47
Protein Structure Prediction Xiaole Shirley Liu And Jun Liu STAT115
| Date post: | 30-Dec-2015 |
| Category: |
Documents |
| Upload: | ross-woodard |
| View: | 78 times |
| Download: | 0 times |

Protein Structure Prediction
Xiaole Shirley Liu
And
Jun Liu
STAT115
2
Protein Structure PredictionRam Samudrala
University of Washington

STAT1153
Outline
• Motivations and introduction
• Protein 2nd structure prediction
• Protein 3D structure prediction– CASP– Homology modeling– Fold recognition– ab initio prediction– Manual vs automation
• Structural genomics

STAT1154
Protein Structure
• Sequence determines structure, structure determines function
• Most proteins can fold by itself very quickly
• Folded structure: lowest energy state

5
Protein Structure• Main forces for considerations
– Steric complementarity– Secondary structure preferences (satisfy H
bonds)– Hydrophobic/polar patterning– Electrostatics

6
Rationale for understanding protein structure and function
Protein sequence
-large numbers of sequences, including whole genomes
Protein function
- rational drug design and treatment of disease- protein and genetic engineering- build networks to model cellular pathways- study organismal function and evolution
?
structure determination structure prediction
homologyrational mutagenesisbiochemical analysis
model studies
Protein structure
- three dimensional- complicated- mediates function

STAT1157
Protein Databases
• SwissProt: protein knowledgebase
• PDB: Protein Data Bank, 3D structure

8
View Protein Structure
• Free interactive viewers
• Download 3D coordinate file from PDB
• Quick and dirty:– VRML– Rasmol– Chime
• More powerful– Swiss-PdbViewer

9
Compare Protein Structures
• Structure is more conserved than sequence• Why compare?
– Detect evolutionary relationships– Identify recurring structural motifs– Predicting function based on structure– Assess predicted structures
• Protein structure comparison and classification– Manual: SCOP– Automated: DALI

10
Compare protein structures
• Need ways to determine if two protein structures are related and to compare predicted models to experimental structures
• Commonly used measure is the root mean square deviation (RMSD) of the Cartesian atoms between two structures after optimal superposition (McLachlan, 1979):
• Usually use C atoms
2 2 2
1
N
i i iidx dy dz
N
3.6 Å 2.9 Å
NK-lysin (1nkl) Bacteriocin T102/as48 (1e68) T102 best model
• Other measures include contact maps and torsion angle RMSDs

STAT11511
SCOP• Compare protein
structure, identify
recurring structural
motifs, predict function• A. Murzin et al, 1995
– Manual classification
– A few folds are highly
populated
– 5 folds contain 20% of all homologous superfamilies
– Some folds are multifunctional

STAT11512
Determine Protein Structure
• X-ray crystallography (gold standard)– Grow crystals, rate limiting, relies on the repeating
structure of a crystalline lattice
– Collect a diffraction pattern
– Map to real space electron density, build and refine structural model
– Painstaking and time consuming

STAT11513
Protein Structure Prediction
• Since AA sequence determines structure, can we predict protein structure from its AA sequence?= predicting the three angles, unlimited DoF!
• Physical properties that determine fold– Rigidity of the protein backbone
– Interactions among amino acids, including• Electrostatic interactions
• van der Waals forces
• Volume constraints
• Hydrogen, disulfide bonds
– Interactions of amino acids with water

14
unfolded
Protein folding landscape
Large multi-dimensional space of changing conformationsfr
ee e
nerg
y
folding reaction
molten globule
J=10-8 s
native
J=10-3 s
G**
barrierheight
15
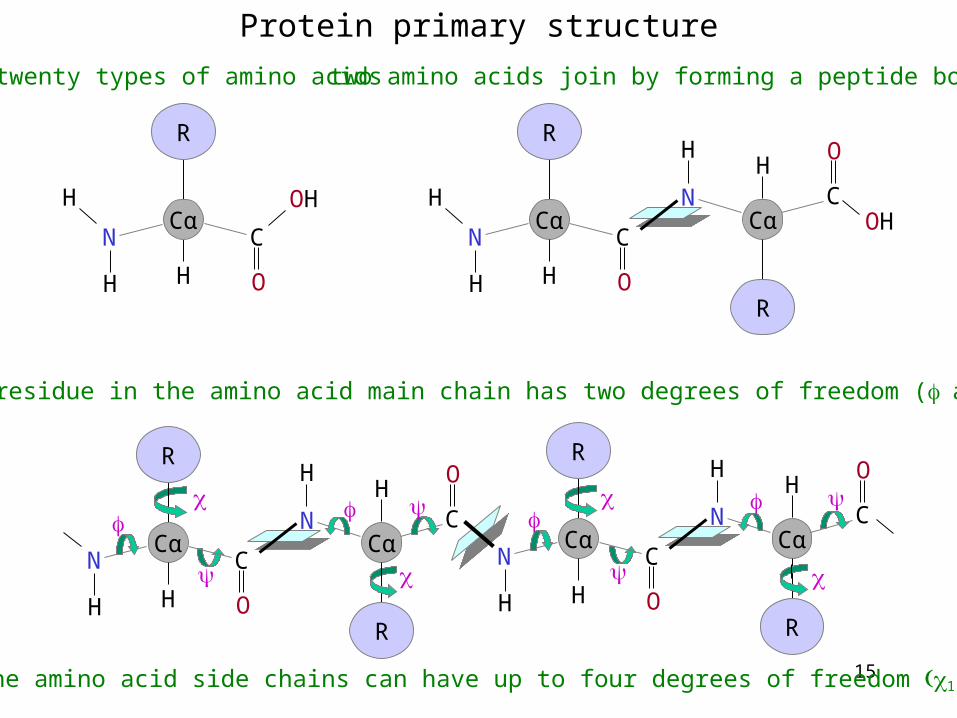
Protein primary structure
twenty types of amino acids
R
H
C
OH
O
N
H
HCα
two amino acids join by forming a peptide bond
R
Cα
H
C
O
N
H
H NCα
H
C
O
OH
R
H
R
Cα
H
C
O
N
H
NCα
H
C
O
R
HR
Cα
H
C
O
N
H
NCα
H
C
O
R
H
each residue in the amino acid main chain has two degrees of freedom (and
the amino acid side chains can have up to four degrees of freedom 1-4

STAT11516
2nd Structure Prediction helix, sheet, turn/loop

STAT11517
2nd Structure Prediction
• Chou-Fasman 1974• Base on 15 proteins (2473 AAs) of known
conformation, determine P, P from 0.5-1.5
• Empirical rules for 2nd struct nucleation– 4 H or h out of 6 AA, extends to both dir,
P > 1.03, P > P, no breakers– 3 H or h out of 5 AA, extends to both dir, P
> 1.05, P > P, no breakers
• Have ~50-60% accuracy
)20//( sj
si ff

STAT11518
P and P
STAT11519
2nd Structure Prediction
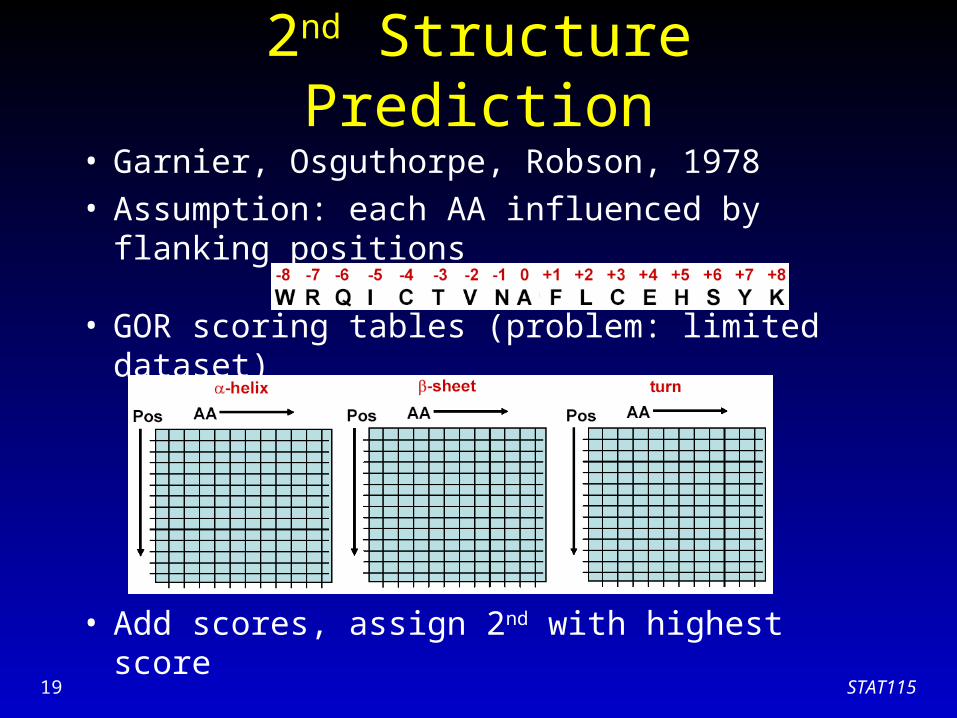
• Garnier, Osguthorpe, Robson, 1978• Assumption: each AA influenced by flanking
positions
• GOR scoring tables (problem: limited dataset)
• Add scores, assign 2nd with highest score

STAT11520
2nd Structure Prediction
• D. Eisenberg, 1986– Plot hydrophobicity as function of sequence
position, look for periodic repeats– Period = 3-4 AA, (3.6 aa / turn)– Period = 2 AA, sheet
• Best overall JPRED by Geoffrey Barton, use many different approaches, get consensus– Overall accuracy: 72.9%

STAT11521
3D Protein Structure Prediction
• CASP contest: Critical Assessment of Structure Prediction
• Biannual meeting since 1994 at Asilomar, CA• Experimentalists: before CASP, submit sequence
of to-be-solved structure to central repository• Predictors: download sequence and minimal
information, make predictions in three categories• Assessors: automatic programs and experts to
evaluate predictions quality

STAT11522
CASP Category I• Homology Modeling (sequences with high
homology to sequences of known structure)
• Given a sequence with homology > 25-30% with known structure in PDB, use known structure as starting point to create a model of the 3D structure of the sequence
• Takes advantage of knowledge of a closely related protein. Use sequence alignment techniques to establish correspondences between known “template” and unknown.

STAT11523
CASP Category II
• Fold recognition (sequences with no sequence identity (<= 30%) to sequences of known structure
• Given the sequence, and a set of folds observed in PDB, see if any of the sequences could adopt one of the known folds
• Takes advantage of knowledge of existing structures, and principles by which they are stabilized (favorable interactions)

STAT11524
CASP Category III• Ab initio prediction (no known homology with any
sequence of known structure)
• Given only the sequence, predict the 3D structure from “first principles”, based on energetic or statistical principles
• Secondary structure prediction and multiple alignment techniques used to predict features of these molecules. Then, some method necessary for assembling 3D structure.

STAT11525
Structure Prediction Evaluation
• Hydrophobic core similar? • 2nd struct identified?• Energy: minimized? H-bond contacts?• Compare with solved crystal structure: gold
standard
N
yxyxNRMSD Ni
ii
...1
2||||),;(

26
Comparative modelling of protein structure
KDHPFGFAVPTKNPDGTMNLMNWECAIPKDPPAGIGAPQDN----QNIMLWNAVIP** * * * * * * * **
… …
scanalign
build initial modelconstruct non-conserved
side chains and main chains
refine

STAT11527
Homology Modeling Results
• When sequence homology is > 70%, high resolution models are possible (< 3 Å RMSD)
• MODELLER (Sali et al)– Find homologous proteins with known
structure and align– Collect distance distributions between atoms in
known protein structures– Use these distributions to compute positions for
equivalent atoms in alignment– Refine using energetics

STAT11528
Homology Modeling Results
• Many places can go wrong:– Bad template - it doesn’t have the same
structure as the target after all– Bad alignment (a very common problem)– Good alignment to good template still gives
wrong local structure– Bad loop construction– Bad side chain positioning

STAT11529
Homology Modeling Results
• Use of sensitive multiple alignment (e.g. PSI-BLAST) techniques helped get best alignments
• Sophisticated energy minimization techniques do not dramatically improve upon initial guess

STAT11530
Fold Recognition Results
• Also called protein threading
• Given new sequence and library of known folds, find best alignment of sequence to each fold, returned the most favorable one

STAT11531
Fold Recognition with Dynamic Programming
• Environmental class for each AA based on known folds (buried status, polarity, 2nd struct)

STAT11532
Protein Folding with Dynamic Programming
• D. Eisenburg 1994• Align sequence to each fold (a string of
environmental classes)
• Advantages: fast and works pretty well• Disadvantages: do not consider AA contacts

STAT11533
Fold Recognition Results
• Each predictor can submit N top hits
• Every predictor does well on something
• Common folds (more examples) are easier to recognize
• Fold recognition was the surprise performer at CASP1. Incremental progress at CASP2, CASP3, CASP4…

STAT11534
Fold Recognition Results
• Alignment (seq to fold) is a big problem

STAT11535
ab initio
• Predict interresidue contacts and then compute structure (mild success)
• Simplified energy term + reduced search space (phi/psi or lattice) (moderate success)
• Creative ways to memorize sequence structure correlations in short segments from the PDB, and use these to model new structures: ROSETTA

36
Ab initio prediction of protein structuresample conformational space such that
native-like conformations are found
astronomically large number of conformations5 states/100 residues = 5100 = 1070
select
hard to design functionsthat are not fooled by
non-native conformations(“decoys”)

37
Sampling conformational space – continuous approaches
• Most work in the field- Molecular dynamics- Continuous energy minimization (follow a valley)- Monte Carlo simulation- Genetic Algorithms
• Like real polypeptide folding process
• Cannot be sure if native-like conformations are sampled
energy

38
Molecular dynamics
• Force = -dU/dx (slope of potential U); acceleration, force = m ×a(t)
• All atoms are moving so forces between atoms are complicated functions of time
• Analytical solution for x(t) and v(t) is impossible; numerical solution is trivial
• Atoms move for very short times of 10-15 seconds or 0.001 picoseconds (ps)
x(t+t) = x(t) + v(t)t + [4a(t) – a(t-t)] t2/6
v(t+t) = v(t) + [2a(t+t)+5a(t)-a(t-t)] t/6
Ukinetic = ½ Σ mivi(t)2 = ½ n KBT
• Total energy (Upotential + Ukinetic) must not change with time
new positionold position
new velocity
old velocity acceleration
n is number of coordinates (not atoms)

39
Energy minimization
For a given protein, the energy depends on thousands of x,y,z Cartesian atomic coordinates; reaching a deep minimum is not trivial
Furthermore, we want to minimize the free energy, not just the potential energy.
energy
number of steps deep minimum
starting conformation

40
Monte Carlo Simulation• Propose moves in torsion or Cartesian conformation
space• Evaluate energy after every move, compute E• Accept the new conformation based on
• If run infinite time, the simulated conformation follows the Boltzmann distribution
• Many variations, including simulated annealing and other heuristic approaches.
ΔEP exp
kT
E(C)( ) exp
kTC

41
Scoring/energy functions
• Need a way to select native-like conformations from non-native ones
• Physics-based functions: electrostatics, van der Waals, solvation, bond/angle terms.
• Knowledge-based scoring functions: – Derive information about atomic properties
from a database of experimentally determined conformations
– Common parameters include pairwise atomic distances and amino acid burial/exposure.

STAT11542
Rosetta
• D. Baker, U. Wash• Break sequence into short segments (7-9 AA)• Sample 3D from library of known segment
structures, parallel computation• Use simulated annealing (metropolis-type
algorithm) for global optimization– Propose a change, if better energy, take; otherwise take
at smaller probability
• Create 1000 structures, cluster and choose one representative from each cluster to submit

STAT11543
Manual Improvements and Automation
• Very often manual examination could improve prediction– Catch errors– Need domain knowledge– A. Murzin’s success at CASP2
• CAFASP: Critical Assessment of Fully Automated Structure Prediction– Murzin Can’t play!!
• MetaServers: combine different methods to get consensus

STAT11544
CAFASP Evaluation

STAT11545
Structural Genomics
• With more and more solved structures and novel folds, computational protein structure prediction is going to improve
• Structural genomics: – Worldwide initiative to high throughput
determine many protein structures– Especially, solve structures that have no
homology

STAT11546
Summary• Protein structures: 1st, 2nd, 3rd, 4th
– Different DB: SwissProt, PDB and SCOP– Determine structure: X-ray crystallography
• Protein structure prediction:– 2nd structure prediction– Homology modeling– Fold recognition– Ab initio– Evaluation: energy, RMSD, etc– CASP and CAFASP contest
• Manual improvement and combination of computational approaches work better
• Structural Genomics, still very difficult problem…

STAT11547
Acknowledgement
• Amy Keating
• Michael Yaffe
• Mark Craven
• Russ Altman

















