Pathogenesis of Street Rabies Virus Infections in Resistant andSusceptible Strains of Mice
DONALD L. LODMELL* AND LARRY C. EWALTLaboratory of Persistent Viral Diseases, Rocky Mountain Laboratories, National Institute of Allergy and Infectious
Diseases, Hamilton, Montana 59840
Received 6 March 1985/Accepted 20 May 1985
Seven strains of mice were examined to determine why susceptibility differences and variations in clinicalcentral nervous system (CNS) disease occurred among these animals after intraperitoneal inoculation of streetrabies virus (SRV). Trace experiments for infectious virus indicated that these differences were associated withrestriction of virus replication within the CNS. Limitation of viral replication appeared to correlate with theantibody response in that prominent serum anti-SRV neutralizing antibody titers were detected in resistantstrains, whereas susceptible strains produced minimal amounts of antibody until their death. The importanceof the immune response was reaffirmed with cyclophosphamide studies in that all resistant SJL/J mice diedafter immunosuppressive treatment. In contrast, cyclophosphamide-treated SJL/J mice whose immune systemswere reconstituted with either unfractionated immune spleen cells or with sera 24 h after SRV inoculationsurvived a lethal dose of SRV. More importantly, immunosuppressed SJL/J and immunodeficient athymic micewere protected when reconstituted with immune serum 72 h after SRV inoculation, a time in which infectiousvirus was detected in the spinal cords of some mice but was not present in the peritoneal cavity. Additionalstudies showed that antibody in the cerebrospinal fluid was unimportant in the resistance of mouse strainswhich remained clinically asymptomatic, but it appeared to be associated with the survival of mice whichdeveloped clinical CNS disease. Furthermore, CNS resistance to intranasal or intracerebral inoculation withchallenge virus standard rabies virus developed as early as 5 days post-intraperitoneal inoculation of SRV.
After introduction of rabies virus by inoculation or animalbite, the virus remains near the site of inoculation, duringwhich time replication may occur. Unless inactivated bynatural or induced defense mechanisms, the virus enters theperipheral nerves and travels centripetally to spinal gangliaand then to the brain. There is no viremia (31). At the onsetof clinical symptoms, virus is widespread throughout thecentral nervous system (CNS) with highest concentrations inthe brainstem, basal ganglion, hippocampus, and cerebel-lum. Furthermore, the bizarre symptomatology and nearly100% fatality rate of this disease are associated with minimalneuropathologic findings (21, 31). After reaching the CNS,rabies virus spreads centrifugally from the CNS and ispresent in neurons throughout the body. Virus may even bedemonstrated by fluorescent staining of corneal cells (23, 38)or of skin biopsies (6, 44). Moreover, the saliva ofpresymptomatic rabid animals may contain virus (5, 39, 48),and intermittent excretion of rabies virus in the saliva ofapparently healthy animals has been reported (12, 14, 52).Although animals infected with street rabies virus (SRV)usually die, there are reports of resistance and of survivalafter onset of clinical CNS disease (1, 2, 4, 13, 18, 25, 27, 28,34, 35). Presently, the mechanisms mediating this resistanceand survival are unknown, but physiological (31), virological(11, 22, 49), nonimmune (3, 17, 19, 45), and immune (10, 26,29, 30, 32, 33, 36, 41, 42, 50) factors alone or in combinationare suggested as possible explanations.
In previous studies, we determined that murine resistanceto intraperitoneally (i.p.) inoculated SRV is genetically con-trolled by the concurrent presence of each of two segregatinggenes (25, 28). Furthermore, three different clinical re-sponses were defined among the inbred mice used in these
* Corresponding author.
studies: (i) SJL/J and CBA/J strains were resistant and didnot develop clinical CNS disease (resistant-asymptomatic),(ii) BALB/cAn and DBA/2J strains developed clinical CNSdisease with irreversible paralysis, but subsequently sur-vived (resistant-recovered), whereas (iii) A/WySnJ, A.SW/SnJ, and athymic mice on the BALB/cAn background died(susceptible) (25). Thus, in contrast to the uniform suscep-tibility of these genetically dissimilar mouse strains tointracerebrally inoculated SRV (25), mechanisms of resist-ance to SRV and of survival after viral invasion of the CNSand onset of clinical disease can be studied following i.p.inoculation.
In an attempt to understand the complex interactionsoccurring during this nonviremic infection, we traced thereplication and spread of SRV in the CNS. In addition, wemeasured the anti-SRV neutralizing antibody response insera and cerebrospinal fluid (CSF), actively intervened in theimmune response of resistant mice with cyclophosphamide(46), passively transferred immune cells and sera to im-munosuppressed and immunodeficient mice, and directlychallenged the CNS of SRV i.p. inoculated mice with rabiesvirus. The results showed that the development of an earlyand markedly efficient immune response protected miceagainst i.p. inoculated SRV. Furthermore, the CNS ofresistant mice was refractory to direct challenge with ahighly virulent rabies virus 5 days after SRV inoculation.
MATERIALS AND METHODS
Mice. SJL/J, CBA/J, A.SW/SnJ, A/WySnJ, and DBA/2Jstrains of mice were maintained as inbred stocks as previ-ously described (25). BALB/cAn (nul+ or +/+) and athymicnude (nulnu) mice were reared as outbread strains by suc-cessive cross-intercrossing onto the BALB/cAn background.Pathogenesis experiments were conducted in 8- to 12-week-
788
MURINE RESISTANCE TO STREET RABIES VIRUS 789
CERVICAL THORACIC
BRAIN CORD
I
LUnR
CORD CORD
91 0 LIW. W
_ _
Day Post-
Inoculation
SRV titer (log10 LD50/0.03 ml)
<0.7 <0.7 <0.7 <0.7
<0.7 <0.7 0.7(±0.20) <0.7
<0.7 0.7 1.3(±0.35) <0.7
6 0.7 2.6(±0.22) 3.8(±0.38)
7 2.4(±0.84) 4.0(±0.43) 5.1(±0.4)
<0.7
0.7(±0.1)
FIG. 1. Early detection of i.p. inoculated SRV in the CNS ofA/WySnJ mice. SRV-susceptible female A/WySnJ mice (8 to 12weeks old) were inoculated i.p. with 5 x 107 mouse intracerebralLD50 of SRV. At different days postinoculation, the brain and0.5-cm sections from the center of the cervical, thoracic, and lumberregions of the spinal cord were removed, prepared as 20% suspen-sions, and assayed for infectious virus by intracerebral inoculationof 21-day-old SW mice. The results represent LD50 mean titers(loglo/0.03 ml) + standard error. The minimal level for virus detec-tion is 100.7 mouse intracerebral LD50/0.03 ml of tissue suspension.No., five mice at each day postinoculation.
old female mice. Outbred Swiss-Webster (SW) mice (3 to 4weeks old) raised at the Rocky Mountain Laboratories wereused to prepare stock viral pools and to titrate SRV in tissuesuspensions.
Rabies viruses. Unless stated otherwise, a wild-type SRVwhich had been isolated from an adult bat (Eptesicusfuscus)and then passaged six times by intracerebral inoculation inSW mice was used. The brain suspension titer was 1065mouse intracerebral 50% lethal doses (LD50/0.03 ml) ascalculated by the method of Reed and Muench (37). Thestock viral pool was prepared, stored, and titrated as previ-ously reported (25).A modified live-virus vaccine (Endurall-R) of canine origin
(HEP-Flury strain) was purchased from Norden Laborato-ries, Inc., Lincoln, Neb., and the laboratory-adapted chal-lenge virus standard (CVS) rabies virus was obtained fromJohn Moore of the Rocky Mountain Laboratories.
Pathogenesis studies. Mice were inoculated i.p. with 0.5 mlof a 10% mouse brain suspension containing 5 x 107 mouseintracerebral LD50 of SRV (25). In initial studies, various0.5-cm sections of spinal cord were assayed to determine thesite at which SRV entered the spinal cord after i.p. inocula-tion. The cervical cord was removed between cervicalvertebrae 3 and 8, the thoracic cord was removed betweenthoracic vertebrae 9 and 12, and lumbar cord was removedbetween lumbar vertebrae 1 and 5. In subsequent studies, atvarious times postinoculation, the entire spinal cord andbrain were removed, prepared as separate 20% suspensions,and assayed for infectious virus by intracerebral inoculationof SW mice. LD50 titers per 0.03 ml of mouse brain suspen-sion were calculated by using 10-fold dilutions and six miceper dilution (37) and were expressed in powers of 10 as thereciprocal of the highest dilution which killed 50% of themice.CSF was obtained by cisternal puncture immediately after
exsanguination. The exposed dura of the cisternal space,under visualization through a magnifying glass, was punc-
tured with a 27-gauge needle. From each mouse, 8 to 12 ,dl ofclear CSF was routinely collected by capillary action in two10-,ul micropipettes joined by Teflon tubing (E. I. du Pont deNemours & Co., Inc., Wilmington, Del.).
Antibody determinations. SRV neutralizing antibody inCSF and serum was quantified with the rapid fluorescent-focus inhibition test (RFFIT) (43) by using chicken embryo-related (CER) cells (40) instead of BHK-21/S13 cells. Insome instances, the RFFIT test was done with SRV-infectedcells. In vivo neutralization tests in SW mice (27) were
performed against SRV. Neutralization titers are expressedas the reciprocal of the highest dilution which reduced by50% the number of fluorescent cells or which protected 50%of the mice.BPL inactivation of virus. SRV and CVS rabies viruses as
10% brain suspensions were inactivated with 3-pro-piolactone (BPL) according to standard procedures (20).Immunosuppression. Mice were inoculated i.p. with
freshly prepared cyclophosphamide (Cytoxan, MeadJohnson & Co., Evansville, Ind.) at a dose of 300 mg/kg ofbody weight.
Passive transfer of sera or cells. SJL/J mice were inoculatedwith 300 mg of cyclophosphamide per kg of body weight andwere challenged i.p. either 2 or 3 days later with 5 x 107mouse intracerebral LD50 of SRV. Treatment was beguneither 24 or 72 h post-SRV inoculation. Unfractionatednormal or immune spleen cells (107) were inoculated intra-venously (i.v.) one time only, whereas 0.2 or 0.3 ml ofnormal or immune serum was inoculated i.v. every thirdday, or every other day, for 18 days. The immune sera andcells were obtained from mice which survived -30 days afteri.p. inoculation of 5 x 107 mouse intracerebral LD50 of SRV.The RFFIT titer of the serum pool was 5,120.
Intranasal challenge. Supernatant fluid (25 ,ul) from a 10%CVS brain suspension was placed on the nares of anesthe-tized mice and inhaled. The intranasal titer in 21-day-old SWmice was 103.9 mouse intranasal LD50/0.025 ml (37).
RESULTSi.p. inoculated SRV entered the CNS of susceptible
A/WySnJ mice in the thoracic region of the spinal cord. Spinalcord sections measuring 0.5 cm were assayed to determinewhere i.p. inoculated SRV initially invaded the CNS. Figure1 shows that at 4 days after inoculation infectious SRV wasfirst detected in the thoracic region of the spinal cord. Onday 5, virus concentration increased in this section, and SRVhad ascended to the cervical region. Subsequently, SRVinitially was detected in the brain on day 6, and then in thelumbar segments of the spinal cord 7 days postinoculation.Thus, SRV entered the CNS in the thoracic region of thespinal cord, replicated at this site, and then rapidly ascendedto the brain. In contrast, there was a more gradual spread ofvirus to the lower lumbar segments.
Replication and spread of SRV within the CNS was differentin resistant and susceptible strains of mice. To compare thepathogeneses of SRV in the CNS of resistant and susceptiblemice, brains and spinal cords of mice of seven murine strainswere assayed for infectious virus. Figure 2 shows that SRVinfected the spinal cords of mice of all seven strains. Theinitial detection of SRV on day 3, compared with day 4 as
previously mentioned (Fig. 1), probably occurred becausethe entire spinal cord was assayed in this experiment (Fig.2), compared with a 0.5-cm midsection of the thoracic regipnin the previous experiment (Fig. 1). Nonetheless, after entry,infectious SRV was present in the spinal cords of mice of allstrains at 5 and 7 days postinoculation (Fig. 2). At this time,
VOL. 55, 1985
790 LODMELL AND EWALT
viral replication in the resistant-asymptomatic SJL/J andCBA/J mice was transient, but clearly detectable. SRV wasnot detected in the spinal cords of these strains on day 10 orthereafter. Virus replication in resistant-recovered BALB/cAn and DBA/2J mice, though transient, manifested peaktiters which were between those of resistant-asymptomaticand susceptible strains of mice (2.5 to 3.3 log1o mouseintracerebral LD50). Viral titers in the resistant-recoveredstrains began to decrease after days 5 (BALB/cAn) and 10(DBA/2J), and infectious SRV was not detected in the spinalcords of mice of either strain at day 15 or thereafter. Incontrast to resistant strains, virus titers in spinal cords ofmice of the susceptible strains steadily increased until themice died between 10 and 15 days postinoculation (Fig. 2).These results illustrated that susceptibility differences
ECO,0
0o
-J.
0
0
-J
I-
cc
Co
0c)
-J
z
C':
DAYS POST INOCULATIONFIG. 2. Comparison of virus titers in spinal cords of genetically
resistant and susceptible strains of mice after i.p. inoculation ofSRV. The 8- to 12-week-old female mice from seven strains wereinoculated i.p. with 5 x 107 mouse intracerebral LD50 of SRV. Ateach postinoculation interval, the brains and spinal cords from miceof each strain were removed, prepared as separate 20% suspensions,and assayed for infectious virus by intracerebral inoculation of21-day-old SW mice. Susceptible mice died between days 10 and 15postinoculation. The minimal level for virus detection is 100-7 mouseintracerebral LD50O0.03 ml of tissue suspension. Each point repre-sents the LD50 mean titer from at least six mice at each interval.Variance among the values used to establish the mean at eachinterval was '20%.
Athymic
Et I9/1A/WySnE::
C,,0
ce 3-tgii
o
I',7 m _/1 aJ2:14571 0 15 2
0 ~~~~~~~DBA/2
I 2
z
0.7-
<0.7L=SJ&DAYS POST INOCULATION
FIG. 3. Comparison of virus titers in brains of genetically resist-ant and susceptible strains of mice after i.p. inoculation of SRV. The8- to 12-week-old female mice from seven strains were inoculatedi.p. with 5 x 107 mouse intracerebral LD50 of SRV. Same procedureand conditions as in the legend to Fig. 2.
among genetically different strains of mice to i,p. inoculatedSRV were not due to failure of virus to infect the spinal cordbut instead were associated with inhibition of viral replica-tion after spinal cord infection.
Similar patterns of virus replication were observed in thebrains of these mice (Fig. 3). However, SRV never wasisolated from brains of mice from the resistant-asymptomaticSJL/J and CBA/J strains. In contrast, maximum, but tran-sient, virus titers of 1.6 to 2.3 log1o mouse intracerebral LD50were attained by day 10 in the resistant-recoveredBALB/cAn and DBA/2J mice. Virus concentrations in thesestrains then decreased, and SRV was not detected on days15 and 21, respectively. As noted in the spinal cord studies,virus concentrations in brains of susceptible mice steadilyincreased until the mice died; at 10 days postinoculation,SRV titers of these mice were 100-fold greater than those ofresistant-recovered BALB/cAn and DBA/2J strains. Theseresults, in conjunction with the spinal cord studies, sug-gested that a resistance mechanism(s) influenced viral repli-cation or spread of virus or both after CNS infection.Serum and CSF anti-SRV neutralizing antibody responses
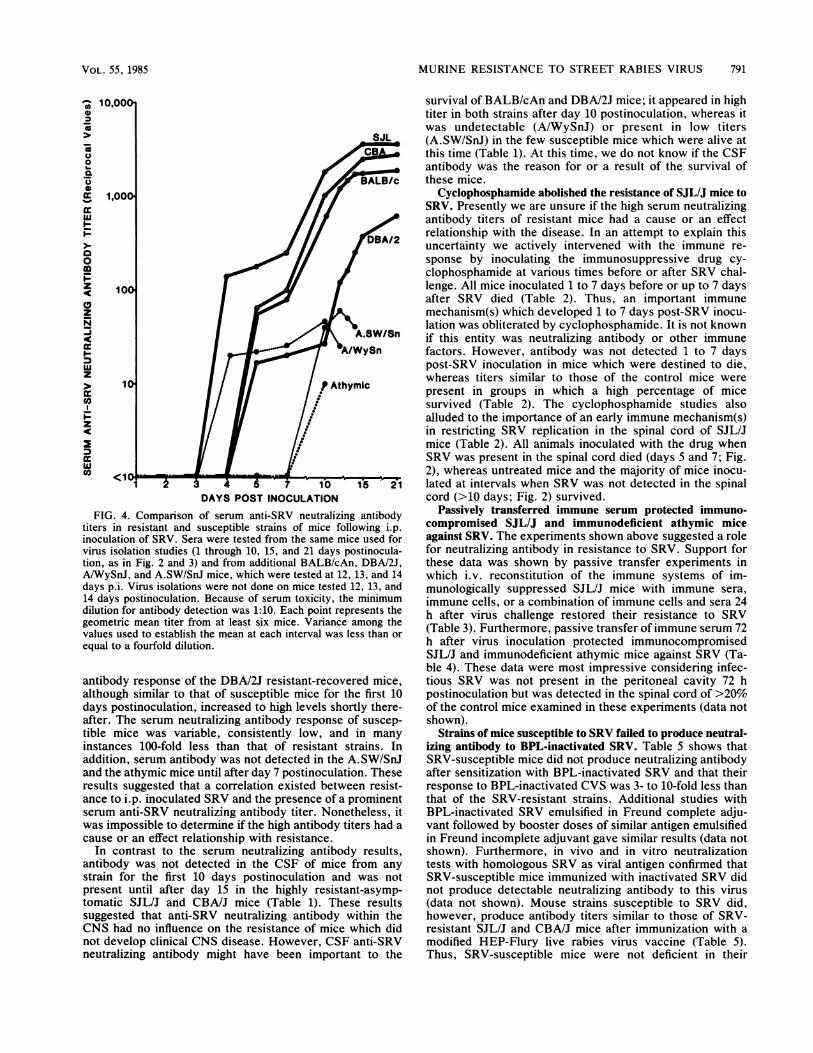
varied among geneticaHly dissimilar strains of mice. Figure 4shows that serum anti-SRV neutralizing antibody titers ofthe resistant-asymptomatic SJL/J and CBA/J strains and oneof the two resistant-recovered strains (BALB/cAn) weregreater than those of susceptible strains at every intervaltested. Furthermore, the antibody titers of these mice stead-ily increased until the experiments were terminated. The
J. VIROL.
MURINE RESISTANCE TO STREET RABIES VIRUS 791
1000
0
0
BsALB/cm 1,00
tion,Fas n Fig. 2and 3) ad from aditionalBALB/cAn DBA/20
z 100
z
A S ahSW/SnA/WySn
w
z
> 1pVP Athyme c
z
7 10b 1-521DAYS POST INOCULATION
FIG. 4. Comparison of serum anti-SRV neutralizing antibody
titers in resistant and susceptible strains of mice following i.p.
inoculation of SRV. Sera were tested from the same mice used for
virus isolation studies (1 through 10, 15, and 21 days postinocula-tion, as in Fig. 2 and 3) and from additional BALB/cAn, DBAI2J,
AiWySnJ, and A.SW/SnJ mice, which were tested at 12, 13, and 14
days p.i. Virus isolations were not done on mice tested 12, 13, and
14 days postinoculation. Because of serum toxicity, the minimum
dilution for antibody detection was 1:10. Each point represents the
geometric mean titer from at least six mice. Variance among the
values used to establish the mean at each interval was less than or
equal to a fourfold dilution.
antibody response of the DBAI2J resistant-recovered mice,
although similar to that of susceptible mice for the first 10
days postinoculation, increased to high levels shortly there-
after. The serum neutralizing antibody response of suscep-
tible mice was variable, consistently low, and in many
instances 100-fold less than that of resistant strains. In
addition, serum antibody was not detected in the A.SW/SnJand the athymic mice until after day 7 postinoculation. Theseresults suggested that a correlation existed between resist-ance to i.p. inoculated SRV and the presence of a prominentserum anti-SRV neutralizing antibody titer. Nonetheless, it
was impossible to determine if the high antibody titers had a
cause or an effect relationship with resistance.In contrast to the serum neutralizing antibody results,
antibody was not detected in the CSF of mice from any
strain for the first 10 days postinoculation and was not
present until after day 15 in the highly resistant-asymp-tomatic SJL/J and CBAIJ mice (Table 1). These results
suggested that anti-SRV neutralizing antibody within the
CNS had no influence on the resistance of mice which did
not develop clinical CNS disease. However, CSF anti-SRVneutralizing antibody might have been important to the
survival of BALB/cAn and DBA/2J mice; it appeared in hightiter in both strains after day 10 postinoculation, whereas itwas undetectable (A/WySnJ) or present in low titers(A.SW/SnJ) in the few susceptible mice which were alive atthis time (Table 1). At this time, we do not know if the CSFantibody was the reason for or a result of the survival ofthese mice.
Cyclophosphamide abolished the resistance of SJL/J mice toSRV. Presently we are unsure if the high serum neutralizingantibody titers of resistant mice had a cause or an effectrelationship with the disease. In an attempt to explain thisuncertainty we actively intervened with the immune re-sponse by inoculating the immunosuppressive drug cy-clophosphamide at various times before or after SRV chal-lenge. All mice inoculated 1 to 7 days before or up to 7 daysafter SRV died (Table 2). Thus, an important immunemechanism(s) which developed 1 to 7 days post-SRV inocu-lation was obliterated by cyclophosphamide. It is not knownif this entity was neutralizing antibody or other immunefactors. However, antibody was not detected 1 to 7 dayspost-SRV inoculation in mice which were destined to die,whereas titers similar to those of the control mice werepresent in groups in which a high percentage of micesurvived (Table 2). The cyclophosphamide studies alsoalluded to the importance of an early immune mechanism(s)in restricting SRV replication in the spinal cord of SJL/Jmice (Table 2). All animals inoculated with the drug whenSRV was present in the spinal cord died (days 5 and 7; Fig.2), whereas untreated mice and the majority of mice inocu-lated at intervals when SRV was not detected in the spinalcord (>10 days; Fig. 2) survived.
Passively transferred immune serum protected immuno-compromised SJL/J and immunodeficient athymic miceagainst SRV. The experiments shown above suggested a rolefor neutralizing antibody in resistance to SRV. Support forthese data was shown by passive transfer experiments inwhich i.v. reconstitution of the immune systems of im-munologically suppressed SJL/J mice with immune sera,immune cells, or a combination of immune cells and sera 24h after virus challenge restored their resistance to SRV(Table 3). Furthermore, passive transfer of immune serum 72h after virus inoculation protected immunocompromisedSJL/J and immunodeficient athymic mice against SRV (Ta-ble 4). These data were most impressive considering infec-tious SRV was not present in the peritoneal cavity 72 hpostinoculation but was detected in the spinal cord of >20%of the control mice examined in these experiments (data notshown).
Strains of mice susceptible to SRV failed to produce neutral-izing antibody to BPL-inactivated SRV. Table 5 shows thatSRV-susceptible mice did not produce neutralizing antibodyafter sensitization with BPL-inactivated SRV and that theirresponse to BPL-inactivated CVS was 3- to 10-fold less thanthat of the SRV-resistant strains. Additional studies withBPL-inactivated SRV emulsified in Freund complete adju-vant followed by booster doses of similar antigen emulsifiedin Freund incomplete adjuvant gave similar results (data notshown). Furthermore, in vivo and in vitro neutralizationtests with homologous SRV as viral antigen confirmed thatSRV-susceptible mice immunized with inactivated SRV didnot produce detectable neutralizing antibody to this virus(data not shown). Mouse strains susceptible to SRV did,however, produce antibody titers similar to those of SRV-resistant SJL/J and CBA/J mice after immunization with amodified HEP-Flury live rabies virus vaccine (Table 5).Thus, SRV-susceptible mice were not deficient in their
VOL. 55, 1985
792 LODMELL AND EWALT
TABLE 1. Anti-SRV neutralizing antibody titers in CSF of resistant and susceptible strains of micea
Day Geometric mean titer in mouse strain:postinoculation SJL/J CBA/J DBA/2J BALB/cAn A.SW/SnJ A/WySnJ Athymic
1 <20 NTb NT NT NT NT NT2 <20 NT NT NT NT NT NT3 <20 <20 <20 <20 <20 <20 <204 <20 <20 <20 <20 <20 <20 <20
14 <20 <20 232 (±25) 820 (±46) 22 (±6) _C _C15 <20 <20 500 (±39) 2,240 (±320) C C C
21 202 (±38) 190 (±40) 1,070 (±160) 360 (±100) C _C C
aCSF was tested from the same mice as those used for data presented in Fig. 4. Because of the minimal volume of CSF available for testing, the minimum levelfor antibody detection was 1:20. Each result is the geometric mean titer (±standard error) for at least six mice.
b NT, Not tested.c, All mice dead.
ability to respond to appropriately presented rabies virusglycoprotein(s).CNS immunity developed 5 days after i.p. inoculation of
SRV. Although it has been shown that normal unsensitizedSJL/J mice are highly susceptible to intracerebrally inocu-lated SRV (25), the data in Tables 2 and 3 strongly suggestedthat the resistance of this strain to i.p. inoculated SRV wasimmunologically mediated. Thus, we questioned whetherprior stimulation of specific anti-rabies virus immune mecha-nisms via i.p. inoculation of SRV would alter the susceptibil-ity of SJL/J mice against direct CNS infection. Mice wereinoculated i.p. with SRV and then challenged at variousintervals either intracerebrally or intranasally with the highlyvirulent CVS rabies virus. The CVS rabies virus was used toensure that 100% of the control mice died. Table 6 showsthat all mice (none of which had serum antibody) died afterCNS challenge at 1 to 3 days post-i.p. inoculation of SRV,whereas a moderate survival rate was detected in mice withlow serum antibody titers challenged at 5 days. In contrast,mice challenged 7 days to 30 weeks post-SRV inoculationwere 100% resistant. Resistance via intranasal inoculationwas most impressive considering that the blood-brain barrierwas not broken during challenge. These results, in conjunc-tion with the cyclophosphamide studies, illustrated that afteri.p. inoculation of SRV, a prompt and efficient immunemechanism(s) became active which protected mice againstthe initial i.p. inoculated SRV and against a subsequentdirect virus challenge to the CNS.
DISCUSSIONThe trace experiments for infectious virus have clearly
demonstrated that differences in mouse strain susceptibilityto SRV and to the development of clinical CNS disease wereassociated with the restriction of virus replication within theCNS. For example, SRV replicated to low titers in the spinalcord; it subsequently ceased replication but did not infect thebrains of resistant-asymptomatic SJL/J and CBA/J mice. Incontrast, in resistant-recovered BALB/cAn and DBA/2Jmice, SRV infected the spinal cords and then the brainsbefore ceasing to replicate in the CNS. In these mousestrains, clinical disease was arrested, but the paralysis wasirreversible. Virus titers continued to increase, however, inCNS tissues of susceptible A/WySnJ, A.SW/SnJ, andathymic mice until their death. Additional experiments are
planned using in situ hybridization and immunoenzymaticstaining for viral antigens to determine if a different permis-sive cell population(s) of the various strains is involved inSRV replication.
TABLE 2. Cyclophosphamide abolished the resistance of SJL/Jmice to SRVa
Serum anti-SRV neutralizingDay of cyclophosphamide antibody geometric mean titer.. . ~~~~~~~~~~~~Survivors/inoculation in relation to 7 da
SRV inoculation postinoculation Termination of total
of SRV eprmn
Days before21 168 (+12) 3,160 (±240) 8/815 180 (±20) 3,320 (±320) 8/87 <10b NAC 0/85 <10 NA 0/83 <10 NA 0/81 <10 NA 0/8
Days after1 <10 NA 0/82 <10 NA 0/83 <10 NA 0/85 <10 NA 0/87 <10 NA 0/810 147 (±45) 1,280 (±255) 6/815 240 (±20) 1,600 (±320) 8/820 181 (±15) 1,368 (±302) 8/8
SRV only 160 (±24) 3,130 (±280) 8/8
Cyclophosphamide only <10 <10 8/8a Mice were inoculated i.p. with 300 mg of cyclophosphamide per kg of
body weight at various intervals before or after i.p. inoculation of 5 x 107mouse intracerebral LD50 of SRV. Seven days after SRV inoculation, 0.1 mlof blood was removed via the retro-orbital plexus to determine serumneutralizing antibody titers. At 41 days after SRV inoculation, the experimentwas terminated.bNone of the mice in groups with geometric mean titers of <10 had
detectable antibody, whereas all mice in groups with titers of >10 possessedantibody. Because of serum toxicity, the minimum dilution for antibodydetection was 1:10. Each result is the geometric mean titer (± standard error)for eight mice.
c NA, All mice died; sera not available.
J. VIROL.
MURINE RESISTANCE TO STREET RABIES VIRUS 793
Nonetheless, limitation of virus replication in the CNSappeared to correlate with the immune response in thathighly susceptible mice produced minimal amounts of serumneutralizing antibody until their death. In contrast, three ofthe four resistant mouse strains produced early and markedserum anti-SRV neutralizing antibody titers which steadilyincreased until the experiments were terminated. The titersof mice of resistant strain DBA/2J were similar to those forthe susceptible mice for the first 10 days but then began toincrease steadily and subsequently became comparable tothe titers of the other resistant strains. Moreover, as theserum antibody titers of the resistant mice increased, theirCNS virus titers decreased. At this time, we are uncertain asto why there was a marked increase in the serum neutralizingantibody titer in DBA/2J mice after day 10. Because previ-ous studies have shown that early immunoglobulin M anti-body is ineffective in protecting mice against rabies (47), itmight be that a shift occurred from an immunoglobulin Mantibody to an immunoglobulin G neutralizing antibodyresponse.The importance of the immune response in resistance to
SRV was corroborated by our cyclophosphamide studies inthat all resistant-asymptomatic SJL/J mice died aftercyclophosphamide treatment. In contrast, cyclophos-phamide-treated SJL/J mice which were passively trans-ferred i.v. with immune sera or with unfractionated immunespleen cells 24 h after i.p. SRV inoculation survived a lethaldose of SRV. Nonetheless, antibody transferred at 24 hpostinoculation might have prevented viral invasion of theCNS by neutralizing SRV in the peritoneal cavity. Tocircumvent this possibility, immune serum was transferredeither to immunocompromised SJL/J mice or to im-munodeficient athymic mice 72 h after i.p. inoculation, a
time at which SRV was no longer detected in the peritonealcavity but was present in the spinal cord. The enhancedsurvival of mice receiving immune sera 72 h post-SRVinoculation emphasized the significance of humoral antibodyas an important immune mechanism in these mice. It is toopremature, however, to conclude that neutralizing antibodywas the sole mechanism of resistance. Interferon (3, 17, 19,45) or such other factors as cytolytic T cells (36, 42, 50),antibody-dependent cellular cytotoxicity (33), or lysis ofinfected cells by antibody and complement (26) might be asimportant as or even supportive of neutralizing antibody.
In contrast to the apparent correlation between serumneutralizing antibody and resistance to rabies virus, neutral-izing antibody in the CSF appeared to be of minimal conse-quence as a resistance mechanism in mice which did notdevelop clinical disease. As shown in Fig. 2 and Table 1,SRV replication ceased in spinal cords of asymptomatic-resistant SJL/J and CBA/J mice during the interval whenantibody was absent in the CSF. Moreover, antibody wasnot detected in the CSF of asymptomatic-resistant mice untilafter day 15 postinoculation. Our data did suggest, however,that CSF neutralizing antibody correlated with the survivalof mice which had developed clinical CNS infections. It waspresent in high titer in the CSF of resistant-recovered miceafter day 10, but was undetectable or present in low titer inthe few susceptible mice which were alive at this time.Moreover, anti-SRV neutralizing antibody titers were higherin CSF than in serum of BALB/cAn and DBA/2J mice in thelate stages of clinical disease (data not shown). These higherCSF titers suggested local CNS antibody production after anextensive antigenic stimulus, as has been demonstrated withother CNS virus infections (16, 24). Furthermore, it has beenshown that animals and humans who have survived rabies
TABLE 3. Passively transferred immune sera or immune cellsrestored the resistance of cyclophosphamide-treated SJL/J mice to
SRVaCyclophos- SRV Treatment Survivors/phamide total
a Mice were inoculated i.p. with 300 mg of cyclophosphamide per kg ofbody weight and 2 days later were inoculated i.p. with 5 x 107 mouseintracerebral LD50 of SRV. After SRV inoculation (24 h), treatment wasbegun. Unfractionated spleen cells were inoculated i.v. one time only,whereas 0.2 ml of serum was inoculated i.v. every third day for 18 days.
b Immune sera and cells were obtained from SJL/J mice which survived.30 days after i.p. inoculation of 5 x 10i mouse intracerebral LD50 of SRV.The RFFIT titer of the serum pool was 5,120.
infections have high CSF anti-rabies virus neutralizing anti-body titers (13, 18).
It is well known that the rabies virus glycoprotein is theonly rabies virus antigen which induces neutralizing anti-body and confers immunity against infection with rabiesvirus (7, 8, 51). Furthermore, it has been shown withmonoclonal antibodies that there is antigenic diversity in theglycoprotein among rabies virus strains (15). At this time, itis unclear why susceptible mouse strains infected with SRVmade an inferior neutralizing antibody response to thisglycoprotein. There are, however, several possibilities. Be-cause previous membrane fluorescence studies have shownthat a variation exists in the expression of viral antigens on
TABLE 4. Passive transfer of immune serum 3 days after virusinoculation protected immunocompromised SJL/J and
immunodeficient athymic mice against SRVa
Experimental Treatment Survivors/ %group total
SJL/J Immune serumb 16/16 lOOcNormal serumb 3/9 33None 1/7 14Cytoxan only 6/6 100SRV only 8/8 100
a SJL/J mice were inoculated i.p. with 300 mg of cyclophosphamide per kgof body weight and 3 days later were inoculated i.p. with 5 x 107 mouseintracerebral LD50 of SRV. Athymic mice were not inoculated withcyclophosphamide. After SRV inoculation (72 h), 0.3 ml of serum wasinoculated i.v. every other day for 18 days. Spinal cords were removed fromfive SJL/J and athymic mice 3 days after virus inoculation; at least 20% ofeach strain was positive for infectious virus as detected by intracranialinoculation of a 20% spinal cord suspension.
b Immune sera were obtained from mice which survived .30 days after i.p.inoculation of 5 x 107 mouse intracerebral LD50 of SRV. The RFFIT titer ofthe serum pool was 5,120. Normal sera was obtained from uninoculated mice.
c Analysis of data by the X2 test for a 2 x 2 contingency table indicated thatthe percent survivors in groups treated with immune serum was significantlydifferent from the controls within their respective groups which receivednormal or no serum.
VOL. 55, 1985
794 LODMELL AND EWALT
different rabies virus infected cells (26), the quantity ofglycoprotein molecules on infected cell surfaces might varyamong these inbred strains of mice. Furthermore, SRVreplication might be confined to the CNS or peripheralnerves of the susceptible mice, thus resulting in ineffectiveprocessing of viral antigens. Susceptible mice also might beresponding to internal or nonstructural virion proteins or tothe rabies soluble glycoprotein (9), all of which lack protec-tive activity and the ability to induce neutralizing antibody.Failure of the susceptible mice to respond to inactivatedSRV, however, was more than likely because of inoculationof insufficient antigenic mass or ineffective processing ofantigen (53). Furthermore, the excellent antibody responseof SRV-susceptible mice to the HEP-Flury modified liverabies vaccine, compared with SRV, suggested that theHEP-Flury strain replicates at sites other than the nervoussystem (53) or that the glycoprotein of different rabiesviruses exists in different conformations (8).We realized that i.p. inoculation of SRV did not mimic a
real host-virus relationship or the natural route of SRVinfection by bite. Furthermore, it was not our intent tooverinterpret this data with regard to the importance ofserum neutralizing antibody in resistance to sylvatic SRVinfections. Nonetheless, we have shown that resistance toi.p. inoculated SRV correlated with the early presence ofserum anti-SRV neutralizing antibody. Furthermore, activeintervention with the immune system followed by passivetransfer of immune serum to immunosuppressed mice start-ing at 72 h postinfection protected them from death. What-ever the mechanism(s), be it neutralizing antibody, cellularimmunity, or antibody in combination with other immunefactors, it is highly impressive because it protected the CNSfrom direct rabies virus challenge very early after i.p.inoculation of SRV. To understand how to generate thisresistance without infection could lead to a highly efficientvaccine. We are currently expanding our model to study thissituation in mice which have been inoculated with physio-logical doses of SRV by more natural routes of infection.
ACKNOWLEDGMENTS
We thank Bruce Chesebro, John Portis, and George Baer forhelpful discussions, Laverne Fadness for excellent technical
TABLE 5. Serum anti-rabies virus neutralizing antibody responseof mice immunized with either inactivated or modified live rabies
viruSa
Virus inoculated:Mousestrain Inactivated Inactivated Modified live
SRV CVS Endurall-R
SJL/J 126 (+20) 1,610 (±320) 5,120
CBA/J 126 (±20) 2,560 (±132) 1,610 (±126)
A/WySnJ <10 254 (±18) 2,560 (±150)
A.SW/SnJ <10 508 (±13) 1,610 (±126)a Mice were inoculated i.v. twice per week for a total of 7 weeks with 0.15
ml of the modified live Endurall-R vaccine or with 0.1 ml of supernatant fluidfrom BPL-inactivated SRV or CVS brain suspensions. After the last inocu-lation (7 days), mice were bled, and the sera were tested for anti-rabies virusneutralizing antibody. Because of serum toxicity, the minimum dilution forantibody detection was 1:10. Each result is the geometric mean titer (+standard error) for five mice.
TABLE 6. CNS immunity to rabies virus developed 5 days afteri.p. inoculation of SRVa
Time after SRV Serum anti-SRVinoculation that the Route of CNS neutralizing antibody Survivors/
CNS was inoculation geometric mean titer totalinoculated with 5 h before CNSCVS rabies virus inoculation
1 day INb <10 0/8ICC <10 0/8
3 days IN <10 (±4) 0/8IC <10 (±3.4) 0/8
5 days IN 50d (±6) 6/8IC 40 (±8) 3/8
7 days IN 160 (±35) 8/8IC 128 (±32) 8/8
14 days IN 3,310 (±120) 8/8IC 2,690 (±123) 8/8
21 days IN 2,930 (±138) 8/8IC 3,330 (±104) 8/8
30 wk IN 640 (±18) 8/8IC 500 (±20) 8/8
Control (no SRV) IN <10 0/8IC <10 0/8
a At various intervals after i.p. inoculation of 5 x 107 mouse intracerebralLD50 of SRV, the CNS of SJL/J mice was inoculated via the intranasal routewith 103.9 mouse intranasal LD50/0.025 ml or via the intracerebral route with108.0 mouse intracerebral LD50/0.03 ml ofCVS rabies virus. The more virulentCVS rabies virus was used for CNS inoculation to ensure that 100% of thecontrol mice died. Before CNS challenge (Sh), 0.1 ml of blood was removedvia the retro-orbital plexus to determine serum anti-SRV neutralizing antibodytiters. Because of serum toxicity, the minimum dilution for antibody detectionwas 1:10. Each result is the geometric mean titer (±standard error) for eightmice.
b IN, Intranasal.c IC, Intracerebral.d All mice had serum antibody titers of -40 5 or more days after inoculation
with SRV.
assistance, Bob Terry for conscientious care of the experimentalanimals, and Helen Blahnik for exceptional secretarial assistance.
LITERATURE CITED1. Arko, R. J., L. G. Schneider, and G. M. Baer. 1973. Nonfatal
canine rabies. Am. J. Vet. Res. 34:937-938.2. Baer, G. M., W. F. Cleary, A. M. Diaz, and D. F. Perl. 1977.
Characteristics of 11 rabies virus isolates in mice: titers andrelative invasiveness of virus, incubation period of infection,and survival of mice with sequelae. J. Infect. Dis. 136:336-345.
3. Baer, G. M., J. H. Shaddock, S. A. Moore, P. A. Yager, S. S.Baron, and H. B. Levy. 1977. Successful prophylaxis againstrabies in mice and rhesus monkeys: the interferon system andvaccine. J. Infect. Dis. 136:286-291.
4. Bell, J. F. 1964. Abortive rabies infection. I. Experimentalproduction in white mice and general discussion. J. Infect. Dis.114:249-257.
5. Bell, J. F., G. J. Moore, and G. H. Raymond. 1969. Protractedsurvival of a rabies-infected insectivorous bat after infectivebite. Am. J. Trop. Med. Hyg. 18:61-66.
6. Blenden, D. C., J. F. Bell, A. T. Tsao, and J. U. Umoh. 1983.Immunofluorescent examination of the skin of rabies-infectedanimals as a means of early detection of rabies virus antigen. J.Clin. Microbiol. 18:631-636.
7. Cox, J. H., B. Dietzschold, and L. G. Schneider. 1977. Rabiesvirus glycoprotein. II. Biological and serological characteriza-
J. VIROL.
MURINE RESISTANCE TO STREET RABIES VIRUS 795
tion. Infect. Immun. 16:754-759.8. Dietzschold, B., T. J. Wiktor, R. Macfarlan, and A. Varrichio.
1982. Antigenic structure of rabies virus glycoprotein: orderingand immunological characterization of the large CNBr cleavagefragments. J. Virol. 44:595-602.
9. Dietzschold, B., T. J. Wiktor, W. H. Wunner, and A. Varrichio.1983. Chemical and immunological analysis of the rabies solubleglycoprotein. Virology 124:330-337.
10. Dubois-Dalcq, M., E. L. Hooghe-Peters, and R. A. Lazzarini.1980. Antibody-induced modulation of rhabdovirus infection ofneurons in vitro. J. Neuropathol. Exp. Neurol. 39:507-522.
11. Faulkner, G., M. Dubois-Dalcq, E. Hooghe-Peters, H, F.McFarland, and R. A. Lazzarini. 1979. Defective interferingparticles modulate VSV infection of dissociated neuron cul-tures. Cell 17:979-991.
12. Fekadu, M. 1972. Atypical rabies in dogs in Ethiopia. Ethiop.Med. J. 10:79-86.
13. Fekadu, M., and G. M. Baer. 1980. Recovery from clinicalrabies of two dogs inoculated with a rabies virus strain fromEthiopia. Am. J. Vet. Res. 41:1632-1634.
14. Fekadu, M., J. H. Shaddock, and G. M. Baer. 1981. Intermittentexcretion of rabies virus in the saliva of a dog two and sixmonths after it had recovered from experimental rabies. Am. J.Trop. Med. Hyg. 30:1113-1115.
15. Flamand, A., T. J. Wiktor, and H. Koprowski. 1980. Use ofhybridoma monoclonal antibodies in the detection of antigenicdifferences between rabies and rabies-related virus proteins. II.
The glycoprotein. J. Gen. Virol. 48:105-109.16. Griffen, D. E., 0. Narayan, J. Bukowski, R. J. Adams, and S. R.
Cohen. 1978. The cerebrospinal fluid in visna, a slow viraldisease of sheep. Ann. Neurol. 4:212-218.
17. Harmon, M. W., and B. Janis. 1975. Therapy of murine rabiesafter exposure: efficacy of polyriboinosinic-polyribocytidylicacid alone and in combination with three rabies vaccines. J.Infect. Dis. 132:241-249.
18. HKattwick, M. A., T. T. Weis, C. J. Stechschulte, G. M. Baer, andM. B. Gregg. 1972. Recovery from rabies-a case report. Ann.Intern. Med. 76:931-942.
19. Hilfenhaus, J., E. Weinmann, M. Majer, R. Barth, and 0.Jaeger. 1977. Administration of human interferon to rabiesvirus-infected monkeys after exposure. J. Infect. Dis. 135:846-849.
20. Hoskins, J. M. 1973. Duck-embryo vaccine. WHO Monogr. Ser.23,248-250.
21. Johnson, R. T. 1982. Rabies, p. 159-167. In R. T. Johnson (ed.),Viral infections of the nervous system. Raven Press, Publishers,New York.
22. Kawai, A., and S. Matsumoto. 1977. Interfering and noninterfer-ing defective particles generated by a rabies small plaque variantvirus. Virology 76:60-71.
23. Larghi, 0. P., E. GonzAlez L., and J. R. Held. 1973. Evaluationof the corneal test as a laboratory method for rabies diagnosis.Appl. Microbiol. 25:187-189.
24. Lipton, H. L., and F. Gongalez-Scarano. 1978. Central nervous
system immunity in mice infected with Theiler's virus. I. Localneutralizing antibody response. J. Infect. Dis. 137:145-151.
25. Lodmell, D. L. 1983. Genetic control of resistance to streetrabies virus in mice. J. Exp. Med. 157:451-460.
26. Lodmell, D. L., Y. T. Arai, and L. C. Ewalt. 1981. Influence ofcell type and virus upon lysis of rabies virus-infected cells byantibody and complement. Arch. Virol. 70:147-155.
27. Lodmell, D. L., J. F. Bell, G. J. Moore, and G. H. Raymond.1969. Comparative study of abortive and nonabortive rabies inmice. J. Infect. Dis. 119:569-580.
28. Lodmell, D. L., and B. Chesebro. 1984. Murine resistance tostreet rabies virus: genetic analysis by testing second-backcrossprogeny and verification of allelic resistance genes in SJL/J andCBA/J mice. J. Virol. 50:359-362.
29. Mifune, K., E. Takeuchi, P. A. Napiorkowski, A. Yamada, andK. Sakamoto. 1981. Essential role ofT cells in the postexposureprophylaxis of rabies in mice. Microbiol. Immunol. 25:895-904.
30. Miller, A., H. C. Morse III, J. Winkelstein, and N. Nathanson.
1978. The role of antibody in recovery from experimentalrabies. I. Effect of depletion of B and T cells. J. Immunol.121:321-326.
31. Murphy, F. A. 1977. Rabies pathogenesis: brief review. Arch.Virol. 54:279-297.
32. Nilsson, M. R., O. A. Sant'anna, M. Siqueira, T. T. Nilsson, andM. Gennari. 1979. Rabies virus immunity in genetically selectedhigh- and low-responder lines of mice. Infect. Immun. 25:23-26.
33. Pereira, C. A., J. N. Nozaki-Renard, J. Schwartz, A. Eyquem,and P. Atanasiu. 1982. Cytotoxicity reactions against target cellsinfected with rabies virus. J. Virol. Methods 5:75-83.
34. Perl, D. P., J. F. Bell, G. J. Moore, and S. J. Stewart. 1977.Chronic recrudescent rabies in a cat (39847). Proc. Soc. Exp.Biol. Med. 155:540-548.
35. Porras, C., J. Jose-Barbosa, E. Fuenzalida, H. Lopez-Adaros,A. M. 0. deDiaz, and J. Furst. 1976. Recovery from rabies inman. Ann. Intern. Med. 85:444 8.
36. Reddehase, M. J., J. H. Cox, and U. H. Koszinowski. 1982.Frequency analysis of cytolytic T cell precursors (CTL-P)generated in vivo during lethal rabies infection of mice. I.Distribution of CTL-P with different interleukin 2 sensitivity.Eur. J. Immunol. 12:519-523.
37. Reed, L. J., and H. Muench. 1938. A simple method of estimat-ing fifty percent endpoints. Am. J. Hyg. 27:493-497.
38. Schneider, L. G. 1969. The cornea test: a new method for theintravitam diagnosis of rabies. Zentralbl. Veterinaermed. ReiheB 16:24-31.
39. Sikes, R. K. 1962. Pathogenesis of rabies in wildlife. 1. Compar-ative effect of varying doses of rabies virus inoculated into foxesand skunks. Am. J. Vet. Res. 23:1041-1047.
40. Smith, A. L., G. H. Tignor, K. Mifune, and T. Motohashi. 1977.Isolation and assay of rabies serogroup viruses in CER cells.Intervirology 8:92-99.
41. Smith, J. S. 1981. Mouse model for abortive rabies infection ofthe central nervous system. Infect. Immun. 31:297-308.
42. Smith, J. S., C. L. McClelland, F. L. Reid, and G. M. Baer.1982. Dual role of the immune response in street rabiesvirusinfection of mice. Infect. Immun. 35:213-221.
43. Smith, J. S., P. A. Yager, and G. M. Baer. 1973. A rapidreproducible test for determining rabies neutralizing antibody.Bull. W.H.O. 48:535-541.
44. Smith, W. B., D. C. Blenden, T. H. Fuh, and L. Hiler. 1972.Diagnosis of rabies by immunofluorescent staining of frozensections of skin. J. Am. Vet. Med. Assoc. 161:1495-1501.
45. Stewart, W. E., II, and S. E. Sulkin. 1966. Interferon productionin hamsters experimentally infected with rabies virus. Proc.Soc. Exp. Biol. Med. 123:650-654.
46. Turk, J. L., and D. Parker. 1979. The effect of cyclo-phosphamide on the immune response. J. Immunopharmacol.1:127-137.
47. Turner, G. S. 1978. Immunoglobulin (IgG) and (IgM) antibodyresponses to rabies vaccine. J. Gen. Virol. 40:596-604.
48. Vaughn, J. B., P. Gerhart, and J. C. S. Paterson, 1963. Excre-tion of street rabies virus in saliva of cats. J. Am. Med. Assoc.184:705-708.
49. Wiktor, T. J., B. Dietzschold, R. N. Leamnson, and H.Koprowski. 1977. Induction and biological properties of defec-tive interfering particles of rabies virus. J. Virol. 21:626-635.
50. Wiktor, T. J., P. C. Doherty, and H. Koprowski. 1977. In vivoevidence of cell-mediated immunity after exposure of mice toboth live and inactivated rabies virus. Proc. Natl. Acad. Sci.U.S.A. 74:334-338.
51. Wiktor, T. J., E. Gyorgy, H, D. Schlumberger, F. Sokol, and H.Koprowski. 1973. Antigenic properties of rabies virus compo-nents. J. Immunol. 110:269-276.
52. Yurkovsky, A. M. 1962. Hydrophobia following the bite ofapparently healthy dogs. J. Hyg. Epidemiol. Microbiol. Immu-nol. 6:73-78.
53. Zinkernagel, R. M. 1981. Recent advances in cellular immunityof infectious disease, p. 103-109. In E. K. Kuwert, T. J. Wiktor,and H. Koprowski (ed.), Cell culture rabies vaccines and theirprotective effect in man. International Green Cross, Geneva.











![Pathogenesis of Bacterial Infections-2009.ppt [Read-Only]ocw.usu.ac.id/.../bbc215_slide_pathogenesis_of_bacterial_infektions.pdf · KOLONISASI BAKTERI DI JARINGAN TUBUH MANUSIA Bakteri](https://static.documents.pub/doc/80x56/5e16eb8b5b015111d57ea171/pathogenesis-of-bacterial-infections-2009ppt-read-onlyocwusuacidbbc215slidepathogenesisofbacterial.jpg)












