Page 1
SYNTHESIS, CHARACTERIZATION AND BIOASSAY OF SOME
NOVEL 6,8-DIOXYGENATED-7-SUBSTITUTED ISOCOUMARINS,
DIHYDROISOCOUMARINS AND RELATED COMPOUNDS
Islamabad
A dissertation submitted to the Department of Chemistry,
Quaid-i-Azam University, Islamabad, in partial fulfillment
of the requirements for the degree of
Doctor of Philosophy
in
Organic Chemistry
by
Muhammad Zaman Ashraf
Department of Chemistry
Quaid-i-Azam University
Islamabad
2010
Page 2
To
MY MOTHER
AND
MY WIFE SAMMAR ZAMAN
Page 3
i
ACKNOWLEDGEMENTS
The entire gamut of creation amplify figures out, in all its dimensions, latent or
manifest, the intricately balanced interplay of its fundamental determinants – knowledge
and practice – which while ordering the elementary constituents, quite distinctly
identifiable in their own right, impacts the whole a fascinating and majestic character that
doubtless evokes the intuitive response of gratitude and indebtedness to the Immanent,
the Transcendent, the Omnipotent – ALLAH.
The human drive to fashion the evolving universal framework in a peculiarly
human perspective, our beloved Prophet MUHAMMD (Peace and Blessing of Allah
be upon him and his descendents) stands for an ever actuating and galvanizing
impulsive force that brings out the divine through the human. Indeed He commands the
highest order amongst the whole creation.
I consider it my first and foremost obligation to express my heartful gratitude to
my respected and inspirational supervisor, Dr. Aamer Saeed. I would not have been able
to complete this assignment without his cordial attitude, kidness, guidance and persistent
interest in my research work.
I am thankful to Chairman Department of Chemistry, Professor Dr. Saqib Ali and
Head of Organic Section, Professor Dr. Nasim Hasan Rama for providing me basic
research facilities. I am thankful to all faculty members, department of chemistry Quaid-
i-Azam University, Islamabad.
I am grateful to Prof. Dr. Peter Langer Rostock, Germany, for cytotoxic studies
of my samples. I am thankful to Dr. Muhammad Ali, HEJ institute, Karachi, for
antimalarial activity and Dr. Imran Arid Agriculture University, Rawalpindi for
antimicrobial assay of my synthesized compounds.
I am thankful to all my lab fellows Ammara Mumtaz, Naeem Abbass, Hummera
Rafique, Rasheed Ahmed Kherra, Madiha Irfan, Aliya Ibrar, Uzma Shaheen, Iram Batool
and Shumaila Bukhari for their cooperation and support. I would like to acknowledge my
friends Shahid Ameen Samra, Latif Hussain, Dr. Muhammad Sher, Latif Shahid, Latif
Shahid, Muhammd Waqas, Yasir Asghar, Safdar sb. Kamran, Imran, my colleagues,
Page 4
ii
Prof. Dr. Saeed, Prof. Dr. Karamat Ali, Prof. Dr. G.A. Miana, Dr. Naghmana Rashid,
Dr. Uzma Yunas, Dr. Imtiaz, Dr. Moazzam, Ziaullah Shah, Aun Muhammad, Tariq
Ismail, Khurram Afzal, Muhammad Imran, Dr. Humaira Nadeem, Miss Kishwar and
Najm-ul-Hassan. I am thankful to all my students at RIPS for their prayers.
I have no words to acknowledge the prayers, support, encouragement and
dedication of my loving parents as the whole I achieved is actually their achievement and
they certainly deserve more than this along with my sister and loving brothers. Their
prayers and encouragement were always with me till the completion of my thesis. My
special love is for my wife who always encouraged and assisted me in completion of this
project along with my adorable kids Hishma Zaman and Muhammad Taha Zaman.
I want to acknowledge my loving Baji Nasim, Baji Shameem, Baji Nasreen,
Mudassir, Ammi and Abba ji who always remember me in their prayers.
At the end I am thankful to all those hands that prayed for my betterment and
success.
(Muhammad Zaman Ashraf)
Page 5
iii
Abstract
The work presented in this thesis has been divided into two parts. Part one deals
with the synthesis, characterization and biological activity of some 7-substituted 6,8-
dioxygenated isocoumarins and 3,4-dihydroisocoumarins. Chapter one includes general
introduction, nomenclature, structural types, biosynthesis, and extensive examples on
pharmacological efficacy of isocoumarins and 3,4-dihydroisocoumarins from literature. It
also provides some of the most significant synthetic routes and the reactions of
isocoumarins and 3,4-dihydroisocoumarins and their interconversion.
The total synthesis of structural analogues of some naturally occurring bioactive
isocoumarins and dihydroisocoumarins viz. Hiburipyranone, Cytogenin, Montroumarin,
Scorzocreticin, Annulatomarin, Thunberginol B, starting from 3,5-dimethoxy-4-methyl
homophthalic acid is the subject of Chapter two. The synthesis of 3,5-dimethoxy-4-
methylhomophthalic acid from simplest precursor p-toluic acid was carried out. The
substituted homophthalic acid was then converted into corresponding anhydride which
was then condensed with various acyl and aroyl chlorides to afford the corresponding 3-
alkyl or 3-arylisocoumarins. The isocoumarins were then converted into corresponding
3,4-dihydroisocoumarins and the latter were then demethylated to afford corresponding
6,8-dihydroxy-3,4-dihydroisocoumarins. The structures of all of the synthesized
compounds were confirmed using FTIR, 1H NMR,
13C NMR and mass spectral data.
Chapter three provides the physical constants and spectroscopic data of the synthesized
compounds.
Chapter four deals with the biological activities of the compounds synthesized.
Antibacterial activity was determined against ten different Gram positive and Gram
negative bacterial strains (Micrococcus luteus, Staphylococcus aureus, Staphylococcus
epidermidis, Lactobacillus bulgaricus, Escherichia coli, Klebsiella pneumonae,
Pasteurella multocida, Proteus vulgaris, Pseudomonas aeruginosa and Salmonella typhi)
using agar well diffusion method. In vitro antimalarial activity was performed against
malarial parasite Plasmodium falciparum. The cytotoxic activity of the synthesized
compounds was determined against human keratinocyte cell lines.
Chapter five depicts total synthesis of a natural product 8-hydroxy-7-
hydoxymethyl-6-methoxy-3,4-dihydroisocoumarin (Stellatin) isolated from mycelium of
Page 6
iv
Aspergillus variecolor. The structures of the precursor compounds and the Stellatin were
determined by FTIR, NMR and mass spectroscopic data. These compounds were
evaluated for their antibacterial activity against ten different gram positive and gram
negative bacterial strains. The cytotoxic activity was performed against human
keratinocyte cell lines.
Part two is related to the synthesis of some 3-(substituted phenyl)isocoumarins, 3-
(substituted phenyl)isocoumarin-1-thiones, 3-(substituted phenyl)isoquinolones and some
1-aryl-7,8-dichloroisochromans. Chapter seven, after general introduction, describes the
synthesis and biological activity of these compounds. The unsubstituted homophthalic
acid was converted into anhydride by treatment with acetic anhydride. The latter was then
converted into 3-(substituted phenyl)isocoumarins by reacting it with suitable acid
chlorides. The isocoumarins were then converted into corresponding 3-(substituted
phenyl)isoquinolones by treatment with formamide. The 3-(substituted phenyl)
isocoumarin-1-thiones were synthesized from isocoumarins using Lawesson’s reagent
under microwave irradiation. Microwave assisted synthesis of some (±)-1-aryl-7,8-
dichloroisochromans was carried out by condensation of 2-(3,4-dimethoxyphenyl)
ethanol with a variety of aromatic aldehydes via an acid catalyzed oxa-Pictet-Spengler
reaction.
All of these synthesized compounds were characterized by IR, 1H,
13C NMR and
mass spectroscopic data. In vitro antibacterial activity of these compounds was
determined against ten different Gram positive and Gram negative bacterial strains using
agar well diffusion method.
The comparative analysis of the antibacterial activity of the 3-(substituted
phenyl)isocoumarins, 3-(substituted phenyl)isocoumarin-1-thiones and 3-(substituted
phenyl)isoquinolones is described. Accordingly, the antibacterial activity increases when
isocoumarins were converted into corresponding isocoumarin-1-thiones but decreases on
conversion into corresponding isoquinolones.
Page 7
v
Table of Contents
Acknowledgement i-ii
Abstract iii-iv
Contents v-xvi
PART I 1-125
CHAPTER ONE 1-35
Introduction
1.1 Nomenclature and Structural Type 1
1.2 Physical Properties 4
1.3 Biosynthesis 4
1.4 Pharmacological Applications 11
1.5 Synthesis of Isocoumarins and 3,4-Dihydroisocoumarins 17
1.5.1 Oxidation of Isochromans 17
1.5.2 Oxidation of Indenes, Indanones and Indones 18
1.5.3 Synthesis involving Metals 20
a) Lithiation 20
b) Thallation-olefination of Arenes 22
c) Silylation 22
d) Organo-mercury catalyzed synthesis 23
e) Palladium catalyzed method 24
f) Iridium catalyzed method 24
g) Rhodium-Catalyzed Oxidative Coupling of Benzoic Acids with
Alkynes via Regioselective C-H Bond Cleavage 25
1.5.4 Aldol-type Condensation between Homophthalic Acids,
Esters or Anhydrides and Carbonyl Compounds 25
a) Stobbe Condensation of Homophthalates with
Aldehydes and Ketones 26
b) Claisen Condensation of Homophthalates with Formates 26
c) Claisen Condensations of Homophthalates with Oxalates 27
d) Condensation of Malonyl Heterocycles with diphenylcarbonate 28
Page 8
vi
e) Condensation of Acid chlorides, Phenols,
Phenol Acids with Homophthalic Acids and Anhydrides 28
1.6 Reactions of Isocoumarins and 3,4-dihydroisocoumarins 30
1.6.1 Hydrolysis 30
1.6.2 Reaction with Ammonia and Amines 31
1.6.3 Reaction with Phosphorus Pentasulfide 32
1.6.4 Nitration 32
1.6.5 Reaction with Grignard Reagents 33
1.6.6 Oxidation 33
1.6.7 Reduction 33
1.7 Interconverision of Isocoumarins and 3,4-Dihydroisocumarins 33
1.7.1 Conversion of 3,4-Dihydroisocoumarins to Isocoumarins 34
a) Alkaline Hydrolysis Followed by Oxidation and Recyclization 34
b) Benzylic Bromination Followed by Dehydrobromination 34
1.7.2 Conversion of Isocoumarins to 3,4-Dihydroisocoumarins 34
a) Alkaline Hydrolysis Followed by Reduction and Recyclization 34
b) Catalytic Reduction 35
CHAPTER TWO 36-55
Experimental
2.1 Purification of Solvents 36
2.2 Instrumentation 36
2.3 Synthesis of methyl 4-methylbenzoate (1) 36
2.4 Synthesis of 3,5-dibromo-4-methylbenzoate (2) 36
2.5 3,5-Dimethoxy-4-methyl benzoic acid (3) 37
2.6 Methyl 3,5-dimethoxy-4-methyl benzoate (4) 37
2.7 (3,5-Dimethoxy-4-methyl phenyl)methanol (5) 37
2.8 3,5-Dimethoxy-4-methyl benzyl bromide (6) 38
2.9 (3,5-Dimethoxy-4-methylphenyl) acetonitril (7) 38
2.10 (3,5-Dimethoxy-4-methyl phenyl) acetic acid (8) 38
2.11 Methyl (3,5-dimethoxy-4-methyl phenyl) acetate (9) 38
2.12 Methyl (2-formyl-3,5-dimethoxy-4-methyl phenyl) acetate (10) 39
Page 9
vii
2.13 2,4-Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid(11) 39
2.14 6-(Carboxymethyl)-2,4-dimethoxy-3-methylbenzoic acid (12) 40
2.15 6,8-Dimethoxy-7-methyl-1H-isochromene-1,3(4H)-dione (13) 40
2.16 General procedure for 6,8-dimethoxy-7-methyl-3-alkyl/arylisocoumarins
(16a-j) 41
2.17 3-Propyl-6,8-dimethoxy-7-methylisocoumarin (16a) 41
2.18 3-Pentyl-6,8-dimethoxy-7-methylisocoumarin (16b) 42
2.19 3-Heptyl-6,8-dimethoxy-7-methylisocoumarin (16c) 42
2.20 3-Chloromethyl-6,8-dimethoxy-7-methylisocoumarin (16d) 42
2.21 3-Hydroxymethyl-6,8-dimethoxy-7-methylisocoumarin (16e) 43
2.22 3-Phenyl-6,8-dimethoxy-7-methylisocoumarin (16f) 43
2.23 3-(2-Chlorophenyl)-6,8-dimethoxy-7-methylisocoumarin (16g) 43
2.24 3-(4-Methoxyphenyl)-6,8-dimethoxy-7-methylisocoumarin (16h) 43
2.25 3-(3,4-Dimethoxyphenyl)-6,8-dimethoxy-7-methylisocoumarin(16i)44
2.26 3-(3,4,5-Trimethoxyphenyl)-6,8-dimethoxy-7-methylisocoumarin (16j) 44
2.27 General procedure for 2,4-dimethoxy-3-methyl-6-(2-oxoalkyl/aryl)benzoic
acid (17a-j) 44
2.28 2,4-Dimethoxy-3-methyl-6-(2-oxopentyl)benzoic acid (17a) 45
2.29 2,4-Dimethoxy-3-methyl-6-(2-oxoheptyl)benzoic acid (17b) 45
2.30 2,4-Dimethoxy-3-methyl-6-(2-oxononyl)benzoic acid (17c) 45
2.31 6-(3-Chloro-2-oxopropyl)-2,4-dimethoxy-3-methylbenzoic
acid(17d) 45
2.32 6-(3-Hydroxy-2-oxopropyl)-2,4-dimethoxy-3-methylbenzoic
acid (17e) 46
2.33 2,4-Dimethoxy-3-methyl-6-(2-oxo-2-phenylethyl)benzoic
acid (17f) 46
2.34 6-[2-(2-Chlorophenyl)-2-oxoethyl]-2,4-dimethoxy-
-3-methylbenzoic acid (17g) 46
2.35 2,4-Dimethoxy-6-[2-(4-methoxyphenyl)-2-oxoethyl]
-3-methylbenzoic acid (17h) 47
Page 10
viii
2.36 2,4-Dimethoxy-6-[2-(3,4-dimethoxyphenyl)-2-oxoethyl]-3-methylbenzoic
acid (17i) 47
2.37 2,4-dimethoxy-6-[2-(3,4,5-trimethoxyphenyl)-2-oxoethyl]
-3-methylbenzoic acid (17j) 47
2.38 General procedure for 6,8-dimethoxy-7-methyl-3-
alkyl/aryl-3,4- dihydroisocoumarins (18a-j) 48
2.39 6,8-Dimethoxy-7-methyl-3-propyl-3,4-dihydroisocoumarins (18a) 48
2.40 6,8-Dimethoxy-7-methyl-3-pentyl-3,4-dihydroisocoumarins (18b) 48
2.41 6,8-Dimethoxy-7-methyl-3-heptyl-3,4-dihydroisocoumarins (18c) 49
2.42 6,8-Dimethoxy-7-methyl-3-chloromethyl-3,4- 49
dihydroisocoumarins (18d)
2.43 6,8-Dimethoxy-7-methyl-3-hydroxymethyl-3,4- 49
dihydroisocoumarins (18e)
2.44 6,8-Dimethoxy-7-methyl-3-phenyl-3,4-dihydroisocoumarins (18f) 50
2.45 6,8-Dimethoxy-7-methyl-3-(2-chlorophenyl)-3,4-
dihydroisocoumarins (18g) 50
2.46 6,8-Dimethoxy-7-methyl-3-(4-methoxyphenyl)-3,4-
dihydroisocoumarins (18h) 50
2.47 6,8-Dimethoxy-7-methyl-3-(3,4-dimethoxyphenyl)-3,4-
dihydroisocoumarins (18i) 51
2.48 6,8-Dimethoxy-7-methyl-3-(3,4,5-trimethoxyphenyl)-3,4-
dihydroisocoumarins (18j) 51
2.49 General procedure for 6,8-dihydroxy-7-methyl-3-alkyl/aryl-3,4-
dihydroisocoumarins (19a-j) 51
2.50 6,8-Dihydroxy-7-methyl-3-propyl-3,4-dihydroisocoumarins (19a) 52
2.51 6,8-Dihydroxy-7-methyl-3-pentyl-3,4-dihydroisocoumarins (19b) 52
2.52 6,8-Dihydroxy-7-methyl-3-heptyl-3,4-dihydroisocoumarins (19c) 52
2.53 6,8-Dihydroxy-7-methyl-3-chloromethyl-3,4-
-dihydroisocoumarins (19d) 53
2.54 6,8-Dihydroxy-7-methyl-3-hydroxymethyl-3,4-
dihydroisocoumarins (19e) 53
Page 11
ix
2.55 6,8-Dihydroxy-7-methyl-3-phenyl-3,4-dihydroisocoumarins (19f) 53
2.56 6,8-Dihydroxy-7-methyl-3-(2-chlorophenyl)-3,4- 53
dihydroisocoumarins (19g)
2.57 6,8-Dihydroxy-7-methyl-3-(4-methoxyphenyl)-3,4-
dihydroisocoumarins (19h) 54
2.58 6,8-Dihydroxy-7-methyl-3-(3,4-dimethoxyphenyl)-3,4-
dihydroisocoumarins 54
2.59 6,8-Dihydroxy-7-methyl-3-(3,4,5-trimethoxyphenyl)-3,4-
dihydroisocoumarins (19j) 54
CHAPTER THREE 56-87
Results and Discussion
3.1 Synthesis of 3,5-dimethoxy-4-methylhomophthalic acid (12) 56
3.2 Synthesis of 6,8-dimethoxy-7-methyl-3-
alkyl/aryl isocoumarins (16a-j) 65
3.3 Synthesis of 6,8-dimethoxy-7-methyl-3-alkyl/aryl-
3,4-dihydroisocoumarins (18a-j) 72
3.4 Synthesis of 6,8-dihydroxy-7-methyl-3-alkyl/aryl-
3,4-dihydroisocoumarins (19a-j) 82
CHAPTER FOUR 88-106
Biological Activities
4.1 Antibacterial Activity 88
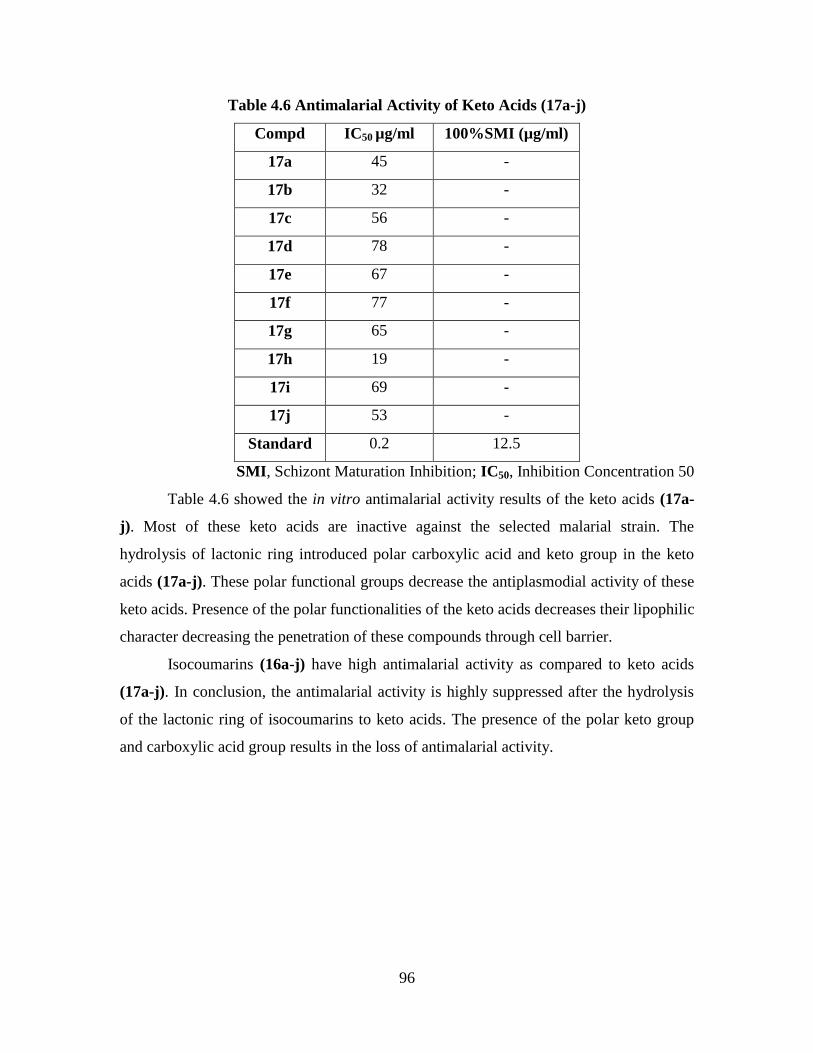
4.2 Antimalarial Activity 94
4.3 Cytotoxicity 100
4.3.1 Cytotoxic Activity of the Isocoumarins (16a-j) 100
4.3.2 Cytotoxic Activity of the Keto Acids (17a-j) 102
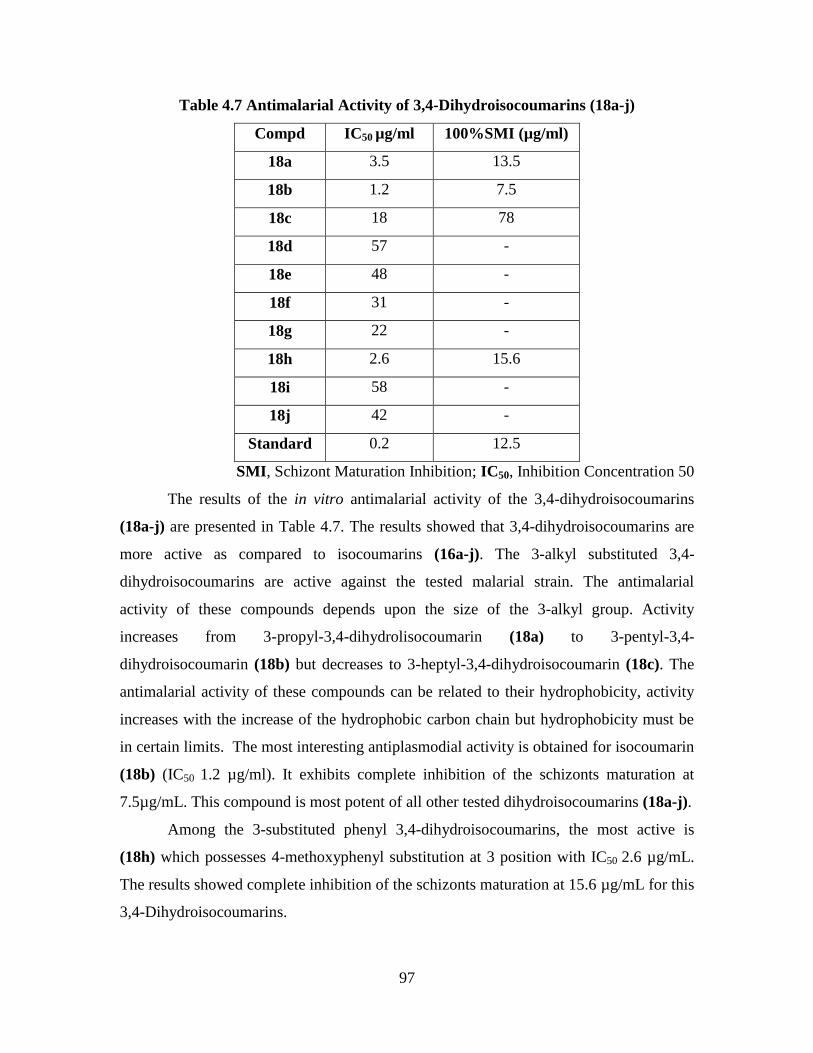
4.3.3 Cytotoxic Activity of the 3,4-Dihydroisocoumarins (18a-j) 103
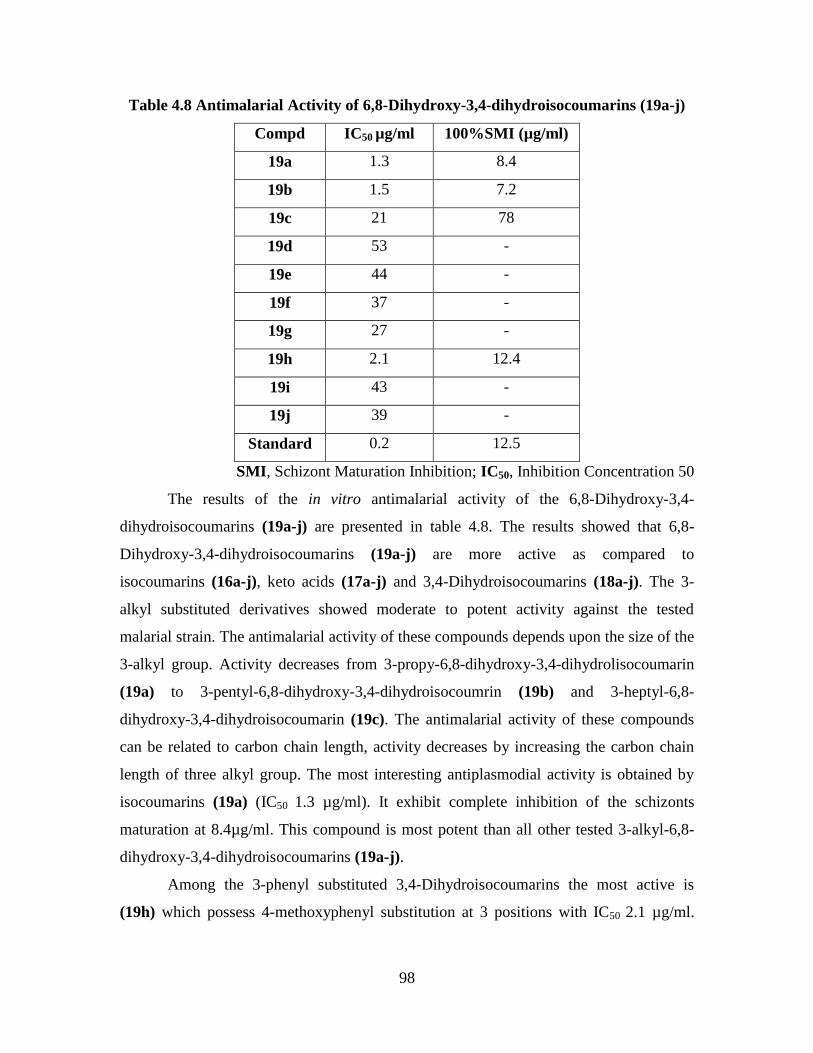
4.3.4 Cytotoxic Activity of the 6,8-Dihydroxy-
3,4-Dihydroisocoumarins (19a-j) 104
CHAPTER FIVE 107-118
5.1 Synthesis of Stellatin 107
Page 12
x
5.1.1 Methyl (3, 5-dimethoxy-4-methyl phenyl) acetate (1) 107
5.1.2 Methyl (2-formyl-3,5-dimethoxy-4-methyl phenyl) acetate (2) 107
5.1.3 2,4-Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid (3) 108
5.1.4 2,4-dimethoxy-6-(2-hydroxyethyl)-3-methylbenzoic acid (4) 108
5.1.5 6,8-dimethoxy-7-methyl-3,4-dihydro-1H-isochromen-1-one (5) 109
5.1.6 6,8-dimethoxy-7-(bromomethyl)-3,4-dihydro-
1H-isochromen-1-one (6) 109
5.1.7 7-(hydroxymethyl)-6,8-dimethoxy-3,4-dihydro-
1H-isochromen-1-one (7) 110
5.1.8 8-hydroxy-7-(hydroxymethyl)-6-methoxy-
3,4-dihydro-1H-isochromen-1-one (8) 110
5.2 Results and Discussion 111
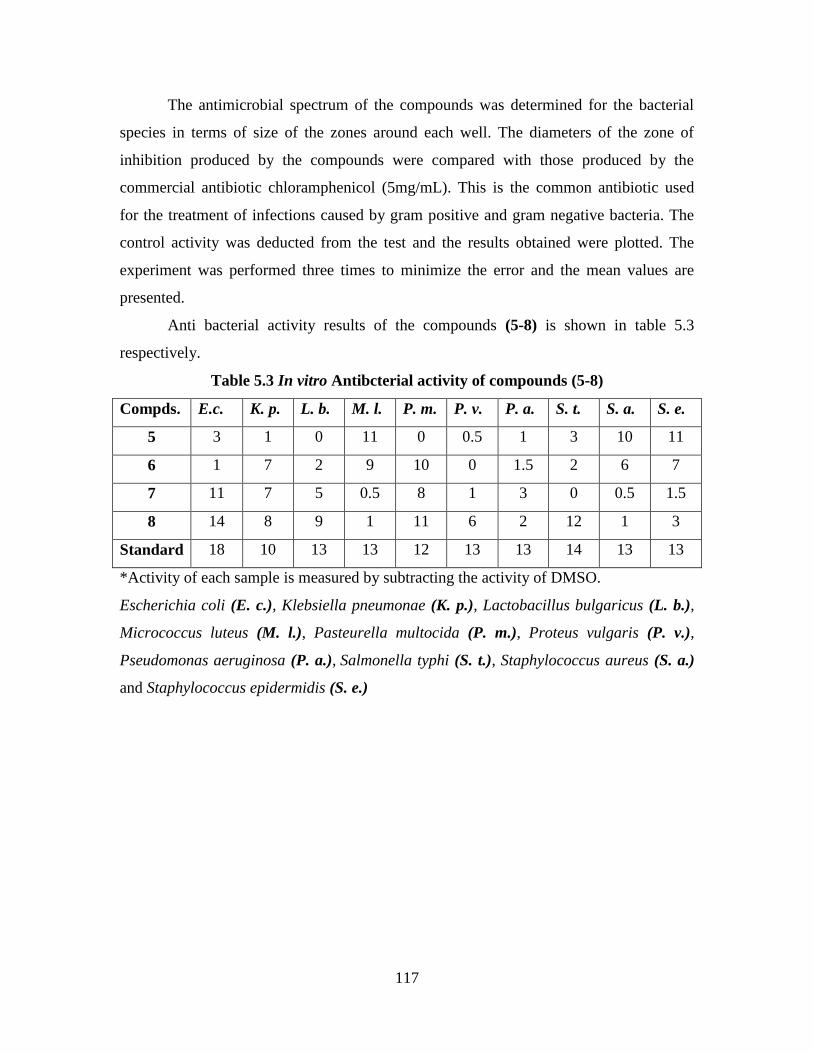
5.3 Antibacterial Activity 116
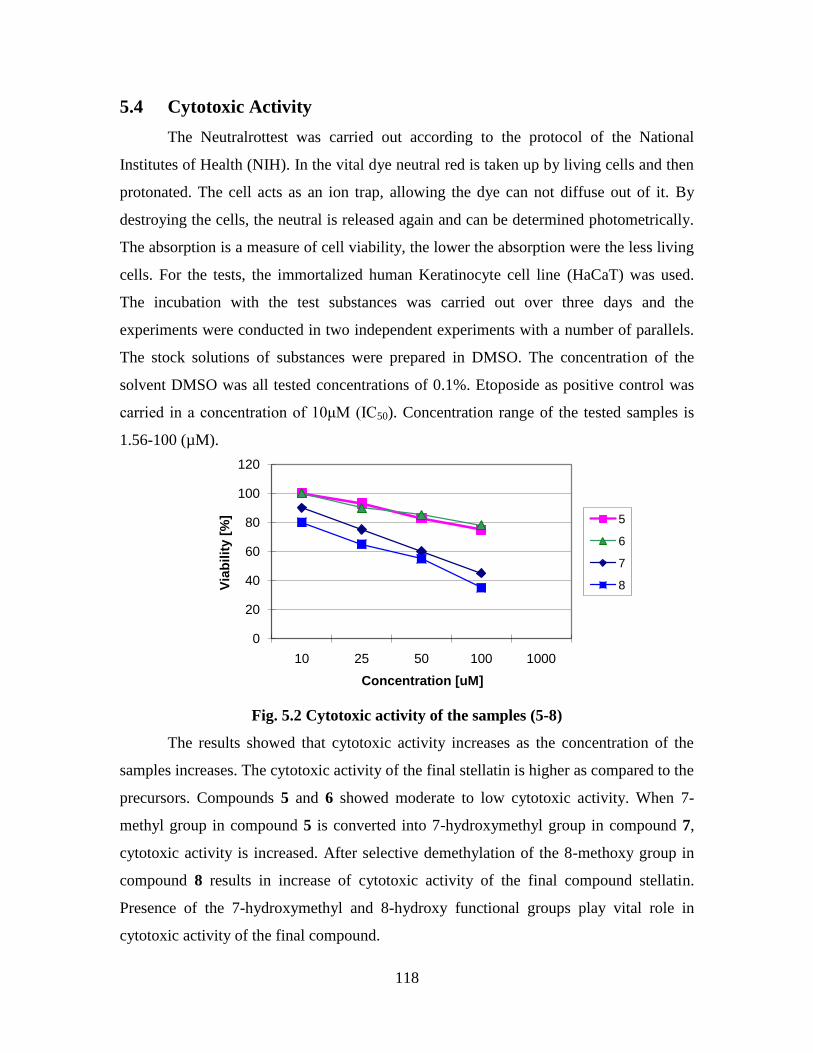
5.4 Cytotoxic Activity 118
CHAPTER SIX References Part I 119-125
Part II 126-162
CHAPTER SEVEN
7.1 Introduction 126
7.2 Experimental 127
Synthesis of homophthalic anhydride (1)
General procedure for 3-alkyl/arylisocoumarins (4a-j) 127
3-(3-Fluorophenyl)isocoumarin (4a) 128
3-(4-Fluorophenyl)isocoumarin (4b) 128
3-(2-Chlorobenzyl)isocoumarin (4c) 128
3-(2-Bromophenyl)isocoumarin (4d) 129
3-(3-Iodophenyl)isocoumarin (4e) 129
3-(2,4-Dichlorophenyl)isocoumarin (4f) 129
3-(2-Chloro-4-fluorophenyl)isocoumarin (4g) 129
3-(3-Nitrophenyl)isocoumarin (4h) 130
3-(2-Chloropyridyl)isocoumarin (4i) 130
Page 13
xi
3-Pentadecylisocoumarin (4j) 130
General procedure for the conversion of isocoumarins
into 1(2H)-isoquinolones (5a–j) 131
3-(3-Fluorophenyl)isoquinolin-1(2H)-one (5a) 131
3-(4-Fluorophenyl)isoquinolin-1(2H)-one (5b) 131
3-(2-Chlorobenzyl)isoquinolin-1(2H)-one (5c) 131
3-(2-Bromophenyl)isoquinolin-1(2H)-one (5d) 132
3-(3-Iodophenyl)isoquinolin-1(2H)-one (5e) 132
3-(2,4-Dichlorophenyl)isoquinolin-1(2H)-one (5f) 132
3-(2-Chloro-4-fluorophenyl)isoquinolin-1(2H)-one (5g) 133
3-(3-Nitrophenyl)isoquinolin-1(2H)-one (5h) 133
3-(2-Chloropyridyl)isoquinolin-1(2H)-one (5i) 133
3-Pentadecylisoquinolin-1(2H)-one (5j) 134
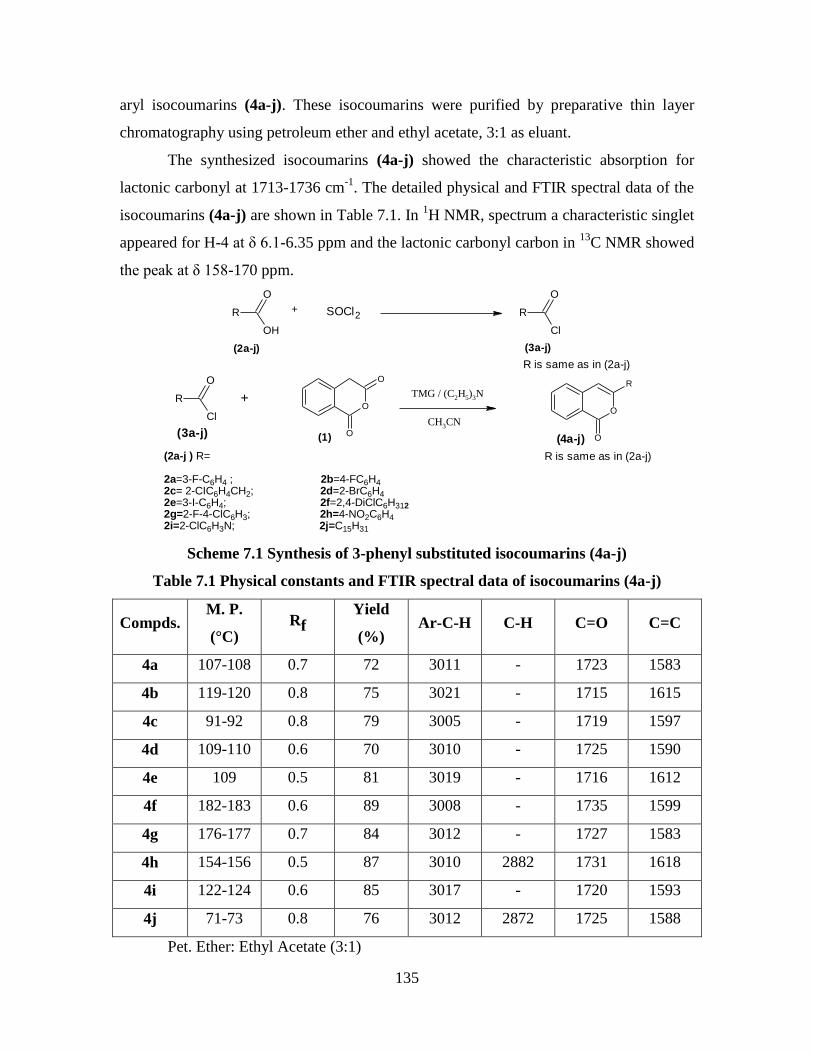
7.3 Results and Discussion 134
Synthesis of 3-phenyl substituted-1H-isochromen
-1-thiones 138
7.4 Experimental 138
General procedure for the conversion of isocoumarins
into 1-1H-isochromene-1-thiones (2a–j) 138
3-(3-Fluorophenyl)-1H-isochromene-1-thione (7a) 138
3-(4-Fluorophenyl)-1H-isochromene-1-thione (7b) 138
3-(4-Chlorophenyl)-1H-isochromene-1-thione (7c) 139
3-(2-Bromophenyl)-1H-isochromene-1-thione (7d) 139
3-(3-Iodophenyl)-1H-isochromene-1-thione (7e) 139
3-(2,4-Dichlorophenyl)-1H-isochromene-1-thione (7f) 140
3-(2-Chloro-4-fluorophenyl)-1H-isochromene-1-thione (7g) 140
3-(4-Methoxyphenyl)-1H-isochromene-1-thione (7h) 140
3-(4-Fluorobenzyl)-1H-isochromene-1-thione (7i) 140
3-(Pentadecyl)-1H-isochromene-1-thione (7j) 141
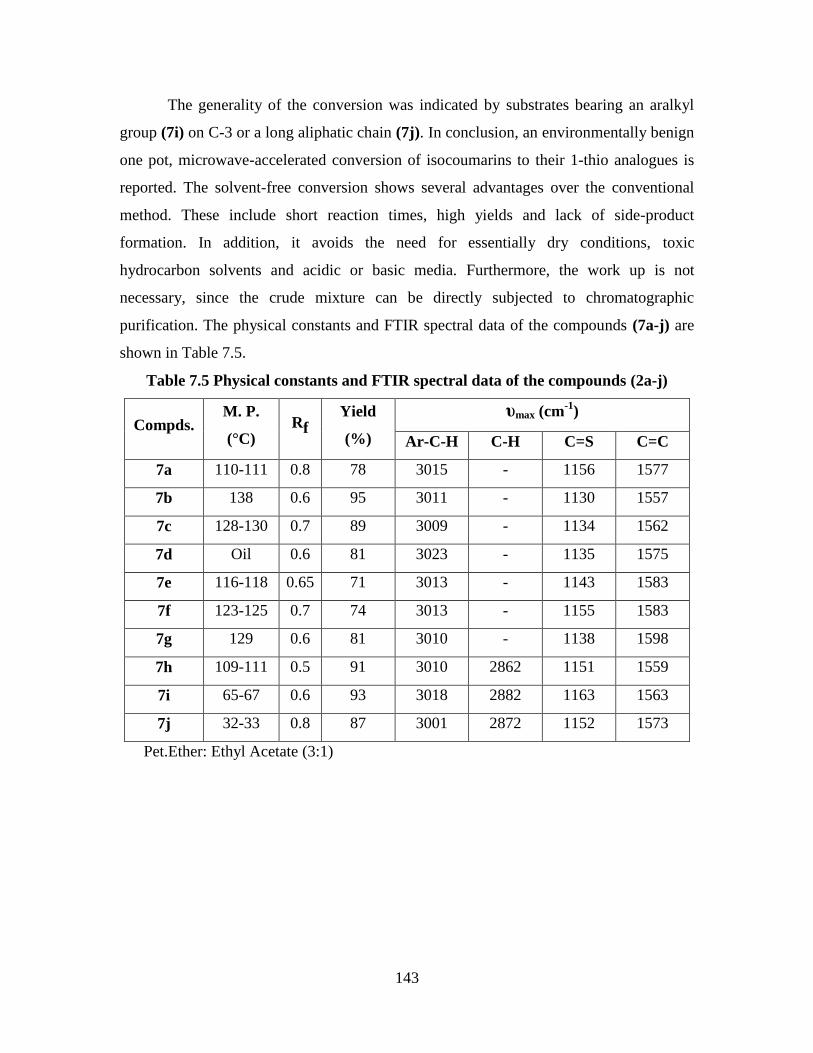
7.5 Results and Discussion 141
Page 14
xii
7.6 Biological Activities 144
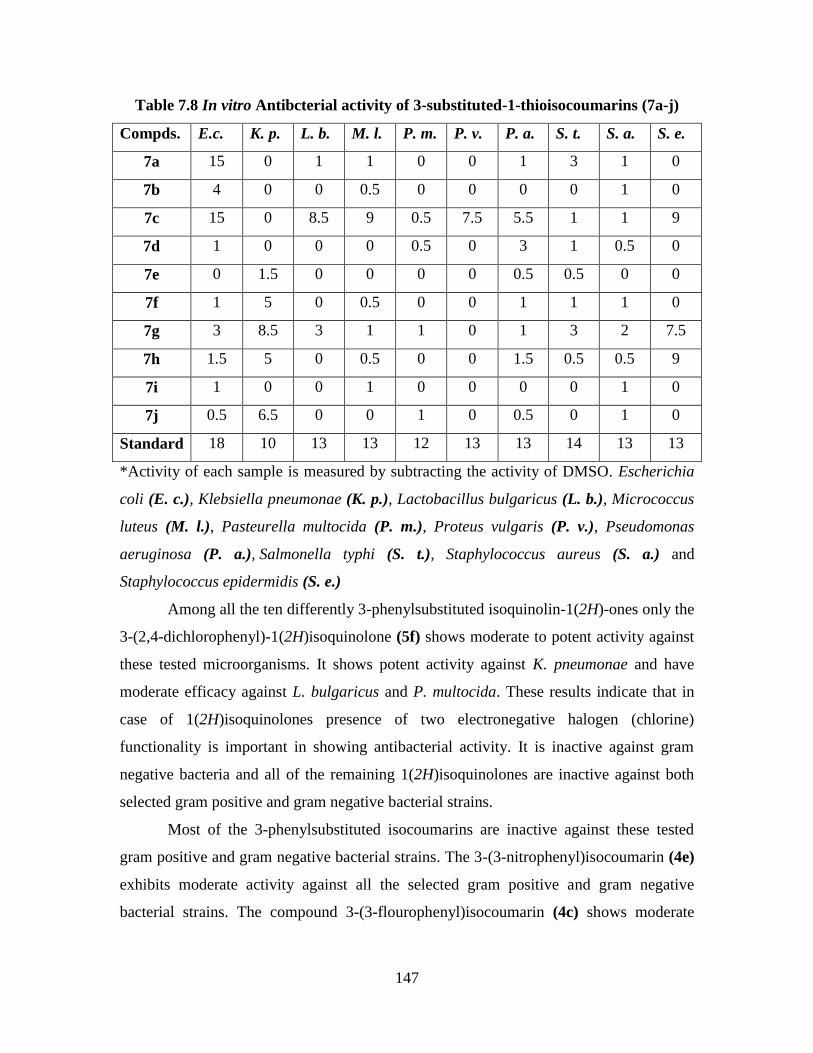
7.7 Antibacterial Activity 144
Synthesis of (±)-1-aryl-7,8-dichloro-3,4-dihydro-
1H-isochromenes 150
7.8 Introduction 150
7.9 Experimental 151
General procedure for the synthesis of (±)-1-aryl-7,8-
dichloro-3,4-dihydro-1H-isochromenes (2a-g) 151
7,8-Dichloro-1-phenyl-3,4-dihydro-1H-isochromene (2a) 151
7,8-Dichloro-1-(2-chlorophenyl)-3,4-dihydro-1H-isochromene(2b) 152
7,8-Dichloro-1-(4-chlorophenyl)-3,4-dihydro-1H-isochromene (2c) 152
7,8-Dichloro-1-(3-methoxyphenyl)-3,4-dihydro-1H-isochromene(2d)152
7,8-Dichloro-1-(3-methoxy-4-hydroxyphenyl)-3,4-dihydro-
1H-isochromene (2e) 153
7,8-Dichloro-1-(3,4,5-trimethoxyphenyl)-3,4-dihydro-
1H-isochromene (2f) 153
7,8-Dichloro-1-(5-nitrobenzo[d] [1,3]dioxol-6-yl)-3,4-dihydro-
1H-isochromene (2g) 153
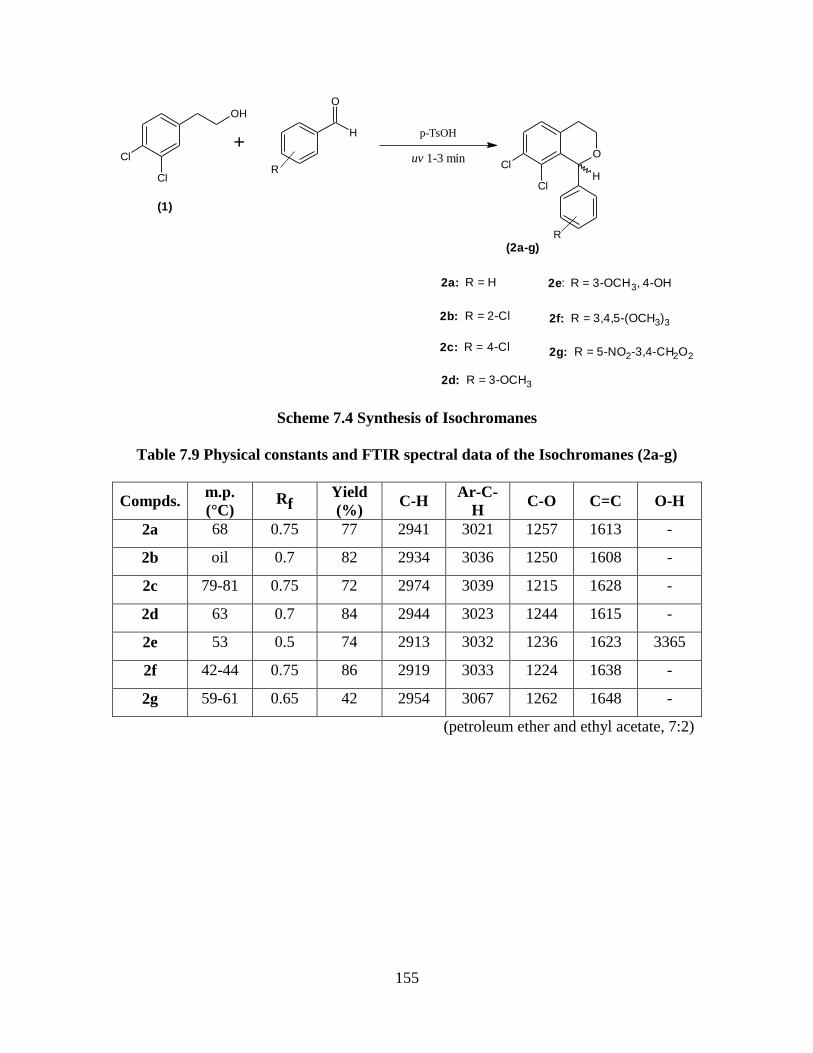
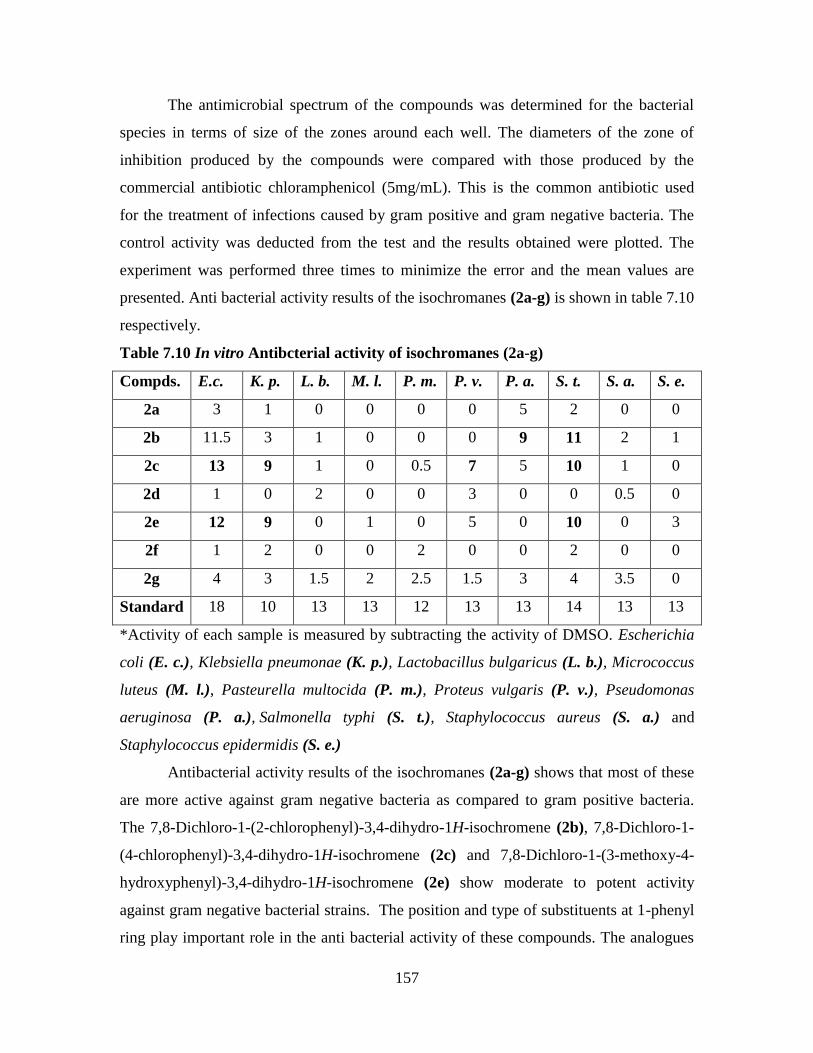
7.10 Results and Discussion 154
7.11 Antibacterial Activity 156
References Part II 159
Page 15
xiii
List of Tables
Table 1.1 Comparison of the melting points of isocoumarins
and dihydroisocoumarins 4
Table 3.1 Physical constants and FTIR spectral data of the compounds (1-12) 59
Table 3.2 1H and
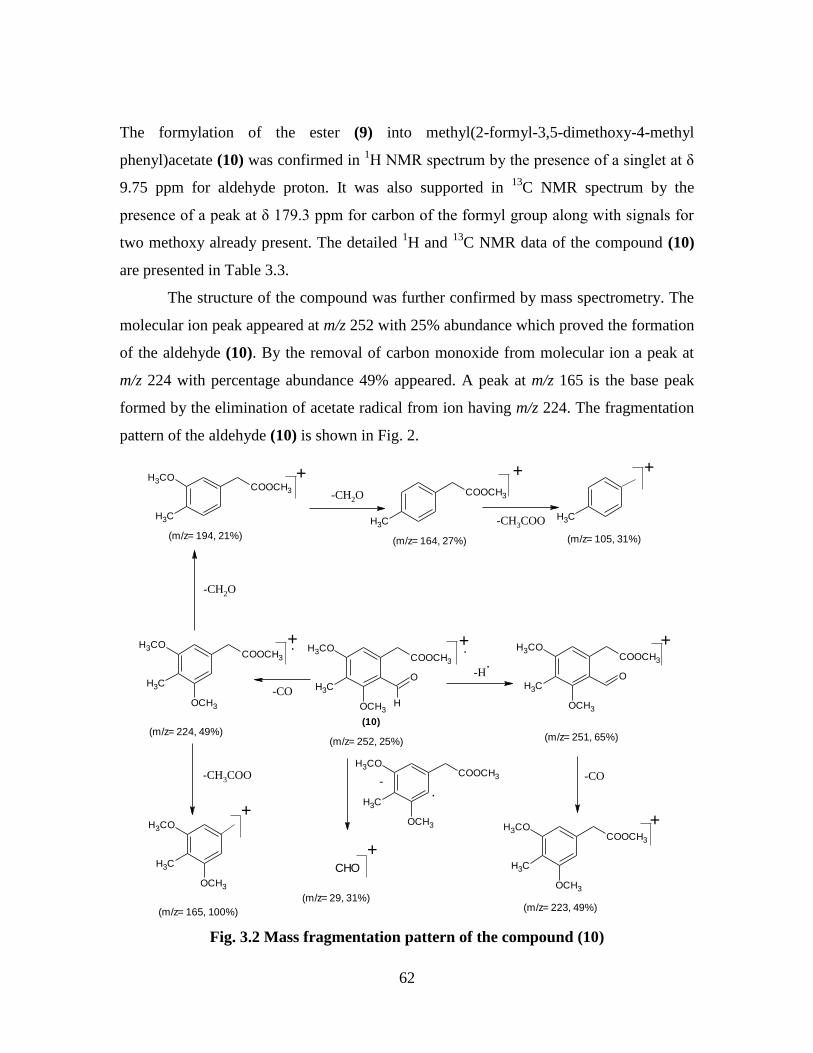
13C NMR data of the compound (9) 60
Table 3.3 1H and
13C NMR data of the compound (10) 61
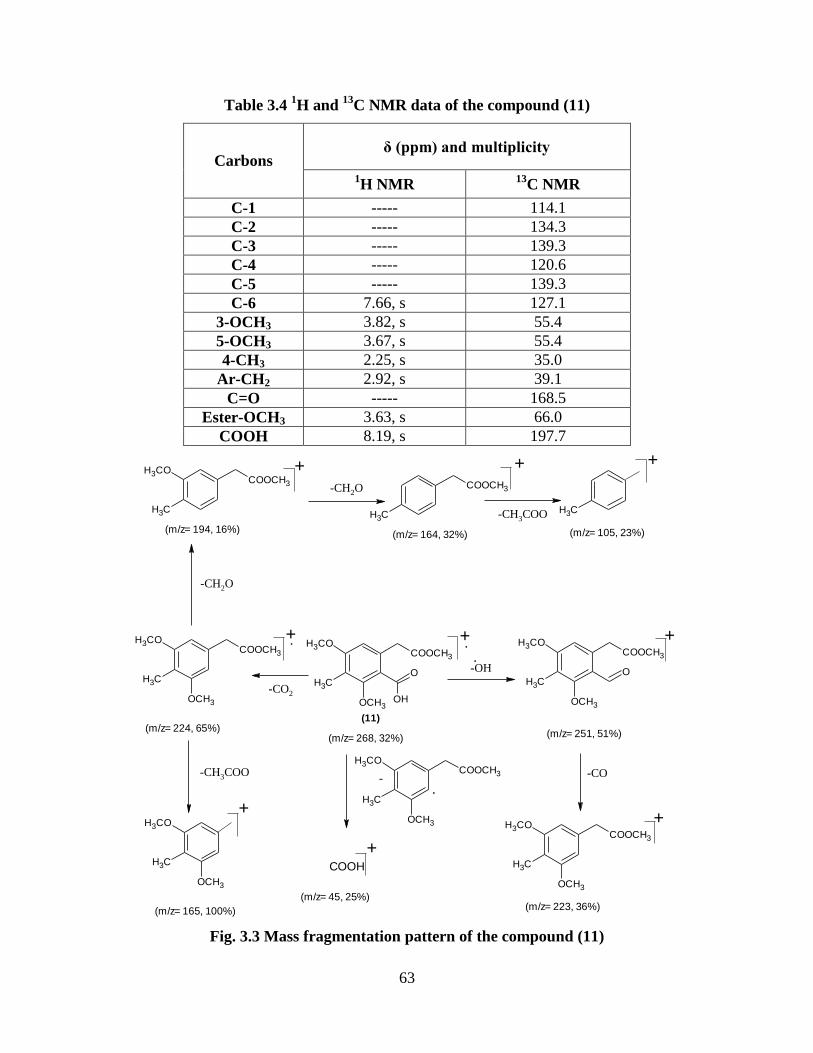
Table 3.4 1H and
13C NMR data of the compound (11) 63
Table 3.5 1H and
13C NMR data of the compound (12) 64
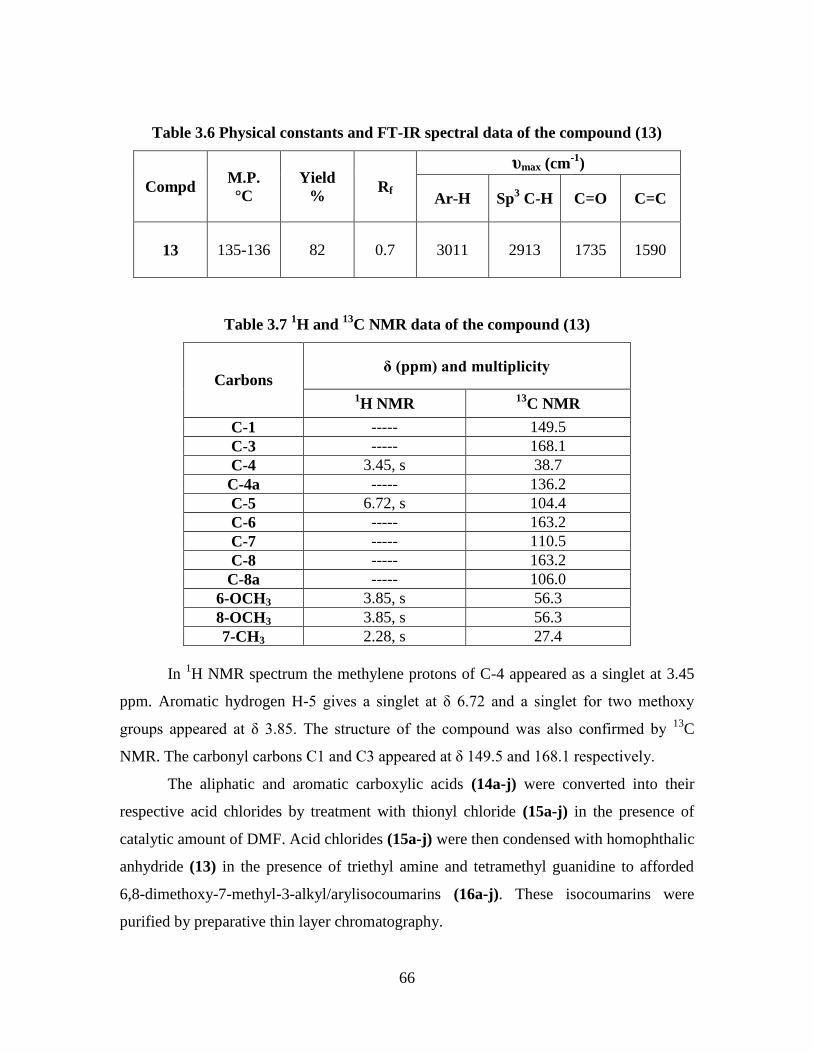
Table 3.6 Physical constants and FT-IR spectral data of the compound (13) 66
Table 3.7 1H and
13C NMR data of the compound (13) 66
Table 3.8 Physical constants and FTIR spectral data of the compounds (16a-j)68
Table 3.9 Elemental analysis data of the compounds (16a-j) 68
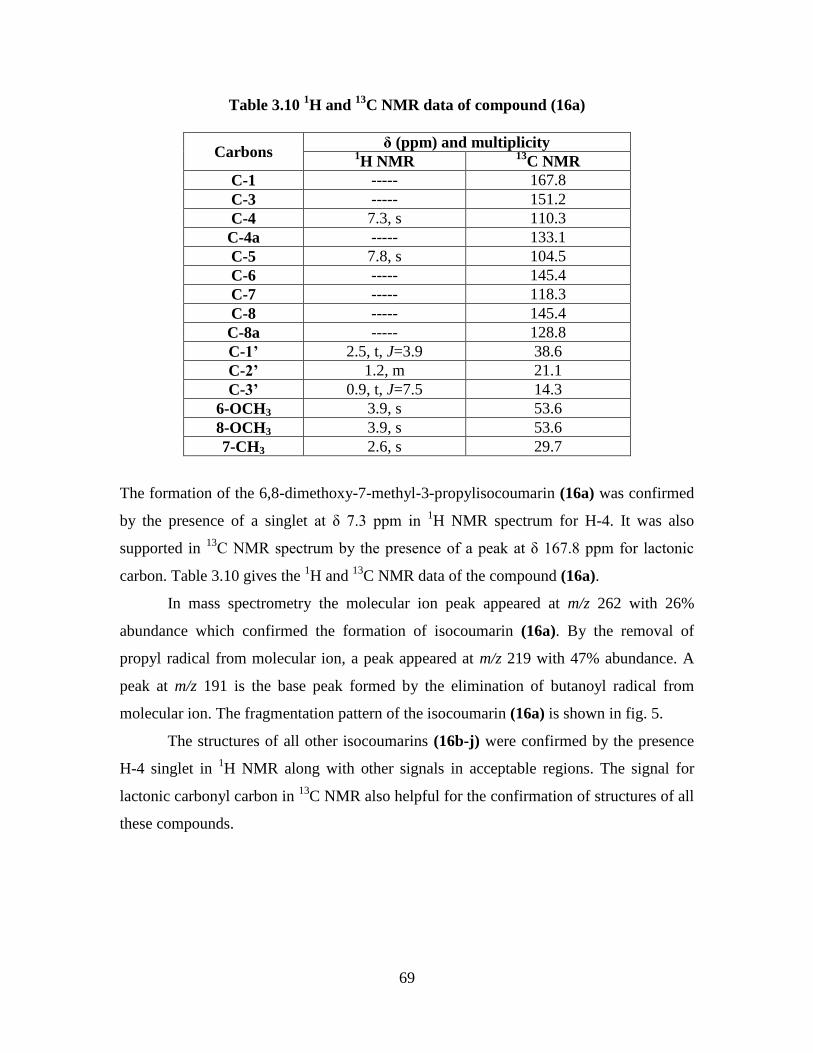
Table 3.10 1H and
13C NMR data of compound (16a) 69
Table 3.11 1H and
13C NMR data of compound (16e) 70
Table 3.12 1H and
13C NMR data of compound (16h) 71
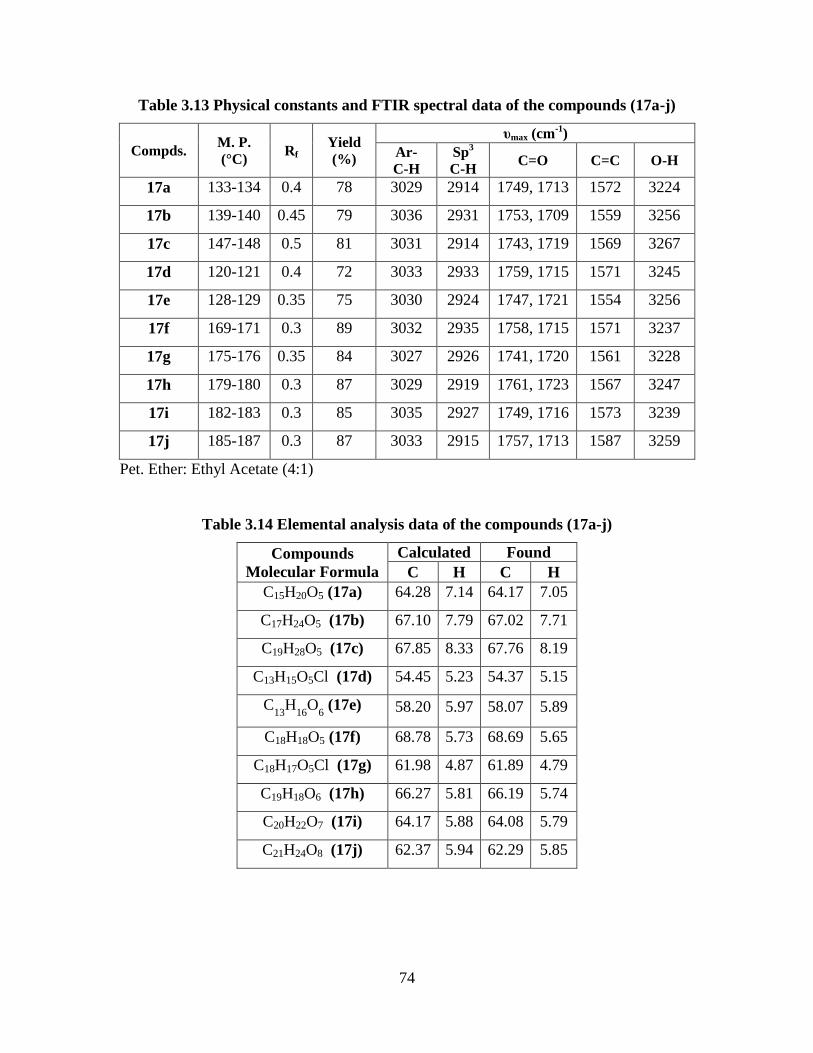
Table 3.13 Physical constants and FTIR spectral data of the
compounds (17a-j) 74
Table 3.14 Elemental analysis data of the compounds (17a-j) 74
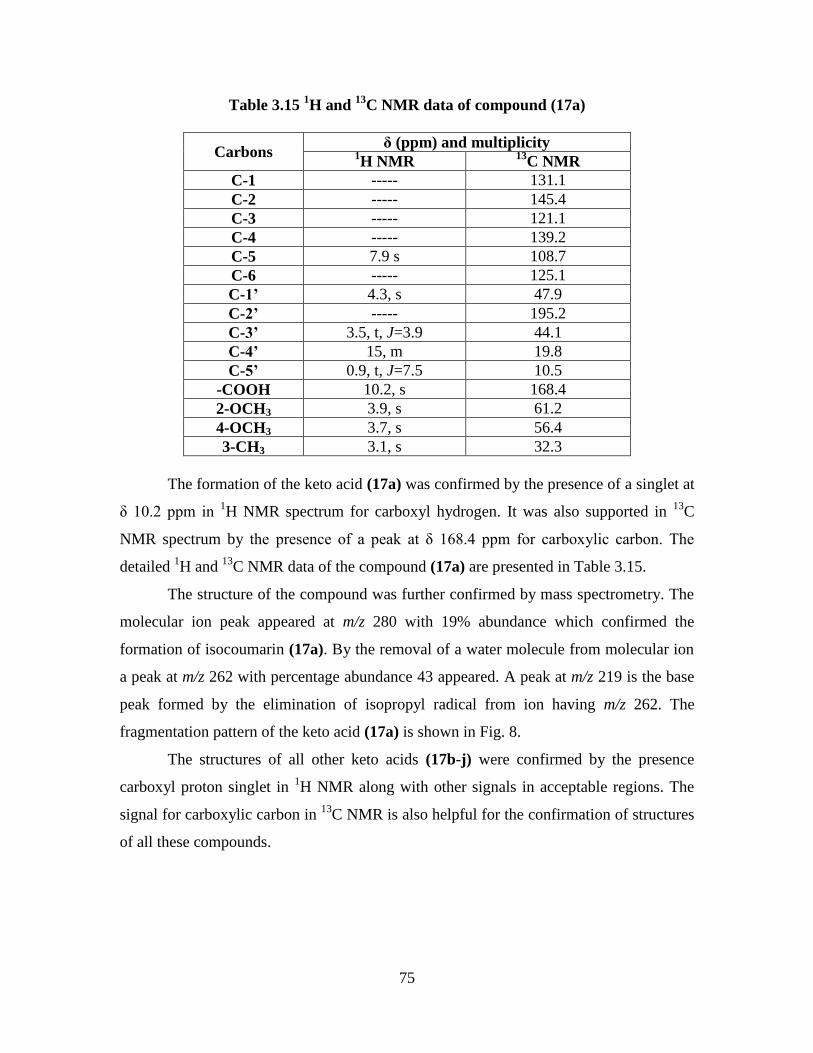
Table 3.15 1H and
13C NMR data of compound (17a) 75
Table 3.16 1H and
13C NMR data of compound (17f) 76
Table 3.17 Physical constants and FTIR spectral data of the compounds (18a-j)78
Table 3.18 Elemental analysis data of the compounds (18a-j) 78
Table 3.19 1H and
13C NMR data of compound (18a) 79
Table 3.20 1H and
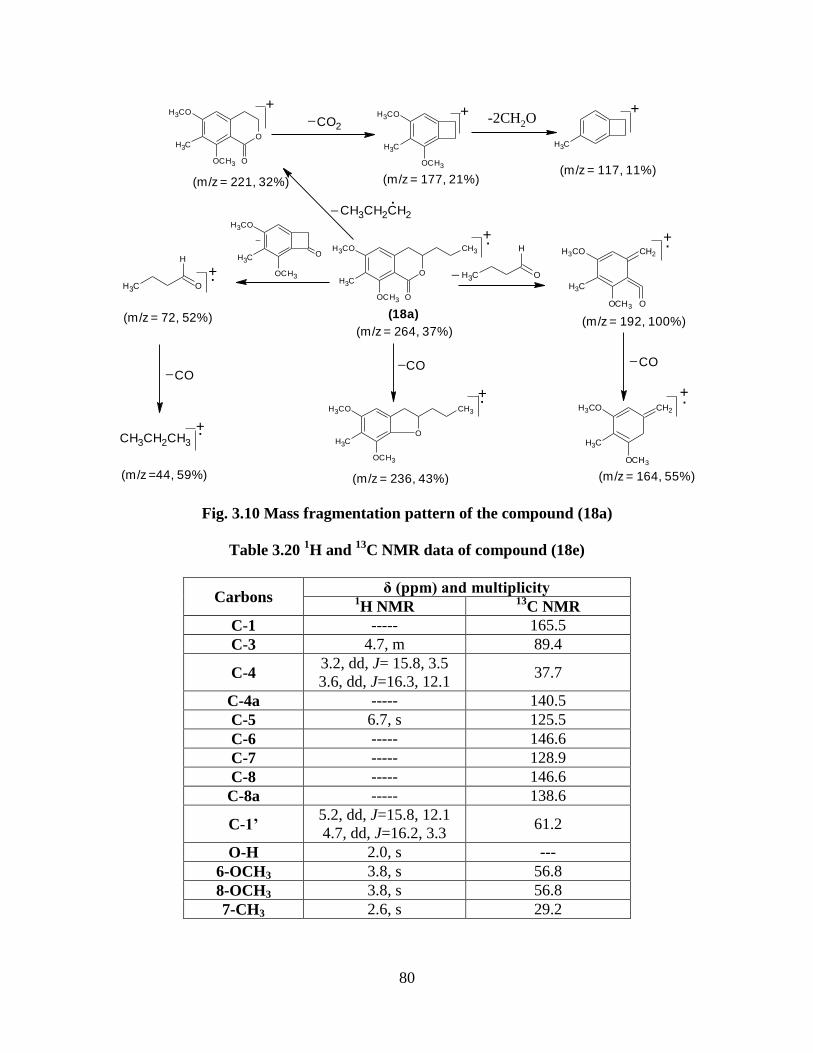
13C NMR data of compound (18e) 80
Table 3.21 1H and
13C NMR data of compound (18f) 81
Table 3.22 Physical constants and FTIR spectral data of the
compounds (19a-j) 83
Table 3.23 Elemental analysis data of the compounds (19a-j) 84
Table 3.24 1H and
13C NMR data of compound (19a) 84
Table 3.25 1H and
13C NMR data of compound (19e) 86
Table 3.26 1H and
13C NMR data of compound (19f) 87
Page 16
xiv
Table 4.1 In vitro Antibacterial Activity of Isocoumarins (16a-j) 90
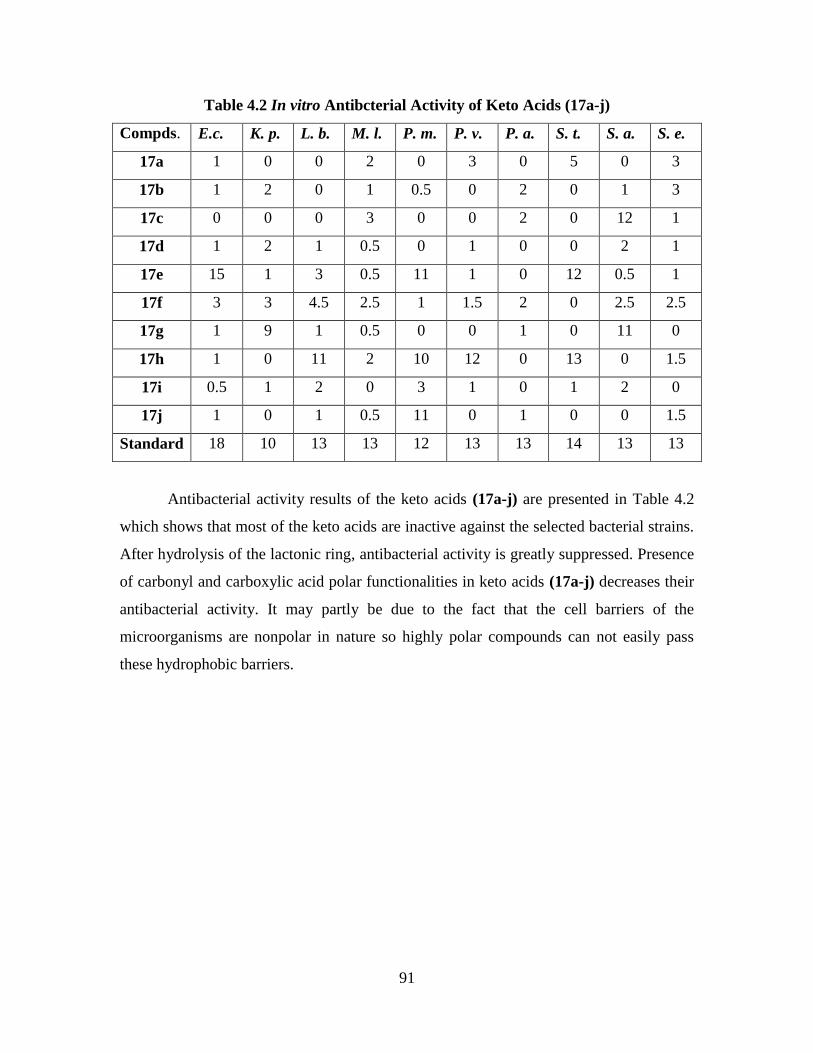
Table 4.2 In vitro Antibacterial Activity of Keto acids (17a-j) 91
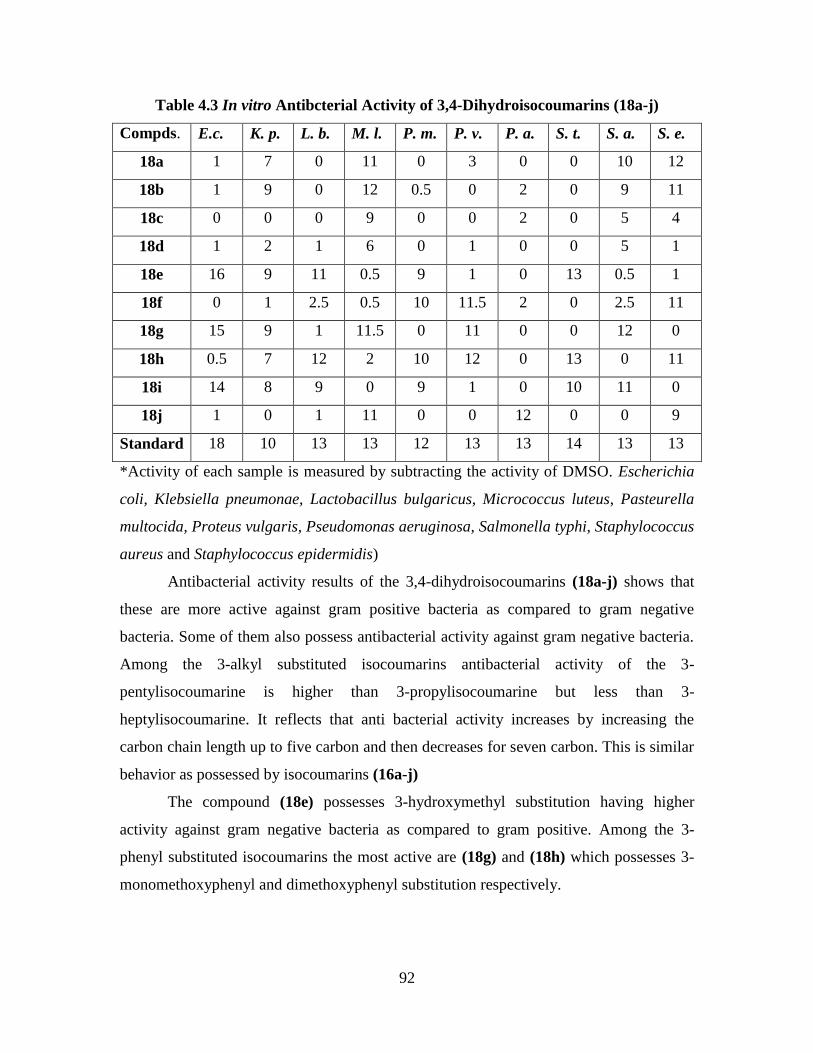
Table 4.3 In vitro Antibacterial Activity of 3,4-Dihydroisocoumarins (18a-j) 92
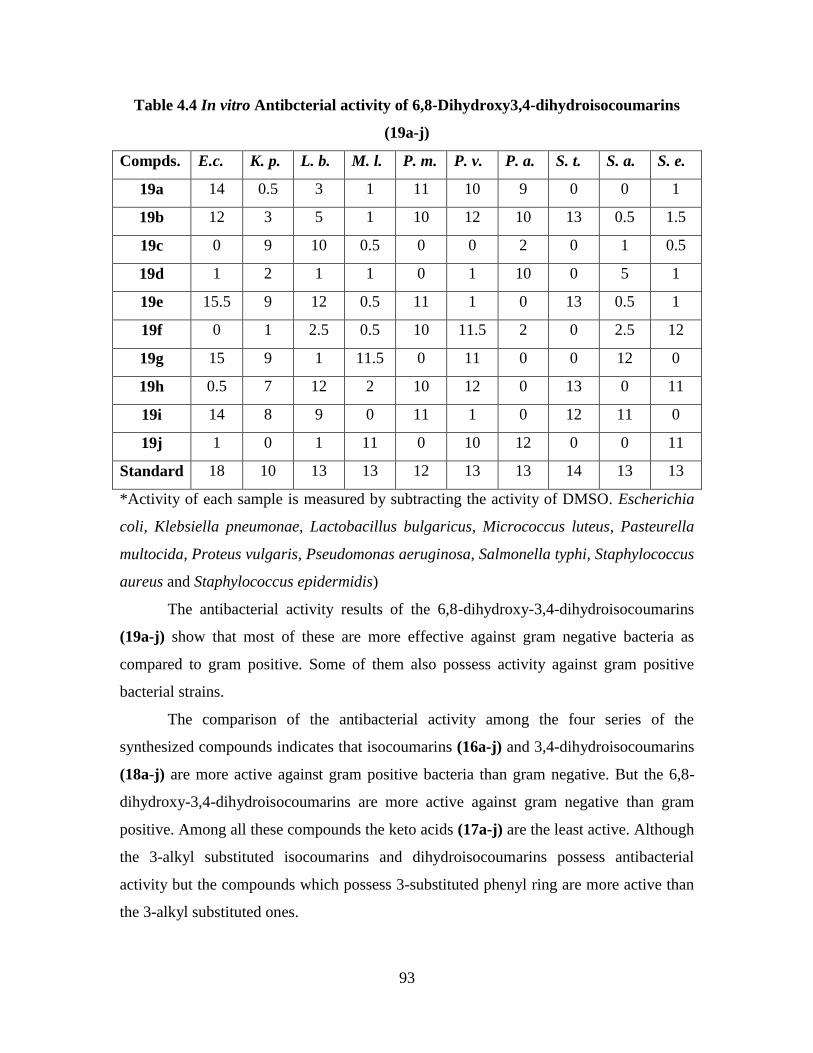
Table 4.4 In vitro Antibacterial activity of 6,8-Dihydroxy3,4-
dihydroisocoumarins (19a-j) 93
Table 4.5 Antimalarial Activity of Isocoumarins (16a-j) 95
Table 4.6 Antimalarial Activity of Keto Acids (17a-j) 96
Table 4.7 Antimalarial Activity of 3,4-Dihydroisocoumarins (18a-j) 97
Table 4.8 Antimalarial Activity of 6,8-Dihydroxy-3,4-
Dihydroisocoumarins (19a-j) 98
Table 5.1 Physical and FTIR spectral data of the compounds (1-8) 113
Table 5.2 1H and
13C NMR data of Stellatin (8) 114
Table 5.3 In vitro antibacterial activity of compounds (5-8) 117
Table 7.1 Physical constants and FTIR spectral data of isocoumarins (4a-j) 135
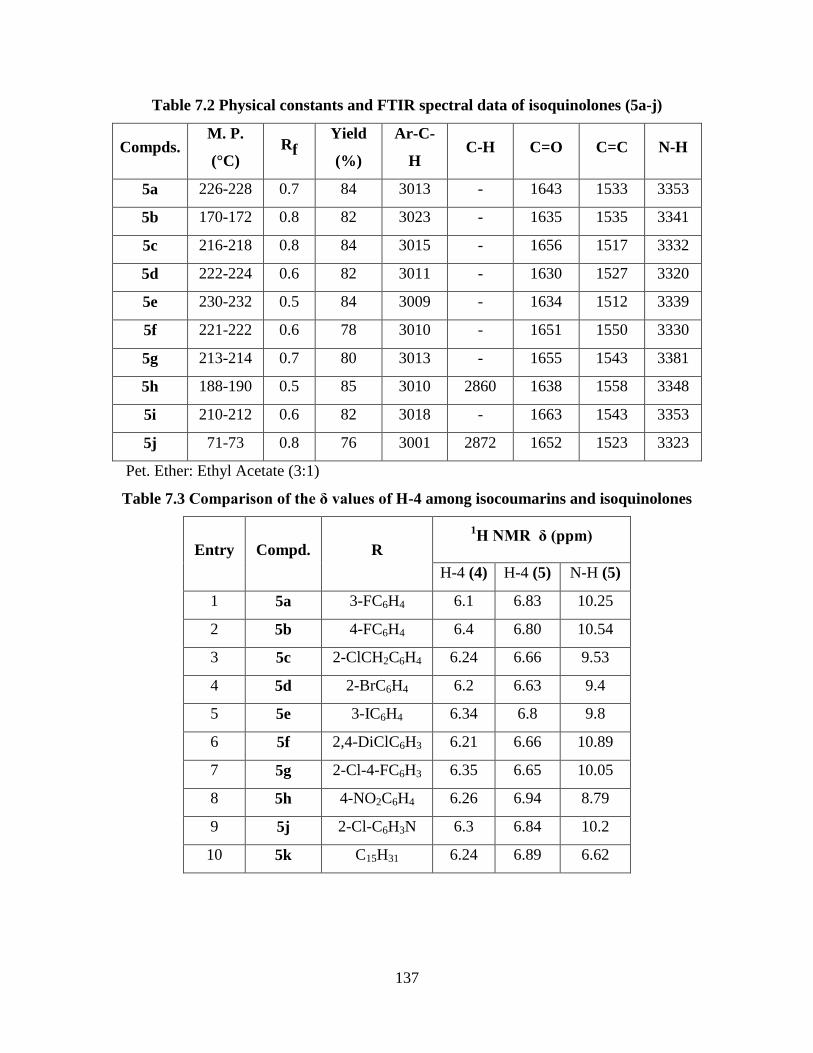
Table 7.2 Physical constants and FTIR spectral data of isoquinolones (5a-j) 137
Table 7.3 Comparison of the δ values of H-4 among isocoumarins
and isoquinolones 137
Table 7.4 Comparison of the chemical shifts of H-4 and C-1
in compounds (6a-j) and (7a-j) 142
Table 7.5 Physical constants and FTIR spectral data of the compounds (2a-j) 143
Table 7.6 In vitro antibacterial activity of 3-substituted isocoumarins (4a-j) 146
Table 7.7 In vitro antibacterial activity of 3-substituted isoquinolones (5a-j) 146
Table 7.8 In vitro antibacterial activity of 3-substituted-1-
thioisocoumarins (7a-j) 147
Table 7.9 Physical constants and FTIR spectral data of the isochromans (2a-g)155
Table 7.10 In vitro antibacterial activity of isochromanes (2a-g) 157
Page 17
xv
List of Figures
Fig. 3.1 Mass fragmentation pattern of the compound (9) 61
Fig. 3.2 Mass fragmentation pattern of the compound (10) 62
Fig. 3.3 Mass fragmentation pattern of the compound (11) 63
Fig. 3.4 Mass fragmentation pattern of the compound (12) 65
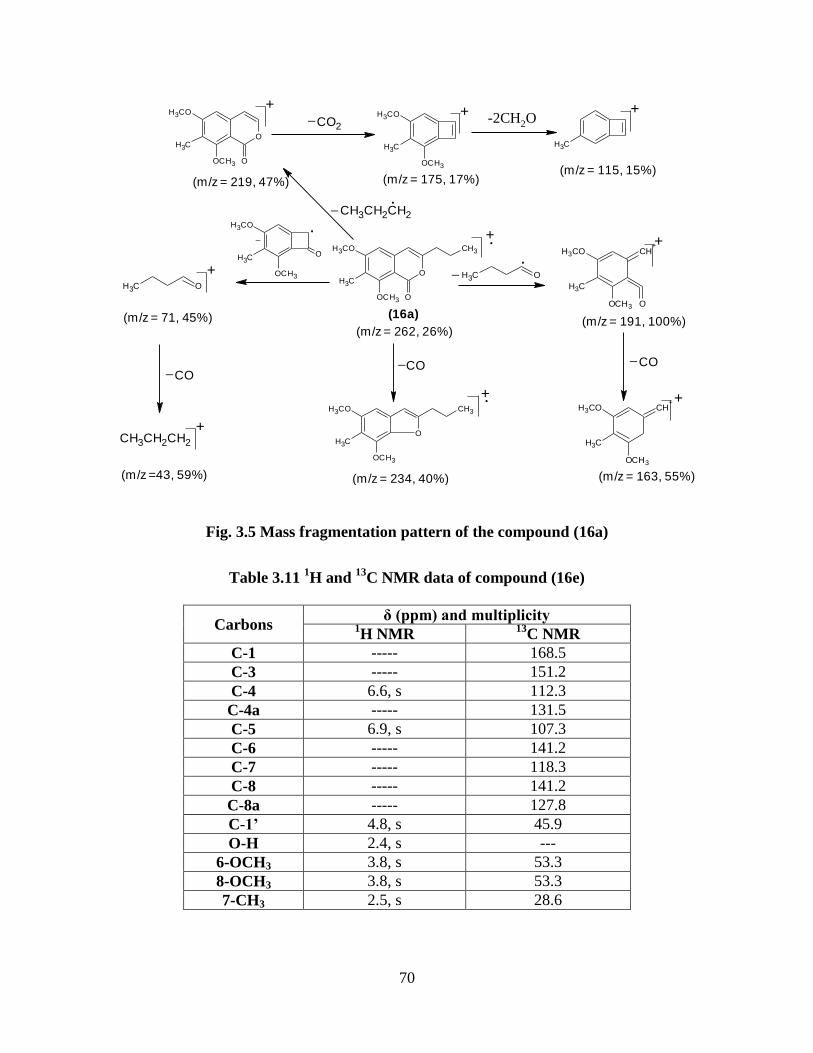
Fig. 3.5 Mass fragmentation pattern of the compound (16a) 70
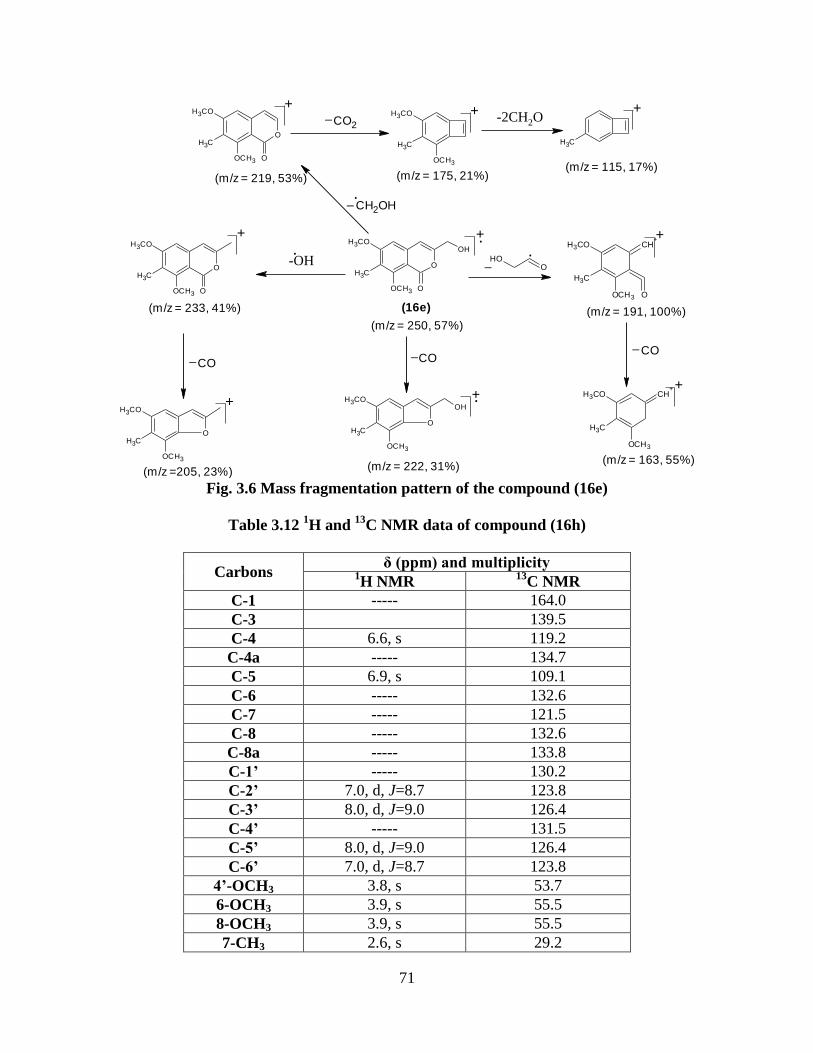
Fig. 3.6 Mass fragmentation pattern of the compound (16e) 71
Fig. 3.7 Mass fragmentation pattern of the compound (16h) 72
Fig. 3.8 Mass fragmentation pattern of the compound (17a) 76
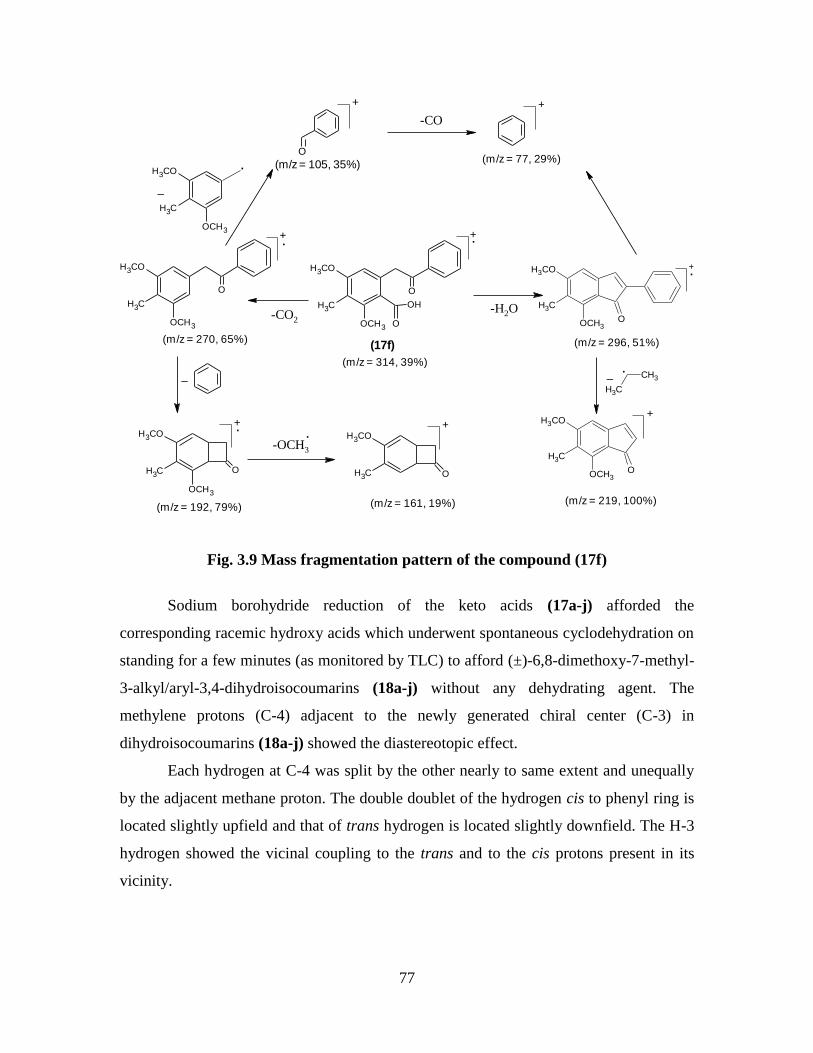
Fig. 3.9 Mass fragmentation pattern of the compound (17f) 77
Fig. 3.10 Mass fragmentation pattern of the compound (18a) 80
Fig. 3.11 Mass fragmentation pattern of the compound (18e) 81
Fig. 3.12 Mass fragmentation pattern of the compound (18f) 82
Fig. 3.13 Mass fragmentation pattern of the compound (19a) 85
Fig. 3.14 Mass fragmentation pattern of the compound (19e) 86
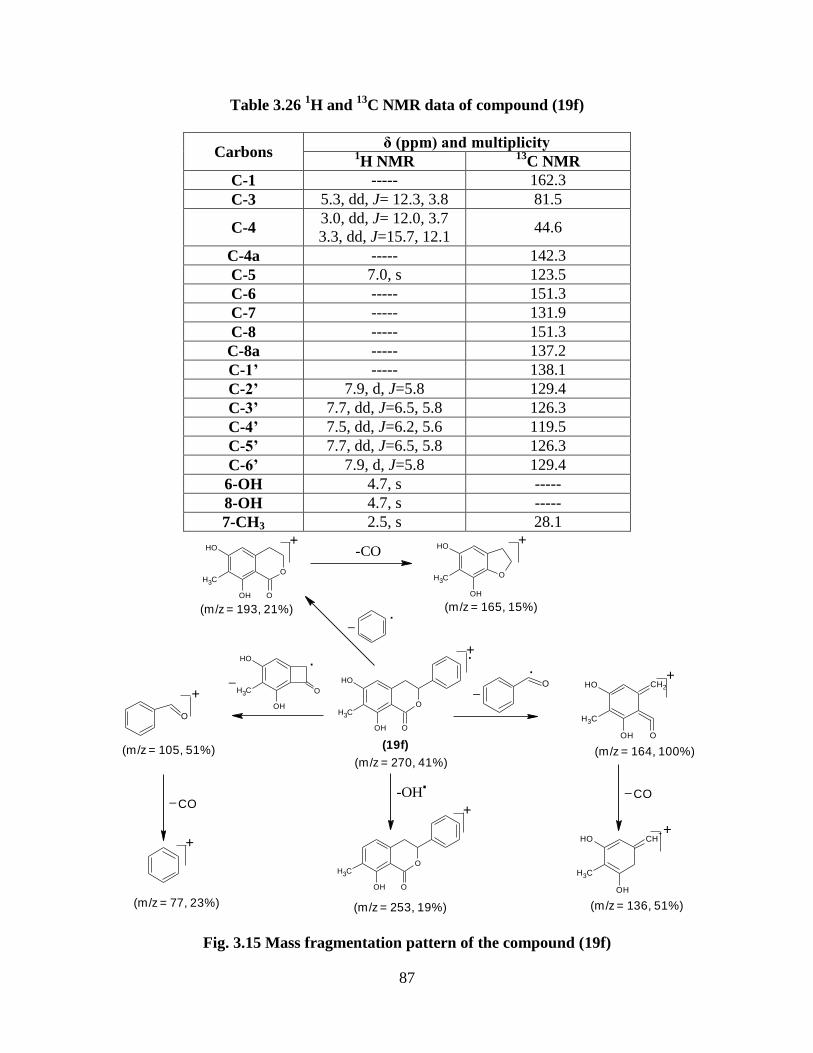
Fig. 3.15 Mass fragmentation pattern of the compound (19f) 87
Fig. 4.1 Cytotoxic activity results of the isocoumarins (16a-e) 101
Fig. 4.2 Cytotoxic activity results of the isocoumarins (16f-j) 101
Fig. 4.3 Cytotoxic activity results of the keto acids (17f-j) 102
Fig. 4.4 Cytotoxic activity results of the keto acids (17f-j) 102
Fig. 4.5 Cytotoxic activity results of the 3,4-dihydroisocoumarins (18a-e) 103
Fig. 4.6 Cytotoxic activity results of the 3,4-dihydroisocoumarins (18f-j) 103
Fig. 4.7 Cytotoxic activity results of the 6,8-dihydroxy-
3,4-Dihydroisocoumarins (19a-e) 104
Fig. 4.8 Cytotoxic activity results of the 6,8-dihydroxy-
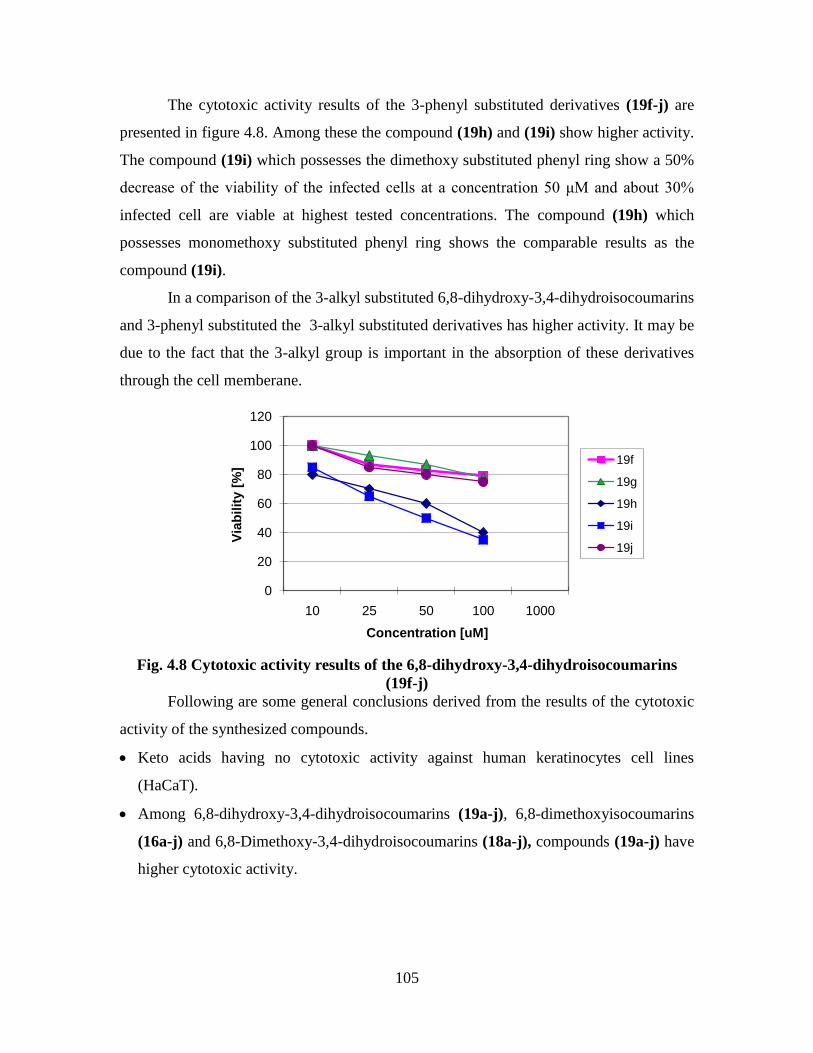
3,4-Dihydroisocoumarins (19f-j) 105
Fig. 5.1 Mass fragmentation pattern of the Stellatin (8) 115
Fig. 5.2 Cytotoxic activity of the samples (5-8) 118
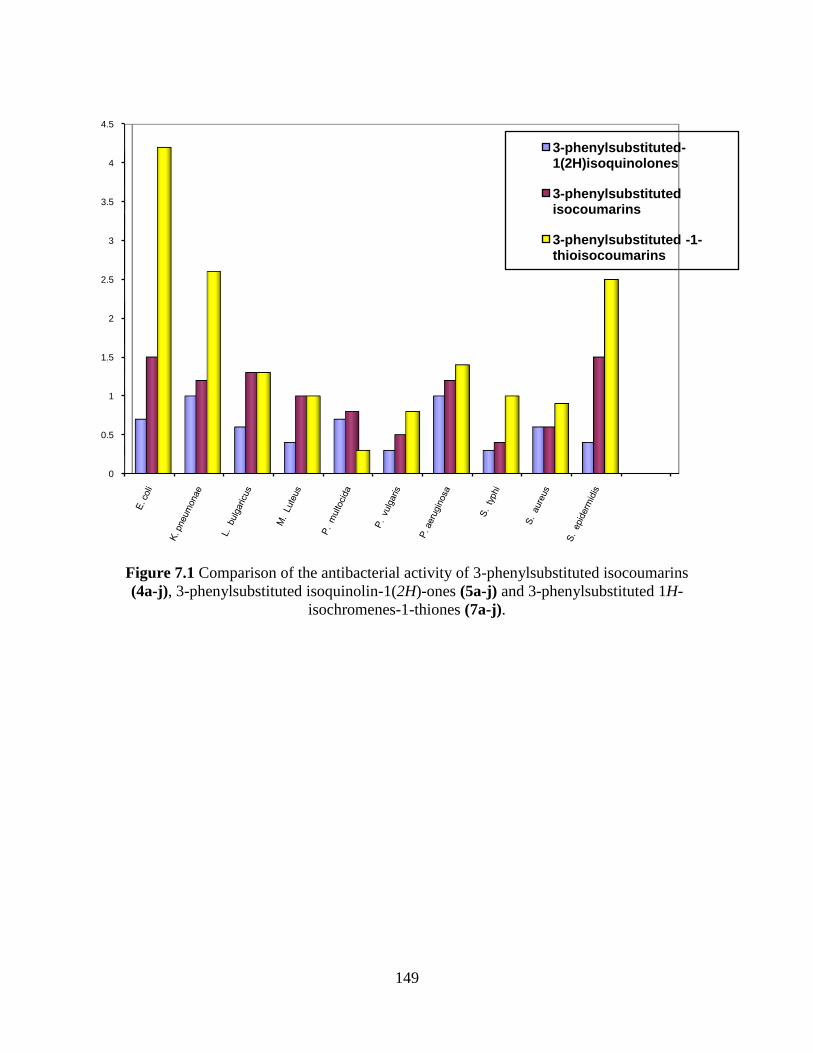
Fig. 7.1 Comparison of the antibacterial activity of 3-phenylsubstituted
isocoumarins (4a-j), 3-phenylsubstituted isoquinolin-
1(2H)-ones (5a-j) and 3-substituted phenyl-1H-isochromenes-
Page 18
xvi
1-thiones (7a-j) 149
SCHEMES
Scheme 3.1 Synthesis of methyl 3,5-dimethoxy-4-methylphenyl acetate (9) 58
Scheme 3.2 Synthesis of 3,5-dimethoxy-4-methyl homophthalic acid (12) 59
Scheme 3.3 Synthesis of 6,8-dimethoxy-7-methyl-3-alkyl/arylisocoumarins (16a-j)67
Scheme 3.4 Synthesis of 6,8-dimethoxy-7-methyl-3-alkyl/aryl-
3,4-dihydroisocoumarins (18a-j) 73
Scheme 3.5 Synthesis of 6,8-dihydroxy-7-methyl-3-alkyl/aryl-
3,4-dihydroisocoumarins (19a-j) 83
Scheme 5.1 Synthesis of Stellatin 112
Scheme 7.1 Synthesis of 3-phenyl substituted isocoumarins (4a-j) 135
Scheme 7.2 Synthesis of 3-phenyl substituted isoquinolones (5a-j) 136
Scheme 7.3 Solvent-free conversion of isocoumarin into 1-thioisocoumarins 142
Scheme 7.4 Synthesis of isochromanes 155
Page 19
1
INTRODUCTION
Isocoumarins and 3,4-dihydroisocoumarins are the secondary metabolites1 of a
wide variety of fungi, lichens, molds, bacteria, higher plants and insects. Majority of
isocoumarins have been isolated from various species of fungal genera Artemisia,
Aspergillus, Ceratocystis, Fusarium, Penicillum, Streptomyces, etc. A number of them
are constituents of a few higher plant families, e. g., Bignoniaceae, Compositae,
Leguminoseae, Myricaceae, Saxifragaceae. Literature reviews published on isocoumarins
include the review by R. D. Barry2 (1964), W. B. Turner and Aldridge
3 (1983), M.
Yamato4 (1983), R. A. Hill
5 (1986), E. Napolitano
6 (1997) and Bin
7 et al. (2000).
1.1 Nomenclature and Structural Type
The name isocoumarin (1) is derived from the fact that these compounds are
isomeric to coumarin (2). Coumarin8 was isolated (1820) from tonka tree formerly known
as Coumarouna odorata. In an isocoumarin, a lactonic pyran ring is fused to a benzene
ring. The IUPAC and Chemical Abstract name for isocoumarin is 1H-2-benzopyran-1-
one, numbered as shown and its 3,4-dihydro analogue (3) is named as 3,4-
dihydroisocoumarin rather than isochroman-1-one.
O
O
R2
R1
R3
R4
R5
R6
O O1
2
34
4a5
6
7
8 8aO
O
R2
R1
R3
R4
R5
R6
12
34
4a5
6
7
8 8a
(1) (2) (3)
As in case of other classes of the natural products (alkaloids, flavonoids, etc.) no
systematic nomenclature exists for isocoumarins. Majority of naturally occurring
isocoumarins and 3,4-dihydroisocoumarins have been assigned trivial names9 which are
derived from generic or specific names of source plant and fungi. Examples of the names
derived from those of parent genera are: agrimonolide (Agrimonia pilosa), fusamarin
(Fusarium spp.), alternariol (Alternaria spp.), artemidin (Artemisia glauca), peniolactol
(Peniophora sanguinea), cladosporin (Cladosporium spp.), homalicine (Homalium
zeylancum), oosponol (Oospora astringes), etc. Those names derived from species are
found in mellein (Aspergillus melleus), ustic acid (A. ustus), duclauxin (P. duclauxi),
Page 20
2
ochratoxin A, B and C (A. ochraceus), capillarin (Artemisia capillaris), viridotoxin (A.
virinutans), moncerin (H. monoceros), etc.
Trivial names of a large number of isocoumarins end in the suffix "-in" for
example artemidin, bergenin, bactobolin A, B and C, actinobolin, baciphelacin,
coriandrin, asperentin, canescin, fusamarin, mellein, stellatin, etc. However, isocoumarin
names ending in other suffixes like “-ol, -one, -ide. -oic acid, anhydride” indicating their
chemical class are also common. Examples are altenuisol, hydrangenol, oosponol,
oospoglycol, peniolactol, reticulol, oospolactone, agrimonolide, feralolide, monocerolide,
ustic acid, β-callatolic acid, β-alectoronic acid, ardisic acid B, chebulic acid, lamellicolic
anhydride, naphthalic anhydride, etc.
Isocoumarin (1) itself (R1-R
6=H) has never been found to occur naturally,
however, its simple derivatives are found in nature. Isocoumarin may be substituted
either on lactone ring or the aromatic ring or on both. Thus R1-R
6 in (1) or (3) may be
alkyl, aryl, heterocyclyl, halo, nitro or any other substituent.
A number of naturally occurring isocoumarins possess a C-3 carbon substituent
and all isocoumarins, biogenetically derived from acetate have C-8 oxygenation and
some have retained the C-6 oxygen. Hydrangenol, phyllodulcin, chebulic acid,
dihydrohomalicine and blepherigenin are isocoumarins found in plants, lack C-6
oxygenation and are not acetate derived. Isocoumarins having a C-4, C-5 or C-7
substituents are relatively uncommon in nature, nevertheless, C-7 oxygenation is fairly
uncommon.
Mellein (4), the 3,4-dihydro-8-hydroxy-3-methylisocoumarin has been taken as
the parent compound for simple isocoumarins. Thus, 3,4-dihydro-8-hydroxy-6-methoxy-
3-methylisocoumarin (5) is known as 6-methoxymellein. Similarly, the compounds (6,
R1=H, R
2=COOH) and (6, R
1=CHO, R
2=H) are called as 7-carboxymellein and 5-
formylmellein, respectively.
O
O
CH3
OH
(4)
O
O
CH3
OH
H3CO
(5)
O
O
CH3
OH
R2
R1
(6)
Page 21
3
Peniolactol (7a) and 3-alkyl-3-hydroxy-3,4-dihydroisocoumarins such as ustic
acid (8a) and its derivatives exist in tautomeric equilibrium between their keto acid forms
(7a & 8a) and lactol forms (7b & 8b) respectively.
O
O
C15H31
OH
OH
OH
(7a)
O
O
C15H31
OH
OHOH
(7b)
O
O
CH3
OH
OH
OH
OHOCH3
(8a)
O
O
CH3
OH
OHOH
OHOCH3
(8b)
The lactam analogue of isocoumarin, 1-(2H)-isoquinolinone (9a), trivially known
as isocarbostyril, exists in equilibrium with its tautomeric form (9b). A large number of
variously substituted isocarbostyrils10
and tetrahydroisoquinolinones (10) can also exist
as their other tautomers have been prepared.
NH
O
(9a)
RN
OH
(9b)
RNH
O
(10)
R
Sulphur analogues have also been known since long and a number of substituted
1-thio- (11, Z=S), 1-hydrazino-(11, Z=NNH2), 1-phenylhydrazino- (11, Z=NNHC6H5), 2-
thio- (12), and 1,2-dithioisocoumarins11
(13) have been prepared.
O
Z
OR
(11)
S
O
(12)
S
S
(13) In 1980, a three-step synthesis of 2-seleno- and 2-telluroisocoumarins was
reported12
. Regiospesific nucleophilic β-addition of methaneselenolate or -tellurolate
Page 22
4
anion to the triple bond of ethyl-2-ethenylbenzoate (14) afforded the chalcogenated esters
(14a). Saponification afforded the corresponding acids (14b) which were electrophilically
cyclized via the acid chlorides to 1H-2-seleno-(15) and 1H-2-telluro- (16)-3-benzopyran-
1-ones.
O
OR X
O
14a) R= C2H5, Y= Se or Te
14b) R= H, Y= Se or Te
15) X= Se
16) X= Te
Y
1.2 Physical Properties
Isocoumarins are usually crystalline solids having higher melting points as
compared to corresponding 3,4-dihydroisocoumarins. Some of the isocoumarins like 3-
propylisocoumarin, 3,5-dimethyl-6,7-dimethoxy-8-hydroxyisocoumarin, etc. are oils.
Isocoumarins have melting points ranging from 49-50 °C (trans-artemidin) to 350 °C
(alternariol) but most of them melt point in the range: 120-180 °C. A comparison of the
melting points of isocoumarins and 3,4-dihydroisocoumarins is presented in Table 1.1.
Table 1.1 Comparison of the melting points of isocoumarins and
dihydroisocoumarins
Substitution
Melting points (°C)
Isocoumarin Dihydroisoumarin
6-methoxy 98 68
8-hydroxy 123-124 56-57
6,7-dimethoxy 122-123 140-141
6,8-dimethoxy-3-methyl 154-156 125-128
6,8-dimethoxy-3-pentadecyl 101 51-53
1.3 Biosynthesis:
The biosynthesis of simple dihydroisocoumarins such as mellein (4) and
derivatives has been firmly established by studies with 13
C-labeled acetate13
and 1H,
13C
doubly labeled acetate14
.
Page 23
5
O
OH O
CH3
(4)
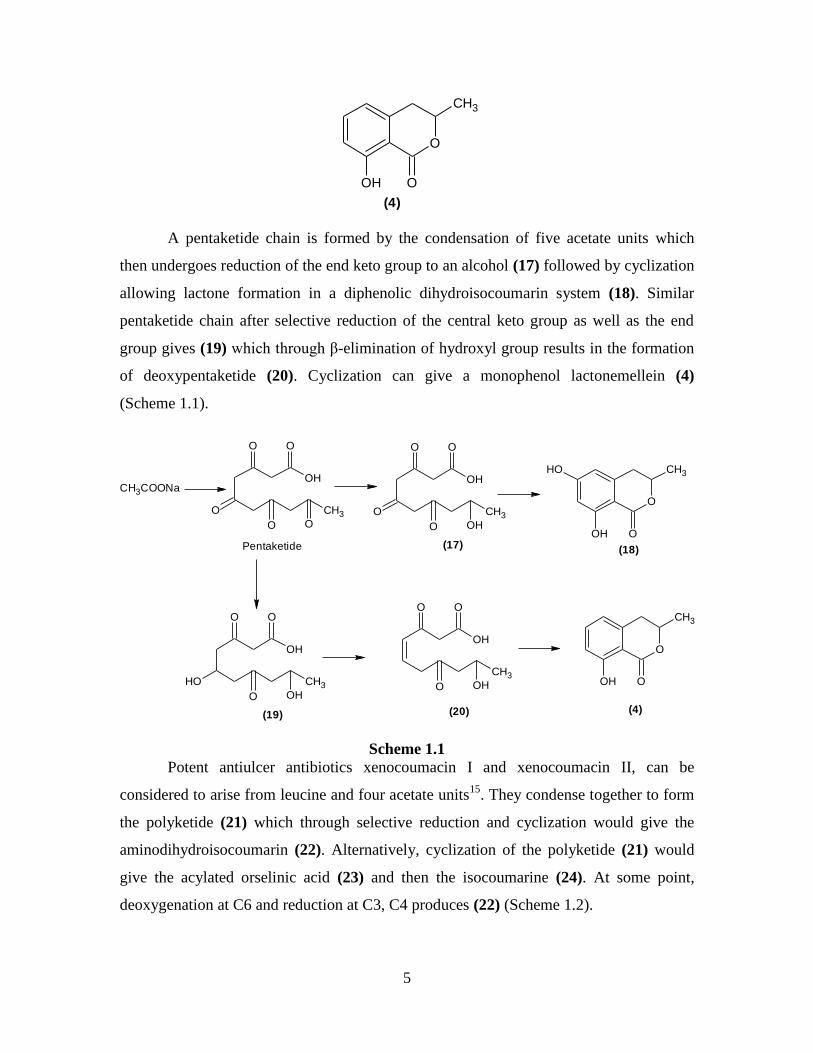
A pentaketide chain is formed by the condensation of five acetate units which
then undergoes reduction of the end keto group to an alcohol (17) followed by cyclization
allowing lactone formation in a diphenolic dihydroisocoumarin system (18). Similar
pentaketide chain after selective reduction of the central keto group as well as the end
group gives (19) which through β-elimination of hydroxyl group results in the formation
of deoxypentaketide (20). Cyclization can give a monophenol lactonemellein (4)
(Scheme 1.1).
O
OH O
CH3
CH3COONaOH
OO
O CH3
O O
O
OH O
CH3OHOH
OO
O CH3
O OH
Pentaketide (17) (18)
OH
OO
OH CH3
O OH
(19)
OH
OO
CH3
O OH
(20) (4)
Scheme 1.1
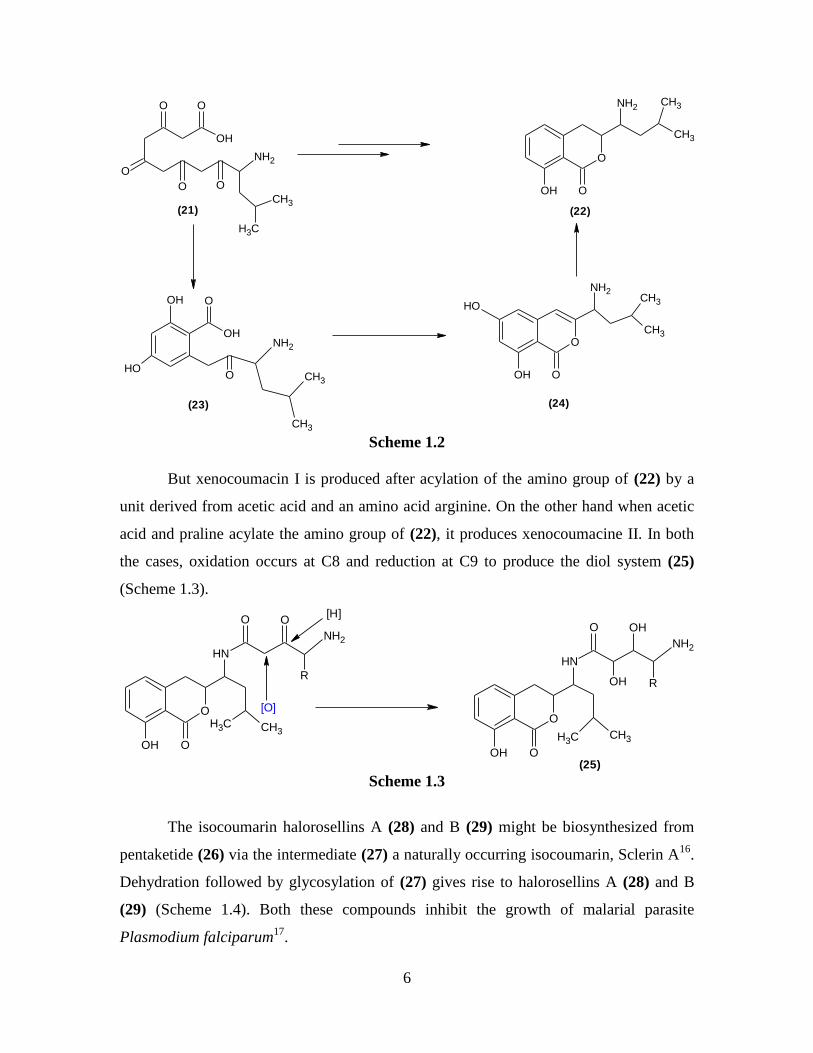
Potent antiulcer antibiotics xenocoumacin I and xenocoumacin II, can be
considered to arise from leucine and four acetate units15
. They condense together to form
the polyketide (21) which through selective reduction and cyclization would give the
aminodihydroisocoumarin (22). Alternatively, cyclization of the polyketide (21) would
give the acylated orselinic acid (23) and then the isocoumarine (24). At some point,
deoxygenation at C6 and reduction at C3, C4 produces (22) (Scheme 1.2).
Page 24
6
OH
OO
O
O O
NH2
CH3
CH3
(21)
OH
OH O
OH
CH3
CH3
NH2
O
(23)
O
OH O
CH3
CH3
NH2
OH
(24)
O
OH O
CH3
CH3NH2
(22)
Scheme 1.2
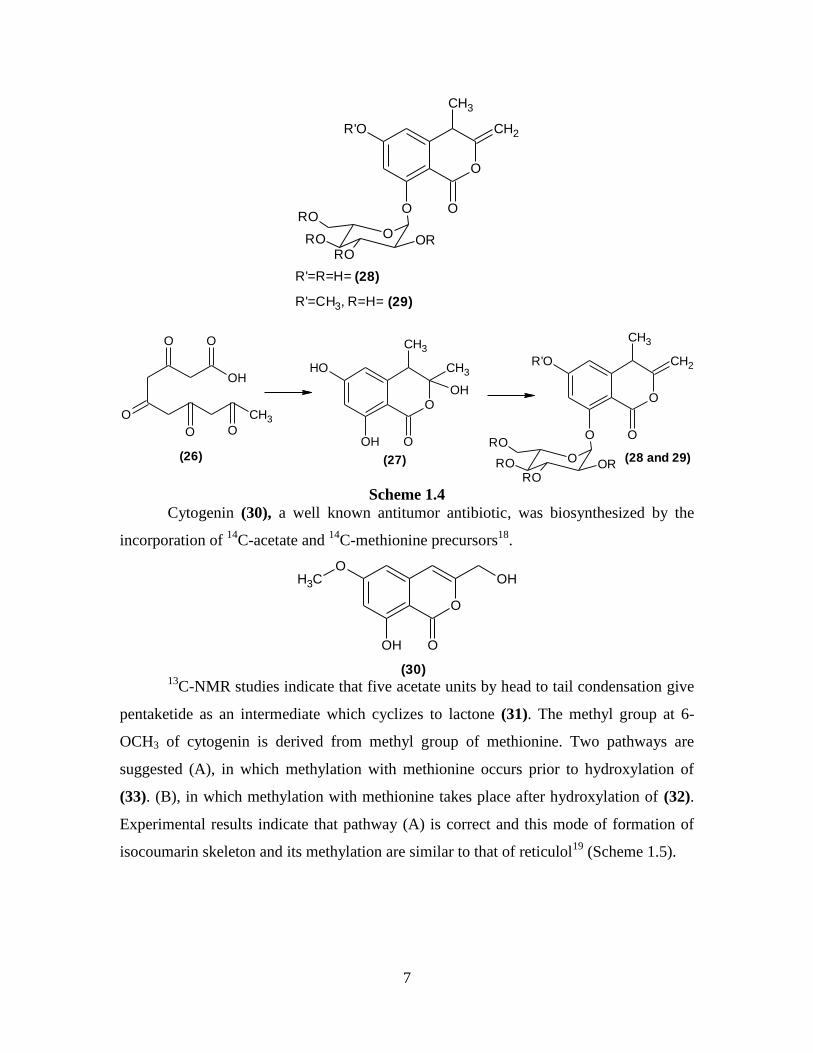
But xenocoumacin I is produced after acylation of the amino group of (22) by a
unit derived from acetic acid and an amino acid arginine. On the other hand when acetic
acid and praline acylate the amino group of (22), it produces xenocoumacine II. In both
the cases, oxidation occurs at C8 and reduction at C9 to produce the diol system (25)
(Scheme 1.3).
O
OH O
NH
CH3CH3
O O
NH2
R
[H]
[O]O
OH O
NH
CH3CH3
O OH
NH2
ROH
(25) Scheme 1.3
The isocoumarin halorosellins A (28) and B (29) might be biosynthesized from
pentaketide (26) via the intermediate (27) a naturally occurring isocoumarin, Sclerin A16
.
Dehydration followed by glycosylation of (27) gives rise to halorosellins A (28) and B
(29) (Scheme 1.4). Both these compounds inhibit the growth of malarial parasite
Plasmodium falciparum17
.
Page 25
7
O
O
O O
CH2R'O
CH3
RO
RORO
OR
R'=R=H= (28)
R'=CH3, R=H= (29)
OH
OO
O CH3
O O
O
OH O
CH3OH
CH3
OH
(26) (27) (28 and 29)O
O
O O
CH2R'O
CH3
RO
RORO
OR
Scheme 1.4
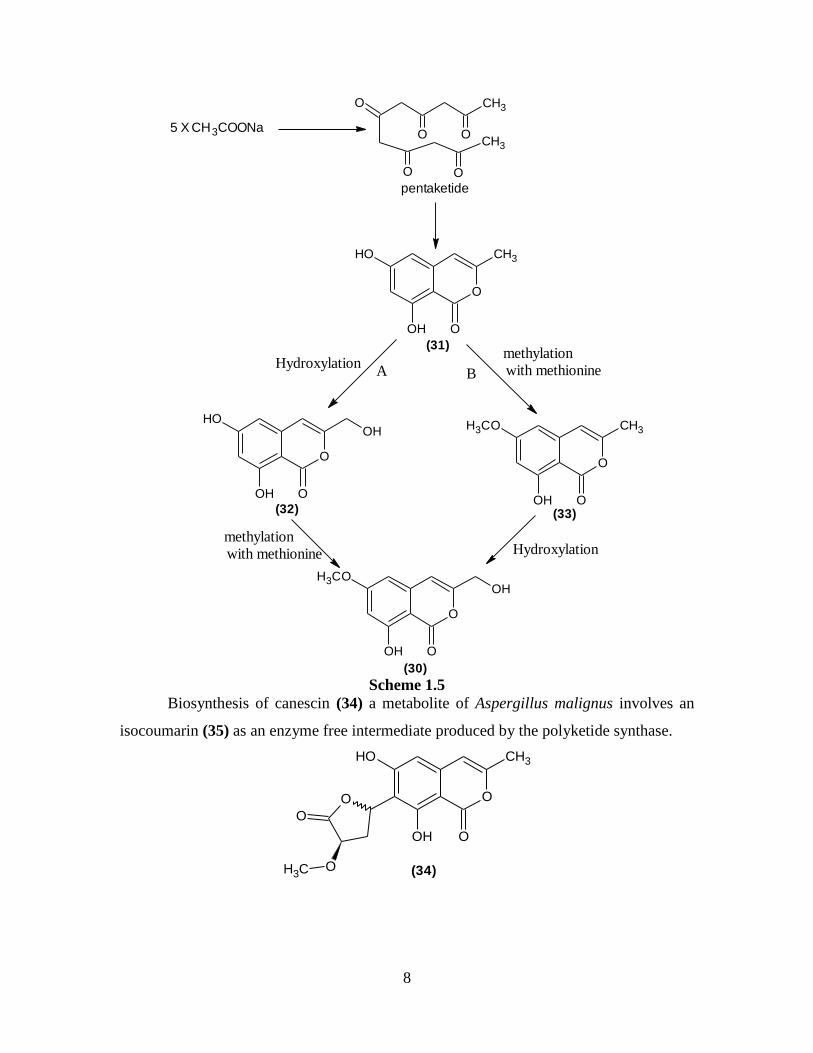
Cytogenin (30), a well known antitumor antibiotic, was biosynthesized by the
incorporation of 14
C-acetate and 14
C-methionine precursors18
.
O
OH O
OCH3 OH
(30) 13
C-NMR studies indicate that five acetate units by head to tail condensation give
pentaketide as an intermediate which cyclizes to lactone (31). The methyl group at 6-
OCH3 of cytogenin is derived from methyl group of methionine. Two pathways are
suggested (A), in which methylation with methionine occurs prior to hydroxylation of
(33). (B), in which methylation with methionine takes place after hydroxylation of (32).
Experimental results indicate that pathway (A) is correct and this mode of formation of
isocoumarin skeleton and its methylation are similar to that of reticulol19
(Scheme 1.5).
Page 26
8
CH3
OOCH3
O O
O
O
OOH
OH CH3
(31)
pentaketide
5 X CH3COONa
O
OOH
OHOH
O
OOH
H3CO CH3
O
OOH
H3COOH
methylation
with methionine Hydroxylation
methylation
with methionineHydroxylation
A B
(32) (33)
(30) Scheme 1.5
Biosynthesis of canescin (34) a metabolite of Aspergillus malignus involves an
isocoumarin (35) as an enzyme free intermediate produced by the polyketide synthase.
O
O
CH3
OH
OH
O
O
OCH3 (34)
Page 27
9
Oxidation of one of the methyl group of (35) gives the aldehyde (36) which is the
later intermediate of this biosynthetic pathway. The aldehyde (36) then undergoes an
aldol condensation with oxaloacetate to give (37). Two pathways A and B were
suggested in the conversion of (37) to (40) by decarboxylation and then lactonization.
A degree of uncertainty is created by the existence of a possible cross linking
between potential intermediate (38) and (39). The alcohol (41) produced as a result of
reduction of keto group of (40). The alcohol then undergoes O-methylatation to give the
canescin (34) (Scheme 1.6). The alcohol (41) has been isolated from the culture medium
which suggests that methylation is a late step in this biosynthetic pathway20
.
O
O
CH3
OH
OH
O
O
OCH3(34)
CH3
OOSEnz
CH3
O O
O
O
OOH
OH CH3
CH3O
OOH
OH CH3
O
H
O
OOH
OH CH3
OH
O
O
OH
O
OH
O
OOH
OH CH3
O
O
OH
O
OOH
OH CH3
OH
O
O
OH
A
B
O
O
CH3
OH
OH
O
O
O(40) (39)
(37)(38)
(36)(35)
O
O
CH3
OH
OH
O
O
OH (41)
polyketide synthase
Scheme 1.6
Page 28
10
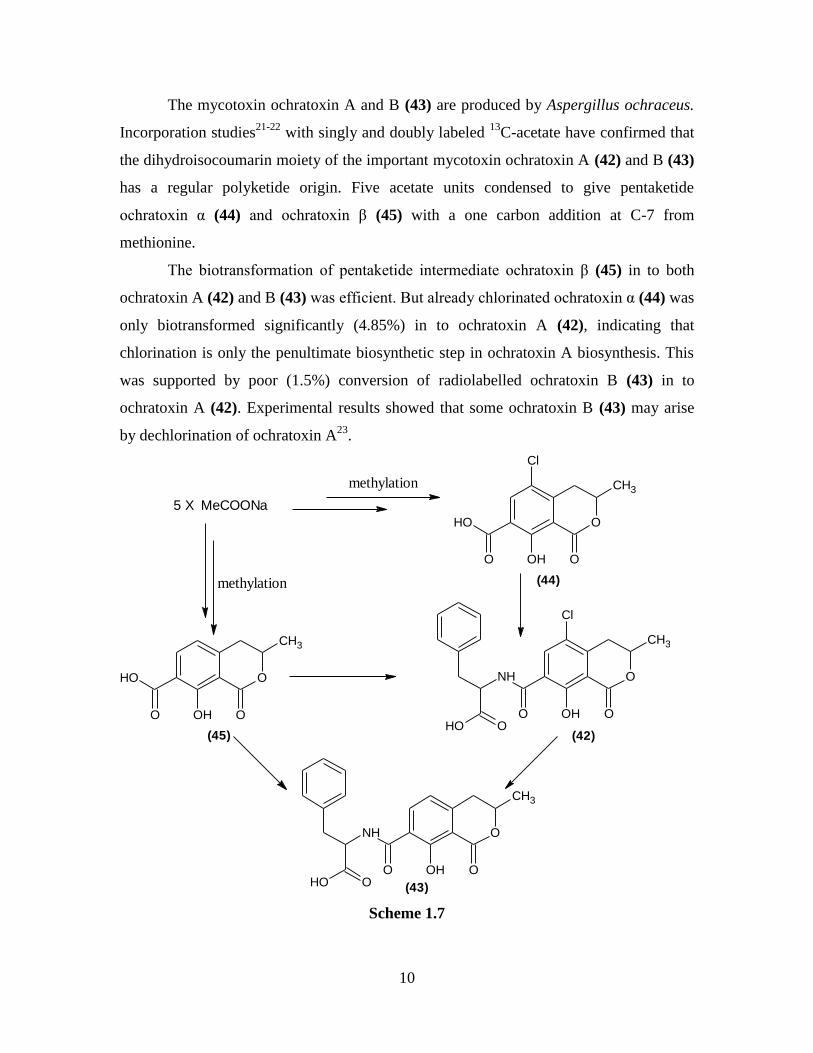
The mycotoxin ochratoxin A and B (43) are produced by Aspergillus ochraceus.
Incorporation studies21-22
with singly and doubly labeled 13
C-acetate have confirmed that
the dihydroisocoumarin moiety of the important mycotoxin ochratoxin A (42) and B (43)
has a regular polyketide origin. Five acetate units condensed to give pentaketide
ochratoxin α (44) and ochratoxin β (45) with a one carbon addition at C-7 from
methionine.
The biotransformation of pentaketide intermediate ochratoxin β (45) in to both
ochratoxin A (42) and B (43) was efficient. But already chlorinated ochratoxin α (44) was
only biotransformed significantly (4.85%) in to ochratoxin A (42), indicating that
chlorination is only the penultimate biosynthetic step in ochratoxin A biosynthesis. This
was supported by poor (1.5%) conversion of radiolabelled ochratoxin B (43) in to
ochratoxin A (42). Experimental results showed that some ochratoxin B (43) may arise
by dechlorination of ochratoxin A23
.
O
OOHO
NH
CH3
Cl
OOH(42)
O
OOHO
NH
CH3
OOH (43)
5 X MeCOONa
methylation
O
OOHO
OH
CH3
Cl
(44)methylation
O
OOHO
OH
CH3
(45)
Scheme 1.7
Page 29
11
1.4 Pharmacological Applications
Isocoumarins and 3,4-Dihydroisocoumarins are the secondary metabolites of
fungi, bacteria, plants and are insect venoms and pheromones. A huge number of them
have been isolated from fungi, lichens and bacteria. Some higher plants, insect and
marine organisms are also the rich source of these secondary metabolites. They exhibit a
broad range of pharmacological activities including antiallergic, antimicrobial,
immunomodulatory, antifungal, antiinflammatory, cytotoxic, and antiangiogenic24-30
.
The insecticides which selectively act on the insect GABA receptor are toxic to
insect but not mammals31
. A new 3,4-dihydroisocoumarin derivative (44) isolated from a
fungal culture extract (which was screened for its ability to inhibit the specific binding of
the noncompetitive antagonist [3H]EBOB to housefly head membrane) of Neosartorya
quadricincta. Compound (44) at 2.2μM inhibited [3H]EBOB binding by 65%. This novel
GABA receptor ligand might prove to be lead compound for the identification of new
insecticides acting at the insect GABA receptor32
.
O
OOH
OOH
O
O
OH(44)
Two new isocoumarin derivatives, stoloniferol A (45) and B (46) were isolated
from the ethyl acetate extract of the sea squirt-derived fungus, penicillium stoloniferum
and a halophilic fungus, penicillium notatum. Their cytotoxicities were evaluated against
the P388, BEL-7402, A-549 and HL-60 cell lines using the MTT method33
.
(46)
O
OOH
OH
(45)
O
OOH
OHOMe
A novel isocoumarin (47) was obtained from a marine fungus Alternaria tenuis
possesses an unusual 7-membered ring in the side chain. This compound exhibited
cytotoxicity against human malignant A375-S2 and human cervicial cancer Hela cells34
.
Page 30
12
It shows structural similarities to amicoumacins and the xenocoumacins which displayed
antibacterial, antitumor and potent antiulcer activities35-37
.
O
OH O
NH
O OH
OH
NH
O
OH
O(47)
An isocoumarin antibiotic (48) was isolated from the culture broth of Bacillus sp.
The strain was isolated from a soil sample collected at Iriomoto Island. Compound (48) is
a basic substituted isocoumarin, active against Gram-positive and Gram-negative
bacteria. It also showed a strong cytotoxic activity against the lymphoid leukemia cell
lines L1210 and P388. The antitumor activity was determined in mice against P388 cells.
It exhibited weak antitumor activity in vivo38
.
O
OOH
NH
O
OH
OH
NH2
(48)
Endophytic fungi, now recognized as potential producers of novel secondary
metabolites, can be used as possible biocontrol agents and drugs39
.
Two new
isocoumarins avicennin A (49) and B (50) with two derivatives (51) and (52) were
isolated from the mangrove endophytic fungus from the South China Sea40
.
O
O
Cl
OH
O
OH
(49)
O
O
O
OH
O
(50)
O
O
O
OH
OH
(51)
O
O
O
OH
(52)
Page 31
13
7-Hydroxyartemidin (53) isolated from the ethanol/water (50:50, V/V) extract of
Artemisia drucunculus L. leaves. This table vegetable has long been used in folk
medicine as a natural food cure for cleaning and diluting of blood and treatment of
dizziness and headache41
.
O
O
OH
(53)
Dihydroisocoumarin (54) isolated from aerial parts of a small shrubs Xyris
pterygoblephara showed aromatase inhibitory activity42
. Aromatase enzyme is a well-
established target for the chemoprevention of breast cancer.
O
O
O
O
O
(54)
A number of natural products with pharmacological activity required structural
modification to reduce their toxicity. Three natural isocoumarins paepalantine (55),
paepalantine 9-O-β-D-glucopyranoside (56), paepalantine9-O-β-D-allopyranosyl(1→6)
glucopyranoside (57) and two semisynthetic paeoalantine 9,10 acylated (58) and
paepalantine 9-OH-10-Methylated derivative (59) obtained from the capitula of
paepalantus bromelioides. The compound (55) has antimicrobial activity as well as
significant in vitro cytotoxic effects in the McCoy cell line. It was observed that the side
effects are reduced by substitution of the 9 and/or 10-OH group43
.
O
O
O
OR1 OR2
O
(55) = R1 = R2 = H (56) = R1 = glc, R2 = H
(57) = R1 = -glc 6 all, R2 = H (58) = R1 = R2 = Ac
(59) = R1 = H, R2 = Me
Page 32
14
Five isocoumarin derivatives, paraphaeosphaerins A-C (60-62) and
chaetochiversin A (63) and chaetochiversin B (64) have been isolated from solid agar
cultures of two fungal strains Paraphaeosphaeria quadriseptata and Chaetomium
chiversii living in association with the Sonoran desert plants, Opuntia leptocaulis, and
Ephedra fasciculate. These compounds are biogenetically related to monocillin I and
radicicol44
.
(60) (61)
O
O
OH
OH
OH
HOH H
O
O
OH
OHO
HHOHH
(63)
(64)
O
O
OH
OH
OH
HOH H
Cl
(62)
O
O
OH
OH
H OHO
H HO
O
OH
OH
Cl
O
HHOHH
Acrosin is a serine-dependent proteolytic enzyme which is responsible in the
dispersal of acrosomal matrix and also helps the sperm in the fertilization of oocytes45
. In
human it has also been studied that high acrosin activity of semen is associated with
improved fertility46-48
. Biotinylated isocoumarin suicide inhibitors were found to be
useful in the determination of activable proacrosin/acrosin levels in cryopreserved bull
semen49
.
An isocoumarin derivative Cytogenin (65) isolated from Streptoverticillium
eurocidicum is a well known antibiotic. It shows antitumor activity50
against Ehrlich
carcinoma at 6.3 to 100mg/Kg/day. It has also been demonstrated that cytogenin is
considerably effective as an immunological regulator51
.
Page 33
15
O
O
OH
OH
O
(65)
Ascomycetes and basidiomycetes are the rich source of chlorine containing
secondary metabolites; most of them are pharmacologically active52
. Lachnum
papyraceum produce mycorrhizins and lachnumon type antibiotics. Production of these
antibiotics was strongly inhibited when the culture media of Lachnum papyraceum
treated with CaBr2 and six isocoumarins derivatives (66-71) having nematicidal and
antimicrobial activity were isolated53
.
O
OOH
R1O
R2 R3
R1 = R2 = R3 = H = (66) R1 = R2 = H, R3 = Cl = (67)
R1 = R2 = H, R3 = Br = (68) R1 = CH3, R2 = R3 = H = (69)
R1 = CH3, R2 = H, R3 = Cl = (70) R1 = H, R2 = OH, R 3 = Cl = (71)
Three novel dihydroisocoumarin derivatives (72-74) with alkyl substitution at
position 7 have been isolated from an endophytic fungus, Geotrichum sp., collected from
Crassocephalum crepidioides. All of these compounds possess antimalarial, antifungal
and antituberculous activity54
.
O
OOH
OH
(72)
O
OOH
OH
(73)
O
OOH
OH
(74)
2-(8-hydroxy-6-methoxy-1-oxo-1H-2-benzopyran-3-yl)propionic acid (NM-3)
(75) is a novel synthetic analogue of cytogenin, an isocoumarin isolated from culture
Page 34
16
filtrate of Streptoverticillium eurocidium55-56
. NM-3 potently inhibits endothelial cell
proliferation, migration, sprouting, tube formation in vitro, and tumor growth in vivo57
. A
phase I clinical study of NM-3 in patients with cancer has demonstrated that it is a highly
orally bioavailable and well tolerated drug in humans58
.
O
OOH
O O
OH
(75)
One of the mechanisms involved in the progression of diabetic nephropathy, the
most common cause of end stage renal failure, is angiogenic phenomenon associated with
the increase of angiogenic factors such as vascular endothelial growth factor (VEGF)-A
and angiopoietin (Ang)-2, an antagonist of Ang-1. NM-3 significantly suppressed the
increase of VEGF induced by high glucose in cultured podocytes and also suppressed the
increase of VEGF and TGF-β induced by high glucose in cultured mesangial cells. This
reflects the potential use of NM-3 as a novel therapeutic agent for renal alterations in type
2 diabetes59
.
NM-3 also induces lethality of human carcinoma cells by both apoptotic and
nonapoptotic mechanism and potentiates the effects of cytotoxic chemotherapeutic
agents. NM-3 potentiates dexamethasone-induced killing of both dexamethasone-
sensitive multiple myeloma (MM1.S) and dexamethasone-resistant RPMI8226 and U266
multiple myeloma cells60
.
Urokinase-type plasminogen activator (uPA) is an attractive target for the
development of new compounds for its inhibition because uPA plays a major role in
extracellular proteolytic events associated with tumor cell growth, migration and
angiogenesis. uPA catalyzed the hydrolysis of extracellular plasminogen to plasmin. The
increased production of plasmin leads to the degradation of extracellular matrix, thereby
assisting the directional migration of cancer cells61-62
. uPA in complex with its receptor
uPAR also affects other boplogical processes including signaling pathways that
influence cell proliferation63
. Potent uncharged inhibitors of uPA can be developed based
upon isocoumarin scaffold. Bromine in the three position and an aromatic group in the
Page 35
17
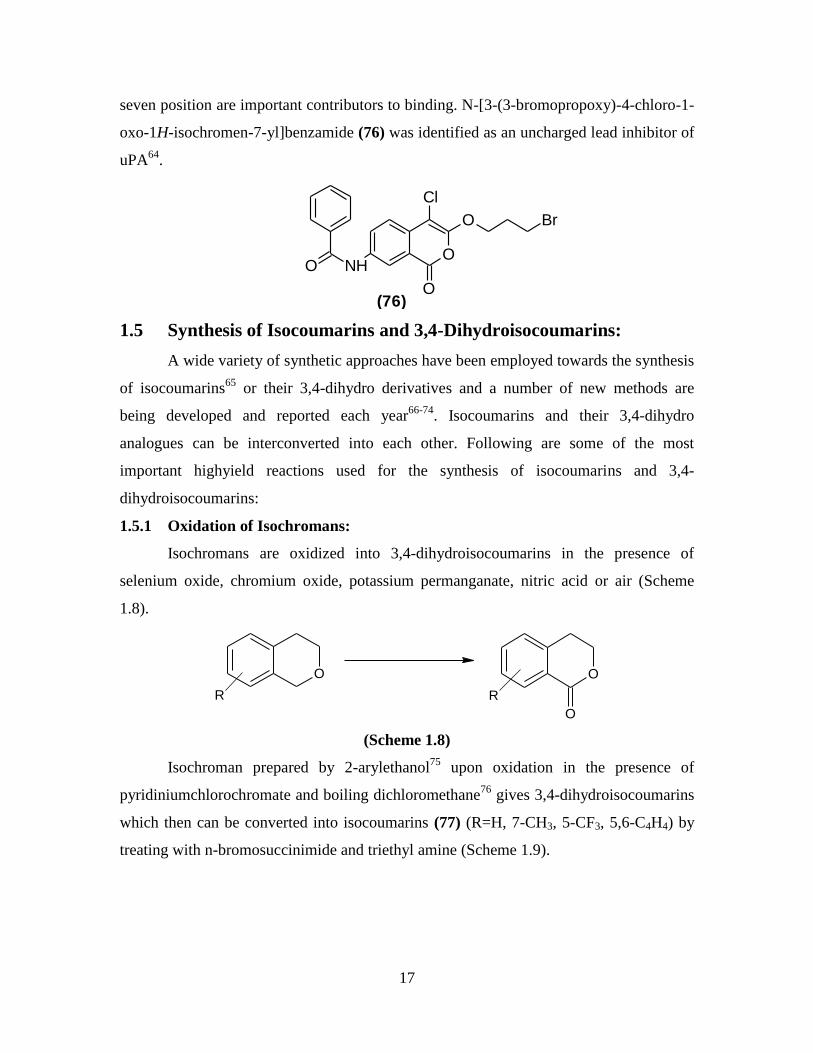
seven position are important contributors to binding. N-[3-(3-bromopropoxy)-4-chloro-1-
oxo-1H-isochromen-7-yl]benzamide (76) was identified as an uncharged lead inhibitor of
uPA64
.
O
O
O Br
Cl
NHO
(76)
1.5 Synthesis of Isocoumarins and 3,4-Dihydroisocoumarins:
A wide variety of synthetic approaches have been employed towards the synthesis
of isocoumarins65
or their 3,4-dihydro derivatives and a number of new methods are
being developed and reported each year66-74
. Isocoumarins and their 3,4-dihydro
analogues can be interconverted into each other. Following are some of the most
important highyield reactions used for the synthesis of isocoumarins and 3,4-
dihydroisocoumarins:
1.5.1 Oxidation of Isochromans:
Isochromans are oxidized into 3,4-dihydroisocoumarins in the presence of
selenium oxide, chromium oxide, potassium permanganate, nitric acid or air (Scheme
1.8).
O
R
O
O
R
(Scheme 1.8)
Isochroman prepared by 2-arylethanol75
upon oxidation in the presence of
pyridiniumchlorochromate and boiling dichloromethane76
gives 3,4-dihydroisocoumarins
which then can be converted into isocoumarins (77) (R=H, 7-CH3, 5-CF3, 5,6-C4H4) by
treating with n-bromosuccinimide and triethyl amine (Scheme 1.9).
Page 36
18
R'O
(R' = H, MEM)
RO
R
O
O
RO
O
R
a) TiCl4 b) PCC c) NBS, (C 2H5)3N
(77)
(Scheme 1.9)
1.5.2 Oxidation of Indenes, Indanones and Indones:
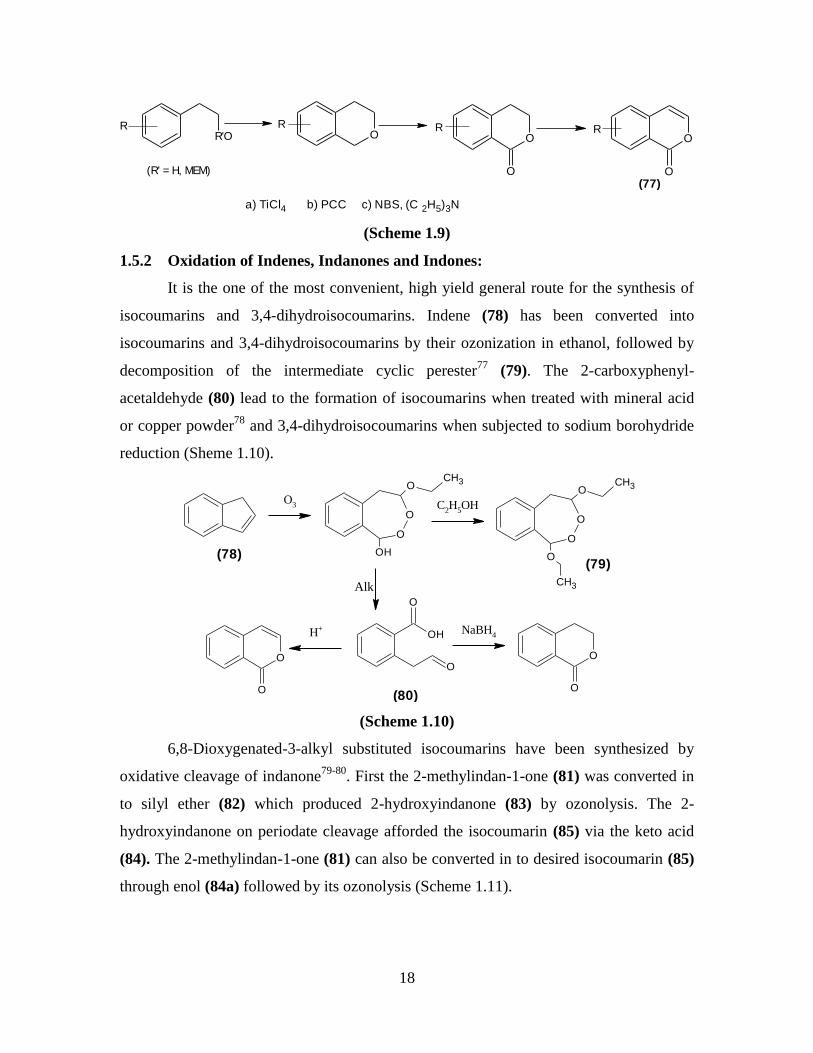
It is the one of the most convenient, high yield general route for the synthesis of
isocoumarins and 3,4-dihydroisocoumarins. Indene (78) has been converted into
isocoumarins and 3,4-dihydroisocoumarins by their ozonization in ethanol, followed by
decomposition of the intermediate cyclic perester77
(79). The 2-carboxyphenyl-
acetaldehyde (80) lead to the formation of isocoumarins when treated with mineral acid
or copper powder78
and 3,4-dihydroisocoumarins when subjected to sodium borohydride
reduction (Sheme 1.10).
O
O
OH
OCH3
O
O
O
OCH3
CH3Alk
O
O
OH
O
O
O
O
C2H
5OH
NaBH4
O3
H+
(78)(79)
(80)
(Scheme 1.10)
6,8-Dioxygenated-3-alkyl substituted isocoumarins have been synthesized by
oxidative cleavage of indanone79-80
. First the 2-methylindan-1-one (81) was converted in
to silyl ether (82) which produced 2-hydroxyindanone (83) by ozonolysis. The 2-
hydroxyindanone on periodate cleavage afforded the isocoumarin (85) via the keto acid
(84). The 2-methylindan-1-one (81) can also be converted in to desired isocoumarin (85)
through enol (84a) followed by its ozonolysis (Scheme 1.11).
Page 37
19
OO
CH3
R
CH3
(81)
OSi(CH 3)3OCH3
R
CH3
(82)
OSi(CH 3)3OCH3
R
CH3
O
OCH3
R
CH3
OH
O
(85)
OCOCF 3OCH3
R
CH3
(48a)
OCH3
R
O
CH3
O
OH
(84)OCH3
R
O
CH3
O
(83)
(Scheme 1.11)
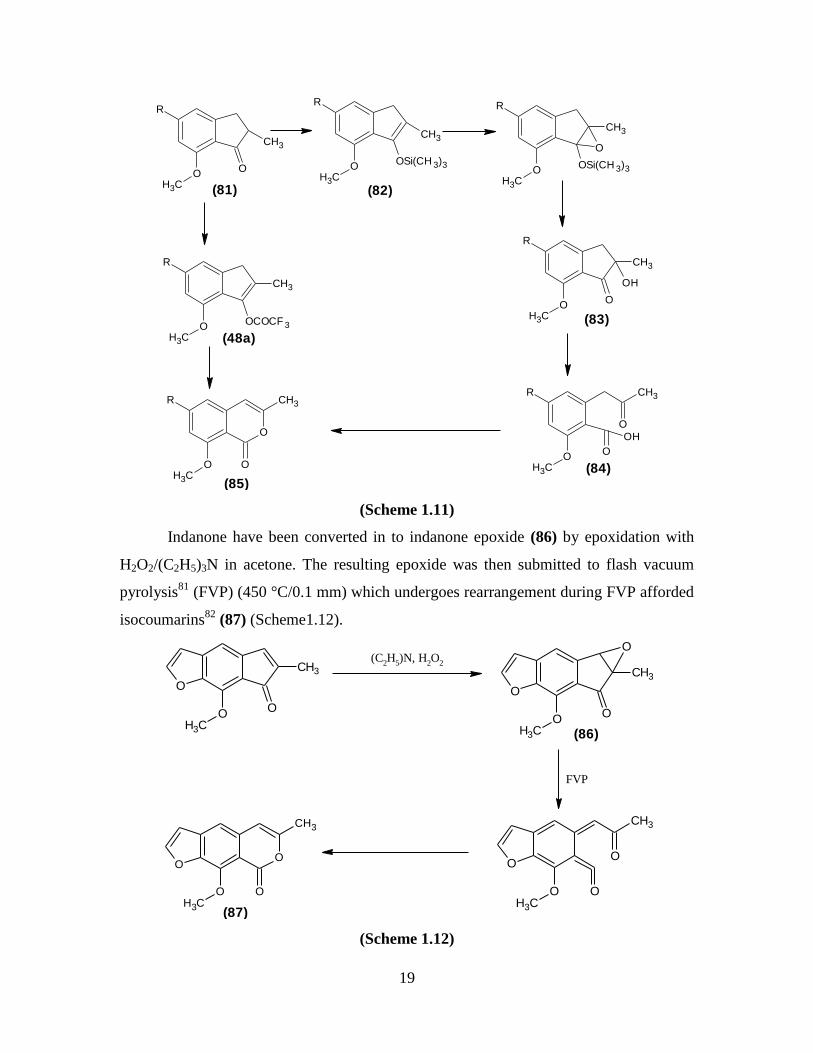
Indanone have been converted in to indanone epoxide (86) by epoxidation with
H2O2/(C2H5)3N in acetone. The resulting epoxide was then submitted to flash vacuum
pyrolysis81
(FVP) (450 °C/0.1 mm) which undergoes rearrangement during FVP afforded
isocoumarins82
(87) (Scheme1.12).
O
O
CH3
OCH3
(C2H
5)N, H
2O
2
O
O
CH3
OCH3
O
FVP
(86)
O
CH3
O
OOCH3
O
OO
CH3
CH3
O
(87)
(Scheme 1.12)
Page 38
20
1.5.3 Synthesis involving metals:
A number of methods have been reported in literature which involve synthesis of
isocoumarins and 3,4-dihydroisocoumarins by metallation (Lithiation, Silylation and
Thallation) at specific positions.
a) Lithiation:
This method was first discovered by Hauser83
and then extensively studied by
Narasimhan and Bhide84
. Benzoic acid derivatives are important precursors of
isocoumarins. Among the methods available for introducing a β-functionalized carbon
substituent ortho to the carboxyl group, those involving ortho-metallation of the benzene
ring have enjoyed a great popularity.
This approach has been thoroughly reviewed85-87
. Summarizing the general
concepts, carboxylic acid derivatives suitable for promoting ortho lithiation88-89
are
tertiary amides (4,4-dimethyl)oxazolin-2-yl group and secondary amides. Lithiated
tertiary amides are readily and generally ortho-lithiated using n-butyllithium and
tetramethylethylenediamine, but their reaction with alkylating agents other than methyl
iodide gives low yields because of a poor nucleophilicity.
Allylation of lithiated tertiary benzamides has, however, been accomplished in
high yields by previous trans-metallation to a magnesium or (better) to a copper
derivative; the allyl group thus introduced has been converted to the β-hydroxyalkyl
group required to complete the lactone ring in the conditions of the acid hydrolysis of the
benzamide, leading to racemic 3,4-dihydroisocoumarins directly, apparently without the
possibility of isolating the intermediate allylbenzoic acids.
Alternatively, asymmetric hydroxylation of the double bond followed by
treatment with acids has been used to obtain 3,4-dihydroisocoumarins with a high degree
of enantiomeric purity, as demonstrated by the enantioselective synthesis of the
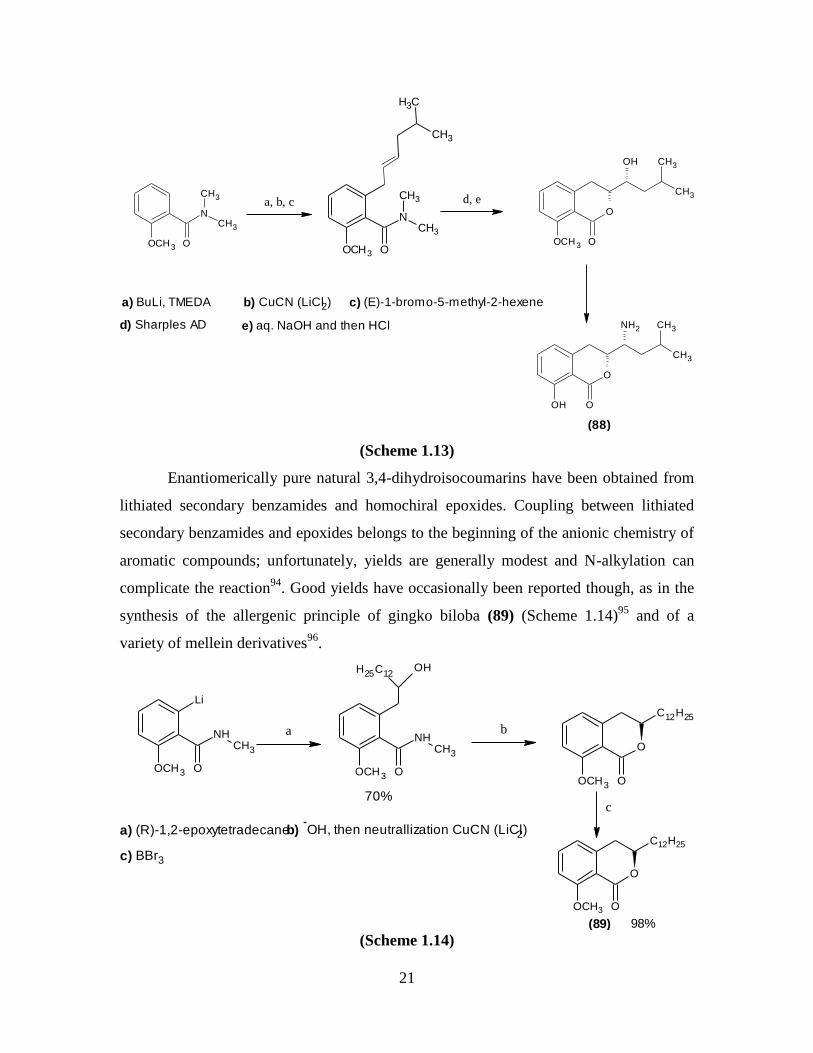
isocoumarin portion of AI77B (88) (Scheme 1.13)90-93
.
Page 39
21
O
NCH3
CH3
OCH3 O
NCH3
CH3
OCH3
CH3
CH3
O
O
OCH3
CH3
CH3OH
O
O
OH
CH3
CH3NH2
(88)
a) BuLi, TMEDA b) CuCN (LiCl2) c) (E)-1-bromo-5-methyl-2-hexene
d) Sharples AD e) aq. NaOH and then HCl
a, b, c d, e
(Scheme 1.13)
Enantiomerically pure natural 3,4-dihydroisocoumarins have been obtained from
lithiated secondary benzamides and homochiral epoxides. Coupling between lithiated
secondary benzamides and epoxides belongs to the beginning of the anionic chemistry of
aromatic compounds; unfortunately, yields are generally modest and N-alkylation can
complicate the reaction94
. Good yields have occasionally been reported though, as in the
synthesis of the allergenic principle of gingko biloba (89) (Scheme 1.14)95
and of a
variety of mellein derivatives96
.
O
NHCH3
Li
OCH3 O
NHCH3
OCH3
OHH25C12
a b
O
O
OCH3
C12H25
c
O
O
OCH3
C12H25
(89)
70%
98%
a) (R)-1,2-epoxytetradecane
c) BBr3
b) -OH, then neutrallization CuCN (LiCl2)
(Scheme 1.14)
Page 40
22
This method can be used for the synthesis of 5-methoxy, 6-methoxy, 8-methoxy,
3-methyl-8-methoxy, 3-methyl-8-hydroxy-3,4-dihydroisocoumarins. Mellein, Kigelin,
Hydrangenol, Phyllodulcin, 3-methyl, 6-methyl, 3,6-dimethyl, 6-chloro, 8-chloro etc
have also been been prepared by this method.
b) Thallation-olefination of Arenes:
Isocoumarins and 3,4-dihydroisocoumarins were prepared in a single pot
reaction97
by reacting a benzoic acid with an electrophilic thallium salt in the presence of
an organic solvent to give O-thalliated benzoic acid followed by reaction with an organic
compound e.g. an alkene in the presence of PdCl2 (Scheme 1.15).
O
OH
TI
CH2=CH2
PdCl2
O
OR R
(Scheme 1.15)
c) Silylation:
Closely related to lithiation is the desilylation of 2-(trimethylsilylmethyl)-
benzamides, which generates carbanions suitable for additions to aldehydes98
. 2-
(Trimethylsilylmethyl)benzoyl chloride (90) also undergo desilylation and addition to
aldehydes to give dihydroisocoumarins (91) through a concerted mechanism involving
ortho-quinodimethanes rather than carbanions as reactive intermediates (Scheme 1.16)99
.
CH3
O
OH
OCH3
a
O
OH
OCH3
Si(CH3)3 b
O
Cl
OCH3
Si(CH3)3 c
O
O
OCH3
Ph
CH2
OOCH3
a) n-BuLi, (CH3)3SiCl b) SOCl2 c) CsF, ArCHO
(90)
(91)
(Scheme 1.16)
Page 41
23
To this class of reactive intermediates belongs the products of UV irradiation of
ortho-toluyl cyanides which add to aliphatic and aromatic acyl cyanides to give 3-cyano-
3-phenyl-8-methoxy-3,4-dihydroisocoumarins which are converted to isocoumarins by
treatment with strong bases (Scheme 1.17)100
.
CH3
OOCH3
CH3
OOCH3
CN
a, b cCH2
OHOCH3
CN
OOCH3
O
Ph
CN
a) (CH3)3SiCN b) PCC c) hv, PhCOCN
(Scheme 1.17)
d) Organo-mercury catalyzed synthesis:
A facile synthesis of 3-substituted arylisocoumarins involve the reaction of ester
(92) (R=H, Br, Cl, I, Ac)101
with mercuric acetate to give isocoumarin mercurials which
undergo substitution reactions to afford the isocoumarins (93) (R1=H, CH3, Cl, Br; R=H,
Br, Cl, I, Ac) (Scheme 1.18).
R
CO2Me
Hg(OAc)2
O
O
RR
1
(92) (93)
(Scheme 1.18)
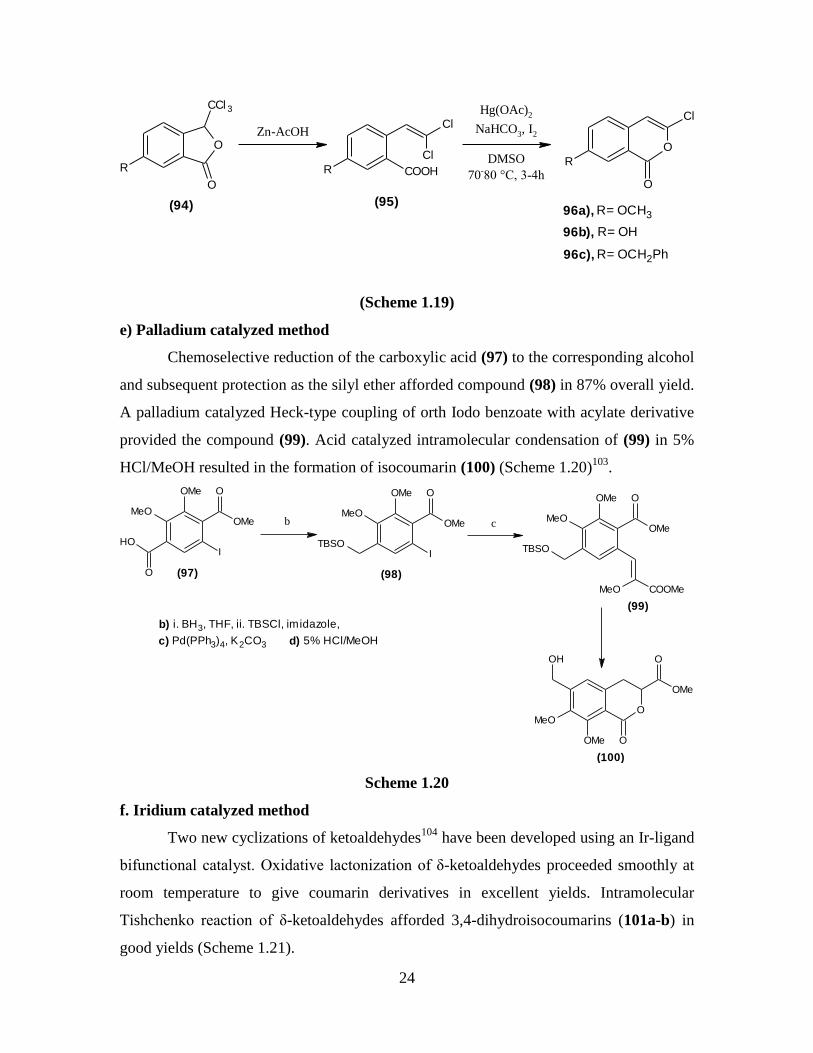
Sulphuric acid-catalyzed chloralhydrate condensation with different m-substituted
benzoic acids formed trichlorophthalides (94), from which Zn+AcOH reduction afforded
various dichloro derivatives (95). These derivatives on treatment with alkaline Hg(OAc)2
+ I2 furnished different substituted isocoumarins (96a-c) (Scheme 1.19)102
.
Page 42
24
O
O
R
CCl 3
Zn-AcOH
COOHR
Cl
Cl
Hg(OAc)2
NaHCO3, I2
DMSO
70-80 °C, 3-4hR
Cl
O
O
96a), R= OCH3
96b), R= OH
96c), R= OCH2Ph
(94) (95)
(Scheme 1.19)
e) Palladium catalyzed method
Chemoselective reduction of the carboxylic acid (97) to the corresponding alcohol
and subsequent protection as the silyl ether afforded compound (98) in 87% overall yield.
A palladium catalyzed Heck-type coupling of orth Iodo benzoate with acylate derivative
provided the compound (99). Acid catalyzed intramolecular condensation of (99) in 5%
HCl/MeOH resulted in the formation of isocoumarin (100) (Scheme 1.20)103
.
I
OMe
MeO
O
OH
O
OMe b
I
OMe
MeO
TBSO
O
OMe c
OMe
MeO
TBSO
O
OMe
COOMeMeO
MeOO
O
OH
OMe
O
OMe
(97) (98)
(99)
(100)
b) i. BH3, THF, ii. TBSCl, imidazole,
c) Pd(PPh3)4, K2CO3 d) 5% HCl/MeOH
Scheme 1.20
f. Iridium catalyzed method
Two new cyclizations of ketoaldehydes104
have been developed using an Ir-ligand
bifunctional catalyst. Oxidative lactonization of δ-ketoaldehydes proceeded smoothly at
room temperature to give coumarin derivatives in excellent yields. Intramolecular
Tishchenko reaction of δ-ketoaldehydes afforded 3,4-dihydroisocoumarins (101a-b) in
good yields (Scheme 1.21).
Page 43
25
O
R
O
R= CH3
R= Ph
O
R
O
OH
R
O+
O
R
O
O
R
O+
O
R
O
101a) R= CH3
101b) R= Ph
Ir Cat (5mol%)
t-BuOH, reflux
(5mol%), cooxidant
base, rt, 16h
NHIr
+
O
Ph
Ph
CH3
CH3CH3
CH3
CH3
Scheme 1.21
g. Rhodium-Catalyzed Oxidative Coupling of Benzoic Acids with Alkynes via
Regioselective C-H Bond Cleavage
The oxidative coupling of benzoic acids with internal alkynes effectively proceeds
in the presence of [Cp*RhCl2]2 and Cu (OAc)2 H2O as catalyst105
and oxidant
respectively to produce the corresponding isocoumarin derivatives. The copper salt can
be reduced to a catalytic quantity under air (Scheme 1.22).
O
OH
H
+ R RRh-Cat
Cu-salt O
R
R
O
Scheme 1.22
1.5.4 Aldol-type Condensation between Homophthalic Acids, Esters or
Anhydrides and Carbonyl Compounds
Isocoumarins and 3,4-dihydroisocoumarins are most commonly synthesized by
using this type of condensation. The most important methods of aldol type condensation
are discussed in four main groups.
Page 44
26
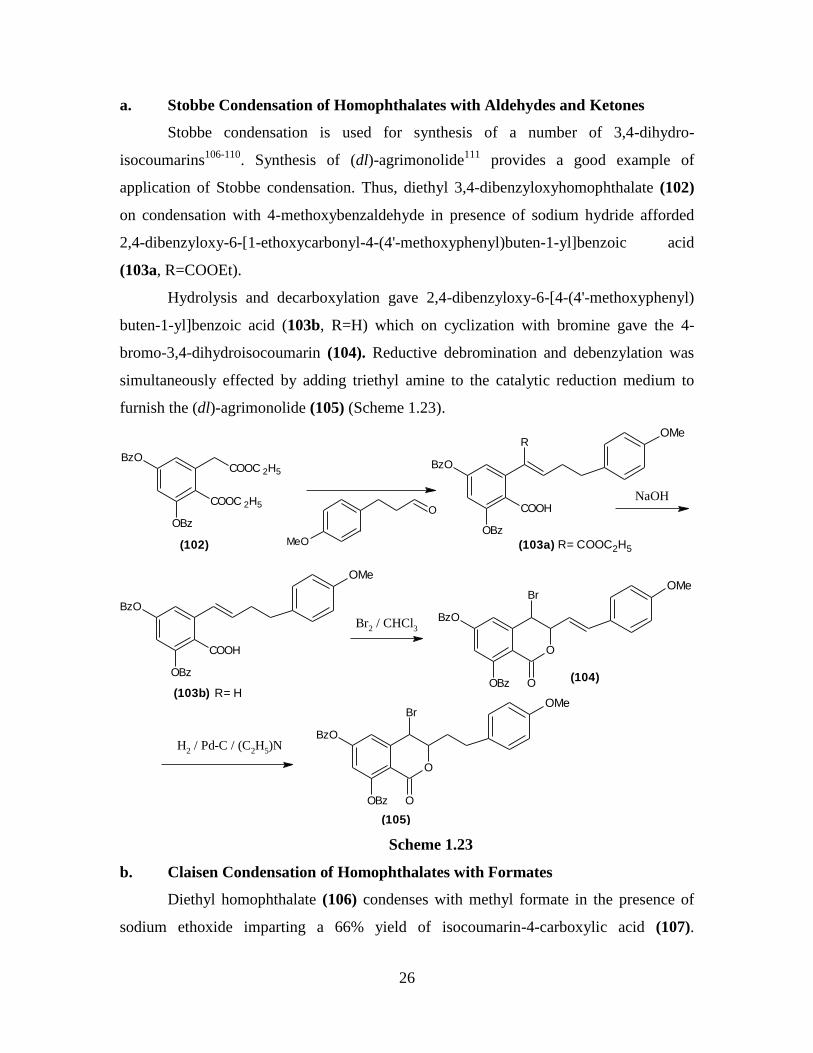
a. Stobbe Condensation of Homophthalates with Aldehydes and Ketones
Stobbe condensation is used for synthesis of a number of 3,4-dihydro-
isocoumarins106-110
. Synthesis of (dl)-agrimonolide111
provides a good example of
application of Stobbe condensation. Thus, diethyl 3,4-dibenzyloxyhomophthalate (102)
on condensation with 4-methoxybenzaldehyde in presence of sodium hydride afforded
2,4-dibenzyloxy-6-[1-ethoxycarbonyl-4-(4'-methoxyphenyl)buten-1-yl]benzoic acid
(103a, R=COOEt).
Hydrolysis and decarboxylation gave 2,4-dibenzyloxy-6-[4-(4'-methoxyphenyl)
buten-1-yl]benzoic acid (103b, R=H) which on cyclization with bromine gave the 4-
bromo-3,4-dihydroisocoumarin (104). Reductive debromination and debenzylation was
simultaneously effected by adding triethyl amine to the catalytic reduction medium to
furnish the (dl)-agrimonolide (105) (Scheme 1.23).
COOC 2H5
COOC 2H5
BzO
OBz
MeO
O COOH
BzO
OBz
ROMe
NaOH
COOH
BzO
OBz
OMe
Br2 / CHCl
3
BzO
OBz
OMe
O
O
Br
H2 / Pd-C / (C
2H
5)N
BzO
OBz
OMe
O
O
Br
(102) (103a)
(103b)
(104)
(105)
R= COOC2H5
R= H
Scheme 1.23
b. Claisen Condensation of Homophthalates with Formates
Diethyl homophthalate (106) condenses with methyl formate in the presence of
sodium ethoxide imparting a 66% yield of isocoumarin-4-carboxylic acid (107).
Page 45
27
Decarboxylation with phosphoric acid furnishes isocoumarin (108) (Scheme 1.24)112
.
COOC 2H5
COOC 2H5
(106)
O
O
COOH
(107)
O
O(108)
HCOOCH3
C2H
5ONa
H3PO
4
-CO2
Scheme 1.24
6,7-Dimethoxyisocoumarin and 5,7-dimethoxyisocoumarin were also prepared by
the above procedure. Ethyl 5,6,7-trimethoxyisocoumarin-4-carboxylate was prepared
from corresponding homophthalate and ethyl formate in the presence of potassium
ethoxide in good yield113
.
c. Claisen Condensations of Homophthalates with Oxalates
Metallic sodium in ether or better without a solvent effects ready condensation
between diethyl homophthalate (109) and diethyl oxalate giving a 67% yield of the
triester (110). This triester loses ethanol when heated yielding diethyl isocoumarin-3,4-
dicarboxylate (111). Under different hydrolysis conditions different products are formed.
Thus heating (111) at 68-72°C for 3hr. gives ethyl isocoumarin-3-(carboxylic
acid)-4-carboxylate (112) and prolonged heating yields isocoumarin-3-carboxylic acid
(113). Boiling hydrochloric acid or heating in a sealed tube at 180-190°C converts (111)
to isocoumarin-3-carboxylic acid in 84% yield114
. These results indicate that the ester at
position 3 in (111) is hydrolyzed first, but the acid at position 4 is more easily
decarboxylated (Scheme 1.25).
Page 46
28
COOC 2H5
COOC 2H5
(109)
O
O
COOC 2H5
COOC 2H5
(111)
NaO
OC2H5O
H5C2O
+COCOOC 2H5
COOC 2H5
O OC2H5
(110)
O
O
COOH
COOC 2H5
(112)
O
O
COOH
(113)
Scheme 1.25
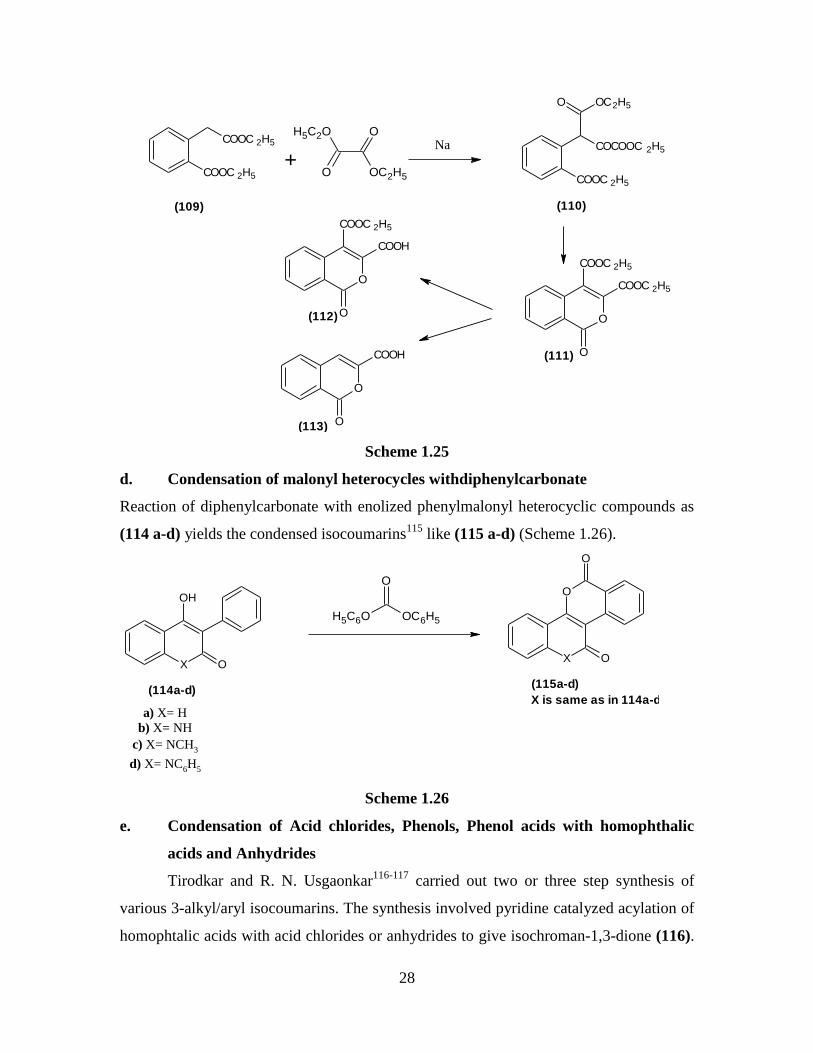
d. Condensation of malonyl heterocycles withdiphenylcarbonate
Reaction of diphenylcarbonate with enolized phenylmalonyl heterocyclic compounds as
(114 a-d) yields the condensed isocoumarins115
like (115 a-d) (Scheme 1.26).
X
OH
O
(114a-d)
O
OC6H5H5C6O
X
O
O
O
(115a-d)
a) X= H
b) X= NH
c) X= NCH3
d) X= NC6H
5
X is same as in 114a-d
Scheme 1.26
e. Condensation of Acid chlorides, Phenols, Phenol acids with homophthalic
acids and Anhydrides
Tirodkar and R. N. Usgaonkar116-117
carried out two or three step synthesis of
various 3-alkyl/aryl isocoumarins. The synthesis involved pyridine catalyzed acylation of
homophtalic acids with acid chlorides or anhydrides to give isochroman-1,3-dione (116).
Page 47
29
Treatment of (116) with conc. sulphuric acid at room temperature gave the 3-alkyl/aryl
isocoumarin-3-carboxylic acid whereas on treatment with 90% sulphuric acid at 90°C
directly gave the isocoumarins (Scheme 1.27).
COOH
COOH
R
(R'CO)2O / Py
R'
O
O
COOH
R
r.t
O
O
O
COR'
R
R'
O
O
R
Conc. H2SO4
90% H2SO
4
90 °C
(116)
Scheme 1.27
S. Nakajima et. al. synthesized various 3-arylisocoumarins (118, Ar =Ph, p-
anisyl, p-(OH)phenyl etc.) and later on 3-alkylisocoumarins in high yields (80%) by
heating directly the homophthalic acids (117, R, R1, R
2=H, OH, OMe, Cl) with aryl or
acyl chlorides at 190°C. These isocoumarins were converted into corresponding 3,4-
dihydroisocoumarins (Scheme 1.28).
COOH
COOH
R
R1
R2
Ar/R
O
R
R1
R2
O
Ar / RCOCl
190 °C
(117) (118)
Scheme 1.28
A. Rose118
and later on H. Yoshikawa119
prepared a large number of 3-
(hydroxyphenyl)isocoumarins by condensation of various phenols with substituted
homophthalic acids in moderate yields in presence of polyphosphoric acid or the
Page 48
30
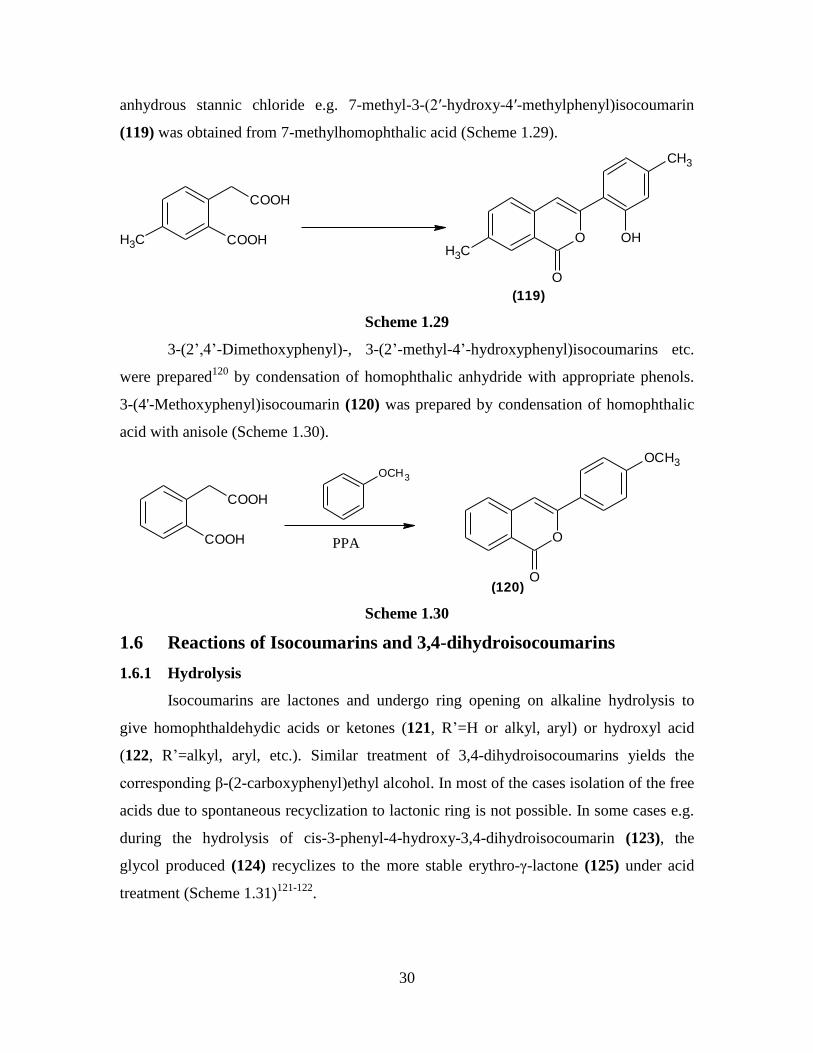
anhydrous stannic chloride e.g. 7-methyl-3-(2′-hydroxy-4′-methylphenyl)isocoumarin
(119) was obtained from 7-methylhomophthalic acid (Scheme 1.29).
COOH
COOH
CH3O
O
CH3
CH3
OH
(119)
Scheme 1.29
3-(2’,4’-Dimethoxyphenyl)-, 3-(2’-methyl-4’-hydroxyphenyl)isocoumarins etc.
were prepared120
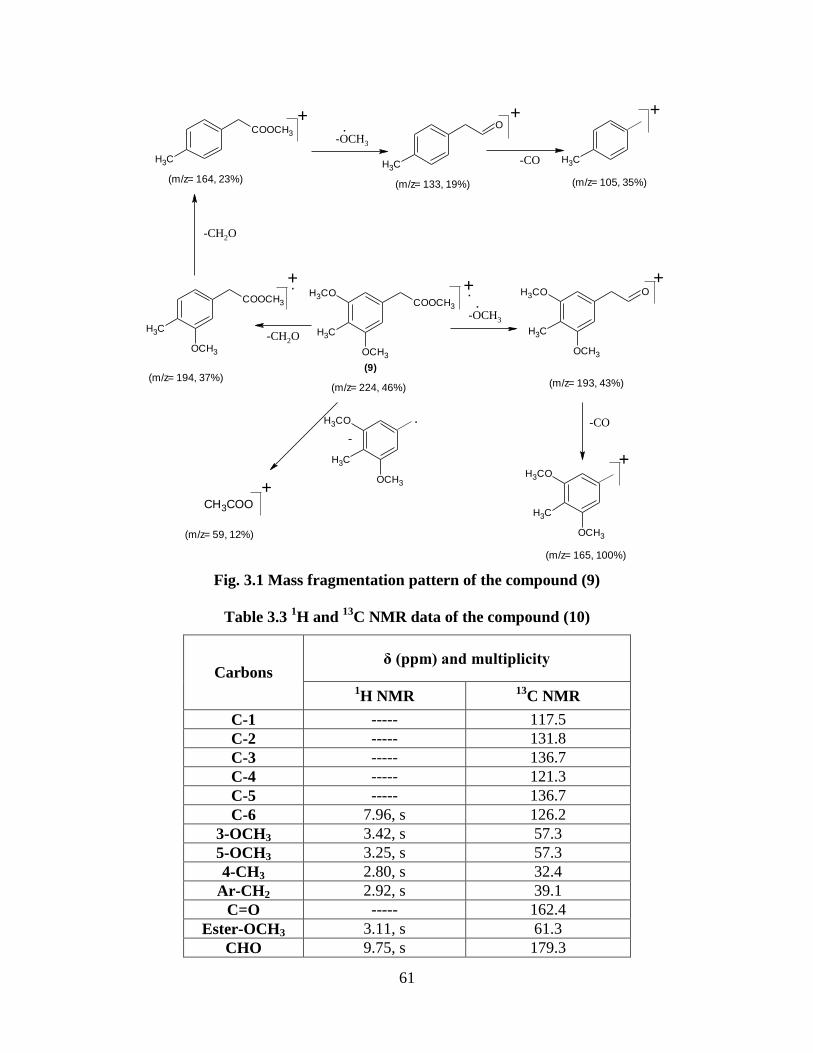
by condensation of homophthalic anhydride with appropriate phenols.
3-(4'-Methoxyphenyl)isocoumarin (120) was prepared by condensation of homophthalic
acid with anisole (Scheme 1.30).
COOH
COOH
O
O
OCH3
(120)
PPA
OCH3
Scheme 1.30
1.6 Reactions of Isocoumarins and 3,4-dihydroisocoumarins
1.6.1 Hydrolysis
Isocoumarins are lactones and undergo ring opening on alkaline hydrolysis to
give homophthaldehydic acids or ketones (121, R’=H or alkyl, aryl) or hydroxyl acid
(122, R’=alkyl, aryl, etc.). Similar treatment of 3,4-dihydroisocoumarins yields the
corresponding β-(2-carboxyphenyl)ethyl alcohol. In most of the cases isolation of the free
acids due to spontaneous recyclization to lactonic ring is not possible. In some cases e.g.
during the hydrolysis of cis-3-phenyl-4-hydroxy-3,4-dihydroisocoumarin (123), the
glycol produced (124) recyclizes to the more stable erythro-γ-lactone (125) under acid
treatment (Scheme 1.31)121-122
.
Page 49
31
COOH
R'
OR
COOH
R'
OHR
(121) (122)
Ph
O
O
OHHH
(123)
COOH
Ph
OH
OHHH
Ph
O
O
H
HOH
(125)(124)
Scheme 1.31
1.6.2 Reaction with Ammonia and Amines
Ammonia and amines add to isocoumarins furnishing isocarbostyrils123
(126), a
reaction typical of esters (Scheme 1.32).
O
O
NR
O
NH3
or
RNH2
(126)
Scheme 1.31
For example isocoumarin and 3-carboxylic acid have been condensed with
tryptamine, and the product subsequently converted to yobyrine (127) and other
derivatives (Scheme 1.32)124
.
O
O
NR
ON
(127)
+N
CH3
NH2
N
O
NH
Scheme 1.32
Page 50
32
It is reported that 3,4-dihydroisocoumarin with ammonia gives the corresponding
tetrahydroisoquinolinones e.g. heating agrimonolide with ammonia at 100°C gave the
isoquinolinone analogue (128) (Scheme 1.33).
O
O
OMe
OH
OH
(128)
NH3
100 °CNH
O
OMe
OH
OH
Scheme 1.33
1.6.3 Reaction with Phosphorus Pentasulfide
Isocoumarin can be converted to 1-thioisocoumarin (129) with phosphorus
pentasulfide and treatment of 1-thioisocoumarin with ammonium sulfide or aniline yields
isoquinolins (Scheme 1.34). Analogously, 3-phenylisocoumarin has been converted to 1-
thio-3-phenylisocoumarin, and treatment with aniline produced (130)125
.
O
O
(129)
N
P2S
5
O
S
(130)
N
O
Ph
Ph
Scheme 1.34
1.6.4 Nitration
The only report of the nitration of an isocoumarin is that of 3-phenyl-3,4-
dihydroisocoumarin (131), in which nitric acid in sulfuric acid gives 3-(4-nitrophenyl)-7-
nitro-3,4-dihydroisocoumarin (132) (Scheme 1.35)126
.
O
O
(131)
HNO3
O
O
NO2
(132)
Scheme 1.35
Page 51
33
1.6.5 Reaction with Grignard Reagents
Addition of phenylmagnesium bromide127-132
to 3-phenylisocoumarin followed by
perchloric acid, anhydrous hydrochloric acid, ferric chloride or ferric bromide yields the
isobenzopyrilium salt (133) (Scheme 1.35).
O
O
Ph
C6H
5MgBr
O
OH
Ph
Ph
O
Ph
Ph
(133)
HY
+Y
-
Y = perchlorate, chloride, ferric chloride or ferric bromide
Scheme 1.35
1.6.6 Oxidation
Chromium trioxide oxidation of 3,4,6,7-tetraphenylisocoumarin (134) produce 2-
benzoyl-4,5-diphenylbenzoic acid (135) (Scheme 1.36).
O
O
Ph
Ph
Ph
Ph
(134)
COOH
O
Ph
Ph
Ph
CrO3
(135)
Scheme 1.36
1.6.7 Reduction
The double bond present between C-3 and C-4 of isocoumarin can be reduced
readily with hydrogen and palladium on charcoal or with other catalyst133-134
. Catalytic
reduction also has been used to remove the halogen from cis- and trans-3-phenyl-4-halo-
3,4-dihydroisocoumarin135-136
.
1.7 Interconverision of isocoumarins and 3,4-dihydroisocumarins
It has been observed that some methods directly afford the isocoumarins while
others produce dihydroisocoumarins. Their interconversion is carried out depending upon
whether the synthesis of isocoumarin is easier or that of its dihydro derivative.
Page 52
34
1.7.1 Conversion of 3,4-Dihydroisocoumarins to Isocoumarins
There are two routs mainly used for conversion of 3,4-dihydroisocoumarins to
isocoumarins:
a. Alkaline Hydrolysis Followed by Oxidation and Recyclization
Alkaline hydrolysis of 3,4-dihydroisocoumarins137
yields the hydroxy acids which
could be oxidized to corresponding keto-acids. Since the hydroxy acids on standing
recyclize to parent dihydroisocoumarins, the oxidation should be carried out immediately.
The keto-acids are readily cyclized e.g. by heating with acetic anhydride to corresponding
isocoumarins (Scheme 1.37).
O
O
R
R COOHOH
RCrO
3
R COOHO
R
R
O
O
R
R
Scheme 1.37
b. Benzylic Bromination Followed by Dehydrobromination
Isocoumarins can be prepared from 3,4-dihydroisocoumarin138-139
via benzylic
bromination with N-bromosuccinimide (NBS), followed by dehydrohalogenation with
triethylamine (Scheme 1.38).
O
O
MeO
MeO
O
O
MeO
MeO
Br
NBS
UV O
O
MeO
MeO(C
2H
5)
3N
Scheme 1.38
1.7.2 Conversion of Isocoumarins to 3,4-Dihydroisocoumarins
Two different methods of reduction mainly used for conversion of isocoumarins
to 3,4-dihydroisocoumarins:
a. Alkaline Hydrolysis Followed by Reduction and Recyclization
Alkaline hydrolysis of isocoumarins with dilute aqueous alkali affords the keto-
acids, which upon reduction with sodium borohydride are converted into corresponding
Page 53
35
hydroxy-acids. Cyclodehydration of the latter affords the dihydroisocoumarins (Scheme
1.39).
O
OOMe
Ar/R
COOHO
Ar/R
OMe
KOH
MeOH
NaBH4
COOHOH
Ar/R
OMe
O
OOMe
Ar/R
-H2O
Scheme 1.39
b. Catalytic Reduction
Hydrogenation140-141
in the presence of palladium charcoal or some other catalyst
has been used to reduce the 3,4-double bond of isocoumarins thereby converting them
directly into 3,4-dihydroisocoumarins.
Page 54
36
2. EXPERIMENTAL
2.1 Purification of Solvents
The solvents were purified and dried according to the standard procedures before
use. The dried solvents were stored under molecular sieves (4 Å).
2.2 Instrumentation
Melting points were recorded using a digital Gallenkamp (SANYO) model MPD
BM 3.5 apparatus and are uncorrected. FTIR spectra were recorded using an FTS 3000
MX spectrophotometer, 1H NMR and
13C NMR spectra were determined as CDCl3
solutions at 300 MHz on a Bruker AM-300 spectrophotometer, mass spectra (EI, 70eV)
on a GCMS instrument, and elemental analyses with a LECO-183 CHNS analyzer
Aigilent Technologies USA. All the compounds were purified by thin layer
chromatography using silica gel HF-254 from Merck.
Synthesis of compounds
2.3 methyl 4-methylbenzoate (1)
A stirred solution of p-toluic acid (13.6g, 100mmol) in dry methanol (75ml) was
treated drop wise with conc. H2SO4 (5ml). The mixture was refluxed for 8-9hrs. The
reaction was monitored by TLC. After the completion of reaction, mixture was
concentrated to 55ml and extracted with ethyl acetate (3x50ml). The extract was washed
with saturated brine, dried and concentrated to afford methyl 4-methylbenzoate (1) as
thick oil (14.21g, 94.79%); Rf: 0.7 (petroleum ether and ethyl acetate, 4:1); m. p. 34°C;
IR (KBr): 3012 (C-H), 1742 (C=O), 1562 (C=C) cm-1
.
2.4 3, 5-Dibromo-4-methylbenzoate (2)
Aluminum chloride anhydrous (11.92g, 89mmol) was added portion wise to
stirred, cooled methyl 4-methyl benzoate (1) (5g, 33.30mmol). Bromine (3.56ml) was
then added to the stirred mixture for 45min at such a rate to keep the temperature at or
below 20°C. Stirring was continued at room temperature for 30min and at 80-85°C for
1h. The mixture was cooled to 30°C and treated with methanol (55.5ml) during 30min
and then stirred overnight. The crude product was collected by filtration, washed with
cooled (10°C) methanol and crystallized from methanol at 10°C to afford methyl 3,5-
dibromo-4-methylbenzoate (2) (5.99g, 58.38%) as colorless crystals, Rf: 0.6 (petroleum
Page 55
37
ether and ethyl acetate, 4:1); m. p. 82-84 °C; IR (KBr): 3021 (C-H), 1719 (C=O), 1559
(C=C) cm-1
.
2.5 3, 5-Dimethoxy-4-methyl benzoic acid (3)
A pyridine solution of methyl 3,5-dibromo-4-methylbenzoate (2) (4.62g,
15.0mmol) was added to a solution of sodium methoxide (2.07g, 90.0mmol) in dry
methanol and freshly prepared anhydrous copper (I) chloride (0.212g, 1.25mmol). The
reaction mixture was refluxed under nitrogen for 15h, cooled to room temperature and
filtered. The filtered cake was washed with warm methanol. The solution was refluxed
for 1h, cooled to room temperature and diluted with saturated brine (30ml). The mixture
was extracted with ethyl acetate (30ml), the extract discarded and the aqueous phase
acidified with cold, concentrated HCl (10ml) and then extracted with ethyl acetate (3x
20ml). The extract was washed with saturated brine, dried and evaporated. Crystallization
of residue from aqueous methanol (1:2) yielded 3,5-dimethoxy-4-methyl benzoic acid (3)
(2.5g, 85%); Rf: 0.4 (petroleum ether and ethyl acetate, 4:1); m. p. 210-212 °C; IR (KBr):
3213 (O-H), 3029 (C-H), 1731 (C=O), 1569 (C=C) cm-1
.
2.6 Methyl 3,5-dimethoxy-4-methylbenzoate (4)
3,5-dimethoxy-4-methyl benzoic acid (3) (2.5g, 12.5mmol) was dissolved in dry
methanol (15ml) and then concentrated sulphuric acid (1-2ml) was added. The mixture
was refluxed for 13-14hrs. The reaction was monitored by TLC. After the completion of
reaction, mixture was concentrated and extracted with ethyl acetate (3x15ml). The extract
was washed with saturated brine, dried and evaporated to afford methyl 3, 5-dimethoxy-
4-methyl benzoate (4) (2.25g, 84%) as colorless crystals, Rf: 0.65 (petroleum ether and
ethyl acetate, 4:1); m. p. 76-78 °C; IR (KBr): 3019 (C-H), 1732 (C=O), 1571 (C=C) cm-1
.
2.7 (3, 5-Dimethoxy-4-methyl phenyl)methanol (5)
Methyl 3, 5-dimethoxy-4-methyl benzoate (4) (4.2g, 0.02 mol) and sodium
borohydride (4.5g, 0.12 mol) were suspended in freshly distilled THF (150ml). The
reaction mixture was stirred for 15min at 65°C and then added methanol (150 ml) drop
wise for 30min. The mixture was refluxed for 4hrs then cooled to room temperature and
treated with saturated ammonium chloride solution (150ml). Stirring was continued for
1hr then acidified with dilute hydrochloric acid and extracted with ethyl acetate
(3x20ml). The extract was dried, evaporated and (3,5-dimethoxy-4-methyl
Page 56
38
phenyl)methanol (5) product was recryatallized with petroleum ether to afford prism like
crystals (3.0g, 82.41%); Rf: 0.5 (petroleum ether and ethyl acetate, 4:1); m. p. 45-47 °C,
IR (KBr): 3442 (O-H), 3023 (C-H), 1554 (C=C) cm-1
.
2.8 3, 5-Dimethoxy-4-methyl benzyl bromide (6)
3, 5-Dimethoxy-4-methyl benzyl alcohol (5) (10.01g, 55.0mmol) was dissolved in
dry benzene (40-50ml). The solution was treated with phosphorous tribromide (14.89g,
5.2ml, 55.0mmol) and stirred the resulting mixture for 4hrs. Then poured the reaction
mixture onto ice cold water, separated the organic layer and evaporated to afford crude 3,
5-Dimethoxy-4-methyl benzyl bromide (6). Prism like crystals were obtained after
recrystallization in petroleum ether (11.2g, 84.21%); Rf: 0.6 (petroleum ether and ethyl
acetate, 4:1); m. p. 68-69 °C, IR (KBr): 3013 (C-H), 1571 (C=C) cm-1
.
2.9 (3, 5-Dimethoxy-4-methylphenyl)acetonitrile (7)
3, 5-Dimethoxy-4-methyl benzyl bromide (6) (7.0g, 28.6mmol) was dissolved in
a mixture of ethyl alcohol (120ml) and water (120ml). Potassium cyanide (2.6g,
40.7mmol) was then added to reaction flask and refluxed for 4hrs. Reaction mixture was
poured onto ice cold water and extracted with ethyl acetate (3x20ml). The extract was
dried over anhydrous Na2SO4, evaporated to afford (3,5-dimethoxy-4-
methylphenyl)acetonitrile (7) and recrystallized in petroleum ether to get prism like
crystals (4.6g, 84.09%); Rf: 0.5 (petroleum ether and ethyl acetate, 4:1); m. p. 48-49 °C,
IR (KBr): 3007 (C-H), 1561 (C=C) cm-1
.
2.10 (3, 5-Dimethoxy-4-methylphenyl) acetic acid (8)
(3, 5-Dimethoxy-4-methylphenyl)acetonitrile (7) (4.0g, 20.9mmol) was dissolved
in a mixture of water (17.8ml) and dioxane (17.8ml). Then added potassium hydroxide
(14.56g in 15ml H2O) and refluxed the mixture for 10-12hrs. Reaction mixture was
poured onto ice cold water and extracted with ethyl acetate (20ml). The extract was
discarded and aqueous layer was acidified with dilute hydrochloric acid. Precipitates
were filtered to afford (3, 5-dimethoxy-4-methylphenyl) acetic acid (8) (3.1g, 70.61%);
Rf: 0.4 (petroleum ether and ethyl acetate, 4:1); m. p. 129-130 °C; IR (KBr): 3242 (O-H),
3013 (C-H), 1707 (C=O), 1567 (C=C) cm-1
.
Page 57
39
2.11 Methyl (3, 5-dimethoxy-4-methyl phenyl) acetate (9)
A stirred solution of (3, 5-dimethoxy-4-methylphenyl) acetic acid (8) (5.0g,
23.8mmol) in dry methanol (30ml) was treated dropwise with conc. H2SO4 (5ml). The
mixture was refluxed for 8-9hrs. The reaction was monitored by TLC. After the
completion of reaction, mixture was concentrated to 55ml and extracted with ethyl
acetate (3x50ml). The extract was washed with saturated brine, dried and concentrated to
give crude oil which was distilled to afford methyl (3, 5-dimethoxy-4-methyl phenyl)
acetate (9) (4.7g, 88.18%), Rf: 0.7 (petroleum ether and ethyl acetate, 4:1); m. p. 38-40
°C; IR (KBr): 3023 (C-H), 1734 (C=O), 1573 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm):
7.45 (2H, s, H-2, H-6), 3.96 (6H, s, 2OCH3), 3.54 (2H, s, Ar-CH2), 3.47 (3H, s,
COOCH3), 2.55 (3H, s, Ar-CH3); 13
C NMR (CDCl3 δ ppm): 168.23 (C=O), 132.54 (C3,
C5), 128.37 (C2, C6), 119.42 (C4), 112.21 (C1), 68.55 (Ester OCH3), 55.34 (Ar-OCH3),
36.91 (CH2), 28.63 (Ar-CH3); MS (70eV): m/z (%); 224 [M]+.
(46), 193 (43), 165 (100),
59 (12); Anal. calcd for C12H16O4: C, 64.28 H, 7.14 O, 28.57 Found: C, 64.02 H, 6.96 O,
28.35.
2.12 Methyl (2-formyl-3, 5-dimethoxy-4-methyl phenyl) acetate (10)
Phosphorus oxychloride (1.61g, 10.0mmol) was added dropwise in to a stirred
solution of methyl (3, 5-dimethoxy-4-methyl phenyl) acetate (9) (2.0g, 8.9mmol) in dry
DMF (10ml) at 55 °C. Reaction mixture was heated at about 100 °C for 2hrs and stirred
overnight at room temperature. Then poured the reaction mixture into aqueous solution of
sodium acetate (10%, 10ml) and shake vigorously. Methyl (2-formyl-3, 5-dimethoxy-4-
methyl phenyl) acetate (10) was precipitated as yellowish solid (1.9g, 84%); Rf: 0.55
(petroleum ether and ethyl acetate, 4:1); m. p. 51-53 °C; IR (KBr): 3029 (C-H), 1722
(C=O), 1690 (CHO), 1545 (C=C) cm-1
; 1H NMR, (CDCl3, δ ppm ): 9.75 (1H, s CHO),
7.96 (1H, s, H-6), 3.42 (3H, s, 3-OCH3), 3.25 (3H, s, 5-OCH3), 3.11 (3H, s, CO2CH3),
2.92 (2H, s, Ar-CH2), 2.80 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm ): 179.32
(Aldehyde C=O), 162.43 (Ester C=O), 136.76 (C3, C5), 131.89 (C2), 126.21 (C6),121.33
(C4), 117.54 (C1), 61.63 (Ester OCH3), 57.34 (Ar-OCH3), 39.12 (Ar-CH2), 32.08 (Ar-
CH3); MS (70eV): m/z (%); 252 [M]+.
(25), 251 (65), 224 (49), 223 (34), 165 (100), 29
(31); Anal. calcd for C13H16O5: C, 61.90 H, 6.34 O, 31.74 Found: C, 61.67 H, 6.16 O,
31.56.
Page 58
40
2.13 2,4-Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid (11)
Methyl (2-formyl-3, 5-dimethoxy-4-methyl phenyl) acetate (10) (6.3g, 25.0m
mol) and sulfamic acid (8.3g, 86.0mmol) in 150ml H2O:THF:DMSO (20:1:1) at 0°C was
treated with NaClO2 (7.24g, 80.0mmol) in 20ml H2O. The reaction mixture was stirred
for 20min at 0°C and then diluted with ethyl acetate (100ml), washed with saturated
aqueous ammonium chloride (2 x 130ml) and then with saturated aqueous sodium
chloride (130ml). Organic layer was dried over anhydrous sodium sulfate and evaporated
to afford 2,4-dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid (11) (6.6g,
79%); Rf: 0.4 (petroleum ether and ethyl acetate, 4:1); m. p. 164-166 °C; IR (KBr): 3265
(O-H), 3037 (C-H), 1734 (C=O), 1715 (COOH), 1562 (C=C) cm-1
; 1H NMR (CDCl3, δ
ppm ): 8.19 (1H, s, COOH), 7.66 (1H, s, H-6), 3.82 (3H, s, 3-OCH3), 3.67 (3H, s, 5-
OCH3), 3.63 (3H, s, CO2CH3), 2.54 (2H, s, Ar-CH2), 2.25 (3H, s, Ar-CH3); 13
C NMR
(CDCl3, δ ppm): 197.78 (Carboxylic C=O), 168.56 (Ester C=O), 139.32 (C3, C5), 134.37
(C2), 127.13 ( C6), 120.62 (C4), 114.17 (C1), 66.09 (Ester OCH3), 55.41 (Ar-OCH3),
35.04 (Ar-CH2), 29.88 (Ar-CH3); MS (70eV): m/z (%); 268 [M]+.
(32), 251 (51), 224
(65), 165 (100), 45 (25); Anal. calcd for C13H16O6: C, 58.20 H, 5.97 O, 35.82 Found: C,
58.04 H, 5.76 O, 35.59.
2.14 6-(carboxymethyl)-2,4-dimethoxy-3-methylbenzoic acid (12)
2,4-Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid (11) (8.4g,
31.3mmol) was dissolved in ethanol (75ml) and treated with KOH (5%, 125ml). The
reaction mixture was refluxed for 1h and the ethanol was rotary evaporated. The aqueous
layer was acidified with dilute hydrochloric acid to afford 6-(carboxymethyl)-2,4-
dimethoxy-3-methylbenzoic acid (12) (6.95g, 87.31%); Rf: 0.4 (petroleum ether and ethyl
acetate, 4:1); m. p. 180-182 °C; IR (KBr): 3195 (O-H), 3013 (C-H), 1741 (C=O), 1587
(C=C) cm-1
: 1H NMR (CDCl3, δ ppm): 10.91(1H, s, Ar-COOH), 10.70 (1H, s, CH2-
COOH), 7.60 (1H, s, H-6), 3.85 (3H, s, 3-OCH3), 3.67 (3H, s, 5-OCH3), 2.53 (2H, s,
CH2), 2.25 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 205.37 (Ar-COOH-C=O), 171.14
(CH2-C=O), 136.25 (C3, C5), 135.25 (C2), 133.07 (C6), 125.04 (C4), 124.41 (C1), 55.45
(2OCH3), 39.73 (Ar-CH2), 29.51 (Ar-CH3); MS (70eV): m/z (%); 254 [M]+.
(46), 237
(57), 210 (34), 165 (100); Anal. calcd for C12H14O6: C, 57.14 H, 5.55 O, 38.09 Found: C,
56.94 H, 5.36 O, 37.92.
Page 59
41
2.15 6,8-dimethoxy-7-methyl-1H-isochromene-1,3(4H)-dione (13)
A solution of 6-(carboxymethyl)-2,4-dimethoxy-3-methylbenzoic acid (12) (2.5g,
9.84mmol) in dry toluene (35ml) was treated with acetic anhydride (1.1g, 10ml,
10.8mmol). The reaction mixture was refluxed for 1h and then added onto ice cold water.
The organic layer was separated, dried over anhydrous sodium sulfate and toluene was
rotary evaporated to get 6,8-dimethoxy-7-methyl-1H-isochromene-1,3(4H)-dione (13)
(1.9g, 81.89%); Rf: 0.7 (petroleum ether and ethyl acetate, 4:1); m. p. 135-136 °C; IR
(KBr): 3011 (C-H), 1735 (C=O), 1590 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 6.72 (1H,
s, H-5), 3.85 (6H, s, 2OCH3), 3.45 (2H, s, H-4), 2.28 (3H, s, Ar-CH3); 13
C NMR (CDCl3,
δ ppm): 149.57 (C1), 168.14 (C3), 163.25 (C6, C8), 136.25 (C4a), 110.51 (C7), 106.04
(C8a), 104.41 (C5), 56.35 (2OCH3), 38.73 (C4), 27.41 (Ar-CH3); MS (70eV): m/z (%);
236 [M]+.
(23), 208 (57), 192 (100), 164 (19); Anal. calcd for C12H12O5: C, 61.01 H, 5.08
O, 33.89 Found: C, 60.91 H, 4.99 O, 33.82.
2.16 General procedure for 6,8-dimethoxy-7-methyl-3-alkyl/arylisocoumarins
(16a-j)
A mixture of aliphatic/aromatic carboxylic acids (14a-j) (1mmol) and thionyl
chloride (1.2mmol) was refluxed for 1hr in the presence of a drop of DMF. The
completion of reaction was determined by stoppage of evolution of gas. Excess of the
thionyl chloride was rotary evaporated to afford acid chlorides (15a-j).
A solution of homophthalic acid anhydride (13) (2.00 mmol) in acetonitril (12ml)
was added to a solution of N, N, N’, N’-tetramethylguanidine (TMG) (2.20 mmol) in
acetonitril (5ml) over 36 min. maintaining an internal temperature of 0°C. Triethylmine
(4.0 mmol) was added in one portion. Acid chlorides (15a-j) (3.20 mmol) were added
over 3 min. and the mixture was stirred an additional 18 min. After the completion of
reaction the cooling bath was removed and reaction was allowed to warm to room
temperature. The reaction mixture was quenched by the addition of HCl (1M, 5ml). The
two phases were separated, and the organic layer was washed with saturated sodium
chloride solution and then dried (Na2SO4) prior to removal of solvent under reduced
pressure to dryness. Isocoumarins (16a-j) were then purified by preparative thin layer
chromatography using (petroleum ether and ethyl acetate, 7:3) as eluant.
Page 60
42
2.17 3-Propyl-6,8-dimethoxy-7-methylisocoumarin (16a): Yield: 72%; Oil; IR
(KBr): 3031 (C-H), 1713 (C=O), 1572 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.8 (1H, s,
H-5), 7.3 (1H, s, H-4), 3.9 (6H, s, 6-OCH3,8-OCH3), 2.6 (3H, s, Ar-CH3), 2.5 (2H, t,
J=3.9, H-1’), 1.2 (2H, m, H-2’), 0.9 (3H, t, J=7.5, H-3’); 13
C NMR (CDCl3, δ ppm):
167.8 (C1), 151.2 (C3), 145.4 (C6, C8), 133.1 (C4a), 128.8 (C8a), 118.3 (C7), 110 (C4),
104.5 (C5), 53.6 (2OCH3), 38.6 (C1’), 29.7 (Ar-CH3), 21.1 (C2’), 14.3 (C3’); MS
(70eV): m/z (%); 262 [M]+.
(26), 191 (100), 71 (45), 43 (59); Anal. calcd for C15H18O4:
C, 68.70 H, 6.87; Found: C, 68.57 H, 6.69.
2.18 3-Pentyl-6,8-dimethoxy-7-methylisocoumarin (16b): Yield: 75%; Oil; IR
(KBr): 3037 (C-H), 1731 (C=O), 1559 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.9 (1H, s,
H-5), 7.3 (1H, s, H-4), 3.6 (6H, s, 6-OCH3,8-OCH3), 2.6 (3H, s, Ar-CH3), 2.3 (2H, t,
J=7.9, H-1’), 1.2-1.4 (6H, m, H-2’,H-3’,H-4’), 0.9 (3H, t, J=6.9, H-5’); 13
C NMR
(CDCl3, δ ppm): 167.8 (C1), 149.6 (C3), 141.6 (C6, C8), 133.5 (C4a), 130.9 (C8a), 118.3
(C7), 109.2 (C4), 105.3 (C5), 53.6 (2OCH3), 38.7 (C1’), 29.1 (Ar-CH3), 25.3 (C2’), 19.3
(C3’), 15.6 (C4’), 10.2 (C-5’); MS (70eV): m/z (%); 290 [M]+.
(36), 191 (100), 99 (49),
71 (34); Anal. calcd for C17H22O4: C, 70.34 H, 7.58; Found: C, 70.19 H, 7.42.
2.19 3-Heptyl-6,8-dimethoxy-7-methylisocoumarin (16c): Yield: 79%; m. p. 88-90
ºC; IR (KBr): 3021 (C-H), 1719 (C=O), 1569 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.3
(1H, s, H-5), 6.8 (1H, s, H-4), 3.9 (6H, s, 6-OCH3,8-OCH3), 2.5 (3H, s, Ar-CH3), 1.4 (2H,
t, J=6.9, H-1’), 1.2-1.3 (10H, m, H-2’,H-3’,H-4’,H-5’,H-6’), 0.9 (3H, t, J=7.2, H-7’); 13
C
NMR (CDCl3, δ ppm): 163.7 (C1), 152.9 (C3), 143.8 (C6, C8), 133.2 (C4a), 132.7 (C8a),
122.8 (C7), 114.7 (C4), 104.7 (C5), 56.2 (2OCH3), 38.7 (C1’), 29.1 (Ar-CH3), 27.8 (C2’),
21.7 (C3’), 16.8 (C4’), 11.7 (C-5’), 10.8 (C6’), 9.7 (C7’); MS (70eV): m/z (%); 318 [M]+.
(46), 191 (100), 127 (52), 99 (23); Anal. calcd for C19H25O4: C, 71.69 H, 8.17; Found: C,
71.54 H, 8.01.
2.20 3-Chloromethyl-6,8-dimethoxy-7-methylisocoumarin (16d): Yield: 70%; Oil;
IR (KBr): 3033 (C-H), 1722 (C=O), 1571 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.8
(1H, s, H-5), 6.4 (1H, s, H-4), 3.7 (6H, s, 6-OCH3,8-OCH3), 5.2 (2H, s, H-1’), 2.3 (3H, s,
Ar-CH3); 13
C NMR (CDCl3, δ ppm): 172.1 (C1), 158.3 (C3), 147.6 (C6, C8), 132.3
(C4a), 131.3 (C8a), 127.9 (C7), 114.7 (C4), 104.7 (C5), 60.0 (2OCH3), 39.6 (C1’), 30.6
Page 61
43
(Ar-CH3); MS (70eV): m/z (%); 268.5 [M]+.
(36), 270.5 [M+2] (27), 191 (100), 77.5 (49),
49 (34); Anal. calcd for C13H13O4Cl: C, 58.10 H, 4.84; Found: C, 57.95 H, 4.69.
2.21 3-Hydroxymethyl-6,8-dimethoxy-7-methylisocoumarin (16e): Yield: 81%; Oil;
IR (KBr): 3345 (O-H), 3023 (C-H), 1729 (C=O), 1554 (C=C) cm-1
; 1H NMR (CDCl3, δ
ppm): 8.1 (2H, d, J=7.5, H-2’,H-6’), 8.0 (1H, s, H-5), 7.8 (7.6, dd, J=7.5), 7.4 (2H, dd,
J=7.8), 6.9 (1H, s, H-4), 3.9 (6H, s, 6-OCH3,8-OCH3), 2.6 (3H, s, Ar-CH3); 13
C NMR
(CDCl3, δ ppm): 168.5 (C1), 151.2 (C3), 141.2 (C6, C8), 131.5 (C4a), 127.8 (C8a), 118.3
(C7), 112.3 (C4), 107.3 (C5), 55.3 (2OCH3), 45.9 (C1’), 28.6 (Ar-CH3); MS (70eV): m/z
(%); 250 [M]+.
(57), 191 (100), 59 (65), 31 (54); Anal. calcd for C13H14O5: C, 62.40 H,
5.60; Found: C, 62.27 H, 5.42.
2.22 3-Phenyl-6,8-dimethoxy-7-methylisocoumarin (16f): Yield: 89%; m. p. 109-
111 oC; IR (KBr): 3033 (C-H), 1715 (C=O), 1571 (C=C) cm
-1; 1
H NMR (CDCl3, δ ppm):
8.1 (2H, d, J=7.5, H-2’,H-6’), 8.0 (1H, s, H-5), 7.6 (1H, dd, J=7.5, H-4’), 7.4 (2H, dd,
J=7.8, H-3’,H-5’), 6.9 (1H, s, H-4), 3.9 (6H, s, 6-OCH3,8-OCH3), 2.6 (3H, s, Ar-CH3);
13C NMR (CDCl3, δ ppm): 172.7 (C1), 143.7 (C3), 138.7 (C6, C8), 133.9 (C4a), 132.6
(C8a), 130.6 (C1’), 128.5 (C3’,C5’), 127.7 (C4’), 125.2 (C2’,C6’), 118.7 (C7), 112.3
(C4), 107.3 (C5), 60.8 (2OCH3), 29.7 (Ar-CH3); MS (70eV): m/z (%); 296.5 [M]+.
(63),
191 (100), 105 (51), 77 (43); Anal. calcd for C18H16O4: C, 72.79 H, 5.40; Found: C, 72.83
H, 5.26.
2.23 3-(2-chlorophenyl)-6,8-dimethoxy-7-methylisocoumarin (16g): Yield: 84%; m.
p. 119-121 o
C; IR (KBr): 3017 (C-H), 1727 (C=O), 1561 (C=C) cm-1
; 1H NMR (CDCl3, δ
ppm): 7.9 (1H, d, J=6.9, H-3’), 7.7 (1H, dd, J=3.5, H-4’), 7.6 (1H, d, J=5.5, H-6’), 7.5
(1H, dd, J=3.3, H-5’), 7.4 (1H, s, H-5), 6.8 (1H, s, H-4), 3.8 (6H, s, 6-OCH3,8-OCH3),
2.5 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 158.2 (C1), 140.4 (C3), 136.2 (C6, C8),
134.1 (C4a), 133.5 (C8a), 132.5 (C2’), 130.2 (C1’), 129.8 (C3’), 126.3 (C4’), 125.8
(C6’), 125.1 (C5’), 118.3 (C7), 114.6 (C4), 103.9 (C5), 67.3 (2OCH3), 30.2 (Ar-CH3);
MS (70eV): m/z (%); 330.5[M]+.
(59), 332.5 []M+2] (44), 191 (100), 139.5 (71), 111.5
(48); Anal. calcd for C18H15O4Cl: C, 65.35 H, 4.53; Found: C, 65.19 H, 4.39.
2.24 3-(4-methoxyphenyl)-6,8-dimethoxy-7-methylisocoumarin (16h): Yield: 87%;
m. p. 154-156 oC; IR (KBr): 3023 (C-H), 1723 (C=O), 1567 (C=C) cm
-1;
1H NMR
(CDCl3, δ ppm): 8.0 (2H, d, J=9.0, H-3’,H-5’), 7.8 (1H, s, H-5), 7.0 (1H, d, J=8.7, H-
Page 62
44
2’,H-6’), 6.8 (1H, s, H-4), 3.9 (6H, s, 6-OCH3,8-OCH3), 3.8 (3H, s, 4’-OCH3), 2.6 (3H, s,
Ar-CH3); 13
C NMR (CDCl3, δ ppm): 164.0 (C1), 139.5 (C3), 137.6 (C6, C8), 134.7
(C4a), 133.8 (C8a), 131.5 (C4’), 130.2 (C1’), 126.4 (C3’,C5’), 123.8 (C2’,C6’), 121.5
(C7), 119.2 (C4), 109.1 (C5), 55.5 (6-OCH3,8-OCH3), 53.7 (4’-OCH3), 29.2 (Ar-CH3);
MS (70eV): m/z (%); 326 [M]+.
(57), 191 (100), 135 (67), 107 (47); Anal. calcd for
C19H18O5: C, 69.93 H, 5.52; Found: C, 69.74 H, 5.36.
2.25 3-(3,4-dimethoxyphenyl)-6,8-dimethoxy-7-methylisocoumarin (16i): Yield:
85%; m. p. 122-124 o
C; IR (KBr): 3029 (C-H), 1736 (C=O), 1573 (C=C) cm-1
; 1
H NMR
(CDCl3, δ ppm): 7.8 (1H, s, H-2’), 7.7 (1H, d, J=3.3, H-5’), 7.5 (1H, d, J=3.3, H-6’), 7.4
(1H, s, H-5), 7.3 (1H, s, H-4), 3.9 (6H, s, 6-OCH3,8-OCH3), 3.8 (6H, s, 3’-OCH3, 4’-
OCH3), 2.5 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 167.7 (C1), 141.1 (C3), 136.6
(C6, C8), 134.5 (C4a), 133.1 (C8a), 131.1 (C3’,C4’), 130.5 (C1’), 128.94 (C2’), 127.2
(C5’), 124.5 (C6’), 119.5 (C7), 114.8 (C4), 103.7 (C5), 68.1 (6-OCH3,8-OCH3), 56.6 (3’-
OCH3,4’-OCH3), 29.7 (Ar-CH3); MS (70eV): m/z (%); 356 [M]+.
(64), 191 (100), 165
(58), 137 (44); Anal. calcd for C19H18O5: C, 67.41 H, 5.61; Found: C, 67.28 H, 5.45.
2.26 3-(3,4,5-Trimethoxyphenyl)-6,8-dimethoxy-7-methylisocoumarin (16j): Yield:
87%; m. p. 135-137 oC; IR (KBr): 3013 (C-H), 1713 (C=O), 1587 (C=C) cm
-1;
1H NMR
(CDCl3, δ ppm): 7.5 (2H, s, H-2’,H-6’), 7.4 (1H, s, H-5), 6.9 (1H, s, H-4), 3.9 (6H, s, 6-
OCH3,8-OCH3), 3.8 (9H, s, 3’-OCH3, 4’-OCH3, 5’-OCH3), 2.5 (3H, s, Ar-CH3); 13
C
NMR (CDCl3, δ ppm): 167.8 (C1), 140.3 (C3), 135.6 (C6, C8), 134.5 (C4a), 133.1 (C8a),
132.4 (C3’,C4’,C5’), 129.6 (C1’), 122.5 (C2’,C6’), 119.5 (C7), 116.9 (C4), 107.1 (C5),
68.1 (6-OCH3,8-OCH3), 55.5 (3’-OCH3,4’-OCH3,5’-OCH3), 29.3 (Ar-CH3); MS (70eV):
m/z (%); 386 [M]+.
(79), 195 (57), 191 (100), 167 (51); Anal. calcd for C21H22O7: C,
65.28 H, 5.69; Found: C, 65.14 H, 5.57.
2.27 General procedure for 2,4-dimethoxy-3-methyl-6-(2-oxoalkyl/aryl)benzoic
acid (17a-j) A stirred solution of 6,8-Dimethoxy-7-methyl-3-alkyl/aryl Isocoumarins
(16a-j) (1.42mmol) in ethanol (20 mL) was treated with 5% KOH (40 mL) and the
mixture refluxed for four hrs. After cooling the reaction mixture, most of the ethanol was
evaporated under reduced pressure. Cold water (20 mL) was added and the mixture
acidified with dilute hydrochloric acid when solid was precipitated. Filtration followed by
drying under vacuum afforded (17a-j).
Page 63
45
2.28 2,4-Dimethoxy-3-methyl-6-(2-oxopentyl)benzoic acid (17a): Yield: 78%; m. p.
133-134 oC; IR (KBr): 3224 (O-H), 3029 (C-H), 1749 (Carboxylic C=O), 1713 (Carbonyl
C=O), 1572 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 10.2 (1H, s, COOH), 7.9 (1H, s, H-
5), 4.3 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.7 (3H, s, 4-OCH3), 3.5 (2H, t, J=3.9 H-3’),
3.1 (3H, s, Ar-CH3), 1.5 (2H, m, H-4’), 0.9 (3H, t, J=7.5, H-5’); 13
C NMR (CDCl3, δ
ppm): 195.2 (C2’), 168.4 (COOH), 145.4 (C2), 139.2 (C4), 131.1 (C1), 125.1 (C6), 121.1
(C3), 108.7 (C5), 61.2 (2-OCH3), 56.4 (4-OCH3), 47.9 (C1’), 44.1 (C3’), 32.3 (Ar-CH3),
19.8 (C4’), 10.5 (C5’); MS (70eV): m/z (%); 280 [M]+ (19), 262 (43), 236 (54), 219
(100); Anal. calcd for C15H20O5: C, 64.28 H, 7.14; Found: C, 64.17 H, 7.05.
2.29 2,4-dimethoxy-3-methyl-6-(2-oxoheptyl)benzoic acid (17b): Yield: 79%; m. p.
139-140 oC; IR (KBr): 3256 (O-H), 3036 (C-H), 1753 (Carboxylic C=O), 1709 (Carbonyl
C=O), 1559 (C=C) cm-1
; 1
H NMR (CDCl3, δ ppm): 9.7 (1H, s, COOH), 8.0 (1H, s, H-5),
4.5 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.8 (3H, s, 4-OCH3), 3.7 (2H, t, J=3.7 H-3’), 3.2
(3H, s, Ar-CH3), 1.2-1.5 (6H, m, H-4’,H-5’,H-6’), 0.9 (3H, t, J=6.7, H-7’); 13
C NMR
(CDCl3, δ ppm): 197.1 (C2’), 169.2 (COOH), 143.1 (C2), 138.1 (C4), 135.2 (C1), 127.7
(C6), 119.8 (C3), 105.2 (C5), 59.5 (2-OCH3), 55.1 (4-OCH3), 49.2 (C1’), 45.5 (C3’), 30.5
(Ar-CH3), 21.5 (C4’), 18.7 (C5’), 15.7 (C6’), 10.2 (C7’); MS (70eV): m/z (%); 308 [M]+
(25), 290 (51), 264 (64) 219 (100); Anal. calcd for C17H24O5: C, 67.10 H, 7.79; Found: C,
67.02 H, 7.71.
2.30 2,4-Dimethoxy-3-methyl-6-(2-oxononyl)benzoic acid (17c): Yield: 81%; m. p.
147-148 oC; IR (KBr): 3267 (O-H), 3031 (C-H), 1743 (Carboxylic C=O), 1719 (Carbonyl
C=O), 1569 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 10.5 (1H, s, COOH), 8.2 (1H, s, H-
5), 4.1 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.8 (3H, s, 4-OCH3), 3.6 (2H, t, J=3.7 H-3’),
3.1 (3H, s, Ar-CH3), 1.3-1.6 (10H, m, H-4’,H-5’,H-6’,H-7’,H-8’), 0.9 (3H, t, J=7.3, H-
9’); 13
C NMR (CDCl3, δ ppm): 190.9 (C2’), 165.4 (COOH), 141.5 (C2), 137.1 (C4),
134.5 (C1), 125.3 (C6), 119.8 (C3), 107.4 (C5), 60.3 (2-OCH3), 52.9 (4-OCH3), 46.1
(C1’), 42.7 (C3’), 29.7 (Ar-CH3), 19.2 (C4’), 16.1 (C5’), 13.5 (C6’), 12.9 (C7’), 12.5
(C8’), 10.5 (C9’); MS (70eV): m/z (%); 336 [M]+ (25), 318 (31), 292 (51) 219 (100);
Anal. calcd for C19H28O5: C, 67.85 H, 8.33; Found: C, 67.76 H, 8.19.
2.31 6-(3-Chloro-2-oxopropyl)-2,4-dimethoxy-3-methylbenzoic acid (17d): Yield:
72%; m. p. 120-121 oC; IR (KBr): 3245 (O-H), 3033 (C-H), 1759 (Carboxylic C=O),
Page 64
46
1715 (Carbonyl C=O), 1571 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 10.8 (1H, s, COOH),
8.2 (1H, s, H-5), 5.2 (2H, s, H-3’), 4.7 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.7 (3H, s, 4-
OCH3), 2.9 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 195.7 (C2’), 169.2 (COOH),
143.5 (C2), 139.2 (C4), 135.2 (C1), 132.5 (C6), 123.0 (C3), 110.6 (C5), 61.7 (2-OCH3),
56.9 (4-OCH3), 51.2 (C3’), 43.5 (C1’), 28.1 (Ar-CH3); MS (70eV): m/z (%); 286.5 [M]+
(16), 288.5 [M+2] (12), 268.5 (41), 242.5 (56), 219 (100), 49.5 (17); Anal. calcd for
C13H15O5Cl: C, 54.45 H, 5.23; Found: C, 54.37 H, 5.15.
2.32 6-(3-Hydroxy-2-oxopropyl)-2,4-dimethoxy-3-methylbenzoic acid (17e): Yield:
75%; m. p. 128-129 oC; IR (KBr): 3256 (O-H), 3030 (C-H), 1747 (Carboxylic C=O),
1721 (Carbonyl C=O), 1554 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 10.9 (1H, s, COOH),
7.6 (1H, s, H-5), 5.9 (2H, s, H-3’), 4.6 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.7 (3H, s, 4-
OCH3), 2.7 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 201.2 (C2’), 168.5 (COOH),
146.7 (C2), 137.5 (C4), 133.1 (C1), 131.8 (C6), 119.5 (C3), 109.5 (C5), 71.5 (C3’), 58.5
(2-OCH3), 55.1 (4-OCH3), 47.9 (C1’), 31.0 (Ar-CH3); MS (70eV): m/z (%); 268 [M]+
(45), 250 (57), 224 (61) 219 (100); Anal. calcd for C13H16O6: C, 58.20 H, 5.97; Found: C,
58.07 H, 5.89.
2.33 2,4-Dimethoxy-3-methyl-6-(2-oxo-2-phenylethyl)benzoic acid (17f): Yield: 89%;
m. p. 169-171 oC; IR (KBr): 3224 (O-H), 3037 (C-H), 1758 (Carboxylic C=O), 1715
(Carbonyl C=O), 1571 (C=C) cm-1
; 1
H NMR (CDCl3, δ ppm): 10.9 (1H, s, COOH), 8.3
(2H, d, J=7.5, H-2’’,H-6’’), 7.9 (1H, s, H-5), 7.7 (1H, dd, J=7.2, H-4’’), 7.5 (2H, dd,
J=7.5, H-3’’,H-5’’), 4.3 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.8 (3H, s, 4-OCH3), 2.5 (3H,
s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 194.2 (C2’), 167.5 (COOH), 143.6 (C1), 138.7
(C2), 137.6 (C1’’), 135.9 (C4), 131.7 (C6), 126.2 (C2’’,C6’’), 123.5 (C3’’,C5’’), 122.7
(C4’’), 121.3 (C3), 105.3 (C5), 62.5 (2-OCH3), 57.3 (4-OCH3), 45.3 (C1’), 29.2 (Ar-
CH3); MS (70eV): m/z (%); 314 [M]+ (39), 296 (51), 270 (65) 219 (100); Anal. calcd for
C18H18O5: C, 68.78 H, 5.73; Found: C, 65.69 H, 5.65.
2.34 6-[2-(2-Chlorophenyl)-2-oxoethyl]-2,4-dimethoxy-3-methylbenzoic acid (17g):
Yield: 84%; m. p. 175-176 oC; IR (KBr): 3228 (O-H), 3027 (C-H), 1741 (Carboxylic
C=O), 1720 (Carbonyl C=O), 1561 (C=C) cm-1
; 1
H NMR (CDCl3, δ ppm): 11.1 (1H, s,
COOH), 8.0 (1H, d, J=7.5, H-3’’), 7.7 (1H, dd, J=6.7, H-4’’), 7.6 (1H, s, H-5), 7.5 (1H,
d, J=7.2, H-6’’), 7.3 (1H, dd, J=6.9, H-5’’), 4.6 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.7
Page 65
47
(3H, s, 4-OCH3), 2.7 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 201.3 (C2’), 168.1
(COOH), 145.7 (C1), 139.3 (C2), 138.2 (C2’’), 137.3 (C1’’), 136.7 (C4), 130.2 (C6),
127.7 (C3’’), 126.9 (C6’’), 124.3 (C4’’), 123.6 (C5’’), 119.2 (C3), 107.2 (C5), 61.4 (2-
OCH3), 56.7 (4-OCH3), 46.8 (C1’), 30.3 (Ar-CH3); MS (70eV): m/z (%); 348.5 [M]+
(35), 350.5 [M+2] (26), 330.5 (71), 304.5 (48), 219 (100); Anal. calcd for C18H17O5Cl: C,
61.98 H, 4.87; Found: C, 61.89 H, 4.79.
2.35 2,4-Dimethoxy-6-[2-(4-methoxyphenyl)-2-oxoethyl]-3-methylbenzoic acid (17h):
Yield: 87%; m. p. 179-180 oC; IR (KBr): 3247 (O-H), 3035 (C-H), 1761 (Carboxylic
C=O), 1723 (Carbonyl C=O), 1567 (C=C) cm-1
; 1
H NMR (CDCl3, δ ppm): 10.7 (1H, s,
COOH), 8.1 (2H, d, J=7.7, H-3’’,H-5’’), 7.9 (1H, s, H-5), 7.6 (2H, d, J=7.2, H-2’’,H-6’’),
4.3 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.8 (3H, s, 4-OCH3), 2.7 (3H, s, Ar-CH3); 13
C
NMR (CDCl3, δ ppm): 193.5 (C2’), 165.7 (COOH), 143.5 (C1), 140.7 (C2), 135.2 (C1’’),
133.3 (C4), 129.7 (C2’’,C6’’), 124.3 (C4’’), 120.3 (C3), 119.5 (C3’’,C5’’), 120.3 (C3),
104.3 (C5), 60.5 (2-OCH3), 58.5 (4’’-OCH3), 55.9 (4-OCH3), 44.7 (C1’), 28.5 (Ar-CH3);
MS (70eV): m/z (%); 344 [M]+ (49), 326 (57), 330 (69), 219 (100); Anal. calcd for
C19H20O6: C, 66.27 H, 5.81; Found: C, 66.19 H, 5.74.
2.36 2,4-Dimethoxy-6-[2-(3,4-dimethoxyphenyl)-2-oxoethyl]-3-methylbenzoic
acid (17i): Yield: 85%; m. p. 182-183 oC; IR (KBr): 3239 (O-H), 3035 (C-H), 1749
(Carboxylic C=O), 1716 (Carbonyl C=O), 1573 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm):
10.9 (1H, s, COOH), 8.0 (1H, s, H-5), 7.6 (1H, s, H-2’’), 7.5 (1H, d, J=7.1, H-5’’), 7.3
(1H, d, J=6.8, H-6’’), 4.5 (2H, s, H-1’), 3.9 (3H, s, 2-OCH3), 3.8 (3H, s, 4-OCH3), 3.7
(6H, s, 3’’-OCH3, 4’’-OCH3), 2.9 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 195.4
(C2’), 167.5 (COOH), 140.3 (C1), 139.2 (C2), 137.5 (C4), 136.6 (C1’’), 130.3 (C6’’),
128.9 (C2’’), 128.2 (C6), 125.5 (C4’’), 124.3 (C3’’), 120.4 (C3), 106.5 (C5), 61.3 (2-
OCH3), 56.2 (4-OCH3), 55.9 (3’’-OCH3), 55.6 (4’’-OCH3), 43.5 (C1’), 29.1 (Ar-CH3);
MS (70eV): m/z (%); 374 [M]+ (46), 356 (52), 330 (69), 219 (100); Anal. calcd for
C20H22O7: C, 64.17 H, 5.88; Found: C, 64.08 H, 5.79.
2.37 2,4-Dimethoxy-6-[2-(3,4,5-trimethoxyphenyl)-2-oxoethyl]-3-methylbenzoic
acid (17j): Yield: 87%; m. p. 185-187 oC; IR (KBr): 3259 (O-H), 3033 (C-H), 1757
(Carboxylic C=O), 1713 (Carbonyl C=O), 1587 (C=C) cm-1
; 1
H NMR (CDCl3, δ ppm):
10.7 (1H, s, COOH), 7.8 (1H, s, H-5), 7.7 (2H, s, H-2’’,H-6’’), 4.7 (2H, s, H-1’), 3.9 (3H,
Page 66
48
s, 2-OCH3), 3.8 (3H, s, 4-OCH3), 3.7 (9H, s, 3’’-OCH3, 4’’-OCH3, 5’’-OCH3), 2.9 (3H,
s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 193.7 (C2’), 166.3 (COOH), 141.1 (C1), 139.6
(C2), 137.5 (C4), 136.7 (C1’’), 129.3 (C6), 127.5 (C2’’,C6’’), 126.7 (C3’’,C5’’), 123.4
(C4’’), 127.4 (C3), 109.1 (C5), 59.8 (2-OCH3), 57.3 (4-OCH3), 55.7 (3’’-OCH3), 55.6
(5’’-OCH3), 55.1 (4’’-OCH3), 46.2 (C1’), 28.3 (Ar-CH3); MS (70eV): m/z (%); 404 [M]+
(39), 386 (45), 360 (57), 219 (100); Anal. calcd for C21H24O8: C, 62.37 H, 5.94; Found:
C, 62.29 H, 5.85.
2.38 General procedure for 6,8-dimethoxy-7-methyl-3-alkyl/aryl-3,4-
dihydroisocoumarins (18a-j)
Sodium borohydride (18 mmol) was added portion wise to a stirred solution of
keto acids (17a-j) (0.66 mmol) in ethanol (25 mL) and water (75 mL). The reaction
mixture was stirred for 2h at room temperature, diluted with water (150 mL), acidified
with conc. HCl and stirred for a further 2h. It was then saturated with ammonium sulfate,
and extracted with ethyl acetate (3 x 100 mL). The layers were separated and the organic
layer was dried over anhydrous magnesium sulfate and concentrated. 6,8-dimethoxy-7-
methyl-3-alkyl/aryl dihydroisocoumarins (18a-j) were purified by preparative thin layer
chromatography using (petroleum ether and ethyl acetate 7:3) as eluent.
2.39 6,8-Dimethoxy-7-methyl-3-propyl-3,4-dihydroisocoumarins (18a): Yield:
75%; Oil; IR (KBr): 3029 (C-H), 1719 (C=O), 1582 (C=C) cm-1
; 1H NMR (CDCl3, δ
ppm): 6.8 (1H, s, H-5), 5.2 (1H, m, H-3), 3.9 (6H, s, 6-OCH3,8-OCH3), 3.4 (1H, dd,
Jgem=15.5, Jtrans=12.2 H-4), 3.1 (1H, dd, Jgem=15.5, Jcis=3.5 H-4), 2.9 (3H, s, Ar-CH3), 2.5
(2H, m, H-1’), 1.2 (2H, m, H-2’), 0.9 (3H, t, J=7.5, H-3’); 13
C NMR (CDCl3, δ ppm):
165.8 (C1), 147.4 (C6, C8), 143.1 (C4a), 138.8 (C8a), 129.2 (C5), 118.3 (C7), 81.1 (C3),
56.6 (6-OCH3,8-OCH3), 43.5 (C4), 35.6 (C1’), 28.7 (Ar-CH3), 19.1 (C2’), 11.2 (C3’);
MS (70eV): m/z (%); 264 [M]+ (49), 192 (100), 71 (35), 43 (65); Anal. calcd for
C15H20O4: C, 68.18 H, 7.57; Found: C, 68.09 H, 7.49.
2.40 6,8-Dimethoxy-7-methyl-3-pentyl-3,4-dihydroisocoumarins (18b): Yield:
79%; Oil; IR (KBr): 3017 (C-H), 1723 (C=O), 1579 (C=C) cm-1
; 1
H NMR (CDCl3, δ
ppm): 6.9 (1H, s, H-5), 4.9 (1H, m, H-3), 3.9 (6H, s, 6-OCH3,8-OCH3), 3.3 (1H, dd,
Jgem=15.8, Jtrans=12.6 H-4), 3.0 (1H, dd, Jgem=16.1, Jcis=3.7 H-4), 2.7 (3H, s, Ar-CH3), 2.4
(2H, m, H-1’), 1.2-1.5 (6H, m, H-2’,H-3’,H-4’), 0.9 (3H, t, J=5.9, H-5’); 13
C NMR
Page 67
49
(CDCl3, δ ppm): 168.2 (C1), 149.7 (C6, C8), 145.5 (C4a), 139.9 (C8a), 127.3 (C5), 123.3
(C7), 79.3 (C3), 55.2 (6-OCH3,8-OCH3), 44.6 (C4), 38.3 (C1’), 29.3 (Ar-CH3), 20.2
(C2’), 15.3 (C3’), 13.3 (C4’), 10.6 (C5’); MS (70eV): m/z (%); 292 [M]+ (53), 192 (100),
99 (51), 71 (23); Anal. calcd for C17H24O4: C, 69.86 H, 8.21; Found: C, 69.79 H, 8.12.
2.41 6,8-Dimethoxy-7-methyl-3-heptyl-3,4-dihydroisocoumarins (18c): Yield: 81%;
m. p. 73-75 ºC; IR (KBr): 3025 (C-H), 1719 (C=O), 1569 (C=C) cm-1
; 1
H NMR (CDCl3,
δ ppm): 6.7 (1H, s, H-5), 5.3 (1H, m, H-3), 3.8 (6H, s, 6-OCH3,8-OCH3), 3.5 (1H, dd,
Jgem=15.3, Jtrans=11.8 H-4), 3.1 (1H, dd, Jgem=16.3, Jcis=3.7 H-4), 2.5 (3H, s, Ar-CH3), 2.4
(2H, m, H-1’), 1.1-1.5 (10H, m, H-2’,H-3’,H-4’,H-5’,H-6’), 0.9 (3H, t, J=5.9, H-7’); 13
C
NMR (CDCl3, δ ppm): 164.7 (C1), 151.4 (C6, C8), 142.9 (C4a), 136.3 (C8a), 121.8 (C5),
123.3 (C7), 83.7 (C3), 58.3 (6-OCH3,8-OCH3), 46.8 (C4), 37.7 (C1’), 30.1 (Ar-CH3),
19.5 (C2’), 13.6 (C3’), 12.8 (C4’), 12.0 (C5’), 11.2 (C6’), 9.8 (C7’); MS (70eV): m/z (%);
320 [M]+ (49), 192 (100), 127 (53), 99 (19); Anal. calcd for C19H28O4: C, 71.25 H, 8.75;
Found: C, 71.14 H, 8.66.
2.42 6,8-Dimethoxy-7-methyl-3-chloromethyl-3,4-dihydroisocoumarins (18d): Yield:
73%; Oil; IR (KBr): 3020 (C-H), 1728 (C=O), 1571 (C=C) cm-1
; 1
H NMR (CDCl3, δ
ppm): 6.9 (1H, s, H-5), 5.6 (1H, dd, Jgem=16.5, Jtrans=11.9 H-1’), 4.9 (1H, dd, Jgem=16.5,
Jcis=3.5 H-1’), 4.7 (1H, m, H-3), 3.7 (6H, s, 6-OCH3,8-OCH3), 3.5 (1H, dd, Jgem=15.9,
Jtrans=12.5 H-4), 3.1 (1H, dd, Jgem=16.2, Jcis=3.7 H-4), 2.7 (3H, s, Ar-CH3); 13
C NMR
(CDCl3, δ ppm): 169.3 (C1), 147.5 (C6, C8), 142.9 (C4a), 136.3 (C8a), 121.8 (C5), 127.8
(C7), 87.3 (C3), 55.5 (6-OCH3,8-OCH3), 45.7 (C1’), 40.1 (C4), 28.7 (Ar-CH3); MS
(70eV): m/z (%); 270.5 [M]+ (32), 272.5 [M+2] (24), 192 (100), 77.5 (54), 49.5 (37);
Anal. calcd for C13H15O4Cl: C, 57.67 H, 5.54; Found: C, 57.59 H, 5.49.
2.43 6,8-Dimethoxy-7-methyl-3-hydroxymethyl-3,4-dihydroisocoumarins (18e):
Yield: 85%; Oil; IR (KBr): 3367 (O-H), 3013 (C-H), 1723 (C=O), 1554 (C=C) cm-1
; 1
H
NMR (CDCl3, δ ppm): 6.7 (1H, s, H-5), 5.2 (1H, dd, Jgem=15.8, Jtrans=12.1 H-1’), 4.7
(1H, dd, Jgem=16.2, Jcis=3.3 H-1’), 4.7 (1H, m, H-3), 3.8 (6H, s, 6-OCH3,8-OCH3), 3.6
(1H, dd, Jgem=16.3, Jtrans=12.1 H-4), 3.2 (1H, dd, Jgem=15.8, Jcis=3.5 H-4), 2.6 (3H, s, Ar-
CH3), 2.0 (1H, s, -OH); 13
C NMR (CDCl3, δ ppm): 165.5 (C1), 146.6 (C6, C8), 140.5
(C4a), 138.8 (C8a), 128.9 (C7), 125.5 (C5), 89.4 (C3), 56.8 (6-OCH3,8-OCH3), 61.2
Page 68
50
(C1’), 37.7 (C4), 29.2 (Ar-CH3); MS (70eV): m/z (%); 252 [M]+ (59), 192 (100), 59 (61),
31 (51); Anal. calcd for C13H16O5: C, 61.90 H, 6.34; Found: C, 61.81 H, 6.26.
2.44 6,8-Dimethoxy-7-methyl-3-phenyl-3,4-dihydroisocoumarins (18f): Yield:
84%; m. p. 92-94 ºC; IR (KBr): 3031 (C-H), 1714 (C=O), 1571 (C=C) cm-1
; 1
H NMR
(CDCl3, δ ppm): 7.9 (2H, d, J=5.8, H-2’,H-6’), 7.7 (2H, dd, J=7.9, H-3’,H-5’), 7.5 (1H,
dd, J=6.7, H-4’), 6.9 (1H, s, H-5), 5.4 (1H, dd, Jtrans=12.1, Jcis=3.3 H-3), 3.9 (6H, s, 6-
OCH3,8-OCH3), 3.4 (1H, dd, Jgem=15.7, Jtrans=12.1 H-4), 3.1 (1H, dd, Jgem=12.0, Jcis=3.7
H-4), 2.8 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 167.3 (C1), 147.3 (C6, C8), 142.3
(C4a), 139.1 (C1’), 137.2 (C8a), 131.9 (C7), 131.5 (C5), 130.0 (C2’,C6’), 123.3
(C3’,C5’), 119.5 (C4’), 83.5 (C3), 56.8 (6-OCH3,8-OCH3), 41.4 (C4); MS (70eV): m/z
(%); 298 [M]+ (67), 192 (100), 164 (37), 105 (54), 77 (47); Anal. calcd for C18H18O4: C,
72.48 H, 6.04; Found: C, 72.39 H, 5.96.
2.45 6,8-Dimethoxy-7-methyl-3-(2-chlorophenyl)-3,4-dihydroisocoumarins (18g):
Yield: 86%; m. p. 105-107 ºC; IR (KBr): 3019 (C-H), 1720 (C=O), 1561 (C=C) cm-1
; 1H
NMR (CDCl3, δ ppm): 7.9 (1H, d, J=7.4, H-3’), 7.8 (1H, d, J=6.4, H-6’), 7.6 (1H, dd,
J=6.1, H-4’), 7.5 (1H, dd, J=7.1, H-5’), 6.8 (1H, s, H-5), 5.2 (1H, dd, Jtrans=12.3, Jcis=3.7
H-3), 3.9 (6H, s, 6-OCH3,8-OCH3), 3.2 (1H, dd, Jgem=16.4, Jtrans=12.1 H-4), 3.0 (1H, dd,
Jgem=12.3, Jcis=3.6 H-4), 2.5 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 162.5 (C1),
145.7 (C6, C8), 145.5 (C4a), 139.6 (C1’), 137.6 (C8a), 136.7 (C2’), 132.3 (C5), 130.2
(C6’), 129.4 (C3’), 128.3 (C7), 126.6 (C4’), 124.5 (C5’), 88.2 (C3), 52.7 (6-OCH3,8-
OCH3), 44.7 (C4), 28.8 (Ar-CH3); MS (70eV): m/z (%); 332.5 [M]+ (59), 334.5 [M+2]
(44), 192 (100), 164 (41), 139.5 (63), 111.5 (48); Anal. calcd for C18H17O4Cl: C, 64.96 H,
5.11; Found: C, 64.89 H, 5.02.
2.46 6,8-Dimethoxy-7-methyl-3-(4-methoxyphenyl)-3,4-dihydroisocoumarins
(18h): Yield: 87%; m. p. 141-143 ºC; IR (KBr): 3027 (C-H), 1727 (C=O), 1567 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm): 7.8 (2H, d, J=6.4, H-3’,H-5’), 7.5 (2H, d, J=6.9, H-2’,H-
6’), 6.9 (1H, s, H-5), 5.3 (1H, dd, Jtrans=12.0, Jcis=3.5 H-3), 3.8 (6H, s, 6-OCH3,8-OCH3),
3.5 (1H, dd, Jgem=16.1, Jtrans=11.6 H-4), 3.1 (1H, dd, Jgem=11.8, Jcis=3.8 H-4), 2.8 (3H, s,
Ar-CH3); 13
C NMR (CDCl3, δ ppm): 164.3 (C1), 148.1 (C6, C8), 144.5 (C4a), 138.3
(C1’), 135.6 (C8a), 133.7 (C4’), 132.3 (C2’,C6’), 130.1 (C5), 129.7 (C3’,C5’), 129.3
(C7), 81.3 (C3), 56.6 (6-OCH3,8-OCH3), 52.2 (4’-OCH3), 41.6 (C4), 29.9 (Ar-CH3); MS
Page 69
51
(70eV): m/z (%); 328 [M]+.
(52), 192 (100), 164 (37), 135 (57), 107 (37); Anal. calcd for
C19H20O5: C, 69.51 H, 6.09; Found: C, 69.43 H, 5.99.
2.47 6,8-Dimethoxy-7-methyl-3-(3,4-dimethoxyphenyl)-3,4-dihydroisocoumarins
(18i): Yield: 85%; m. p. 115-117 ºC; IR (KBr): 3022 (C-H), 1711 (C=O), 1573 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm): 7.8 (1H, s, H-2’), 7.7 (1H, d, J=6.8, H-5’), 7.6 (1H, d,
J=5.4, H-6’), 6.8 (1H, s, H-5), 5.1 (1H, dd, Jtrans=12.3, Jcis=3.7 H-3), 3.9 (6H, s, 6-
OCH3,8-OCH3), 3.7 (6H, s, 3’-OCH3,4’-OCH3), 3.4 (1H, dd, Jgem=15.6, Jtrans=11.4 H-4),
3.0 (1H, dd, Jgem=11.6, Jcis=3.5 H-4), 2.5 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm):
168.5 (C1), 144.5 (C6, C8), 140.6 (C4a), 137.5 (C1’), 136.9 (C8a), 133.7 (C3’), 133.2
(C4’), 131.3 (C5), 127.7 (C7), 124.5 (C6’), 123.4 (C2’), 120.2 (C5’), 83.4 (C3), 52.9 (6-
OCH3,8-OCH3), 51.3 (3’-OCH3,4’-OCH3), 45.5 (C4), 29.9 (Ar-CH3); MS (70eV): m/z
(%); 358 [M]+.
(58), 192 (100), 165 (51), 164 (45), 137 (24); Anal. calcd for C20H22O6: C,
67.03 H, 6.4; Found: C, 66.94 H, 6.06.
2.48 6,8-Dimethoxy-7-methyl-3-(3,4,5-trimethoxyphenyl)-3,4-dihydroisocoumarins
(18j): Yield: 82%; m. p. 126-127 ºC; IR (KBr): 3013 (C-H), 1732 (C=O), 1587 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm): 7.9 (2H, s, H-2’,H-6’), 6.9 (1H, s, H-5), 5.3 (1H, dd,
Jtrans=12.1, Jcis=3.5 H-3), 3.8 (6H, s, 6-OCH3,8-OCH3), 3.7 (9H, s, 3’-OCH3,4’-OCH3, 5’-
OCH3), 3.5 (1H, dd, Jgem=15.9, Jtrans=11.8 H-4), 3.1 (1H, dd, Jgem=12.0, Jcis=3.7 H-4), 2.7
(3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 168.5 (C1), 149.6 (C6, C8), 143.3 (C4a),
138.1 (C8a), 136.6 (C1’), 134.3 (C3’,C5’), 132.2 (C4’), 130.5 (C5), 127.9 (C2’,C6’),
127.7 (C7), 83.4 (C3), 56.8 (6-OCH3,8-OCH3), 54.5 (3’-OCH3,4’-OCH3, 5’-OCH3), 45.5
(C4), 30.3 (Ar-CH3); MS (70eV): m/z (%); 388 [M]+.
(69), 195 (39), 192 (100), 167 (25),
164 (19); Anal. calcd for C21H24O7: C, 64.94 H, 6.18; Found: C, 64.87 H, 6.09.
2.49 General procedure for 6,8-dihydroxy-7-methyl-3-alkyl/aryl-3,4-
dihydroisocoumarins (19a-j)
6,8-Dimethoxy-7-methyl-3-alkyl/aryl-3,4-dihydroisocoumarins (18a-j) (1.25
mmol) was dissolved in ethanethiol (3.5 mL) and solution was cooled on ice. Aluminium
chloride (3.8 mmol) was added as 3 portions with an interval of 30 min. After all the
aluminium chloride was added, the reaction mixture was stirred on ice for 1 h. The
reaction was quenched with water, alkalinized (10 % NaHCO3) and extracted with ethyl
acetate, added sodium chloride to enhance layer separation. The combined organic layers
Page 70
52
were washed with brine once, dried over sodium sulfate and concentrated to give 6,8-
dihydroxy-7-methyl-3-alkyl/aryl-3,4-dihydroisocoumarins (19a-j).
2.50 6,8-Dihydroxy-7-methyl-3-propyl-3,4-dihydroisocoumarins (19a): Yield: 71%;
m. p. 93-95 °C; IR (KBr): 3474 (O-H), 3019 (C-H), 1723 (C=O), 1572 (C=C) cm-1
; 1H
NMR (CDCl3, δ ppm): 6.9 (1H, s, H-5), 5.0 (1H, m, H-3), 4.6 (2H, s, 6-OH,8-OH), 3.5
(1H, dd, Jgem=15.9, Jtrans=12.1 H-4), 3.0 (1H, dd, Jgem=15.8, Jcis=3.7 H-4), 2.7 (3H, s, Ar-
CH3), 2.4 (2H, m, H-1’), 1.2 (2H, m, H-2’), 0.9 (3H, t, J=7.5, H-3’); 13
C NMR (CDCl3, δ
ppm): 167.7 (C1), 149.4 (C6, C8), 143.1 (C4a), 138.8 (C8a), 129.2 (C5), 118.3 (C7), 81.6
(C3), 44.3 (C4), 37.3 (C1’), 29.4 (Ar-CH3), 18.1 (C2’), 10.2 (C3’); MS (70eV): m/z (%);
236 [M]+ (32), 164 (100), 136 (17), 71 (37), 43 (5); Anal. calcd for C13H16O4: C, 66.10 H,
6.77; Found: C, 66.02 H, 6.69.
2.51 6,8-Dihydroxy-7-methyl-3-pentyl-3,4-dihydroisocoumarins (19b): Yield: 73%;
m. p. °C; IR (KBr): 3456 (O-H), 3021 (C-H), 1719 (C=O), 1559 (C=C) cm-1
; 1
H NMR
(CDCl3, δ ppm): 7.1 (1H, s, H-5), 5.3 (1H, m, H-3), 4.9 (2H, s, 6-OH,8-OH), 3.4 (1H, dd,
Jgem=16.0, Jtrans=12.4 H-4), 3.1 (1H, dd, Jgem=16.3, Jcis=4.2 H-4), 2.5 (3H, s, Ar-CH3), 2.5
(2H, m, H-1’), 1.2-1.6 (6H, m, H-2’,H-3’,H-4’), 0.9 (3H, t, J=5.9, H-5’); 13
C NMR
(CDCl3, δ ppm): 163.7 (C1), 151.4 (C6, C8), 145.5 (C4a), 139.9 (C8a), 127.3 (C5), 123.3
(C7), 79.3 (C3), 42.5 (C4), 39.5 (C1’), 30.1 (Ar-CH3), 21.3 (C2’), 18.5 (C3’), 12.9 (C4’),
11.6 (C5’); MS (70eV): m/z (%); 264 [M]+ (53), 164 (100), 136 (21), 99 (57), 71 (20);
Anal. calcd for C15H20O4: C, 68.18 H, 7.57; Found: C, 68.11 H, 7.49.
2.52 6,8-Dihydroxy-7-methyl-3-heptyl-3,4-dihydroisocoumarins (19c): Yield: 79%;
m. p. 115-117 ºC; IR (KBr): 3467 (O-H), 3017 (C-H), 1727 (C=O), 1569 (C=C) cm-1
; 1
H
NMR (CDCl3, δ ppm): 6.9 (1H, s, H-5), 4.9 (1H, m, H-3), 4.5 (2H, s, 6-OH,8-OH), 3.6
(1H, dd, Jgem=15.7, Jtrans=11.9 H-4), 3.2 (1H, dd, Jgem=16.1, Jcis=3.9 H-4), 2.4 (3H, s, Ar-
CH3), 2.4 (2H, m, H-1’), 1.1-1.4 (10H, m, H-2’,H-3’,H-4’,H-5’,H-6’), 0.9 (3H, t, J=5.4,
H-7’); 13
C NMR (CDCl3, δ ppm): 166.9 (C1), 148.7 (C6, C8), 142.9 (C4a), 136.3 (C8a),
119.8 (C5), 123.3 (C7), 80.7 (C3), 43.8 (C4), 38.7 (C1’), 29.4 (Ar-CH3), 19.5 (C2’), 13.6
(C3’), 12.9 (C4’), 12.1 (C5’), 11.4 (C6’), 10.4 (C7’); MS (70eV): m/z (%); 292 [M]+ (39),
164 (100), 136 (5), 127 (43), 99 (43); Anal. calcd for C17H24O4: C, 69.86 H, 8.21; Found:
C, 69.78 H, 8.13.
Page 71
53
2.53 6,8-Dihydroxy-7-methyl-3-chloromethyl-3,4-dihydroisocoumarins (19d):
Yield: 73%; m. p. 73-75 °C; IR (KBr): 3445 (O-H), 3010 (C-H), 1717 (C=O), 1571
(C=C) cm-1
; 1
H NMR (CDCl3, δ ppm): 6.9 (1H, s, H-5), 5.5 (1H, dd, Jgem=15.8,
Jtrans=12.1 H-1’), 5.1 (1H, dd, Jgem=15.6, Jcis=4.1 H-1’), 4.8 (1H, m, H-3), 4.7 (2H, s, 6-
OH,8-OH), 3.5 (1H, dd, Jgem=15.9, Jtrans=12.5 H-4), 3.1 (1H, dd, Jgem=16.2, Jcis=3.7 H-4),
2.7 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 168.9 (C1), 150.5 (C6, C8), 142.9 (C4a),
136.3 (C8a), 125.1 (C5), 127.8 (C7), 85.8 (C3), 48.7 (C1’), 41.8 (C4), 27.4 (Ar-CH3);
MS (70eV): m/z (%); 242.5 [M]+ (32), 244.5 [M+2] (24), 164 (100), 77.5 (49), 49.5 (31);
Anal. calcd for C11H11O4Cl: C, 54.43 H, 4.53; Found: C, 54.35 H, 4.35.
2.54 6,8-Dihydroxy-7-methyl-3-hydroxymethyl-3,4-dihydroisocoumarins (19e):
Yield: 81%; m. p. 89-90 °C; IR (KBr): 3481 (O-H), 3009 (C-H), 1720 (C=O), 1554
(C=C) cm-1
; 1
H NMR (CDCl3, δ ppm): 6.7 (1H, s, H-5), 5.4 (1H, dd, Jgem=16.3,
Jtrans=12.1 H-1’), 4.8 (1H, dd, Jgem=15.7, Jcis=3.8 H-1’), 4.6 (1H, m, H-3), 4.5 (2H, s, 6-
OH,8-OH), 3.6 (1H, dd, Jgem=16.3, Jtrans=12.1 H-4), 3.2 (1H, dd, Jgem=15.8, Jcis=3.5 H-4),
2.9 (3H, s, Ar-CH3), 2.1 (1H, s, -OH); 13
C NMR (CDCl3, δ ppm): 165.5 (C1), 149.2 (C6,
C8), 140.5 (C4a), 138.8 (C8a), 128.9 (C7), 121.5 (C5), 88.7 (C3), 65.3 (C1’), 39.1 (C4),
30.2 (Ar-CH3); MS (70eV): m/z (%); 224 [M]+ (34), 164 (100), 136 (31), 59 (51), 31
(43); Anal. calcd for C11H12O5: C, 58.92 H, 5.35; Found: C, 58.83 H, 5.26.
2.55 6,8-Dihydroxy-7-methyl-3-phenyl-3,4-dihydroisocoumarins (19f): Yield:
86%; m. p. 125-126 ºC; IR (KBr): 3477 (O-H), 3020 (C-H), 1722 (C=O), 1571 (C=C)
cm-1
; 1
H NMR (CDCl3, δ ppm): 7.9 (2H, d, J=5.8, H-2’,H-6’), 7.7 (2H, dd, J=7.9, H-
3’,H-5’), 7.5 (1H, dd, J=6.7, H-4’), 7.0 (1H, s, H-5), 5.3 (1H, dd, Jtrans=12.3, Jcis=3.8 H-
3), 4.7 (2H, s, 6-OH,8-OH), 3.3 (1H, dd, Jgem=15.7, Jtrans=12.1 H-4), 3.0 (1H, dd,
Jgem=12.0, Jcis=3.7 H-4), 2.5 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 162.3 (C1),
151.3 (C6, C8), 142.3 (C4a), 138.1 (C1’), 137.2 (C8a), 131.9 (C7), 129.4 (C2’,C6’),
126.3 (C3’,C5’), 123.5 (C5), 119.5 (C4’), 81.5 (C3), 44.6 (C4); MS (70eV): m/z (%); 270
[M]+ (41), 164 (100), 136 (51), 105 (51), 77 (23); Anal. calcd for C16H14O4: C, 71.11 H,
5.18; Found: C, 71.04 H, 5.11.
2.56 6,8-Dihydroxy-7-methyl-3-(2-chlorophenyl)-3,4-dihydroisocoumarins (19g):
Yield: 84%; m. p. 129-131 ºC; IR (KBr): 3488 (O-H), 3023 (C-H), 1720 (C=O), 1561
(C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.9 (1H, d, J=7.1, H-3’), 7.8 (1H, d, J=6.4, H-6’),
Page 72
54
7.6 (1H, dd, J=6.5, H-4’), 7.5 (1H, dd, J=7.3, H-5’), 6.9 (1H, s, H-5), 5.5 (1H, dd,
Jtrans=12.1, Jcis=3.4 H-3), 4.9 (2H, s, 6-OH,8-OH), 3.4 (1H, dd, Jgem=16.1, Jtrans=12.1 H-
4), 3.0 (1H, dd, Jgem=12.3, Jcis=3.6 H-4), 2.9 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm):
166.5 (C1), 148.2 (C6, C8), 145.5 (C4a), 139.6 (C1’), 138.6 (C8a), 137.7 (C2’), 132.3
(C5), 130.2 (C6’), 129.4 (C3’), 128.3 (C7), 125.6 (C4’), 124.5 (C5’), 83.2 (C3), 40.7
(C4), 29.8 (Ar-CH3); MS (70eV): m/z (%); 304.5 [M]+ (35), 306.5 [M+2] (26), 164 (100),
136 (21), 139.5 (53), 111.5 (48); Anal. calcd for C16H13O4Cl: C, 63.05 H, 4.27; Found: C,
62.97 H, 4.18.
2.57 6,8-Dihydroxy-7-methyl-3-(4-methoxyphenyl)-3,4-dihydroisocoumarins (19h):
Yield: 83%; m. p. 161-162 ºC; IR (KBr): 3467 (O-H), 3018 (C-H), 1724 (C=O), 1567
(C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.7 (2H, d, J=6.9, H-3’,H-5’), 7.4 (2H, d, J=6.5,
H-2’,H-6’), 6.9 (1H, s, H-5), 5.2 (1H, dd, Jtrans=12.2, Jcis=3.7 H-3), 4.8 (2H, s, 6-OH,8-
OH), 3.5 (1H, dd, Jgem=15.8, Jtrans=11.6 H-4), 3.1 (1H, dd, Jgem=12.1, Jcis=3.9 H-4), 2.8
(3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 161.3 (C1), 150.1 (C6, C8), 144.5 (C4a),
138.3 (C1’), 135.6 (C8a), 133.7 (C4’), 132.3 (C2’,C6’), 131.7 (C5), 129.7 (C3’,C5’),
128.3 (C7), 83.5 (C3), 55.4 (4’-OCH3), 45.6 (C4), 29.9 (Ar-CH3); MS (70eV): m/z (%);
300 [M]+ (39), 164 (100), 136 (27), 135 (52), 107 (19); Anal. calcd for C17H16O5: C,
68.00 H, 5.33; Found: C, 67.93 H, 5.26.
2.58 6,8-Dihydroxy-7-methyl-3-(3,4-dimethoxyphenyl)-3,4-dihydroisocoumarins
(19i): Yield: 85%; m. p. 153-154 ºC; IR (KBr): 3479 (O-H), 3017 (C-H), 1719 (C=O),
1573 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.9 (1H, s, H-2’), 7.8 (1H, d, J=6.5, H-5’),
7.6 (1H, d, J=5.4, H-6’), 6.9 (1H, s, H-5), 5.3 (1H, dd, Jtrans=12.1, Jcis=3.4 H-3), 4.6 (2H,
s, 6-OH,8-OH), 3.7 (6H, s, 3’-OCH3,4’-OCH3), 3.4 (1H, dd, Jgem=15.6, Jtrans=11.4 H-4),
3.0 (1H, dd, Jgem=11.6, Jcis=3.5 H-4), 2.9 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm):
167.5 (C1), 146.5 (C6, C8), 140.6 (C4a), 137.5 (C1’), 136.9 (C8a), 133.7 (C3’), 133.2
(C4’), 131.3 (C5), 127.7 (C7), 126.5 (C6’), 123.4 (C2’), 120.2 (C5’), 80.4 (C3), 55.4 (3’-
OCH3,4’-OCH3), 41.5 (C4), 29.9 (Ar-CH3); MS (70eV): m/z (%); 330 [M]+ (44), 164
(100), 165 (48), 137 (20), 136 (25),; Anal. calcd for C18H18O6: C, 65.45 H, 5.45; Found:
C, 65.37 H, 5.36.
2.59 6,8-Dihydroxy-7-methyl-3-(3,4,5-trimethoxyphenyl)-3,4-dihydroisocoumarins
(19j): Yield: 82%; m. p. 145-147 ºC; IR (KBr): 3459 (O-H), 3010 (C-H), 1716 (C=O),
Page 73
55
1587 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.8 (2H, s, H-2’,H-6’), 6.9 (1H, s, H-5), 5.1
(1H, dd, Jtrans=11.8, Jcis=3.7 H-3), 4.5 (2H, s, 6-OH,8-OH), 3.7 (9H, s, 3’-OCH3,4’-OCH3,
5’-OCH3), 3.5 (1H, dd, Jgem=15.9, Jtrans=11.8 H-4), 3.1 (1H, dd, Jgem=12.0, Jcis=3.7 H-4),
2.7 (3H, s, Ar-CH3); 13
C NMR (CDCl3, δ ppm): 166.5 (C1), 149.6 (C6, C8), 143.3 (C4a),
138.1 (C8a), 136.6 (C1’), 134.3 (C3’,C5’), 132.2 (C4’), 130.5 (C5), 127.9 (C2’,C6’),
127.7 (C7), 84.5 (C3), 54.7 (3’-OCH3,4’-OCH3, 5’-OCH3), 42.5 (C4), 27.9 (Ar-CH3); MS
(70eV): m/z (%); 360 [M]+ (32), 195 (34), 167 (155), 164 (100); Anal. calcd for
C19H20O7: C, 63.33 H, 5.55; Found: C, 63.26 H, 5.47.
Page 74
56
3. RESULTS AND DISCUSSION
Total synthesis of a natural 3,4-dihydroisocoumarins (Stellatin) and those of the
structural analogues of some naturally occurring bioactive isocoumarins and 3,4-
dihydroisocoumarins viz. Hiburipyranone, Cytogenin, Montroumarin, Scorzocreticin,
Annulatomarin, Thunberginol B, Stoloniferol A) has been carried out.
3,5-Dimethoxy-4-methylhomophthalic was synthesized starting from commercially
available inexpensive 4-methylbenzoic acid (p-toluic acid). It was then converted into
3,5-dimethoxy-4-methyl homophthalic anhydride. The latter was then condensed with
various acyl and aroyl chlorides to afford 6,8-dimethoxy-7-methyl-3-alkyl/arylisoco-
umarins. The synthesized isocoumarins were then converted into corresponding 6,8-
dimethoxy-7-methyl-3-alkyl/aryl-3,4-dihydroisocoumarins via hydrolysis and reduction.
Finally, 3,4-dihydroisocoumarins were demethylated to give 6,8-dihydroxy-7-methyl-3-
alkyl/aryl-3,4-dihydroisocoumrins.
3.1 Synthesis of 3,5-dimethoxy-4-methylhomophthalic acid (12)
4-Methylbenzoic acid was converted into methyl 4-methylbenzoate (1) using
methanol in presence of conc. sulfuric acid as catalyst, which showed C=O stretching at
1742 cm-1
in IR spectrum.
Nuclear halogenation of the acid derivative (1) to methyl 3,5-dibromobenzoate (2)
was carried out by using “swamping catalyst method”. It involves the use of an excess of
anhydrous aluminum chloride or aluminum bromide as catalyst and no solvent is used.
The aluminum chloride complexes with the carbonyl group of the acid derivative, thus
suppressing the side chain halogenation. One mole of catalyst complexes with the
carbonyl, while the second mole increases the activity of attacking reagent, by producing
the highly electrophilic free Br+
ion or the ion pair Br+ AlCl3 Br
.
The 4-methyl group in (1) is ideally situated for directing the incoming bromides
to C-3 and C-5 positions since the dibromination of methyl benzoate affords the methyl
3,5-dibromobenzoate.
Nucleophilic substitution of a pyridine solution of methyl 3,5-dibromobenzoate
(2) with sodium methoxide was carried out in presence of freshly prepared copper (I)
chloride as catalyst to afford methyl 3,5-dimethoxy-4-methylbenzoate. In this reaction
3,5-dimethoxy-4-methylbenzoic acid (3) was directly obtained due to concurrent
Page 75
57
hydrolysis, which was confirmed by the presence of a broader stretching at 3212 cm-1
for
(O-H) in IR spectrum. In 1H NMR the six methoxy protons appeared as a singlet at δ 3.89
ppm and carboxyl showed a singlet at δ 8.95 ppm. 13
C NMR showed a signal for
carboxyl carbon at δ 168.6 ppm and the methoxy carbons appeared at δ 56.3 ppm.
The acid (3) was converted into its methyl ester (4) by using methanol in the
presence of conc. sulfuric acid as catalyst which showed the disappearance of a broad
band for (O-H) in IR spectrum. The methyl ester (4) was then reduced to 3,5-dimethoxy-
4-methylbenzyl alcohol (5) using sodium borohydride-methanol system refluxed in THF.
The IR showed a broad band at 3421 cm-1
for the hydroxyl group and also the
disappearance of carbonyl absorption was noted. The reduction of esters and similar
functional groups using sodium borohydride is relatively difficult to obtain and it has not
been widely used. However, the reactivity of the sodium borohydride can be enhanced by
carrying out the reaction in the presence of NaBH4-CH3OH. This methodology is simple,
safe, inexpensive, and general and the reduction of methyl esters was completed after
refluxing in THF.
The alcohol (5) was converted to 3,5-dimethoxy-4-methylbenzyl bromide (6) in
the presence of phosphorous tribromide (PBr3) and dry benzene in 84% yield. In IR
spectrum the absence of signals due to hydroxyl group was noticed. Nucleophilic
substitution of the bromide (6) by the cyanide, using potassium cyanide and ethanol
furnished the 3,5-dimethoxy-4-methylbenzyl cyanide (7) which showed the nitrile
absorption at 2284 cm-1
in IR spectrum.
The alkaline hydrolysis of the nitrile (7) was carried out using aqueous methanolic
potassium hydroxide in dioxane to afford the 3,5-dimethoxy-4-methylphenyl acetic acid
(8) in 71% yield. The IR showed a strong absorption at 1707 cm-1
for carbonyl and a
broad band at 3242 cm-1
for hydroxyl group. The phenylacetic acid (8) was then
converted into its methyl ester (9). In IR spectrum absorption for carbonyl appeared at
1734 cm-1
and disappearance of a broad band for hydroxyl group was observed. In 1H
NMR a singlet for methoxy protons of ester appeared at δ 3.45 ppm.
Page 76
58
CH3
O
OH
CH3
O
OCH3
NaBH4/THF
CH3OH
(1)
CH3
O
OH
OCH3
H3CO
(3)
CH3
O
OCH3
Br
Br
(2)
CH3
O
OCH3
OCH3
H3CO
(4)
CH3
OCH3
H3COOH
(5)
i) Br2 /AlCl
3 (Anhd.)
ii) CH3OH
CH3OH / H+
CH3OH / H+NaOCH3, Pyridine
Cu2Cl
2 (Anhd.)
Reflux for 15 h under nitrogen
CH3
OCH3
H3CO Br
CH3
OCH3
H3CO O
OH
CH3
OCH3
H3CO O
OCH3
(6)
(8) (9)
CH3
OCH3
H3COCN
(7)
KCN
C2H5OH / H2O
CH3OH
H+
KOH / H2O
Dioxane / CH3OH
PBr3
Benzene (dry)
Scheme 3.1 Synthesis of methyl 3,5-dimethoxy-4-methylphenyl acetate (9)
Vilsmeier Haack formylation of the acetate (9) using phosphorus oxychloride in
N, N-dimethylformamide (DMF) afforded the methyl (2-formyl-3, 5-dimethoxy-4-methyl
phenyl)acetate (10). The IR showed very strong new carbonyl absorption at 1690 cm-1
for
aldehydic carbonyl in addition to the 1722 cm-1
peak for ester carbonyl already present.
The 1H NMR showed the singlet for aldehydic proton at δ 9.75 and the characteristic
changes in the chemical shifts of the benzylic protons. 13
C NMR showed a peak at δ
179.3 for aldehydic carbon and a peak at 162.4 for ester carbon already present. The
molecular ion peak appeared at m/z 252 and the base peak at m/z 165.
The aldehyde (10) was oxidized to 2,4-dimethoxy-6-(2-methoxy-2-oxoethyl)-3-
methylbenzoic acid (11) using sulfamic acid and sodium chlorite at 0°C in 79% yield.
The carbonyl absorption in IR shifted from 1690 cm-1
to 1715 cm-1
due to oxidation of
aldehydic function into carboxyl one. The absorption at 3265 cm-1
for (O-H) is also
Page 77
59
present in IR spectrum. In 1H NMR, a singlet at δ 8.19 appeared for carboxyl proton and
downfield shift from δ 179.3 to δ 197.7 for carboxylic carbon was also observed in 13
C
NMR.
The alkaline hydrolysis of the ester acid (11) to 3,5-dimethoxy-4-methylhomo-
phthalic acid (12) was carried out by using 10% potassium hydroxide and ethanol in 87%
yield. The physical and FT-IR spectral data of the compounds (1-12) are shown in Table
3.1.
CH3
OCH3
H3CO O
OCH3CH3
OCH3
H3CO O
OCH3O
H
CH3
OCH3
H3CO O
OHO
OH
10%KOH
C2H5OH
(9)(10)
(12)
CH3
OCH3
H3CO O
OCH3O
OH
(11)
POCl3 / Freshly distilled DMF
CH3COO- Na
+
NH2SO3H / NaClO2 / 0 °C
H2O:THF:DMSO
Scheme 3.2 Synthesis of 3,5-dimethoxy-4-methyl homophthalic acid (12)
Table 3.1 Physical constants and FTIR spectral data of the compounds (1-12)
Compds m. p.
(°C) Rf
Yield
(%) υmax (cm
-1)
Ar-H Sp3 C-H C=O C=C O-H
1 34 0.7 95 3012 2904 1742 1562 -
2 82-84 0.6 58 3021 2917 1719 1559 -
3 210-212 0.4 74 3029 2931 1702 1569 3213
4 76-78 0.65 84 3019 2913 1732 1571 -
5 45-47 0.5 81 3023 2943 - 1554 3421
6 68-69 0.6 84 3013 2958 - 1571 -
7 48-49 0.5 84 3007 2924 - 1561 -
8 106-107 0.4 71 3013 2914 1707 1567 3242
9 38-40 0.7 88 3023 2925 1734 1573 -
10 51-53 0.55 84 3029 2917 1690, 1722 1545 -
11 164-166 0.4 79 3037 2928 1734, 1715 1562 3265
12 180-182 0.4 87 3013 2934 1741 1587 3195
Pet.Ether: Ethyl Acetate (4:1)
Page 78
60
Table 3.2 1H and
13C NMR data of the compound (9)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 112.2
C-2 7.45, s 128.3
C-3 ----- 132.5
C-4 ----- 119.4
C-5 ----- 132.5
C-6 7.45, s 128.3
3-OCH3 3.96, s 55.3
5-OCH3 3.96, s 55.3
4-CH3 2.55, s 28.6
Ar-CH2 3.54, s 36.9
C=O ----- 168.2
Ester-OCH3 3.47, s 68.5
The conversion of 3,5-dimethoxy-4-methyl phenyl acetic acid (8) to methyl ester
(9) was confirmed in 1H NMR spectrum by the presence of a singlet at δ 3.47 ppm for
methoxy protons of ester and the disappearance of the signal for hydroxyl group. It was
also supported in 13
C NMR spectrum by the presence of a peak at δ 68.5 ppm for carbon
of the ester methoxy along with signals for two methoxy already present. The detailed 1H
and 13
C NMR data of the compound (9) are presented in Table 3.2.
The structure of the compound was further confirmed by mass spectrometry. The
molecular ion peak appeared at m/z 224 with 46% abundance which established the
formation of the ester (9). A peak at m/z 165 is the base peak formed by the elimination
of methoxy and carbon monoxide. The fragmentation pattern of the ester (9) is shown in
Fig. 1.
Page 79
61
OCH3
H3CO
CH3
COOCH3
(m/z= 224, 46%)
-OCH3
(9)
+
..
+
OCH3
H3CO
CH3
O+
(m/z= 193, 43%)
-CO
OCH3
H3CO
CH3
+
(m/z= 165, 100%)
-CH2OOCH3
CH3
COOCH3
(m/z= 194, 37%)
-
OCH3
H3CO
CH3
CH3COO
.
(m/z= 59, 12%)
+
-CH2O
CH3
COOCH3
(m/z= 164, 23%)
+
CH3
O
(m/z= 133, 19%)
+
-OCH3
.
-CO CH3
(m/z= 105, 35%)
.+
Fig. 3.1 Mass fragmentation pattern of the compound (9)
Table 3.3 1H and
13C NMR data of the compound (10)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 117.5
C-2 ----- 131.8
C-3 ----- 136.7
C-4 ----- 121.3
C-5 ----- 136.7
C-6 7.96, s 126.2
3-OCH3 3.42, s 57.3
5-OCH3 3.25, s 57.3
4-CH3 2.80, s 32.4
Ar-CH2 2.92, s 39.1
C=O ----- 162.4
Ester-OCH3 3.11, s 61.3
CHO 9.75, s 179.3
Page 80
62
The formylation of the ester (9) into methyl(2-formyl-3,5-dimethoxy-4-methyl
phenyl)acetate (10) was confirmed in 1H NMR spectrum by the presence of a singlet at δ
9.75 ppm for aldehyde proton. It was also supported in 13
C NMR spectrum by the
presence of a peak at δ 179.3 ppm for carbon of the formyl group along with signals for
two methoxy already present. The detailed 1H and
13C NMR data of the compound (10)
are presented in Table 3.3.
The structure of the compound was further confirmed by mass spectrometry. The
molecular ion peak appeared at m/z 252 with 25% abundance which proved the formation
of the aldehyde (10). By the removal of carbon monoxide from molecular ion a peak at
m/z 224 with percentage abundance 49% appeared. A peak at m/z 165 is the base peak
formed by the elimination of acetate radical from ion having m/z 224. The fragmentation
pattern of the aldehyde (10) is shown in Fig. 2.
OCH3
H3CO
CH3
COOCH3
O
H
(m/z= 252, 25%)
(10)
+
.
.
+
OCH3
H3CO
CH3
COOCH3
O
+
(m/z= 251, 65%)
-CO
OCH3
H3CO
CH3
COOCH3
+
(m/z= 223, 49%)
OCH3
CH3
COOCH3
H3CO
(m/z= 224, 49%)
.+
-
OCH3
H3CO
CH3
COOCH3
CHO
.
(m/z= 29, 31%)
+
-CH2O
CH3
COOCH3
H3CO
(m/z= 194, 21%)
+
CH3
COOCH3
(m/z= 164, 27%)
CH3
(m/z= 105, 31%)
-H
-CO
-CH2O
-CH3COO
OCH3
CH3
H3CO
(m/z= 165, 100%)
+
+
-CH3COO
Fig. 3.2 Mass fragmentation pattern of the compound (10)
Page 81
63
Table 3.4 1H and
13C NMR data of the compound (11)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 114.1
C-2 ----- 134.3
C-3 ----- 139.3
C-4 ----- 120.6
C-5 ----- 139.3
C-6 7.66, s 127.1
3-OCH3 3.82, s 55.4
5-OCH3 3.67, s 55.4
4-CH3 2.25, s 35.0
Ar-CH2 2.92, s 39.1
C=O ----- 168.5
Ester-OCH3 3.63, s 66.0
COOH 8.19, s 197.7
OCH3
H3CO
CH3
COOCH3
O
OH
(m/z= 268, 32%)
(11)
+
.
.
+
OCH3
H3CO
CH3
COOCH3
O
+
(m/z= 251, 51%)
-CO
OCH3
H3CO
CH3
COOCH3
+
(m/z= 223, 36%)
OCH3
CH3
COOCH3
H3CO
(m/z= 224, 65%)
.+
-
OCH3
H3CO
CH3
COOCH3
COOH
.
(m/z= 45, 25%)
+
-CH2O
CH3
COOCH3
H3CO
(m/z= 194, 16%)
+
CH3
COOCH3
(m/z= 164, 32%)
CH3
(m/z= 105, 23%)
-CH2O
-CH3COO
OCH3
CH3
H3CO
(m/z= 165, 100%)
+
+
-CH3COO
-OH
-CO2
Fig. 3.3 Mass fragmentation pattern of the compound (11)
Page 82
64
Table 3.5 1H and
13C NMR data of the compound (12)
Carbons
δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 124.4
C-2 ----- 135.2
C-3 ----- 136.2
C-4 ----- 125.0
C-5 ----- 136.2
C-6 7.60, s 133.0
3-OCH3 3.85, s 55.4
5-OCH3 3.67, s 55.4
4-CH3 2.25, s 29.5
Ar-CH2 2.53, s 39.7
Ar-COOH 10.91, s 205.3
COOH 10.70, s 171.1
The formation of the 3,5-dimethoxy-4-methylhomophthalic acid (12) was
confirmed by the presence of two singlets at δ 10.91 ppm and 10.70 ppm in 1H NMR
spectrum for two protons of two carboxylic acids. It was also supported by 13
C NMR
spectrum due to the presence of two peaks at δ 205.3 and 171.1 ppm for two carboxylic
groups. The detailed 1H and
13C NMR data of the compound (12) are presented in Table
3.5.
In mass spectrometry, the molecular ion peak appeared at m/z 254 with
percentage abundance 46% which confirmed the formation of homophthalic acid (12). By
the removal of water from molecular ion, a peak at m/z 236 with percentage abundance
48% appeared. A peak at m/z 192 is the base peak formed by the elimination of carbon
dioxoide from the ion having m/z 236. The fragmentation pattern of the homophthalic
acid (12) is shown in Fig. 4.
Page 83
65
OCH3
H3CO
CH3
COOH
O
OH
(m/z= 254, 46%)
(12)
.
.
+
OCH3
H3CO
CH3
COOH
O
+
(m/z= 237, 57%)
-CO
OCH3
H3CO
CH3
COOH+
(m/z= 209, 23%)
OCH3
CH3
COOHH3CO
(m/z= 210, 34%)
.+
-
(m/z= 236, 48%)
CH3
CH3H3CO
OCH3
(m/z= 166, 29%)
+
CH3
CH3H3CO
(m/z= 136, 18%)
-CH2O
(m/z= 192, 100%)
+
+
-OH
-CO2
H2O
+
OCH3
H3CO
CH3
O
O
OCH2
OCH3
CH3
CH3
-CO2
-CO2
.
-CH2O
CH3
CH3
(m/z= 106, 25%)
+
Fig. 3.4 Mass fragmentation pattern of the compound (12)
3.2 Synthesis of 6,8-dimethoxy-7-methyl-3-alkyl/aryl isocoumarins (16a-j)
3,5-Dimethoxy-4-methylhomophthalic acid (12) was converted into
corresponding anhydride (13) by refluxing it with acetic anhydride in the presence of dry
toluene as solvent. The formation of the anhydride (13) was confirmed by the presence of
absorption at 1735 cm-1
and disappearance of the absorption for hydroxyl groups in IR
spectrum. The 1H NMR and
13C NMR data of the homophthalic anhydride (13) are
presented in Table 3.7.
O
O
OCH3
H3CO
CH3
OHOH
(12)
O
O
O
OCH3
H3CO
CH3
(13)
Acetic Anhydride
Dry Toluene
Page 84
66
Table 3.6 Physical constants and FT-IR spectral data of the compound (13)
Compd M.P.
°C
Yield
% Rf
υmax (cm-1
)
Ar-H Sp3 C-H C=O C=C
13 135-136 82 0.7 3011 2913 1735
1590
Table 3.7 1H and
13C NMR data of the compound (13)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 149.5
C-3 ----- 168.1
C-4 3.45, s 38.7
C-4a ----- 136.2
C-5 6.72, s 104.4
C-6 ----- 163.2
C-7 ----- 110.5
C-8 ----- 163.2
C-8a ----- 106.0
6-OCH3 3.85, s 56.3
8-OCH3 3.85, s 56.3
7-CH3 2.28, s 27.4
In 1H NMR spectrum the methylene protons of C-4 appeared as a singlet at 3.45
ppm. Aromatic hydrogen H-5 gives a singlet at δ 6.72 and a singlet for two methoxy
groups appeared at δ 3.85. The structure of the compound was also confirmed by 13
C
NMR. The carbonyl carbons C1 and C3 appeared at δ 149.5 and 168.1 respectively.
The aliphatic and aromatic carboxylic acids (14a-j) were converted into their
respective acid chlorides by treatment with thionyl chloride (15a-j) in the presence of
catalytic amount of DMF. Acid chlorides (15a-j) were then condensed with homophthalic
anhydride (13) in the presence of triethyl amine and tetramethyl guanidine to afforded
6,8-dimethoxy-7-methyl-3-alkyl/arylisocoumarins (16a-j). These isocoumarins were
purified by preparative thin layer chromatography.
Page 85
67
The synthesized isocoumarins (16a-j) showed the characteristic absorption for
lactonic carbonyl at 1713-1736 cm-1
in IR spectra. The physical constants and FTIR
spectral data of the compounds (16a-j) is shown in table 3.8 and the elemental analysis
data of these isocoumarins is presented in table 3.9. The isocoumarins showed the
characteristic 1H singlet of isocoumarin moiety (H-4) at δ 6.4-7.3 ppm in 1H NMR
spectrum and the lactonic carbonyl carbon in 13
C NMR showed the peak at δ 158-170
ppm.
R=
14a = 14b = 14c =
14d = -CH2Cl 14e = -CH2OH
14f = 14g = 14h = 14i = 14j =Cl
OCH3
OCH3
OCH3
OCH3
OCH3
H3CO
R
O
OH
+ SOCl2 R
O
Cl
(15a-j)
CH2 CH3_
CH2 CH3_
CH2 CH3_
(14a-j)
R is same as in (14a-j)
R
O
Cl
+O
O
O
OCH3
H3CO
CH3
(13)
O
OOCH3
H3CO
CH3
R
(16a-j)(15a-j)
TMG / (C2H5)3N
CH3CN
R is same as in (14a-j)
Scheme 3.3 Synthesis of 6,8-dimethoxy-7-methyl-3-alkyl/arylisocoumarins (16a-j)
Page 86
68
Table 3.8 Physical constants and FTIR spectral data of the compounds (16a-j)
Compds. M. P.
(°C) Rf
Yield
(%) υmax (cm
-1)
Ar-C-H Sp3 C-H C=O C=C
16a oil 0.7 72 3031 2924 1713 1572
16b oil 0.8 75 3037 2933 1731 1559
16c 88-90 0.8 79 3021 2924 1719 1569
16d oil 0.6 70 3033 2931 1722 1571
16e oil 0.5 81 3023
3345(O-H) 2914 1729 1554
16f 109-111 0.6 89 3033 2931 1715 1571
16g 119-121 0.7 84 3017 2924 1727 1561
16h 154-156 0.5 87 3023 2914 1723 1567
16i 122-124 0.6 85 3029 2925 1736 1573
16j 135-137 0.7 87 3013 2934 1713 1587
Pet. Ether: Ethyl Acetate (4:1)
Table 3.9 Elemental analysis data of the compounds (16a-j)
Compounds
Molecular Formula
Calculated Found
C H C H
C15H18O4 (16a) 68.70 6.87 68.57 6.69
C17H22O4 (16b) 70.34 7.58 70.19 7.42
C19H26O4 (16c) 71.69 8.17 71.54 8.01
C13H13O4Cl (16d) 58.10 4.84 57.95 4.69
C13H14O5 (16e) 62.40 5.60 62.27 5.42
C18H16O4 (16f) 72.97 5.40 72.83 5.26
C18H15O4Cl (16g) 65.35 4.53 65.19 4.39
C19H18O5 (16h) 69.93 5.52 69.74 5.36
C20H20O6 (16i) 67.41 5.61 67.28 5.45
C21H22O7 (16j) 65.28 5.69 65.14 5.57
Page 87
69
Table 3.10 1H and
13C NMR data of compound (16a)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 167.8
C-3 ----- 151.2
C-4 7.3, s 110.3
C-4a ----- 133.1
C-5 7.8, s 104.5
C-6 ----- 145.4
C-7 ----- 118.3
C-8 ----- 145.4
C-8a ----- 128.8
C-1’ 2.5, t, J=3.9 38.6
C-2’ 1.2, m 21.1
C-3’ 0.9, t, J=7.5 14.3
6-OCH3 3.9, s 53.6
8-OCH3 3.9, s 53.6
7-CH3 2.6, s 29.7
The formation of the 6,8-dimethoxy-7-methyl-3-propylisocoumarin (16a) was confirmed
by the presence of a singlet at δ 7.3 ppm in 1H NMR spectrum for H-4. It was also
supported in 13
C NMR spectrum by the presence of a peak at δ 167.8 ppm for lactonic
carbon. Table 3.10 gives the 1H and
13C NMR data of the compound (16a).
In mass spectrometry the molecular ion peak appeared at m/z 262 with 26%
abundance which confirmed the formation of isocoumarin (16a). By the removal of
propyl radical from molecular ion, a peak appeared at m/z 219 with 47% abundance. A
peak at m/z 191 is the base peak formed by the elimination of butanoyl radical from
molecular ion. The fragmentation pattern of the isocoumarin (16a) is shown in fig. 5.
The structures of all other isocoumarins (16b-j) were confirmed by the presence
H-4 singlet in 1H NMR along with other signals in acceptable regions. The signal for
lactonic carbonyl carbon in 13
C NMR also helpful for the confirmation of structures of all
these compounds.
Page 88
70
(m/z = 262, 26%)(m/z = 191, 100%)
CH+
O
H3CO
OCH3
CH3
._
(m/z = 71, 45%)
(m/z = 163, 55%)
CH+
H3CO
OCH3
CH3
CO_
CO_
(m/z =43, 59%)
.
O
OCH3
CH3
H3CO
_
_ .
O
O
H3CO
OCH3
CH3
(m/z = 219, 47%)
CO2_ H3CO
OCH3
CH3
(m/z = 175, 17%)
_CO
O
OCH3
CH3
CH3H3CO
(m/z = 234, 40%)
+.
++
+
+
+
+
+
O
O
H3CO
OCH3
CH3
CH3
OCH3OCH3
CH3CH2CH2
CH3CH2CH2
.
CH3
(m/z = 115, 15%)
+-2CH2O
(16a)
Fig. 3.5 Mass fragmentation pattern of the compound (16a)
Table 3.11 1H and
13C NMR data of compound (16e)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 168.5
C-3 ----- 151.2
C-4 6.6, s 112.3
C-4a ----- 131.5
C-5 6.9, s 107.3
C-6 ----- 141.2
C-7 ----- 118.3
C-8 ----- 141.2
C-8a ----- 127.8
C-1’ 4.8, s 45.9
O-H 2.4, s ---
6-OCH3 3.8, s 53.3
8-OCH3 3.8, s 53.3
7-CH3 2.5, s 28.6
Page 89
71
(m/z = 250, 57%)(m/z = 191, 100%)
CH+
O
H3CO
OCH3
CH3
._
(m/z = 233, 41%)
(m/z = 163, 55%)
CH+
H3CO
OCH3
CH3
CO_
CO_
(m/z =205, 23%)
.
_.
O
O
H3CO
OCH3
CH3
(m/z = 219, 53%)
CO2_ H3CO
OCH3
CH3
(m/z = 175, 21%)
_CO
O
OCH3
CH3
OHH3CO
(m/z = 222, 31%)
+.
++
+
++
+
+
O
O
H3CO
OCH3
CH3
OH
OOH
CH2OH
.
CH3
(m/z = 115, 17%)
+-2CH2O
(16e)
-OHO
O
H3CO
OCH3
CH3
O
H3CO
OCH3
CH3
Fig. 3.6 Mass fragmentation pattern of the compound (16e)
Table 3.12 1H and
13C NMR data of compound (16h)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 164.0
C-3 139.5
C-4 6.6, s 119.2
C-4a ----- 134.7
C-5 6.9, s 109.1
C-6 ----- 132.6
C-7 ----- 121.5
C-8 ----- 132.6
C-8a ----- 133.8
C-1’ ----- 130.2
C-2’ 7.0, d, J=8.7 123.8
C-3’ 8.0, d, J=9.0 126.4
C-4’ ----- 131.5
C-5’ 8.0, d, J=9.0 126.4
C-6’ 7.0, d, J=8.7 123.8
4’-OCH3 3.8, s 53.7
6-OCH3 3.9, s 55.5
8-OCH3 3.9, s 55.5
7-CH3 2.6, s 29.2
Page 90
72
O
O
H3CO
OCH3
CH3
OCH3
(m/z = 326, 47%)(m/z = 191, 100%)
CH+
O
H3CO
OCH3
CH3
O
H3CO
.
_
O
H3CO
(m/z = 135, 57%)
(m/z = 163, 55%)
CH+
H3CO
OCH3
CH3
CO_
CO_
H3CO
m/z = 77, 23%
.
O
OCH3
CH3
H3CO
_
OCH3
_
.
O
O
H3CO
OCH3
CH3
(m/z = 219, 39%)
CO2_ H3CO
OCH3
CH3
(m/z = 175, 21%)
_CO.
O
OCH3
CH3
OCH3
(m/z = 298, 31%)
+ .
++
+
+
+
+
+
(16h)
CH3
(m/z = 115, 17%)
+-2CH2O
Fig. 3.7 Mass fragmentation pattern of the compound (16h)
3.3 Synthesis of 6,8-dimethoxy-7-methyl-3-alkyl/aryl-3,4-dihydroisocoumarins
(18a-j)
Alkaline hydrolysis of the 6,8-dimethoxy-7-methyl-3-alkyl/arylisocoumarins
(16a-j) to furnish the 2,4-dimethoxy-3-methyl-6-(2-oxoalkyl/aryl)benzoic acid (17a-j)
was accomplished in 75-85% yield. The ketonic and carboxylic carbonyl absorptions
were observed in IR spectra at 1743-1759 and 1709-1719 cm-1
, respectively. The physical
constants and the IR spectral data of the keto acids (17a-j) are given in Table 3.13 and the
elemental analysis data in Table 3.14. The keto acids (17a-j) showed the characteristic
2H singlet at δ 4.3-4.7 (H-4, Ar-CH2) in the 1H NMR spectrum. The C-1 and C-4
appeared at δ 165-169 and δ 43-47, respectively, in 13
C NMR.
Page 91
73
R=
16a = 16b = 16c =
16d = -CH2Cl 16e = -CH2OH
16f = 16g = 16h = 16i = 16j =Cl
OCH 3
OCH3
OCH3
OCH3
OCH3
H3CO
CH2 CH3_
CH2 CH3_
CH2 CH3_
O
OOCH3
H3CO
CH3
R
(16a-j)
OOCH3
H3CO
CH3
R
O
OH
(17a-j)
OOCH3
H3CO
CH3
R
OHOH
H
5%KOH(aq)
C2H
5OH, 4h reflux
NaBH4
C2H
5OH, 2h R.T
OOCH3
H3CO
CH3
R
O
H
(18a-j)
R is same as in (16a-j)
R is same as in (16a-j)
Scheme 3.4 Synthesis of 6,8-dimethoxy-7-methyl-3-alkyl/ary-3,4-
dihydroisocoumarins (18a-j)
Page 92
74
Table 3.13 Physical constants and FTIR spectral data of the compounds (17a-j)
Compds. M. P.
(°C) Rf
Yield
(%)
υmax (cm-1
)
Ar-
C-H
Sp3
C-H C=O C=C O-H
17a 133-134 0.4 78 3029 2914 1749, 1713 1572 3224
17b 139-140 0.45 79 3036 2931 1753, 1709 1559 3256
17c 147-148 0.5 81 3031 2914 1743, 1719 1569 3267
17d 120-121 0.4 72 3033 2933 1759, 1715 1571 3245
17e 128-129 0.35 75 3030 2924 1747, 1721 1554 3256
17f 169-171 0.3 89 3032 2935 1758, 1715 1571 3237
17g 175-176 0.35 84 3027 2926 1741, 1720 1561 3228
17h 179-180 0.3 87 3029 2919 1761, 1723 1567 3247
17i 182-183 0.3 85 3035 2927 1749, 1716 1573 3239
17j 185-187 0.3 87 3033 2915 1757, 1713 1587 3259
Pet. Ether: Ethyl Acetate (4:1)
Table 3.14 Elemental analysis data of the compounds (17a-j)
Compounds
Molecular Formula
Calculated Found
C H C H
C15H20O5 (17a) 64.28 7.14 64.17 7.05
C17H24O5 (17b) 67.10 7.79 67.02 7.71
C19H28O5 (17c) 67.85 8.33 67.76 8.19
C13H15O5Cl (17d) 54.45 5.23 54.37 5.15
C13
H16
O6 (17e) 58.20 5.97 58.07 5.89
C18H18O5 (17f) 68.78 5.73 68.69 5.65
C18H17O5Cl (17g) 61.98 4.87 61.89 4.79
C19H18O6 (17h) 66.27 5.81 66.19 5.74
C20H22O7 (17i) 64.17 5.88 64.08 5.79
C21H24O8 (17j) 62.37 5.94 62.29 5.85
Page 93
75
Table 3.15 1H and
13C NMR data of compound (17a)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 131.1
C-2 ----- 145.4
C-3 ----- 121.1
C-4 ----- 139.2
C-5 7.9 s 108.7
C-6 ----- 125.1
C-1’ 4.3, s 47.9
C-2’ ----- 195.2
C-3’ 3.5, t, J=3.9 44.1
C-4’ 15, m 19.8
C-5’ 0.9, t, J=7.5 10.5
-COOH 10.2, s 168.4
2-OCH3 3.9, s 61.2
4-OCH3 3.7, s 56.4
3-CH3 3.1, s 32.3
The formation of the keto acid (17a) was confirmed by the presence of a singlet at
δ 10.2 ppm in 1H NMR spectrum for carboxyl hydrogen. It was also supported in
13C
NMR spectrum by the presence of a peak at δ 168.4 ppm for carboxylic carbon. The
detailed 1H and
13C NMR data of the compound (17a) are presented in Table 3.15.
The structure of the compound was further confirmed by mass spectrometry. The
molecular ion peak appeared at m/z 280 with 19% abundance which confirmed the
formation of isocoumarin (17a). By the removal of a water molecule from molecular ion
a peak at m/z 262 with percentage abundance 43 appeared. A peak at m/z 219 is the base
peak formed by the elimination of isopropyl radical from ion having m/z 262. The
fragmentation pattern of the keto acid (17a) is shown in Fig. 8.
The structures of all other keto acids (17b-j) were confirmed by the presence
carboxyl proton singlet in 1H NMR along with other signals in acceptable regions. The
signal for carboxylic carbon in 13
C NMR is also helpful for the confirmation of structures
of all these compounds.
Page 94
76
OOCH3
H3CO
CH3
CH3
OH
O
(17a)
+.
-H2OOOCH3
H3CO
CH3
CH3
+ .
-CO2OCH3
H3CO
CH3
CH3
O
+.
CH3
O
+
-COCH3 CH3
+
OCH3
H3CO
CH3
_
OOCH3
H3CO
CH3
+
OCH3
H3CO
CH3
CH3
O
_
+
CH3
CH3.
_
H3CO
CH3
+
-OCH3
..
(m/z = 280, 19%)
.
(m/z = 236, 54%)
(m/z = 165, 61%)(m/z = 134, 31%) (m/z = 219, 100%)
(m/z = 262, 43%)
(m/z = 43, 67%)(m/z = 71, 39%)
Fig. 3.8 Mass fragmentation pattern of the compound (17a)
Table 3.16 1H and
13C NMR data of compound (17f)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 143.6
C-2 ----- 138.7
C-3 ----- 121.3
C-4 ----- 131.7
C-5 7.9, s 105.3
C-6 ----- 135.9
C-1’ ----- 137.6
C-2’ 8.3, d, J=7.5 126.2
C-3’ 7.5, dd, J=7.1, 6.9 123.5
C-4’ 7.9, dd, J=7.2, 6.7 122.7
C-5’ 7.5, dd, J=7.1, 6.9 123.5
C-6’ 8.3, d, J=7.5 126.2
-CH2 4.3, s 45.3
C=O ----- 194.2
-COOH 10.9, s 167.5
2-OCH3 3.9, s 62.5
4-OCH3 3.8, s 57.3
3-CH3 2.5, s 29.2
Page 95
77
OOCH3
H3CO
CH3OH
O
(17f)
+.
-H2OOOCH3
H3CO
CH3
+.
-CO2OCH3
H3CO
CH3
O
+.
O
+
-CO
+
OCH3
H3CO
CH3
_
OOCH3
H3CO
CH3
+
OCH3
H3CO
CH3O
_
+
CH3
CH3.
_
H3CO
CH3 O
+
-OCH3
.
(m/z = 314, 39%)
.
(m/z = 270, 65%)
(m/z = 192, 79%) (m/z = 161, 19%) (m/z = 219, 100%)
(m/z = 296, 51%)
(m/z = 77, 29%)(m/z = 105, 35%)
.
Fig. 3.9 Mass fragmentation pattern of the compound (17f)
Sodium borohydride reduction of the keto acids (17a-j) afforded the
corresponding racemic hydroxy acids which underwent spontaneous cyclodehydration on
standing for a few minutes (as monitored by TLC) to afford (±)-6,8-dimethoxy-7-methyl-
3-alkyl/aryl-3,4-dihydroisocoumarins (18a-j) without any dehydrating agent. The
methylene protons (C-4) adjacent to the newly generated chiral center (C-3) in
dihydroisocoumarins (18a-j) showed the diastereotopic effect.
Each hydrogen at C-4 was split by the other nearly to same extent and unequally
by the adjacent methane proton. The double doublet of the hydrogen cis to phenyl ring is
located slightly upfield and that of trans hydrogen is located slightly downfield. The H-3
hydrogen showed the vicinal coupling to the trans and to the cis protons present in its
vicinity.
Page 96
78
Table 3.17 Physical constants and FTIR spectral data of the compounds (18a-j)
Compds. M. P.
(°C) Rf
Yield
(%) υmax (cm
-1)
Ar-C-H Sp3 C-H C=O C=C
18a oil 0.7 75 3029 2933 1719 1582
18b oil 0.8 79 3017 2923 1723 1579
18c 73-75 0.8 81 3025 2924 1719 1569
18d oil 0.6 73 3020 2932 1728 1571
18e oil 0.5 85 3013
3367(O-H) 2923 1723 1554
18f 92-94 0.6 84 3031 2919 1714 1571
18g 105-107 0.7 86 3019 2921 1720 1561
18h 141-143 0.5 87 3027 2917 1727 1567
18i 115-117 0.6 85 302 2923 1711 1573
18j 126-127 0.7 82 3013 2934 1732 1587
Pet. Ether: Ethyl Acetate (4:1)
Table 3.18 Elemental analysis data of the compounds (18a-j)
Compounds
Molecular Formula
Calculated Found
C H C H
C15H18O4 (18a) 68.18 7.57 68.09 7.49
C17H22O4 (18b) 69.86 8.21 69.79 8.12
C19H26O4 (18c) 71.25 8.75 71.14 8.66
C13H13O4Cl (18d) 57.67 5.54 57.59 5.49
C13H14O5 (18e) 61.90 6.34 61.81 6.26
C18H16O4 (18f) 72.48 6.04 72.39 5.96
C18H15O4Cl (18g) 64.96 5.11 64.89 5.02
C19H18O5 (18h) 69.51 6.09 69.43 5.99
C20H20O6 (18i) 67.03 6.14 66.94 6.06
C21H22O7 (18j) 64.94 6.18 64.87 6.09
Page 97
79
Table 3.19 1H and
13C NMR data of compound (18a)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 165.8
C-3 5.2, m 81.3
C-4 3.1, dd, J= 15.5, 3.5
3.4, dd, J=15.5, 12.2 43.5
C-4a ----- 143.1
C-5 6.8, s 129.2
C-6 ----- 147.4
C-7 ----- 118.3
C-8 ----- 145.4
C-8a ----- 138.8
C-1’ 2.5, m 35.6
C-2’ 1.2, m 19.1
C-3’ 0.9, t, J=7.5 11.2
6-OCH3 3.9, s 56.6
8-OCH3 3.9, s 56.6
7-CH3 2.9, s 28.7
The formation of 6,8-dimethoxy-7-methyl-3-propyl-3,4-dihydroisocoumarin
(18a) was confirmed by the presence of two double doublets at δ 3.1 and 3.4 ppm in 1H
NMR spectrum for H-4 hydrogens, a multiplet at δ 5.2 ppm for H-3 hydrogen and
disappearance of singlet for carboxylic proton. It was also supported in 13
C NMR
spectrum by the presence of a peak at δ 165.8 ppm for lactonic carbonyl carbon. The
detailed 1H and
13C NMR data of the compound (18a) are presented in Table 3.19. The
molecular ion peak appearing at m/z 264 with percentage abundance 37, confirmed the
formation of dihydroisocoumarin (18a). By the removal of a molecule of butanal from
molecular ion, a peak arose at m/z 192 (base peak). The fragmentation pattern of the
dihydroisocoumarin (18a) is shown in Fig. 10.
The detailed 1H and
13C NMR data of the dihydroisocoumarins (18e) and (18f)
are presented in Tables 3.20 and 3.21 respectively. The mass fragmentation pattern of the
(18e) and (18f) are shown in Fig. 11 and 12, respectively.
Page 98
80
(m/z = 264, 37%)(m/z = 192, 100%)
O
H3CO
OCH3
CH3
CH2
_
(m/z = 72, 52%)
(m/z = 164, 55%)
H3CO
OCH3
CH3
CH2
CO_
CO_
(m/z =44, 59%)
.
O
OCH3
CH3
H3CO
_
_ .
O
O
H3CO
OCH3
CH3
(m/z = 221, 32%)
CO2_ H3CO
OCH3
CH3
(m/z = 177, 21%)
_CO
O
OCH3
CH3
CH3H3CO
(m/z = 236, 43%)
+.
++
+
+
+
+
+
O
O
H3CO
OCH3
CH3
CH3
OCH3
H
OCH3
H
CH3CH2CH3
CH3CH2CH2
.
CH3
(m/z = 117, 11%)
+-2CH2O
(18a)
.
.
.
Fig. 3.10 Mass fragmentation pattern of the compound (18a)
Table 3.20 1H and
13C NMR data of compound (18e)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 165.5
C-3 4.7, m 89.4
C-4 3.2, dd, J= 15.8, 3.5
3.6, dd, J=16.3, 12.1 37.7
C-4a ----- 140.5
C-5 6.7, s 125.5
C-6 ----- 146.6
C-7 ----- 128.9
C-8 ----- 146.6
C-8a ----- 138.6
C-1’ 5.2, dd, J=15.8, 12.1
4.7, dd, J=16.2, 3.3 61.2
O-H 2.0, s ---
6-OCH3 3.8, s 56.8
8-OCH3 3.8, s 56.8
7-CH3 2.6, s 29.2
Page 99
81
(m/z = 252, 61%)(m/z = 192, 100%)
CH2
O
H3CO
OCH3
CH3
.
_
(m/z = 235, 37%)
(m/z = 164, 48%)
CH2H3CO
OCH3
CH3
CO_
CO_
(m/z =207, 29%)
.
_.
O
O
H3CO
OCH3
CH3
(m/z = 221, 57%)
CO2_ H3CO
OCH3
CH3
(m/z = 177, 17%)
_CO
O
OCH3
CH3
OHH3CO
(m/z = 224, 34%)
+.
++
+
++
+
+
O
O
H3CO
OCH3
CH3
OH
OOH
H
CH2OH
.
CH3
(m/z = 117, 23%)
+-2CH2O
(18e)
-OHO
O
H3CO
OCH3
CH3
O
H3CO
OCH3
CH3
.
Fig. 3.11 Mass fragmentation pattern of the compound (18e)
Table 3.21 1H and
13C NMR data of compound (18f)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 167.3
C-3 5.4, dd, J= 12.1, 3.3 83.5
C-4 3.1, dd, J= 12.0, 3.7
3.4, dd, J=15.7, 12.1 41.4
C-4a ----- 142.3
C-5 6.9, s 131.5
C-6 ----- 147.3
C-7 ----- 131.9
C-8 ----- 147.3
C-8a ----- 137.2
C-1’ ----- 139.1
C-2’ 7.9, d, J=5.8 130.0
C-3’ 7.7, dd, J=7.9, 6.1 123.3
C-4’ 7.5, dd, J=6.7, 5.1 119.5
C-5’ 7.7, dd, J=7.9, 6.1 123.3
C-6’ 7.9, d, J=5.8 130.0
6-OCH3 3.9, s 56.8
8-OCH3 3.9, s 56.8
7-CH3 2.8, s 30.1
Page 100
82
O
O
H3CO
OCH3
CH3
(m/z = 298, 56%)(m/z = 192, 100%)
O
H3CO
OCH3
CH3
CH2
.
_
(m/z = 105, 51%)
(m/z = 164, 55%)
CH2H3CO
OCH3
CH3
CO_
CO_
(m/z = 77, 23%)
_.
O
O
H3CO
OCH3
CH3
m/z = 221, 19%
H3CO
OCH3
CH3O
m/z = 193, 13%
_OCH3
.
O
OOCH3
CH3
(m/z = 267, 39%)
+.
++
+
+
+
+
+
O
-CO
O
H3CO
OCH3
CH3 O
_
.
(18f)
Fig. 3.12 Mass fragmentation pattern of the compound (18f)
3.4 Synthesis of 6,8-dihydroxy-7-methyl-3-alkyl/aryl-3,4-dihydroisocoumarins
(19a-j)
Demethylation of the compounds (18a-j) was achieved using ethanethiol
(C2H5SH) and anhydrous aluminum chloride in good yield to furnish the corresponding
6,8-dihydroxy-7-methyl-3-alkyl/aryl-3,4-dihydroisocoumarins (19a-j). The 6,8-
dihydroxy-3,4-dihydroisocoumarins were characterized by the absence of both methoxy
singlets and presence of singlets for hydroxyl protons in 1H NMR spectrum. In
13C NMR
spectrum, the signals for methoxy carbons are also absent. The IR spectrum showed the
(O-H) absorption bands at 3445-3481 cm-1
. The physical constants and the IR spectral
data of the 6,8-dihydroxy-3,4-dihydroisocoumarins (19a-j) are shown in Table 3.22 and
the elemental analysis data in Table 3.23.
Page 101
83
Scheme 3.5 Synthesis of 6,8-dihydroxy-7-methyl-3-alkyl/aryl-3,4-
dihydroisocoumarins (19a-j)
Table 3.22 Physical constants and FTIR spectral data of the compounds (19a-j)
Compds. M. P.
(°C) Rf
Yield
(%) υmax (cm
-1)
Ar-C-H Sp3 C-H C=O C=C O-H
19a 93-95 0.4 71 3019 2914 1723 1572 3474
19b 101-103 0.45 73 3021 2931 1719 1559 3456
19c 115-117 0.5 79 3017 2914 1727 1569 3467
19d 73-75 0.4 73 3010 2933 1717 1571 3445
19e 89-90 0.3 81 3009 2924 1720 1554 3481
19f 125-126 0.4 86 3020 2935 1722 1571 3477
19g 129-131 0.45 84 3023 2926 1720 1561 3488
19h 161-162 0.4 83 3018 2919 1724 1567 3467
19i 153-154 0.35 85 3017 2927 1719 1573 3479
19j 145-147 0.4 82 3010 2915 1716 1587 3459
Pet. Ether: Ethyl Acetate (4:1)
R=
18a = 18b = 18c =
18d = -CH2Cl 18e = -CH2OH
18f = 18g = 18h = 18i = 18j =Cl
OCH3
OCH3
OCH3
OCH3
OCH3
H3CO
CH2 CH3_
CH2 CH3_
CH2 CH3_
O
OOCH3
H3CO
CH3
R
H
(18a-j)
O
OOH
OH
CH3
RH
(19a-j)
C2H
5SH / 0-5 °C, 1h
AlCl3(Anhyd)
R is same as in (18a-j)
Page 102
84
Table 3.23 Elemental analysis data of the compounds (19a-j)
Compounds
Molecular Formula
Calculated Found
C H C H
C13H16O4 (19a) 68.18 7.57 68.09 7.49
C15H20O4 (19b) 69.86 8.21 69.79 8.12
C17H24O4 (19c) 71.25 8.75 71.14 8.66
C11H11O4Cl (19d) 57.67 5.54 57.59 5.49
C11H12O5 (19e) 61.90 6.34 61.81 6.26
C16H14O4 (19f) 72.48 6.04 72.39 5.96
C16H13O4Cl (19g) 64.96 5.11 64.89 5.02
C17H16O5 (19h) 69.51 6.09 69.43 5.99
C18H8O6 (19i) 67.03 6.14 66.94 6.06
C19H20O7 (19j) 64.94 6.18 64.87 6.09
Table 3.24 1H and
13C NMR data of compound (19a)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 167.7
C-3 5.0, m 81.6
C-4 3.0, dd, J= 15.8, 3.7
3.5, dd, J=15.9, 12.1 44.3
C-4a ----- 143.1
C-5 6.9, s 129.2
C-6 ----- 149.4
C-7 ----- 118.3
C-8 ----- 149.4
C-8a ----- 138.8
C-1’ 2.4, m 37.3
C-2’ 1.2, m 18.1
C-3’ 0.9, t, J=7.5 10.2
6-OH 4.6, s -----
8-OH 4.6, s -----
7-CH3 2.7, s 29.4
The formation of 6,8-dihydroxy-7-methyl-3-propyl-3,4-dihydroisocoumarin (19a)
was confirmed by the presence of singlet at δ 4.6 ppm in 1H NMR spectrum for 6 and 8-
Page 103
85
hydroxy hydrogens and disappearance of signals for methoxy hydrogens. It was also
supported in 13
C NMR spectrum by the presence of a peak at δ 167.7 ppm for lactonic
carbonyl carbon. The detailed 1H and
13C NMR data of the compound (19a) are presented
in Table 3.24.
The molecular ion peak which appeared at m/z 236 with 41% abundance,
confirmed the formation of dihydroisocoumarin (19a). By the removal of a molecule of
butanal from molecular ion, base peak at m/z 164 appeared. The fragmentation pattern of
the dihydroisocoumarin (19a) is shown in Fig. 13.
The detailed 1H and
13C NMR data of the dihydroisocoumarins (19e) and (19f)
are presented in Tables 3.25 and 3.26, respectively. The mass fragmentation pattern of
the (19e) and (19f) is shown in Fig. 14 and 15, respectively.
(m/z = 236, 41%)(m/z = 164, 100%)
O
OH
OH
CH3
CH2
_
(m/z = 72, 39%)
(m/z = 136, 47%)
OH
OH
CH3
CH2
CO_
CO_
(m/z =44, 62%)
.
O
OH
CH3
OH
_
_ .
O
O
OH
OH
CH3
(m/z = 193, 26%)
CO2_ OH
OH
CH3
(m/z = 149, 19%)
_CO
O
OH
CH3
CH3OH
(m/z = 208, 34%)
+.
++
+
+
+
+
+
O
O
OH
OH
CH3
CH3
OCH3
H
OCH3
H
CH3CH2CH3
CH3CH2CH2
.
CH3
(m/z = 115, 15%)
+
(19a)
.
.
.
-2OH.
Fig. 3.13 Mass fragmentation pattern of the compound (19a)
Page 104
86
Table 3.25 1H and
13C NMR data of compound (19e)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 165.5
C-3 4.9, m 88.7
C-4 3.2, dd, J= 15.8, 3.5
3.6, dd, J=16.3, 12.1 39.1
C-4a ----- 140.5
C-5 6.7, s 121.5
C-6 ----- 149.2
C-7 ----- 128.9
C-8 ----- 149.2
C-8a ----- 138.6
C-1’ 5.4, dd, J=15.7, 12.1
4.8, dd, J=15.7, 3.8 65.3
O-H 2.1, s -----
6-OH 4.5, s -----
8-OH 4.5, s -----
7-CH3 2.9, s 30.2
(m/z = 224, 58%)(m/z = 164, 100%)
CH2
O
OH
OH
CH3
.
_
(m/z = 207, 28%)
(m/z = 136, 39%)
CH2OH
OH
CH3
CO_
CO_
(m/z =179, 31%)
.
_.
O
O
OH
OH
CH3
(m/z = 193, 45%)
CO2_ OH
OH
CH3
(m/z = 149, 23%)
_CO
O
OH
CH3
OHOH
(m/z = 196, 28%)
+.
++
+
++
+
+
O
O
OH
OH
CH3
OH
OOH
H
CH2OH
.
CH3
(m/z = 115, 13%)
+
(19e)
-OHO
O
OH
OH
CH3
O
OH
OH
CH3
.
.-2OH
Fig. 3.14 Mass fragmentation pattern of the compound (19e)
Page 105
87
Table 3.26 1H and
13C NMR data of compound (19f)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 162.3
C-3 5.3, dd, J= 12.3, 3.8 81.5
C-4 3.0, dd, J= 12.0, 3.7
3.3, dd, J=15.7, 12.1 44.6
C-4a ----- 142.3
C-5 7.0, s 123.5
C-6 ----- 151.3
C-7 ----- 131.9
C-8 ----- 151.3
C-8a ----- 137.2
C-1’ ----- 138.1
C-2’ 7.9, d, J=5.8 129.4
C-3’ 7.7, dd, J=6.5, 5.8 126.3
C-4’ 7.5, dd, J=6.2, 5.6 119.5
C-5’ 7.7, dd, J=6.5, 5.8 126.3
C-6’ 7.9, d, J=5.8 129.4
6-OH 4.7, s -----
8-OH 4.7, s -----
7-CH3 2.5, s 28.1
O
O
OH
OH
CH3
(m/z = 270, 41%)(m/z = 164, 100%)
O
OH
OH
CH3
CH2
.
_
(m/z = 105, 51%)
(m/z = 136, 51%)
CH+
OH
OH
CH3
CO_
CO_
(m/z = 77, 23%)
_.
O
O
OH
OH
CH3
(m/z = 193, 21%)
OH
OH
CH3O
(m/z = 165, 15%)
.
O
OOH
CH3
(m/z = 253, 19%)
+.
++
+
+
+
+
+
O
-CO
O
OH
OH
CH3 O
_
.
-OH
(19f)
Fig. 3.15 Mass fragmentation pattern of the compound (19f)
Page 106
88
4. BIOLOGIOCAL ACTIVITIES
A rapid advance in the development of new techniques for determining the
biological activity of synthetic and natural compounds has triggered a renaissance in the
drug development. Primary bioassay screening plays a very important role in the drug
development program. These screenings act as a tool to conduct activity directed
isolation of bioactive compounds for curing humans and animals. Primary screenings
provide first indication of bioactivities and thus help in the selection of lead compounds
for secondary screening for detailed pharmacological evaluation.
Isocoumarins, keto acids, 3,4-dihydroisocoumarins and 6,8-dihydroxy-3,4-
dihydroisocoumarins were tested for the following activities:
1. Antibacterial activity against ten different gram positive and gram
negative bacterial strains.
2. Antimalarial activity against Plasmodium falciparum
3. Cytotoxicity against human keratinocyte cell lines
4.1 Antibacterial Activity
Bacterial infections constitute one of the most serious situations in infectious
diseases. The detection and identification of these bacteria is one of the most important
functions of clinical microbiology. Isolation of an infectious agent from the patient with
disease is often not sufficient for determining proper therapy. Since the susceptibility of
many bacteria to antimicrobial agents cannot be predicted, testing individual pathogens,
against appropriate agent (with the most activity against the pathogen, the least toxicity to
the host, the least important on normal flora, appropriate pharmacologic characteristics
and most economical) can then be chosen allowing a more certain therapeutic outcome.
Antibacterial activity of the synthesized isocoumarins (16a-j), keto acids (17a-j),
3,4-dihydroisocoumarins (18a-j) and 6,8-dihydroxy-3,4-dihydroisocoumarins (19a-j) was
determined against various gram positive and gram negative bacterial strains by using
agar well diffusion. The purified samples were dissolved in DMSO 5mg/mL. DMSO is
the negative control and antibiotic chloramphenicol is the positive control in this in vitro
antibacterial study.
Ten bacterial strains Escherichia coli (E. c.), Klebsiella pneumonae (K. p.),
Lactobacillus bulgaricus (L. b.), Micrococcus luteus (M. l.), Pasteurella multocida (P.
Page 107
89
m.), Proteus vulgaris (P. v.), Pseudomonas aeruginosa (P. a.), Salmonella typhi (S. t.),
Staphylococcus aureus (S. a.) and Staphylococcus epidermidis (S. e.) were selected for
this antibacterial assay. Micrococcus luteus, Staphylococcus aureus and Staphylococcus
epidermidis are the example of Gram positive and the remaining seven are Gram negative
bacteria. All of the tested microorganisms were maintained on nutrient agar at 4°C and
sub-cultured before use. The bacteria studied are clinically important ones causing
several infections and it is essential to overcome them through some active therapeutic
agents.
The antibacterial assay was performed by agar well diffusion method against
different bacterial strains142
. Each tested bacterium was sub-cultured in nutrient broth at
37°C for 24h. One hundred micro liters of each bacterium was spread with the help of
sterile spreader onto a sterile Muller-Hinton agar plate so as to achieve a confluent
growth. The plates were allowed to dry and wells (6mm diameter) were punched in the
agar with the help of cork borer. 0.1mL of each compound solution (5mg/mL) in DMSO
was introduced into the well and the plates were incubated overnight at 37°C.
The antimicrobial spectrum of the compounds was determined for the bacterial
species in terms of size of the zones around each well. The diameters of the zone of
inhibition produced by the compounds were compared with those produced by the
commercial antibiotic chloramphenicol (5mg/mL). This is the common antibiotic used
for the treatment of infections caused by gram positive and gram negative bacteria. The
control activity was deducted from the test and the results obtained were plotted. The
experiment was performed thrice to minimize the error and the mean values are
presented.
Antibacterial activity results of the isocoumarins (16a-j), keto acids (17a-j), 3,4-
dihydroisocoumarins (18a-j) and 6,8-dihydroxy3,4-dihydroisocoumarins are shown in
Tables 4.1, 4.2, 4.3 and 4.4, respectively.
Page 108
90
Table 4.1 In vitro Antibcterial Activity of Isocoumarins (16a-j)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
16a 1 5 0 11 0 3 0 0 9 9
16b 1 8 0 12 0.5 0 2 0 8 11
16c 0 0 0 7 0 0 2 0 9 6
16d 1 2 1 8 0 1 0 0 8 1
16e 16 8 7 0.5 6 1 0 12 0.5 1
16f 3 3 4.5 2.5 5 1.5 2 0 2.5 2.5
16g 13 9 1 11 0 0 10 0 11 0
16h 1 0 11 2 10 12 0 13 0 11
16i 14 8 9 0 9 1 0 10 11 0
16j 1 0 1 10 0 0 11 0 0 11
Standard 18 10 13 13 12 13 13 14 13 13
Antibacterial activity results of the isocoumarins (16a-j) show that most of these
are more active against gram positive bacteria as compared to gram negative bacteria.
Some of them also possess activity against gram negative bacteria. Among the 3-alkyl
substituted isocoumarins, antibacterial activity of the 3-pentylisocoumarin is higher than
3-propylisocoumarin but less than 3-heptylisocoumarin. It reflects that antibacterial
activity increases by increasing the carbon chain length up to five carbons and then
decreases for seven carbons. Antibacterial activity does not directly correlate to
hydrophobicity.
The compound (16e) which possesses 3-hydroxymethyl substitution shows higher
activity against gram negative bacteria as compared to gram positive. Among the 3-
phenyl substituted isocoumarins, the most active are (16g) and (16h) which possess 3-
monomethoxyphenyl and dimethoxyphenyl substitution, respectively.
Page 109
91
Table 4.2 In vitro Antibcterial Activity of Keto Acids (17a-j)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
17a 1 0 0 2 0 3 0 5 0 3
17b 1 2 0 1 0.5 0 2 0 1 3
17c 0 0 0 3 0 0 2 0 12 1
17d 1 2 1 0.5 0 1 0 0 2 1
17e 15 1 3 0.5 11 1 0 12 0.5 1
17f 3 3 4.5 2.5 1 1.5 2 0 2.5 2.5
17g 1 9 1 0.5 0 0 1 0 11 0
17h 1 0 11 2 10 12 0 13 0 1.5
17i 0.5 1 2 0 3 1 0 1 2 0
17j 1 0 1 0.5 11 0 1 0 0 1.5
Standard 18 10 13 13 12 13 13 14 13 13
Antibacterial activity results of the keto acids (17a-j) are presented in Table 4.2
which shows that most of the keto acids are inactive against the selected bacterial strains.
After hydrolysis of the lactonic ring, antibacterial activity is greatly suppressed. Presence
of carbonyl and carboxylic acid polar functionalities in keto acids (17a-j) decreases their
antibacterial activity. It may partly be due to the fact that the cell barriers of the
microorganisms are nonpolar in nature so highly polar compounds can not easily pass
these hydrophobic barriers.
Page 110
92
Table 4.3 In vitro Antibcterial Activity of 3,4-Dihydroisocoumarins (18a-j)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
18a 1 7 0 11 0 3 0 0 10 12
18b 1 9 0 12 0.5 0 2 0 9 11
18c 0 0 0 9 0 0 2 0 5 4
18d 1 2 1 6 0 1 0 0 5 1
18e 16 9 11 0.5 9 1 0 13 0.5 1
18f 0 1 2.5 0.5 10 11.5 2 0 2.5 11
18g 15 9 1 11.5 0 11 0 0 12 0
18h 0.5 7 12 2 10 12 0 13 0 11
18i 14 8 9 0 9 1 0 10 11 0
18j 1 0 1 11 0 0 12 0 0 9
Standard 18 10 13 13 12 13 13 14 13 13
*Activity of each sample is measured by subtracting the activity of DMSO. Escherichia
coli, Klebsiella pneumonae, Lactobacillus bulgaricus, Micrococcus luteus, Pasteurella
multocida, Proteus vulgaris, Pseudomonas aeruginosa, Salmonella typhi, Staphylococcus
aureus and Staphylococcus epidermidis)
Antibacterial activity results of the 3,4-dihydroisocoumarins (18a-j) shows that
these are more active against gram positive bacteria as compared to gram negative
bacteria. Some of them also possess antibacterial activity against gram negative bacteria.
Among the 3-alkyl substituted isocoumarins antibacterial activity of the 3-
pentylisocoumarine is higher than 3-propylisocoumarine but less than 3-
heptylisocoumarine. It reflects that anti bacterial activity increases by increasing the
carbon chain length up to five carbon and then decreases for seven carbon. This is similar
behavior as possessed by isocoumarins (16a-j)
The compound (18e) possesses 3-hydroxymethyl substitution having higher
activity against gram negative bacteria as compared to gram positive. Among the 3-
phenyl substituted isocoumarins the most active are (18g) and (18h) which possesses 3-
monomethoxyphenyl and dimethoxyphenyl substitution respectively.
Page 111
93
Table 4.4 In vitro Antibcterial activity of 6,8-Dihydroxy3,4-dihydroisocoumarins
(19a-j)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
19a 14 0.5 3 1 11 10 9 0 0 1
19b 12 3 5 1 10 12 10 13 0.5 1.5
19c 0 9 10 0.5 0 0 2 0 1 0.5
19d 1 2 1 1 0 1 10 0 5 1
19e 15.5 9 12 0.5 11 1 0 13 0.5 1
19f 0 1 2.5 0.5 10 11.5 2 0 2.5 12
19g 15 9 1 11.5 0 11 0 0 12 0
19h 0.5 7 12 2 10 12 0 13 0 11
19i 14 8 9 0 11 1 0 12 11 0
19j 1 0 1 11 0 10 12 0 0 11
Standard 18 10 13 13 12 13 13 14 13 13
*Activity of each sample is measured by subtracting the activity of DMSO. Escherichia
coli, Klebsiella pneumonae, Lactobacillus bulgaricus, Micrococcus luteus, Pasteurella
multocida, Proteus vulgaris, Pseudomonas aeruginosa, Salmonella typhi, Staphylococcus
aureus and Staphylococcus epidermidis)
The antibacterial activity results of the 6,8-dihydroxy-3,4-dihydroisocoumarins
(19a-j) show that most of these are more effective against gram negative bacteria as
compared to gram positive. Some of them also possess activity against gram positive
bacterial strains.
The comparison of the antibacterial activity among the four series of the
synthesized compounds indicates that isocoumarins (16a-j) and 3,4-dihydroisocoumarins
(18a-j) are more active against gram positive bacteria than gram negative. But the 6,8-
dihydroxy-3,4-dihydroisocoumarins are more active against gram negative than gram
positive. Among all these compounds the keto acids (17a-j) are the least active. Although
the 3-alkyl substituted isocoumarins and dihydroisocoumarins possess antibacterial
activity but the compounds which possess 3-substituted phenyl ring are more active than
the 3-alkyl substituted ones.
Page 112
94
4.2 Antimalarial Activity
Malaria is the worldwide most important parasitic disease with an incidence of
almost 300 million clinical cases and over one million deaths per year. Malaria indirectly
contributes to illness and deaths from respiratory infections, diarrhoeal disease and
malnutrition. Plasmodium falciparum, the potentially lethal malarial parasite, has shown
itself capable of developing resistance to nearly all used antimalarial drugs and resistant
strains have rapid extension143
.
The lost of effectiveness of chemotherapy constitute the greatest threat to the
control of malaria. Therefore, to overcome malaria, new knowledge, products and tools
are urgently needed; especially new drugs are required144
. All the synthesized compounds
are evaluated for their potential against malarial parasite Plasmodium falciparum.
In vitro antimalarial assay was performed in duplicate in a 96 well microtiter
plate. The assay was based on assessing the inhibition of schizont maturation.
For the cultivation of Plasmodium falciparum, RPMI 1640 supplemented with 25 mM
HEPES, 1% D-glucose, 0.23% sodium bicarbonate and 10% heat inactivated human
serum.
A stock solution of 5mg/ml of each of the test samples was prepared in DMSO
and subsequent dilutions were prepared with culture medium. The final concentrations
ranged from 1.5 µg/mL to 100 µg/mL. The diluted samples in 20µL volume were added
to the test well and the culture plates were incubated at 37 °C for 36 to 40 hrs. After
incubation, contents of the wells were harvested and stained for 30min in a 2% Giemsa
solution pH 7.2, after that the developed schizonts were counted.
The antimalarial activity of the synthesized compounds was expressed by the
inhibitory concentrations 50 (IC50), representing the concentration of the drug that
induced a 50% parasitaemia decrease and 100% schizonts maturation inhibition.
Chloroquine phosphate is the positive control with IC50 value 0.2 µg/mL and 12.5 µg/mL
values for 100% schizont maturation inhibition.
Page 113
95
Table 4.5 Antimalarial Activity of isocoumarins (16a-j)
Compds. IC50 µg/ml 100%SMI (µg/ml)
16a 5 15
16b 2 9.5
16c 16 89
16d 65 -
16e 53 -
16f 34 -
16g 25 -
16h 4 19
16i 62 -
16j 36 -
Standard 0.2 12.5
SMI, Schizont Maturation Inhibition; IC50, Inhibition Concentration 50
The results of the in vitro antimalarial activity of the isocoumarins (16a-j) are
presented in table 4.5. The results showed that isocoumarins possess moderate to potent
antimalarial activity. The 3-alkyl substituted isocoumarins are active against the tested
malarial strain. The antimalarial activity of these compounds depends upon the size of the
3-alkyl group. Activity increases from 3-propylisocoumarin (16a) to 3-pentylisocoumrin
(16b) but decreases to 3-heptylisocoumarin (16c). The antimalarial activity of these
compounds can be correlated to their hydrophobicity, the activity increases with the
increase of the hydrophobic carbon chain but hydrophobicity must be in certain limits.
The most interesting antiplasmodial activity is obtained by isocoumarins (16b) (IC50 2
µg/ml). It exhibit complete inhibition of the schizonts maturation at 9.5µg/ml. This
compound is most potent than all other tested isocoumarins (6a-j).
Among the 3-phenyl substituted isocoumarins the most active is (16h) which
possess 4-methoxyphenyl substitution at 3 positions with IC50 4 µg/ml. The results
showed complete inhibition of the schizonts maturation at 19 µg/ml for this
isocoumarins.
Page 114
96
Table 4.6 Antimalarial Activity of Keto Acids (17a-j)
Compd IC50 µg/ml 100%SMI (µg/ml)
17a 45 -
17b 32 -
17c 56 -
17d 78 -
17e 67 -
17f 77 -
17g 65 -
17h 19 -
17i 69 -
17j 53 -
Standard 0.2 12.5
SMI, Schizont Maturation Inhibition; IC50, Inhibition Concentration 50
Table 4.6 showed the in vitro antimalarial activity results of the keto acids (17a-
j). Most of these keto acids are inactive against the selected malarial strain. The
hydrolysis of lactonic ring introduced polar carboxylic acid and keto group in the keto
acids (17a-j). These polar functional groups decrease the antiplasmodial activity of these
keto acids. Presence of the polar functionalities of the keto acids decreases their lipophilic
character decreasing the penetration of these compounds through cell barrier.
Isocoumarins (16a-j) have high antimalarial activity as compared to keto acids
(17a-j). In conclusion, the antimalarial activity is highly suppressed after the hydrolysis
of the lactonic ring of isocoumarins to keto acids. The presence of the polar keto group
and carboxylic acid group results in the loss of antimalarial activity.
Page 115
97
Table 4.7 Antimalarial Activity of 3,4-Dihydroisocoumarins (18a-j)
Compd IC50 µg/ml 100%SMI (µg/ml)
18a 3.5 13.5
18b 1.2 7.5
18c 18 78
18d 57 -
18e 48 -
18f 31 -
18g 22 -
18h 2.6 15.6
18i 58 -
18j 42 -
Standard 0.2 12.5
SMI, Schizont Maturation Inhibition; IC50, Inhibition Concentration 50
The results of the in vitro antimalarial activity of the 3,4-dihydroisocoumarins
(18a-j) are presented in Table 4.7. The results showed that 3,4-dihydroisocoumarins are
more active as compared to isocoumarins (16a-j). The 3-alkyl substituted 3,4-
dihydroisocoumarins are active against the tested malarial strain. The antimalarial
activity of these compounds depends upon the size of the 3-alkyl group. Activity
increases from 3-propyl-3,4-dihydrolisocoumarin (18a) to 3-pentyl-3,4-
dihydroisocoumarin (18b) but decreases to 3-heptyl-3,4-dihydroisocoumarin (18c). The
antimalarial activity of these compounds can be related to their hydrophobicity, activity
increases with the increase of the hydrophobic carbon chain but hydrophobicity must be
in certain limits. The most interesting antiplasmodial activity is obtained for isocoumarin
(18b) (IC50 1.2 µg/ml). It exhibits complete inhibition of the schizonts maturation at
7.5µg/mL. This compound is most potent of all other tested dihydroisocoumarins (18a-j).
Among the 3-substituted phenyl 3,4-dihydroisocoumarins, the most active is
(18h) which possesses 4-methoxyphenyl substitution at 3 position with IC50 2.6 µg/mL.
The results showed complete inhibition of the schizonts maturation at 15.6 µg/mL for this
3,4-Dihydroisocoumarins.
Page 116
98
Table 4.8 Antimalarial Activity of 6,8-Dihydroxy-3,4-dihydroisocoumarins (19a-j)
Compd IC50 µg/ml 100%SMI (µg/ml)
19a 1.3 8.4
19b 1.5 7.2
19c 21 78
19d 53 -
19e 44 -
19f 37 -
19g 27 -
19h 2.1 12.4
19i 43 -
19j 39 -
Standard 0.2 12.5
SMI, Schizont Maturation Inhibition; IC50, Inhibition Concentration 50
The results of the in vitro antimalarial activity of the 6,8-Dihydroxy-3,4-
dihydroisocoumarins (19a-j) are presented in table 4.8. The results showed that 6,8-
Dihydroxy-3,4-dihydroisocoumarins (19a-j) are more active as compared to
isocoumarins (16a-j), keto acids (17a-j) and 3,4-Dihydroisocoumarins (18a-j). The 3-
alkyl substituted derivatives showed moderate to potent activity against the tested
malarial strain. The antimalarial activity of these compounds depends upon the size of the
3-alkyl group. Activity decreases from 3-propy-6,8-dihydroxy-3,4-dihydrolisocoumarin
(19a) to 3-pentyl-6,8-dihydroxy-3,4-dihydroisocoumrin (19b) and 3-heptyl-6,8-
dihydroxy-3,4-dihydroisocoumarin (19c). The antimalarial activity of these compounds
can be related to carbon chain length, activity decreases by increasing the carbon chain
length of three alkyl group. The most interesting antiplasmodial activity is obtained by
isocoumarins (19a) (IC50 1.3 µg/ml). It exhibit complete inhibition of the schizonts
maturation at 8.4µg/ml. This compound is most potent than all other tested 3-alkyl-6,8-
dihydroxy-3,4-dihydroisocoumarins (19a-j).
Among the 3-phenyl substituted 3,4-Dihydroisocoumarins the most active is
(19h) which possess 4-methoxyphenyl substitution at 3 positions with IC50 2.1 µg/ml.
Page 117
99
The results showed complete inhibition of the schizonts maturation at 12.4 µg/ml for this
analogue.
• Keto acids are inactive against tested strains of malarial parasite.
• 6,8-Dihydroxy-3,4-dihydroisocoumarins (19a-j) are most effective as compared
to isocoumarins (16a-j) and 3,4-dihydroisocoumarins (18a-j).
• 6,8-Dihydroxy-3,4-dihydroisocoumarins having 3-alkyl substitution are more
effective as compared to 3-aryl substituted derivatives.
• The size of carbon chain length at position 3 also plays important role in
antimalarial activity of these compounds. Activity increases when n-propyl and n-
pentyl group are present.
Page 118
100
4.3 Cytotoxicity
The neutralrottest was in line with the protocol of the National Institute of Health
(NIH) implemented. It is of vital dye Neutral from living cells and then protonated. The
cell acts as an ion trap, whereby the dye is no longer out of can diffuse. By destroying the
cells, the neutral released and can be determined photometrically. It represents the
absorption is a measure of the vitality of the cells. The lower the absorption, the fewer
living cells were present.
For testing immortalize human keratinocyte cell line (HaCaT) was used. The
incubation with the test substances was made over three days and the experiments were
carried out in two independent experiments performed with several parallels.
The stock solutions of the substances in DMSO were prepared. The tested
concentration range was between 1.56 to 100 µM. the concentration of the solvent
DMSO was tested at all concentrations at 0.1%. Etoposide was used as a positive control
in a concentration of 10 µM with IC50 0.8 µM and DMSO as a negative control.
4.3.1 Cytotoxic Activity of the isocoumarins (16a-j)
The Cytotoxic activity results of the isocoumarins (16a-e) and (16f-j) are
presented in figures 4.1 and 4.2 respectively. The activity of all these compounds
increases as the concentration of the compounds increases and they show the maximum
activity at highest concentration. Among the 3-alkyl substituted isocoumarins the most
active is the isocoumarins (16b) having 3-pentyl substitution.
The isocoumarin (16e) possess 3-hydroxymethyl group is the most active which
show only the 20% viability of the infected cells at concentration of 100 μM. The 3-
hydroxymethyl group plays vital role in the activity of this compound and may be
important for the receptor binding. This derivative has the structural similarities with the
well known antitumor agent cytogenin.
Page 119
101
Fig. 4.1 Cytotoxic activity results of the isocoumarins (16a-e)
The isocoumarins (16f-j) show moderate to potent cytotoxic activity against the
immortalized human keratinocyte cell lines. When we compare the cytotoxicity of the 3-
phenyl substituted isocoumarins (16f-j) the isocoumarins (16h) and (16i) have higher
activity than the others. These two derivatives possess monomethoxy and dimethoxy
phenyl ring at 3-position.
The compound (16h) having 4-methoxyphenyl substitution at 3-position show a
40% viability of the infected cells at a concentration of 50μM and 20% viability at
highest concentration 100μM. The isocoumarin (16i) show a 50% decrease of the
viability of the infected cells at highest tested concentration 100 μM and is less active
than the isocoumarin (16h).
Fig. 4.2 Cytotoxic activity results of the isocoumarins (16f-j)
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bilit
y [
%]
Concentration [uM]
16a
16b
16c
16d
16e
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bil
ity [
%]
Concentration [uM]
16f
16g
16h
16i
16j
Page 120
102
4.3.2 Cytotoxic Activity of the Keto Acids (17a-j)
The cytotoxicity of the keto acids (17a-e) and (17f-j) are summarized in figures 3
and 4 respectively. From the graphs shown in figures 4.3 and 4.4 it is clear that the keto
acids show only 20-30% decrease of the viability of the infected cells. The results
indicates that the keto acids (17a-j) are inactive against the immortalized human
keratinocyte cell lines (HaCaT). The presence of the carboxylic acid and keto
functionalities decreases their penetration through the plasma membrane due to which
they show no activity against the immortalized human keratinocyte cell lines.
Fig. 4.3 Cytotoxic activity results of the keto Acids (17f-j)
Fig. 4.4 Cytotoxic activity results of the Keto Acids (17f-j)
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bilit
y [
%]
Concentration [uM]
17a
17b
17c
17d
17e
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bilit
y [
%]
Concentration [uM]
17f
17g
17h
17i
17j
Page 121
103
4.3.3 Cytotoxic Activity of the 3,4-Dihydroisocoumarins (18a-j)
The Cytotoxic activity results of the 3,4-Dihydroisocoumarins (18a-e) and (18f-j)
are presented in figures 4.5 and 4.6 respectively. They show moderate to potent
cytotoxicity against the infected cell lines. The activity of all these compounds increases
as the concentration of the compounds increases and they show the maximum activity at
highest concentration. Among the 3-alkyl substituted isocoumarins the most active is the
3-pentyl-3,4-dihydroisocoumarin (18b) which show 205 viability of the infected cells at
100 μM.
The 3,4-Dihydroisocoumarin (18e) possess 3-hydroxymethyl group is the most
active which show less than 20% viability of the infected cells at concentration of 100
μM. The reduction of the double bond between C-3, C-4 and 3-hydroxymethyl group
plays vital role in the activity of this compound and may be important for the receptor
binding.
Fig. 4.5 Cytotoxic activity results of the 3,4-Dihydroisocoumarins (18a-e)
Fig. 4.6 Cytotoxic activity results of the 3,4-Dihydroisocoumarins (18f-j)
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bil
ity [
%]
Concentration [uM]
18a
18b
18c
18d
18e
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bil
ity [
%]
Concentration [uM]
18f
18g
18h
18i
18j
Page 122
104
Among the 3,4-Dihydroisocoumarins (18f-j) the compound (18h) and (18i) have
higher activity than the others. These two derivatives possess monomethoxy and
dimethoxy phenyl ring at 3-position.
The compound (18h) having 4-methoxyphenyl substitution at 3-position show a
50% decrease of the viability of the infected cells at a concentration of 27 μM and less
than 20% viability at highest concentration 100 μM. The 3,4-dihydroisocoumarin (18i)
show a 50% decrease of the viability of the infected cells at highest tested concentration
100 μM and is less active than the isocoumarin (18h).
4.3.4 Cytotoxic Activity of the 6,8-dihydroxy-3,4-Dihydroisocoumarins (19a-j)
The figure 4.7 presented the cytotoxicity of the 6,8-Dihydroxy-3,4-
dihydroisocoumarins (19a-e). The compounds (19a), (19b) and (19e) show higher
activity. Among the 3-alkyl substituted compounds the activity decreases as size of the
alkyl group increases. The compounds (19a) which possess 3-propyl substituent show a
50 % decrease of the viability of the infected cells at a concentration 10 μM. The other
two which possess 3-pentyl and 3-heptyl substituents show a 50% decrease at higher
concentrations. Activity of these derivatives can be related to their hydrophobicity,
greater the hydrophobicity lesser will be the cytotoxic activity. The compound (19e)
which possesses 3-hydroxymethyl group show a 50% decrease of the viability of the
infected cells at 15 μM and less than 10% viability at highest tested concentration.
Fig. 4.7 Cytotoxic activity results of the 6,8-Dihydroxy-3,4-Dihydroisocoumarins
(19a-e)
0
20
40
60
80
100
10 25 50 100 1000
Via
bil
ity [
%]
Concentration [uM]
19a
19b
19c
19d
19e
Page 123
105
The cytotoxic activity results of the 3-phenyl substituted derivatives (19f-j) are
presented in figure 4.8. Among these the compound (19h) and (19i) show higher activity.
The compound (19i) which possesses the dimethoxy substituted phenyl ring show a 50%
decrease of the viability of the infected cells at a concentration 50 μM and about 30%
infected cell are viable at highest tested concentrations. The compound (19h) which
possesses monomethoxy substituted phenyl ring shows the comparable results as the
compound (19i).
In a comparison of the 3-alkyl substituted 6,8-dihydroxy-3,4-dihydroisocoumarins
and 3-phenyl substituted the 3-alkyl substituted derivatives has higher activity. It may be
due to the fact that the 3-alkyl group is important in the absorption of these derivatives
through the cell memberane.
Fig. 4.8 Cytotoxic activity results of the 6,8-dihydroxy-3,4-dihydroisocoumarins
(19f-j)
Following are some general conclusions derived from the results of the cytotoxic
activity of the synthesized compounds.
Keto acids having no cytotoxic activity against human keratinocytes cell lines
(HaCaT).
Among 6,8-dihydroxy-3,4-dihydroisocoumarins (19a-j), 6,8-dimethoxyisocoumarins
(16a-j) and 6,8-Dimethoxy-3,4-dihydroisocoumarins (18a-j), compounds (19a-j) have
higher cytotoxic activity.
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bilit
y [
%]
Concentration [uM]
19f
19g
19h
19i
19j
Page 124
106
• 3-alkyl substituted analogous are more active as compared to 3-phenyl
substituted. Compound having 3-(4’-methoxyphenyl) substitution have high
potency.
• Among 6,8-Dihydroxy-3,4-dihydroisocoumarins, the derivative possess 3-
hydroxymethyl group is more potent as compared to all other members, as this
member is the structural analogue of a well known cytotoxic agent cytogenin.
Page 125
107
5.1 Synthesis of Stellatin
The solvents were purified and dried according to the standard procedures before
using. The dried solvents were stored under molecular sieves (4 Å). Standard procedures
were employed for the purification and drying of solvents. Melting points were recorded
using a digital Gallenkamp (SANYO) model MPD BM 3.5 apparatus and are
uncorrected. FTIR spectra were recorded using an FTS 3000 MX spectrophotometer, 1H
NMR and 13
C NMR spectra were determined as CDCl3 solutions at 300 MHz on a Bruker
AM-300 spectrophotometer, mass spectra (EI, 70eV) on a GCMS instrument, and
elemental analyses with a LECO-183 CHNS analyzer. All the compounds were purified
by thin layer chromatography using silica gel HF-254 from Merck.
5.1.1 Methyl (3, 5-dimethoxy-4-methylphenyl) acetate (1)
A stirred solution of (3,5-dimethoxy-4-methyl phenyl) acetic acid (5.0g, 23.8
mmol) in dry methanol (30 mL) was treated drop wise with conc. H2SO4 (5mL). The
mixture was refluxed for 8-9h. The reaction was monitored by TLC. After the completion
of reaction, mixture was concentrated to 55mL and extracted with ethyl acetate (3x50
mL). The extract was washed with saturated brine, dried and concentrated to give crude
oil which was distilled to afford Methyl (3,5-dimethoxy-4-methyl phenyl) acetate (1)
(4.7g, 88.18%); Rf: 0.7 (petroleum ether and ethyl acetate, 4:1); m. p. 38-40 °C, IR
(KBr): 3023 (C-H), 1734 (C=O), 1573 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm): 7.45 (2H,
s, H-2, H-6), 3.96 (6H, s, 3-OCH3, 5-OCH3), 3.54 (2H, s, Ar-CH2), 3.47 (3H, s,
COOCH3), 2.55 (3H, s, 4-CH3); 13
C NMR (CDCl3 δ ppm): 168.23 (C=O), 132.54 (C3,
C5), 128.37 (C2, C6), 119.42 (C4), 112.21 (C1), 68.55 (Ester OCH3), 55.34 (Ar-OCH3),
36.91 (CH2), 28.63 (Ar-CH3); MS (70eV): m/z (%); 224 [M]+.
(46), 193 (43), 165 (100),
59 (12); Anal. calcd for C12H16O4: C, 64.28 H, 7.14 O, 28.57 Found: C, 64.02 H, 6.96
O, 28.35.
5.1.2 Methyl (2-formyl-3,5-dimethoxy-4-methylphenyl) acetate (2)
Phosphorus oxychloride (1.61g, 10.0mmol) was added dropwise in to a stirred
solution of methyl (3,5-dimethoxy-4-methyl phenyl) acetate (1) (2.0g, 8.9mmol) in
freshly distilled DMF (10mL) at 55 °C. Reaction mixture was heated at about 100 °C for
2 hr and stirred overnight at room temperature. Then poured the reaction mixture into
aqueous solution of sodium acetate (10%, 10mL) and shake vigorously. Methyl (2-
Page 126
108
formyl-3,5-dimethoxy-4-methyl phenyl) acetate (2) was precipitated out as yellowish
precipitates (1.9g, 84%); Rf: 0.55 (petroleum ether and ethyl acetate, 4:1); m. p. 51-53
°C; IR (KBr): 3029 (C-H), 1722 (C=O), 1690 (CHO), 1545 (C=C) cm-1
; 1H NMR,
(CDCl3, δ ppm ): 9.75 (1H, s, CHO), 7.96 (1H, s, H-6), 3.42 (3H, s, 3-OCH3), 3.25 (3H,
s, 5-OCH3), 3.11 (3H, s, CO2CH3), 2.92 (2H, s, Ar-CH2), 2.80 (3H, s, 4-CH3); 13
C NMR
(CDCl3, δ ppm ): 179.32 (Aldehyde C=O), 162.43 (Ester C=O), 136.76 (C3, C5), 131.89
(C2), 126.21 (C6), 121.33 (C4), 117.54 (C1), 61.63 (Ester OCH3), 57.34 (Ar-OCH3),
39.12 (Ar-CH2), 32.08 (4-CH3); MS (70eV): m/z (%); 252 [M]+ (25), 251 (65), 224 (49),
223 (34), 165 (100), 29 (31); Anal. calcd. for C13H16O5: C, 61.90 H, 6.34 O, 31.74
Found: C, 61.67 H, 6.16 O, 31.56.
5.1.3 2,4-Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid (3)
Methyl (2-formyl-3, 5-dimethoxy-4-methyl phenyl) acetate (2) (6.3g, 25.0 mmol)
and sulfamic acid (8.3g, 86.0 mmol) in 150ml H2O:THF:DMSO (20:1:1) at 0°C was
treated with NaClO2 (7.24g, 80.0 mmol) in 20mL H2O. The reaction mixture was stirred
for 20min at 0°C and then diluted with ethyl acetate (100mL), washed with saturated
aqueous ammonium chloride (2 x 130mL) saturated aqueous sodium chloride (130mL).
Organic layer was dried over anhydrous sodium sulfate and evaporated to afford 2,4-
Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid (3) (6.6g, 79%); Rf: 0.4
(petroleum ether and ethyl acetate, 4:1); m. p. 164-166 °C; IR (KBr): 3265 (O-H), 3037
(C-H), 1734 (C=O), 1715 (COOH), 1562 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm ): 8.19
(1H, s, COOH), 7.66 (1H, s, H-6), 3.82 (3H, s, 3-OCH3), 3.67 (3H, s, 5-OCH3), 3.63 (3H,
s, CO2CH3), 2.54 (2H, s, Ar-CH2), 2.25 (3H, s, 3-CH3); 13
C NMR (CDCl3, δ ppm ):
197.78 (Carboxylic C=O), 168.56 (Ester C=O), 139.32 (C3, C5), 134.37 (C2), 127.13
(C6), 120.62 (C4), 114.17 (C1), 66.09 (Ester OCH3), 55.41 (Ar-OCH3), 35.04 (Ar-CH2),
29.88 (3-CH3); MS (70eV): m/z (%); 268 [M]+ (32), 251 (51), 224 (65), 165 (100), 45
(25); Anal. calcd for C13H16O6: C, 58.20 H, 5.97 O, 35.82 Found: C, 58.04 H, 5.76 O,
35.59.
5.1.4 2,4-Dimethoxy-6-(2-hydroxyethyl)-3-methylbenzoic acid (4)
2,4-Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-methylbenzoic acid (3) (0.5g, 1.86
mmol) and sodium borohydride (0.84g, 22.32 mmol) were suspended in freshly distilled
THF (10mL). The reaction mixture was stirred for 15min at 65 °C and then added
Page 127
109
methanol (10 mL) dropwise during 30min. The mixture was refluxed for 4 h then cooled
to room temperature and treated with saturated ammonium chloride solution (10 mL).
Stirring was continued for 1 h then acidified with dilute hydrochloric acid and extracted
with ethyl acetate (3 x 20mL). The extract was dried over anhydrous sodium sulfate and
evaporated to afford 2,4-dimethoxy-6-(2-hydroxyethyl)-3-methylbenzoic acid (4) (0.35g,
78%); Rf: 0.3 (petroleum ether and ethyl acetate, 4:1); m. p. 72-74 °C, IR (KBr): 3481
(O-H), 3009 (C-H), 1710 (C=O), 1574 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm ): 8.22 (1H,
s, COOH), 7.48 (1H, s, H-5), 4.21 (2H, t, J=3.8, H-1’), 3.90 (3H, s, 2-OCH3), 3.75 (3H,
s, 4-OCH3), 2.65 (2H, t, J=3.8Hz, H-2’ ) 2.51 (3H, s, 3-CH3); 13
C NMR (CDCl3, δ ppm):
190.5 (COOH), 166.2 (C2), 162.4 (C4), 141.3 (C6), 108.1 (C3), 105.6 (C1), 101.2 (C5),
63.2 (C2’), 56.4 (2-OCH3,4-OCH3), 32.3 (C1’), 28.8 (3-CH3); MS (70eV): m/z (%); 240
[M]+ (32), 223 (24), 196 (43), 165 (100), 45 (19), 31 (37); Anal. Calcd. for C12H16O5: C,
60.00 H, 6.66 Found: C, 59.87 H, 6.57.
5.1.5 6,8-Dimethoxy-7-methyl-3,4-dihydro-1H-isochromen-1-one (5)
2,4-dimethoxy-6-(2-hydroxyethyl)-3-methylbenzoic acid (4) (1.0g, 4.16 mmol)
was dissolved in acetic anhydride (5mL) and refluxed for 1 h. Then reaction mixture was
poured into ice cold water and extracted with ethyl acetate (3 x 20mL). The combined
ethyl acetate extract was washed with (1% NaHCO3) and then with water. The ethyl
acetate was evaporated under reduced pressure to afford 6,8-dimethoxy-7-methyl-3,4-
dihydro-1H-isochromen-1-one (5). Yield 78% Rf: 0.6 (petroleum ether and ethyl acetate,
4:1); m. p. 145-147°C; IR (KBr): 3010 (C-H), 1702 (C=O), 1591 (C=C) cm-1
; 1H NMR
(CDCl3, δ ppm ): 7.48 (1H, s, H-5), 4.25 (2H, t, J=3.6, H-3), 3.90 (6H, s, 6-OCH3, 8-
OCH3), 2.66 (3H, s, 7-CH3), 2.56 (2H, t, J=3Hz, H-4); 13
C NMR (CDCl3, δ ppm ) 163.9
(C1), 152.3 (C6, C8), 140.8 (C4a), 134.6 (C8a), 108.9 ( C7), 103.7 (C5), 65.9 (C3), 56.4
(6-OCH3, 8-OCH3), 27.4 (C4), 26.2 (7-CH3); MS (70eV): m/z (%); 222 [M]+ (52), 194
(45), 192 (100), 164 (31), 30 (21); Anal. Calcd. for C12H14O4 C, 64.86 H, 6.30 Found C,
64.75 H, 6.19 O.
5.1.6 6,8-Dimethoxy-7-(bromomethyl)-3,4-dihydro-1H-isochromen-1-one (6)
To a stirred solution of 6,8-dimethoxy-7-methyl-3,4-dihydro-1H-isochromen-1-
one (5) (0.5g, 2.25 mmol) in dry carbon tetrachloride (10mL) was added N-
bromosuccinimide (0.6g, 3.37 mmol) and benzoyl peroxide (7.5mg). The reaction
Page 128
110
mixture was refluxed for 6 h, then cooled, filtered and washed with a little carbon
tetrachloride. Solvent was rotary evaporated to get 6,8-dimethoxy-7-(bromomethyl)-3,4-
dihydro-1H-isochromen-1-one (6). (Yield 81%); Rf: 0.6 (petroleum ether and ethyl
acetate, 4:1); m. p. 91-93°C; IR (KBr): 3013 (C-H), 2926 (Ar-C-H), 1718 (C=O), 1582
(C=C) cm-1
; 1H NMR (CDCl3, δ ppm ): 7.50 (1H, s, H-5), 4.85 (2H, s, CH2-Br), 4.24 (2H,
t, J=3.6, H-3), 3.90 (6H, s, 6-OCH3, 8-OCH3), 2.58 (2H, t, J=3.6, H-4); 13
CNMR
(CDCl3, δ ppm ): 169.2 (C1), 155.4 (C6, C8), 144.1 (C4a), 110.7 ( C7), 106.2 (C8a),
104.6 (C5), 67.4 (C3), 53.7 (6-OCH3, 8-OCH3), 39.5 (CH2-Br), 26.4 (C4). MS (70eV):
m/z (%); 200 [M]+ (24), 302 [M+2] (24), 272 (39), 221 (19), 191 (100), 30 (28); Anal.
calcd. for C12H13O4Br C, 47.84 H, 4.31 Found C, 47.72 H, 4.23.
5.1.7 7-(Hydroxymethyl)-6,8-dimethoxy-3,4-dihydro-1H-isochromen-1-one (7)
6,8-Dimethoxy-7-(bromomethyl)-3,4-dihydro-1H-isochromen-1-one (6) (1.0g,
3.32 mmol) was dissolved in a mixture of water and acetone (10 mL, 1:1). The reaction
mixture was refluxed for 1 h then most of the acetone was rotary evaporated. Then
poured into ice cold water and the solid was filtered, washed with water and dried to
afford 7-(hydroxymethyl)-6,8-dimethoxy-3,4-dihydro-1H-isochromen-1-one (7), (Yield
77%); Rf: 0.4 (petroleum ether and ethyl acetate, 4:1); m. p. 136-138°C; IR (KBr): 3467
(O-H), 3010 (C-H), 2919 (Ar-C-H), 1714 (C=O), 1587 (C=C) cm-1
; 1H NMR (CDCl3, δ
ppm ): 7.62 (1H, s, H-5), 4.88 (2H, s, CH2-OH), 4.20 (2H, t, J=3.7, H-3), 3.90 (6H, s, 6-
OCH3, 8-OCH3), 2.56 (2H, t, J=3.7, H-4), 2.36 (1H, s, O-H); 13
C NMR (CDCl3, δ ppm ):
168.6 (C1), 157.3 (C6, C8), 142.1 (C4a), 115.2 ( C7), 105.2 (C8a), 102.6 (C5), 64.5 (C3),
55.7 (6-OCH3, 8-OCH3), 48.6 (CH2-OH), 27.4 (C4); MS (70eV): m/z (%); 238 [M]+ (49),
221 (31), 210 (55), 208 (100), 30 (19); Anal. calcd. for C12H14O5 C, 60.50 H, 5.88 Found
C, 60.38 H, 5.76.
5.1.8 8-Hydroxy-7-(hydroxymethyl)-6-methoxy-3,4-dihydro-1H-isochromen-1-one (8)
7-(Hydroxymethyl)-6,8-dimethoxy-3,4-dihydro-1H-isochromen-1-one (7) (1.5g,
6.3mmol) was dissolved in dry THF (20mL) and treated with magnesium (0.2g, 7.58
mmol) and iodine 1.0g, 8.38 mmol) in dry benzene (30mL). The resulting mixture was
refluxed for 30min. and then poured into water and organic layer was separated and
washed with water. Evaporation of the solvent under reduced pressure afforded 8-
hydroxy-7-(hydroxymethyl)-6-methoxy-3,4-dihydro-1H-isochromen-1-one (8), (Yield
Page 129
111
47%); Rf: 0.5 (petroleum ether and ethyl acetate 4:1); m. p. 127-128 °C; IR (KBr): 3559
(O-H), 3001 (C-H), 2920 (Ar-C-H), 1710 (C=O), 1593 (C=C) cm-1
; 1H NMR (CDCl3, δ
ppm ): 10.90 (1H, s, 8-OH), 7.06 (1H, s, H-5), 4.54 (2H, s, CH2-OH), 4.12 (2H, t, J=3.7,
H-3), 3.90 (3H, s, 6-OCH3), 2.86 (2H, t, J=3.7, H-4), 2.29 (1H, s, CH2O-H); 13
C NMR
(CDCl3, δ ppm): 166.5 (C1), 158.7 (C8), 151.4 (C6), 143.1 (C4a), 116.9 (C7), 105.2
(C8a), 102.6 (C5), 63.9 (C3), 56.9 (6-OCH3), 47.1 (CH2-OH), 28.4 (C4); MS (70eV): m/z
(%); 224 [M]+.
(59), 222 (21), 207 (42), 194 (100), 30 (19); Anal. Calcd. for C11H10O5 C,
59.45 H, 4.50 Found C, 59.37 H, 4.39 O.
5.2 Results and Discussion
Vilsmeier Haack formylation of the acetate (1) with phosphorous oxychloride in
N, N-dimethylformamide (DMF) afforded the methyl (2-formyl-3, 5-dimethoxy-4-methyl
phenyl) acetate (2). The IR showed very strong new carbonyl absorption at 1690 cm-1
for
aldehydic carbonyl in addition to the 1722 cm-1
peak for ketonic carbonyl already
present. The 1H NMR showed the singlet for aldehydic proton at δ 9.75 ppm and the
characteristic changes in the chemical shifts of the benzylic protons. 13
C NMR showed a
peak at δ 179.3 ppm for aldehydic carbon and a peak at 162.4 for ester carbon already
present. The molecular ion peak appeared at m/z 252 with 25% bundance and the base
peak at m/z 165.
The aldehyde (2) was oxidized to 2,4-Dimethoxy-6-(2-methoxy-2-oxoethyl)-3-
methylbenzoic acid (3) by using sulfamic acid and sodium chlorite at 0°C in 79% yield.
The carbonyl absorption in IR shifted from 1690 cm-1
to 1715 cm-1
due to oxidation of
aldehydic function into carboxyl one. The absorption at 3265 cm-1
for (O-H) is also
present in IR spectrum. In 1H NMR a singlet at δ 8.19 ppm appeared for carboxylic
proton and downfield shift from δ 179.3 to δ 197.7 ppm for carboxylic carbon was also
appeared in 13
C NMR.
The ester acid (3) was then reduced to 2,4-dimethoxy-6-(2-hydroxyethyl)-3-
methylbenzoic acid (4) using sodium borohydride-methanol system refluxed in freshly
distilled THF. The IR showed a broad band at 3481 cm-1
for the hydroxyl group and also
the disappearance of ester carbonyl absorption was noticed. In 1H NMR a singlet for
hydroxyl proton appeared at δ 2.03 ppm and two methylenes showed two triplet at δ 4.23
ppm, J=3.8Hz and δ 2.65 ppm, J=3.8Hz. The reduction of esters and similar functional
Page 130
112
groups using sodium borohydride is relatively difficult to obtain and it has not been
widely used. However, the reactivity of the sodium borohydride can be enhanced by
carrying out the reaction in the presence of NaBH4-CH3OH. This methodology is simple,
safe, inexpensive, and general and the reduction of methyl esters was completed after
refluxing in THF.
Then cyclization of the alcohol acid (4) was carried out in the presence of acetic
anhydride refluxed in dry toluene to afford 6,8-dimethoxy-7-methyl-3,4-dihydro-1H-
isochromen-1-one (5). In IR spectrum the absorption band for hydroxyl disappeared and
the lactonic carbonyl showed absorption at 1702 cm-1
. Similarly in 1H NMR the singlet
for hydroxyl and carboxyl protons were also disappeared. The lactonic carbonyl carbon
showed peak at δ 163.9 ppm in 13
C NMR spectrum.
OCH3
H3CO
CH3
O
OH
CH3OH
Conc.H2SO
4 OCH3
H3CO
CH3
O
OCH3
OCH3
H3CO
CH3 COOH
OHNaBH
4/ THF
CH3OH
(1)
(4)
O
O
CH3
OCH 3
H3CO
Acetone/H2O Mg/I2
THF/Benzene
O
O
CH2
OCH3
H3CO
Br
(5) (6)
O
O
CH2
OCH3
H3CO
OH
(7)
O
O
CH2
OH
H3CO
OH
(8)
(CH3CO)
2O
1h reflux
CH3
OCH3
H3CO O
OCH3O
H
(2)
CH3
OCH3
H3CO O
OCH3O
OH
(3)
POCl3 / Freshly distilled DMF
CH3COO- Na
+
NH2SO3H / NaClO2 / 0 °C
H2O:THF:DMSO
Benzoylperoxide /CCl4
N-bromosuccinimide
Scheme 5.1 Synthesis of Stellatin
Page 131
113
The benzylic bromination of the 6,8-dimethoxy-7-methyl-3,4-dihydro-1H-
isochromen-1-one (5) in the presence of N-bromosuccinimide and catalytic amount of
benzoyl peroxide refluxed in dry CCl4 afforded 6,8-dimethoxy-7-(bromomethyl)-3,4-
dihydro-1H-isochromen-1-one (6). In 1H NMR, the singlet for 7-methyl hydrogens
shifted downfield after bromination from δ 2.66 ppm to δ 4.85 ppm. Then 6,8-dimethoxy-
7-(bromomethyl)-3,4-dihydro-1H-isochromen-1-one (6) was converted into 7-
(hydroxymethyl)-6,8-dimethoxy-3,4-dihydro-1H-isochromen-1-one (7) by nucleophilic
substitution using acetone and H2O. IR showed a broad band at 3467 cm-1
for hydroxyl
group. In 1H NMR, a singlet for hydroxyl proton appeared at δ 2.36 ppm.
The selective demethylation of the 8-hydroxyl group in 7-(hydroxymethyl)-6,8-
dimethoxy-3,4-dihydro-1H-isochromen-1-one (7) using Mg/I2 refluxed in THF/benzene
afforded 8-hydroxy-7-(hydroxymethyl)-6-methoxy-3,4-dihydro-1H-isochromen-1-one
(Stellatin) (8). In 1H NMR, a singlet at δ 10.90 ppm appeared for 8-hydroxy and 7-
hydroxy proton appeared as a singlet at δ 2.29 ppm. The physical constants and FTIR
spectral data of the compounds (1-8) are presented in Table 5.1.
Table 5.1 Physical and FTIR spectral data of the compounds (1-8)
Compds M. P.
(°C)
Rf Yield
(%) Ar-C-H Sp
3 C-H C=O C=C O-H
1 38-40 0.7 87 3023 2925 1734 1583 -
2 51-53 0.55 85 3015 2917 1722,
1690 1595 -
3 164-166 0.4 79 3011 2928 1734,
1715 1582 3265
4 72-174 0.3 83 3009 2924 1710 1574 3481,
3034
5 145-147 0.65 78 3010 2935 1702 1591 -
6 91-93 0.6 81 3013 2926 1718 1582 -
7 136-138 0.4 77 3010 2919 1714 1587 3467
8 127-128 0.5 45 3001 2920 1710 1593 3559,
3178
Pet. Ether: Ethyl Acetate (4:1)
Page 132
114
Table 5.2 1H and
13C NMR data of Stellatin (8)
Carbons δ (ppm) and multiplicity
1H NMR
13C NMR
C-1 ----- 166.5
C-3 4.12, t, J=3.7Hz 63.9
C-4 2.86, t, J=3.7Hz 28.7
C-4a ----- 143.1
C-5 7.06, s 102.6
C-6 ----- 15.4
C-7 ----- 116.9
C-8 ----- 158.7
C-8a ----- 105.2
6-OCH3 3.90, s 56.9
7-CH2 4.56, s 47.1
8-OH 10.90, s -----
CH2-OH 2.29, s -----
The formation of the 8-hydroxy-7-(hydroxymethyl)-6-methoxy-3,4-dihydro-1H-
isochromen-1-one (8) was confirmed by the presence of two triplets at δ 4.12 ppm with
coupling constant 3.7Hz for H-4 and 2.86 ppm with coupling constant 3.7Hz for H-3
hydrogens in 1H NMR spectrum. A singlet at δ 3.90 ppm appeared for 6-methoxy
hydrogens. The 8-hydroxy appeared as a singlet at δ 10.90 ppm in 1H NMR spectrum. It
was also supported in 13
C NMR spectrum by the presence of a peak at δ 166.5 ppm for
lactonic carbon C-1. The detailed 1H and
13C NMR data of the compound (8) are
presented in Table 5.2.
Page 133
115
The structure of the Stellatin (8) was further confirmed by mass spectrometry.
The molecular ion peak appeared at m/z 224 with 59% abundance which confirmed the
formation of isocoumarin (8). By the removal of hydroxyl radical from molecular ion, a
peak at m/z 207 with 42% abundance appeared. A peak at m/z 194 is the base peak
formed by the elimination of formaldehyde fragment from molecular ion. The
fragmentation pattern of the Stellatin (8) is shown in Fig. 5.1.
m/z = 224 [M]+.
59%m/z = 207, 42%m/z = 30, 19%
m/z = 179, 51%
CO_
_
CH2
O
H3CO
OH
OH
m/z = 194, 100%
m/z = 196, 47%
+.
+
+
+
H3CO
OH
OOH
O
O
H3CO
OHOH
-OH.
+
O
O
H3CO
OH
+
O
H3CO
OH
O
OH
H3CO
OH
CO_
.
CH2O
.
CH2
O
_.
m/z = 206, 63%
+.
O
O
H3CO
O
-H2O
-H2
m/z = 222, 21%
+.
O
O
H3CO
OO
Fig. 5.1 Mass fragmentation pattern of the Stellatin (8)
Page 134
116
5.3 Antibacterial Activity
Bacterial infections constitute one the most serious situations in infectious
disease. The detection and identification of these bacteria is one of the most important
functions of clinical microbiology. Isolation of an infectious agent from the patient with
disease is often not sufficient for determining proper therapy. Since the susceptibility of
many bacteria to antimicrobial agents cannot be predicted testing individual pathogens,
against appropriate agent (with the most activity against the pathogen, the least toxicity to
the most, the least important on normal flora, appropriate pharmacologic characteristics
and most economical) can then be chosen allowing a more certain therapeutic outcome.
Antibacterial activity of the compounds (5-8) was determined against various
gram positive and gram negative bacterial strains by using agar well diffusion. The
purified samples were dissolved in DMSO 5mg/ml. DMSO is the negative control and
antibiotic chloramphenicol is the positive control in this invitro antibacterial study.
Ten bacterial strains Escherichia coli (E. c.), Klebsiella pneumonae (K. p.),
Lactobacillus bulgaricus (L. b.), Micrococcus luteus (M. l.), Pasteurella multocida (P.
m.), Proteus vulgaris (P. v.), Pseudomonas aeruginosa (P. a.), Salmonella typhi (S. t.),
Staphylococcus aureus (S. a.) and Staphylococcus epidermidis (S. e.) were selected in
this antibacterial assay. Micrococcus luteus, Staphylococcus aureus and Staphylococcus
epidermidis are the example of Gram positive and the remaining seven are gram negative
bacteria. All of the tested microorganisms were maintained on nutrient agar at 4°C and
sub-cultured before use. The bacteria studied are clinically important ones causing
several infections and it is essential to overcome them through some active therapeutic
agents.
The antibacterial assay was performed by agar well diffusion method against
different bacterial strains. Each tested bacterium was sub-cultured in nutrient broth at
37°C for 24h. One hundred micro liters of each bacterium was spread with the help of
sterile spreader on to a sterile Muller-Hinton agar plate so as to achieve a confluent
growth. The plates were allowed to dry and wells (6mm diameter) were punched in the
agar with the help of cork borer. 0.1mL of the each compound solution (5mg/mL) in
DMSO was introduced in to the well and the plates were incubated overnight at 37°C.
Page 135
117
The antimicrobial spectrum of the compounds was determined for the bacterial
species in terms of size of the zones around each well. The diameters of the zone of
inhibition produced by the compounds were compared with those produced by the
commercial antibiotic chloramphenicol (5mg/mL). This is the common antibiotic used
for the treatment of infections caused by gram positive and gram negative bacteria. The
control activity was deducted from the test and the results obtained were plotted. The
experiment was performed three times to minimize the error and the mean values are
presented.
Anti bacterial activity results of the compounds (5-8) is shown in table 5.3
respectively.
Table 5.3 In vitro Antibcterial activity of compounds (5-8)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
5 3 1 0 11 0 0.5 1 3 10 11
6 1 7 2 9 10 0 1.5 2 6 7
7 11 7 5 0.5 8 1 3 0 0.5 1.5
8 14 8 9 1 11 6 2 12 1 3
Standard 18 10 13 13 12 13 13 14 13 13
*Activity of each sample is measured by subtracting the activity of DMSO.
Escherichia coli (E. c.), Klebsiella pneumonae (K. p.), Lactobacillus bulgaricus (L. b.),
Micrococcus luteus (M. l.), Pasteurella multocida (P. m.), Proteus vulgaris (P. v.),
Pseudomonas aeruginosa (P. a.), Salmonella typhi (S. t.), Staphylococcus aureus (S. a.)
and Staphylococcus epidermidis (S. e.)
Page 136
118
5.4 Cytotoxic Activity
The Neutralrottest was carried out according to the protocol of the National
Institutes of Health (NIH). In the vital dye neutral red is taken up by living cells and then
protonated. The cell acts as an ion trap, allowing the dye can not diffuse out of it. By
destroying the cells, the neutral is released again and can be determined photometrically.
The absorption is a measure of cell viability, the lower the absorption were the less living
cells. For the tests, the immortalized human Keratinocyte cell line (HaCaT) was used.
The incubation with the test substances was carried out over three days and the
experiments were conducted in two independent experiments with a number of parallels.
The stock solutions of substances were prepared in DMSO. The concentration of the
solvent DMSO was all tested concentrations of 0.1%. Etoposide as positive control was
carried in a concentration of 10μM (IC50). Concentration range of the tested samples is
1.56-100 (µM).
Fig. 5.2 Cytotoxic activity of the samples (5-8)
The results showed that cytotoxic activity increases as the concentration of the
samples increases. The cytotoxic activity of the final stellatin is higher as compared to the
precursors. Compounds 5 and 6 showed moderate to low cytotoxic activity. When 7-
methyl group in compound 5 is converted into 7-hydroxymethyl group in compound 7,
cytotoxic activity is increased. After selective demethylation of the 8-methoxy group in
compound 8 results in increase of cytotoxic activity of the final compound stellatin.
Presence of the 7-hydroxymethyl and 8-hydroxy functional groups play vital role in
cytotoxic activity of the final compound.
0
20
40
60
80
100
120
10 25 50 100 1000
Via
bilit
y [
%]
Concentration [uM]
5
6
7
8
Page 137
119
6. REFERENCES PART I
1. Bu’Lock, J. D. The Biosynthesis of Natural Product, an introduction to secondary
metabolite. Mcgraw-Hill, New York, London, 1965.
2. Barry, R. D. Chem. Rev.1964, 64, 229.
3. Turner, W. B.; Aldridge, D. C. Fungal metabolites II; 1983, Academic Press,
London.
4. Yamato, V.; Yuki Gosei kagaku kyokaishi., 1983, 41, 958.
5. Hill, R. A. Progress in the Chemistry of Organic Natural Products. 1986, 49, 1-
78.
6. Napolitano, E. Organic Preparations and Procedures Int., 1997, 26, 631.
7. Bin, Y.; Song, L.; Xiaohui, G. Tianran Chanwu Yanjiu Yu Kaifa., 2000, 12, 95.
8. Filho, R. B.; De Moraes, M. P. L.; Gottieb, O. R. Phytochem. 1980, 19, 2003.
9. Vogel, A. Gilbert’s Ann. Phy., 1820, 64,161.
10. Henderson, G. B.; Hill, R. A. J. Chem. Soc. Perkin Trans. I., 1982, 1111.
11. Cantello, B. C. C.; Buckle, D. R.; Smith, H. UK patent, GB1480737, 1977.
12. Bailey, D. M.; DE Grazia, C. G. J. Org. Chem., 1970, 35, 4088.
13. Turner, W. B. Fungal metabolites, 1971, Academic Press London.
14. Suzuki, Y. Agric. Biol. Chem; (Japan), 1970, 34, 760.
15. Mcinerney, B. V.; Taylor, W. C.; Lacey, R. J.; Akhurst, R. J.; Gregson, R. P. J.
Nat. Prod., 1991, 785-795.
16. a) Curtis, R. F.; Hassall, C. H.; Nazer, M. J. Chem. Soc. (C), 1968, 85. b) Barber,
J.; Garson, J. Stanton, J. J. chem. Soc., Perkin Trans. 1, 1981, 2584. c) Fujii, I.;
Watanebe, A.; Sankawa, U.; Ebiauka, Y. Chem. Biol., 2001, 8, 189.
17. Maneekarn, C.; Prasat, K.; masahiko, I.; Ratchanda, C.; Morako, T.; Yodhathai,
T. J. chem. Soc., Perkin Trans. 1, 2002, 2473-2476.
18. Hiroyuki, K.; Masahide, A.; Hiroshi, N,; Tsutomu, S.; masaaki, I.; Tomio, T.
J.Antibiotics, 1994, 47, 4, 440-446.
19. Furutani, Y.; Tsuchiya, I.; Naganawa, H.; Takeuchi, T.; Umezawa, H. Agric. Biol.
Chem. 1977, 41, 1581-1585.
20. Christopher, N. L.; James, S.; David, C. S. J. chem. Soc., Perkin Trans. I, 1988,
747-754.
Page 138
120
21. De Jesus, A. E.; Steyn, P. S.; Vieggaar, R.; Wessels, P. L. J. chem. Soc., Perkin
Trans. 1, 1980, 52.
22. Wiesleder, D.; Lillehoj, E. Tetrahedron Lett., 1980, 21, 993.
23. Jonathan, P. H.; Peter, G. M. Phytochemistry, 2001, 58, 709-716.
24. Matsuda, H.; Shimoda, H.; Yoshikawa, M. Bioorg. Med. Chem., 1999, 7, 1445-
1450.
25. Yoshikawa, M.; Harada, E.; Naitoh, Y.; Inoue, K.; Matsuda, H.; Shimoda, H.;
Yamahara, J.; Murakami, N. Chem. Pharm. Bull., 1994, 42, 2225-2230.
26. a) Matsuda, H.; Shimoda, H.; Yamahara, J.; Yoshikawa, M.; Bioorg. Med. Chem.
Lett., 1998, 8, 215-220. b) Shimoda, H.; Matsuda, H.; yamahara, J.; Yoshikawa,
M.; Biol. Pharm. Bull., 1998, 21, 809-813.
27. Nozawa, K.; Yamada, M.; Tsuda, Y.; kawai, K.; Nakajima, S.; Chem. Pharm.
Bull., 1981, 29, 2689-2691.
28. Furuta, T.; Fukuyama, Y.; Asakawa, Y. Phytochemistry, 1986, 25, 517-520.
29. Whyte, A. C.; Gloer, J. B.; Scott, J. A.; Mallock, D. J. Nat. Prod., 1996, 59, 765-
769.
30. Lee, J. H.; Park, Y. J.; Kim, H. S.; Hong, Y. S.; kim, K. W.; Lee, J. J.; J. Antibiot.,
2001, 54, 463-466.
31. Bloomquist, J. R. Arch. Insect Biochem. Physiol., 2003, 54, 145-156.
32. Yoshihisa, O.; Tadahiko, K.; Yuji, T.; Kenzo, H.; Nobutoshi, T.; Takashi, Y.;
Emiko, S.; Kei-ichi, I.; Kazuhiko, O. J. Pestic. Sci., 2004, 29, 4, 328-331.
33. Zhi-Hong, X.; Li-Tian,; Tian-jiao, Z.; Wen-Liang, W.; Lin, D.; Yu-chun, F.;
Qian-Qun, G, Wei-Ming, Z. Arch. Pharm. Res. 2007, 30,7, 816-819.
34. Yong-Fu, H.; Lin-Hao, L.; Li, T.; Li, Q.; Hui-Ming, H.; Yue-Hu, P. J. Antibiot.
2006, 59, 6, 355-357.
35. Shimojima, Y.; Hayashi, H.; Ooka, T.; Shibukawa, M. Agric. Biol. Chem., 1982,
46, 1823-1829.
36. Itoh, J.; Omoto, S.; Shomura, T.; Nishizawa, N.; Miyado, S.; Yuda, Y.; Shibata,
U.; Inouye, S. Agric. Biol. Chem., 1982, 46, 1255-1259.
37. Mcinerney, B. V.; Taylor, W. C.; Lacey, M. J.; Akhurst, R. J.; Gergson, R. P. J.
Nat. Prod., 1991, 54, 785-795.
Page 139
121
38. Tutomu, S.; koji, N.; Kenichi, S.; Moto, M.; Takeshi, S. The Journal of
Antibiotics, 1992, 45, 12, 1949-1952.
39. Rodrigues, K. F.; Petrini, O. Biodiversity of Tropical Microfungi (ed. K. D. Hyde),
Hong Kong, 1997, 57-69.
40. Lin, Y.; wan, J.; Zhou, S.; Gareth, J. CJI, 2001, 3, 7, 30.
41. Razieh, Y.; Hamid, R. A.; Ataback, B. DARU, 2000, 8, 1 & 2, 42-44.
42. Danise, C. E.; Keller, G. G.; Tamara, P. K.; john, M. P.; Fernao, C. B. J. Nat.
Prod., 2008, 71, 6, 1082-1084.
43. Karina, F. D.; Maria, S. G. R.; Eliana, A. V.; wagner, V. Z. Naturforsch., 2002,
57c, 85-88.
44. Kithsiri, E. M. W.; Priyani, A. P.; Leslie, A. A. G. Tetrahedron, 2006, 62, 34,
8439-8446.
45. Perreault, S. D.; Zirkin, B. R. J. Exp. Zool., 1982, 224, 253-257.
46. Tummon, I. S.; Yuzpe, A. A.; Daniel, S. A.; Duetsch, A. Fertil. Steril., 1991, 56,
933-938.
47. Francavilla, S.; Palermo, G.; Gabriele, A.; Cordeschi, G.; Poccia, G. Fertil. Steril.,
1992, 57, 1311-1316.
48. De Jonge, C. J.; Tarchala, S. M.; Rawlins, R. G.; Binor, Z.; Radwariska, E. Hum.
Reprod., 1993, 8, 253-257.
49. Palencia, D. D.; Garner, D. L.; Hudig, D.; Holcombe, D. W.; Burner, C. A.;
Redelman, D.; Fernandez, G. C. J.; Abuelyaman, A. S.; Kam, C. M.; Powers, J. C.
Biology of Reproduction, 1996, 55, 536-542.
50. Kumagai, H. et al., Journal of Antibiotics, 1990, 72, 1505.
51. Shin-ichi, H.; Toshiyuki, M.; Naoki, A.; Hiroshi, I.; Naoki, M.; Takeo, Y.;
Hiroshi, T.; Hiroyuki, K.; Massaki, I.; Tomio, T. US Pat. Appl., 2000, US
6020363.
52. Neidleman, S. L.; Geigert, J. Biohalogenation, 1986, 13-15.
53. Marc, S.; Heidrun, A. The Journal of Antibiotics, 1995, 48, 3, 261-266.
54. Kongsaeree, P.; Prabpai, S.; Sriubolmas, N.; Vongvein, C.; Wiyakrutta, S. J. Nat.
Prod., 2003, 66, 5, 709-711.
Page 140
122
55. Oikawa, T.; Sasaki, M.; Inose, M.; Shimamura, M.; Kobuki, H.; Hirano, S.;
Kumagai,H.; Ishizuka, M.; Takeuchi, T. Anticancer Res., 1997, 17, 1881-1886.
56. Nakashima, T.; Hirano, S.; Agata, N.; Kumagai, H.; Isshiki, K.; Yoshioka, T.;
Ishizuka, M.; Maeda, K.; Takeuchi, T. J. Antibiot., 1999, 52, 426-428.
57. Reimer, C. L.; Agata, N.; Tammam, J. G.; Bamberg, M.; Dickerson, W. M.;
Kamphaus, G. D.; Rook, S. L.; Milhollen, M.; Fram, R.; Kalluri, R.; Kufe, D.;
Kharbanda, S. Cancer Res. 2002, 62, 789-795.
58. Agata, N.; Nogi, H.; Bamberg, M.; Milhollen, M.; Pu, M.; Weitman, S.;
Kharbanda, S.; Kufe, D. Cancer Chemother. Pharmacol. 2005, 56, 610-614.
59. Kunihiro, I.; Yohei, M.; Yoshihiko, Y.; Masaru, K.; Kumiko, H.; Hiroyuki, K.;
Yuki, T.; Hitoshi, S.; Yasushi, Y.; Naoki, A.; Hirofumi, M. Diabetes, 2006, 55,
1232-1242.
60. Agata, N.; Nogi, H.; Milhollen, M.; Kharbanda, S.; Kufe, D. Cancer Res. 2004,
64, 8512-8516.
61. Wang, Y. Med. Res. Rev., 2001, 21, 146-170.
62. Romer, J.; Neilsen, B.; Ploug, M. Curr. Pharm. Des., 2004, 10, 2359-2376.
63. Jo, M.; Thomas, K.; Marozkina, N.; Amin, T.; Silva, C.; Parsons, S.; Gonias, S. J.
Biol. Chem., 2005, 280, 17449-17457.
64. Justin, J. H.; Lucy, A. H.; Thomas, A. V. J.; Lorraine, M. D.; David, L. V. J. BMC
Chemical Bilogy, 2006, 6, 1472.
65. Narasimhan, N. S.; Mali, R. S. Topics in current chemistry, 1987, 138, 63.
66. Woon, E. C. Y.; Dhami, A.; Mahon, M. F.; Threadgill, M. D. Tetrahedron.
2006, 62, 4829.
67. Subramanian, V.; Batchu, V. R.; Barange, D.; Pal, M. J. Org. Chem. 2005,
70, 4778.
68. Roy, H.; Sarkar, M. Synth. Commun. 2005, 35, 2177.
69. Cherry, K.; Parrain, J. L.; Thibonnet, J.; Duchene, A.; Abarbri, M. J. Org. Chem.
2005, 70, 6669.
70. Suzuki, T.; Yamada, T.; Watanabe, K.; Katoh, T. Bioorg. Med. Chem. Lett.
2005, 15, 2583.
71. Opatz, T.; Ferenc, D. Eur. J. Org. Chem. 2005, 817.
Page 141
123
72. Martinez, A.; Fernandez, M.; Estevez, J. C.; Estevez, R. J.; Castedo, L.
Tetrahedron 2005, 61, 485.
73. Yao, T.; Larock, R. C. J. Org. Chem., 2003, 68, 5936.
74. Liao, H.-Y.; Cheng, C. H. J. Org. Chem., 1995, 60, 3711.
75. Bonadies, F.; DiFabio, R. J. Org. Chem., 1984, 49, 1647.
76. Kinder, M. A.; Kopf, J.; Margaretha, P. Tetrahedron, 2000, 56, 6763.
77. Colonge, J.; Boisde, P. Bull. Soc. Chem. France, 1956, 1337.
78. Carter, R. H.; Colyer, R. M.; Hill, R. A.; Staunton, J. J. Chem. Soc. Perkin. Trans.
I., 1982, 1438.
79. Kendall, J. K.; Fisher, T. H. J. Org. Chem., 1989, 54, 4218.
80. Mal, D.; Bandyopadhyay, M.; Datta, K.; Murty, K. V. S. N. Tetrahedron, 1998,
54, 7525.
81. Mal, D.; Bandyopadhyay, M.; Sujit, K.; Datta, K. Tetrahedron Lett., 2000, 41, 1.
82. Kinder, M. A.; Kopf, J.; Margaretha, P. Tetrahedron, 2000, 56, 6763.
83. a) Vaulx, R. L.; Puterbauth, W. H.; Hauser, C. R. J. Org. Chem., 1964, 29, 3514.
b) Mao, C. L.; Barnish, I. T.; Hauser, C. R. J. Heterocyle. Chem., 1969, 6, 83.
84. Narasimhan, N. S.; Bhide, B. H. Tetrahederon, 1971, 27, 6171.
85. Narasimhan, N.S.; Mali, R. S. Synthesis, 1983, 63.
86. Snieckus, V. Chem. Rev., 1990, 90, 879.
87. Lee, D.; Still, W.C. J. Org. Chem., 1989, 54, 4715.
88. Reitz, D. B.; Massey, S. M. J. Org. Chem., 1990, 55, 1375.
89. Superchi, S.; Minutolo, F.; Pini, D.; Salvadori, P. J. Org. Chem., 1996, 61, 3183.
90. Mroady, S. M.; Rexhausen, J. E.; Thomas, E. J. J. Chem. Soc. Perkin Trans. 1.,
1999, 1083.
91. Pini, D.; Superchi, S.; Salvadori, P. J. Organometallic. Chem., 1993, 452.
92. Gruniwald, G. L.; Dahanukar, V. H. J. Heterocyclic Chem., 1994, 31, 1609.
93. Bestmann, H. J.; Kern, F.; Schafer, D.; Witschel, M. C. Angew. Chem., Int. Ed.
Engl., 1992, 31, 795.
94. Choukchou-Braham, N.; Asakawa, Y.; Lepoittevin, J. P. Tetrahedron Lett., 1994,
35, 3949.
95. Bhide, B. H.; Akolkar, V. D.; Brahmbhat, D. I. Ind. J. Chem., 1992, 31(B), 116.
Page 142
124
96 Kurosaki, Y.; Fukuda, T.; Iwao, M. Tetrahedron, 2005, 61, 3289–3303.
97. Zenner, J. M.; Larock, R.C. J. Org. Chem., 1999, 64, 7312.
98. Kessar, S. V.; Singh, P.; Vohra, R.; Kaur, N. P.; Venugopra, D. J. Org. Chem.,
1992, 57, 6716.
99. Conners, R.; Tran, E.; Durst, T. Can. J. Chem., 1996, 74, 221.
100. Larock, C. L.; Varaprath, S. US Pat. Appl., 1987, US 4650881.
101. Nagarjaan, A.; Balasubramanian, R.T. Indian J. Chem., Sect.B, 1987, 26B, 917.
102. Roy, H. N.; Sarkar, M. S. Synthetic Communications, 2005, 35, 2177.
103. Stephen, P. W.; Marisa, C. K. Tetrahedron Letters., 2001, 42, 3567-3570.
104. Suzuki,T.; Yamada, T.; Watanabe, K.; Katoh, T. Biorg. Med.Chem. Lett. 2005,
15, 2583-2585.
105. Ueura, K., Satoh, T., Miura, M., J. Org. Chem. 2007, 72, 5362.
106. Loewenthal, H. J. E.; Pappo, R. J. Chem. Soc., 1992, 4799.
107. Chatterjea, J. N.; Mukherjea, H. Experietia, 1960, 16, 439.
108. Chatterjea, J. N.; Mukherjee, H. J. Indian chem. Soc., 1960, 37, 379.
109. Chatterjea, J. N.; Mukherjee, H. J. Indian chem. Soc., 1960, 37, 443.
110. Yamato, M.; Hashigaki, K. Chem. Pharm. Bull., 1976, 24, 200.
111. Kabayashi, T. Sci. Rept. Tohoku Univ., First Ser., 1942, 31, 73; C. A., 1950, 44,
4013.
112. Haworth, R. D.; Pindred, H. K.; Jafferies, P. R. J. Chem. Soc., 1954, 3617.
113. Chatterjea, J. N. J. Indian chem. Soc., 1953, 30, 103.
114. Vorozhtsov, N. N.; Petushova, A. T. J. Gen. Chem. USSR, 1957, 27, 2282.
115. Tirdkar, R. B.; Usgoankar, R. N. Indian J. Chem., 1970, 8, 123.
116. Modi, R.; Usgoankar, R. N. Indian J. Chem., 1979, 17B, 360.
117. Rose, A.; Buu-Hoi, N. P.; Jacquinon, P. J. Chem. Soc., 1965, 6100.
118. Yoshikawa, H.; Taniguchi, E.; Maekawa, K. J. Pesticide Sci., 1980, 5, 1.
119. Sarkhel, B. K.; Srivasta, J. N. J. Indian chem. Soc., 1976, 53, 915; Ibid., 1977, 54,
925.
120. Tuanli Yao and Richard C. Larock, Tetrahedron Lett., 2002, 43, 7401.
121. Berti, G. Tetahedron, 1958, 4, 393.
122. Muller, E. Chem. Ber., 1909, 42, 423.
Page 143
125
123. Ribbens, C.; Koninkl, N. V.; Fabrieken, P. v/h brocades-Stheeman Pharmacia,
1960-61, 10, 9. C. A. 1962, 56, 7378.
124. Stadlbauer, W.; Ghobrial, N.; Kappe T. Z. Naturforsch, 1980, 35b, 892.
125. Prey, V.; Kerres, B.; Berbalk, H. Monatsh. Chem., 1960, 91, 774.
126. Berti, G.; Marsili, A.; Mini, V. Ann. Chim.(Rome), 1960, 50, 669.
127. Birk, A. J.; Donovan, F. W. Australian J. Chem., 1953, 6, 360.
128. Colonge, J.; Boisde, P. Bull. Soc. Chim. France, 1956, 1337.
129. Maitte, P. Compt. Rend., 1954, 239, 1508.
130. Siegel, S.; Colburn, S. K.; Levering, D. R. J. Am. Chem. Soc., 1951, 73, 3163.
131. Shriner, R. L.; Knox, W. R. J. Org. Chem., 1951, 16, 1064.
132. Alder, E.; Magnusson, R.; Berggren, B. Acta Chem, Scand., 1960, 14, 539.
133. Grimshaw, J.; Haworth, R. D.; Pindred, H. K. J. Chem. Soc., 1955, 833.
134. Yamamoto, I. Agri. Biol. Chem. (Tokyo), 1961, 25, 400; C. A., 1961, 55, 670.
135. Thomas, O.L.; Jens, B. J. Nat. Prod., 1999, 62, 1182.
136. Stadler, M.; Anke, H.; Sterner, O. J. Antibiot., 1995, 48, 261.
137. Yamato, M.; Hashigaki, K.; Honda, E.; Sato, K.; Koyama, T. Chem. Pharm. Bull.,
1977, 25, 695.
138. Liu, D. Zhonghua Yixue Zashi, 1982, 62, 336.
139. Haworth, R. D.; Pindred, H. K.; Jafferies, P. R. J. Chem. Soc., 1954, 3617.
140. Nozawa, K.; Yamada, M.; Tsuda, Y.; Kawai, K. I.; Nakajima, S. Chem. Pharm.
Bull., 1981, 29, 2491.
141. Berti, G. J. Org. Chem., 1959, 24, 934.
142. Okeke, M. I.; Iroegbu, C. U.; Eze, E. N.; Okoli, A. S.; Esimone, C. O. Journal of
Ethnopharmacology, 2001, 78, 119-127.
143. Plowe, C. V.; Djimde, A.; Bouare, M.; Doumbo, O.; Wellens, T. E. Am. J. Trop.
Med. Hyg. 1995, 52, 6, 565-568.
144. Omulokoli, E.; Khan, B.; Chhabra, S. C. J. Ethnopharmacol. 1997, 56, 133-137.
Page 144
126
7.1 INTRODUCTION
Isocoumarins (1H-2-benzopyran-1-ones) are the secondary metabolites of an
extensive variety of fungi, bacteria, higher plants, marine organisms and are also
among insect venoms and pheromones; exhibiting a wide range of structural diversity and
biological activities1-3
. Important examples of natural bioactive isocoumarins include
the furoisocoumarin coriandrin phototoxic to RNA-virus Sindbis virus, DNA-virus
murine cytomegalo-virus and HIV4, thunberginol, phyllodulcin and hydrangenol having
differentiation inducing, antiallergic, and immunomodulatory effects5, ochratoxins A &
B, nephratoxic, hepatotoxic6, hiburipyranone, cytotoxic
7, duclauxin, antitumor
8,
cytogenin and its synthetic analogues antitumor, antidiabetic anticancer9 and Sg17-1-4,
possessing potent cytotoxic activities10
.
Majority of the natural isocoumarins being of polyketide origin are derived
biogenetically from acetate-polymalonate pathway, hence most of them possess a C-3
alkyl / aryl substituent. Although, more than two hundred isocoumarins and
dihydroisocoumarins have been isolated and the number is still increasing dramatically,
1-thioisocoumarins are thus far unknown in nature. A review of literature reveals that
while, the thio analogues of a number of associated natural products viz., chromones,11
flavones,12
isoflavones13
and coumarins14
have been prepared, the reports of synthetic 1-
thioisocoumarins (1H-isochromene-1-thiones) are exceptional15
.
1(2H)-isoquinolones (isocarbostyrils) are the nitrogen analogues of isocoumarins
(1H-2-benzopyran-1-ones). Various 1(2H)-isoquinolone derivatives are found in several
bioactive natural products such as thalifoline, doryphorine16
, uprechstyril
17, narciclasine
18,
pancratistatin, lycoricidine19
, the alkaloids coryaldine20
, dorianine21
hydroxyhydrastinine
and thalflavine21
. Isoquinolone nucleus is also an integral part of complex isoquinoline
alkaloids and is a useful building block in organic synthesis.
The isoquinolone skeleton biogenetically derived from amino acid phenylalanine,
exhibits biomimetic characteristics22
. Substituted isoquinolones are orally effective
antagonists of receptors 5-HT3, which have shown higher efficacy in the control of cancer
models23
, thymidylate synthase (TS) inhibitors24
, human Tumor Necrosis Factor (TNF)
inhibitors, and tachykinin receptors25
. Substituted isocarbostyrils showing
Page 145
127
antidepressant26
, anti-inflammatory27
, analgesic28
and hypolipidemic29
activities have also
been reported.
7.2 EXPERIMENTAL
Melting points were recorded using a digital Gallenkamp (SANYO) model MPD
BM 3.5 apparatus and are uncorrected. 1H NMR spectra were determined as CDCl3
solutions at 300 MHz on a Bruker AM-300 spectrophotometer. FT IR spectra were
recorded using an FTS 3000 MX spectrophotometer; Mass Spectra (EI, 70eV) on a GC-
MS instrument and elemental analyses with a LECO-183 CHNS analyzer. All
compounds were purified by thick layer chromatography using silica gel from Merck.
Synthesis of homophthalic anhydride (1)
A solution of homophthalic acid (2.0g, 12.34 mmol) in dry toluene (35 mL) was
treated with acetic anhydride (1.1g, 10mL, 10.8 mmol). The reaction mixture was
refluxed for 1 hr and then poured into ice cold water. The organic layer was separated,
dried over anhydrous sodium sulfate and toluene was rotary evaporated to get
homophthalic acid anhydride (1). Yield 82%; Rf: 0.7 (petroleum ether and ethyl acetate,
4:1); m. p. 140-142°C; IR (KBr): 3011 (C-H), 1735 (C=O), 1590 (C=C) cm-1
; 1H NMR
(CDCl3, δ ppm): 7.85 (1H, d, J=3.7, H-8), 7.3-7.4 (2H, m, H-6, H-7), 6.97 (1H, d, J=3.4,
H-5), 3.47 (2H, s, H-4); 13
C NMR (CDCl3, δ ppm): 165.5 (C3), 147.1 (C1), 137.2 (C4a),
134.4 (C6), 131.5 (C8a), 130.7 (C8), 129.7 (C5), 127.5 (C7), 38.2 (C4); MS (70eV): m/z
(%) ; 162 [M+] (25), 134 (43), 118 (100), 90 (32); Anal. Calcd. for C9H6O3: C, 66.66 H,
3.70; Found: C, 66.53 H, 3.59.
General procedure for 3-alkyl/arylisocoumarins (4a-j)
A mixture of aliphatic/aromatic carboxylic acids (2a-j) (1 mmol) and thionyl
chloride (1.2 mmol) was refluxed for 1 hr in the presence of a drop of DMF. The
completion of reaction was determined by stoppage of evolution of gas. Excess of the
thionyl chloride was rotary evaporated to afford acid chlorides (3a-j).
A solution of homophthalic anhydride (1) (2.00 mmol) in acetonitril (12mL) was
added to a solution of N,N,N’,N’-tetramethylguanidine (TMG) (2.20 mmol) in acetonitril
(5mL) over 36 min maintaining an internal temperature of 0°C. Triethylmine (4.0 mmol)
was added in one portion. Acid chlorides (3a-j) (3.20 mmol) were added over 3 min and
the mixture was stirred for an additional 18 min. After the completion of reaction the
Page 146
128
cooling bath was removed and reaction was allowed to warm to room temperature. The
reaction mixture was quenched by the addition of HCl (1M, 5mL). The two phases were
separated and organic layer was washed with saturated sodium chloride solution and then
dried (Na2SO4) prior to removal of solvent under reduced pressure to dryness.
Isocoumarins (4a-j) were then purified by preparative thin layer chromatography using
(petroleum ether and ethyl acetate, 7:3) as eluant.
3-(3-Fluorophenyl)isocoumarin (4a)
Yield 79%; m. p. 91-92 °C; Rf. 0.8; IR (KBr): 2980 (C-H), 1730 (C=O), 1615 (C=C) cm-
1;
1H NMR (CDCl3, δ ppm) 8.14 (1H, s, H-2’), 7.56-7.70 (2H, m, H-4’, H-5’), 7.4 (1H, d,
J=2.1, H-6’), 7.3 (2H, d, J=7.8, H-5, H-8), 7.22 (1H, dd, J=1.8, 2.1, H-6), 7.15 (1H, dd,
J=2-4, 2-4, H-7), 6.87 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 164.5 (C1), 159.2 (C3’),
143.2 (C3), 138.4 (C4a), 134.7 (C6), 132.2 (C1’), 130.5 (C8), 129.4 (C5’), 128.1 (C8a),
127.6 (C7), 126.2 (C5), 122.1 (C6’), 116.2 (C4’), 112.5 (C2’), 104.2 (C4); MS (70eV):
m/z (%) 240 [M+] (100), 212 (45), 145 (53), 117 (27); Anal. Calcd. for C15H9FO2: C,
75.00 H, 3.75 Found: C, 74.93 H, 3.69.
3-(4-Fluorophenyl)isocoumarin (4b)
Yield 70%; m. p. 109-110 °C; Rf. 0.6; IR (KBr): 3020 (C-H), 1725 (C=O), 1590 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm) 8.70 (2H, d, J=7.8, H-3’, H-5’), 7.77 (2H, d, J=3, H-2’,
H-6’), 7.72 (1H, d, J=1.2, H-5), 7.51 (3H, m, H-6, H-7, H-8), 6.95 (1H, s, H-4); 13
C NMR
(CDCl3, δ ppm) 168.2 (C1), 158.7 (C4’), 141.7 (C3), 136.3 (C4a), 133.5 (C6), 131.9
(C8), 129.5 (C8a), 128.2 (C2’, C6’), 127.6 (C7), 126.3 (C5), 125.7 (C1’), 119.6 (C3’,
C5’), 106.4 (C4); MS (70eV): m/z (%) 240 [M+] (100), 212 (45), 145 (53), 117 (27);
Anal. Calcd. for C15H9FO2: C, 75.00 H, 3.75 Found: C, 74.93 H, 3.69.
3-(2-Chlorobenzyl)isocoumarin (4c)
Yield 87%; m. p. 154-156 °C; Rf. 0.5; IR (KBr): 2860 (C-H), 1738 (C=O), 1558 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm) 7.48-7.51 (2H, m, H-6, H-7), 7.58 (1H, d, J=1.6, H-5),
7.65 (1H, d, J=1.5, H-8), 7.81 (1H, dd, J=1.5, H-5’), 7.85 (1H, d, J=1.5, H-6’), 8.1 (1H,
dd, J=1.5, 1.8, H-4’), 8.9 (1H, d, J=8.1), 2.0 (2H, s, CH2), 6.52 (1H, s, H-4); 13
C NMR
(CDCl3, δ ppm) 165.4 (C1), 143.7 (C3), 138.4 (C4a), 137.5 (C1’), 135.6 (C2’), 133.2
(C6), 131.6 (C8), 130.4 (C6’), 129.1 (C8a), 128.5 (C3’), 127.4 (C7), 126.1 (C4’), 125.5
(C5’), 123.3 (C5), 106.7 (C4), 35.2 (CH2); MS (70eV): m/z (%) 270 [M+] (100), 272
Page 147
129
[M+2] (70), 242 (49), 117 (23); Anal. Calcd. for C16H11ClO2: C, 70.97 H, 4.06 Found: C,
70.89 H, 3.97.
3-(2-Bromophenyl)isocoumarin (4d)
Yield 75%; m. p. 119-120 °C; Rf. 0.8; IR (KBr): 3025 (C-H), 1710 (C=O), 1590 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm) 7.81 (1H, d, J= 2.4, H-3
’), 7.55-7.65 (3H, m, H-4
’,H-5
’,
H-6’), 7.2-7.3 (4H, m, H-5, H-6, H-7, H-8), 6.75 (1H, s, H-4);
13C NMR (CDCl3, δ ppm)
158.7 (C1), 141.1 (C3), 136.8 (C4a), 135.5 (C1’), 134.6 (C6), 131.6 (C3’), 130.7 (C4’),
129.6 (C8), 128.5 (C8), 127.6 (C6’), 126.3 (C7), 125.5 (C5’), 124.2 (C5), 118.6 (C2’),
102.7 (C4); MS (70eV): m/z (%) 300 [M+] (100), 302 [M+2] (98), 272 (43), 226 (40),
145 (60), 117 (13); Anal. calcd. for C15H9BrO2: C, 59.80 H, 2.99 Found: C, 59.73 H,
2.91.
3-(3-Iodophenyl)isocoumarin (4e)
Yield 75%; m. p. 107-108 °C; Rf. 0.7; IR (KBr): 3010 (C-H), 1725 (C=O), 1580 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm) 8.15 (1H, s, H-2’), 7.96 (1H, d, J=9, H-4’), 7.67 (1H, d,
J=8.1, H-6’), 7.61 (1H, dd, J=4.8, 3.3, H-5’), 7.51-7.55 (4H, m, H-5, H-6, H-7, H-8), 6.96
(1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 163.7 (C1), 145.6 (C3), 139.2 (C4a), 136.2 (C4’),
135.7 (C6), 134.5 (C2’), 133.1 (C1’), 130.8 (C8), 129.4 (C5’), 128.6 (C8a), 127.3 (C7),
126.8 (C5), 125.3 (C6’), 112.5 (C3’), 104.2 (C4); MS (70eV): m/z (%) 348 [M+] (100),
320 (54), 145 (64), 117 (19); Anal. calcd. for C15H9IO2: C, 51.72 H, 2.58 Found: C, 51.64
H, 2.51.
3-(2,4-Dichlorophenyl)isocoumarin (4f)
Yield 89%; m. p. 182-183 °C; Rf. 0.6; IR (KBr): 2970 (C-H), 1705 (C=O), 1620 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm) 7.6 (1H, d, J=0.9, H-3’), 7.5 (1H, d, J=13.2, H-5’), 7.4
(1H, d, J=8.5,H-6’), 7.1-7.35 (4H, m, H-5, H-6, H-7, H-8), 6.95 (1H, s, H-4); 13
C NMR
(CDCl3, δ ppm) 164.6 (C1), 141.5 (C3), 138.6 (C4a), 136.5 (C4’), 135.6 (C2’), 134.2
(C6), 132.8 (C8), 131.5 (C3’), 130.9 (C1’), 128.5 (C8a), 127.8 (C6’), 126.5 (C7), 125.7
(C5’), 123.6 (C5), 103.7 (C4); MS (70eV): m/z (%) 291 [M+] (100), 293 [M+2] (70), 295
[M+4] (13), 263 (49), 117 (23); Anal. calcd. for C15H8Cl2O2: C, 61.85 H, 2.74 Found: C,
61.77 H, 2.67.
Page 148
130
3-(2-Chloro-4-fluorophenyl)isocoumarin (4g)
Yield 84%; m. p. 176-177 °C; Rf. 0.7; IR (KBr): 2990 (C-H), 1705 (C=O), 1595 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm) 7.80 (1H, s, H-3
’), 7.78 (1H, d, J=2.7 H-5
’), 7.19 (1H, d,
J=2.4, H-6’), 7.15 (4H, m, H-5, H-6, H-7, H-8), 6.77 (1H, s, H-4);
13C NMR (CDCl3, δ
ppm) 167.3 (C1), 159.5 (C4’), 143.6 (C3), 137.5 (C4a), 135.4 (C2’), 134.5 (C6), 132.1
(C8), 131.0 (C8a), 130.4 (C6’), 129.4 (C1’), 127.3 (C7), 126.2 (C5), 121.3 (C3’), 117.4
(C5’), 105.3 (C4); MS (70eV): m/z (%) 274.5 [M+] (100), 276.5 (70), 246.5 (44), 145
(62), 117 (19); Anal. calcd. for C15H8ClFO2: C, 65.57 H, 2.91 Found: C, 65.49 H, 2.84.
3-(3-Nitrophenyl)isocoumarin (4h)
Yield 81%; m. p. 109 °C; Rf. 0.5; IR (KBr): 2893 (C-H), 1734 (C=O), 1512 (C=C) cm-1
;
1H NMR (CDCl3, δ ppm) 8.4 (1H, s, H-2’), 8.2 (1H, d, J=8.2,H-4’), 7.7-7.5 (2H, m, H-5’,
H-6’), 7.2-7.4 (4H, m, H-5, H-6, H-7, H-8), 7.1 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm)
162.3 (C1), 149.3 (C3’), 141.4 (C3), 137.5 (C4a), 135.1 (C6), 132.5 (C1’), 131.6 (C6’),
130.5 (C5’), 129.1 (C8), 128.4 (C8a), 127.3 (C7), 126.1 (C5), 123.5 (C2’), 121.8 (C4’),
105.1 (C4); MS (70eV): m/z (%) 267 [M+] (100), 239 (59), 145 (61), 117 (21); Anal.
calcd. for C15H9NO4: C, 67.41 H, 3.37 N, 5.24 Found: C, 67.33 H, 3.31 N, 5.15.
3-(2-Chloropyridyl)isocoumarin (4i)
Yield 85%; m. p. 122-124 °C; Rf. 0.6; IR (KBr): 2882 (C-H), 1743 (C=O), 1543 (C=C)
cm-1
; 1H NMR (CDCl3, δ ppm) 8.7 (1H, d, J=1.6, H-3’), 8.5 (1H, d, J=2.1, H-5’), 8.0
(1H, dd, J=2.2, 2.4, H-4’), 7.6 (1H, d, J=1.5, H-8), 7.2-7.5 (3H, m, H-5, H-6, H-7), 6.95
(1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 168.2 (C1), 157.8 (C1’), 148.6 (C3’), 146.3 (C3),
140.8 (C5’), 138.6 (C4a), 134.2 (C6), 131.5 (C8), 129.5 (C8a), 127.9 (C7), 126.3 (C5),
124.1 (C4’), 121.6 (C6’), 104.3 (C4); MS (70eV): m/z (%) 257.5 [M+], (100), 259.5
[M+2] (70), 229.5 (34), 145 (63), 117 (17); Anal. calcd. for C14H8ClNO2: C, 65.24 H,
3.10 N, 5.43 Found: C, 65.17 H, 3.04 N, 5.37.
3-Pentadecylisocoumarin (4j)
Yield 76%; m. p. 71-73 °C; Rf. 0.55; IR (film): 2918 (C-H), 2849 (Sp3 C-H), 1728
(C=O), 1604 (C=C), cm-1
; 1H NMR (CDCl3, δ ppm) 8.25 (1H, d, J = 8.16, H-8), 7.65
(1H, m, H-6), 7.49 (1H, td, J = 0.88, 7.28, H-7), 7.34 (1H, d, J = 8.16, H-5), 6.24 (1H, s,
H-4), 2.52 (2H, t, J = 7.08, 2H, H-1’), 1.70 (2H, p, J = 8.4, H2’), 1.28 (24H, brs, H3’-
H14’), 0.87 (3H, t, J = 6.28, H-15’); 13
C NMR (CDCl3, δ ppm): 165.4 (C1), 152.3 (C3),
Page 149
131
138.5 (C4a), 134.3 (C6), 131.5 (C8), 129.4 (C8a), 128.7 (C7), 126.2 (C5), 105.3 (C4),
33.9 (C1’), 24.2 (C2’), 29.8 (C3’), 29.6 (C4’), 29.4 (C5’), 28.8 (C6’), 28.6 (C7’), 28.2
(C8’), 27.9 (C9’), 27.6 (C10’), 26.8 (C11’), 25.4 (C12’), 24.6 (C13’), 14.3 (C14’), 11.7
(C15’); MS (70eV): m/z (%) 356 [M+] (100), 328 (34), 145 (37), 117 (21); Anal. calcd.
for C24H36O2: C, 80.89 H, 10.11 Found: C, 80.81 H, 10.04.
General procedure for the conversion of isocoumarins into 1(2H)-
isoquinolones (5a–j)
A mixture of isocoumarins (4a-j) (10 mmol) and formamide (10 mmol) was
refluxed for 2-4 h. On completion of the reaction, followed by TLC, the solution was
poured into water (300 ml). The resulting precipitates were filtered and recrystallized
from ethyl acetate to afford 1(2H)-isoquinolones (5a–j).
3-(3-Flourophenyl) isoquinolin-1(2H)-one (5a)
Yield 84%; m. p. 216-218 °C; Rf. 0.65; IR (KBr): 3332 (NH), 2825 (C-H), 1656 (C=O),
1517 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 10.3 (1H, s, NH), 8.4 (1H, d, J=1.2, H-4’),
7.72 (1H, d, J=0.9, H-2’), 7.70 (1H, d, J=1.5, H-6’), 7.54 (1H, dd, J=1.8, 1.6, H-5’), 7.23
(1H, d, J=2.4, H-8), 7.1-7.2 (3H, m, H-5, H-6, H-7), 6.83 (1H, s, H-4); 13
C NMR (CDCl3,
δ ppm) 168.2 (C1), 156.2 (C3’), 139.4 (C3), 137.5 (C4a), 135.4 (C6), 131.7 (C1’), 130.8
(C8), 129.9 (C5’), 128.5 (C8a), 127.2 (C7), 126.7 (C5), 123.4 (C6’), 117.4 (C4’), 113.7
(C2’), 103.6 (C4); MS (70eV): m/z (%) 239 [M+] (100), 211 (41), 144 (47), 117 (19);
Anal. calcd. for C15H10FNO: C, 75.31 H, 4.18 N 5.85 Found: C, 75.24 H, 4.11 N, 5.78.
3-(4-Flourophenyl)isoquinolin-1(2H)-one (5b)
Yield 82%; m. p. 222-224 °C; Rf. 0.55; IR (KBr): 3320 (NH), 2870 (C-H), 1630 (C=O),
1527 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 10.54 (1H, s, NH), 8.4 (2H, d, J=8.4, H-3’,
H-5’), 7.7 (2H, d, J=1.8, H-2’,H-6’), 7.63 (1H, d, J=1.2, H-8), 7.5-7.6 (2H, m, H-6, H-7),
7.4 (1H, dd, J=1.2, 1.1, H-5), 6.8 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 166.5 (C1),
159.4 (C4’), 142.3 (C3), 137.4 (C4a), 134.2 (C6), 131.6 (C8), 129.8 (C8a), 128.5 (C2’,
C6’), 127.1 (C7), 126.6 (C5), 125.2 (C1’), 119.2 (C3’, C5’), 105.4 (C4); MS (70eV): m/z
(%) 239 [M+] (100), 211 (41), 144 (47), 117 (19); Anal. calcd. for C15H10FNO: C, 75.31
H, 4.18 N 5.85 Found: C, 75.24 H, 4.11 N, 5.78.
Page 150
132
3-(2-Chlorobenzyl)isoquinolin-1(2H)-one (5c)
Yield 85%; m. p. 188-190 °C; Rf. 0.6; IR (KBr): 3348 (NH), 2860 (C-H), 1638 (C=O),
1558 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 9.53 (1H, s, NH), 9.1 (1H, d, J=8.1, H-3’),
8.3 (1H, dd, J=1.5, 1.8, H-4’), 7.89 (1H, d, J=1.5, H-6’), 7.85 (1H, dd, J=1.5, 1.6, H-5’),
7.7 (1H, d, J=1.5, H-8), 7.6 (1H, d, J=1.5,H-5), 7.5-7.55 (2H, m, H-6,H-7), 6.66 (1H,
s,H-4), 2.19 (2H, s, CH2); 13
C NMR (CDCl3, δ ppm) 167.5 (C1), 141.3 (C3), 137.5 (C4a),
136.4 (C1’), 135.2 (C2’), 134.5 (C6), 132.8 (C8), 131.3 (C6’), 129.7 (C8a), 128.8 (C3’),
127.5 (C7), 126.5 (C4’), 124.9 (C5’), 122.8 (C5), 104.6 (C4), 37.6 (CH2); MS (70eV):
m/z (%) 269 [M+] (100), 271 [M+2] (70), 241 (37), 117 (23); Anal. calcd. for
C16H12ClNO: C, 71.24 H, 4.45 N, 5.19 Found: C, 71.19 H, 4.37 N, 5.13.
3-(2-Bromophenyl)isoquinolin-1(2H)-one (5d):
Yield 82%; m. p. 170-172 °C; Rf. 0.65; IR (KBr): 3341 (NH), 2864 (C-H), 1635 (C=O),
1535 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 9.4 (1H, s, NH), 9.12 (1H, d, J=8.4, H-3’),
8.38 (1H, d, J=1.5, H-6’), 7.66-7.75 (2H, m, H-4’,H-5’), 7.63 (1H, d, J=2.7, H-8), 7.45-
7.55 (2H, m, H-6, H-7), 7.34 (1H, d, J=1.8, H-5), 6.63 (1H, s, H-4); 13
C NMR (CDCl3, δ
ppm) 163.6 (C1), 143.1 (C3), 137.3 (C4a), 134.6 (C1’), 133.8 (C6), 132.3 (C3’), 130.3
(C4’), 129.5 (C8), 128.4 (C8), 127.6 (C6’), 126.7 (C7), 125.2 (C5’), 123.9 (C5), 117.3
(C2’), 104.5 (C4); MS (70eV): m/z (%) 299 [M+] (100), 301 [M+2] (98), 271 (43), 144
(60), 117 (19); Anal. calcd. for C15H10BrNO: C, 60.00 H, 3.33 N, 4.66 Found: C, 59.93
H, 3.26 N, 4.61.
3-(3-Iodophenyl)isoquinolin-1(2H)-one (5e):
Yield 84%; m. p. 226-228 °C; Rf. 0.7; IR (KBr): 3353 (NH), 2892 (C-H), 1643 (C=O),
1533 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 10.6 (1H, s ,NH), 8.5 (1H, s, H-2’), 8.2 (1H,
d, J=13.8, H-4’), 8.1 (1H, t, J=1.8, H-5’), 7.7 (1H, d, J=1.2, H-6’), 7.6 (1H, d, J=1.2, H-
8), 7.5 (1H, d, J=1.2, H-5), 7.5-7.54 (2H, m, H-6, H-7), 6.8 (1H, s, H-4); 13
C NMR
(CDCl3, δ ppm) 166.3 (C1), 143.5 (C3), 138.3 (C4a), 136.7 (C4’), 135.3 (C6), 134.5
(C2’), 133.4 (C1’), 131.7 (C8), 129.7 (C5’), 128.2 (C8a), 127.5 (C7), 126.5 (C5), 125.7
(C6’), 113.6 (C3’), 105.3 (C4); MS (70eV): m/z (%) 347 [M+] (100), 319 (47), 144 (34),
117 (13); Anal. calcd. for C15H10INO: C, 51.87 H, 2.88 N, 4.03 Found: C, 51.81 H, 2.82
N, 3.96.
Page 151
133
3-(2,4-Dichlorophenyl)isoquinolin-1(2H)-one (5f):
Yield 78%; m. p. 221-222 °C; Rf. 0.6; IR (KBr): 3330 (NH), 2862 (C-H), 1650 (C=O),
1550 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 10.9 (1H, s, NH), 9.0 (1H, d, J=8.1, H-5’),
8.4 (1H, d, J=7.8, H-6’), 7.88 (1H, s, H-3’), 7.7 (1H , d, J=6.9, H-8), 7.61-7.67 (2H, m
,H-6, H-7), 7.5 (1H, d, J= 5.7, H-5), 6.6 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 167.3
(C1), 142.4 (C3), 137.3 (C4a), 136.1 (C4’), 135.2 (C2’), 134.5 (C6), 133.2 (C8), 132.9
(C3’), 131.2 (C1’), 129.8 (C8a), 128.7 (C6’), 126.2 (C7), 125.4 (C5’), 124.1 (C5), 104.2
(C4); MS (70eV): m/z (%) 290 [M+] (100), 292 [M+2] (70), 294 [M+4] (13), 262 (38),
117 (27); Anal. calcd. for C15H9Cl2NO: C, 60.06 H, 3.10 N, 4.82 Found: C, 59.98 H,
3.03 N, 4.74.
3-(2-Chloro-4-Flourophenyl)isoquinolin-1(2H)-one (5g):
Yield 80%; m. p. 213 °C; Rf. 0.55; IR (KBr): 3381 (NH), 2876 (C-H), 1655 (C=O), 1550
(C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 10.0 (1H, s, NH), 8.4 (1H, d, J=8.1, H-5’), 8.2
(1H, s, H-3’), 7.9 (1H, d, J=1.2, H-6’), 7.8 (1H, d, J=1.2, H-8), 7.7 (1H, d, J =1.2, H-5),
7.5-7.6 (2H, m, H-6, H-7), 6.65 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 168.4 (C1), 158.4
(C4’), 141.5 (C3), 138.2 (C4a), 136.2 (C2’), 135.1 (C6), 132.3 (C8), 131.5 (C8a), 130.8
(C6’), 129.6 (C1’), 128.2 (C7), 127.8 (C5), 124.2 (C3’), 118.8 (C5’), 104.8 (C4); MS
(70eV): m/z (%) 273.5 [M+] (100), 275.5 (70), 245.5 (56), 144 (61), 117 (21); Anal.
calcd. for C15H9ClFNO: C, 65.81 H, 3.29 N, 5.11 Found: C, 65.75 H, 3.22 N, 5.04.
3-(3-Nitrophenyl)isoquinolin-1(2H)-one (5h):
Yield 84%; m. p. 230-232 °C; Rf. 0.7; IR (KBr): 3339 (NH), 2821 (C-H), 1634 (C=O),
1512 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 8.75 (1H, s, NH), 8.6 (1H, s, H-2’), 8.4 (1H,
d, J=8.4, H-4’), 7.7-7.2 (2H, m, H-5’, H-6’), 7.4-7.6 (4H, m, H-5, H-6, H-7, H-8), 6.9
(1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 164.7 (C1), 151.5 (C3’), 143.5 (C3), 138.2 (C4a),
136.2 (C6), 132.9 (C1’), 131.8 (C6’), 130.7 (C5’), 129.5 (C8), 128.6 (C8a), 127.7 (C7),
126.2 (C5), 124.1 (C2’), 122.3 (C4’), 104.2 (C4); MS (70eV): m/z (%) 266 [M+] (100),
238 (46), 144 (51), 117 (14); Anal. calcd. for C15H10N2O3: C, 67.66 H, 3.75 N, 10.52
Found: C, 67.59 H, 3.69 N, 19.45.
3-(2-Chloropyridyl)isoquinolin-1(2H)-one (5i):
Yield 82%; m. p. 210-212 °C; Rf. 0.6; IR (KBr): 3353 (NH), 2882 (C-H), 1663 (C=O),
1543 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 10.2 (1H, s, NH), 8.8 (1H, d, J=1.8, H-3’),
Page 152
134
8.7 (1H, d, J=2.2, H-5’), 8.1 (1H, dd, J=2.4, 2.3, H-4’), 7.8 (1H, d, J=1.5, H-8), 7.5-7.7
(3H, m, H-5, H-6, H-7), 6.84 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm) 169.5 (C1), 159.1
(C1’), 149.5 (C3’), 144.5 (C3), 141.5 (C5’), 138.2 (C4a), 134.3 (C6), 132.1 (C8), 129.8
(C8a), 128.2 (C7), 127.1 (C5), 125.2 (C4’), 122.4 (C6’), 105.7 (C4); MS (70eV): m/z (%)
256.5 [M+], (100), 258.5 [M+2] (70), 228.5 (31), 144 (58), 117 (26); Anal. calcd. for
C14H9ClN2O: C, 65.49 H, 3.50 N, 10.91 Found: C, 65.42 H, 3.43 N, 10.84.
3-Pentadecylisoquinolin-1(2H)-one (5j):
Yield 76%; m. p. 71-73 °C; Rf. 0.55; IR (KBr): 3323 (NH), 2876 (C-H), 1652 (C=O),
1513 (C=C) cm-1
; 1H NMR (CDCl3, δ ppm) 9.62 (1H, s, NH), 8.2 (1H, d, J=13.5, H-8),
7.7-7.74 (2H, m, H-6, H-7), 7.6 (1H, d, J=1.2, H-5), 6.9 (1H, s, H-4), 2.3 (2H, t, J=7.5,
H-1’), 1.5-1.7 (26H, m, H-2’-H-14’), 0.9 (3H, t, J=5.1, H-15’); 13
C NMR (CDCl3, δ
ppm): 167.5 (C1), 151.7 (C3), 137.9 (C4a), 135.1 (C6), 132.2 (C8), 129.7 (C8a), 128.3
(C7), 126.3 (C5), 104.5 (C4), 33.2 (C1’), 32.3 (C2’), 29.7 (C3’), 29.5 (C4’), 29.3 (C5’),
28.6 (C6’), 28.1 (C7’), 27.9 (C8’), 27.6 (C9’), 27.1 (C10’), 26.8 (C11’), 25.4 (C12’), 24.6
(C13’), 14.3 (C14’), 11.7 (C15’); MS (70eV): m/z (%) 355 [M+] (100), 327 (39), 144
(49), 117 (18); Anal. calcd. for C24H37NO: C, 81.12 H, 10.42 N, 3.94 Found: C, 81.05
H, 10.35 N, 3.88.
7.3 RESULTS AND DISCUSSION
Homophthalic acid was converted into corresponding anhydride (1) by refluxing
homophthalic acid with acetic anhydride in the presence of dry toluene as solvent. The
formation of the anhydride (1) was confirmed by the presence of absorption at 1735cm-1
and disappearance of the absorption for hydroxyl groups in IR spectrum.
O
O
OHOH O
O
O
(1)
Acetic Anhydride
Dry Toluene
The substituted aromatic carboxylic acids (2a-j) were converted into their
respective acid chlorides by reacting them with thionyl chlorides in the presence of
catalytic amount of DMF. Acid chlorides (3a-j) were then condensed with homophthalic
anhydride (1) in the presence of triethyl amine and tetramethyl guanidine to afforded 3-
Page 153
135
aryl isocoumarins (4a-j). These isocoumarins were purified by preparative thin layer
chromatography using petroleum ether and ethyl acetate, 3:1 as eluant.
The synthesized isocoumarins (4a-j) showed the characteristic absorption for
lactonic carbonyl at 1713-1736 cm-1
. The detailed physical and FTIR spectral data of the
isocoumarins (4a-j) are shown in Table 7.1. In 1H NMR, spectrum a characteristic singlet
appeared for H-4 at δ 6.1-6.35 ppm and the lactonic carbonyl carbon in 13
C NMR showed
the peak at δ 158-170 ppm.
R
O
OH
+ SOCl2 R
O
Cl
(3a-j)(2a-j)
R is same as in (2a-j)
R
O
Cl
+O
O
O
(1)
O
O
R
(4a-j)(3a-j)
TMG / (C2H5)3N
CH3CN
R is same as in (2a-j)(2a-j ) R=
2a=3-F-C6H4 ; 2b=4-FC6H42c= 2-CIC6H4CH2; 2d=2-BrC6H42e=3-I-C6H4; 2f=2,4-DiClC6H3122g=2-F-4-ClC6H3; 2h=4-NO2C6H42i=2-ClC6H3N; 2j=C15H31
Scheme 7.1 Synthesis of 3-phenyl substituted isocoumarins (4a-j)
Table 7.1 Physical constants and FTIR spectral data of isocoumarins (4a-j)
Compds. M. P.
(°C) Rf
Yield
(%) Ar-C-H C-H C=O C=C
4a 107-108 0.7 72 3011 - 1723 1583
4b 119-120 0.8 75 3021 - 1715 1615
4c 91-92 0.8 79 3005 - 1719 1597
4d 109-110 0.6 70 3010 - 1725 1590
4e 109 0.5 81 3019 - 1716 1612
4f 182-183 0.6 89 3008 - 1735 1599
4g 176-177 0.7 84 3012 - 1727 1583
4h 154-156 0.5 87 3010 2882 1731 1618
4i 122-124 0.6 85 3017 - 1720 1593
4j 71-73 0.8 76 3012 2872 1725 1588
Pet. Ether: Ethyl Acetate (3:1)
Page 154
136
An equimolar mixture of the isocoumarin (4a-j) and methanamide was refluxed
for 2-4 hours to afford corresponding 1(2H)-isoquinolones (5a-j). The products were
obtained in 76-85 % yields in high purity. The progress of the reaction was followed by
TLC. The successful substitution was initially indicated by appearance of a fluorescent
blue spot under longer wave length of UV lamp, having Rf values lower than that of the
parent isocoumarin.
The products were further characterized by comparison of their m. p., IR, 1H,
13C
NMR and mass spectral data with those of the corresponding isocoumarins. Thus, a shift
of lactonic carbonyl absorption from 1710-1730 cm-1
to 1630-1650 cm-1
and appearance
of absorption at 3220-3380 cm-1
for NH was noted in the IR spectra. The physical
constants and FTIR spectral data of the compounds (5a-j) is shown in table 7.2. In the 1H
NMR a downfield shift of the characteristic H-4 proton of the isocoumarins at δ 6.0-6.2
to δ 6.6-6.9 ppm in isoquinolones was observed besides, appearance of NH absorption at
δ 9.4-10.8 ppm shown in table 7.3. A variety of substituents on the aryl ring are well-
tolerated, and the reaction leads to completion in all the cases. The generality of the
conversion was indicated by substrates bearing an aralkyl group (5c), heterocyclyl (5i) or
a long aliphatic chain (5j) at C-3 position.
O
O
R
NH
O
R
2-4h; 76-85 %
(4a-j ) R=
4a=3-F-C6H4 ; 4b=4-FC6H44c= 2-CIC6H4CH2; 4d=2-BrC6H44e=3-I-C6H4; 4f=2,4-DiClC6H3124g=2-F-4-ClC6H3; 4h=4-NO2C6H44i=2-ClC6H3N; 4j=C15H31
(5a-j)
R= same as in 4a-j
OH2N
Scheme 7.2 Synthesis of 3-phenyl substituted isoquinolones (5a-j)
Page 155
137
Table 7.2 Physical constants and FTIR spectral data of isoquinolones (5a-j)
Compds. M. P.
(°C) Rf
Yield
(%)
Ar-C-
H C-H C=O C=C N-H
5a 226-228 0.7 84 3013 - 1643 1533 3353
5b 170-172 0.8 82 3023 - 1635 1535 3341
5c 216-218 0.8 84 3015 - 1656 1517 3332
5d 222-224 0.6 82 3011 - 1630 1527 3320
5e 230-232 0.5 84 3009 - 1634 1512 3339
5f 221-222 0.6 78 3010 - 1651 1550 3330
5g 213-214 0.7 80 3013 - 1655 1543 3381
5h 188-190 0.5 85 3010 2860 1638 1558 3348
5i 210-212 0.6 82 3018 - 1663 1543 3353
5j 71-73 0.8 76 3001 2872 1652 1523 3323
Pet. Ether: Ethyl Acetate (3:1)
Table 7.3 Comparison of the δ values of H-4 among isocoumarins and isoquinolones
Entry Compd. R
1H NMR δ (ppm)
H-4 (4) H-4 (5) N-H (5)
1 5a 3-FC6H4 6.1 6.83 10.25
2 5b 4-FC6H4 6.4 6.80 10.54
3 5c 2-ClCH2C6H4 6.24 6.66 9.53
4 5d 2-BrC6H4 6.2 6.63 9.4
5 5e 3-IC6H4 6.34 6.8 9.8
6 5f 2,4-DiClC6H3 6.21 6.66 10.89
7 5g 2-Cl-4-FC6H3 6.35 6.65 10.05
8 5h 4-NO2C6H4 6.26 6.94 8.79
9 5j 2-Cl-C6H3N 6.3 6.84 10.2
10 5k C15H31 6.24 6.89 6.62
Page 156
138
Synthesis of 3-phenyl substituted-1H-isochromen-1-thiones
7.4 EXPERIMENTAL
Melting points were recorded using a digital Gallenkamp (SANYO) model MPD
BM 3.5 apparatus and are uncorrected. 1H NMR and the
13C NMR spectra were
determined as CDCl3 solutions at 300 MHz and 100 MHz respectively, on a Bruker AM-
300 machine. FT IR spectra were recorded using an FTS 3000 MX spectrophotometer;
Mass Spectra (EI, 70eV) on a GCMS instrument and elemental analyses with a LECO-
183 CHNS analyzer. The reactions were carried out in an unmodified domestic
microwave oven (MW 900 W, frequency 2450 MHz, Power level 1, Dawlance,
Pakistan). The analytical TLC was carried out using recoated plated from Merck and
thick layer chromatography using silica gel from Merck.
General procedure for the conversion of isocoumarins into 1-1H-
isochromene-1-thiones (2a–j)
A homogenized mixture of isocoumarin (6a-j) (1 mmol) and Lawesson’s reagent
(0.5-0.6 mmol) was irradiated for 1-3 min in an alumina bath inside the microwave oven.
The progress of the reaction was followed by TLC examination using hexane/ethyl
acetate (9:1). On completion, the reaction mixture was diluted with ethyl acetate and
subjected to thick layer chromatography using same solvent system. Elution using ethyl
acetate followed by concentration afforded the products (7a–j) which crystallized on
standing as yellow needles or plates.
3-(3-Fluorophenyl)-1H-isochromene-1-thione (7a)
Yield: 78%; m. p. 109-113 °C; Rf 0.8; IR (KBr): 2980 (C-H), 1615 (C=C), 1190 (C=S)
cm-1
. 1H NMR (CDCl3, δ ppm): 8.34 (1H, s, H-2’), 7.66-7.75 (2H, m, H-4’, H-5’), 7.62
(1H, d, J=2.1, H-6’), 7.53 (2H, d, J=7.8, H-5, H-8), 7.44 (1H, dd, J=1.8, 2.1, H- 6), 7.15
(1H, dd, J=2.4, 2.4, H-7), 6.98 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm): 203 (C1), 164
(C3), 162 (C3’), 152 (C8a), 137 (C1’), 135 (C6), 134 (C5), 130 (C2’,C4’), 129 (C8), 126
(C7), 120 (C6’,C5’), 112 (C 4a), 102 (C4); MS (70eV): m/z (%); 256 [M+] (100), 161
(67), 95 (48). Anal. calcd. For C15H9OSF: C, 70.31 H, 3.51 S, 12.50. Found. C, 70.25 H,
3.45 S, 11.45.
Page 157
139
3-(4-Fluorophenyl)-1H-isochromene-1-thione (7b)
Yield: 95%; m. p. 138 °C; Rf. 0.6, IR (KBr): 3020 (C-H), 1590 (C=C), 1195 (C=S) cm-1
.
1H NMR (CDCl3, δ ppm): 8.73 (2H, d, J=7.8, H-3’, H-5’), 7.97 (2H, d, J=3, H-2’, H-6’),
7.72 (1H, d, J=1.2, H-5), 7.51 (3H, m, H-6, H-7, H-8), 7.08 (1H, s, H-4); 13
C NMR
(CDCl3, δ ppm): 200 (C-1), 165 (C-3), 162 (C-4’), 155 (C-8a), 135 (C-1’), 132 (C-3’,C-
5’), 130 (C-8,C-5), 129 (C-7), 127 (C-2’,C-6’), 116 (C-4a), 104 (C-4); MS (70eV): m/z
(%) 256 [M+] (100), 161 (52), 95 (38). Anal. calcd. For C15H9OSF: C, 70.31 H, 3.51 S,
12.50. Found. C, 70.19 H, 3.41 S, 11.41.
3-(4-Chlorophenyl)-1H-isochromene-1-thione (7c)
Yield: 89%; m. p. 128-130 °C; Rf. 0.7, IR (KBr) 3025 (C-H), 1615 (C=C), 1171 (C=S)
cm-1
; 1H NMR (CDCl3, δ ppm): 7.85 (2H, d, J=1.8, H-3’,H-5’), 7.83 (2H, d, J=2.1, H-2’-
H-6’), 7.75 (1H, d, J=1.5, H-5), 7.73 (1H, d, J=1.2, H-8), 7.40 (2H, m, H-6, H-7), 6.96
(1H, s, H-4); 13
C NMR (CDCl3, δ ppm): 195 (C-1), 162 (C-3), 152 (C-4’), 152 (C-8a),
137 (C-6), 136 (C-5), 135 (C-3’,C-5’), 130 (C-2’,C-6’), 129 (C-1’), 128 (C-8), 126 (C-
7), 120 (C-4a), 102 (C-4); MS (70eV): m/z (%) 272.5 [M+] (100), 161 (67), 111.5 (48).
Anal. calcd. For C15H9OSCl: C, 66.05 H, 3.30 S, 11.74. Found. C, 65.76 H, 3.22 S,
11.66.
3-(2-Bromophenyl)-1H-isochromene-1-thione (7d)
Yield: 81%; m. p. Oil; Rf. 0.6, IR (KBr): 3025 (C-H), 1590 (C=C), 1079 (C=S) cm-1
; 1H
NMR (CDCl3, δ ppm): 8.03 (1H, d, J= 2.4, H-3’), 7.61-7.69 (3H, m, H-4’,5’,6’), 7.30-
7.40 (4H, m, H-5, 6, 7, 8), 6.89 (1H, s, H-4); 13
C NMR (CDCl3, δ ppm): 194 (C- 1), 164
(C-3), 152 (C-8a), 141 (C-2’), 136 (C-6), 134 (C-5), 133.8 (C-1’), 133 (C-3’), 132 (C-
4’,C-6’), 131 (C-5’), 129 (C-8), 127 (C-7), 113 (C-4a), 107 (C-4); MS (70eV): m/z (%)
316 [M+] (100), 161 (68), 155 (52). Anal. calcd. For C15H9OSBr: C, 56.96 H, 2.84 S,
10.12. Found. C, 56.87 H, 2.78 S, 10.05.
3-(3-Iodophenyl)-1H-isochromene-1-thione (7e)
Yield: 71%; m. p. 116-118 °C; Rf. 0.65; IR (KBr): 3010 (C-H), 1580 (C=C), 1085 (C=S)
cm-1
; 1H NMR (CDCl3, δ ppm): 8.25 (1H, s, H-2’), 8.06 (1H, d, J=9, H-4’), 7.77 (1H, d,
J=8.1, H-6’), 7.73 (1H, dd, J=4.8, 3.3, H-5’), 7.52-7.57 (4H, m, H-5-H-8), 6.97 (1H, s,
H-4); 13
C NMR (CDCl3, δ ppm): 197 (C-1), 158 (C-3), 155 (C-8a), 152 (C-3’), 137 (C-6),
135 (C-5), 132 (C-8), 130 (C-7), 128 (C-2’,C-4’), 109 (C-4), 125 (C- 1’), 113 (C-5’), 112
Page 158
140
(C-6’), 109 (C-4a); MS (70eV): m/z (%) 364 [M+] (100), 203 (48), 161 (67). Anal. calcd.
For C15H9OSI: C, 49.45 H, 2.47 S, 8.79. Found. C, 49.37 H, 2.39 S, 8.71.
3-(2,4-Dichorophenyl)-1H-isochromene-1-thione (7f)
Yield: 74%; m. p. 123-125 °C; Rf. 0.7 IR (KBr): 2970 (C-H), 1620 (C=C), 1128 (C=S),
cm-1
; 1H NMR (CDCl3, δ ppm): 7.80 (1H, d, J=0.9, H-3’), 7.74 (1H, d, J=13.2, H-5’),
7.71 (1H, d, J=8.5, H-6’), 7.51-7.61 (4H, m, H-5-H-8), 7.03 (1H, s, H-4); 13
C NMR
(CDCl3, δ ppm): 208 (C-1), 150 (C-3), 137 (C-2’,C-4’), 136 (C-8a), 135 (C-3’), 133 (C-
1’), 131 (C-5’), 130 (C-6), 129 (C-5), 128 (C-8), 127 (C-7), 126 (C-6’), 108 (C-4a), 106
(C-4); MS (70eV): m/z (%) 307 [M+] (100), 161 (62), 146 (52). Anal. calcd. For
C15H9OSCl2: C, 58.63 H, 2.60 S, 10.42. Found. C, 58.55 H, 2.52 S, 10.37.
3-(2-Chloro-4-fluorophenyl)-1H-isochromene-1-thione (7g)
Yield: 81%; m. p. 129 °C; Rf. 0.6; IR (KBr): 2990 (C-H), 1595 (C=C), 1275 (C=S) cm-1
;
1H NMR (CDCl3, δ ppm): 7.83 (1H, s, H-3’), 7.81 (1H, d, J=2.7 H-5’), 7.29 (1H, d,
J=2.4, H-6’), 7.28 (4H, m, H-5-8), 6.97 (1H, s, H-4). 13
C NMR (CDCl3, δ ppm): 200
(C-1), 164 (C-3), 161 (C-4’), 153 (C-2’), 153 (C-8a), 135 (C-6), 134 (C-5), 133 (C-3’),
132 (C-5’), 131 (C-1’), 130 (C-6’), 129 (C-8), 127 (C-7), 114 (C-4a), 107 (C-4), MS
(70eV): m/z (%) 290.5 [M+] (100), 161 (75), 129.5 (34); Anal. calcd. For C15H9OSClF:
C, 61.96 H, 2.75 S, 11.01; Found. C, 61.85 H, 2.68 S, 10.96.
3-(4-Methoxyphenyl)-1H-isochromene-1-thione (7h)
Yield: 91%; m. p. 109-111 °C; Rf. 0.5; IR (KBr): 3015 (C-H), 1575 (C=C), 1205 (C=S)
cm-1
; 1H NMR (CDCl3, δ ppm): 7.85 (2H, d, J=2.1, H-3’, H-5’), 7.83 (2H, d, J=2.1, H-
2’-H-6’), 7.74 (1H, d, J=1.5, H-5), 7.69 (1H, d, J=1.5, H-8), 7.40-7.50 (2H, m, H-6, H-7),
6.86 (1H, s, H-4), 3.88 (3H, s, H-4’-OCH3); 13
C NMR (CDCl3, δ ppm): 203 (C-1),
162 (C-3), 161 (C-4’), 153 (C-8a), 137 (C-6), 134 (C-5), 130 (C-3’,C-5’), 129 (C-1’), 128
(C-8), 127 (C-7), 126 (C-2’,C-6’), 114 (C-4a), 100 (C-4), 55.0 (C-OCH3), MS (70eV):
m/z (%) 268 [M+] (100), 161 (63), 107 (72); Anal. calcd. For C16H12O2S: C, 71.64 H,
4.47 S, 11.94. Found. C, 71.57 H, 4.39 S, 11.87.
3-(4-Fluorobenzyl)-1H-isochromene-1-thione (7i)
Yield: 93%; m. p. 65-67 °C; Rf. 0.6; IR (KBr): 2960 (C-H), 1610 (C=C), 1194 (C=S) cm-
1;
1H NMR (CDCl3, δ ppm): 7.73 (2H, d, J = 3.1, H-3’, 5’), 7.71 (2H, d, J=3.3 H-2’, H-
6’), 7.30 (4H, m, H-5-H-8), 7.01 (1H, s, H-4), 3.64 (2H, s, CH2); 13
C NMR (CDCl3, δ
Page 159
141
ppm): 176 (C-1), 167 (C-3), 164 (C-4’), 153 (C-8a), 136 (C-6), 135 (C-5), 134 (C-
3’,5’), 131 (C-2’,6’), 130 (C-1’), 129 (C-8), 127 (C-7), 114 (C-4a), 107 (C-4), 68 (CH2);
MS (70eV): m/z (%) 270 (M+] (100), 161 (62), 109 (37); Anal. calcd. for C16H11OSF: C,
71.11 H, 4.07 S, 11.85. Found. C, 71.05 H, 4.01 S, 11.78.
3-(Pentadecyl)-1H-isochromene-1-thione (7j)
Yield: 87%; m. p. 32-33 °C; Rf. 0.8; IR (KBr): 3010 (C-H), 1605 (C=C), 1272 (C=S); 1H
NMR (CDCl3, δ ppm): 8.26 (1H, d, J=8.1, H-8), 7.66-7.68 (2H, m, H-6, H-7), 7.36
(1H, d, J=7.8, H-5), 6.27 (1H, s, H-4), 2.50 (2H, t, J=7.5, H-1’), 1.73 (2H, p, J=6.6, H-
2’), 1.27-1.38 (24H, m, H-3’-H-14’), 0.89 (3H, t, J=5.4, H-15’). 13
C NMR (CDCl3, δ
ppm): 201 (C-1), 167 (C-3), 158 (C-8a), 137 (C-6), 132 (C-8), 129 (C-7), 128 (C-5), 125
(C-4a), 112 (C-4), 68 (C-1’), 55 (C-2’), 38 (C-3’), 33 (C-4’), 31 (C-5’), 30 (C-6’), 29 (C-
7’), 29 (C-8’), 29 (C-9’), 28 (C-10’), 26 (C-11’), 23 (C-12’), 22 (C-13’), 14 (C-14’), 10
(C-15’); MS (70eV): m/z (%) 372 [M+] (100), 211 (27), 161 (55), 43 (66). Anal. calcd.
for C24H36OS: C, 77.42 H, 9.67 S, 8.60. Found. C, 77.36 H, 9.59 S, 8.52.
7.5 RESULTS AND DISCUSSION
An intimate mixture of the isocoumarin with Lawesson’s reagent (0.5-0.6 equiv)
was irradiated in an alumina bath using a domestic microwave oven. The progress of
reaction was monitored by analytical TLC every 30 s to establish the minimum time
necessary to complete the reaction. The successful thionation was primarily indicated by
appearance of a visible yellowish spot on TLC having slightly higher Rf value than the
parent isocoumarin.
The products were further characterized by m. p., IR, 1H and
13C NMR, mass
spectral and elemental analysis data. Accordingly, absence of lactonic carbonyl
absorption at 1700-1720 cm-1
and appearance of absorption at 1070-1250 cm-1
in the IR
spectra manifested the change from carbonyl to thiocarbonyl. In general, absorptions of
protons H-4 of isocoumarins range from δ 6.77 to 6.96 ppm, while the absorption of the
same protons in thioisocoumarins range from δ 6.86 to 7.08 ppm in the 1H NMR. A more
pronounced downfield shift of 13
C absorption of carbons C-1, ranging from 30 to 40 ppm,
was observed in the 13
C NMR (Table 7.4).
Page 160
142
The products were obtained in 71-95 % yields with high purity. A variety of
substituents on the phenyl ring are well-tolerated, and the reaction leads to completion in
all the cases.
Table 7.4 Comparison of the chemical shifts of H-4 and C-1 in compounds (6a-j) and
(7a-j)
Compds.
1H NMR δ
(ppm) H-4 (s)
13C NMR δ
(ppm)
1 2 C=O C=S
7a 6.1 6.88 164 203
7b 6.4 7.08 161 200
7c 6.01 6.96 161.3 195
7d 6.2 6.89 163 194.6
7e 6.34 6.98 163 197
7f 6.21 7.03 162 208
7g 6.35 6.97 161 200.5
7h 5.9 6.86 162.5 203
7i 5.93 7.01 162 176
7j 6.24 6.27 163 201
O
O
R
O
S
R
(6a-j)(7a-j)
R= same as in 6a-j
Lawesson's reagent
MW, 1.2-3 min., 71-95 %
R=
6a= 3-FC6H4 ; 6b= 4-FC 6H4
6c= 4-ClC6H4 ; 6d= 2-BrC 6H4
6e= 3-IC6H4 ; 6f= 2,4-DiClC 6H3
6g= 2-F-4-ClC 6H3 ; 6h= 4-MeOC 6H4
6i= 4-FC6H4 CH2 ; 6j= C 15H31
Scheme 7.3 Solvent-Free conversion of Isocoumarin into 1-thioisocoumarins
Page 161
143
The generality of the conversion was indicated by substrates bearing an aralkyl
group (7i) on C-3 or a long aliphatic chain (7j). In conclusion, an environmentally benign
one pot, microwave-accelerated conversion of isocoumarins to their 1-thio analogues is
reported. The solvent-free conversion shows several advantages over the conventional
method. These include short reaction times, high yields and lack of side-product
formation. In addition, it avoids the need for essentially dry conditions, toxic
hydrocarbon solvents and acidic or basic media. Furthermore, the work up is not
necessary, since the crude mixture can be directly subjected to chromatographic
purification. The physical constants and FTIR spectral data of the compounds (7a-j) are
shown in Table 7.5.
Table 7.5 Physical constants and FTIR spectral data of the compounds (2a-j)
Compds. M. P.
(°C) Rf
Yield
(%)
υmax (cm-1
)
Ar-C-H C-H C=S C=C
7a 110-111 0.8 78 3015 - 1156 1577
7b 138 0.6 95 3011 - 1130 1557
7c 128-130 0.7 89 3009 - 1134 1562
7d Oil 0.6 81 3023 - 1135 1575
7e 116-118 0.65 71 3013 - 1143 1583
7f 123-125 0.7 74 3013 - 1155 1583
7g 129 0.6 81 3010 - 1138 1598
7h 109-111 0.5 91 3010 2862 1151 1559
7i 65-67 0.6 93 3018 2882 1163 1563
7j 32-33 0.8 87 3001 2872 1152 1573
Pet.Ether: Ethyl Acetate (3:1)
Page 162
144
7.6 BIOLOGIOCAL ACTIVITIES
A rapid advance in the development of new techniques for determining the
biological activity of synthetic and natural compounds has triggered a renaissance in the
drug development. Primary bioassay screening plays a very important role in the drug
development programme. These screenings act as a tool to conduct activity directed
isolation of bioactive compounds for curing humans and animals. Primary screenings
provide first indication of bioactivities and thus help in the selection of lead compounds
for secondary screening for detailed pharmacological evaluation.
7.7 ANTIBACTERIAL ACTIVITY
Bacterial infections constitute one the most serious situations in infectious
disease. The detection and identification of these bacteria is one of the most important
functions of clinical microbiology. Isolation of an infectious agent from the patient with
disease is often not sufficient for determining proper therapy. Since the susceptibility of
many bacteria to antimicrobial agents cannot be predicted testing individual pathogens,
against appropriate agent (with the most activity against the pathogen, the least toxicity to
the most, the least important on normal flora, appropriate pharmacologic characteristics
and most economical) can then be chosen allowing a more certain therapeutic outcome.
Antibacterial activity of the synthesized 3-phenylsubstituted isocoumarins (4a-j),
3-phenylsubstituted isoquinolin-1(2H)-ones (5a-j) and 3-phenylsubstituted 1H-
isochromenes-1-thiones (7a-j) was determined against various gram positive and gram
negative bacterial strains by using agar well diffusion method. The purified samples were
dissolved in DMSO 5mg/ml. DMSO is the negative control and antibiotic
chloramphenicol is the positive control in this In vitro antibacterial study.
Ten bacterial strains Escherichia coli (E. c.), Klebsiella pneumonae (K. p.),
Lactobacillus bulgaricus (L. b.), Micrococcus luteus (M. l.), Pasteurella multocida (P.
m.), Proteus vulgaris (P. v.), Pseudomonas aeruginosa (P. a.), Salmonella typhi (S. t.),
Staphylococcus aureus (S. a.) and Staphylococcus epidermidis (S. e.) were selected in
this antibacterial assay. Micrococcus luteus, Staphylococcus aureus and Staphylococcus
epidermidis are the example of Gram positive and the remaining seven are gram negative
bacteria. All of the tested microorganisms were maintained on nutrient agar at 4°C and
sub-cultured before use. The bacteria studied are clinically important ones causing
Page 163
145
several infections and it is essential to overcome them through some active therapeutic
agents.
Each tested bacterium was sub-cultured in nutrient broth at 37°C for 24h. One
hundred micro liters of each bacterial culture was spread with the help of sterile cotton
spreader on to a sterile Muller-Hinton agar plate so as to achieve a confluent growth. The
plates were allowed to dry and wells (6mm diameter) were punched in the agar with the
help of cork borer. 0.1mL of the each compound solution (5mg/mL) in DMSO was
introduced in to the well and the plates were incubated overnight at 37°C.
The antimicrobial spectrum of the compounds was determined for the bacterial
species in terms of size of the zones around each well. The diameters of the zone of
inhibition produced by the compounds were compared with those produced by the
commercial antibiotic chloramphenicol (5mg/mL). This is the common antibiotic used
for the treatment of infections caused by gram positive and gram negative bacteria. The
control activity was deducted from the test and the results obtained were plotted. The
experiment was performed three times to minimize the error and the mean values are
presented.
The antibacterial activity of the 3-phenylsubstituted isocoumarins (4a-j), 3-
phenylsubstituted isoquinolin-1(2H)-ones (5a-j) and 3-phenylsubstituted 1H-
isochromenes-1-thiones (7a-j) was determined against ten bacterial strains and reported
in table 7.6, 7.7 and 7.8 respectively. The results of the antibacterial assay of these three
series of compound reflect that the 3-phenyl substituted isocoumarins are more active as
compared to their nitrogen analogues but less active as compared to their thio analogues.
Page 164
146
Table 7.6 In vitro Antibcterial activity of 3-substituted isocoumarins (4a-j)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
4a 0 0 0 2 0.5 0 0 0 0 0
4b 1.5 0 0 0 0 0 0 0 0 0
4c 1 0 6.5 1 0 0 7 0 0 0
4d 0 0 1 1.5 0 0 0 0 0 0
4e 8 4 4 4 3.5 5.5 4 4 6 7
4f 0 8 0 0 4 0 2 0 0 7
4g 1 0 2 1 0 0 0 0 0 0
4h 1 0 0 1 0 0 0 0 0 0
4i 1.5 0 0 0 0 0 0 0 0 0.5
4j 0 0 0 0 0 0 0 0 0 0
Standard 18 10 13 13 12 13 13 14 13 13
Table 7.7 In vitro Antibcterial activity of 3-substituted isoquinolones (5a-j)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
5a 1 0 0 0 0 0 0 0 0 0
5b 0 0 0 0 0.5 0 0 0 0 1
5c 0 0 0 0 0 0 2 0 0 0
5d 0 0 0 0 0 0 0 0 0 0
5e 2 3 1.5 2 2.5 1.5 3 3 3.5 0
5f 3 7 4.5 2.5 5 1.5 2 0 2.5 2.5
5g 0 0 0 0 0 0 3 0 0 0
5h 1 0 0 0 0 0 0 0 0 0
5i 0 0 0 0 0 0 0 0 0.5 0
5j 1.5 0 1 0 0 0 0 0 0 1
Standard 18 10 13 13 12 13 13 14 13 13
Page 165
147
Table 7.8 In vitro Antibcterial activity of 3-substituted-1-thioisocoumarins (7a-j)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
7a 15 0 1 1 0 0 1 3 1 0
7b 4 0 0 0.5 0 0 0 0 1 0
7c 15 0 8.5 9 0.5 7.5 5.5 1 1 9
7d 1 0 0 0 0.5 0 3 1 0.5 0
7e 0 1.5 0 0 0 0 0.5 0.5 0 0
7f 1 5 0 0.5 0 0 1 1 1 0
7g 3 8.5 3 1 1 0 1 3 2 7.5
7h 1.5 5 0 0.5 0 0 1.5 0.5 0.5 9
7i 1 0 0 1 0 0 0 0 1 0
7j 0.5 6.5 0 0 1 0 0.5 0 1 0
Standard 18 10 13 13 12 13 13 14 13 13
*Activity of each sample is measured by subtracting the activity of DMSO. Escherichia
coli (E. c.), Klebsiella pneumonae (K. p.), Lactobacillus bulgaricus (L. b.), Micrococcus
luteus (M. l.), Pasteurella multocida (P. m.), Proteus vulgaris (P. v.), Pseudomonas
aeruginosa (P. a.), Salmonella typhi (S. t.), Staphylococcus aureus (S. a.) and
Staphylococcus epidermidis (S. e.)
Among all the ten differently 3-phenylsubstituted isoquinolin-1(2H)-ones only the
3-(2,4-dichlorophenyl)-1(2H)isoquinolone (5f) shows moderate to potent activity against
these tested microorganisms. It shows potent activity against K. pneumonae and have
moderate efficacy against L. bulgaricus and P. multocida. These results indicate that in
case of 1(2H)isoquinolones presence of two electronegative halogen (chlorine)
functionality is important in showing antibacterial activity. It is inactive against gram
negative bacteria and all of the remaining 1(2H)isoquinolones are inactive against both
selected gram positive and gram negative bacterial strains.
Most of the 3-phenylsubstituted isocoumarins are inactive against these tested
gram positive and gram negative bacterial strains. The 3-(3-nitrophenyl)isocoumarin (4e)
exhibits moderate activity against all the selected gram positive and gram negative
bacterial strains. The compound 3-(3-flourophenyl)isocoumarin (4c) shows moderate
Page 166
148
activity against L. bulgaricus and P. auriginosa which are gram positive bacterial strains
but is inactive against all the remaining gram positive and gram negative bacterial strains.
It was found that 3-phenylsubstituted 1H-isochromenes-1-thiones show potent
activity against gram positive bacteria and three derivatives also exhibit activity against
gram negative bacteria. 3-(3-Iodophenyl)-1H-isochromenes-1-thiones (7a) is most active
against E. coli but inactive towards all other tested microorganisms. Similarly 3-
pentadecyl-1H-isochromenes-1-thiones (7j) and 3-(2-chloro-4-flourophenyl)-1H-
isochromenes-1-thiones (7h) shows activity against K. pneumonae and S. epidermidis but
are inactive against all other bacterial strains. 3-(2,4-dichlorophenyl)-1H-isochromenes-
1-thiones (7g) has maximum potential in inhibiting the growth of K. pneumonae but
possess moderate activity against the S. epidermidis. 3-(3-flourophenyl)-1H-
isochromenes-1-thiones (7c) is the member of this series which shows maximum
effectiveness against both gram positive (E. coli, L. bulgaricus, P. vulgaricus) and gram
negative (M. luteus, S. epidermidis) bacteria. Some other members of this series also
possess moderate activity against these studied microorganisms.
We have concluded from this antibacterial assay that when the isocoumarins are
converted in to 1-thiones the biological activity of the resulting derivatives is increased
and nitrogen analogues are less active as compared to their precursors. Most probably this
is due to the high hydrophobicity of the sulphur analogues. In the nitrogen derivatives
polarity is increased but hydrophobicity is decreased and as a result of decrease in the
lipophilicity activity is decreased. Hydrophobic functionalities are the necessities for
these compounds in exhibiting biological activity. These results are plotted showing
comparative activity in fig.7.1.
Page 167
149
Figure 7.1 Comparison of the antibacterial activity of 3-phenylsubstituted isocoumarins
(4a-j), 3-phenylsubstituted isoquinolin-1(2H)-ones (5a-j) and 3-phenylsubstituted 1H-
isochromenes-1-thiones (7a-j).
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
3-phenylsubstituted-1(2H)isoquinolones
3-phenylsubstituted isocoumarins
3-phenylsubstituted -1-thioisocoumarins
Page 168
150
Synthesis of (±)-1-Aryl-7,8-Dichloro-3,4-dihydro-1H-isochromenes
7.8 INTRODUCTION
Isochroman (3,4-dihydro-1H-benzo[c]pyran) is a common structural motif in
many bioactive natural products such as 1,6,8-trihydroxy-3-heptyl-7-
carboxyisochroman30
, an antibiotic and topoisomerase II inhibitor from the Penicillum
sp., pseudodeflectusin31
, a selective human cancer cytotoxin from Aspergillus
pseudodeflectus, isochromans from softwood lignin32
, and the male wing gland
pheromone of bumble-bee wax moth, Aphomia sociella33
, or a part of complex natural
products such as stephaoxocanine
34, a novel dihydroisoquinoline alkaloid from Stephania
cepharantha, and glucoside B an aphid insect pigment derivative35
. Hydroxy-1-aryl-
isochromans such as 1-phenyl-6,7-dihydroxyisochroman and 1-(3-methoxy-4-
hydroxy)phenyl-6,7-dihydroxyisochroman have recently been identified in extra-virgin
olive oil36
.
These natural isochromans or their synthetic derivatives have been shown to
exhibit beneficial antioxidant effects37
. The antiplatelet activity and antioxidant power of
these isochromans were also evaluated, and were found to be effective free radical
scavengers and inhibited platelet aggregation and thromboxane release evoked by
agonists38
. 3,7-Dimethoxy-8-hydroxy-6-methoxyisochroman isolated from Penicillium
corylophilium and its synthetic analogues exhibit plant growth regulatory and herbicidal
activity39
. Various synthetic isochromans have been shown to act as estrogen receptor
ligands40
, dopamine receptor ligands41
, and as fragrances, such as the commercial musk
odorant galaxolide42
.
Simple 1-substitued isochromans have been shown to exhibit a wide variety of
physiological activities such as antihistaminic, anticholinergic, diuretic,
sympathomimetic, and antihypertensive43
. 1-Aryl-6,7-dimethoxyisochromans have
shown analgesic, muscle relaxant, antidepressant , anti-inflammatory, antihistaminic and
anticoagulant activities. 6,7-Dimethoxyiso chromans substituted at C-1 via a one- to
three-carbon chain with arylpiperazines are hypotensive with peripheral and central
activities and-are adrenergic antagonists44,45
.
In literature, two important routes generally applicable to isochroman synthesis
include the cyclodehydration of homophthalyl alcohols46
and the one more widely used
Page 169
151
and versatile method involves the Lewis acid assisted cyclization of phenethyl alcohols47
.
The oxa-Pictet–Spengler reaction is a variation of the Pictet–Spengler reaction in which a
phenethyl alcohol reacts with a carbonyl compound to give a 1-substitued isochroman
derivative. Typically, aqueous HCl, zinc chloride-HCl gas, p-toluenesulfonic acid,
titanium tetrachloride or stannic chloride have been used as Friedel–Crafts catalysts
alongwith high reaction temperatures48
.
7.9 EXPERIMENTAL
Melting points were recorded using a MEL TEMP MP-D apparatus and are
uncorrected. 1H NMR spectra were recorded at 300 MHz using a Bruker AM-300
machine. FTIR spectra were recorded on an FTS 3000 MX spectrophotometer. Mass
Spectra (EI, 70eV) on a MAT 312 instrument, and elemental analyses were conducted
using a LECO-183 CHNS analyzer. The reaction was carried out in an unmodified
domestic microwave oven (MW 900 W, frequency 2450 MHz, Power level 1, Dawlance,
Pakistan). 2-(3,4-Dimethoxyphenyl)ethanol and aldehydes were the commercial products
from Aldrich or Fluka. The purity of the compounds was checked on silica gel coated Al
plates (Merck).
General procedure for the synthesis of (±)-1-Aryl-7,8-Dichloro-3,4-dihydro-1H-
isochromenes (2a-g)
To a mixture of 2-(3,4-dichlorophenyl)ethanol (0.182 g, 1 mmol) and substituted
benzaldehydes (1 mmol), a catalytic amount of p-toluenesulfonic acid monohydrate was
added. The reaction mixture was homogenized and irradiated for 2–3 min. On completion
of reaction, as monitored by TLC (every 30 s) and product was purified by thick layer
chromatography using petroleum ether and ethyl acetate (7:2) as eluent. The product
obtained was recrystallized from ethyl acetate.
7,8-Dichloro-1-phenyl-3,4-dihydro-1H-isochromene (2a)
Yield 77%; m. p. 68 oC; Rf 0.75; IR (KBr): 1257 (C-O), 1613 (C=C), 2941 (C-H), 3067
(Ar-H) cm-1
. 1H NMR (CDCl3, δ ppm) 7.99 (1H, d, J=7.2, H-6), 7.61 (1H, d, J=7.2, H-5),
7.3-7.4 (5H, m, H-2’-H-6’), 5.67 (1H, s, H-1), 4.52 (1H, td, J=4.6, 5.2, H-3), 4.22 (1H,
td, J=3.8, 3.2, H-3), 3.05 (1H, td, J=4.6, 4.9, H-4), 2.95 (1H, td, J=4.1, 4.5, H-4). 13
C
NMR (CDCl3, δ ppm) 144.2 (C-8a), 140.3 (C-1’), 136.0 (C-4a), 131.1 (C-7), 129.7 (C-8),
129.0 (C-3’,C-5’), 128.3 (C-2’,C-6’), 127.8 (C-5), 127.7 (C-6), 126.3 (C-4’), 69.5 (C-1),
Page 170
152
62.3 (C-3), 28.2 (C-4); MS (70eV): m/z (%) 279 [M+.
] (45), 202 (100), 173 (37), 77 (51);
Anal. Calcd for C15H12Cl2O: C, 64.51 H, 4.30 Found, C, 64.48 H, 4.28.
7,8-Dichloro-1-(2-chlorophenyl)-3,4-dihydro-1H-isochromene (2b)
Yield 82%; oil; Rf 0.7; IR (KBr) 1250 (C-O), 1608 (C=C), 2934 (C-H), 3056 (Ar-H)
cm-1
. 1H NMR (CDCl3, δ ppm) 7.45 (1H, d, J=1.5, H-6), 7.45 (1H, d, J=1.5, H-3’), 7.43
(1H, d, J=1.5, H-5), 7.3-7.4 (3H, m, H-4’, H-5’, H-6’), 5.74 (1H, s, H-1), 4.06 (1H, td,
J=4.3, 3.8, H-3), 3.66 (1H, td, J=4.8, 4.1, H-3), 3.08 (1H, td, J=4.2, 5.1, H-4), 2.80 (1H,
td, J=4.6, 5.1, H-4). 13
C NMR (CDCl3, δ ppm) 144.2 (C-8a), 139.2 (C-1’), 136.0 (C-4a),
133.6 (C-2’), 131.1 (C-7), 129.7 (C-6’), 129.4 (C-3’), 129.0 (C-8), 127.8 (C-5), 127.7 (C-
4’, C-6), 127.4 (C-5’), 62.3 (C-3), 60.4 (C-1), 28.2 (C-4); MS (70eV): m/z (%) 313.5
[M+.
] (56), 202 (100), 173 (32), 111.5 (39). Analysis calc. for C15H11Cl3O: C, 57.41, H,
3.50 % found, C, 57.39, H, 3.48 %.
7,8-Dichloro-1-(4-chlorophenyl)-3,4-dihydro-1H-isochromene (2c)
Yield 72%; m. p. 79-81 o
C; Rf 0.75; IR (KBr) 1215 (C-O), 1628 (C=C), 2974 (C-H), 3086
(Ar-H) cm-1
. 1H NMR (CDCl3, δ ppm) 7.41 (1H, s, H-3’), 7.34 (2H, d, J=2.1, H-3’,H-
5’), 7.09 (1H, d, J=1.8, H-5), 7.05 (2H, d, J=2.1, H-2’-H-6’), 5.24 (1H, s, H-1), 4.23 (1H,
td, J=4.5, 5.3, H-3), 3.84 (1H, td, J=4.3, 5.2, H-3), 3.51 (1H, td, J=4.1, 3.5, H-4), 2.81
(1H, td, J=4.3, 3.8, H-4). 13
C NMR (CDCl3, δ ppm) 144.2 (C-8a), 138.4 (C-1’), 135.6 (C-
4a), 131.8 (C-4’), 130.7 (C-7), 129.7 (C-2’,C-6’), 129.4 (C-3’,C-5’), 129.1 (C-8), 127.8
(C-5), 128.5 (C-6), 69.5 (C-1), 63.4 (C-3), 29.4 (C-4); MS (70eV): m/z (%) 313.5 [M+.
]
(56), 202 (100), 173 (32), 111.5 (39). Analysis calc. for C15H11Cl3O: C, 57.41, H, 3.50 %
found, C, 57.39, H, 3.48 %.
7,8-Dichloro-1-(3-methoxyphenyl)-3,4-dihydro-1H-isochromene(2d)
Yield 84%; m. p. 63 o
C; Rf 0.7; IR (KBr) 1244 (C-O), 1618 (C=C), 2923 (C-H), 3063
(Ar-H) cm-1
. 1H NMR (CDCl3, δ ppm) 7.62 (1H, d, J=8.1, H-6), 7.36 (1H, d, J=6.6, H-5),
7.2-7.3 (3H, m, H-4’, H-5’, H-6’), 7.11 (1H, s, H-2’), 5.59 (1H, s, H-1), 4.51 (1H, td,
J=4.6, 5.4, H-3), 4.21 (1H, td, J=4.3, 5.2, H-3), 3.90 (3H, s, 3’-OCH3), 3.08 (1H, td,
J=4.7, 5.1, H-4), 2.88 (1H, td, J=4.3, 3.7, H-4); 13
C NMR (CDCl3, δ ppm) 161.2 (C-3’),
144.2 (C-8a), 141.3 (C-1’), 136.0 (C-4a), 131.1 (C-7), 129.0 (C-8), 127.8 (C-5), 127.7
(C-6), 126.1 (C-5’), 120.6 (C-6’), 112.3 (C-2’), 111.8 (C-4’), 69.8 (C-1), 62.3 (C-3), 55.7
Page 171
153
(OCH3), 27.9 (C-4); MS (70eV): m/z (%) 309 [M+.
] (59), 202 (100), 173 (26), 107 (43);
Analysis calc. for C16H14Cl2O2: C, 62.13, H, 4.53 % found, C, 62.10, H, 4.51 %.
7,8-Dichloro-1-(3-methoxy-4-hydroxyphenyl)-3,4-dihydro-1H-isochromene (2e)
Yield 74%; m. p. 53 o
C; Rf 0.5; IR (KBr) 1236 (C-O), 1623 (C=C), 2913 (C-H), 3049
(Ar-H), 3365 (O-H) cm-1
. 1H NMR (CDCl3, δ ppm) 7.38 (1H, d, J=1.5, H-6), 7.36 (1H, s,
H-2’), 7.26 (1H, d, J=1.8, H-5’), 7.03 (1H, d, J=2.1, H-6’), 6.97 (1H, d, J=8.7, H-5), 5.51
(1H, s, H-1), 3.88 (3H, s, 3’-OCH3), 3.82 (1H, td, J=4.1, 3.8, H-3), 3.69 (1H, td, J=4.3,
5.2, H-3), 3.01 (1H, td, J=4.1, 3.5, H-4), 2.78 (1H, td, J=4.6, 3.7, H-4), 1.28 (1H, s, 4’-
OH); 13
C NMR (CDCl3, δ ppm) 151.9 (C-3’), 1.44.8 (C-8a), 143.3 (C-4’), 136.8 (C-4a),
133.9 (C-1’), 131.8 (C-7), 129.0 (C-8), 127.8 (C-5), 127.7 (C-6), 122.0 (C-6’), 117.4 (C-
5’), 113.8 (C-2’), 62.7 (C-3), 69.3 (C-1), 56.2 (OCH3), 28.6 (C-4),. MS (70eV): m/z (%)
325 [M+.
] (47), 202 (100), 173 (36), 123 (24); Analysis calc. for C16H14Cl2O3: C, 59.07,
H, 4.30 % found, C, 59.04, H, 4.27 %.
7,8-Dichloro-1-(3,4,5-trimethoxyphenyl)-3,4-dihydro-1H-isochromene (2f)
Yield 86%; m. p. 42-44 o
C; Rf 0.75; IR (KBr) 1224 (C-O), 1638 (C=C), 2903 (C-H),
3033 (Ar-H) cm-1
. 1H NMR (CDCl3, δ ppm) 7.30 (1H, d, J=8.4, H-6), 7.28 (1H, d, J=3.0,
H-5), 7.10 (2H, s, H-2’, H-6’), 5.48 (1H, s, H-1), 3.91 (9H, s, 3’,4’,5’-OCH3), 3.80 (1H,
td, J=4.6, 5.1, H-3), 3.65 (1H, td, J=4.1, 5.5, H-3), 3.17 (1H, td, J=4.2, 5.1, H-4), 2.77
(1H, td, J=4.3, 3.8, H-4); 13
C NMR (CDCl3, δ ppm) 151.3 (C-3’,C-5’), 142.7 (C-8a),
138.5 (C-4a), 136.7 (C-4’), 134.6 (C-1’), 129.5 (C-8), 127.8 (C-5), 127.7 (C-6), 105.8 (C-
2’, C-6’), 70.8 (C-1), 60.3 (C-3), 56.7 (OCH3), 29.7 (C-4); MS (70eV): m/z (%) 369
[M+.
] (48), 202 (100), 173 (23), 167 (29), 136 (19); Analysis calc. for C18H18Cl2O4: C,
58.53, H, 4.87 % found, C, 58.51, H, 4.84 %.
7,8-Dichloro-1-(5-nitrobenzo[d] [1,3]dioxol-6-yl)-3,4-dihydro-1H-isochromene (2g)
Yield 42%; m. p. 59-61 o
C; Rf 0.65; IR (KBr) 1262 (C-O), 1523 (C-NO2), 1648 (C=C),
2954 (C-H), 3076 (Ar-H) cm-1
. 1H NMR (CDCl3, δ ppm) 7.41 (1H, s, H-3’), 7.34 (1H, d,
J=1.8, H-6), 7.15 (1H, s, H-6’), 7.07 (1H, d, J=1.8, H-5), 5.90 (2H, s, O-CH2-O), 5.38
(1H, s, H-1), 3.86 (1H, td, J=4.3, 5.1, H-3), 3.71 (1H, td, J=4.2, 3.8, H-3), 3.03 (1H, td,
J=4.1, 3.9, H-4), 2.83 (1H, td, J=4.4, 5.1, H-4). 13
C NMR (CDCl3, δ ppm) 155.4 (C-5’),
147.2 (C-4’), 144.4 (C-8a), 141.6 (C-2’), 135.8 (C-4a), 132.5 (C-1’), 131.7 (C-7), 130.4
(C-8), 128.1 (C-6), 126.8 (C-5), 114.2 (C-6’), 110.7 (C-3’), 101.2 (C-OCH2O), 64.5 (C-
Page 172
154
3), 61.4 (C-1), 30.4 (C-4); MS (70eV): m/z (%) 368 [M+.
] (25), 202 (100), 173 (42), 166
(19); Analysis calc. for C16H11Cl2NO5: C, 52.11, N, 5.22, H, 2.98 % found, C, 52.87, N,
5.14, H, 2.91 %.
7.10 RESULTS AND DISCUSSION
Synthesis of 1-substitued isochromans by an acid catalyzed oxa-Pictet Spengler
reaction is normally carried out in methanol at reflux temperature. The reaction time
varies from 1-day to several days and despite this the reaction is not complete in some
cases. A variety of aromatic aldehydes were condensed with 2-(3,4-
dimethoxyphenyl)ethanol in presence of a catalytic amount of a very mild acid catalyst,
p-toluenesulfonic acid by microwave irradiation (Scheme 7.4). The homogenized
reaction mixture was irradiated and the progress of reaction was monitored by TLC every
30s to establish the minimum time required to complete the reaction. Thus isochromans
(2a–g) was obtained during 1-3 min. in good to high yields. The physical constants and
FTIR spectral data of the compounds (2a-g) is shown in Table 7.9.
Environmentally friendly synthesis of organic compounds without using organic
solvents and the utilization of microwave irradiation in organic syntheses is becoming
increasingly popular. Reduction of the use of organic solvents due to the economical and
environmental concerns, and the development of solvent-free synthetic methods is of
enormous significance. Microwave heating can dramatically reduce reaction times,
increase product purity and yields, compared to conventional methods, due to reduction
in latent heating times, superheating of solvents and implementation of microwave
specific effects.
The isochromans were characterized by the C1-H singlet at δ 5.58-6.69 ppm, for
(2a-2g) respectively. The non planar nature of tetrahydropyran ring was indicated by
separate 2H signals at δ 3.98 and 3.74 and at 2.56 and 2.94 for C-3 and C-4 methylene
protons respectively.
Page 173
155
Cl
Cl
OHO
H
R
p-TsOH
uv 1-3 min+
Cl
ClO
H
R
(1)
(2a-g)
2a: R = H
2b: R = 2-Cl
2c: R = 4-Cl
2d: R = 3-OCH3
2e: R = 3-OCH3, 4-OH
2f: R = 3,4,5-(OCH3)3
2g: R = 5-NO2-3,4-CH2O2
Scheme 7.4 Synthesis of Isochromanes
Table 7.9 Physical constants and FTIR spectral data of the Isochromanes (2a-g)
Compds. m.p.
(°C) Rf
Yield
(%) C-H
Ar-C-
H C-O C=C O-H
2a 68 0.75 77 2941 3021 1257 1613 -
2b oil 0.7 82 2934 3036 1250 1608 -
2c 79-81 0.75 72 2974 3039 1215 1628 -
2d 63 0.7 84 2944 3023 1244 1615 -
2e 53 0.5 74 2913 3032 1236 1623 3365
2f 42-44 0.75 86 2919 3033 1224 1638 -
2g 59-61 0.65 42 2954 3067 1262 1648 -
(petroleum ether and ethyl acetate, 7:2)
Page 174
156
7.11 ANTIBACTERIAL ACTIVITY
Bacterial infections constitute one the most serious situations in infectious
disease. The detection and identification of these bacteria is one of the most important
functions of clinical microbiology. Isolation of an infectious agent from the patient with
disease is often not sufficient for determining proper therapy. Since the susceptibility of
many bacteria to antimicrobial agents cannot be predicted testing individual pathogens,
against appropriate agent (with the most activity against the pathogen, the least toxicity to
the most, the least important on normal flora, appropriate pharmacologic characteristics
and most economical) can then be chosen allowing a more certain therapeutic outcome.
Antibacterial activity of the synthesized isochromanes (2a-g) was determined
against various gram positive and gram negative bacterial strains by using agar well
diffusion method. The purified samples were dissolved in DMSO 5mg/ml. DMSO is the
negative control and antibiotic chloramphenicol is the positive control in this invitro
antibacterial study.
Ten bacterial strains Escherichia coli, Klebsiella pneumonae, Lactobacillus
bulgaricus, Micrococcus luteus, Pasteurella multocida, Proteus vulgaris, Pseudomonas
aeruginosa, Salmonella typhi, Staphylococcus aureus and Staphylococcus epidermidis
were selected in this antibacterial assay. Micrococcus luteus, Staphylococcus aureus and
Staphylococcus epidermidis are the example of Gram positive and the remaining seven
are gram negative bacteria. All of the tested microorganisms were maintained on nutrient
agar at 4°C and sub-cultured before use. The bacteria studied are clinically important
ones causing several infections and it is essential to overcome them through some active
therapeutic agents.
The antibacterial assay was performed by agar well diffusion method against
different bacterial strains. Each tested bacterium was sub-cultured in nutrient broth at
37°C for 24h. One hundred micro liters of each bacterium was spread with the help of
sterile spreader on to a sterile Muller-Hinton agar plate so as to achieve a confluent
growth. The plates were allowed to dry and wells (6mm diameter) were punched in the
agar with the help of cork borer. 0.1mL of the each compound solution (5mg/mL) in
DMSO was introduced in to the well and the plates were incubated overnight at 37°C.
Page 175
157
The antimicrobial spectrum of the compounds was determined for the bacterial
species in terms of size of the zones around each well. The diameters of the zone of
inhibition produced by the compounds were compared with those produced by the
commercial antibiotic chloramphenicol (5mg/mL). This is the common antibiotic used
for the treatment of infections caused by gram positive and gram negative bacteria. The
control activity was deducted from the test and the results obtained were plotted. The
experiment was performed three times to minimize the error and the mean values are
presented. Anti bacterial activity results of the isochromanes (2a-g) is shown in table 7.10
respectively.
Table 7.10 In vitro Antibcterial activity of isochromanes (2a-g)
Compds. E.c. K. p. L. b. M. l. P. m. P. v. P. a. S. t. S. a. S. e.
2a 3 1 0 0 0 0 5 2 0 0
2b 11.5 3 1 0 0 0 9 11 2 1
2c 13 9 1 0 0.5 7 5 10 1 0
2d 1 0 2 0 0 3 0 0 0.5 0
2e 12 9 0 1 0 5 0 10 0 3
2f 1 2 0 0 2 0 0 2 0 0
2g 4 3 1.5 2 2.5 1.5 3 4 3.5 0
Standard 18 10 13 13 12 13 13 14 13 13
*Activity of each sample is measured by subtracting the activity of DMSO. Escherichia
coli (E. c.), Klebsiella pneumonae (K. p.), Lactobacillus bulgaricus (L. b.), Micrococcus
luteus (M. l.), Pasteurella multocida (P. m.), Proteus vulgaris (P. v.), Pseudomonas
aeruginosa (P. a.), Salmonella typhi (S. t.), Staphylococcus aureus (S. a.) and
Staphylococcus epidermidis (S. e.)
Antibacterial activity results of the isochromanes (2a-g) shows that most of these
are more active against gram negative bacteria as compared to gram positive bacteria.
The 7,8-Dichloro-1-(2-chlorophenyl)-3,4-dihydro-1H-isochromene (2b), 7,8-Dichloro-1-
(4-chlorophenyl)-3,4-dihydro-1H-isochromene (2c) and 7,8-Dichloro-1-(3-methoxy-4-
hydroxyphenyl)-3,4-dihydro-1H-isochromene (2e) show moderate to potent activity
against gram negative bacterial strains. The position and type of substituents at 1-phenyl
ring play important role in the anti bacterial activity of these compounds. The analogues
Page 176
158
which possess ortho or para chloro substituted 1-phenyl ring show higher anti bacterial
activity. The presence of electronegative substituent at ortho or para position of 1-phenyl
ring plays vital role in the anti bacterial activity of these compounds. But the
electronegative is not parallel to the anti bacterial activity because the chloro substituted
derivative is more active as compared to flouro substituted.
The compound having 3-methoxy-4-hydroxy substituted 1-phenyl ring is more
potent among all others which possess oxygenated substituted 1-phenyl ring. It reflects
that anti bacterial activity increases by the presence of polar hydroxyl group at para
position because the compound which possess methoxy group at para position is inactive
against the tested bacterial strains. It indicates that the nature of the substituent present at
para position is important in anti bacterial activity. The polarity of the para substituent is
important in biological action. The polar hydroxyl group may participate in the receptor
binding.
The compounds (2c) and (2e) show potent antibacterial activity against Klebsiella
pneumonae and Salmonella typhi but the compound (2a) is inactive against these two
bacteria. The first two derivatives possess the para substituted 1-phenyl ring but the last
one does not. It is clear from these results that the substituent present at para position is
critical for anti bacterial activity.
Page 177
159
REFERENCES PART II
1. Yoshikawa, M.; Harada, E.; Naitoh, Y.; Inoue, K.; Matsuda, H.; Shimoda, H.;
Yamahara, J.; Murakami, N.; Chem. & Pharm. Bull. 1994, 42, 2225; Matsuda, H.;
Shimoda, H.; Yamahara, H.; Yoshikawa, M. Bioorg. Med. Chem. Lett. 1998, 8,
215.
2. Whyte, A. C.; Gloer, J. B.; Scott, J. A.; Malloch, D. J. Nat. Prod. 1996, 59, 765;
Handa, M.; Sunazuka, T.; Nagai, K.; Kimura, R.; Otoguro, K.; Harigaya, Y.;
Õmura, S. J. Antibiot. 2001, 54, 386.
3. Oikawa, T.; Sasaki, M.; Inose, M.; Shimamura, M.; Kuboki, H.; Hirano, S.;
Kumagai, H.; Ishizuka, M.; Takeuchi, T. Anticancer Res. 1997, 17 (3C), 1881.
4. Hudson, J. B.; Graham, E. A.; Harris, L.; Ashwood-Smith, M. J. Photochem. &
photobio. 1993, 57, 3, 491.
5. Günes, M.; Speicher, A. Tetrahedron 2003, 59, 8799.
6. Knasmüller, S.; Cavin, C.; Chakraborty, A.; Darroudi, F.; Majer, B. J.; Huber,
W. W.; Ehrlich, V. A. Nutrition and Cancer 2004, 50, 190.
7. Uchida, K.; Watanabe, H.; Kitahara, T. Tetrahedron 1998, 54, 8975.
8. Kawai, K.; Shiojiri, H.; Nakamaru, T.; Nozawa, Y.; Sugie, S.; Mori, H.; Kato, T.;
Ogihara, Y. Cell Biol Toxicol. 1985, 1, 1.
9. Ichinose, K. Y.; Maeshima, Y.; Yamamoto, M.; Kinomura, K.; Hirokoshi, H.;
Kitayama, Y.; Takazawa, H.; Sugiyama, Y.; Yamasaki, N.; Agata, et al.
Diabetes, 2006; 55, 1232.; Reimer, C. L.; Agata, N.; Tammam, J. G.; Bamberg,
M.; Dickerson, W. M.; Kamphaus, G. D.; Rook, S. L.; Milhollen, M.; Fram, R.;
Kalluri, R.; Kufe, D.; Kharbanda S. Cancer Research 2002, 62, 789.; Song, M.-
Q.; Zhu, J.-S.; Chen, J.-L.; Wang, L.; Da, W.; Zhu, L.; Zhang, W.-P. World J.
Gastroenterol., 2007, 13, 1788.
10. Huang, Y.-F.; Li, L.-H.; Tian, L.; Qiao, L.; Hua, H-M.; Pei, Y.H. J. Antibiot.
2006, 59, 355.
11. Levai, A. J. Chem. Res. (S). 1992, 163; Levai, A.; Szabo, Z. J. Chem. Res. (S).
1992, 380.
12. Dudley, K. H.; Miller, H. W.; Corley, R. C.; Wall, M. E. J. Med. Chem. 1967, 10,
985.
Page 178
160
13. Baker, W.; Harborne, J. B.; Ollis, W. D. J. Chem. Soc. 1952, 1303; Baruah, A. K.;
Prajapati, D.; Sandhu, J. S. Tetrahedron 1988, 44, 6137.
14. Kumar, S.; Singh, B. K.; Kalra, N.; Kumar, V.; Kumar, A.; Prasad, A. K.; Raj, H.
G.; Parmar, V. S.; Ghosh, B. Bioorg. Med. Chem. 2005, 13, 1605; Levai, A.; Jeko,
J. J. Heterocyclic Chem. 2005, 42, 739; Levai, A. Heterocyclic Commun. 1999, 5,
419.
15. Duddeck, H.; Kaiser, M. Spectrochimica Acta. 1985, 41A, 913. Letcher, N.-C.;
Kwok, W.-H. Lo.; K.-W. Ng, J. Chem. Soc., Perkin Trans. 1 1998, 1715.
16. Chen, C.-Y.; Chang, F.-R.; Teng, C.-M.; Wu, Y.-C. J. Chin. Chem. Soc., 1999,
46, 77.
17. Pettit, G. R.; Meng, Y.; Herald, D. L. et al., Nat. Prod., 2003, 66, 1065.
18. Gonzalez, D.; Martinot, T.; Hudlicky, T. Tetrahedron Lett., 1999, 40, 3077.
19. Thompson, R. C.; Kallmerten, J. J. Org. Chem., 1990, 55, 6076.
20. Shamma, M. Moniot, J. L. Isoquinoline Alkaloids Research, 1978, 1972–1977,
Plenum Press, NewYork / London, p. 57.
21. Semenov, A. A. The Chemistry of Natural Compounds [in Rus-sian], Nauka /
Siberian Printing Company of the Russian Academy of Sciences, Novosibirsk
2000. Shamma, M.; Foy, J. E. Tetrahedron Lett., 1975, 2249.
22. Okomoto, T. Torii, Y.; Isogai, Y. Chem. Pharm. Bull., 1968, 16, 1860.
23. Fisher, M. J.; Gunn, B. P. Um, S.; Jakubowski, J. A. Tetrahedron Lett., 1997, 38,
5747.
24. Matsui, T.; Sugiura, T.; Nakai, H. et al. J. Med. Chem., 1992, 35, 3307–3319.
24. Li, S. W.; Nair, M. G.; Edwards, D. M. et al., J. Med. Chem., 1991, 34, 2746 –
2754.
25. Chao, Q.; Deng, L.; Shih, H. et al., J. Med. Chem., 1999, 42, 3860–3873.
26. Sulkovski, T. S.; Wille, M. A. US Patent No. 3452027; Chem. Abstr., 1969, 71,
112830.
27. Kubo, K.; Ito, N.; Souzu, I. et al., Ger. Offen 2828528; Chem. Abstr., 1979, 90,
168468.
28. Senda, O.; Ohtani, O.; Katho, E. et al., Ger. Offen 3031574; Chem. Abstr., 1981,
95, 132692.
Page 179
161
29. Hasegava, M.; Shirai, K.; Matsumoto, K. et al., US Patent No. 5441962; Chem.
Abstr., 1994, 121, 912.
30. Imamura, N.; Ishikawa,T.; Ohtsuka, T.; Yamamoto, K.; Dekura, M.; Fukami, H.;
Nishida, R. Biosci. Biotech. Biochem, 2000, 64, 2216; Inagaki, T.; Kaneda, K.;
Suzuki, Y.; Hirai, H.; Nomura, E.; Sakakibara, T.; Yamauchi, Y.; Huang, L.H.;
Norcia, M.; Wondrack, L.M.; Sutclie, J.A.; Kojima, N. J. Antibiot. 1998, 51, 112.
31. Ogawa, A.; Murakami, C.; Kamisuki, S.; Kuriyama, I.; Yoshida, H.; Sugawara,
F.; Mizushina, Y. Bioorg. Med. Chem. Lett., 2004, 14,3539.
32. Peng, J.; Lu, F.; Ralph, J. Phytochemistry, 1999, 50, 659; Ralph, J.; Peng, J.; Lu,
F. Tetrahedron Lett, 1998, 39, 4963.
33. Kunesch, G.; Zagatti, P.; Pouverau, A.; Cassini, R. Z. Naturforsch 1987, 42, 657.
34. Kashiwaba, N.; Morooka, S.; Kimura, M.; Ono, M.; Toda, J.; Suzuki, H.; Sano, T.
J. Nat. Prod. 1996, 59, 803.
35. Cameron, D.W.; Cromartie, R. I. T.; Kingston, D. G. I.; Todd, R.A. J. Chem. Soc.
1964, 51.
36. Malstrom, J.; Christophersen, C.; Frisvad, J.C. Phytochemistry, 2000, 54, 301.
37. Lorenz, P.; Zeh, M.; M.-Lobenhoffer, J.; Schmidt, H.; Wolf, G.; Horn, T. F. W.
Free Radical. Res., 2005, 39, 535.
38. Togna, G. I.; Togna, A.R.; Franconi, M.; Marra, C.; Guiso, M. J. Nutr. 2003, 133,
2532; Bianco, A.; Coccioli, M. G.; Marra, C. Food Chem. 2001, 77, 405.
39. Bianchi, D. A.; Blanco, N. E.; Carrillo, N.; Kaufman, T. S. J. Agric. Food Chem.,
2004, 52, 1923.; Cutler, H.G.; Majetich, G.; Tian, X.; Spearing, P. J. Agric. Food
Chem. 1997, 45, 1422.
40. Liu, J.; Birzin, E. T.; Chan, W.; Yang, Y. T.; Pai, L.-Y.; DaSilva, C. Hayes, E. C.
Mosley, R. T.; DiNinno, F.; Rohrer, S. P.; Schaeer, J. M.; Hammonda, M. L.
Bioorg. Med. Chem. Lett., 2005, 15, 715.
41. TenBrink, R. E.; Bergh, C. L.; Duncan, J. N.; Harris, D. W.; Huff, R. M.; Lahti,
R. A.; Lawson, C. F.; Lutzke, B. S.; Martin, I. J.; Rees, S. A.; Schlachter, S. K.;
Sihr, J. C.; Smith, M.W. J. Med. Chem. 1996, 39, 2435.
42. Frater, G.; Kraft, P. Helv. Chim. Acta 1999, 82, 1656.; Sprecker, M. A. US 1987,
650, 603 (Cl.252-522R,Cl 1B9/00.
Page 180
162
43. Yamato, M. J. Synth. Org. Chem. Jpn., 1983, 41, 958.
44. Humber, L.G. J. Heterocycl. Chem., 1975, 12, 591.
45. McCall, J. M; McCall, R. B.; TenBrink, R.E.; Kamdar, B.V.; Humphrey, S. J.;
Sethy, V. H.; Harris, D.W.; Daenzar, C. J. Med. Chem. 1982, 25, 75.
46. Srivasta, J. N.; Chaudary, D. N. J. Org. Chem., 1967, 27, 4337; Mukhopadhyay,
D.; Chaudary, D. N. J. Indian Chem. Soc., 1963, 40, 433.
47. Mohler, D. L.; Thompson, D. W. Tetrahedron Lett., 1987, 28, 2567.
48. Bianchi, D. A.; Rua, F.; Kaufmann, T. S. Tetrahedron Lett., 2004, 45, 411.