: 105–116.
11. Jha N, Bapat S. Chronobiology and chronotherapeutics,Kathmandu University Medical Journal 2004; 2(8): 384-
388.12. Khan Z, Pillay V, Choonara YE, du Toit LC. Drug delivery
technologies for chronotherapeutic applications. Pharm
Dev Technol 2009; 14:602-12.
13. Singh R, Pramod Kumar Sharma and Rishabha M.Review on Chronotherapeutics - A New Remedy in theTreatment of Various Diseases. Eur J Bio Sci 2010; 2(3): 67-76.
14. Smolensky M. H, Scott P. H, Harrist R. B. Administrationtime dependecy of the pharamocokinetic behaviorand Therapeutic effect of a once-a-day theophylline inasthmatic children. Chronobiol. Int .1987; 4(3): 4
15. Preparation, in the treatment of nocturnal asthma, Am.J. Med 1988; 85:60-63 35-47.
16. Drayer JI, Automated ambulatory blood pressuremonitoring: a study in age-matched normotensiveand hypertensive men. Am Heart J 1985; 109: 1334–1338.
17. Lemmer B. Cardiovascular chronobiology andchronopharmacology. Biological Rhythms in Clinical andLaboratory Medicine 1992; 418– 427.
18. Bellary N, Sothern R. B, Campbell J. Rhythmic variationsin pain perception in osteoarthritis of knee. Journal of
Rheumatology. 1990; 17: 364-372.
19. Swannell A. J. Biological rhythms and their effect inassessment of disease activity in rheumatoid arthritis.British Journal of Clinical Practice. 1983; 38 (Supplement33): 16-19.
20. Rupali Singh, Pramod Kumar Sharma and Rishabha M.Review on Chronotherapeutics - A New Remedy in theTreatment of Various Diseases. Eur J Bio Sci 2010; 2(3): 67-76.
21. Waldhäusl W. Circadian rhythms of insulin needs andactions, Diabetes Research and Clinical Practice 1989;6 (4).
22. Hulcher FH, Reynolds J, Rose JC. Circadian rhythmof HMG-CoA reductase and insulin in African greenmonkeys. Biochem Int 1985; 10: 177– 185.
23. Stein EA, Davidson MH, Dobs AS, Schrott H, DujovneCA, Bays H, Weiss SR, Melino MR, Mitchel ME, MitchelYB. Efcacy and safety of simvastatin 80 mg/day in
hypercholesterolemic patients. Am J Cardiol 1998; 82:311- 316.
24. Moore JG, Englert Jr E. Circadian rhythm of gastric acidsecretion in man. Nature 1970; 226:1261–1262.
25. Dalvadi H., Patel J. K., Chronpharmaceutics, pulsatiledrug delivery system as current trend, Asian J. Pharm.
27. Sharma GS, Srikanth MV, Uhumwangho MU, PhaniKumar KS and Ramana Murthy KV. Recent trends inpulsatile drug delivery systems - A review. InternationalJournal of Drug Delivery, 2010; 2: 200-212.
28. Krögel I, Bodmeier R. Pulsatile drug release from aninsoluble capsule body controlled by an erodible plug.Pharm. Res. 1998; 15(3): 474-481.
29. Developing a modied pulsincap system. Pharm Tech
Europe 2009; 21.
30. Swanson J, Gupta S, Gunita D, Flynn D, Agler D, LennerM, William L, Shoulson I, S Wigal. Acute tolerance tomethylphenidate in the treatment of attention decit
hyperactivity disorder in children. Clin Pharmacol Ther 66:295–305, 1999.
31. Linkwitz A, Magruder JA, Merrill S. OsmoticallyDriven Delivery Device with Expandable Orifice for
Pulsatile Delivery Effect , US Patent No. 5,318,558,1994.
32. Pollock DC, Dong L, Wong P. A new system to deliver adelayed bolus of liquid drug formulation, Proceed InternSymp. Control. Rel. Bioact. Mater 2001; 28: 6033.
33. Balaban SM, Pike JB, Smith JP, Baile CA. OsmoticallyDriven Delivery Devices with Pulsatile Effect. US PatentNo. 5209746; 1993.
34. Magruder PR, Barclay B, Wong PSL, TheeuwesF. Composiition comprising salbutamol. US PatentNo.4751071; 1988.
35. Magruder PR, Barclay B, Wong PSL, Theeuwes F.Composition Comprising a Therapeutic Agent and aModulating Agent. US Patent No. 4851229; 1989.
36. Gazzaniga A, Busetti C, Moro L, Crimella T, SangalliME, Giordano F. Evaluation of low viscosity HPMC as
retarding coating material in the preparation of a time-based oral colon specic delivery system. Proceed Intern
Symp Control Rel Bioact Mater. 1995; 22:242-243.
37. Wilding IR, Davis SS, Pozzi F, Furlani P, Gazzaniga A.Enteric coated timed release systems for colonic targeting.Int J Pharm 1994; 111: 99-102.
38. Conte U, Giunchedi P, Maggi L, Sangalli ME, GazzanigaA, Colombo P, Manna A. Ibuprofen delayed releasedosage forms: a proposal for the preparation of an invitro/in vivo pulsatile system, Eur. J. Pharm . 1992; (6):209-212.
39. Krögel I, Bodmeier R. Floating or pulsatile drug deliverysystems based on coated effervescent cores. Int. J.
44. Beckert TE, Pogarell K, Hack I, Petereit HU. Pulsed drugrelease with lm coatings of Eudragit & Mac226; RS 30D.
Proceed Int'l Symp Control Rel Bioact45. Soppimath K. S., Aminabhavi T. M., Dave A. M., et al.
Stimulus-responsive ‘smart’ hydrogels as novel drugdeli- very systems. Drug Dev. Ind. Pharm., 2002, 28:957-974.
46. Gutowska A, Bark JS, Kwon IC, Bae YH, Kim SW.Squeezing hydrogels for controlled oral drug delivery.J Control Rel 1997; 48: 141-148.
47. Yui N, Okano T, Sakurai Y. Inammation Responsive
Degradation of Cross-Linked Hyaluronic-Acid Gels.J Control Rel 1992; 22:105-116
48. Y.H. Bae, T. Okano, S.W. Kim. ‘On–off’ thermocontrol
of solute transport. I. Temperature dependence ofswelling of N-isopropylacrylamide networks modied
with hydrophobic components in water, Pharm. Res.1991; 8 (4): 531–537.
49. I.C. Kwon, Y.H. Bae, T. Okano, B. Berner, S.W. Kim.Stimuli sensitive polymers for drug delivery systems,Makro mol. Chem. Macromol. Symp 1990; 33: 265–277.
50. T. Y. Liu, S. H. Hu, T. Y. Liu, et al. Magnetic-sensitive
behavior of intelligent ferrogels for controlled release ofdrug. Langmuir, 2006, 22: 5974-5978.
51. Nyborg Wesley L. Biological effects of ultrasound:Development of safety guidelines. Part II: General review.Ultrasound Med Biol 2001, 27: 301-333.
52. A. C. Richards Grayson, R. S. Shawgo, Y. Li, et al.Electronic MEMS for triggered delivery. Adv. Drug Del.
Reviews, 2004, 56: 173-184.
53. Y. Qiu, K. Park. Environment-sensitive hydrogels for drugdelivery. Adv. Drug Del. Reviews, 2001, 53: 321-339.
54. Jao F, Wong P, Huynh H, et al. Inter. Jou. Pha (1992):17
55. Percel P, Vishnupad KS, and Venkatesh GM. Timed
pulsatile drug deliverysystems. US Patent 6,627,2231.56. Katstra WE, Palazzolo RD, Rowe CW, et al. Oral dosage
forms fabricated by three dimensional printing. J. Control.
Rel 2000; 66: 1-9.
57. Stevens HNE, Wilson CG, Welling PG, et al. Evaluationof pulsincap to provide regional delivery of dofetilide tothe human G.I. tract. Int. J. pharm 2002; 236 :27-34.
58. Sougata Jana, Arijit Gandhi. Dendrimers: Synthesis,Properties, Biomedical and Drug Delivery Applications.Am J PharmTech Res 2012; 2(1)
59. Pawar VK, Awasthi R. Chronotherapy: an approach tosynchronize drug delivery with circadian rhythm. JChrDD 2010; 1(1): 1-8
60. Roy P. Aliasgar Shahiwala, Multiparticulate formulationapproach to pulsatile Drug Delivery: Currentperspectives. J Contr Re2009;134;7480;DOI:10.1016/j. jconrel.2008.11.011 61)Rowe CW, Katstra WE,Pa lazzo lo RD, G i r i t l i og lu B , Teung P ,Cima MJ. Multimechanism oral dosage forms fabricatedby three dimensional printing. J Control Release 2000;66:11-7; PMID: 10708874; DOI: 10.1016/S0168-3659(99)00224-2.
62. Panoz DE. Geoghegan, Edward J, inventors. 1989 Sept.15/ Controlled absorption pharmaceutical composition,US Patent 4863742.
63. Conte U, Maggi L. Modulation of the dissolution prolesfrom Geomatrix® multilayer matrix tablets containing
drugs of different solubility. Biomoterials 1996; 17:889-96.
(Pinaceae), commonly known as chir pine, is a talltree with a spreading crown found in the Himalayanfrom Kashmir to Bhutan, Afghanistan and in southernIndian hills. The tapping of the stem produces a clear,transparent oleo-resin with a pungent and bittertaste. Distillation of the turpentine oil from the oleo-resin leaves faintly aromatic and transparent rosin(colophony)1. It is used in preparation of ointments andplasters and in many products such as chewing gum,
polishes, and varnishes, but is a common cause ofcontact allergy. The resin is applied to cure boils2 andadministered orally to combat gastric troubles3.
Native Americans have used pine resin totreat rheumatism because of its anti-inammatory
properties. The resin acts to remove the jointinammation caused by rheumatism, which helps to
restore movement and to alleviate pain. A traditional
use for pine resin has been as an external treatmentfor burns and sores. The pine resin has stimulant,diuretic and laxative properties. In China, the resin
from a particular pine tree is used to treat abscesses.Resin from the spruce tree was used by colonialAmericans as a cold and cough remedy, as well asstraight from the tree as a cancer treatment4.
Different parts of the plant are prescribed to treatcough, colds, inuenza, tuberculosis, bronchitis,
as antiseptic, diaphoretic, diuretic, rubefacient,
stimulant and febrifuge5,6. Rosin consists mainlya mixture of diterpenic acids. The principal acid is
abietic acid (37.5%) followed by isopimaric (20.9%),neoabietic (15.1%), levopimaric (13.5%), pimaricand dihydroabietic acids. In this paper, we report theisolation and structure elucidation of four triterpenoicacids linked with dehydroabietic acid derivativesobtained from the colophony ofPinus roxburghii Sarg.,collected from Haldwani (Uttarakhand).
melting apparatus (Ambala, Haryana, India) andare uncorrected. UV spectra were measured with aLambda Bio 20 spectrophotometer (Perkin-Elmer-Rotkreuz, Switzerland) in methanol. Infra red spectrawere recorded on Bio-Rad FTIR 5000 (FTS 135,Kawloon, Hong Hong) spectrophotometer usingKBr pellets; γ
max values are given in cm-1. 1H and 13C
NMR spectra were screened on advance DRX 400,Bruker spectrospin 400 and 100 MHz instrument in 5mm spinning tubes at 27oC, respectively (Karlsruhe,Germany) using TMS as an internal standard. Mass
spectra were scanned by effecting FAB ionization at70 eV on a JEOL-JMS-DX 303 spectrometer (Japan)equipped with direct inlet probe system. Columnchromatography was performed on silica gel (60-120mesh; Qualigen, Mumbai, India). TLC was run on silicagel G (Qualigen). Spots were visualised by exposing
to iodine vapours, UV radiation, and spraying withceric sulphate solution.
Plant material
The oleo-resin was procured from a Rosin factory,Haldwani, Uttarakhand. The sample was identied
on the basis of exomorphic characters, chemical
reactions and reviews of literature by Dr. H.B. Singh,Taxonomist, NISCAIR, CSIR, New Delhi. A voucher
specimen of the sample (No. N/R/C/2007/08/851/35)was deposited in the RHM Division, NISCAIR, NewDelhi-110012.
Extraction and isolation
The air dried oleo-resin (220 g) was coarsely
powdered and dissolved in methanol. The concentratedsolution was adsorbed on silica gel particles. It wasdried in the air and pulverized to get uniform particlesize and chromatographed over silica gel (60-120mesh) column packed in petroleum ether (b.p. 60-80 ºC). The column (1.6 m × 16 mm × 2 mm) waseluted successively with petroleum ether, mixture of
petroleum ether and chloroform (9:1, 3:1, 1:1, and1:3), chloroform and nally the mixture of chloroform
and methanol (99:1, 97:3, 19:1, 23:2, 9:1, 3:1, 1:1,1:3). Various fractions were collected separatelyand matched by TLC to check homogeneity. Similarfractions having same R
f values were combined
and crystallized. The isolated compounds wererecrystallized to get pure compounds. The followingcompounds were isolated:
Dehydroabietic acid (1)
Elution of the column with petroleum etherproduced light brown amorphous powder of 1,recrystallized with methanol-acetone (1:1), 0.24 g(0.109 % yield). R
Further elution of the column with petroleum ether-chloroform (1:1) gave dark yellow sticky mass of 3,recrystallized from methanol-acetone (1:1), 14.99 g(6.81% yield); R
Further elution of the column with petroleumether-chloroform (1:3) furnished brown sticky massof 5, recrystallized from methanol, 4.73 g (2.15%yield); R
with sodium bicarbonate solution. Its IR spectrumdisplayed characteristic absorption bands for carbonylgroup (1701 cm-1), carboxyl functions (3420, 1696
cm-1), unsaturation (1645 cm-1) and aromatic ring
(1529 cm-1
). On the basis of +ve FAB mass and13
CNMR spectra its molecular ion peak was determinedat m/z 766 consistent with the molecular formula ofa triterpenoid linked with diterpenoid, C
50H
70O
6. The
important fragment ion peaks generated at m/z 434[C
30H
42O
2]+ and 330 [C
20H
26O
4]+ were due to ether
linkage cleavage. The subsequent ion fragments ofthe diterpenic acid moiety arising at m/z 287 [330-C
3H
7]+, 272 [287-Me]+, 226 [272-HCOOH]+, 195 [226-
CH2OH]+, 284 [330-HCOOH]+, 299 [330-CH
2OH]+,
253 [299-HCOOH]+ and 238 [253-Me]+ suggested the
presence of one each isopropyl, carboxylic, ketonicand oxygenated methylene groups in the molecule.
The ion fragments of the triterpenic moiety generatingat m/z 137 [C
8H
9O
2, side chain, SC]+, 297 [434-SC]+
and 255 [297-ring D]+ suggested the presence of onevinylic linkage in the triterpenic molecules and theexistence of tri-unsaturated C
8-side chain with one
carboxylic group. The 1H NMR spectrum of 2 displayedtwo doublets at δ 7.19 (J= 9.4 Hz) and 6.96 (J= 2.7Hz) and a double-doublet at δ 7.15 (J= 7.8, 2.7 Hz),one-proton each, ascribed to ortho-coupled H-11’,meta-coupled H-14’ and ortho-meta-coupled H-12’aromatic protons, respectively. A two-proton broadsignal at δ 3.41 was ascribed to oxygenated H
2-19’
methylene protons. Two doublets at δ 1.17 (J= 6.3Hz), 1.22 (J= 6.1 Hz) and a broad singlet at δ 1.28,three-protons each, were assigned correspondingly tosecondary methyl Me-16', Me-17' and tertiary methylMe-20' protons, respectively. Two doublets at δ 5.31(J= 5.3 Hz) and 5.87 (J= 10.8 Hz) and a triple doubletat δ 5.70 (J= 8.4, 6.6, 10.8 Hz), one-proton each, were
ascribed correspondingly to cis -oriented vinylic H-6,H-22, and H-23 protons of the triterpenic unit. Fourone-proton broad singlets at δ 4.87, 4.86 and at δ 4.93, 4.92 were assigned to unsaturated methyleneprotons H
2-21 and H
2-27, respectively. A one-proton
double doublet atδ 4.33 [J =9.1, 5.2 Hz] was attributedto α-oriented oxygenated methine H-3 proton. Five
three-proton broad singlets at δ 0.75, 1.02, 1.19, 0.84and 0.89 were ascribed to tertiary Me-18, Me-19, Me-
at δ 183.22 (C-26), 181.83 (C-18'), aromatic carbons
betweenδ 123.41-149.70; vinylic carbons at δ 145.03(C-5), 120.74 (C-6), 137.38 (C-20), 112.27 (C-21),134.02 (C-22), 128.06 (C-23), 145.70 (C-25), 108.88(C-27), oxygenated methine carbon at δ 75.13 (C-3)and oxygenated methylene carbon at 67.44 (C-19′).The shifting of H
2-19 methylene signal at δ 3.41in
the 1H NMR spectrum and its 13C NMR signal at δ67.44 suggested linkage of the triterpenic moietyC-3 with diterpenic residue at C-19′. The 1H and13C NMR spectral data of 2 were compared with thevalues of the reported lanostene type triterpenoids.
The spectral data of the dehydroabietic acid unitwere compared with the reported values of thesimilar compounds10-12. On the basis of the foregoingaccount the structure of 2 has been established aslanost-5,20(21),22,25(27)-tetraen-26-oic acid-3β-olyl(3→19')-3'-oxodehydroabietic acid (Fig.1). This is
a new lanostenoic acid linked with dehydroabieticacid moiety.
Compound 3, designated as roxburghianoic
acid B, was obtained as a yellow sticky mass from
petroleum ether-chloroform (1:1) eluents. It producedeffervescences with sodium bicarbonate solutionand exhibited IR characteristic absorption bands for
carboxylic groups (3441, 1694 cm1-), aromatic (1524cm-1) and unsaturation (1642 cm-1). Its +ve FABmass and 13C NMR spectra suggested a molecularion peak at m/z 748 consistent with the molecularformula of a triterpenic acid linked with a diterpene,C
50H
68O
5. The important fragment ions peaks arose
at m/z 432 [C30
H40
O2]+ and 316 [C
20H
28O
3]+ and at
m/z 449 [C30H41O3]+
due to ether linkage ssion. Thesubsequent fragments of the diterpenic unit arisingat m/z 270 [316-HCOOH]+, 255 [270-Me]+, 224 [255-CH
2OH]+, 285 [316-CH
2OH]+, 239 [285-HCOOH]+,
301 [316-Me]+ and 258 [301-C3H
7]+ suggested the
presence of oxygenated methylene, carboxylic and
isopropyl groups in the diterpenic units. The ionfragments of the triterpenic unit generating at m/z 137 [C
indicated the presence of two double bonds in thetriterpenic unit with tri-unsaturated C
8 side chain and
carboxylic group. The ion peaks also indicated the
existence of oxygenated methine carbon in ring A
which was placed at C-3 on the basis of biogeneticconsideration. The 1H NMR spectrum of 3 exhibited
three one-proton downeld signals consisting of a
double doublet at δ 7.15 (J= 2.5, 9.0 Hz) and twodoublets at δ 6.98 (J= 2.5 Hz) and 7.23 (J= 9.0 Hz)assigned correspondingly to ortho-, meta- coupledH-12’, meta-coupled H-14’ and ortho-coupled H-11’aromatic protons. A two-proton broad singlet at δ 3.66was ascribed to H
2-19’ oxygenated methylene protons.
Two doublets at δ 1.19 (J= 6.6 Hz) and 1.23 (J= 8.0Hz) and a broad signal at δ 1.25, each integrating
for three protons, were assigned correspondingly tosecondary Me-16', Me-17' and tertiary Me-20' methylprotons of the diterpenic unit. A one-proton doublet atδ 5.83 (J =9.5 Hz) and three one-proton multiplets atδ 5.72 (w
½ =10.3 Hz), 5.34 and 5.37 were ascribed to
cis -oriented vinylic H-6, H-7, H-22 and H-23 protons,respectively. Four one-proton broad singlets at δ
4.95, 4.90 and at δ 4.88 and 4.85 were attributed toH
2-21 and H
2-27 methylene protons of the triterpenic
unit. A one-proton double doublet at δ 4.30 (J= 9.0,5.3 Hz) was attributed to H-3α-carbinol proton. Theremaining methylene and methine protons resonatedbetweenδ 1.48-2.84. Five three-proton broad singletsat δ 0.77, 1.01, 0.91, 0.85 and 0.83 were accountedcorrespondingly to tertiary methyl Me-18, Me-19,Me-28, Me-29 and Me-30 protons of triterpenicmoiety. The 13C NMR spectrum of 3 displayed 50carbon signals and important signals appeared forcarboxylic carbons atδ 182.91 (C-26), 184.33 (C-18');aromatic carbons between δ 123.67-149.98; vinyliccarbons at δ 144.37 (C-5), 120.35 (C-6), 146.61
carbon at 68.23 (C-19'). The shifting of the C-19′ oxygenated methylene proton signal at δ 3.66 in the1H NMR spectrum and carbon signal at δ 68.23 inthe 13C NMR spectrum suggested the linkage of thetriterpenic unit at this carbon. The 1H and 13C NMRspectral data of 3 were compared with the values of
the reported lanostene type triterpenoids10-12. Thespectral data of the dehydroabietic acid unit werecompared with the reported values of the similarcompounds10-12. On the basis of above discussion
the structure of 3 was formulated as lanost-5, 9(11),20(21), 22, 25(27)-pentaen-26-oic acid-3β-olyl(3→19')-3'-oxodehydroabietic acid (Fig. 1). This is
a new triterpenoic acid linked with dehydroabieticacid derivative.
Compound 4, designated as roxburghianoic
acid C, was obtained as a dark yellow crystallinepowder from petroleum ether-chloroform (1:3)eluants. It produced effervescences with sodiumbicarbonate solution and displayed characteristic
IR absorption bands for carboxyl group (3260, 1694cm-1), unsaturation (1645 cm-1) and aromatic ring(1527 cm-1). On the basis of +ve FAB mass and 13CNMR spectra, its molecular ion peak was determinedat m/z 748 consistent with the molecular formula oftriterpenoid with abietic acid C
50H
68O
5. The important
fragment ion peaks arose at m/z 449 [C30
H41
O3]+ and
299 [C20
H27
O2]+ due to C
3-C
19′ bond cleavage. The
subsequent ion fragments of the diterpenic acid moietyarising at 256 [299-C
3H
7]+ and 254 [299-COOH]+
indicated the presence of one each of carboxylic
and isopropyl groups in the diterpenic unit. The ionfragments of the triterpenic unit generating at m/z 137 [C
8H
9O
2, side chain, SC]+, 312 [449-SC]+, 432
[449-OH]+ and 295 [432-SC]+ suggested the presenceof two vinylic linkages in triterpenic skeleton andthe presence of a tri-unsaturated C
8-side chain with
carboxylic group in the compound. The 1H NMRspectrum of 4 displayed two downeld doublets at δ
correspondingly to H-11’, H-14’ and H-12’ aromaticprotons, respectively. A two-proton broad singlet at δ3.52 was attributed to oxygenated methylene H
2-19′
protons. Two three-proton doublets at δ 1.16 (J =6.6Hz) and 1.26 (J =6.6 Hz) were accounted to secondaryC-16′ and C-17′ methyl protons, respectively. Threeone-proton doublets at δ 5.79 (J= 4.8 Hz), 5.76 (J= 4.8Hz), 5.84 (J= 4.5 Hz) and one-proton multiplet at δ5.67 (w
cis -oriented vinylic H-6, H-7, H-22 and H-23 protonsof triterpenic unit. Four one-proton broad singlets atδ 4.97, 4.96 and at δ 4.90 and 4.89, were assignedto methylene protons H
2-21 and H
2-27, respectively.
A one-proton double doublet at δ 4.21 (J =5.1, 8.8Hz) was due to oxygenated methine H-3α proton.Five three-proton broad signals at δ 0.78, 0.92,0.87, 0.84 and 0.81 were attributed to tertiary Me-18,Me-19, Me-28, Me-29 and Me-30 methyl protons,respectively. The 13C NMR spectrum of 4 displayedsignals for carboxylic carbons at δ 185.46 (C-26),185.24 (C-18'); vinylic and aromatic carbons betweenδ 150.18-109.23, oxygenated methine carbon at δ
75.69 (C-3) and oxygenated methylene carbon at
δ 67.58 (C-19′). The 1H and 13C NMR spectral data
of 4 were compared with the values of the reportedlanostene type triterpenoids13. The spectral data ofthe dehydroabietic acid unit were compared with thereported values of the similar compounds10-12,14. On thebasis of these results the structure of 3 was elucidatedas lanost-5,7,20(21),22,25(27)-pentaenyl-26-oic acid(3→19')-3'-oxodehydroabietic acid (Fig. 1). This is a
new triterpenoic acid linked with dehydroabietic acidderivative.
Compound 5, designated as roxburghianoic
acid D, was obtained as a brown sticky mass frompetroleum ether-chloroform (1:3) eluants. It producedeffervescences with sodium bicarbonate solution dueto the presence of carboxylic group and showed IR
absorption bands for carboxylic groups (3485, 1695
cm-1) and unsaturation (1645 cm-1). On the basisof 13C NMR and +ve FAB mass spectra, molecularweight of the 5 was established at m/z 778 consistentwith the molecular formula of a lanostenoic acidlinked with abietatrienoic dioic acid, C
50H
66O
7. The
important fragment ion peaks arose at m/z 432[C30
H40
O2]+ and 346 [C
20H
26O
5]+. The subsequent
fragment ion peaks of the diterpenic unit appearingat m / z 300 [346-HCOOH]+, 331 [346-Me]+, 273 [346– CH(CH
3)COOH]+ and 315 [346-CH
2OH]+ suggested
the presence of one oxygenated methylene and two
carboxylic groups in diterpenic moiety. The fragment
ions of the triterpenic unit generating at m/z 137[C
8H
9O
2,side chain, SC]+and 295 [432-SC]+ indicated
the presence of two double bonds in ring A/B and atri-unsaturated C
8-side chain with carboxylic group
and oxygenated methine carbon in ring A at C-3. The1H NMR spectrum of 5 displayed three one-proton
downeld signals consisting of two doublets at δ 6.99(J= 8.1 Hz), 6.86 (J= 2.5 Hz) and a double doublet atδ 7.16 (J= 8.1, 2.5 Hz) ascribed correspondingly toortho-coupled H-11’, meta-coupled H-14’ and ortho-,meta-coupled H-12’ aromatic protons. A two-protonbroad signal at δ 3.44 was assigned to oxygenated
methylene H2-19’ protons and its shifting in the lower
eld indicated its linkage to C-3 of the triterpenic
moiety. The remaining methylene protons of thediterpene unit resonated between δ 1.33-2.71. Adoublet at δ 1.21 (J= 6.6 Hz), and a broad singlet at δ
1.25, three protons each, were ascribed to secondaryH-17' and tertiary H-20' methyl protons, respectively.Three one-proton doublets at δ 5.79 (J= 5.6 Hz),5.73 (J= 5.6 Hz), 5.81 (J= 5.8 Hz) and a one-protonmultiplet at δ 5.63 were ascribed correspondinglyto vinylic H-6, H-7, H-22 and H-23 protons of thetriterpenic unit. Four one-proton broad signals atδ 4.93, 4.92 and at δ 4.88, 4.87 were attributed tomethylene H
2-21 and H
2-27 protons, respectively.
A one-proton double doublet at δ 4.16 (J= 5.5, 9.0Hz) was accounted to H-3 α-carbinol proton. Theremaining methylene and methine protons resonatedbetweenδ 1.42-2.86. Five three-proton broad signalsat δ 0.75, 1.19, 0.98, 0.89 and 0.85 were associatedwith C-18, C-19, C-28, C-29 and C-30 methyl protons,respectively. The 13C NMR spectrum of 5 exhibited
signals for carboxylic carbons at δ 185.01 (C-26),185.23 (C-16'), 183.34 (C-18'), aromatic carbonsbetween δ 123.87-150.23 and vinylic carbonsbetween 112.86-153.01. The oxygenated methine and
oxygenated methylene carbons appeared at δ 75.19
(C-3) and 65.01 (C-19'), respectively. The shifting ofthe oxygenated methylene carbon at δ 65.01 in thedeshielded eld supported linkage of the triterpenic
moiety at C19′
. The 1H and 13C NMR spectral dataof 5 were compared with the values of the reportedlanostene type triterpenoids13. The spectral data ofthe dehydroabietic acid unit were compared with thereported values of the similar compounds10-12,14. Onthe basis of above discussion the structure of 5 was
acid (Fig. 1). This is a new triterpenic linked with
dehydroabietic acid derivative.
Fig. 1: Structures of abietatrien-18-oic acid (1), roxburghianoic acid A (2), roxburghianoic acid B (3),roxburghianoic acid C (4) and roxburghianoic acid D (5)
New phytoconstituents are reported from theoleo-resin of Pinus roxburghii Sarg. for the firsttime which may be used as chromatographic
marker of the drug and for medicinal importanceof the drug.
ACKNOWLEDGEMENT
The authors are thankful to the Head, SophisticatedAnalytical Instrumentation Facility, Central DrugResearch Institute, Lucknow, for recording massspectra of the compounds.
REFERENCES1. Anonymous, The Wealth of India, A Dictionary of
Indian Raw Materials and Industrial Products, RawMaterials, National Institute of Science Communicationand Information Resources, CSIR, New Delhi, 2003,
69-78.2. Rajbhandari K.R.: Ethnobotany of Nepal. Ethnobotanical
Society of Nepal, Kathmandu, 2001.
3. Manandhar N.P.: Plants and people of Nepal. TimberPress Inc. Portland, Oregon, 2002.
12. Georges P., Legault J., Lavoie S., Grenon C., Pichette,
A.: Diterpenoids from the Buds of Pinus banksiana Lamb.,Molecules. 2012, 17, 9716-9727.
13. Chung I.-M., Ali M., Chun S.-C., Lee O.K., Ahmad A.:Sativalanosteronyl glycoside and oryzatriacontolideconstituents from hulls of Oriza sativa, Asian J. Chem. 2007, 19(2), 1535-1543.
14. Ramirez-Macias I., Marin C., Chahboun R., Olmo F.,Messouri I., Huertas O., Rosales M.J., Gutierrez-SanchezR., Alvarez-Manzaneda E., Sanchez-Moreno M.: Invitro evaluation of new terpenoid derivatives againstLeishmania infantum and Leishmania braziliensis. Mem.
Inst. Oswaldo Cruz, Rio de Janeiro. 2012, 107(3), 370-376.
ANALYTICAL METHOD DEVELOPMENT AND VALIDATION OF PERINDOPRILERBUMINE AND AMLODIPINE BESYLATE IN BULK AND TABLET DOSAGE FORM
BY HPLC
Pattan S. R.*, Patni A.C., Mali R.A., Patni C.J. ,
Godge R.K., Bhawar H.S., Merekar A.N. and Marathe R.P.
(Received 24 December 2012) (Accepted 23 February 2013)
ABSTRACT
The objective of this present work was to develop and validate analytical method for quantitativedetermination of perindopril erbumine and amlodipine besylate in bulk as well as in tablet formulation. Thechromatographic separation of the two drugs was achieved on a Varian Microsorb-MV 100-5 C18 column(150×4.6mm, 10 µm). The mobile phase constituted of acetonitrile: buffer (65:35) and pH adjusted to 2.6with ortho- phosphoric Acid was delivered at the ow rate 1mL/min. Detection was performed at 210 nm.
Separation was completed within 6 min calibration curves were linear with correlation coefcient between
0.99 to 1.0 over the concentration range of 2.5 to 15 µg/mL of perindopril erbumine and 10 to 60 µg/mL
of amlodipine besylate The relative standard deviation (R.S.D.) was found <2.3%. The proposed methodis precise, accurate, selective and rapid for the simultaneous determination of perindopril erbumine andamlodipine besylate.
Perindopril erbumine (PE) and amlodipinebesylate (AB) are antihypertensive drugs. Perindopril
erbumine is an angiotensin converting enzymeinhibitor. It is used in the treatment of hypertension.It may be used alone or in the combination withother antihypertensive agents. Amlodipine besylateis a calcium channel blocker. It is also used asantihypertensive agent and in the treatment ofangina. It lowers the blood pressure and relaxes the
heart muscles and dilates the heart blood vessels toprevent the spasm.
The chemical name of perindopril erbumine is2 – methyl propane-2- amine (2S, 3As, 7As)-1-[(2S)-2-2[[(1S)-1-(ethoxycarbonyl)butyl]amine]propanoyl]
P-4000 Pump, SP- thermosepration products AS-3000 Auto-sampler Spectra Physics analytical UV-2000 detector and Column used is Varian Microsorb-MV 100-5 C18 Column (150×4.6mm, 10 µm).
Buffer Preparation
6.8 gm of potassium dihydrogen phosphate isdissolve in 1000 mL of Milli Q water and mixed.
Mobile Phase Preparation
Mix of Acetonitrile: Buffer in the ratio of 65:35 V/V.
Adjust the pH to 2.6 with orthro-phosphoric acid.
Standard Preparation
The gift samples of the drugs perindoprilerbumine and amlodipine besylate were receivedfrom Kaytross ACG Life Sciences Pharmaceuticals(Nashik), India. Standard stock solution of 400 µg/mLand 1000µg/mL were prepared by dissolving 40mgof perindopril erbumine and 100 mg of amlodipinebesylate in 100 mL of mobile phase respectively.From this stock solution working standard solutionhaving concentration 40µg/mL and 100µg/mL wereprepared by appropriate dilution with mobile phasefor perindopril erbumine and amlodipine besylate
respectively.
Sample Preparation
Weight accurately tablets powder equivalent toabout 40 mg and 100 mg of perindopril erbumine andamlodipine besylate respectively. Mix it with 100 mL
of mobile phase to prepare stock solution. Workingsample solutions having concentration of 40µg/mL and100µg/mL were prepared by appropriate dilution withmobile phase for perindopril erbumine and amlodipinebesylate respectively.
Linearity
Several aliquots of standard solutions ofperindopril erbumine and amlodipine besylate weretaken in different 10mL volumetric asks and diluted
up to the mark with mobile phase such that the nal
concentrations were 2.5-15 µg/mL and 10-60 µg/ mL respectively. Evaluation of the two drugs was
performed with UV detector at 210 nm, peak arearecorded for all the peaks. The slope and interceptvalue for calibration curve R2 = 0.9997 for perindoprilerbumine and R2 = 0.9999 for amlodipine besylate.
The results show that an excellent correlation existsbetween peak area and concentration of drugs withinthe concentration range indicated.
Assay
50 µL of each standard and sample solutions wereinjected and from the peak area of PE and AB, eachamount of drug in samples were computed.
Method Validation8
Limit of Detection and Limit of Quantication
The limit of detection (LOD) and limit ofquantification (LOQ) of the developed methodwere determined by injecting progressively lowconcentrations of the standard solutions using thedeveloped HPLC method. The LOD for perindoprilerbumine and amlodipine besylate were found to be0.04 and 0.28 µg /mL respectively. The LOQ was0.25 and 0.90 µg/mL for perindopril erbumine andamlodipine besylate respectively.
Ruggedness and Robustness
The ruggedness of the method was determined bycarrying out the experiment on different instruments by
different operators using different columns of similartypes. Robustness of the method was determinedby making slight changes in the chromatographicconditions. It was observed that there were no markedchanges in the chromatograms, which demonstratedthat the HPLC method so developed is rugged androbust.
Recovery Studies
To study the accuracy and reproducibility of theproposed method recovery experiments were carried
out. A xed amount of preanalyzed sample was taken
and standard drug was added at 80%, 100% and120% levels. Each level was repeated three times.The contents of perindopril erbumine and amlodipinebesylate per tablet found by proposed method are
Linearity range 2.5-15 µg/mL 10-60 µg/mLCorrelationcoefcient
0.9997 0.9999
LOD 0.04 0.28
LOQ 0.25 0.90
Recovery(n=3) 100.98 100.19
Precission(% RSD)
Intra-day(n=3) 0.90 1.00
Inter-day(n=3) 0.81 0.75
shown in Table III the lower values of RSD of assayindicate the method is accurate. The mean recoveriesof PE and AB were in range of 100.98 % and 100.19%that shows there is no interference from excipients.
RESULTS AND DISCUSSION
The HPLC procedure was optimized with aview to develop accurate assay method in a tabletdosage form using Varian Microsorb-MV 100-5 C18
Column (150×4.6mm, 10 µm) in isocratic mode,with mobile phase composition of acetonitrile: buffer(65:35V/V). The ow rate was 1mL/min and both the
components were measured with UV Detector at210nm. The retention time of PE and AB was 2.77and 5.67 min respectively. The method was linearin the range of 2.5 – 15 µg/mL for PE and 10 – 60μg/mL for AB with correlation coefcient 0.9997 and
0.9999 respectively with good linearity responsegreater than 0.998. The % recovery was found tobe 100.98% for PE and 100.19% for AB. The %RSDfor intra-day and Inter-day precision is shown inTable III. The sensitivity of method LOD and LOQ
is shown in Table III. Typical chromatograms of thesample and the standard is shown in Fig. 1 & 2. Theassay procedures were repeated for Five times andthe percentage of individual rugs and the results werefound to give 101% of PE and 101.75% of AB.
Linearity of Perindopril Erbumine
Regression analysis of the cal ibrationcurve for perindopril erbumine showed a linear
Fig. 2: Typical chromatogram of the standard ofPerindopril Erbumine and Amlodipine Besylate
Fig. 1: Typical chromatogram of sample containingPerindopril Erbumine and Amlodipine Besylate
relationship between the Concentration andResponse factor, with correlation coefficientshigher than 0.9997.
Linearity of Amlodipine Besylate
Regression analysis of the calibration curve forAmlodipine Besylate showed a linear relationshipbetween the Concentration and Response factor, withcorrelation coefcients higher than 0.9999 .
CONCLUSION
Proposed study describes HPLC method for theestimation of perindopril erbumine and amlodipinebesylate in bulk drugs as well as in tablet formulation.
The method was validated and found to be simple,sensitive, accurate and precise. Therefore theproposed method can be used for quantication of
perindopril erbumine and amlodipine besylate inbulk drugs as well as for routine analysis in qualitycontrol.
ACKNOWLEDGEMENT
The authors wish to thank Planning andDevelopment Department University of Pune for
nancial assistance for this project and Dr. ShashankDalvi Hon. Vice-Chancellor Pravara Institute ofMedical Sciences, Deemed University Loni, for kindsupport and encouragement.
REFERENCES1. K.D. Tripathi’s Essentials of medical Pharmacology, 5th
edition, Jaypee Brothers Medical Publishers (P) Ltd.,2003, p 488, 496, 497.
2. O’Neil M. J., Edn The Merck Index- An Encyclopedia of
Chemicals, Drug and biological, 14th
edition, Merck & co.,Inc, 2006, p 6836
3. British Pharmacopoeia the department of health, London2008.
4. Goodman and Gilman’s. The pharmacological basis ofTherapeutics. 8th edition, New York: McGraw Hill; 1992.P. 718-721
5. Indian Pharmacopeia 6th edition. New Delhi: The IndianPharmacopial Commission; 2010.
6. Williams A. 2004. Foye’s Principles of medicinal chemistry5th edition, published by, B. I. Publications Pvt. Ltd.
7. Sethi P. D. Quantitative analysis of drug in a pharmaceuticalformulation. 3rd edition New Delhi: CBS publishers anddistributers, 1997 p 50-57
8. Part-2 Validation of analytical procedure: MethodologyQ2B, ICH Harmonized Tripartite Guidelines, 1996;6-12.
9. Safeer K, Anabarasi B. N. Senthil kumar, analyticalmethod development and validation of amlodipine andhydrochlorothiazide in combined dosage form by RP-HPLC Int. Journal of Chem tech research, 2010, vol.2,No.01, p 21-25.
10. Patil P. S., More H. N., Pishwikar S. A., RP- HPLC methodfor simultaneous estimation of Amlodipine Besylate andOlmesartan Medoxomil from tablet, Intr. Journal of
pharmacy and pharmaceutical sciences, Vol. 3, suppl3, 2011, 146-149.
11. Prameela Rani A., Bala Sekaran C., A validated RP-HPLCmethod for the determination of perindopril Erbumine inpharmaceutical formulation. Int. J.Chem tech research,2009,vol.1, No. 3, p 575-578.
Available DVD of IP – 2010
DVD of IP – 2010 is available from the ofce of the Secretary-cum-Scientic Director,I.P.Commission, Sector-23, Raj Nagar, Ghaziabad – 201002, (U.P.), @ Rs.25,000/-perDVD.For more details please visit the website: www.ipc.gov.in; E.mail Id: [email protected] contact to The Publication Division, I.P.Commission, Tel.Nos.: 0120-2783392, 2783400,Extn.309, 308 & 112.
DEVELOPMENT OF HPTLC METHOD FOR SIMULTANEOUS ESTIMATION OFCIPROFLOXACIN AND ORNIDAZOLE IN COMBINED DOSAGE FORM
Dighade N. R.*, Shende M. D. and Kasture A. V.
(Received 11 October 2012) (Accepted 23 February 2013)
ABSTRACT
A simple and accurate high performance thin layer chromatographic (HPLTC) method has beendeveloped and validated as per ICH guidelines for estimations of Ciprooxacin (CP) and Ornidazole
(ORN) in combined dosage form. The mobile phase was acetonitrile: toluene: water and triethylamine(5.5:1.8:1.5:1.6 V/V) was found to be best which gave high resolution with R f 0.16 and 0.84 forciprooxacin and ornidazole respectively. The linearity of ciprooxacin and ornidazole was found to be
in the range of 0.4 to 0.8 μg/mL and 0.4 to 0.8 μg/mL, respectively. The coefcient of correlation (r2) wasfound to be greater than 0.989 for both the components by this method. The tablet analyses result (n =5) were found to be > 100.84 % by HPTLC for both the components. The proposed method was found
to be simple, accurate and suitable for routine quality control of marketed formulations containing thesedrugs.
Keywords: High Performance Thin LayerChromatography, Ciprooxacin and Ornidazole.
INTRODUCTION
Ciprofloxacin, (1-cyclopropyl-6-fluoro-1,
4-dihydro-4-oxo-7-(piperazin-1-yl) quinoline-3-
carboxylic acid (CP), a synthetic uroquinolone, has a
bactericidal mode of action which is achieved throughinhibition of DNA gyrase, an essential component ofthe bacterial DNA replication system. Ornidazole,(RS)-1-chloro-3-(2-methyl-5-nitroimidazol-1-yl)propan-2-ol (ORN), is an antiamoebic and is indicatedin intestinal and extraintestinal amoebiasis, as
prophylaxis after abdominal surgery. The structures
of these drugs are given in Fig. 1.
Methods such as spectrophotometric1-3,NMR4, spectrouorimetric assay5, capillary zone
electrophoresis 7 and TLC- uorimetry8 are reportedfor determination of ciprofloxacin alone or in
combination with other agents. The literature surveyrevealed methods for determination of ornidazole
obtained form Zim Laboratories, Kalmeshwar,Nagpur and Vapi Care Pharma Pvt. Ltd., Gujarat.Analytical and HPLC grade chemicals and solventsof Merck and Qualigens Fine Chemicals were used.Ocimix tablets, were procured from the local market
containing ciprooxacin (500 mg) and ornidazole
(500 mg) per tablet.
Instrumentation
CAMAG HPTLC system consisting of CAMAGLINOMAT-IV as sample applicator was used forHPTLC analysis. CAMAG TLC SCANNER III(densitometer) with CAT’s 4.0 version software forscanning and documentation, UV cabinet tted with
dual wavelength 254/366 nm, sw, UV lamp for visualinspection of HPTLC plates and CAMAG TWINTROUGH glass chamber with stainless steel lidswere used for chromatographic development.
Preparation of mobile phase: Mobile phaseconsisted of acetonitrile, toluene, water andtriethylamine in the ratio of 5.5: 1.8: 1.5: 1.6 V/V.
Preparation of standard solution
i) Solution A: An accurately weighed equivalentquantity of about 25 mg of CP was transferred to25 mL volumetric ask (29 mg of Ciprooxacin
hydrochloride ≅ 25 mg of Ciprooxacin) and
was dissolved in 1 mL of water and 10-15 mLof methanol. Volume was made up to the markwith methanol (conc. 1 mg/mL). 1 mL of abovesolution was transferred to 10 mL volumetricask and volume was made up to the mark with
methanol (conc. 0.1 mg/mL).
ii) Solution B: An accurately weighed quantity ofabout 25 mg of ORN was transferred to 25 mLvolumetric ask and was dissolved in 10-15 mL
methanol and volume was made up to the mark
with methanol (conc. 1 mg/mL). 1 mL of abovesolution was transferred to 10 mL volumetricask and volume was made up to the mark with
methanol (conc. 0.1 mg/mL).
iii) Solution C: An accurately weighed equivalentquantity of about 25 mg of CP and 25 mg of ORNwas transferred to 25 mL volumetric ask and
was dissolved in 1 mL of water and 10-15 mLof methanol. Volume was made up to the markwith methanol (conc. CP 1 mg/mL and ORN 1mg/mL). 1 mL of above solution was transferredto 10 mL volumetric ask and volume was made
up to the mark with methanol (conc. 0.1 mg/mLof CP and ORN).
Chromatographic conditions
The following chromatographic conditions wereoptimized by trial and error for effective separationand densitometric evaluation of drugs.
Stationary phase Aluminium platesprecoated with silica gel60 F
254 (Merck)
Mobile phase Acetonitrile:Toluene:Water:Triethylamine
(5.5:1.8:1.5:1.6 V/V)Plate size 10 cm x 10 cm,
Thickness: 200 µmMode of application BandBand size 4 mmDistance between
two bands
6 mm
Sample volume 4 µL
Developing chamber Twin-trough glasschamber, 10 cm x 10 cm
with stainless steel lid.Saturation time 10 minutes
After the application of standard solution C withthe help of CAMAG LINOMAT-IV applicator, the
plate was chromatographed in twin-trough glasschamber saturated with mobile phase for 10 minutes.After chromatographic development, the plate wasremoved and air dried. The separated bands on theTLC plates were scanned over the wavelength rangeof 200-360 nm. The wavelength 299 nm was selectedfor densitometric evaluation of separated bands. Theoverlay spectrum obtained is depicted in Fig. 2.
Preparation of calibration curve
The standard solution C containing mixture ofCP and ORN was applied on the TLC plate in therange of 1-10 µL with the help of microsyringe usingLINOMAT-IV automatic sample applicator. The platewas then developed and scanned under the abovementioned chromatographic conditions.
Peak height and peak area were recorded foreach drug concentration and the calibration curves ofconcentration vs peak height/ area were constructedfor both the drugs.
The calibration curve for CP and ORN are depictedin Fig. 3 and 4, respectively.
Analysis of laboratory mixture
i) Standard solution:Standard solution C was used(conc. CP 0.1 mg/mL and ORN 0.1 mg/mL).
ii) Preparation of laboratory mixture (sample):
Five samples of laboratory mixture were prepared
as follows:
Sample solution: An accurately weighedquantity equivalent of about 25 mg of CP and 25mg of ORN was transferred to 25 mL volumetricask and dissolved in 1 mL of water and 10-15 mL
of methanol. The volume was made up to the markwith methanol. 1 mL of above solution was transferred
Fig. 2: Overlay Spectra of Ciprooxacin and Ornidazole
Fig. 3: Calibration Curves for CP Fig. 4: Calibration Curves for ORN
to 10 mL of volumetric ask and volume was made
up to mark with methanol. This solution was used assample solution.
On TLC plate two bands of standard and eightbands of sample solution, 4 µL each, were appliedand the plate was developed and scanned under
the optimized chromatographic conditions. Afterscanning, the chromatograms obtained for standardand sample were integrated.
Typical densitograms obtained for standard andlaboratory mixture (sample) are shown in Fig. 5.
The amount of CP and ORN present in appliedvolume of standard solution was fed to the computer.
Amount of both the drugs present in applied volumeof sample solution was obtained by comparisonbetween peak height and peak area of standard andsample bands.
The total amount of CP and ORN estimated inlaboratory mixture was calculated by using following
formula:
The results of estimation of CP and ORN inlaboratory mixture are shown in Table I.
The results obtained indicate that CP and ORNcan be estimated accurately in laboratory mixture.
Thus, it was thought worthwhile to extend the same
method to marketed formulation.
Application of proposed method to marketed
formulation
Standard solution:Standard solution C was used(conc. CP 0.1 mg/mL and ORN 0.1 mg/mL).
Sample preparation
Twenty tablets were accurately weighed andaverage weight was calculated. The tablets werethen crushed to obtain ne powder. An accurately
weighed quantity of tablet powder equivalent to about25 mg of CP was transferred to 25 mL of volumetricask and shaken with 1 mL of water and 10-15 mL of
methanol for 10 minutes. The volume was made up tothe mark with methanol and then solution was ltered
through Whatman grade 1 lter paper. 1 mL of above
ltrate was transferred to 10 mL volumetric ask and
volume was made up to the mark with methanol. Thissolution was used as a sample solution.
On TLC plate two bands of standard and eightbands of sample solution, 4 µL each, were appliedand the plate was developed and scanned underthe optimized chromatographic conditions. Afterscanning, the chromatograms obtained for standardand sample were integrated.
The amount of each drug present in appliedvolume of standard solution was fed to the computer.Amount of both the drugs present in applied volume
of sample solution was obtained by comparisonbetween peak height and peak area of standard andsample bands.
Amount of each drug estimated in average weightof tablet was calculated by following formula:
The results of estimation of CP and ORN inmarketed formulation are shown in Table I.
System Suitability
The system suitability test was performedby few repeated application for the standardsolution containing 100 μg/mL of ciprooxacin and
ornidazole. The results obtained by repeating theestimation procedure ve times were observed to
be good.
Validation
It was carried out for the following parameters asper USP guidelines.
A. Accuracy: Accuracy of proposed method wasascertained on the basis of recovery studiesperformed by standard addition method.Accurately known amounts of standard drugs
Fig. 5: Densitograms of Ciprooxacin (1) and
Ornidazole (2) for Standard and Marketed Formulation(Ocimix)
were added to known amount of pre analyzedtablet powder and it was analyzed by the proposedmethod to ascertain if there are positive ornegative interferences from excipients present in
formulation. The per cent recovery was calculatedby using following formula.
A - B% Recovery = ---------- x 100
C
Where A = total drug estimated in mg; B= amountof drug contributed by tablet powder (as perproposed method); C= amount of pure drugadded.
The results of accuracy studies are shown inTable II.
B. Precision: Precision of an analytical methodis expressed as S.D. or R.S.D. of series of
measurements. It is ascertained by replicateestimation of the drug by proposed method. Theresults are shown in Table II.
C. Specicity: The specicity studies were carried
out by attempting deliberate degradation to thetablet sample with exposure to stress conditions
like acidic (0.1 M HCl), alkaline (0.1 M NaOH),
oxidizing (3% H2O2), heat (60
o
C), and UV.i) Standard solution: Solution C was prepared
as described under preparation of standardsolutions (conc. CP 0.1 mg/mL and ORN0.1 mg/mL).
ii) Sample solution: An accurately weighedquantities of preanalyzed tablet powderequivalent to 25 mg of CP were taken in ve
different 25 mL volumetric aks and were
stored for 24 hours under different conditionsas follows.
i) At 50 oC, in 1 mL 0.1 M HCl (Acid) – Acidic
Hydrolysisii) At 50oC, in 1 mL 0.1 M NaOH (Alkali) –
Alkaline Hydrolysis
iii) At 50oC, in 1 mL 3% H2O
2 (Oxide) -
Oxidation
iv) At 60oC (Heat) – Thermal degradation
v) UV-chamber - Photolysis
After 24 hrs, all the asks were removed and
allowed to cool. The samples were then analyzed
in similar manner as described under marketedformulation. The results of specicity studies are
shown in Table II.
Linearity and Range: The study was performedby applying different volumes of standard solutionon TLC plate, developing and scanning the plateas per the optimized chromatographic conditions.The concentration vs response curves wereplotted as per area and height. The results arerecorded in tabular form in Table III.
D. Ruggedness: The ruggedness of the proposedmethod was ascertained by carrying out theanalysis of marketed formulation in the similarmanner as described earlier, under three differentconditions i.e., on different times (morning,afternoon and evening) on the same day, ondifferent days and by different analysts.
The results are shown in Table II.
Table III: Results of Linearity Studies
Drug
Linearity range (µg)Coefcient
correlationSlope Y-Intercept
BY Height By Area BY Height By Area BY Height By Area BY Height By Area
The mobile phase consisting of acetonitrile:toluene: water: triethylamine in the ratio 5.5: 1.8: 1.5:1.6 V/V was found suitable as it gave good resolution
of both drugs. The Rf values were found to be 0.16and 0.84 for CP and ORN respectively.
The densitometric evaluation of chromatogramwas carried out at 299 nm as both the drugs havesufcient absorbance showing better sensitivity.
CONCLUSION
The proposed HPTLC method was simple, rapid,precise and accurate and can be conveniently adoptedfor the routine quality control analysis of ciprooxacin
and ornidazole from its pharmaceutical formulationand bulk drug. The highlight of the method is short runtime which signicantly reduces the analysis time.
ACKNOWLEDGEMENT
The author gratefully acknowledge ZimLaboratories and Vapi Care Pharma Pvt. Ltd. Forproviding gift samples of pure drugs and Departmentof Pharmaceutical Sciences, Rashtrasant TukadojiMaharaj Nagpur University, Nagpur for providing
the necessary facilities for carrying out this researchwork.
REFERENCES1. Sharma R., Pathodiya G., Mishra G. P., Jitendra S.,
Spectrophotometric method for quantitative determinationof ciprooxacin hydrochloride and tinidazole in tablets
using hydrotropic solubilizing agent, J. Pharm. Res., 2011,4(3),859-861.
2. Patel S. A., Patel N. M., Patel M. M., Simultaneousspectrophotometric estimation of ciprooxacin and
ornidazole in tablets , Indian J. Pharm. Sci., 2006,10,
665-667.3. Jadhav V., Kashew J. K., Mishra P., Spectrophotometric
method for estimation of ciprooxacin hydrochloride in
tablets, Indian J. Pharm. Sci., 2005, 8, 482-484.
4. Reinscheid U. M., Direct determination of ciprooxacin
in admixtures with metronidazole and ampicillin by NMR,
J. Pharm. Biomed. Anal., 2005, p. 22.
5. Durmus Z., Canel E. Kiliq E., Spectrouorimetric assay
of ciprooxacin hydrochloride in tablets, Anal. Quan.
Cytol. Histol., 2005, 27(3),162-66.6. Shervington L.A., Abba M Hussain B, Donnelly J.,Simultaneous separation and determination of ve
quinoline antibiotics using isocratic RP-HPLC: Applicationto stability studies on an ciprooxacin tablet formulation,
J. Pharm. Biomed. Anal., 2005,39 (3-4),769-775.
7. Michalska K., Pajchel G, Tyski S. J., Determinationof ciprooxacin and its impurities by capillary zone
electrophoresis, Chromatogr. A., 2004,1051(1-2),267-272.
8. Feng Y. L., Dong C. J., Simultaneous determination oftrace of loxacin, ciprooxacin and sparoxacin by micelle
TLC-uorimetry, Chromatogr., 2004, 42 (9),474-477.
9. Patel P.U., Suhagia B.N., Patel C.M., Patel M.M., PatelG.C., Patel G.M., Simultaneous spectrophotometricestimation of gatioxacin and orindazole in mixture,
Indian J. Pharm. Sci.,2005, 3, 356-357.
10. Reddy M., Narayanan R., Satyamargan GUH., Rao,KNV., Quantitative determination of ornidazole inpharmaceutical dosage form by spectrophotometry , J.
Inst. Chem., 2000, 72, 157-158.
11. Manjunath S., Ravi Raju KUM, Appala S., Simultaneousestimation of ornidazole and tinidazole , Indian Drugs.,1990, 25, 346-371.
12. Toral Inesm, Soto Cesar., Estimation of azomycin andornidazole by derivative spectroscopy , Quim Sociedad
Chilenade Quimica., 1998, 43, 349-357.
13. Soma Shekhar M., Vidya Sagar J., Narsaiah N., AnandKumar R., Krishna D R., Validated HPLC method for thedetermination of ornidazole in human serum and urine, Indian J. Pharm. Sci., 2005, 3, 302-306.
14. Heizmann P., Geschke, R. Zinapold, K. Determinationof ornidazole and its main metabolites in biological uid,
J. of Chrom. B., 1990, 534, 233-240.
15. Kale V. N., Naidu K R., Shingare M .S., Simultaneousdetermination of norfloxacin and ornidazole in
pharmaceutical dosage by RP-HPLC, Indian Drugs.,2003, 40,397-400.
16. Padhye V. V., Kachhwaha S.J., Dhaneshwar S.R.,Electrochemical method for the estimation ofmetronidazole, ornidazole and tinidazole, East. Pharm.,1999, 42 (503), 121-122.
ANTIBACTERIAL ACTIVITY OF BARK EXTRACTS OF FICUS GLOMERATA ROXBAGAINST SOME GRAM POSITIVE AND GRAM NEGATIVE BACTERIA
Bhinge S. D.*, Hogade M. G., Savali A. S., Chitapurkar H. R., and Magdum C. S.
(Received 23 November 2012) (Accepted 16 March 2013)
ABSTRACT
Petroleum ether, ethanol and aqueous extracts of the bark of Ficus glomerata (Urticaceae) werescreened for their antibacterial activity using agar diffusion method. They were tested against six bacteria;
three Gram-positive bacteria (Bacillus subtilis, Bacillus aureus and Staphylococcus aureus ) and threeGram-negative bacteria (Escherichia coli , Klebsiella pneumoniae , and Pseudomonas aeruginosa) . Thesusceptibility of the microorganisms to the extracts of these plants was compared with each other and
Urticaceae), locally known as Gular, is abundantlyavailable in India. It has been used since olden timesin the ethnomedicine for many varied medicinalpurposes including as a remedy of diabetes mellitus1.In addition, they are known to possess tonic,expectorant, emollient, stomachic and carminative
properties2. The sap extracted from trunk of the treeis also considered curative in diabetes. Moreover,the powdered seeds mixed with pure bee-honey
are prescribed in diabetes in folklore medicine3, 4. Itis used in dental preparations5, 6. It is also useful intreatment of asthma, piles and skin diseases. Thetree is found to be stomachic, carminative and usefulin the treatment of menorrhagia. The bark is usefulin treating diabetes7.
MATERIALS AND METHODS
Plant materials
The bark of Ficus glomerata (Family -Urticaceae) was collected from Atpadi, Dist
Sangli (Maharashtra, in Dec 2009); and a voucherspecimen no BHISOFIR2 has been deposited atthe herbarium by Botanical Survey of India, Pune(Maharashtra).
Preparation of the crude extracts
The plant material was washed with distilledwater, dried in shade and powdered with the help ofan electric grinder. The coarse material was extracted
successively with petroleum ether, ethanol andwater. The extracts were dried at 50°C in a water
bath. The percentage yields obtained of the differentsuccessive extracts were 10.20% and 8.50%, w/w
respectively.
Preparation of test samples
Stocks of petroleum ether (60 to 80oC fraction),ethanol and aqueous extracts of Ficus glomerata
were prepared in sterile distilled water (1mg/mL).Further test dilutions were made ranging from 10µg/ mL to 100µg/mL in sterile distilled water8.
Test Bacteria
The bacterial cultures employed in this study areBacillus subtilis, Bacillus aureus, Staphylococcus
aureus , Escherichia coli , Klebsiella pneumoniae
and Pseudomonas aeruginosa obtained from thedepartment of Microbiology, Gulbarga University,Gulbarga (Karnataka).
The bacterial suspension was prepared bytransferring a loopful of inoculum into 1mL sterilesaline solution from the stock culture maintained at
40C in 10mL nutrient broth8.
Preparations of Plates
Nutrient agar medium was sterilized at 15lb/cm2 pressure for 20 min in an autoclave. Then 15mL ofmedium was poured in each petri plate under sterileconditions9.
Antibacterial Assay
Each extracts were tested against Gram-
positive bacteria (Bacillus subtilis, Bacillus aureusand Staphylococcus aureus ) and Gram-negativebacteria (Escherichia coli , Klebsiella pneumoniae ,Pseudomonas aeruginosa). Antibacterial activity wasdetermined by pour plate method in sterile nutrientagar medium plate. The 6.0 mm wells were madeon each petri plate. Plate were allowed to stand for1hr and inoculated with 1mL extract. Respective
dilutions ranging from 10µg/mL to 100 µg/mL wereprepared. The applied sample extracts were allowed
to diffuse properly by keeping the petri plates in
refrigerators at 40C for 4hr. Then the petri plates
were transferred to incubation chamber for 24hr at370C. The diameter zone of inhibition in mm wasmeasured10, 11.
RESULTS AND DISCUSSION
The petroleum ether, ethanol and aqueousextracts of the bark of Ficus glomerata were subjectedto a preliminary screening for antimicrobial activityagainst six bacteria i.e. Bacillus subtilis, Bacillus
aureus, Staphylococcus aureus, Escherichia
coli , Klebsiella pneumoniae and Pseudomonas
aeruginosa. It is clear from Tables I, II and III thatpetroleum ether extract of the bark of Ficus glomerata
did not show any activity against all organisms tested.
The ethanol extract showed low activity againstB.
subtilis ,B aureusand S. aureusand signicant activity
against E. coli, K. pneumoniae and P. aeruginosa.
Aqueous extracts shown the low activity against
B aureus and S. aureus and high activity against
E.coli, K. pneumoniae and P. aeruginosa, and it isinactive against B. subtilis . This antibacterial activitymight be supported by these chemicals i.e alkaloids,glycosides, saponins, cardiac glycosides, tannins,xed oils, tannic acid and simple phenol compounds
present in plants material. 100µg/mL ethanolic
Table I: Preliminary screening for antimicrobial activity of Bark of Ficus glomerata against Standard Gram
We acknowledge Prof. Kishore Singh Chatripati,President, RMES’s college of pharmacy, Gulbarga
REFERENCES1. Akhtar M. S., Qureshi A. Q., Phytopharmacological
evaluation ofFicus glomerata, Roxb. fruit for hypoglycaemic
activity in normal and diabetic rabbits, Pak J Pharm Sci.1988, 1(2), 87-96.
2. Satyavati G. V., Raina M. K., Sharma M. Medicinal Plantsof India, New Delhi: Indian Council of Medical Research,1976, 1, 377.
3. Nadkarni A.K., Indian Mated Medica, Popular PrakashanBomaby, 1976, 548-550.
4. Chopra R. N., Glossary of Indian Medicinal Plants; Councilof Scientic and Industrial Research, New Delhi. 1956,
19.
5. The Wealth of India; CSIR. New Delhi. 2006, I, 370.
6. Kitrtikar K. R., Indian Medicinal Plants, International Bookdistributors, Dehradoon, 1999, III, 2327-2329.
7. Nadkarni K. M., Indian Materia Medica, PopularPrakashan, Bombay, 2005, I, 48.
8. Prahalathan P., Bragadeeswaran S., Sasikala R.,
Kanagaraj U., Kumaravel P., Antimicrobial And HemolyticActivities Of Marine Sponge - Halichondria Panicea, J
Herb Med Toxic. 2009, 3(2), 45-48.
9. Anas, K., Jayasree P. R., Vijaykumar T., ManishkumarP R., In vitro antibacterial activity of Psidium guagava Linn. Leaf extract on clinical isolates of multidrug resistant
10. Arthur L. B., Marie B. C., Clyde T. Gerlach E. H., RonaldW. H., Methods of Measuring Zones of Inhibition with theBauer- Kirby Disk Susceptibility Test, J Clini Microbio. 1979, 10(6), 885-889.
11. Devkar S., Jagtap S., Kale Y., Kasote D., Antibacterial
activity of Eulophia Ochreata L. Tubers, J Herb MedToxic. 2009, 3(2), 31-33.
ENVIRONMENTAL MONITORING IN CORRECTIVE/PREVENTIVE ACTIONS
CLEANROOMS (CAPA) GUIDELINE
Copies are available at IDMA Ofce, Mumbai. We do not mail any publications against VPPpayment. All payments to be made in advance as DD only in favour of “INDIAN DRUG
MANUFACTURERS’ ASSOCIATION” at Mumbai.
For More Details Please Contact: PUBLICATIONS DIVISION. Ms. Prachi
DEVELOPMENT OF VALIDATED RP-HPLC METHOD FOR THE QUANTITATIVEANALYSIS OF CHROMIUM PICOLINATE III IN A DIET VINEGAR FORMULATION
Lodhi A., Jain A.* and Biswal B.
(Received 04 January 2013) (Accepted 12 February 2013)
ABSTRACT
A validated high performance liquid chromatographic method was developed for the determination ofchromium picolinate in pharmaceutical dosage forms. The analysis was performed at room temperatureusing a reversed-phase ODS, 5µm (250×4.6) mm column. The mobile phase consisted of acetonitrile:buffer (60:40 V/V) at a ow rate of 0.5 mL/min. The PDA-detector was set at 264 nm. The developed
method showed a good linear relationship in the concentration range from 1.5 – 12.5 µg/mL with acorrelation coefcient from 0.999. The limit of detection and limit of quantication were 0.0540513 and
Chromium picolinate (III) is very well known as anessential mineral. It is suggested as a cofactor in themaintenance of both normal lipid and carbohydratemetabolism by assisting the action insulin on a cellmembrane. Chromium picolinate (III) is lipophilic,
which facilitates its entry in to and through theplasma membrane of cells1, 2. In the skeletal musclecells, the rate of insulin was increased and theuptake of both glucose and lucine was improved.Chromium picolinate (III) as a nutritional supplement(Diet Vinegar Formulation) and weight-loss agenthas been commercially successful and known tohave benecial effects on carbohydrate and lipid
is generally compared with the standard material. Inthe present research, we studied the determinationof chromium picolinate (III) using RP-HPLC anddetermined the chromium picolinate (III) in diet vinegarformulation6.
EXPERIMENTAL
Reagents & Apparatus
Diet vinegar formulation, chromium picolinate wereobtained from ZIM Laboratories Ltd., Nagpur, Acetonitrileused for HPLC analysis was of chromatographicgrade and purchased from merck. Water was puriedby doubled distilled system. The octadecyl bondedcolumn was used. HPLC was performed using WatersHigh Performance Liquid Chromatography with PDA-detector. Optimal conditions of high performance liquidchromatography were found to be; ow rate = 0.5 mL/
min, wavelength = 264 nm, column oven temperature =25-30oC, and mobile phase composition = acetonitrile: buffer = 60:40.
Instrumentation
Waters 600E HPLC system and column was used.Separation and quantitation was done on C
8column
i.e., (250 X 4.6 mm 5 µm particle size) column
Chromatographic Conditions
Using C8 column i.e., (250 X 4.6 mm 5 µm particlesize) column. The mobile phase was prepared by
Standard solution of dipyridamole was prepared atthe concentration of 10 µg mL-1 dissolving appropriatedamount of standard in the mobile phase. This standardsolution was used to quantify active and nal product
taken and diluted to 10 mL with the mobile phase toyield dilution of 10 μg/mL. Then the nal solution was
ltered through 0.45 micron Whatman lter paper and
sonicated for HPLC analysis.
Method Validation
Method validation was done as per ICH guidelinesand accordingly the parameters evaluated were: 7
1. Linearity
2. Precision
3. Robustness
4. Ruggedness
5. Accuracy
6. System suitability
Linearity
The linearity of analytical procedure is its ability(within given range) to obtain test results which aredirectly proportional to concentration in sample. Thiswas studies by analyzing ten concentrations withinthe range of 1.5 – 12.5 µg/mL solution of chromiumpicolinate. A graph was plotted in μg/mL on X axis
versus response on Y axis.
Precision
The precision of analytical procedure express the
closeness of agreement (degree of scatter) betweenseries of measurement obtained from multiplesampling of the same homogeneous sample underprescribed condition. The precision of the methodwas demonstrated by:
System Precision: Standard solution of chromiumpicolinate was injected in 10 replicate injections and%RSD was calculated.
Method Precision: Analyzing six replicate
injections of chromium picolinate standards andsample solution. Percentage assay of sample tothat of label claim was calculated by comparing the
sample solution response to that of standard solutionresponse. %RSD of assay result was calculated.
Intermediate Precision: Intermediate precisionexpress within laboratory variation. Two analyst on
different HPLC system conducted and analyst toanalyst variability study by assaying six different test
preparations of chromium picolinate8.
Robustness
The robustness of analytical procedure is a
measure of its capacity to remain unaffected by small,but deliberate variation in method parameters andprovides an indication of its reliability during normalusage and done by changing,
1. Inuence of variation of pH in the mobile phase
(±0.2).
2. Inuence of variation of ow rates (±10%).
Ruggedness (system to system variation)
Ruggedness of analytical method is degree of
reproducibility of test result obtained by the analysisof the same sample under verity of condition such aslaboratory, analyst and instruments. System to systemvariation was conducted on two HPLC systems by usingthe same column by assaying six different test preparation
of chromium picolinate (10µg/mL) as per method9,10.
Accuracy
Recovery study performed for the accuracy.Recovery spiked placebo was conducted. Sample
solution was analyzed in triplicate for eachconcentration level and assayed as per method. Theresults from recovery study for accuracy determinationare depicted in the Table I.
HPLC method development and optimization offollowing parameters are shown in Fig. 1 & Table I.
Linearity
The data obtained in linearity experiments was
subject to linear regression analysis. The coefcient
of regression (r2) was found to be 0.9997. Linearitydata and plot are reported in the Table I and Fig. 2.
Method Precision
The precision of test method was evaluated byassaying six sample of chromium picolinate (10μg/
mL). %RSD of assay was found to be 0.076885.
Results are given in the Table I.
Intermediate Precision
Two analyst on different HPLC system conductedanalyst to analyst variability study by assaying six different
test preparation of chromium picolinate. The average %
assay obtained by both analysts was found to be 98.7and 98.7 with RSD of 0.4% and 0.3% respectively.Thesystem suitability parameter were evaluated as permethod by both analyst and found to be within limits.The limits are summarized in the Table I.
Robustness
No signicant change in the chromatographic
parameters were observed when change in theoptimized condition like change in the pH and ow
rates. The results are summarized in the Table I.
Fig. 2: Calibration curve of chromium picolinate (III)
Fig. 1: Representative chromatogram of the Chromium picolinate (III) (standard) underoptimized condition, concentration: 10 µg/mL in mobile phase (ACN;buffer = 60:40) ; ow
System to system variability was conductedby two HPLC systems by using same column byassaying six different test preparation of chromium
picolinate in same condition. The system suitabilityparameter was found to be within limits. The averageassay for system was found to be 98.7 and 98.7 withvalues between centrifuged and ltered samples to
be within limits. This study indicates that both lters
are suitable for ltration.
Accuracy
The results from recovery study for accuracydetermination are depicted in the Table I. Recovery
from spiked placebo was conducted. Samplesolution was analyzed in triplicate for eachconcentration level and assayed as per method.The percentage recovery was found to be withinthe limits (97.5-100.4%). The mean recovery ofchromium picolinate should not be less than 97%and not more than 103%.
System Suitability
All the values of parameter i.e. USP plate count,USP tailing (1.3) and %RSD of six replicate injections
(0.1%) of system suitability were found to be within
in the acceptable limits. It is concluded that themethod and systems are adequate for the analysisto be performed.
CONCLUSION
The developed method was validated in termsof specicity, precision, accuracy, LOD and LOQ
as per ICH guidelines and it was found that all theparameter lie within the limit prescribes by ICH. LOD& LOQ was found to be 0.0540513 and 0.1637919
respectively.
The proposed analytical method has beenproved to be simple, specic and accurate which
fulll all the parameters of the validation. Therefore
this method has been successfully applied to dietvinegar formulation.
Authors are very thankful to Jim Laboratory,Nagpur for providing chemicals and this researchproject and to B. R. Nahata college of Pharmacy for
providing all the facility to complete this project.
REFERENCES1. Anderson R.A.: Essential and Toxic Trace Elements in
Human Health and Desease. 1993, 2, 221-234.
2. Anderson R. A: J. Adv. Med. 1995, 5, 8-37
3. Anderson R.A. and Leander O. A.: Selenium, Chromium,Manganese, in Modern Nutrition in Health and Disease.1988, 9, 34-56
4. Linderman R.D.:Minerals in Medical Practice; QuickReference to Clinical Nutrition: A Guide for Physicians.1987:7: 23-67
5. Evan G.W. and Bowman T.D: J. Inorg. Biochem.1992,6,46-243.
6. Broadburst C.L Schmidt W.F. Reeves J.B. PolanskyM.M. Gautschi K. Anderson R.A.:J. Inorg. Biochem.1997,8,66- 119.
7. International Conference on Harmonization (ICH)of Technical Requirements for the registration ofPharmaceuticals for Human Use. Validation of analyticalprocedures: Methodology. ICH-Q2B Step-4. USA, Nov-1996, p. 1-8.
8. Breaux J. John K. Boules P.: Understanding andImplementing efcient Analytical Methods Developmentand Validation. J. Pharma Tech. 2003; 6-13.
9. Sethi PD and Sethi R. High Performance LiquidChromatography: Quantitative Analysis of PharmaceuticalDosage Forms, 1st ed. Vol. 1. 2007; 1-200.
10. Indian Pharmacopoeia. Published by Ministry of Healthand Family Welfare. Government of India. Volume I andII. 2007.
IDMA PUBLICATIONSSr. No. Name of Publications Cost in `
1. IDMA BULLETIN(Published on 7, 14, 21 and 30th of every month)
Each Copy 25/-
ANNUAL SUBSCRIPTION:
Members 1000/- p.a.
Non Members 3000/- p.a.
2. INDIAN DRUGS (Published on 28th of every month)
Each Copy 75/-
ANNUAL SUBSCRIPTION:
Members & Educational institutions 1000/- p.a.
Non Members 3000/- p.a.
3. IDMA APA Forum
Annual Membership 500/-
Life Membership 5000/-
4. TECHNICAL MONOGRAPHS:
NO. 1: STABILITY TESTING OF EXISTING DRUG SUBSTANCES ANDPRODUCTS
400/-
NO. 2: PRIMARY & SECONDARY CHEMICAL REFE RENCE SUBSTANCES 400/-
NO. 3: INVESTIGATION OF OUT OF SPECIFICATION (OOS) TEST RESULTS 400/-
Kindly note: Courier charges are applicable. We do not mail any publication against VPP Payment. All payments to bemade in advance as DD only in favour of “INDIAN DRUG MANUFACTURERS’ ASSOCIATION” payable at Mumbai
For More Details Please Contact: PUBLICATIONS DIVISION. Ms. Prachi
INDIAN DRUG MANUFACTURERS’ ASSOCIATION102-B, “A”- Wing, Poonam Chambers, Dr A B Road, Worli, Mumbai 400018.
A METHOD FOR PREPARATION OF DROSERONE FROM PLUMBAGIN
ABSTRACT
Droserone (2,5-dihyrdoxy-3-methyl-1,4- naphthoquinone) was prepared from naturally occurring
naphthoquinone plumbagin. Plumbagin was isolated from roots of Plumbago zeylanica. Plumbaginwas rst brominated at C-3 which was subsequently substituted with hydroxy group by a nucleophilic
substitution to obtain Droserone. The synthesized compounds were characterized by IR, MS, 1H and 13CNMR spectral data. RP-HPLC was used to ascertain the purity of the obtained compound.
Keywords: P. zeylanica, Plumbagin, Droserone,semi–synthesis.
1, 4-Naphthoquinones can be viewed as derivativesof naphthalene obtained through the replacement oftwo hydrogen atoms by two ketone groups. Naturallyoccurring naphthoquinone derivatives have signicant
pharmacological properties. They are cytotoxic, they
have signicant antibacterial, antifungal, antiviral,insecticidal, anti-inflammatory and antipyreticproperties1. Plants with naphthoquinone contentare widely used in China and the countries of SouthAmerica, where they are used to treat malignant
and parasitic diseases. Plumbagin and droseroneare naturally occurring antifeedant compoundsoccurring in carnivorous plants likeDrosera burmanii 2,Dionaea muscipula3,Triphophyllum peltatum 4 andNephthensis khasiana5. Amongst the two naturallyoccurring quinones, the content of plumbagin is foundto be signicantly high in these plants. Droserone
occurs in a relatively lesser concentration makingit difcult to isolate from plant sources and more
expensive when compared to plumbagin. Because
of its non-availability, it is much less studied whencompared to plumbagin. Hence an attempt wasmade to isolate plumbagin and synthesize droseronefrom the same. Droserone has been reported to besynthesized from plumbagin using epoxidation of C-2
C-3 double bond and subsequent hydrolysis in about31% yields6 and from naturally occurring juglone andchlorplumbagin7.
Plumbago zeylanica L. is a semi-climbing,perennial and sub-scandent shrub with semi woodystem and numerous branches that grows in tropical
and subtropical regions of Asia, Australia and Africaand also found wild in peninsular India, Uttar Pradesh,West Bengal and cultivated in gardens throughoutIndia8,9,10. A wide range of chemical compoundsincluding plumbagin, plumbagic acid, chitranone,elliptinone, isoshinanolone, zeylanone, isozeylanone,plumbazeylanone, seselin, suberosin,β-sitosterol etc.have been isolated from this species in recent times11.The roots of plant are reported to contain about 0.2%w/w of plumbagin and hence was preferred choice as
a natural source for isolation of plumbagin12
.The dried roots of P. zeylanica were obtained
from local market of Jaipur, India and authenticatedusing microscopical and chemical evaluation, whichwas conrmed by Prof. H. M. Pandit, Khalsa College,
Mumbai; a voucher specimen was deposited inMedicinal Natural Products Research Laboratory,ICT, Mumbai. Potassium hydroxide and Light
petroleum ether (60-80°C) were obtained fromS. D. Fine Chemicals Limited, Mumbai. HPLC-grade
Methanol was sourced from Merck (India). Distilledwater was ltered through 0.45 μm lter. The roots
were powdered and used for extraction purpose.
Mass spectrum was recorded on Micromass Q-TOFMS Mass Spectrometer. All 1H and 13C-NMR spectrawere recorded on a JOEL 400-MHz instrument withan internal standard of tetramethylsilane (TMS).UV/Vis Spectrum was recorded on a JASCO V-530Spectrophotometer. TLC was performed on 0.20 mm
(E. Merck) aluminum-supportedplates. Compound was visualized either under UVlight at 254 nm or with Borntrager’s reagent; Reagentgrade solvents were used for extraction; HPLC
grade solvents were employed for chromatographicanalysis. HPLC analysis was performed with aJASCO (Hachioji, Tokyo, Japan) system, using 250mm × 4.6 mm i.d., RP-18 (5-µm particle size) MerckPurospher star column, an intelligent pump (PU-1580,PU-2080), a high-pressure mixer (MX-2080-31), a
manual sample injection valve (Rheodyne 7725i),with a ow rate of 1.00 mL/min, Injection volume
loop: 20 µL., monitoring at 250 nm (UV-1575), andelution program: 15 min isocratic, Methanol : Watercontaining 0.1% V/V of o -phosphoric acid (65 : 35).
Chromatographic data were processed with software(Borwin).
Isolation of plumbagin from roots of Plumbago
zeylanica
Powdered dried roots of P. zeylanica (750g) were subjected to Soxhlet extraction using
chloroform. The chloroform fraction thus obtainedwas concentrated, loaded on silica and subjectedto column chromatography using light petroleum
ether: ethyl acetate as eluent. A 5% fraction of ethylacetate in light petroleum ether yielded 270 mg ofplumbagin.
Preparation of 3-bromo-plumbagin from
plumbagin
Plumbagin (120 mg) was added to a 10 mLsolution of bromine in chloroform and stirred for 30minutes at room temperature. A mixture of 5 mL of
ethanol and 5 mL of glacial acetic acid was added toit and the reaction mixture was reuxed for additional
2 h. The progress of the reaction was monitored usingTLC. The reaction mixture was cooled and transferred
into a separating funnel and partitioned with water;the organic layer was separated and allowed toevaporate. The residue so obtained was loaded onsilica and puried by column chromatography, using
light petroleum ether: ethyl acetate (97: 3) as eluentto yield 80 mg of 3-bromoplumbagin.
Conversion of 3-bromo-plumbagin to
droserone
3-Bromo-plumbagin (80 mg) dissolved in 20 mLof ethanol to which a 5 mL of 10% sodium hydroxide
solution was added. The reaction mixture was reuxedon water bath for 90 min. The reaction mixture was
then allowed to cool and poured in 50 mL of dilutehydrochloric acid solution and kept aside for 2 h.Yellowish brown precipitate was formed. The solutioncontaining the ne precipitate was partitioned with light
petroleum ether. The organic layer was concentratedand the residue obtained was loaded on silica andpuried using column chromatography using light
petroleum ether and ethyl acetate (90:10) as eluentto yield 10 mg droserone.
Spectroscopic data
Plumbagin (5-Hydroxy-2-methyl-1,4-
naphthoquinone) :Infra red (IR) spectrum of theisolated plumbagin showed a broad peak at 3434cm-1(hydroxyl) and 1604, 1641 cm-1 (carbonyl) 1604characteristic of (C=C) in quinones. Molecular ionpeak at 188 m/e gave the molecular weight of thecompound. The UV/Vis maxima in methanol were
found to be at 214, 264, 340,400 and 417 nm. The
1H NMR (400 MHz, CDCl3) shows δ 2.1 (s, 3H,R-CH
3), 5.3 (s, 1H, Ar-OH), 7.2(m, 3H, Ar-H), 7.5(s,
1H, Ar-H); 13C NMR( MHz, CDCl3) 16.4 (C
11), 129
(C9), 133 (C
10), 119 (C
8), 124 (C
6),132 (C
3),136 (C
7),
150 (C2), 161 (C
5), 184 (C
1),190 (C
4). The isolated
compound melts at 76°C. Plumbagin showed 98 %purity by HPLC.
Bromo-plumbagin (3-bromo-5-Hydroxy-
2-methyl-1,4-naphthoquinone): Infra red (IR)spectrum of the isolated plumbagin showed a broad
peak at 3456 cm-1(hydroxyl) and 1662, 1636 cm-1 (carbonyl), 1636 characteristic of (C=C) in quinones,748 (bromo). Molecular ion peak at 266,268 m/e gave the molecular weight of the compound. The 1HNMR (400 MHz, CDCl
naphthoquinone): Infra red (IR) spectrum of thesynthesized droserone shows a broad peak at3454 and 3434 cm-1(hydroxyl) and 1744, 1712 cm-1 (carbonyl), 1625 characteristic of (C=C) in quinones.Molecular ion peak at 204 m/e gave the molecularweight of the compound. The UV/Vis maxima in
bromination of plumbagin at C-3 using bromine inchloroform and glacial acetic acid and subsequentlysubstituted with a hydroxyl group using nucleophilic
substitution. The reaction of plumbagin with bromine
in chloroform in presence of glacial acetic acid anddry chloroform afforded the 3- bromoplumbagin in45-47% yields. The bromine was then substituted byhydroxyl using neucleophilic substitution with sodium
hydroxide to obtain droserone in about 16% yield.
All the reported compounds were puried over silica
gel column and characterized spectroscopically. TheTLC of plumbagin and droserone was carried outusing toluene: formic acid:: 99: 1 and the R
fvalues
were found to be 0.45 and 0.3 respectively. Boththese compounds gave a positive Borntrager’s testindicating the presence of hydroxyquinoid moiety.
The yield of the above method is less when comparedto other methods reported in literature however thismethod can be optimized for enhancement of yield.The spectral data conrmed the formation of desired
products. The purity of compound was analyzedusing HPLC and was found to be 95 %. Droseronethus synthesized can nd various applications inpharmaceutical and other allied elds.
ACKNOWLEDGEMENT
The authors are thankful to UGC-SAP for providingnancial assistance for this project.
REFERENCES1. Babula P., Adam V., Havel L. and Kizek R.;
Naphthoquinones and their pharmacological properties,Ceska a Slovenska farmacie : casopis Ceske
farmaceuticke spolecnosti a Slovenske farmaceuticke
spolecnosti , 2007, 56, 3, 114–20. PMID 17867522.
2. Putalun W., Udomsin O., Yusakul G., JuengwatanatrakulT., Sakamoto S. and Tanaka H., Enhanced plumbaginproduction from in vitro cultures ofDrosera burmanii using
elicitation, Biotechnology Lett, 2010, 32, 5.3. Kreher B., Neszmélyi A., Wagner H., Naphthoquinonesfrom Dionaea muscipula, Phytochemistry, 1990, 29,2,605-606.
4. Bringmann G. and Feinies D., Stress related polyketidemetabolismof Dioncophyllaceae and Ancistrocladaceae,J Exp Bot, 2001, 52, 363, 2015-2022.
5. Eilenberg H., Pnini-Cohen S., Rahamim Y, Sionov E,Segal E, Carmeli S., and Zilberstein A, Induced productionof antifungal naphthoquinones in the pitchers of thecarnivorous plant Nepenthes khasiana, J Exp Bot, 2010,61, 3, 911–922.
6. Ogihara K., Yamashiro R., Higa M., Yogi S., Preparation
of naphthoquinone derivatives from plumbagin and theirichthyototoxicity, Chemical Pharmaceutical Bulletin,1997, 45,3, 437-445.
8. Vijver L. M. V. and Lotter A.P., The constituents in the rootsof Plumbago auriculata LAM and Plumbago zeylanica L.responsible for antibacterial activity. Planta Med, 1971,22, 8–13.
9. Li H. L., Flora of Taiwan. Vol. 4. 2nd Edition. EditorialCommittee of the Flora of Taiwan (Eds.), 1998, pp. 79.
10. Wealth of India, A Dictionary of Indian Raw materials andIndustrial products, Raw Materials Volume 8th :Ph-Re.
National Institute of Science and Industrial Research,CSIR Publication, 2005, pp. 163-64.
11. Lin L. C., Yang L. L., Chou C. J., Cytotoxic naphthoquinones
and plumbagic acid glycosides from Plumbago zeylanica,Phytochemistry, 2003,62, 619-622.
12. Dorni C.AI., Vidyalaxmi K.S., Vasanthi H. R.,
Rajamanickam G.V., HPTLC method for quantication of
plumbagin in three plumbago species, Research Journal
of Phytochemistry, 2007, 1, 46-51.
Department of Pharmaceutical Sciences and Technology Ferreira G.M.* and Laddha K.S.Institute of Chemical Technology
We are pleased to inform you that Indian Drug Manufacturers’ Association (IDMA) and Association of PharmaceuticalAnalysts (APA) have pleasure in announcing the 16th Pharmaceutical Analysts’ Convention (PAC) 2013 on Friday &Saturday, 27th & 28th September 2013, at Hotel Hyatt Regency, Sahar Airport Road, Andheri, Mumbai.
The main theme for this year is: “GENERICS THE GAME CHANGER”
THE ORGANIZER:
IDMA is recognized as the Voice of the National Sector with over 700 members comprising Large, Medium and Small manufacturersspread across the length and breadth of the country. IDMA works with the Government on the industry’s development plans andvarious public matters, also conducts seminars, workshops, training programmes etc.
At this prestigious Convention, Eminent technical personnel from Indian Pharmaceutical Industry, Research and Academic willconverge and get-together to interact on various recent developments.
The topics & speakers would be finalized shortly.
OBJECTIVE:
IDMA-APA has successfully organized Fifteen Pharmaceutical Analysts Convention (PACs) since the year 1997. This year’s conventiontheme is “GENERICS THE GAME CHANGER” in order to update and exceed the current regulatory requirement & Science based
systems approach in Pharmaceutical Industry.
INDIAN DRUG MANUFACTURERS’ ASSOCIATION
“16th PHARMACEUTICAL ANALYSTS’ CONVENTION”on 27th & 28th September 2013
at Hotel Hyatt Regency, Sahar Airport Road, Mumbai
WHO SHOULD ATTEND?
This Convention is designed to attract all those involved in:
` ` ` Early Bird Discounts (IDMA / Non-IDMA Members)
Before 30th July 2013: 15% discount Before 30th August 2013: 10% discount
Group registration benefits (cannot be combined with discounts): For every 3 delegates registered from the same organization, 1 delegate registration is complimentary
SPONSORSHIP OPPORTUNITIES:
Event Sponsor – ` 3,50,000/-
(IDMA Offers
Kit Bags – ` 2,50,000/-
(IDMA Offers
Sponsored Session – ` 2,00,000/-
(IDMA Offers:
Badges Sponsor - ` 1,00,000/-
(IDMA Offers
Page Colour Advertisement in Sovenier)
Spiral Bound Note Books– ` 80,000/-
(IDMA Offers
Pens – ` 60,000/-
(IDMA Offers Cocktails & Dinner – ` 2,00,000/-
Lunch– ` 80,000/- per session (2 Days)
(IDMA Offers
Souvenir Sponsorship – ` 70,000/-
(IDMA Offers
Product brochures would also be distributed)
Tea / Coffee- ` 40,000/- per session (4 Sessions)
(IDMA Offers Distribution of Company Brochures – ` 40,000/-
(IDMA Offers
Complimentary)
EXHIBITION OPPORTUNITIES:
Table Spaces– ` 40,000/- per table for 2 days (IDMA Offers: Table Size: 6ft.x2ft. with a power connection, Lunch for





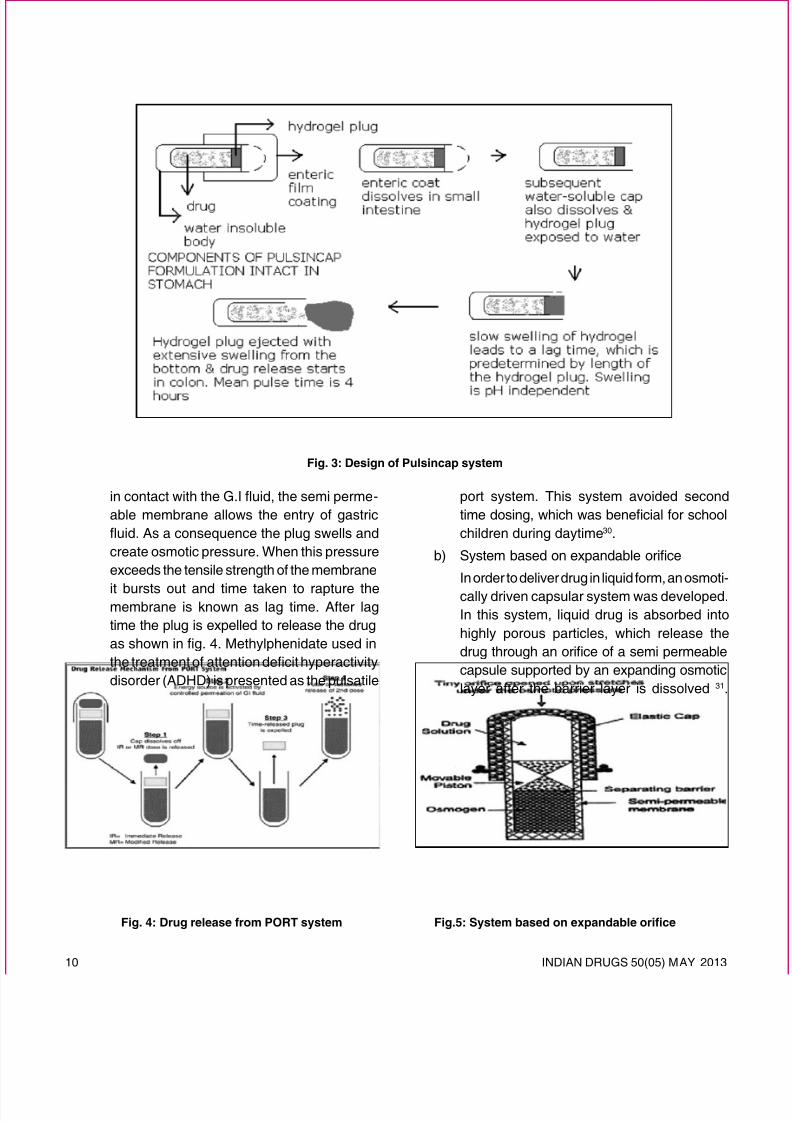




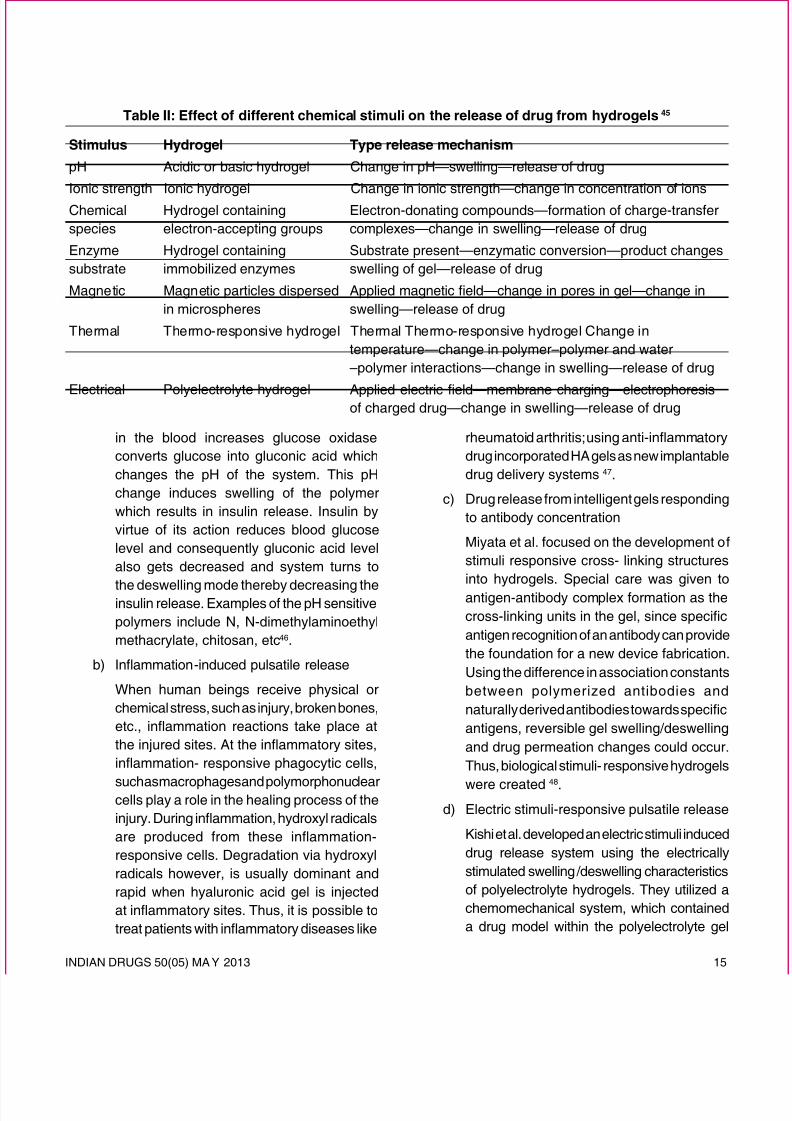

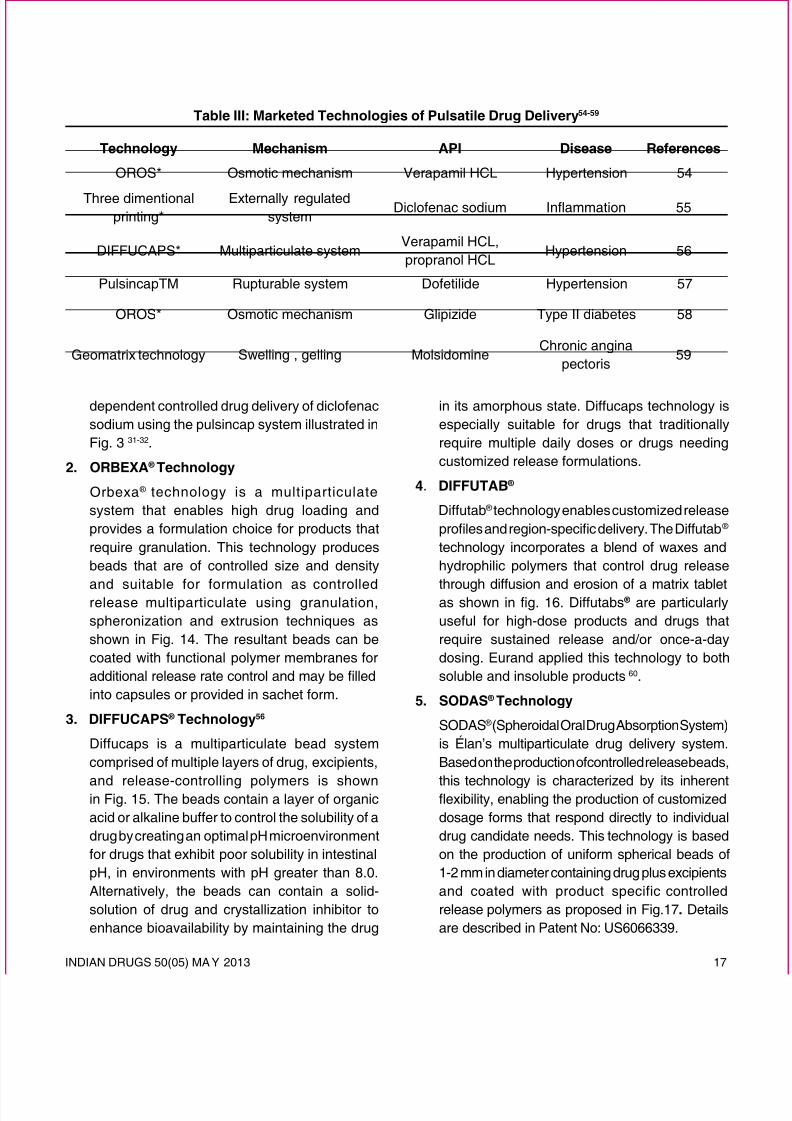
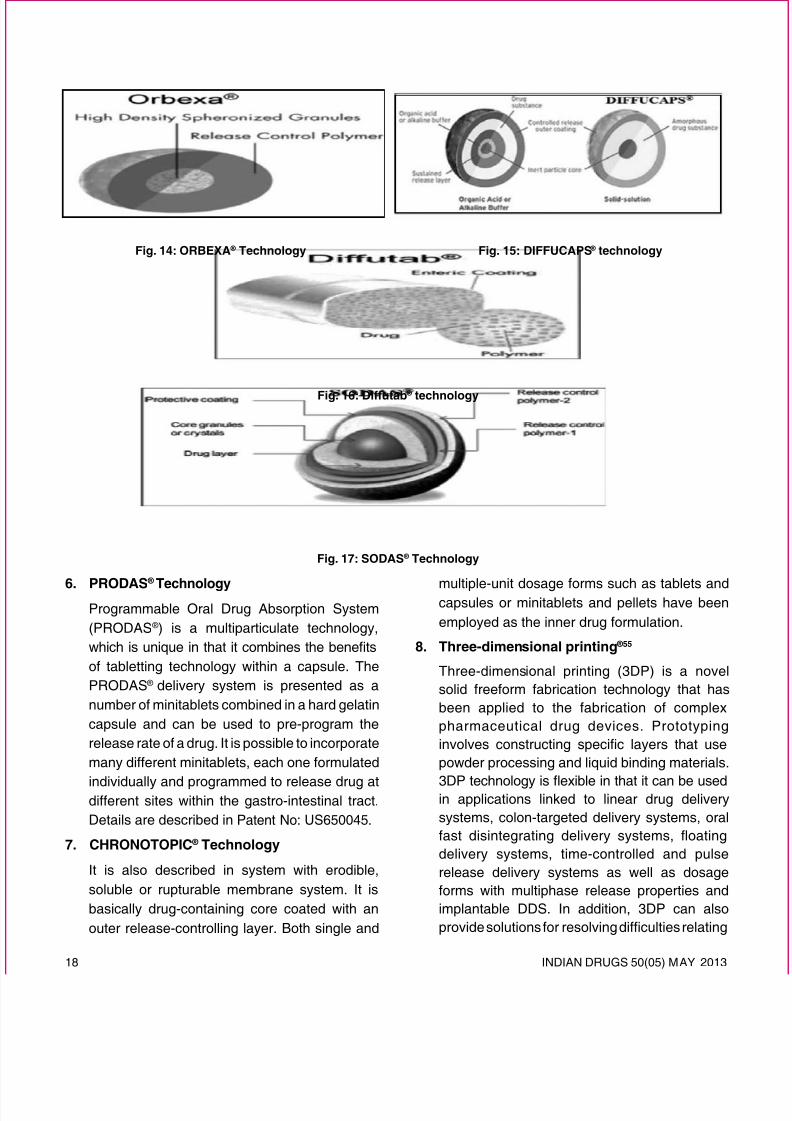










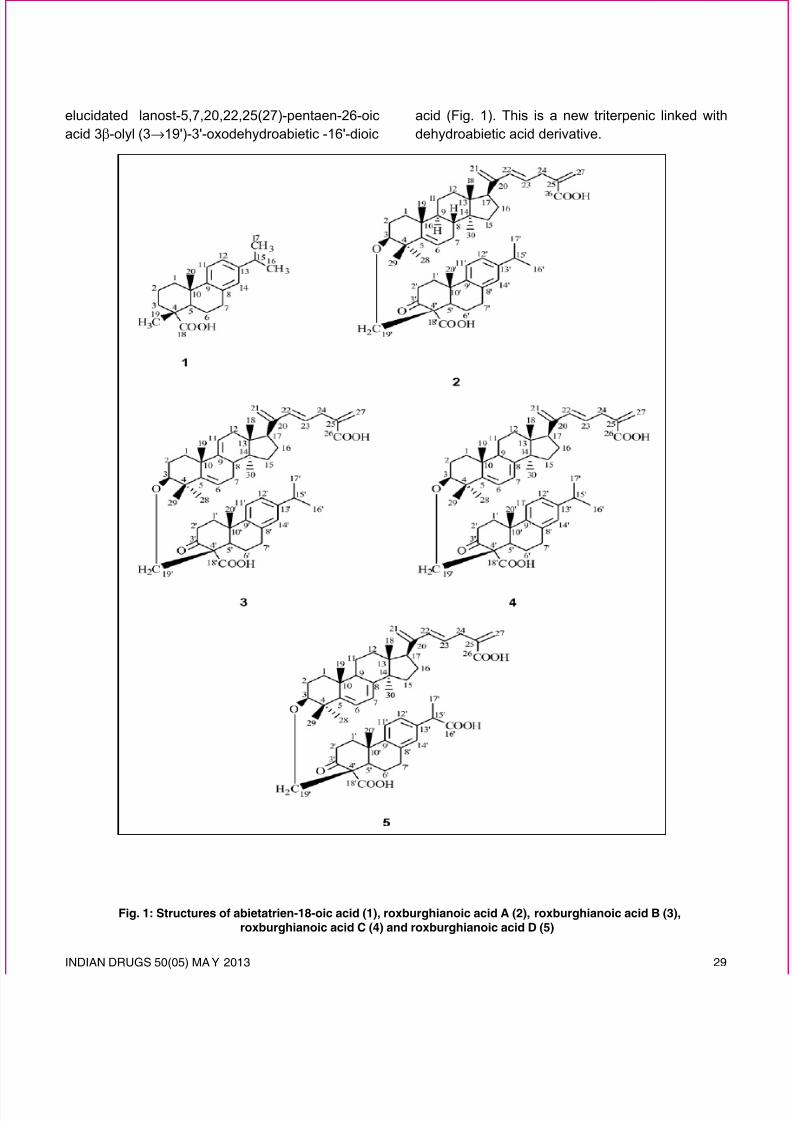
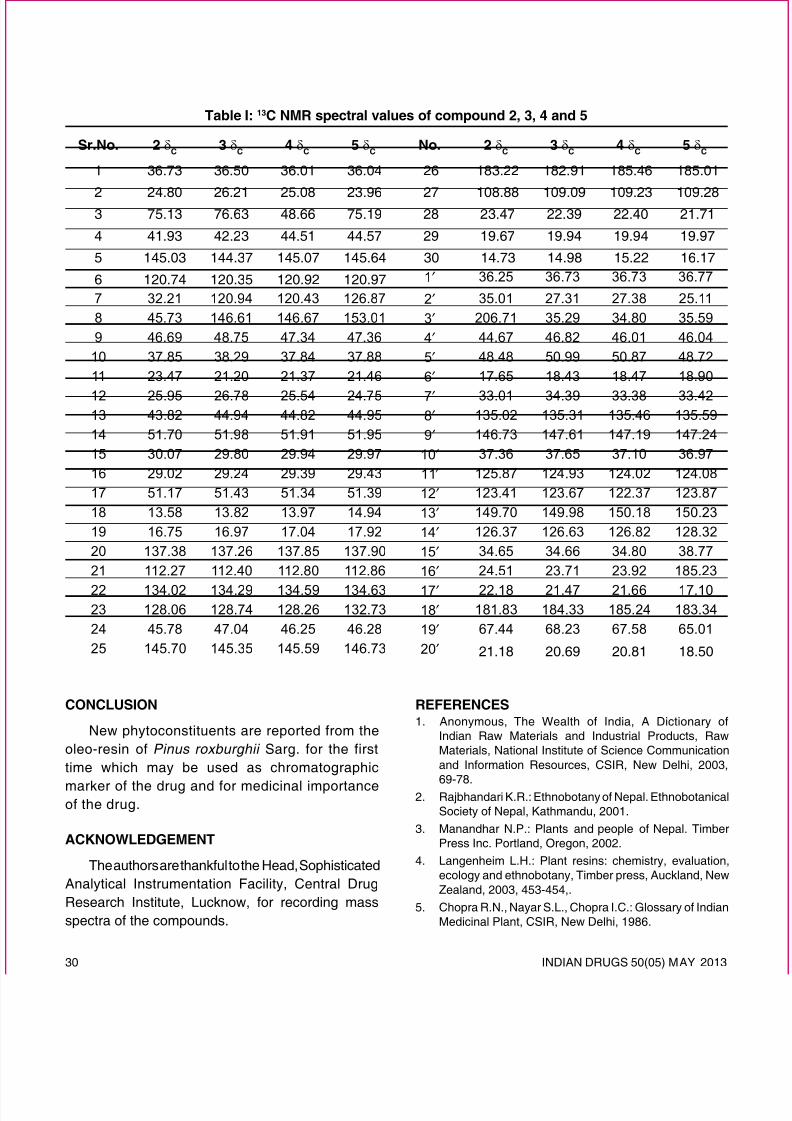


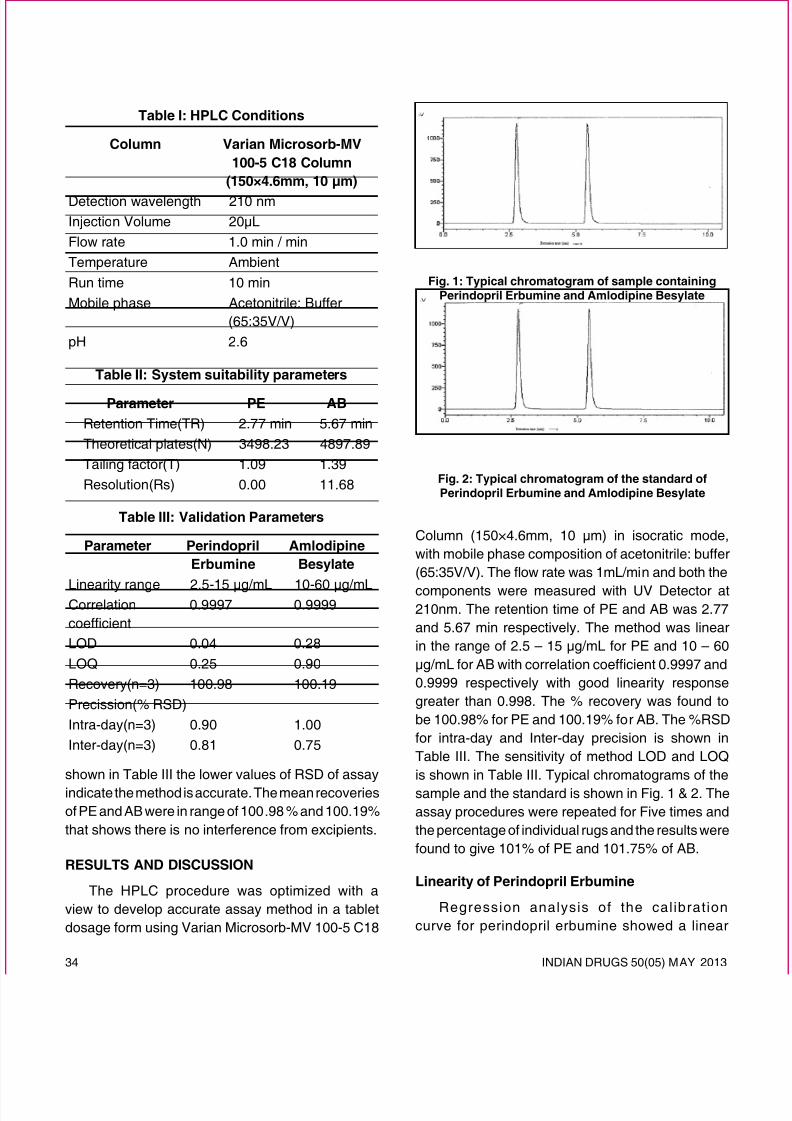



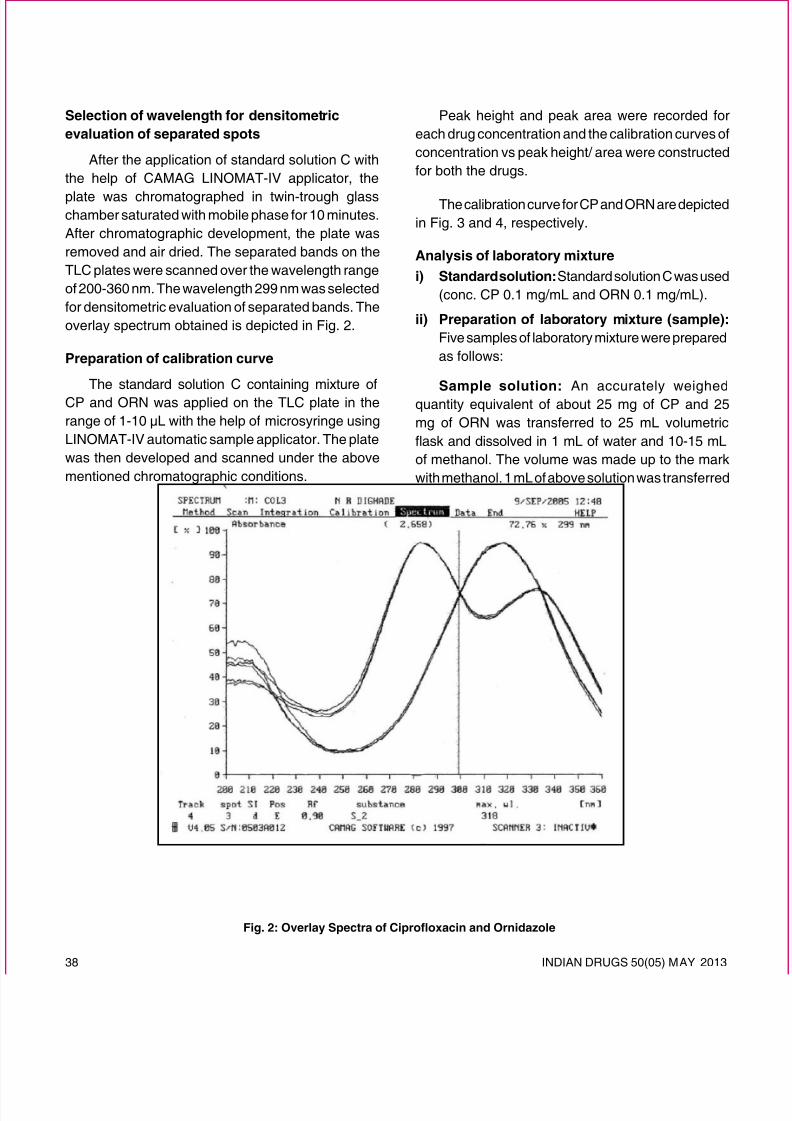




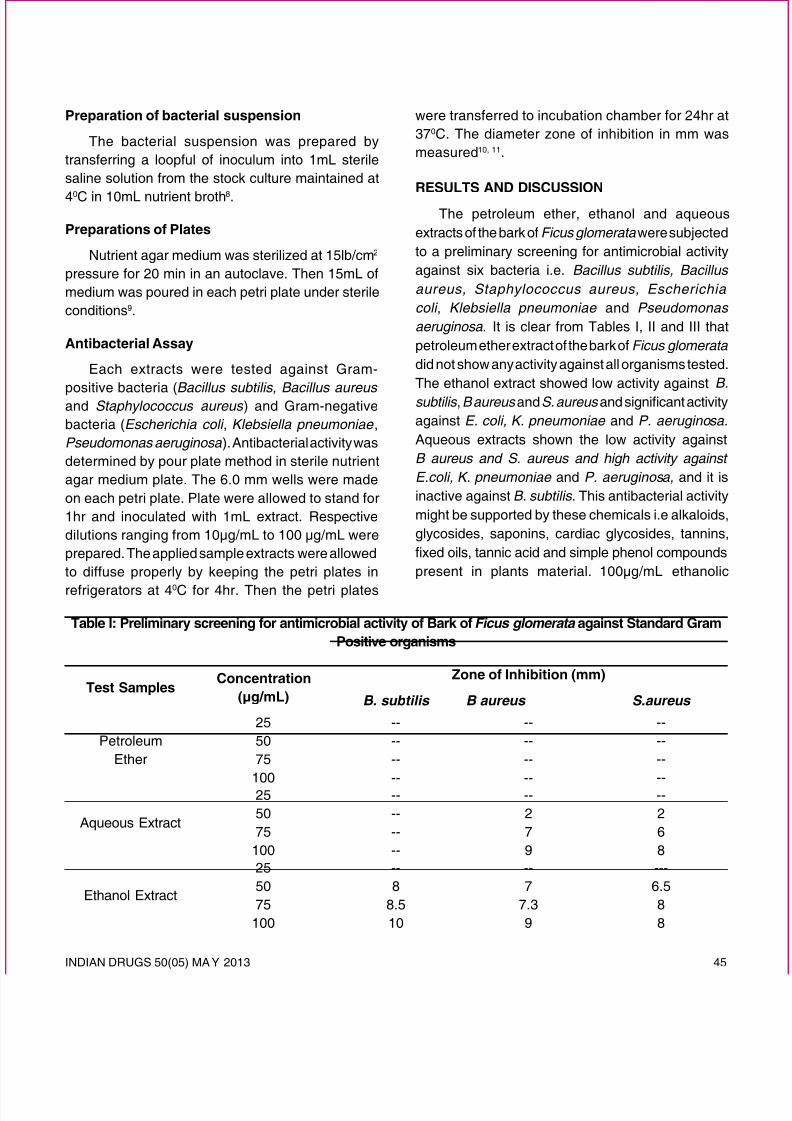
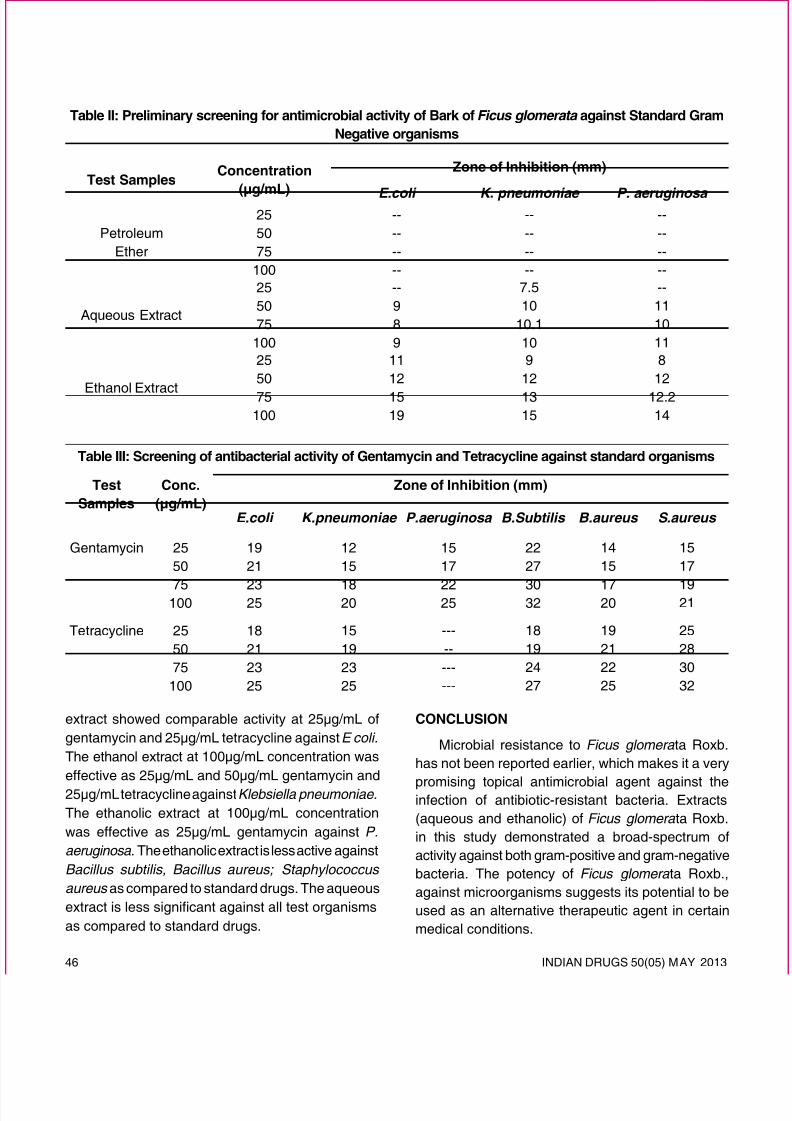






























![1,5, Center of Condensed Matter Sciences, arXiv:1305.6039v1 [cond-mat.str-el] 26 May 2013 · 2018. 6. 4. · arXiv:1305.6039v1 [cond-mat.str-el] 26 May 2013 PbCu3TeO7: An S=1 2 staircase](https://static.documents.pub/doc/80x56/60278efd16ce896590470917/15-center-of-condensed-matter-sciences-arxiv13056039v1-cond-matstr-el-26.jpg)



