42
Overview and Management of Undiagnosed Congenital Malformations Dorothy Gleditsch, MD Staff Neonatologist Freeman Health System Joplin, Missouri Management of Surprise Oops-a- daisies
| Date post: | 17-Dec-2015 |
| Category: |
Documents |
| Upload: | emery-dickerson |
| View: | 216 times |
| Download: | 0 times |

Overview and Management of Undiagnosed Congenital Malformations
Dorothy Gleditsch, MD
Staff Neonatologist
Freeman Health System
Joplin, Missouri
Management of Surprise Oops-a-daisies
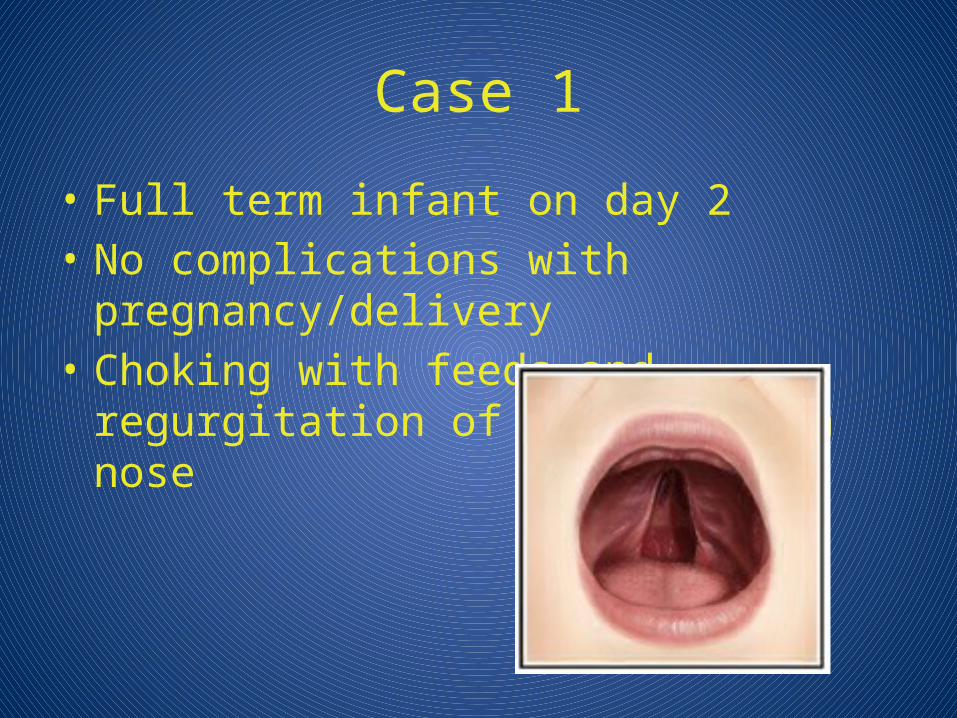
Case 1
• Full term infant on day 2• No complications with pregnancy/delivery• Choking with feeds and regurgitation of
milk through nose

Cleft Lip/Palate
• Definition- anterior cleft-lateral; posterior cleft- midline
• Etiology: Most are isolated entities; midline facial clefts associated with brain abnormalities (Trisomy 13)
• Epidemiology: 1:900 – 1:2000• Delivery room management
– Most without respiratory problems– multiple anomalies (Pierre Robin)-prone/lateral, jaw
thrust, oral airway

Cleft Lip/Palate
• Nursery management– Choking attacks, cyanotic spells esp with
feeds, regurg of milk through nose– Cleft: Keep with mother, lactation consult,
Habermann feeder, staged surgery – PRS-nurse side lying. Keep level, Oral airway

Case 2
• Full term c/section, 1 hour old• During resuscitation, pink and screaming• Now resting quietly in mom’s room and
with increased respiratory distress• When unwrapped and examined, pink and
screaming, improved respiratory distress

Choanal atresia
• Epidemiology: – 1:5000 newborn. – Bilateral to unilateral atresia is 2:1– Bony (90%), soft (10%) tissue
• Etiology: Single or combined anomaly (eg. CHARGE- coloboma, heart disease, choanal atresia, retarded growth/development, ear malformations)

Choanal atresia
• Clinical Presentation: – bilateral atresia
• respiratory distress, retractions; • improved with crying.
– Unilateral atresia • difficult to detect
• Diagnosis in delivery room: – dyspnea when mouth closed, – no condensation on scissors, no breath auscultated
from nares, – failure to pass NG

Choanal atresia: management
• keep airways open with oral airway (Guedel tube, <50mm)
• LMA/intubation rarely indicated
• If suspected, no PO feeds or pacifier
• Scope +/- CT prep- Afrin

Case 3
• SVD, full term• No prenatal care• L&D call about “something on his back”

Myelomeningocele• Definition: protrusion of neural elements through
open vertebral arches • Embryology:
– Defective closure of the vertebral arches in 6th week. – Associated with hydrocephalus due to Arnold-Chiari
malformation in 90%• Epidemiology:
most common type of NTD; very geographic- ranges from 0.5-2 per 1000

Myelomeningocele
• Delivery room management: – use sterile, non-latex gloves– respiratory distress frequent with high lesions– Avoid pressure and contamination of site– Estimate level of lesion before covering – Common for small amount of CSF weeps from edges– Cover with soaked gauze (LR or NS) and sterile transparent
non-adherent plastic or soaked Kerlix wrapped around torso , repeat soak at least every 4h
– Do not change dressing unless obviously soiled– If sac is open, do not use gauze or cotton on defect – Obtain IV for fluids and broad-spectrum antibiotics
(cefotaxime and gentamicin)

Myelomeningocele
• NICU management:– Monitor temperatures closely– Place prone, hips abducted, feet neutral position– Exam for spontaneous activity– HUS (hydrocephalus), Abd US (renal, ureteral dilatations
and reflux), Hip US (dysplasia)– Monitor for meningitis– Surgery for repair at 24-72 hours
• Prognosis: – 10% with isolated defect
have MR. Improved long-term functioning but GU and orthopedic disability still common
Level Implications for mobility
High lumbar-thoracic Limited walking with long brace, most use wheelchairs
Low lumbar Can walk with short leg braces and forearm crutches
High sacral Can walk with gluteal lurch using braces to stabilize

Case 4
Full term infant, SVD
Midline mass on forehead, skin covered, purple hue
Resident called and thinks it’s a hemangioma…..you think otherwise.

Encephalocele: overview
• Definition: skin-covered neural tube defects with a midline mass overlying a skull defect
• Presentation: – 75% mass is occipital, 12% frontal, and 13% parietal. – May be fluid filled, brain filled or a mix. – May be entirely skin covered or leaking CSF
• Differential:– Nuchal edema, cystic hygroma, Dandy-Walker cyst, cephalohematoma,
cranial hemangiomas, and fetal scalp cysts…..all have intact skull
• Prognosis: – dependent on the brain malformation, not the size of the defect. – Microcephaly has poor prognosis.
• Recurrence: Most are sporadic with multifactorial recurrence risk (2-5%)

Encephalocele: management
• most do not require early assistance unless large defect with mostly brain
• Side-lying intubation or BMV for occipital• Special care to avoid injury/pressure to
mass• Potential heat loss• Head imaging

Case 5• Full term, SVD, 24 hours old• Pregnancy c/b polyhydramnios• Tons of secretions, choking with breast feeds• Plan to start NG feeds, but cannot auscultate

TE Fistula/atresia
• Epidemiology: 1/4500 births, most often isolated, 1/3 with additional anomalies, VACTERL
• Etiology: Abnormal formation 4th week of gestation
• Clinical Presentation: Unable to pass NG to stomach, coil in esophagus, cyanosis/aspiration with feeds
• Prognosis: Survival
Without other anomalies Near 100%
With associated anomalies
Near 50%
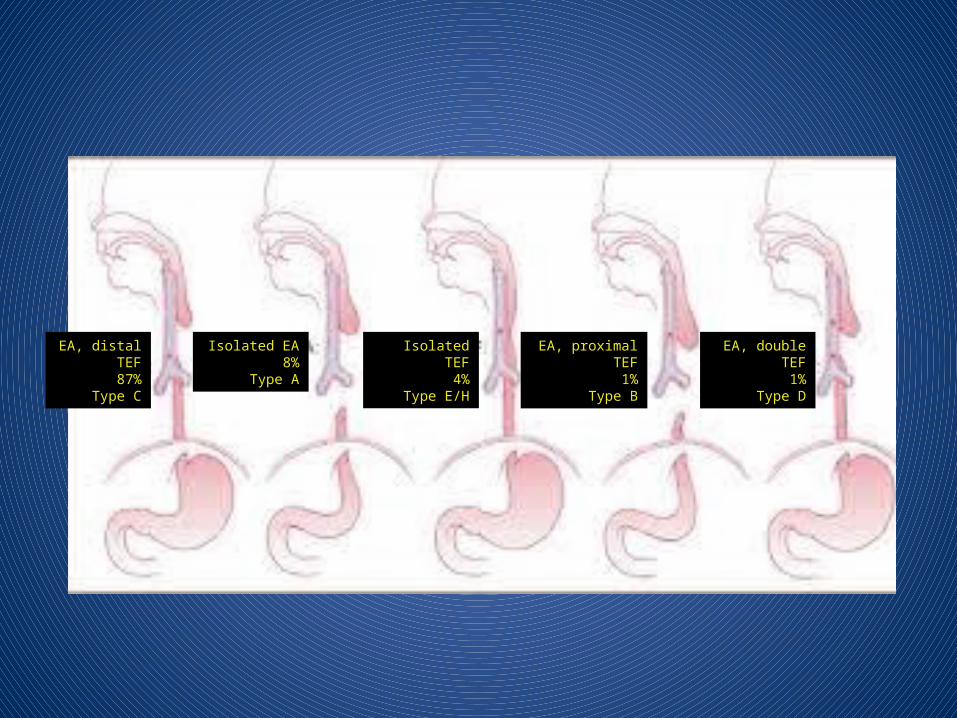
EA, distal TEF87%
Type C
Isolated EA8%
Type A
Isolated TEF4%
Type E/H
EA, proximal TEF1%
Type B
EA, double TEF1%
Type D

TE fistula/atresia: management
• Upright positioning• Catheter placed in upper pouch to
intermittent suction• If respiratory distress, intubation preferred
to BMV• NPO, TPN• Surgical repair: primary vs delayed vs
staged

Case 6
• 37 week, c/section• No respiratory effort at delivery• Intubated to 9cm, unable to hear breath sounds
on L• ETT pulled back, inadvertently removed• PPV with BMV given, still no BS on L, notice
scaphoid abdomen• Reintubated to 8cm, no BS on L, improved sats,
mist in tube, BS on right, CO2 detector yellow


Congenital Diaphragmatic Hernia
• Definition: abdominal contents protrudes into the chest. Left > right
• Presentation: – Based on severity: Severe respiratory failure to accidential
diagnosis– Good prognosis if distress
onset after 10 minutes – Severe CDH:
• decline in apgar• scaphoid abdomen,• breath sounds absent on ipsilateral side, • deviation of trachea to unaffected side,• displacement of heart sounds to the right in most patients (CDH
more common on left)
LCDH liver down LCDH liver up RCDH

CDH: management
• Do not BMV, intubate trachea immediately, • 50-100% FiO2, low PIP and low PEEP; lowest settings to
stablize, HFOV• Pre and post ductal sats • Insert replogle and place on suction • Place central lines (UVC, UAC, or PICC) • Allow permissive hypercapnea but avoid acidosis• CXR to confirm diagnosis, ECHO to exclude CHD • Be prepared to deal with pneumothorax • Minimize stimulation, keep comfortable• Delayed surgical repair 24-72h

CDH: management debate
• Surfactant: – use associated with higher use of ECMO and incidence of
CLD, survival is lower
• Paralysis agents: – allowing spont breathing may be assocaited with improved
survival. Some protocols emphasize early paralysis and sedation
• HFOV: – some reports suggest improved survival
• iNO: – slightly worse outcomes in meta-analysis of randomized
trials with use of iNO

Case 7
• 12 hour old infant• Mild respiratory distress and persistent
cyanosis not responsive to O2
• No murmur• “Doesn’t look good”


Transposition of Great Arteries
• Incidence: 0.2-0.4/1000 births. 4-6% of CHD. Most common CHD presenting in first week
• Recurrence risk: 2-5% risk of recurrence if 1 previous child, if 2 children risk increases to 5-10%
• Presentation: Cyanotic heart diseases- 5Ts + DORV, Ebsteins– 1- truncus arteriosus– 2- transposition of great arteries– 3- tricuspid atresia– 4- tetrology of fallot– 5- total anomalous pulmonary venous return

Transposition of Great Arteries
• Clinical presentation: – severe cyanosis result of separate circulations and
poor mixing, – worse cyanosis if restrictive PFO or ASD, – no murmur unless VDS or PS
• CXR: “egg on a string”, heart size nrl to slightly increased, increased pulmonary markings
• Prognosis: – Operative mortality is 3-5% – 88% survival at 15 years – Long term is unknown

Transposition of Great Arteries- management
• Spontaneous respirations generally present, mechanical ventilation usually not needed
• Secure venous access required +/- arterial catheter • Aim for saturations of 70-85% without metabolic
acidosis• PGE1, (0.05-0.1mcg/kg/min)• Emergent Rashkind procedure if inadequate mixing • Arterial switch within first week when pulmonary
pressures decreasing

Case 8
• At a delivery and get handed this…….


Omphalocele
• Epidemiology: 1/3000 to 1/10,000 births; 3:1 males to female. – Associated defects (present in ~80%)
• Etiology: intestinal loop fails to return to abdominal cavity at 11 weeks or somatic folds fail to complete formation of the abdominal wall by 18 weeks gestation
• Prognosis: – depends on associated anomalies; – worse prognosis with large defects, ruptured sac, LBW,
early respiratory failure. – Survival 90% with isolated

Omphalocele
• Clinical presentation:– amniotic sac and peritoneum protect intestinal
loops – covering sac may be ruptured – umbilical cord inserts onto the amniotic sac– abdominal wall musculature normal– larger (giant >5cm) may contain liver – all have malrotation – rare to have other intestinal anomalies (vs gastro
where common)

Omphalocele: management
• wrapped with a sterile dressing, minimize heat and fluid loss
• Large defects typically have small lungs…be prepared to intubate
• BMV contraindicated• Prompt gastric decompression with
intermittent suctioning

Case 9
• The next day you get this……


Gastroschisis
• Epidemiology- 1/4000 births• Etiology- several proposed-
– Involution of right umbilical vein creates weak spot at junction of the right aspect of the umbilical ring and the abdominal wall; may allow for rupture and bowel herniation
• Presentation– Eviscerated intestinal loops without covering through
abdominal wall defect located to the right of the umbilical cord
– Intact cord– Loops may be thickened, foreshortened, or covered with
fibrous peel

Gastroschisis: management
• Assistance with onset of respirations usually not needed, BMV contraindicated
• Place infant of right side to decrease risk of kinking• Place infant in bowel bag• NS bolus, initial fluid management with total fluid
~150ml/kg/d due to substantial loss• Surgical repair- primary vs delayed• Prognosis- survival >90%; long repair associated
with: prematurity, LBW, staged silo repair, intestinal atresia. 10-15% with long-term developmental disabilities


Case 10
• Full term, SVD, 36 hours• Breast feeding • At report, mentioned that she hasn’t
stooled• When changing her diaper, you are
greeted with this….

Imperforate Anus
• Definition: Congenital absence of anal opening
• Epidemiology: 1/5000 (M3:F2) Associated anomalies present in 90%
• Management:– NPO, IVF/TPN – establish secure venous line – nasogastric decompression – ECHO, chromosomes

Questions??

















