Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2016 Nephroblastome : Praktisch immer in Studien! Bode, Peter K; Moch, Holger Abstract: Nephroblastomas are the most commonly occurring renal neoplasms in childhood and are treated almost exclusively in clinical trials. An important factor for further therapeutic management is the pathological evaluation of the nephrectomy specimen. Tumor stage and risk group classification are the most crucial parameters. An independent assessment of the tumor by a reference pathology center is an essential standard procedure. Although many molecular genetic discoveries have been made in nephroblastomas over recent years, molecular parameters do not (yet) play a role in treatment stratifi- cation. DOI: https://doi.org/10.1007/s00292-016-0148-x Other titles: Nephroblastomas : Almost exclusively in clinical trials! Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-124024 Journal Article Accepted Version Originally published at: Bode, Peter K; Moch, Holger (2016). Nephroblastome : Praktisch immer in Studien! Der Pathologe, 37(2):166-171. DOI: https://doi.org/10.1007/s00292-016-0148-x
Transcript
Zurich Open Repository andArchiveUniversity of ZurichMain LibraryStrickhofstrasse 39CH-8057 Zurichwww.zora.uzh.ch
Year: 2016
Nephroblastome : Praktisch immer in Studien!
Bode, Peter K; Moch, Holger
Abstract: Nephroblastomas are the most commonly occurring renal neoplasms in childhood and aretreated almost exclusively in clinical trials. An important factor for further therapeutic management isthe pathological evaluation of the nephrectomy specimen. Tumor stage and risk group classification arethe most crucial parameters. An independent assessment of the tumor by a reference pathology centeris an essential standard procedure. Although many molecular genetic discoveries have been made innephroblastomas over recent years, molecular parameters do not (yet) play a role in treatment stratifi-cation.
DOI: https://doi.org/10.1007/s00292-016-0148-x
Other titles: Nephroblastomas : Almost exclusively in clinical trials!
Posted at the Zurich Open Repository and Archive, University of ZurichZORA URL: https://doi.org/10.5167/uzh-124024Journal ArticleAccepted Version
Originally published at:Bode, Peter K; Moch, Holger (2016). Nephroblastome : Praktisch immer in Studien! Der Pathologe,37(2):166-171.DOI: https://doi.org/10.1007/s00292-016-0148-x
P. K. Bode · H. MochInstitut für Klinische Pathologie, UniversitätsSpital Zürich, Zürich, Schweiz
NephroblastomePraktisch immer in Studien!
Hintergrund
Das Nephroblastom (Synonym Wilms-Tumor) nimmt innerhalb der WHO-Klassifikation der renalen Neoplasieneine gewisse Sonderstellung ein, da essich in mehrerlei Hinsicht von denanderen Entitäten unterscheidet:4 Nephroblastome treten fast aus-
schließlich im Kindesalter auf.4 Nephroblastome besitzen eine eigene
Tumorstadiumklassifikation, dasTNM-System spielt keine Rolle.
4 Die Patienten werden praktischimmer in Studien behandelt, sodassdie Aufgaben der Pathologie auchim Studienprotokoll explizit definiertsind.
4 Eine referenzpathologische Zweitbe-urteilung ist Standard.
Der vorliegende Artikel fasst die wich-tigen Fakten zum Nephroblastom zu-sammen und soll einen Überblick ins-besondere über die Rolle der Pathologiebeim Management dieser Tumorerkran-kung verschaffen.
Definition, Klinik undBehandlungsprinzipien
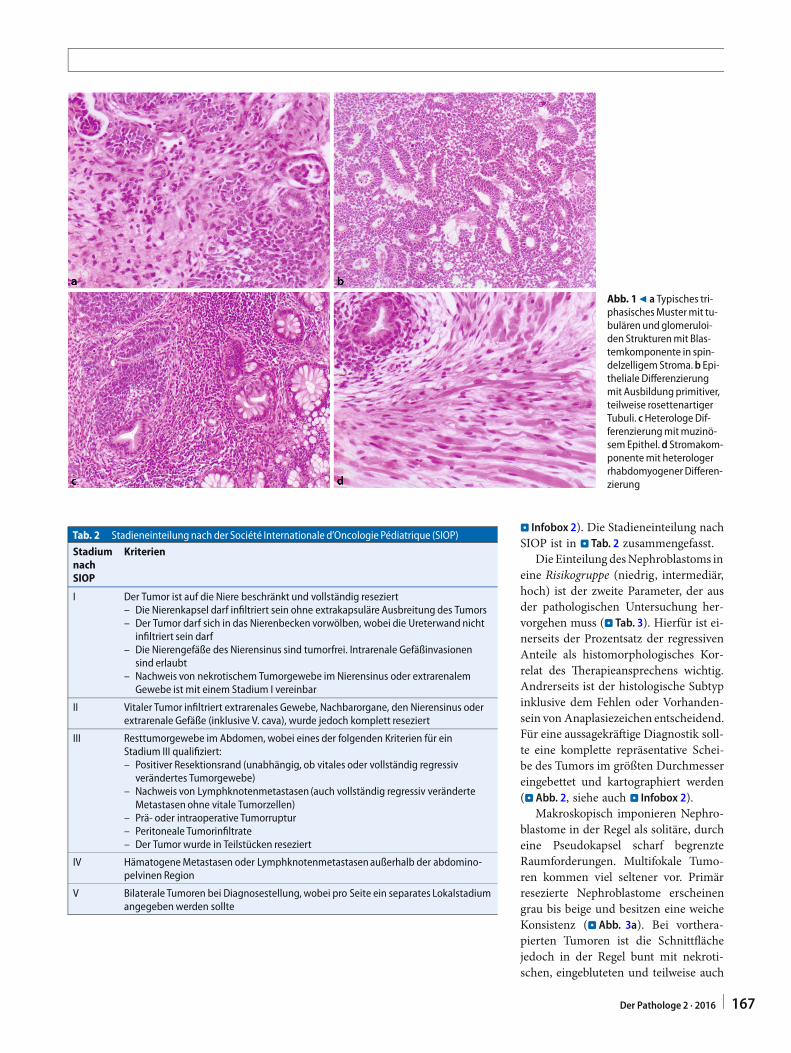
DasNephroblastomgehörtzurKlassedermalignen embryonalen Tumoren (Blas-tome), die vorwiegend im Kindesalterauftreten. Es leitet sich von nephrogenenResten ab und imitiert histomorpho-logisch die Nierenentwicklung unterAusbildung epithelialer, stromaler undblastemreicher Anteile (charakteristi-sches triphasisches Muster, . Abb. 1a).Das Nephroblastom ist mit einem Anteilvon > 80% aller renalen Neoplasien der
häufigste Nierentumor im Kindesalter(. Tab. 1; [1]).
Etwa eines von8000Kindernerkranktan einem Nephroblastom [6]. Mädchenund Jungen sind gleichermaßen betrof-fen. Im Durchschnitt sind die Patientenbei Diagnosestellung etwa 2 bis 3 Jahrealt. Nur ungefähr 2% der Nephroblas-tome treten jenseits des 10. Lebensaltersauf. Innerhalbderersten6Lebensmonateist dieser Tumor höchst selten [5, 8]. Ob-wohl Nephroblastome in der Regel spo-radisch auftreten, sindAssoziationenmitverschiedenenfamiliärenSyndromenbe-kannt (s. unten, Abschn. „Molekularge-netik“).
Klinisch fallen die betroffenen Kinderhäufig durch einen tastbaren Tumor imAbdomen auf. In der Bildgebung impo-niert das Nephroblastom als scharf ab-grenzbare solide und heterogene Raum-forderung, die Nierenparenchym und-becken verdrängt. Der Tumor me-tastasiert typischerweise in regionäreLymphknoten, Leber und Lunge [2]. BeiMetastasen in anderen Organen solltendifferenzialdiagnostisch andere Tumor-entitäten in Betracht gezogen werden.
Im deutschsprachigen Raum werdenNephroblastome fast ausschließlich inStudien gemäß Protokoll der SociétéInternationale d’Oncologie Pédiatrique(SIOP) behandelt. Darin ist eine bi-optische Abklärung vor präoperativerChemotherapie nicht zwingend vorgese-hen. Trotzdem kann die Diagnosesiche-rung mithilfe einer Biopsie sinnvoll sein,damit Patienten mit anderen renalenRaumforderungen einer adäquaten The-rapie zugeführt werden, die sich teilweisedeutlich vom Nephroblastomprotokollunterscheidet. Die meisten Nephroblas-tome zeigen ein gutes Ansprechen auf
die Chemotherapie, was sich positiv aufTumorgröße und -stadium auswirkt.
Rolle der Pathologie imtherapeutischen Management
Nach Abschluss der präoperativen Che-motherapie erfolgt die Resektion des Pri-märtumors, dessen pathologische Unter-suchung direkten Einfluss auf das weite-re Procedere hat. Je nach postoperativemTumorstadium und Risikogruppenzuord-nung schließen sich weitere Therapienunterschiedlicher Intensität und Moda-lität (Chemo- und/oder Radiotherapie)an oder nicht [17].
Die Kriterien für die Tumorstadi-eneinteilung beim Nephroblastom un-terscheiden sich grundlegend von derTNM-Klassifikation der Nierenzellkar-zinome, was beim Zuschnitt beachtetwerden muss (insbesondere die Berück-sichtigung der Hilusregion und allerfraglicher Resektatränder siehe auch
Tab. 1 Häufigkeitsverteilung der Nieren-tumoren im Kindesalter [1]
Abb. 19 a Typisches tri-phasischesMustermit tu-bulären undglomeruloi-den Strukturenmit Blas-temkomponente in spin-delzelligem Stroma.b Epi-theliale Differenzierungmit Ausbildung primitiver,teilweise rosettenartigerTubuli. cHeterologe Dif-ferenzierungmitmuzinö-sem Epithel.d Stromakom-ponentemit heterologerrhabdomyogener Differen-zierung
Tab. 2 Stadieneinteilung nach der Société Internationale d’Oncologie Pédiatrique (SIOP)
StadiumnachSIOP
Kriterien
I Der Tumor ist auf die Niere beschränkt und vollständig reseziert– Die Nierenkapsel darf infiltriert sein ohne extrakapsuläre Ausbreitung des Tumors– Der Tumor darf sich in das Nierenbecken vorwölben, wobei die Ureterwand nicht
infiltriert sein darf– Die Nierengefäße des Nierensinus sind tumorfrei. Intrarenale Gefäßinvasionen
sind erlaubt– Nachweis von nekrotischem Tumorgewebe im Nierensinus oder extrarenalem
Gewebe ist mit einem Stadium I vereinbar
II Vitaler Tumor infiltriert extrarenales Gewebe, Nachbarorgane, den Nierensinus oderextrarenale Gefäße (inklusive V. cava), wurde jedoch komplett reseziert
III Resttumorgewebe im Abdomen, wobei eines der folgenden Kriterien für einStadium III qualifiziert:– Positiver Resektionsrand (unabhängig, ob vitales oder vollständig regressiv
verändertes Tumorgewebe)– Nachweis von Lymphknotenmetastasen (auch vollständig regressiv veränderte
Metastasen ohne vitale Tumorzellen)– Prä- oder intraoperative Tumorruptur– Peritoneale Tumorinfiltrate– Der Tumor wurde in Teilstücken reseziert
IV HämatogeneMetastasen oder Lymphknotenmetastasenaußerhalb der abdomino-pelvinen Region
V Bilaterale Tumoren bei Diagnosestellung, wobei pro Seite ein separates Lokalstadiumangegebenwerden sollte
. Infobox 2). Die Stadieneinteilung nachSIOP ist in . Tab. 2 zusammengefasst.
Die Einteilung desNephroblastoms ineine Risikogruppe (niedrig, intermediär,hoch) ist der zweite Parameter, der ausder pathologischen Untersuchung her-vorgehen muss (. Tab. 3). Hierfür ist ei-nerseits der Prozentsatz der regressivenAnteile als histomorphologisches Kor-relat des Therapieansprechens wichtig.Andrerseits ist der histologische Subtypinklusive dem Fehlen oder Vorhanden-sein von Anaplasiezeichen entscheidend.Für eine aussagekräftige Diagnostik soll-te eine komplette repräsentative Schei-be des Tumors im größten Durchmessereingebettet und kartographiert werden(. Abb. 2, siehe auch . Infobox 2).
Makroskopisch imponieren Nephro-blastome in der Regel als solitäre, durcheine Pseudokapsel scharf begrenzteRaumforderungen. Multifokale Tumo-ren kommen viel seltener vor. Primärresezierte Nephroblastome erscheinengrau bis beige und besitzen eine weicheKonsistenz (. Abb. 3a). Bei vorthera-pierten Tumoren ist die Schnittflächejedoch in der Regel bunt mit nekroti-schen, eingebluteten und teilweise auch
Der Pathologe 2 · 2016 167
(pseudo-)zystischen Arealen (. Abb. 2aund 3b). Histologisch präsentieren sichdie typischen Veränderungen nach Che-motherapie als mehr oder weniger zell-reiches Narbengewebe, dichte Schaum-zellaggregate, Blutungsresiduen in Formvon Siderophagen oder als frische Ne-krosen mit schemenhaft erkennbaremTumorgewebe. Beim regressiven Typ desNephroblastoms sollten die eben be-schriebenen Areale mehr als zwei Drittelausmachen. Das übrige vitale Tumorge-webe kann alle anderen Komponentenenthalten. Komplett nekrotische Nephro-blastome haben eine exzellente Prognoseund bedürfen keiner weiteren Behand-lung. Für diese Diagnose ist allerdingsein extensives Sampling erforderlich [4].
Histologischer Subtyp
Abgesehen vom Ausmaß der RegressionberuhtdieRisikogruppeneinteilungauchauf dem histologischen Subtyp, wobeidie namensgebende Komponente (epi-thelialer Typ, Stromatyp, blastemreicherTyp) mit mehr als zwei Dritteln Anteilmorphologisch dominierend sein muss.Der epitheliale Typ zeigt überwiegendtubuläre bis glomeruloide Strukturen(. Abb. 1a). Auch primitive Rosettenkönnen vorkommen (. Abb. 1b). Teil-weise ist eine heterologe Differenzierungin schleimbildendes oder squamösesEpithel zu beobachten (. Abb. 1c). ImStromatyp dominieren fibroblastenrei-che oder glattmuskuläre Anteile, wo-bei die heterologe Differenzierung einegroße Bandbreite aufweisen kann. Sokann Fett-, Knorpel-, Knochen- undauch neuronales Gewebe vorkommen.Sehr typisch und häufig ist eine rhab-domyogene Komponente – insbeson-dere nach Chemotherapie (. Abb. 1d).Der blastemreiche Typ ist charakterisiertdurch eine solide oder trabekuläre His-toarchitektur (. Abb. 4a, b). Die Zellenweisen eine klein-, blau- und rundzelligeMorphologie auf. Zytoplasma ist prak-tisch nicht erkennbar. Das Chromatinist leicht gekörnt, kleine Chromozentrenkönnen sichtbar sein. Die mitotischeAktivität ist hoch. Beim blastemreichenTyp kann eine Risikogruppeneinteilungnur dann erfolgen, wenn bekannt ist, obeine präoperative Chemotherapie erfolgt
ist oder nicht. Primär resezierte blas-temreiche Nephroblastome werden derintermediärenRisikogruppe zugeordnet,wohingegen vorbehandelte blastemrei-che Nephroblastome als Tumoren derHochrisikogruppe weiter behandelt wer-den.
Ungefähr 5–8% der NephroblastomeweisenAnaplasiezeichenauf.Siekommenim Vergleich zu Nephroblastomen ohneAnaplasie kaum während der ersten 2Lebensjahre vor, sondern betreffen typi-scherweise ältereKinder (Durchschnitts-alter 5 Jahre). Folgende Kriterien müssenfür eine Anaplasie erfüllt sein:4 anaplastische Kerne müssen min-
destens 3-mal so groß sein wie dieübrigen Kerne des Tumorgewebes,
4 die Kerne müssen hyperchromatischsein (. Abb. 4c),
4 atypische tri- oder multipolareMitosefiguren müssen vorliegen(. Abb. 4d).
Anaplasiezeichen können in allen Tu-morkomponenten vorkommen, sindaber in der Blastemkomponente amhäufigsten. Für die Risikogruppeneintei-lung ist die Verteilung der anaplastischenZellen im Tumor entscheidend, da nurNephroblastome mit diffuser Anapla-sie aufgrund der schlechten Prognoseder Hochrisikogruppe zugeteilt werden,während Nephroblastome mit fokalerAnaplasie als Tumoren der intermedi-ären Risikogruppe eingeteilt werden [9].Die Kriterien für eine diffuse Anapla-sie sind streng definiert (. Infobox 1),von denen mindestens eines erfüllt seinmuss.
Molekulargenetik
Etwa 10% aller Nephroblastome ent-stehen im Rahmen von Syndromen mitdefinierter genetischerAberration [7, 11,12, 14, 15]. Das Tumorsuppressor-GenWT1 (lokalisiert auf Chromosom11p13)spielt eine wichtige Rolle während derrenalen und gonadalen Entwicklung undin der Pathogenese zweier Syndrome:Deletionen der gesamten 11p13-Regionliegen dem WAGR-Syndrom (Wilms-Tumor/Aniridie/urogenitale Fehlbildun-gen und mentale Retardierung) zugrun-de und gehen mit einem Risiko von
ZusammenfassungNephroblastome stellen die häufigstenrenalen Neoplasien im Kindesalterdar. Sie werden praktisch immerin Studien behandelt, wobei derpathologischen Beurteilung desTumorpräparats eine wichtige Bedeutunghinsichtlich der Weichenstellung für dasweitere therapeutische Managementbeigemessen wird. Entscheidendhierbei sind das Tumorstadium unddie Risikogruppeneinteilung. Einereferenzpathologische Zweitbeurteilung iststandardmäßig erforderlich. Auch wenn inden letzten Jahren wichtige Entdeckungenim Bereich der Molekulargenetik vonNephroblastomen gemacht wurden, spielenmolekularpathologische Parameter alsTherapiestratifizierung (noch) keine Rolle.
Nephroblastomas. Almostexclusively in clinical trials!
AbstractNephroblastomas are the most commonlyoccurring renal neoplasms in childhood andare treated almost exclusively in clinicaltrials. An important factor for furthertherapeuticmanagement is the pathologicalevaluation of the nephrectomy specimen.Tumor stage and risk group classificationare the most crucial parameters. Anindependent assessment of the tumor by areference pathology center is an essentialstandard procedure. Although manymolecular genetic discoveries have beenmade in nephroblastomas over recent years,molecular parameters do not (yet) play arole in treatment stratification.
KeywordsNephroblastoma · Wilms tumor ·International Society of PaediatricOncology · Cancer · Childhood
168 Der Pathologe 2 · 2016
Abb. 28 aNephroblastommit regressiven Veränderungen (Einblutungen, Nekrosen, Narben) nachpräoperativer Chemotherapie.b Repräsentative Aufarbeitung einer kompletten TumorscheibemitKartographierung.cAbschätzungderregressivenAnteileunddervitalenTumorkomponente(schwarzmarkiert) nach histologischer Aufarbeitung in Korrelationmit der kartographierten Tumorscheibe
30% einher, ein Nephroblastom zu ent-wickeln. Beim Denys-Drash-Syndrom(Trias aus Intersexualität, Mesangioskle-rose und Nephroblastom) hingegen liegtdas Risiko bei 90%. Ursächlich ist in die-sem Fall eine Keimbahnpunktmutationim WT1-Gen. In sporadischen Nephro-blastomen findet man nur in etwa 20%der Fälle eine WT1-Mutation, die imGegensatz zu WT1-Wildtyp-Tumoreninteressanterweisehäufignochzusätzlicheine β-Catenin-Mutation aufweisen.
Beim Beckwith-Wiedemann-Syn-drom kommt es durch fehlreguliertesgenomisches Imprinting von Genender 11p15-Region (insbesondere vonInsulin Growth Factor [IGF]-2) zur ty-
pischen Konstellation von Hemihyper-trophie, Makroglossie, Viszeromegaliemit Bauchwandfehlbildungen, postnata-ler Hypoglykämie und erhöhtem Risikofür Nephroblastome, Hepatoblastomesowie Neuroblastome in den ersten8 Lebensjahren.
Ein Verlust der Heterozygotie („lossof heterocygosity“, LOH) von Chromo-som 1p und 16q [10] und der Zugewinnvon Chromosom 1q [16] wurden jeweilsin der Literatur mit einer schlechterenPrognose in Verbindung gebracht, wobeidiese Daten momentan (noch) keinenEinfluss auf die Therapiestratifizierunghaben. Mutationen im SIX 1/2 Pathwayund DROSHA/DGCR8-miRNA-Mikro-
Tab. 3 RevidierteWorking Classificationof Nephroblastoma der Société Internatio-nale d’Oncologie Pédiatrique (SIOP)
Vorbehandelte FälleI. Tumoren der niedrigen Risikogruppe
– Alle nicht anaplastischenNephro-blastome (inkl. blastemreicher Typ)
– Nephroblastommit fokaler Anapla-sie
III. Tumoren der Hochrisikogruppe
– Nephroblastommit diffuserAnaplasie
prozessorkomplexwurden in blastemrei-chen Nephroblastomen der Hochrisiko-gruppe beschrieben [18]. AnaplastischeNephroblastome sind eng mit Mutatio-nen im TP53-Gen gekoppelt, währendNephroblastome mit günstigem Verlaufkeine TP53-Mutationen aufweisen [3].Möglicherweise wird in Zukunft derTP53-Mutationsstatus in anaplastischenNephroblastomen zu einer besseren Ri-siko- undTherapiestratifizierung führen,da die chemotherapieinduzierte Tumor-apoptose eng mit einem funktionalenp53-Protein verbunden ist, was auch dieChemoresistenz von TP53-mutiertenanaplastischenNephroblastomen erklärt[13].
Der Pathologe 2 · 2016 169
Schwerpunkt: Nierentumoren
Abb. 38 aPrimär reseziertes, bifokalesNephroblastommit homogenerbeigefarbener Schnittfläche.bVortherapiertes Nephroblastommit kleinherdigen Einblutungen und xanthomatösen Veränderun-gen. Beachte die extrarenale Ausbreitung (mindestens Stadium II)
Abb. 49 a Blastemkom-ponentemit solidemWachstumsmuster.b Blas-temkomponentemittrabekuläremWachstums-musterundfibromyxoidemStroma. cAnaplasiezei-chen:Mehr als 3-mal sogroße Tumorzellemit hy-perchromatischemKernundbizarrer Kontur.dAna-plasiezeichen: AtypischemultipolareMitose inBlastemkomponente
170 Der Pathologe 2 · 2016
Infobox 1 Kriterien für diffuseAnaplasie
4 Anaplastische Zellen in verschiedenenHerden des Tumors (auf verschiedenenSchnittpräparaten)
4 Anaplastische Zellen außerhalb derTumorkapsel
4 Anaplastische Zellen in Gefäßen, imNierensinus oder in Metastasen
4 Anaplastische Zellaggregate, die nichtscharf vom nicht anaplastischen Tumorabgrenzbar sind
4 Anaplastische Zellen in einem einzelnenHerd, wobei das übrige Tumorgewebestärker ausgeprägte Atypien aufweist, dieaber die Anaplasiekriterien noch nichterfüllen (sogenannter „nuclear unrest“)
4 AnaplastischeZellen ineinerTumorbiopsie
Infobox 2 Checkliste für dieHandhabung und Beurteilungvon Nephrektomiepräparaten beiNephroblastomen
1. Vorbereitung des frisch eingesandtenPräparats für die weitere Aufarbeitung,insbesondere Überprüfung der Intaktheit(Kapselrupturen?), Markierung derkritischen Resektionsränder, Identifikationder Nierenvene, -arterie sowie des Uretersmit Absetzungsrändern, Asservierunghilärer Lymphknoten
2. Sagittalschnitt und Beurteilung derextrarenalen Ausbreitung, insbesondereim Bereich der Hilusregion
3. Tumorbanking mit Asservierung vonFrischgewebe
4. Fixation des Präparats in 4%igem Formalinfür 24 h
5. Fotodokumentation und Zuschnitteiner kompletten Tumorscheibe mitKartographierung
6. Obligate Parameter im pathologischenBericht: Tumorstadium nach SIOP unddie Risikogruppenzuordnung (. Tab. 2und 3)
7. Versand eines HE-gefärbten Schnitt-satzes der fotodokumentierten undkartographierten Tumorscheibe zurreferenzpathologischen Beurteilung andas Kindertumorregister Kiel
Fazit für die Praxis
4 Nephroblastome sind die häufigstenNierentumoren im Kindesalter.
4 Sie werden in der Regel in Studienbehandelt.
4 Die pathologische Beurteilung desTumorpräparats hat unmittelbarenEinfluss auf das weitere therapeuti-sche Management.
4 Tumorstadium und Risikogruppen-einteilung sind die entscheidendenParameter.
4 Die referenzpathologische Zweitbe-urteilung ist Standard.
Korrespondenzadresse
Dr. P. K. BodeInstitut für Klinische Pathologie,UniversitätsSpital ZürichSchmelzbergstr.12, 8091 Zürich, [email protected]
Einhaltung ethischer Richtlinien
Interessenkonflikt. P. K. BodeundH.Mochgebenan,dass kein Interessenkonflikt besteht.
Dieser Beitragbeinhaltet keine Studien anMenschenoder Tieren.
Literatur
1. IARC (2016)WHO Classification of Tumours of theUrinarySystemandMaleGenitalOrgans, Lyon
2. Argani PBJ (2015) Renal neoplasms of childhood.LippincottWilliams&Wilkins,Philadelphia
4. Boccon-Gibod L, Rey A, Sandstedt B, DelemarreJ, Harms D, Vujanic G, De Kraker J, Weirich A,Tournade MF (2000) Complete necrosis inducedby preoperative chemotherapy in Wilms tumoras an indicator of low risk: report of theinternational societyofpaediatriconcology (SIOP)nephroblastoma trial and study 9. Med PediatrOncol34:183–190
5. Breslow N, Beckwith JB, Ciol M, Sharples K (1988)Age distribution of Wilms’ tumor: report fromthe national Wilms’ tumor study. Cancer Res48:1653–1657
6. BreslowN,OlshanA,BeckwithJB,GreenDM(1993)Epidemiology of Wilms tumor. Med Pediatr Oncol21:172–181
7. Choufani S, Shuman C, Weksberg R (2013)Molecular findings in Beckwith-Wiedemannsyndrome. Am J Med Genet C Semin Med Genet163C:131–140
8. Davidoff AM (2012) Wilms tumor. Adv Pediatr59:247–267
9. Dome JS, Cotton CA, Perlman EJ, BreslowNE, Kalapurakal JA, Ritchey ML, Grundy PE,Malogolowkin M, Beckwith JB, Shamberger RC,HaaseGM,CoppesMJ,CocciaP,KletzelM,WeetmanRM, Donaldson M, Macklis RM, Green DM (2006)Treatment of anaplastic histology Wilms’ tumor:results fromthefifthNationalWilms’ TumorStudy.JClinOncol24:2352–2358
10. Grundy PE, Breslow NE, Li S, Perlman E, BeckwithJB,RitcheyML,ShambergerRC,HaaseGM,D’AngioGJ, Donaldson M, Coppes MJ, Malogolowkin M,Shearer P, Thomas PR, Macklis R, Tomlinson G,Huff V, Green DM, National Wilms Tumor Study G(2005) Loss of heterozygosity for chromosomes1p and 16q is an adverse prognostic factor infavorable-histology Wilms tumor: a report fromthe National Wilms Tumor Study Group. J ClinOncol23:7312–7321
11. Huff V (2011) Wilms’ tumours: about tumoursuppressor genes, an oncogene and a chameleongene.NatRevCancer11:111–121
13. Maschietto M, Williams RD, Chagtai T, PopovSD, Sebire NJ, Vujanic G, Perlman E, AndersonJR, Grundy P, Dome JS, Pritchard-Jones K (2014)TP53 mutational status is a potential marker forrisk stratification in Wilms tumour with diffuseanaplasia.PLOSONE9:e109924
14. Rivera MN, Haber DA (2005) Wilms’ tumour:connecting tumorigenesis and organ develop-ment in thekidney.NatRevCancer5:699–712
15. Scott RH, Stiller CA, Walker L, Rahman N (2006)Syndromes and constitutional chromosomalabnormalities associated with Wilms tumour. JMedGenet43:705–715
16. Segers H, van den Heuvel-Eibrink MM, WilliamsRD, van Tinteren H, Vujanic G, Pieters R, Pritchard-Jones K, BownN, Children’s C, LeukaemiaG (2013)Gain of 1q is a marker of poor prognosis inWilms’tumors. Genes Chromosomes Cancer. UKCCG52:1065–1074
17. Vujanic GM, Sandstedt B, Harms D, KelseyA, Leuschner I, de Kraker J, Committee SNS(2002) Revised International Society of PaediatricOncology (SIOP) working classification of renaltumorsofchildhood.MedPediatrOncol38:79–82
18. Wegert J, Ishaque N, Vardapour R, Georg C, GuZ, Bieg M, Ziegler B, Bausenwein S, Nourkami N,LudwigN, Keller A, GrimmC, Kneitz S,Williams RD,ChagtaiT,Pritchard-JonesK,vanSluisP,VolckmannR, Koster J, Versteeg R, Acha T, O’SullivanMJ, BodePK, Niggli F, Tytgat GA, van Tinteren H, van denHeuvel-EibrinkMM,Meese E, Vokuhl C, LeuschnerI, Graf N, Eils R, Pfister SM, KoolM, GesslerM (2015)Mutations in theSIX1/2pathwayandtheDROSHA/DGCR8 miRNAmicroprocessor complex underliehigh-riskblastemal typeWilms tumors.CancerCell27:298–311










![Novel technique: direct access partial nephrectomy ...nuf.nu/NoRenCa/Novel technique direct access... · of its equivalent oncological results compared to radical nephrectomy [3].](https://static.documents.pub/doc/80x56/606c80ba9bb7de31a926ad1d/novel-technique-direct-access-partial-nephrectomy-nufnunorencanovel-technique.jpg)












