Optimization Optimization Methods for Methods for Reliable Genomic- Reliable Genomic- Based Pathogen Based Pathogen Detection Systems Detection Systems K.M. Konwar, I.I. Mandoiu, A.C. Russell, and A.A. Shvartsman Computer Science & Engineering Department University of Connecticut, Storrs, CT 06269
Transcript
Optimization Methods Optimization Methods for Reliable Genomic-for Reliable Genomic-
Based Pathogen Based Pathogen Detection SystemsDetection Systems
K.M. Konwar, I.I. Mandoiu, A.C. Russell, and A.A. Shvartsman
Computer Science & Engineering DepartmentUniversity of Connecticut, Storrs, CT 06269
AbstractAbstract
Recent advances in genomic technologies have opened the way for the development of Genomic-based Pathogen Detection Systems (GPDSs) that can provide early warning in case of rapidly proliferating outbreaks of new natural pathogens such as the SARS corona-virus or bio-terrorist attacks. A critical step of all GPDS architectures proposed to date is DNA amplification by Multiplexed Polymerase Chain Reaction (MP-PCR).
In this poster we present ongoing theoretical and practical research on the minimum primer set selection problem for MP-PCR. We give algorithms with improved approximation guarantees for this problem and report results of empirical experiments on both synthetic and public genomic database test cases showing that our algorithms are highly scalable and produce better results compared to previous heuristics.
GPDS Components and RequirementsGPDS Components and Requirements Key GPDS components:
Selection of distinguishing DNA oligonucleotides based on available genomic sequences for the pathogens
Selective amplification of collected genetic material Hybridization-based detection of present distinguishers Pathogen identification by comparison with stored
signatures/barcodes of known pathogens
GPDS design requirements High specificity and sensitivity of detection Discrimination between pathogens and non-pathogenic organisms Ability to work with trace amounts of genetic material, and to detect
multiple pathogens at the same time Fully automated operation (should require minimal human
intervention
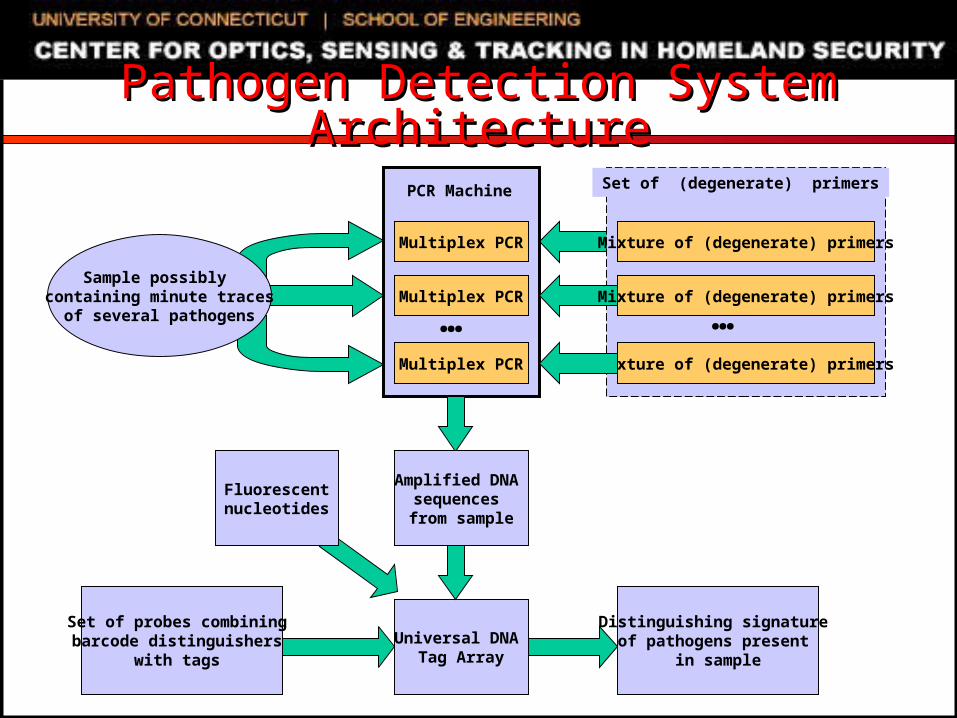
Pathogen Detection System ArchitecturePathogen Detection System Architecture
Multiplex PCR
PCR Machine
Mixture of (degenerate) primers
Set of (degenerate) primers
Mixture of (degenerate) primers
Universal DNA Tag Array
Amplified DNA sequences from sample
Sample possibly containing minute traces
of several pathogens
Set of probes combining barcode distinguishers
with tags
… …
Distinguishing signature of pathogens present
in sample
Fluorescentnucleotides
Multiplex PCR Mixture of (degenerate) primers
Multiplex PCR
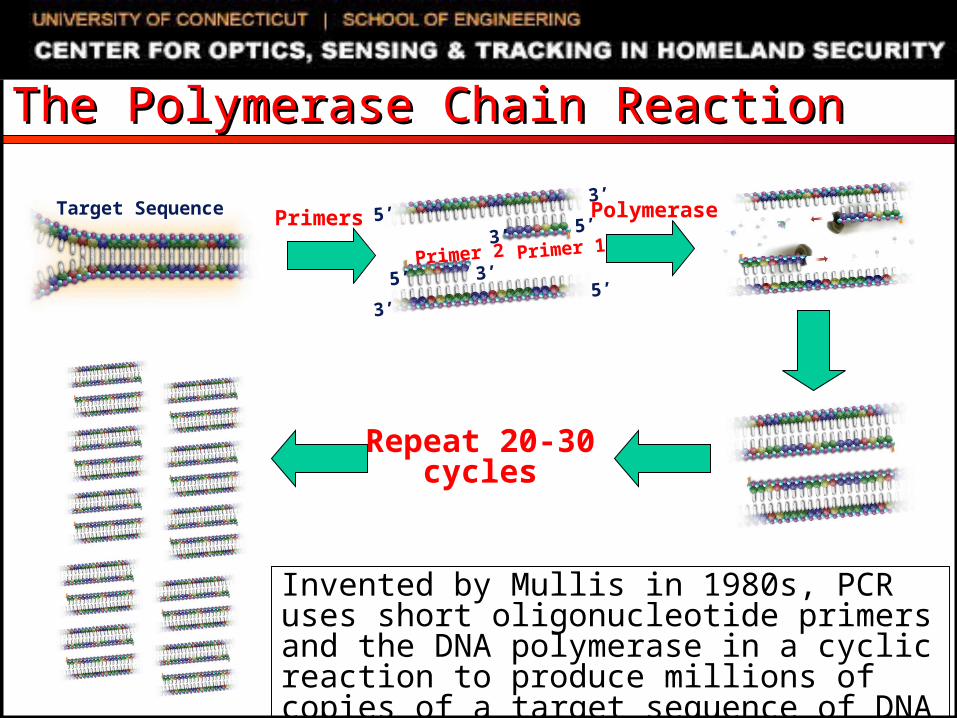
The Polymerase Chain Reaction The Polymerase Chain Reaction
Target Sequence
Primer 1Primer 25’
3’
5’
5’
3’
5’
3’
3’
PolymerasePrimers
Repeat 20-30 cycles
Invented by Mullis in 1980s, PCR uses short oligonucleotide primers and the DNA polymerase in a cyclic reaction to produce millions of copies of a target sequence of DNA
Primer Pair Selection ProblemPrimer Pair Selection Problem
• Given:
• Genomic sequence around amplification locus
• Primer length k
• Amplification upperbound L
• Find: Forward and reverse primers of length k that hybridize within a distance of L of each other and optimize amplification efficiency (melting temperatures, secondary structure, cross hybridization, etc.)
L
Forward primer
Reverse primer
amplification locus
3'
3'
5'
5'
Multiplex PCRMultiplex PCR Multiplex PCR (MP-PCR)
Multiple DNA fragments amplified simultaneously Boundaries of each amplification fragment still defined by
two oligonucleotide primers A primer may participate in the amplification of multiple
targets
Original DNA Template
PCR Products
Primer set selection Typically done by time-consuming
trial and error An important objective is to minimize
the total number of primers Reduced assay cost Higher effective concentration of
primers higher amplification efficiency
Reduced unintended amplification
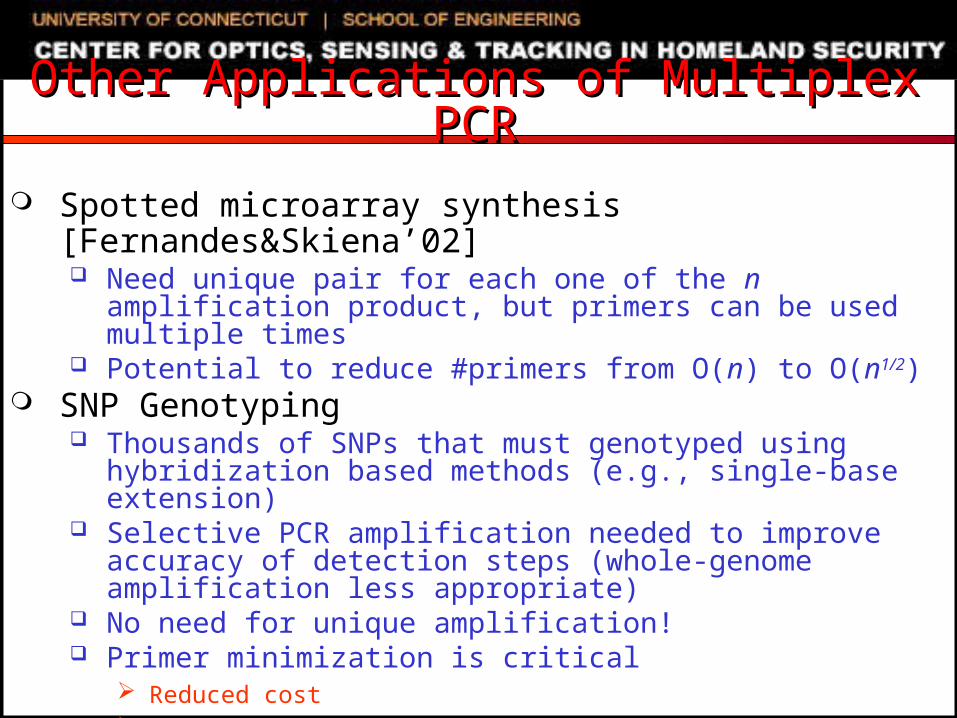
Other Applications of Multiplex PCROther Applications of Multiplex PCR
Spotted microarray synthesis [Fernandes&Skiena’02] Need unique pair for each one of the n amplification
product, but primers can be used multiple times Potential to reduce #primers from O(n) to O(n1/2)
SNP Genotyping Thousands of SNPs that must genotyped using
hybridization based methods (e.g., single-base extension) Selective PCR amplification needed to improve accuracy
of detection steps (whole-genome amplification less appropriate)
No need for unique amplification! Primer minimization is critical
Reduced cost Fewer multiplex PCR reactions
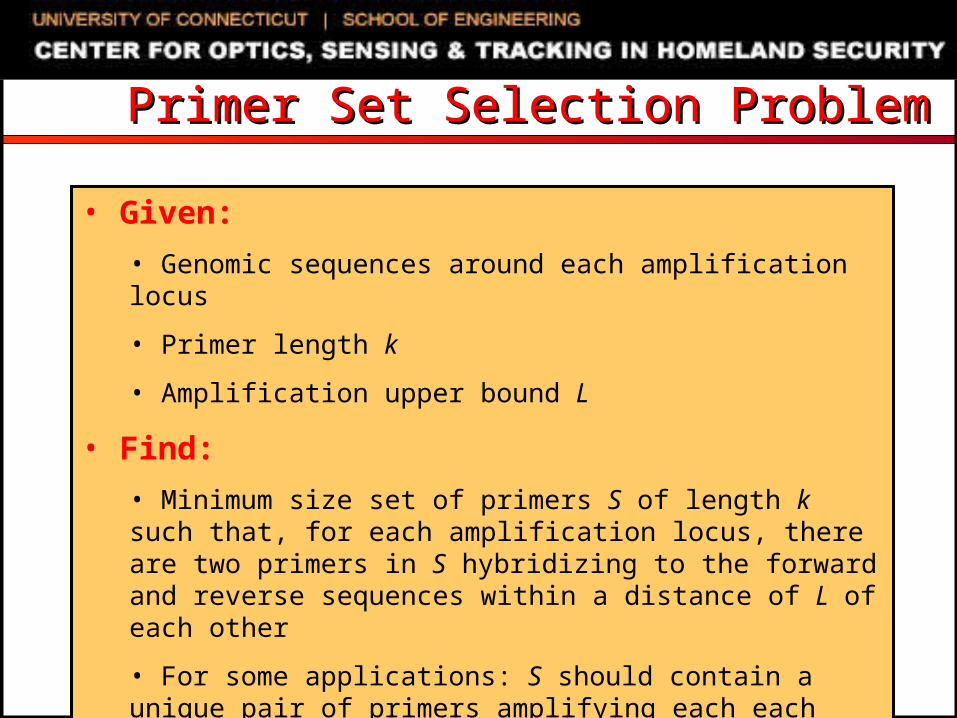
Primer Set Selection ProblemPrimer Set Selection Problem
• Given:
• Genomic sequences around each amplification locus
• Primer length k
• Amplification upper bound L
• Find:
• Minimum size set of primers S of length k such that, for each amplification locus, there are two primers in S hybridizing to the forward and reverse sequences within a distance of L of each other
• For some applications: S should contain a unique pair of primers amplifying each each locus
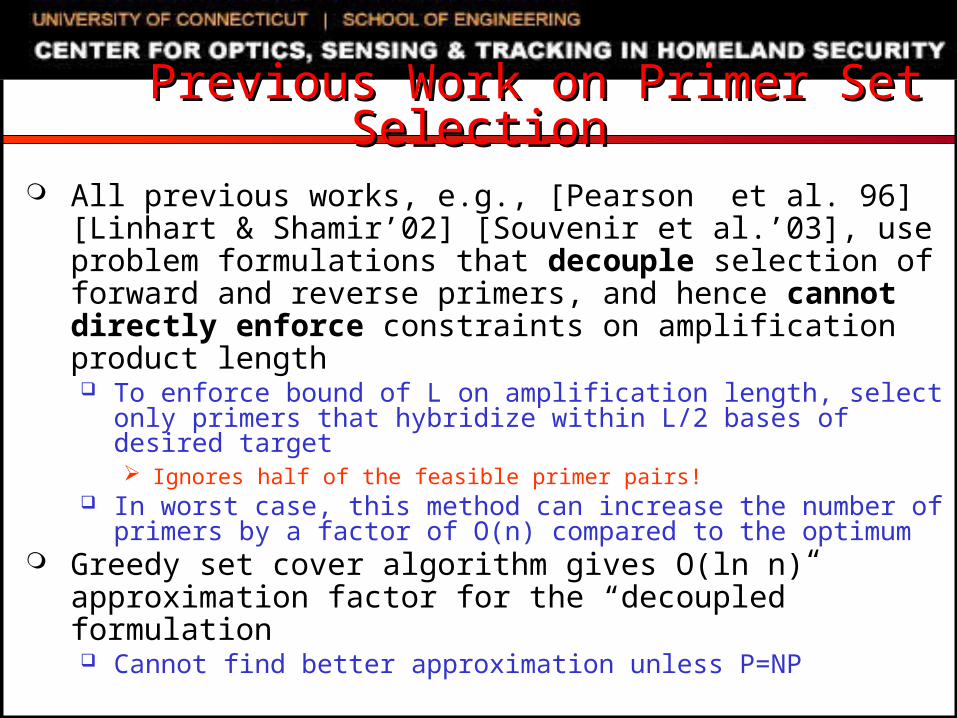
Previous Work on Primer Set SelectionPrevious Work on Primer Set Selection
All previous works, e.g., [Pearson et al. 96][Linhart & Shamir’02] [Souvenir et al.’03], use problem formulations that decouple selection of forward and reverse primers, and hence cannot directly enforce constraints on amplification product length To enforce bound of L on amplification length, select only
primers that hybridize within L/2 bases of desired target Ignores half of the feasible primer pairs!
In worst case, this method can increase the number of primers by a factor of O(n) compared to the optimum
Greedy set cover algorithm gives O(ln n) approximation factor for the “decoupled” formulation Cannot find better approximation unless P=NP
Previous Work (contd.)Previous Work (contd.)
[Fernandes&Skiena’02] model primer selection as a minimum multicolored subgraph problem: Vertices of the graph correspond to candidate primers
There is an edge colored by color i between primers u and v if they hybridize to i-th forward and reverse sequences within a distance of L
Goal is to find minimum size set of vertices inducing edges of all colors
No non-trivial approximation factor known previously
• Can be seen as a “simultaneous set covering” problem:
- The ground set is partitioned into n disjoint sets, each with 2L elements
- The goal is to select a minimum number of sets (== primers) that cover at least half of the elements in each partition
• Naïve modifications of the greedy set cover algo do not work
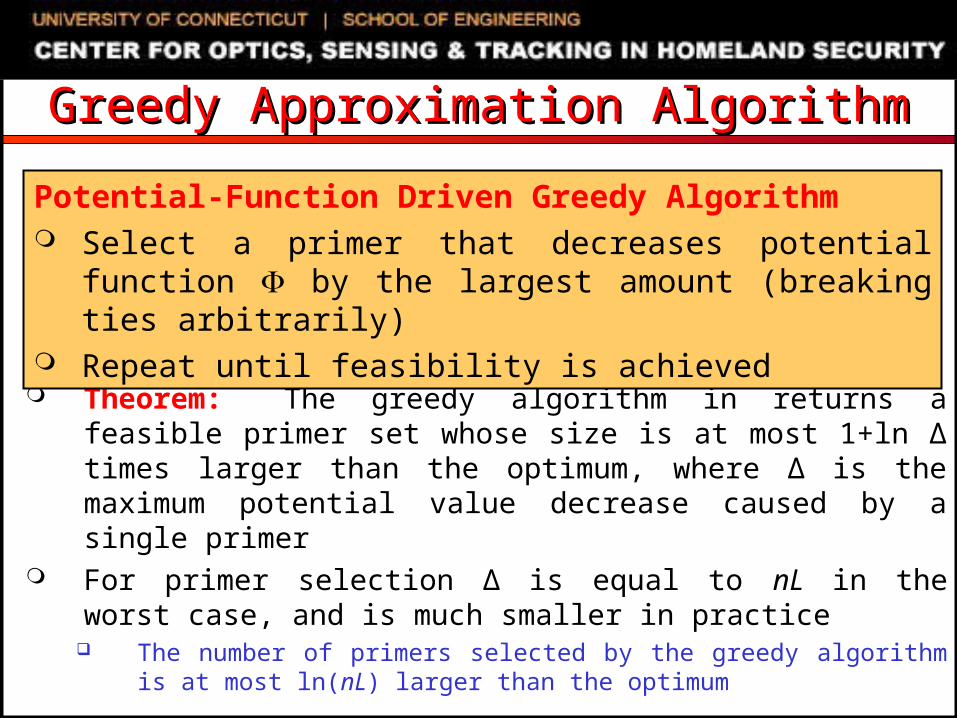
• Key idea: use potential function to measure progress towards fasibility. For primer selection, potential function counts the total number of elements that remain to be covered
Theorem: The greedy algorithm in returns a feasible primer set whose size is at most 1+ln ∆ times larger than the optimum, where ∆ is the maximum potential value decrease caused by a single primer
For primer selection ∆ is equal to nL in the worst case, and is much smaller in practice The number of primers selected by the greedy algorithm is at most
ln(nL) larger than the optimum
Potential-Function Driven Greedy Algorithm Select a primer that decreases potential function by the
largest amount (breaking ties arbitrarily) Repeat until feasibility is achieved
Can be modeled as minimum multicolored sub-graph problem: add edge colored by color i between two primers if they amplify i-th target but do not amplify any other genomic sequence
Trivial approximation algorithm: select 2 primers for each amplification target O(n1/2) approximation since at least n1/2 primers required
by every feasible solution
No non-trivial approximation known previously
Integer Program FormulationInteger Program Formulation
Variable xv for every graph node (candidate primer) v V; xv set to 1 if v is selected, and to 0 otherwise
Variable ye for every graph edge e E; ye set to 1 if corresponding primer pair selected to amplify one of the targets,
Theorem: With probability of at least 1/3, the number of selected nodes is within a factor of O(m1/2lnn) of the optimum, where m is the maximum number of edges sharing the same color and n is the number of nodes (candidate primers).
For primer selection, m L2 approximation factor is O(Llnn)
LP-Rounding Algorithm
• Solve linear programming relaxation
• Select node u with probability xu
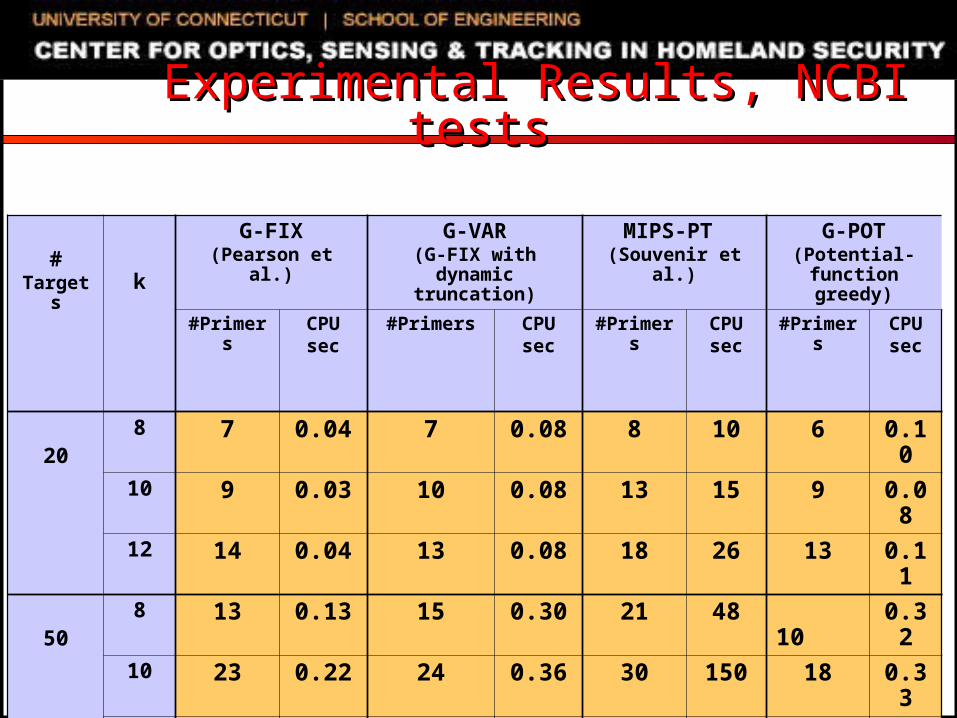
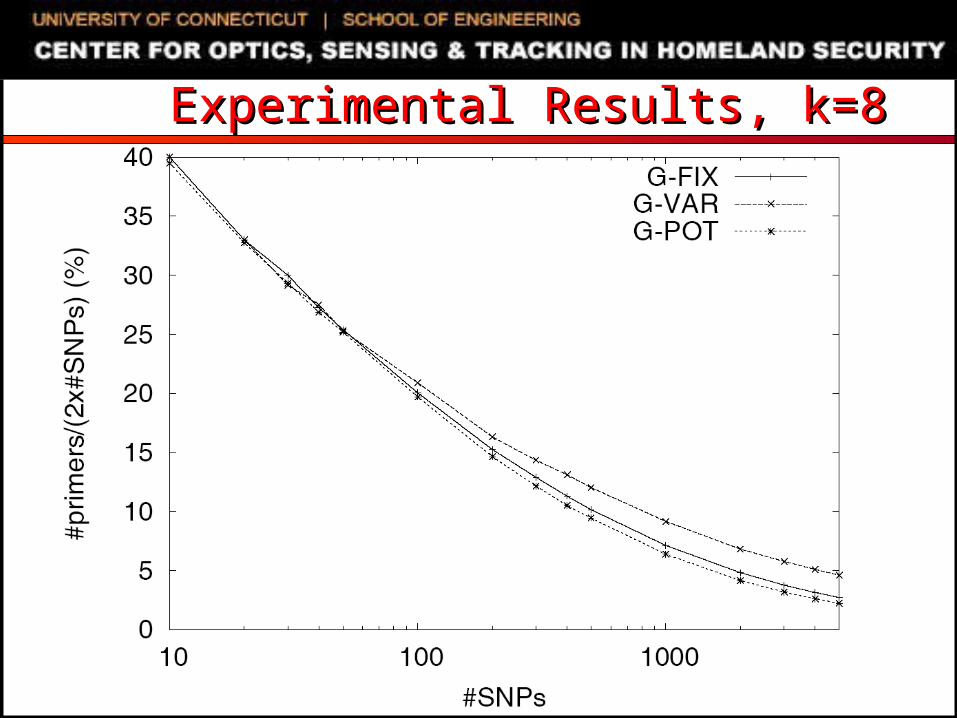
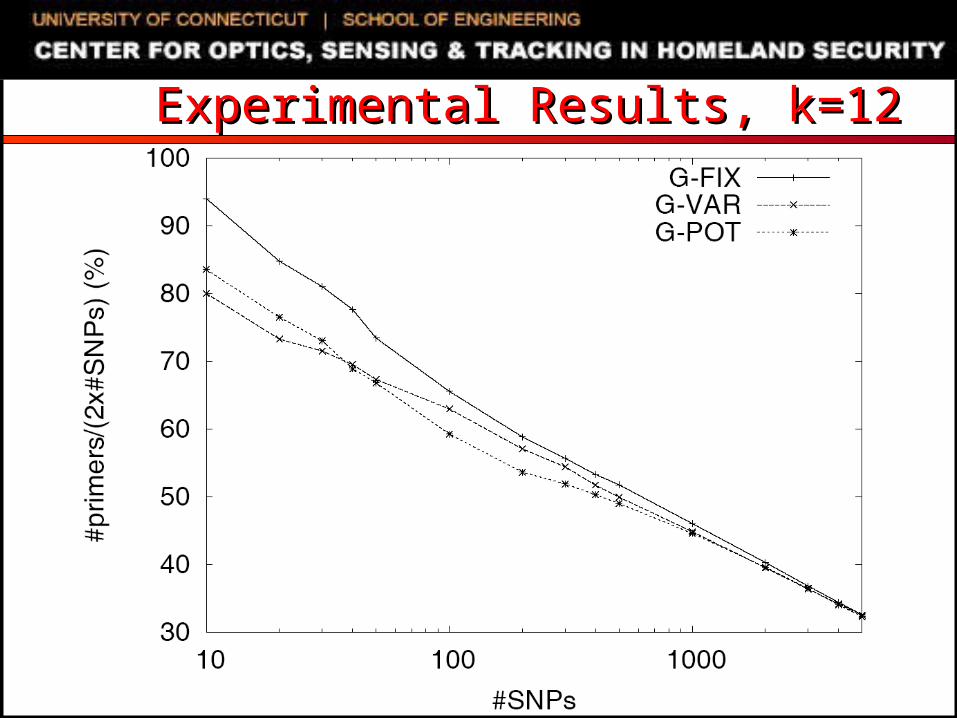
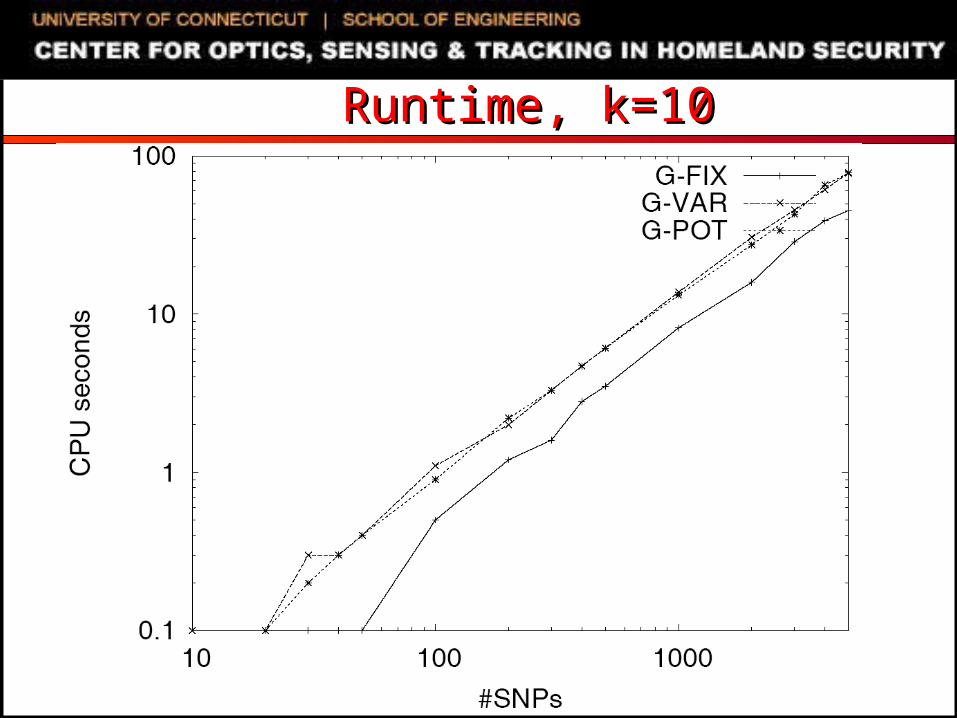
Experimental SettingExperimental Setting SNP genotyping datasets
Extracted from NCBI databases Randomly generated using uniform distribution
C/C++ code, 2.8GHz Dell PowerEdge running Linux Compared algorithms
G-FIX: greedy primer cover algorithm of Pearson et al. Primers restricted to be within L/2 bases of amplified SNPs
G-VAR: naïve modification of G-FIX For each SNP, first selected primer can be up to L bases away
from SNP If first selected primer is L1 bases away from the SNP, opposite
sequence is truncated to a length of L- L1 MIPS-PT: iterative beam-search heuristic of Souvenir et al. G-POT: potential function driven greedy algorithm
Ongoing Work on Primer SelectionOngoing Work on Primer Selection
Extending the greedy algorithm to degenerate primer selection Huge number of feasible candidate primers impractical to find
primer with largest reduction in potential function The greedy algorithm remains provably good if only near-optimal
choices are made in each step
Incorporating improved hybridization models Allow hybridization with mismatches, enforce constraints on melting
temperature, secondary structure, cross hybridization, etc.
Closing gap between O(lnn) inapproximability bound and O(m1/2lnn) approximation factor for the minimum multi-colored subgraph problem
Finding approximation algorithms and practical heuristics for partitioning into multiple multiplexed PCR reactions (Aumann et al. WABI’03)
The String Barcoding ProblemThe String Barcoding Problem
String barcoding is a pathogen identification technique recently proposed by Rash and Gusfield, and Bornemann et al. In this technique, a number of short oligos called distinguishers are spotted or synthesized on a microarray and hybridized with the fluorescently labeled DNA of unknown pathogens. The hybridization pattern can be viewed as a string of 0's and 1’s. The unknown pathogen can be identified by comparing this 0/1 pattern (its ``barcode'') with a set of pre-computed patterns for the pathogens. The main objective is to minimize the number of distinguishers needed to uniquely identify the pathogens.
Given: Genomic sequences g1,…, gn
Find: Minimum number of strings t1,…,tk
Such that: For every gi gj, there exists a string tl which is the Watson-Crick complement for a substring of gi or gj, but not of both
Ongoing Work on String BarcodingOngoing Work on String Barcoding The greedy setcover algorithm, in which pairs of pathogens are viewed as
elements to be covered, and candidate distinguishers are viewed as sets, is known to guarantee an approximation factor of 2lnn
An “information content” greedy algorithm was recently shown by Berman et al. to have an approximation factor of 1+lnn
In ongoing work we explore heuristics for the following important extensions of the string barcoding problem: Probe mixtures as distinguishers. In spotted microarrays, it is feasible
to spot a mixture consisting of a limited number of probes at any given array location. Using probe mixtures can reduce the number of spots on the array - hence barcode length - close to the information theoretical lower-bound of log2n
Robust barcodes. Practical application of string barcoding is complicated by imperfect hybridization, experimental errors, and variability in pathogen genomic sequence. We are exploring robust barcodes using redundant distinguishers and error correcting schemes
ReferencesReferences R.J. Fernandes and S.S. Skiena. Microarray synthesis through multiple-use
PCR primer design. Bioinformatics, 18:S128–S135, 2002. M.T. Hajiaghayi, K. Jain, K.M. Konwar, L.C. Lau, I.I. Mandoiu, A.C. Russell,
A.A. Shvartsman, and V.V. Vazirani. The Minimum k-colored subgraph problem in haplotyping and DNA primer selection, submitted to ACM Symp. on Discrete Algorithms.
K.M. Konwar, I.I. Mandoiu, A.C. Russell, and A.A. Shvartsman, Improved Algorithms for Minimum PCR Primer Set Selection with Amplification Length Constraints, submitted to 3rd Asia Pacific Bioinformatics Conference.
K.M. Konwar, I.I. Mandoiu, A.C. Russell, and A.A. Shvartsman, Approximation algorithms for minimum PCR primer set selection with amplification length and uniqueness constraints. ACM Computing Research Repository, Technical Report cs.DS/0406053, 2004.
W.R. Pearson, G. Robins, D.E. Wrege, and T. Zhang. On the primer selection problem for polymerase chain reaction experiments. Discrete and Applied Mathematics, 71:231–246, 1996.
R. Souvenir, J. Buhler, G. Stormo, and W. Zhang. Selecting degenerate multiplex PCR primers. In Proc. 3rd Intl. Workshop on Algorithms in Bioinformatics (WABI), pages 512–526, 2003.