39
Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery Vishal A Mevada Page 5 2 Review of Literature
| Date post: | 15-Mar-2018 |
| Category: |
Documents |
| Upload: | nguyenkhue |
| View: | 231 times |
| Download: | 6 times |

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 5
2 Review of Literature

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 6
Malaria is an infectious disease caused by the parasite called Plasmodium and is a serious
problem for human health, especially to the so-called ―Third World‖ (Carter & Mendis,
2002). The parasites are spread to people through the bites of
infected Anopheles mosquitoes, called "malaria vectors", which bite mainly between dusk
and dawn. This bite introduces the parasites from the mosquito's saliva into a person's blood
(Organization, 2014a). There are five parasite species that cause malaria in humans:
Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale and
Plasmodium knowlesi (Caraball, 2014). Amongst them, Plasmodium falciparum is the most
deadly. During the ancient time, it was believed that, Malaria was spread by foul-smelling
swamp gasses hence the name malaria which literally means bad air. Towards the end of the
19th
century, Charles Louis Alphonse Laveran, a French army surgeon, noticed parasites in
the blood of a patient suffering from malaria, and Dr. Ronald Ross, a British medical officer
in Hyderabad, India, discovered that mosquitoes transmitted malaria. The Italian professor
Giovanni Battista Grassi subsequently showed that human malaria could only be transmitted
by Anopheles mosquitoes (Tuteja, 2007).
Malaria is an entirely preventable and treatable mosquito-borne illness. However, in 2014,
97 countries and territories had ongoing malaria transmission. An estimated 3.2 billion
people are at risk of malaria (90% of them in sub-Saharan Africa, two thirds of the
remaining cases occur in six countries like India, Brazil, Sri Lanka, Vietnam, Colombia and
Solomon Islands), of whom 1.2 billion are at high risk. While, 198 million cases of malaria
worldwide (range 124–283 million) and an estimated 584 000 deaths (range 367 000–
755 000) in 2013. The WHO 2014 report specified 437 000 African children died before
their fifth birthday due to malaria during year 2013 (WHO, 2014). The geographical
distribution of malaria, according to WHO 2012 report is shown in Figure 2.1. World
Health Organization (WHO) forecasts a 16% growth in malaria cases annually. One child
dies of malaria in Africa every 20 sec., and there is one malarial death every 12 sec
somewhere in the world. Malaria kills in 1 year what AIDS killed in 15 years. In 15 years, if
5 million have died of AIDS, 50 million have died of malaria (Organization, 2009).
In India, the malaria control program was initiated in 1953 with an aim of eradication;
Recent malaria report published by National Vector Borne Diseases Control Program
(NVBDCP), Ministry of Health and Family Welfare, and the government of India revealed
that there were about 0.88 million confirmed malaria cases and 440 deaths attributed to

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 7
malaria in 2013 (NVBDC, 2014). In asia-pacific region, three countries accounted for 96%
of cases in 2013: India (58%), Myanmar (22%) and Indonesia (16%) (WHO, 2014).
Although there has been marked reduction in the number of malaria cases in India under
National Vector Borne Disease Control Programme (NVBDCP), malaria still is the leading
cause of infectious diseases with the development of drug resistant Plasmodium species and
insecticide resistant mosquitoes. The country also faces the most deadly threat of the
malaria strain becoming resistant to the most advanced drugs available till date, thanks to
unregulated selling of banned malaria therapies.
Figure 2.1 : Geographical Distribution of Malaria (WHO 2012 Report)
The overall malaria cases and deaths have decreased in recent years in India, but a study
funded by NIH, USA and published in Lancet argues that more than 0.2 million Indians die
of malaria annually (Dhingra et al., 2010). The study further reports that about 90% of
malaria related deaths were in rural areas while about 86% took place away from the health
centers. A recent study has suggested that there is a certain population in India that does not
have access to medical facilities for one or another reason, which further strengthen the
claim that many deaths might occur at home without reaching to any reporting facility
(Kumar et al., 2011). The actual estimate of malaria mortality in India has been argued in
recent years (Kumar et al., 2011).

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 8
2.1 Life-cycle of the parasite
The malaria parasite exhibits a complex life cycle involving an insect vector (mosquito) and
a vertebrate host (human). Four Plasmodium species infect humans: P. falciparum, P. vivax,
P. ovale and P. malariae. All four species exhibit a similar life cycle with only minor
variations. Generally it includes three stages, (1) Erythrocytic stage, (2) Mosquito stage and
(3) Exothrocytic stage
2.1.1 Life stages within the human blood stream (erythrocytic stages)
1. The merozoite, regardless of its source, attaches and induces the red blood cell to
produce extra membrane and, enclosed in this membrane, enters the red blood cell.
2. Early-stage trophozoites appear ring-shaped under Giemsa stain and are easily
recognized in diagnosis.
3. Late-stage trophozoites grow to fill the host cell. In infections with Plasmodium
falciparum, these trophozoites induce the production of a histidine-rich protein (Pf
HRP II) which causes red blood cells to clump around an infected cell and block
capillaries.
4. Mitotic nuclear division takes place to form a schizont full of merozoites ready to
enter the blood stream upon cell lysis. Toxic hemozoin — the crystallized
accumulation of the haem groups left as a by-product of the parasite‘s hemoglobin
digestion left in the lysed blood cells upon lysis.
5. and 6. Microgametocytes (5, male) and Macrogametocytes (6, female) form from
ring-stage parasites and remain inside un-lysed red blood cells in circulation. They
seen in parasite cultures and the mechanism governing gametogenesis versus
schizogony is not entirely understood.
2.1.2 Life stages of the mosquito
7. 8 and 9. Any parasites ingested by a female mosquito taking a blood meal are
digested except for microgametocyte and microgametocyte which are released from
their surrounding red blood cell and activated by the mosquito‘s digestive tract. For
the female gamete, this activation is simple, and the activated macrogametocyte

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 9
becomes a macrogamete (9). At this point, the activated microgameteocyte begins a
process known as exflagellation, producing a free-living microgamete (8) capable of
fertilizing the macrogamete at which point the combined diploid organism is called a
zygote.
10. The zygote matures to become the mobile ookinete which embeds itself in the
mosquito gut wall. Although exact details remain unclear, it is likely that the zygote
is the only diploid form of Plasmodium 36 with meiosis occurring during its
maturation into the ookinete.
11. The embedded ookinete matures into the oocyst.
12. and 13. The oocysts divide asexually until individual sporozoites are mature.
14. The sporozoites lyse the host cell and circulate within the mosquito until reaching
the salivary gland where they settle, ready to be injected when the mosquito takes it
next blood meal.
2.1.3 Life stage within the human liver (exoerythrocytic stages)
15. Sporozoites injected into the human blood stream settle in the liver where they infect
hepatocytes. At this stage, the parasites are almost invisible to clinicians and are thus
referred to as cryptozoites.
16. Hepatic schizonts form, the direct equivalent of the schizonts in red blood cells.
17. The lysis of hepatic schizonts releases merozoites into the blood stream to commence the
red-blood cycle.
2.2 Pathogenesis
Among all species of Plasmodium, P. falciparum causes severe malaria via sequestration, a
distinctive property not shared by any other human malaria. Within the 48-hour asexual
blood stage cycle, the mature forms change the surface properties of infected red blood
cells, causing them to stick to blood vessels (a process called cytoadherence). This leads to
obstruction of the microcirculation and results in dysfunction of multiple organs, typically
the brain in cerebral malaria (Arjen M. Dondorp et al., 2004).

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 10
Figure 2.2: The multiple life stages and host cells of Plasmodium. Parasite nuclei are shown in dark purple, cytosol
as a lighter purple, as under Giemsa stain
2.3 The P. falciparum genome sequence
In 1995, a consortium, the Malaria Genome Project, was set up to sequence the genome
of P. falciparum. The genome of its mitochondrion was reported in 1995, that of the
nonphotosynthetic plastid known as the apicoplast in 1996 (Wilson et al., 1996) and the
sequence of the first nuclear chromosome (chromosome 2) in 1998. The sequence of
chromosome 3 was reported in 1999. The complete P. falciparum 3D7 nuclear genome
sequence was published in 2002 (Gardner et al., 2002) and contains 14 chromosomes,
ranging in size from 0.643 Mb (chromosome 1) to 3.290 Mb (chromosome 14). Two non-
nuclear genomes, consisting of the 6 kb mitochondrial genome and the 35 kb apicoplast
genome, are also present.
Annotated genome data can now be fully analyzed at several database resources including
the UCSC Malaria Genome Browser, PlasmoDB and GeneDB. The roughly 24-megabase
genome is extremely AT-rich (about 80%) and is organised into 14 chromosomes. The

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 11
22,853,764 bp nuclear genome encodes 5300 predicted genes, implying an average
frequency of one gene every 4338 bp (Gardner et al., 2002).
More than, 3208 (60.9%) protein-coding genes have no assigned function or homology to
genes of other organisms with known genome sequence and have been designated as
‗hypothetical proteins‘. Introns are found in 53.9% of the genes and their AT- composition
rises to 90% when compared to the overall AT-content of the whole genome, which is
80.6%. P. falciparum has the highest AT-content of all the organisms sequenced to date.
The Homo sapiens genome, for example, only has a 59% AT-content (Bunnik et al., 2014;
Lander et al., 2001).
This can be attributed to the presence of numerous codons encoding low complexity regions
found in Plasmodium proteins. These low-complexity regions may be over several hundred
amino acids in length and contain stretches of one or a few amino acids, particularly
asparagine and lysine (Di Girolamo et al., 2005).
Low complexity regions tend to be located on the outer surface of the folded protein, where
they form unstructured soluble stretches that do not interfere with the functions of the
domains or the rest of the protein (Thiele et al., 2013).
2.4 P. falciparum protein identification as drug candidate
Before the 1970s, P. falciparum proteins were isolated directly from malaria-infected
patients. However, this method had limited success and thus the establishment of parasites
in continuous culture by Trager and Jensen (1976) provided a new source of parasite
proteins in the laboratory. Purification of proteins directly from the parasite by conventional
methods was difficult due to the presence of erythrocyte protein contaminants and
insufficient quantities of parasite protein being isolated.
Several parasite surface proteins, for example, MSP-1 (Holder & Freeman, 1982), were
isolated by immunoprecipitation from parasite culture supernatants with human sera
containing parasite-specific antibodies. Hybridoma technology was also applied to produce
antibodies that recognise specific parasite antigens, for example, CSP (Nardin et al., 1982).
The antibodies were subsequently used to purify parasite proteins by affinity
chromatography and for immunolocalization experiments to determine the exact location of
the proteins, such as AMA-1 in the parasite (Peterson et al., 1989).

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 12
Standard methods, such as SDS-PAGE, chromatography and centrifugation, were also
applied to isolate P. falciparum proteins. For example, Kilejian (1979) compared the
erythrocyte membrane protein composition of cultured parasitised and non-parasitised
erythrocytes by SDS-PAGE and isolated KHARP/HRP-I and Perkins (1984) used
immobilized erythrocyte glycophorins to isolate RESA and glycophorin-binding protein
from lysed parasites. Goldberg et al. (1990) isolated P. falciparum food vacuoles by
applying a combination of saponin lysis and Percoll gradient centrifugation and this group
subsequently used anion exchange chromatography and gel filtration to isolate plasmepsin-I
and –II, falcipain-I and falcilysin from these food vacuoles (Eggleson et al., 1999; Gluzman
et al., 1994; Goldberg et al., 1990). Sanders et al. (2005) used sucrose density
centrifugation to isolate erythrocyte detergent-resistant membranes from cells that were
infected with parasites (Sanders et al., 2005). The amino acid sequence of the N-termini of
some isolated proteins, for example falcipain-2 (Shenai et al., 2000) and MSP-6 (Trucco et
al., 2001) were determined by Edman sequencing and the amino acid sequence of peptide
fragments of other proteins, such as RhopH1 (Kaneko et al., 2001), were determined by
tandem mass spectroscopy.
Jia et al., predicted mitochondrial proteins of malaria parasite using bi-profile Bayes feature
extraction (Jia et al., 2011).
Ogouyèmi-Hounto et al., (2013) isolated merozoite surface protein-1 and merozoite surface
protein-2 gone under Genetic polymorphism in Plasmodium falciparum isolates from
children in South of Benin (Ogouyemi-Hounto et al., 2013).
The genes coding for several isolated malaria proteins, for example MSP-2 and the rhoptry
associated membrane antigen (Smythe et al., 1988), were identified by screening a P.
falciparum bacteriophage λ cDNA library with MSP-2 and RAMA-specific antibodies.
Some parasite genes, for example MAEBL (Karine G Le Roch et al., 2003) and TRAP
(Robson et al., 1988), were isolated by hybridization and polymerase chain reaction (PCR)
experiments using specific or degenerate oligonucleotide primers designed from the DNA
and amino acid sequences of previously identified related malaria proteins or other
conserved protein motifs. Primers were also used to screen mRNA, genomic DNA,
expressed sequence tag (EST) libraries and cDNA libraries ultimately to obtain the
complete DNA sequences coding for malaria proteins.

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 13
After the publication of the P. falciparum genome sequence in 2002 and the subsequent
availability of the annotated genome at PlasmoDB (www.plasmodb.org) (Bahl et al., 2003),
researchers probed the genomic and EST databases with conserved regions of known
malaria proteins and motifs of non-malaria proteins to identify several malaria protein
families, for example the plasmepsins (Banerjee et al., 2002) and the rhomboids (Baker et
al., 2006).
Along with that, there were several methods employed to study protein-protein interactions.
Some of these include the yeast two-hybrid (LaCount et al., 2005) and the phage display
system (Enright et al., 1999). Phage display has only been used to study antibody-antigen
interactions, for example, to identify epitopes that interact with AMA-1 antibodies (Coley
et al., 2001). This study was found to use as an important application to identify malaria
proteins that interact with human erythrocyte spectrin.
Cambell et al., characterize an Atypical MAPK Phosphatase of Plasmodium falciparum as a
Suitable Target for Drug Discovery using in silico approach (Campbell et al., 2014).
Armistead et al. recently inhibit the Universal Transmission of Plasmodium falciparum and
Plasmodium vivax by Antibodies to a single, conserved epitope in anopheles APN1
(Armistead et al., 2014).
Stallman et al., identified SERA5 of Plasmodium falciparum plays a non-enzymatic role in
the malarial asexual blood-stage lifecycle (Stallmach et al., 2014).
In same year Keluskar et al., (2014) reported Genetic polymorphism in enzyme lactate
dehydrogenase from Indian isolates of Plasmodium falciparum and Plasmodium vivax
(Keluskar et al., 2014)
2.5 Treatment and chemotherapy for malaria
In 2700 BC, several characteristic symptoms of what would later be named malaria were
described in the Nei Ching in the book ―The Canon of Medicine‖. Nei Ching was edited by
Emperor Huang Ti. Malaria became widely recognized in Greece by the 4th century BCE,
and it was responsible for the decline of many of the city-state populations. After long
period of time, there were extensive references to malaria in the literature and depopulation
of rural areas were recorded.

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 14
In the Susruta, a Sanskrit medical treatise, the symptoms of malarial fever were described
and attributed to the bites of certain insects. A number of Roman writers attributed malarial
diseases to the swamps. But due to lack of effective treatment, until the early 17th
century,
no truly effective cure for malaria had found forcing the patients to die. However, in China,
during the second century BCE, the Qing-hao plant (Artemisia annua) was described in the
medical treatise, 52 Remedies, found in the Mawangdui Tomb to have antimalarial ability.
Spanish Jesuit missionaries, learned from indigenous Indian tribes of a medicinal bark used
for the treatment of fevers. The bark from the tree was then called Peruvian bark and the
tree was named Cinchona. Later on, it was found that, The effective molecule in the bark of
Cincona was Quinine (Druilhe et al., 1988; Sinton et al., 1930).
The active ingredient of Qinghao, known as artemisinin, was isolated by Chinese scientists
in 1971. Derivatives of this extract, known collectively as artemisinins, are today very
potent and effective antimalarial drugs, especially in combination with other medicines. In
last 2 decades, several studies had performed for artemisinin and its derivatives as
antimalarial compound.
A group of scientists had found inhibition against African isolates and clones of
Plasmodium falciparum in their in vitro study for artemisinin derivatives (Basco & Le
Bras, 1993).
Now a days, several different therapeutic agents were used to treat the malarial infection.
However, the parasite developed resistance against the wel-known drugs available in the
market. Quinolines, folate antagonists, antibiotics, artemisinin derivatives and hydroxy-
napthoquinones are the main classes of antimalarial drugs.
The choice of an anti-malarial agent largely depends on the patient‘s level of immunity, the
drug‘s side effect profile, cost and the site where the infection was acquired an indicator of a
particular drug‘s resistance probability (Whitesides, 2004). In critical condicions,
Combinatorial drug therapies play a significant role in the control malaria (Lefevre et al.,
2008). Although drugs in use target different stages of the malaria life cycle, majority of
them act on the asexual, intra-erythrocytic stage: the phase responsible for the clinical
symptoms of the disease.

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 15
2.5.1 Quinolines and Chloroquinone
Quinoline antimalarials are derived from quinine, the active component extracted from the
bark of cinchona trees (Quinona officinalis). The ground tree bark has been used since the
beginning of the 17th
century to treat malaria.
Johann "Hans" Andersag and colleagues synthesized and tested some 12,000 compounds,
eventually producing Resochin®
as a substitute for quinine in the 1930s (Hempelmann,
2007; Jensen & Mehlhorn, 2009; Krafts et al., 2012).
It is chemically related to quinine through the possession of aquinoline nucleus and the
dialkylaminoalkylamino side chain. Resochin (7-chloro-4-4-(diethylamino)-1– methylbutyl
amino quinoline) and a similar compound Sontochin (3-methyl Resochin) were synthesized
in 1934 (COATNEY, 1963). In March 1946, the drug was officially named Chloroquine
(Loeb, 1946). Chloroquine is an inhibitor of hemozoin production
through biocrystallization. Quinine and chloroquine affect malarial parasites only at life
stages when the parasites are forming hematin-pigment (hemozoin) as a byproduct
of hemoglobin degradation.
2.5.2 Artemisinin
Systematic screening of traditional Chinese medical herbs was carried out by Chinese
research teams, consisting of hundreds of scientists in the 1960s and 1970s (Y. Li & Wu,
2010). Qinghaosu, later named artemisinin, was cold-extracted in a neutral milieu (pH 7.0)
from the dried leaves of Artemisia annua (Liao, 2009; Wright et al., 2010). Analogues such
as a water soluble artesunate and the oil soluble artemether are now commonly used in
combination therapies with other antimalarials like mefloquine.
The artemisinin-based combination therapies (ACT) are recommended by the WHO as the
best current treatment of falciparum malaria. Several modes of action have been suggested
for artemisinins, such as inhibition of heme detoxification (Hermjakob et al., 2004;
Meshnick et al., 1996).
Studies in a yeast model suggested that artemisinin affects the membrane potential in the
mitochondria of P. falciparum in a mechanism different to atovaquone and leads to the
generation of reactive oxygen species (L. Li et al., 2010).

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 16
Eckstein-Ludwig et al., (2003) describe artemisinin target SERCA-type ATPase PfATP6 in
the parasites endoplasmic reticulum (Muhia et al., 2003). The artemisinins were considered
to be the only family of drug components fully effective against all strains of P. falciparum.
However, the artemisinin resistance was reported on Thai-Cambodian border with
decreased efficacy of ACT (Lim et al., 2005).
2.5.3 Antifolates
Antifolates such as pyrimethamine and proguanil target the parasites dihydrofolate
reductase (DHFR) (Dahlström et al., 2014; Schneider et al., 2012). Proguanil is a prodrug,
which is converted to the active form cycloguanil by the parasite. Antifolates often used in
combination with sulpha drugs such as sulphonamides and sulphones, which inhibit
dihydropteroate synthase (DHPS). Both enzymes involved in the biosynthesis pathway of
folate, a cofactor essential for DNA synthesis and other metabolic pathways. Combinations
of DHFR and DHPS inhibitors such as sulfadoxine and pyrimethamine (Fansidar) or
chloroproguanil-dapson (LapDap) used as antimalarials. Proguanil is not only useful in the
erythrocytic stages but also on the liver stages of the parasite.
2.6 Emergence of drug resistance
Chloroquine-resistant forms of P. falciparum emerged only 19 years later after the
discovery of chloroquine (Wellems & Plowe, 2001). The first resistant strains were detected
around the Cambodia-Thailand border and in Colombia, in the 1950s (Payne, 1987). In
1989 chloroquine resistance in P. vivax was reported in Papua New Guinea. The resistant
strains spread rapidly, producing a large mortality increase, particularly in Africa during the
1990s (Snow et al., 2001).
Sulphadoxinepyrimethamine (SP), a combination antifolate drug was widely used
inexpensive antimalarial drug, but resistance lead to unacceptable levels of therapeutic
failure in many areas in Asia, South America and now Africa (A M Dondorp et al., 2000).
It is believed that resistance of P. falciparum to chloroquine is related to an increased
capacity for the parasite to expel chloroquine at a rate that does not allow chloroquine to
reach levels required for inhibition of haem polymerization (Foley & Tilley, 1997). This

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 17
chloroquine efflux occurs at a rate of 40 to 50 times faster among resistant parasites than
sensitive ones (Krogstad et al., 1987).
Alleles associated with drug resistance of P. falciparum to chloroquine (pfcrt), and (pfmd-1)
and to pyrimethamine dihydrofolate reductase (dhfr) were seen at a high frequency at the
beginning of treatment and increased further through time following both drug treatments
(Ali et al., 2006).
Although some initial efforts at gene silencing through RNA interference appeared
successful in reducing growth rate (McRobert & McConkey, 2002) further studies have
shown that P. falciparum lacks the genetic machinery required for general application of
RNAi (Baum et al., 2009).
The resistance was reported in PfATP6, also known as PfSERCA or PfATPase6, is a
calcium ATPase gene for Artemsinin resistance in P. falciparum (L. L. Cui et al., 2012).
Takala-Harrison et al., reported mutations in a kelch protein encoded on P. falciparum
chromosome 13 (K13) have been associated with resistance in vitro and in field samples
from Cambodia. P. falciparum infections from artesunate efficacy trials in Bangladesh,
Cambodia, Laos, Myanmar, and Vietnam (Takala-Harrison et al., 2014).
Conrad et al., found polymorphisms in K13 and Falcipain-2 Associated with Artemisinin
Resistance in Plasmodium falciparum Isolated from Ugandan Children (Conrad et al.,
2014).
However, resistance to antifolates and their combinations with sulpha drugs soon emerged
after their introduction as antimalarial drug due to point mutations in both DHFR and DHPS
(Dahlström et al., 2014; Schneider et al., 2012).
2.7 Methods to study parasite
2.7.1 Cell culture of erythrocytic stages
Human malaria parasites had proved impossible to culture until 1976 when Trager and
Jenson realized that the low oxygen environment of coagulated red blood cells (RBCs)
within the capillaries was essential to the progression of the P. falciparum life-cycle
(Desjardins et al., 1979). Just a year later the technique had advanced to a system we would

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 18
recognize today whereby frozen parasitized red blood cells could be thawed and grown in
washed human RBCs in RPMI medium fortified with human serum all within culture flasks
kept with a little oxygen internal environment (Hui et al., 1984). At the time, the less
oxygen environment was provided by burning a candle at the entrance of the culture flask to
lower the oxygen concentration. Modern improvements in these methods have improved
reproducibility; low-oxygen gas of known composition replaced candles and a standard
culture medium of RPMI+AlbuMAX has largely replaced the variable composition of
human serum (Coppens et al., 2010).
2.7.2 Cell culture of liver stages
Limited success in cultivating liver stages of Plasmodium falciparum and Plasmodium
vivax to study their ability to persist as hyponozooites has been achieved (L. Cui et al.,
2009). Furthermore, recent advances with Plasmodium falciparum are making it possible to
culture the hepatic stages of the parasite in human liver cell lines (March et al., 2013).
These techniques are currently very difficult, but may offer a way to study the organism‘s
metabolism more thoroughly and may allow the development of new types of drugs, both
prophylactic and as treatment, that target the liver-stage of the parasite‘s life cycle.
2.7.3 Other in vitro methods of study
In past years, a number of methods have been developed to study cell viability and
proliferation in cell culture (Cook & Mitchell, 1989). The most convenient, modern assays
have been optimized for the use of microtiterplates (96-well format). This miniaturization
allows many samples to be analyzed rapidly and simultaneously. Colorimetric and
luminescence based assays allow samples to be measured directly in the plate by using a
microtiterplate reader or ELISA plate reader. Cytototoxicity assays have been developed
which use different parameters associated with cell death and proliferation.
One parameter for cell death is the integrity of the cell membrane, which can be measured
by the cytoplasmic enzyme activity released by damaged cells. Lactate dehydrogenase is a
stable cytoplasmic enzyme present in all cells. It is rapidly released into the cell culture
supernatant upon damage of the plasma membrane (Korzeniewski & Callewaert, 1983). The
LDH activity is determined in an enzymatic test. The first step is the reduction of NAD+
to
NADH/H+ by the LDH catalyzed conversion of lactate to pyruvate. In a second step, the

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 19
catalyst (diaphorase) transfers H/H+ from NADH/H+ to the tetrazolium salt 2-(4-
iodophenyl)- 3-(4-nitrophenyl)-5-phenyltetrazolium chloride (INT), which is reduced to a
red formazan (Decker & Lohmann-Matthes, 1988; Lappalainen et al., 1994; Nachlas et al.,
1960)
Another parameter used as the basis for colorimetric assays is the metabolic activity of
viable cells. Tetrazolium salts are reduced only by metabolically active cells. Thus, 3-(4,5-
dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) can be reduced to a blue
colored formazan (Smith, 1951).
Some of the basic in vitro drug sensitivity assays include; WHO micro test, isotopic (titrated
hypoxanthine uptake) assay, lactate dehydrogenase (pLDH), histidine-rich protein 2(HRP2),
SYBER Green 1 and other fluorescent dyes.
2.7.3.1 WHO Micro test for Plasmodium
The in vitro micro-test provides information on the quantitative drug response of P.
falciparum irrespective of the patient/s immune status. It is an epidemiological tool for
assessing baseline sensitivity and for monitoring the drug response of P. falciparum over
time and place and therefore can provide background information for the development and
evaluation of drug policies. Changes in parasite drug sensitivity in vitro can be an indicator
of future therapeutic failure. Unlike the in vivo tests, the results of an in vitro test are not
disturbed by ongoing malaria transmission. The in vitro test may be carried out with several
drugs simultaneously and is independent of the patients clinical condition. It also permits
the monitoring of drug response with compounds for which an in vivo test is not yet
available (Organization, 2001).
2.7.3.2 Histidine-rich protein 2(HRP2)
Plasmodium falciparum parasites synthesize several proteins containing large amounts of
the amino acid histidine, which are commonly referred to as histidine-rich proteins
(PfHRP). One of these, PfHRP-2, has been sequenced and shown to contain 34% histidine
and 37% alanine, as deduced from cloned genomic DNA (Howard et al., 1986).
PfHRP-2 is synthesized by the parasite and released from infected erythrocytes as a water-
soluble protein (Parra et al., 1991). It can be detected in in vitro culture supernatants of

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 20
synchronized parasites as early as 2 to 8 h after ring development, indicating that it is
actively secreted from infected cells. The amount of PfHRP-2 released in vitro continues to
increase throughout the erythrocytic cycle, with a large amount being released during
schizont rupture (Howard et al., 1986). One would speculate that PfHRP-2 might circulate
in human serum during acute infection.
2.7.3.3 MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) test
The standard World Health Organization (WHO) method for testing new antimalarial drugs
involves tedious microscopic parasite counting method (MPCM). Other in vitro methods are
haem polymerization inhibition assay (HPIA) (Slater & Cerami, 1992), lactate
dehydrogenase assay (LDHA) (Delhaes et al., 1999; Makler et al., 1993) and the isotopic
microtest (IMT) (Desjardins et al., 1979). All these in vitro tests measure some activity of
the parasite and the endpoints are directly parasite related. Destruction of red blood cells
(RBCs) is the end result of malarial infection. None of the available in vitro assays uses
RBCs-related activity to determine endpoints. Furthermore, the available in vitro assays do
not test for drug selectivity unless a separate assay on RBC toxicity is done. In this paper,
we report on a tetrazolium-based colorimetric selective assay (MTT-based CSA) that uses
indirect protection of RBCs from parasite destruction as an endpoint for measuring the
effect of drugs on parasites and also determines the possible drug toxicity to the RBCs.
Dell`Agli et al., (2008) evaluated cytotoxicity in human fibroblasts from skin biopsies. Cell
proliferation was followed by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-
diphenyltetrazolium bromide) test, in the presence of the MeOH extract and CHCl3 fraction
(0–100 μg/ml) of LC50 achieved. Fibroblasts (8×104/well) were grown in 24 well plates
with DMEM (Dulbecco's modified Eagle's medium) containing 10% fetal calf serum, 1%
penicillin/ streptomycin, and 1% L-glutamine as previously described (Dell‘Agli et al.,
2008).
Dell`Agli et al., used same MTT assay for Antiplasmodial activity of Punica granatum L.
fruit rind (Dell‘Agli et al., 2009).
In year 2012, Prajapati et al., MTT assay for synthesis, characterization and antimalarial
evaluation of new β-benzoylstyrene derivatives of acridine (Prajapati et al., 2012). Ehata et
al., (2012) used the assay for in vitro antiprotozoal and cytotoxic activity of aqueous extract,

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 21
the 80 % methanol extract and its fractions from the seeds of Brucea sumatrana growing in
democratic Republic of Congo (Ehata, 2012).
Whereas, Medes et al.,(2013) used in identification of antimalarial activity of 4-
Metoxychalcones as Falcipain/Plasmepsin Inhibitors in their in vitro assay (Mendes et al.,
2013). In the same year, Mani (2013) used this assay for new substituted Heterocyclic
compounds for their antimicrobial, anticancer, Anti-inflammatory activities and Molecular
docking (Mani, 2013).
Recently, Shekhar et al., utilized same assay method for finding the antimalarial agents
from pyrido quinoxalines (Shekhar et al., 2014). The usage of this tetrazolium-based
selective colorimetric assay was found to be suitable in identifying potential new
antimalarial drugs (Ayisi et al., 2011).
2.7.3.4 in vitro antimalarial drug sensitivity assays
The in vitro drug sensitivity assay is a laboratory tool to monitor trends of antimalarial drug
susceptibility. As many countries to combination therapies to increase treatment efficacy
and delay the emergence of drug resistance using drug combinations by in vitro drug
sensitivity assays and molecular markers is helpful (Organization, 2014b).The stage-
dependent action of antimalarial drugs must be controlled because they cause a significant
impact on the outcome of these assays (Price et al., 1999; Zhu et al., 2008). With respect to
that, Drug sensitivity assay was an important method to study described by Desjardins et al.
with some modifications (Desjardins et al., 1979; Ngemenya et al., 2006). Dose-response
assay was carried out to obtain the 50% inhibitory concentration (IC50) of the individual
drugs. Ring stage infected erythrocytes (100 µL per well with 2% hematocrit and 1%
parasitaemia) inoculation in triplicate with twofold serial dilution of each drug for 48 hours
should be measured calorimetrically.
2.8 Integrative and Systems biology in Drug Discovery
The goal of modern Integrative and Systems Biology is to understand physiology and
disease from the level of molecular pathways, regulatory networks, cells, tissues, organs and
ultimately the whole organism (Eugene C Butcher et al., 2004; Hood & Perlmutter, 2004).

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 22
Systems biology means understanding complex biological systems with the integration of
experimental and computational research (Kitano, 2002a). It describes a number of trends in
bioscience research and draws importance on those trends. Some describe systems biology
as a biology-based inter-disciplinary subject that focuses on complex interactions in
biological systems with a new perspective of holism instead of reduction. The motive of
systems biology is the modeling and discovery of emergent properties of a system, whose
theoretical description is only possible only by using systems biology technology generally
involve cell signaling networks (Bu & Callaway, 2011).
It is almost 12 years since the computational systems biology concept has been proposed in
2002 (Kitano, 2002a). Since, the impact of collaboration and integration of biology, physics,
and mathematics and computer science on biological and medical research in the past
decade was excellent. As a result, this energetic interdisciplinary field, i.e., computational
systems biology keeps making significant progresses to address fundamental questions in
biology and to further lead to practical applications in medicine, drug discovery, bio-
engineering (Wang et al., 2012), neurophysiology and cybernetics. Systems biology
research encompasses the generation of high-throughput datasets of system components,
experimental methods of analysis and data integration, as well as the development and
application of network approaches and computationally derived models. Much of the
academic focus is on developing fundamental computational and informatics tools required
to integrate large amounts of reductionist data into models of regulatory networks and cell
behavior.
2.9 Systems biology in Drug Discovery
The focus of innovation in current drug discovery is on new targets, yet compound efficacy
and safety in biological models of disease not target selection are the criteria that determine
which drug candidates enter the clinic. The biology driven approach to drug discovery that
involves screening compounds by automated response profiling in disease models based on
complex human cell systems. Drug discovery through cell systems biology could
significantly reduce the time and cost of new drug development (E C Butcher, 2007).
In pharmaceutical research, systems biology efforts are directed towards the identification
of drug targets, the development of novel therapeutics and new indications for existing
drugs. Studies tend to be compound-centric, concerned with the identification and

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 23
characterization of small molecules or biologics that selectively inhibit (or activate) specific
molecular targets or pathway mechanisms. Thus, studies related to drug mechanisms of
action and those that support drug development goals, such as clinical indication selection
and patient stratification, are of particular interest.
2.9.1 Role of Systems biology in Controlling Plasmodium falciparum
The first draft of the P. falciparum genome published after seven years of the international
effort. The genome was sequenced using the Sanger method and chromosome shotgun
strategy (Gardner et al., 2002). The size of the genome was initially estimated at 22.8 Mb
separated into 14 chromosomes, and 5300 protein-encoding predicted genes. In addition to
its nuclear genome, the parasite contains 6 and 35kb circular DNA found in its mitochondria
and plant related apicoplast, respectively. Today, the P. falciparum genome remains to be
the most AT-rich genome. The overall (A + T) composition is 80% and can rise to 95% in
introns and intergenic regions. After almost 9 years of coordinated genome curation efforts,
the complete genome sequence is defined as haploid and 23.26 Mb in size. It contains 6372
genes and 5524 protein-coding genes (http:// plasmodb.org/plasmo/). Approximately half of
these genes have no detected sequence homology with any other model organism. Despite
recent access to comparative and functional genomics studies and the completion of genome
sequencing of more than eight Plasmodium species, the cellular function of most of the
parasite genes remains obscure.
Over the past few years, extensive re-sequencing efforts have been successfully undertaken
to identify genes and genetic traits associated with parasite‘s drug resistance and severity of
the clinical outcomes. Initial sequencing surveys of genetic variation across the P.
falciparum genomes published in 2007 (Jeffares et al., 2007; Volkman et al., 2007).
Volkman et al., sequenced high-quality draft genomes of three parasite laboratory clones
isolated from different parts of the world. Their work alone identified 26845 single-
nucleotide polymorphisms (SNPs) at a frequency of one SNP every 780 bases between the
three clones and an additional 37039 insertion–deletions between 3D7 and HB3. They
further extended their genotyping to 12 P. falciparum strains and 20 genomic regions from
54 worldwide P. falciparum isolates. Results were consistent with initial genetic diversity
studies that were performed using whole-genome microarray analysis (Kidgell et al., 2006).
All together, they identified more than 46937 SNPs across the whole genome. High levels
of SNP detected in genes involved in antigenic variation as well as genes involved in drug

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 24
resistance. These data confirmed by the survey of approximately 60% of P. falciparum
predicted genes (Mu et al., 2007) and a shotgun sequencing strategy of a Ghanaian clinical
isolate (Jeffares et al., 2007). All together, these reports identified a high number of rare
SNP variants and suggested that most SNPs have yet to be discovered. As a whole, these
results underscore the importance of creating comprehensive maps of genetic diversity in P.
falciparum field isolates. These SNPs are strongly suspected to be markers for various
phenotypic traits such as virulence or resistance to drugs. Recent advances in next-
generation sequencing (NGS) technologies are enabling fast and affordable production of
large amounts of genome sequence information. These technologies are already opening
new perspectives of functional genomics in the field of primary, applied and clinical malaria
research (K G Le Roch et al., 2012).
Recent advances in next-generation sequencing (NGS) technologies are enabling fast and
affordable production of large amounts of genome sequence information. These
technologies are already opening new perspectives of functional genomics in the field of
primary, applied and clinical malaria research for drug discovery.
2.9.2 High-Throughput Sequencing as backbone to Systems biology for the
antimalarial research
After 30 years of dominance of first-generation ‗Sanger‘ di-deoxy sequencing, the past 5
years have seen the explosion of NGS methods. Next-generation sequencing has
transformed the field of whole-genome sequencing and analysis. The major issue with
cloning based in vitro was developed due to extremely AT-rich genome of P. falciparum.
Four main NGS platforms have been commercialized over the past 5 years: the Roche 454
(Roche Life Sciences, Branford, CT, USA), Applied Biosystems SOLiD (Applied
Biosystems , Carlsbad, CA, USA), Illumina and finally ION Torrent.
2.9.2.1 Genome to function and metabolic network
The essential goal of functional genomics and systems biology in the malaria field is to
discover the properties of the parasite to pinpoint its weaknesses and identify long lasting
antimalarial strategies. The amalgamation of large-scale genome-wide analysis
(microarrays, deep sequencing, quantitative mass spectrometry, epigenome mapping,
computational modelling, etc.) has been used to mine Plasmodium‘s genome in an unbiased

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 25
manner and identify the genetic elements that may be targeted in the fight against malaria
(Figure 2.3). Here, we present significant contributions of the main ‗omics‘ to the malaria
field (K G Le Roch et al., 2012).
Figure 2.3 : A system biology approach to understanding the parasite biology for the design of new drug based on Systems biology
2.9.2.2 Transcriptomics
Microarray-based large-scale analysis of P. falciparum‘s transcripts led to the discovery of
expressed genes and their functional association with the various stages of the parasite life
cycle and their involvement in particular biological processes with a high degree of
accuracy (Bozdech et al., 2008; Karine G Le Roch et al., 2003). More recent sequencing
based studies such as RNAseq confirmed these initial microarray experiments and showed
promising results on the prediction of new splicing events. These studies also allowed the
identification of new open reading frames with their untranslated flanking regions (Sorber et
al., 2008, 2011). Moreover, transcriptome analyzes in P. falciparum field isolate identified
previously unknown factors involved in the pathogenesis and immune evasion (Mackinnon
et al., 2009; Siau et al., 2007). Finally, analysis of transcription profiles of variant surface
antigens identified patterns that are specific to the parasite‘s sexual stages and could be

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 26
relevant for new vaccine interventions (Petter et al., 2008). In addition to mRNA-related
transcriptomics, noncoding protein RNA (ncpRNA) transcriptome has been analyzed
(Raabe et al., 2009). In eukaryotes, structural ncpRNA is known to participate in the
regulation of diverse biochemical pathways, e.g. transcription, translation, epigenetic
regulations, cell differentiation, and proliferation. In P. falciparum, 604 putative ncpRNAs
were detected (Chakrabarti et al., 2007) and were showed to form a complex regulatory
network. Altogether, these latest analyzes suggest that P. falciparum ncpRNAs may play a
critical role in determining antigenic variation and virulence mechanisms (Raabe et al.,
2009).
2.9.2.3 Proteomics
Previous proteomics (Florens et al., 2002; Lasonder et al., 2002) and interactomics
(LaCount et al., 2005) studies have confirmed and complemented the functional annotations
proposed based on transcriptome profiling. Numerous proteomics analyzes surveyed stage-
specific proteins and investigated as potential drug targets, including sex-specific proteins in
male and female gametocytes that could be utilized for transmission blocking strategies
(Khan et al., 2005). Genomics, cell biology and proteomics studies identified a conserved
protein export motif, the PEXEL motif, which has been reported in as many as 400 proteins.
Most of these proteins are expressed during erythrocytic stages. They are thought to be
involved in creating knob structures on the surface of red blood cells and are often
associated with cyto-adherence and antigenic variation. A complete understanding of their
function and regulation will therefore be critical to disrupt one of the most pathological
effects of Plasmodium infections.
2.9.2.4 Metabolomics
In an effort to improve functional annotation and increase our understanding of the
parasite‘s biology, a number of Parasite Immunology research groups have been leveraging
biochemical metabolic profiling and metabolomics strategies (Lakshmanan et al., 2011).
Metabolomics is the study of the entire repertoire of metabolites, i.e. small molecules such
as amino acids, sugars and fatty acids that are known to perform critical functions in various
biological processes. Correlation analysis of transcriptomics, proteomics and metabolomics
data are a powerful way to identify new metabolic pathways as well as genes that encode
for specific enzymatic functions (van Brumelen et al., 2009). While the study of

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 27
metabolomics in Plasmodium is still in its infancy, it has already uncovered important
biological insights with possible implications in terms of adaptation, evolution and host–
pathogen interactions (Lian et al., 2009).
2.9.2.5 Functional genomics
Functional genomics suffers from the lack of tools to analyse the malaria parasite‘s genome.
For example, gene silencing using RNAi cannot be used in Plasmodium because the
machinery does not exist in the parasite; gene knockout experiments are time-consuming
processes not compatible with large-scale high-throughput analysis. However, in the past
few years, a transposon-based mutagenesis approach in Plasmodium has been developed
(Balu et al., 2009). A Plasmodium specific selection cassette was added to lepidopteran
transposon piggyBac and transfected in parasites together with a transposase containing
helper plasmid (Balu et al., 2005). Random insertional mutant obtained by multiple
integrations of the transposon at TTAA recognition sites. Recent studies used piggyBac-
based approaches to validate candidate parasite-specific secreted proteins (Van Ooij et al.,
2008) and also identify genes that are essential for the parasite‘s proliferation (Balu et al.,
2010). It is also used in combination with other genomics and proteomics analysis.
piggyBac-based strategies could provide a better understanding of the parasite‘s biology and
its interactions with its hosts.
2.10 Systems biology approaches for identifying antimicrobial target
With the availability of advanced genomic and bioinformatics techniques and dedicated
databases, a large number of biological networks have been constructed and reported (T. Y.
Kim et al., 2010a). The best established biological networks are the genome-scale metabolic
networks that describe the metabolism of an organism, which can be examined using
constraints based flux analysis (Thiele & Palsson, 2010). At present, more than 80 genome-
scale metabolic network models have been reported, which have undergone manual
curation, and another 130 models have also been generated in an automatic fashion using
the Model SEED (Henry et al., 2010). These genome scale metabolic network models have
been particularly useful in studying bacteria, compared to eukaryotes, due to the lesser
biological complexity in bacteria, e.g., lack of intracellular compartments (H. U. Kim et al.,
2010). However, metabolic network models of eukaryotic pathogens Leishmania major
(Chavali et al., 2008) and Plasmodium falciparum (Plata et al., 2010) are also available.

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 28
Several important drug-targeting approaches have been developed by identifying weak
points in the metabolic network using network topology analysis. While, Chokepoint
analysis (Yeh et al., 2004) identifies enzymes that consume or produce a unique metabolite
in the metabolic network, and is based on the rationale that blockage of such an enzyme
would cripple the pathogen‘s metabolism by accumulating the associated metabolites or
preventing the cell from consuming those metabolites. This method was applied to identify
drug targets in malaria causing P. falciparum (Yeh et al., 2004) and in several other
microbial pathogens including Helicobacter pylori, Mycobacterium tuberculosis and
Staphylococcus aureus (T. Y. Kim et al., 2010b).
Another, yet similar, topological approach is the identification of load points, which are
defined as enzymes or metabolites with a high ratio of k-shortest paths (alternative
metabolic pathways between two metabolites) to the number of the nearest neighbour links
connected to them (Rahman & Schomburg, 2006). A node in the metabolic network, either
metabolite or enzyme, receives the greater load point value if a greater number of shortest
paths go through this node with fewer number of edges connected to this node, which
reveals the topological importance of the metabolite or enzyme in metabolism.
In another topological approach, a drug targeting method was developed by categorizing
metabolites and enzymes into one of several classes based on the nature of their connections
to different functional modules (Guimerà et al., 2007). In this approach, the metabolic
network was first modularized in terms of node (metabolite) connectivity. Then, metabolites
were classified based on their connectivities within and outside their modules and edges
(reactions) were likewise categorized according to their connecting metabolites. This
network-based modularization revealed that reactions in certain edge types are more likely
to be essential in an organism specific manner. More recently, a protein-protein influence
network was derived from a genome-scale metabolic model of M. tuberculosis (Jamshidi &
Palsson, 2007), and various methods that interrogate the network topology were employed
to efficiently disrupt the metabolism of M. tuberculosis. These methods include the number
of connections with other proteins for a given protein or pathway, the characteristic path
length, and combinations of proteins with high connectivity. By such analysis, both
validated and potential drug targets could be identified (Raman et al., 2009). All together,
these network topology analysis provide insights into the metabolic network structure as
well as weak points of the network, which can be used for identifying drug targets.

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 29
Another advantage of constraints based flux analysis is that an initial set of predicted
antimicrobial targets can be further screened using additional criteria, resulting in the
identification of more robust drug targets. Additional criteria include eliminating proteins
having high homologies to human proteins to avoid possible side effects (Raman et al.,
2005), giving priorities to the highly connected metabolites (T. Y. Kim et al., 2010b),
analyzing the sequence, structure and expression patterns of the target enzymes (Raman et
al., 2008), examining the similarity to already known target proteins, and analyzing the
specific physiological roles of the protein in the target pathogen (Hasan et al., 2006).
2.11 Antimicrobial targets to drug discovery
Recent proof-of-concept studies paved the way for the effective applications of genome
scale metabolic network models to antimicrobial discovery beyond simple drug targeting. In
2010, Shen et al. (Shen et al., 2010) combined network model-based drug target prediction,
virtual screening against target enzymes and experimental validation, enabling simultaneous
exploration of drug targets and identification of corresponding effective drug candidates.
Essential metabolic reactions were first predicted using the network models of Escherichia
coli and S. aureus under all growth conditions, and, in conjunction with reported
experimental evidence, enzymes involved in bacterial fatty acid biosynthesis pathways were
pinpointed as final drug targets. Virtual screening was subsequently performed against the
target enzymes for both drug like properties and structural conformation of the active sites
for proper docking of virtual ligands. Finally, a series of experiments were conducted to
validate the in silico identified hit compounds: in vitro enzyme assay for the potency of the
identified compounds, disc inhibition assays against E. coli and S. aureus strains for cell
level viability test, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide cell
viability and trypan blue exclusion assays for possible side effects or cytotoxicity in human
cells.
2.11.1 Application of systems biology in drug discovery in Plasmodium
falciparum
Recent network-based analyses integrate host and pathogen models to describe more
accurately the metabolic interactions and to improve the search of putative drug targets. For
example, the topology of automatically inferred networks of Plasmodium falciparum and its

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 30
human host are studied to identify essential enzymes (Fatumo et al., 2011). The metabolic
network of Mycobacterium tuberculosis is integrated with a human alveolar macrophage to
describe three degrees of infection (Bordbar et al., 2010). Similarly, a metabolic model of
Plasmodium falciparum is embedded in a red blood cell model to simulate its intra-
erythrocytic developmental stage (Huthmacher et al., 2010). Furthermore, the selectivity of
enzymatic drug targets is already extensively predicted with large-scale metabolic networks
of human cancer cells (Folger et al., 2011). Thus, it is reasonable to assess the selectivity of
enzymatic drug targets in host-pathogen metabolic interactions with genome-scale networks
(McKay et al., 2011). The aim of present study was the prediction of selective enzymatic
inhibitions with genome-scale networks for plasmepsin protein of Plasmodium falciparum.
2.12 Reconstruction of Biological networks
A metabolic reconstruction provides a highly mathematical, structured platform on which to
understand the systems biology of metabolic pathways in an organism. The integration of
biochemical, metabolic pathways with rapidly available, unannotated genome sequences,
has developed what are called genome-scale metabolic models (Thiele & Palsson, 2010).
Simply put, these models correspond metabolic genes with metabolic pathways. In general,
the more information about physiology, biochemistry and genetics is available for the target
organism, the better the predictive capacity of the reconstructed models. Mechanically
speaking, the process of reconstructing prokaryotic and eukaryotic metabolic networks is
essentially the same. Having said this, eukaryote reconstructions are typically more
challenging because of the size of genomes, coverage of knowledge, and the multitude of
cellular compartments (Thiele & Palsson, 2010). The first genome-scale metabolic model
was generated in 1995 for Haemophilus influenzae (Fleischmann et al., 1995). The first
multicellular organism, C. elegans was reconstructed in 1998 (C. elegans sequencing
consortium, 1998). Since then, many reconstructions have been formed. For a list of
reconstructions that have been converted into a model and experimentally validated.
2.13 Metabolism in Plasmodium falciparum
2.13.1 Carbohydrate metabolism
During erythrocytic stages, the parasite produces its energy mainly through anaerobic
glycolysis, with pyruvate being converted to lactate (Gardner et al., 2002; Gómez-Arreaza

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 31
et al., 2014). Genes encoding for the TCA cycle enzymes are present in the genome, but it is
unclear whether the TCA cycle is used for oxidation of glycolytic products to be used for
energy production, or for metabolite intermediate biosynthesis. The hypothetic role of TCA
cycle in P. falciparum is for production of succinyl-CoA, to be used in heme biosynthesis in
the parasite. Genes for nearly all of the pentose phosphate pathway enzymes have been
identified from genome sequence using similarity based approach (Gardner et al., 2002).
2.13.2 Protein metabolism
The comparative genomics based analysis proved significant similarity of protein
interactions in the parasite Plasmodium falciparum from conserved interactions in
Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster and
Escherichia coli and pool them with experimental interaction data (Wuchty et al., 2009). It
has been proved that, parasite obtains nearly all of its amino acids by salvaging from the
host or through the degradation of hemoglobin (Bonilla et al., 2007). This is supported by
the fact that genomic analysis has found no enzymes necessary for amino acid biosynthesis,
except for glycine-serine, cysteine-alanine, aspartate-asparagine, proline-ornithine and
glutamine-glutamate interconversions (Kidgell et al., 2006).
2.13.3 Hemoglobin metabolism
During the erythrocytic stage of the parasite's life cycle, it uses intracellular hemoglobin as
a food source for host. The protein broken down into peptides, and heme group is released
and detoxified by biocrystallization in the form of hemozoin (Bonday et al., 2000).
Conversely, the parasite can produce pyrimidines de novo using glutamine, bicarbonate, and
aspartate (Gardner et al., 2002). P. falciparum uses several proteases act in a semi-ordered
pathway to degrade hemoglobin (Banerjee et al., 2003; Sivaraman et al., 2012). The
degradation process appears to follow an ordered pathway (Eggleson et al., 1999;
Humphreys et al., 1999) However, it has been difficult to determine the precise sequence of
events, especially whether a plasmepsin or a falcipain catalyzes the initial cleavage
(Coombs et al., 2001; Gluzman et al., 1994; Sijwali et al., 2001). The general pathway is
outlined in Figure 2.4. Initial cleavage between aPhe (Vennerstrom et al., 2004) and aLeu34
in the hinge region of the domain responsible for holding the native hemoglobin tetramer
together, unravels the protein exposing it to further cleavage. Subsequent degradation into

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 32
smaller peptides can be accomplished by both plasmepsins and falcipains (Bagavan et al.,
2011).
Several enzymes have been shown to be involved in hemoglobin proteolysis. In P.
falciparum these are aspartic proteases (plasmepsin (Plm) I, II, IV (Dame et al., 1994;
Gluzman et al., 1994; Humphreys et al., 1999) and the closely related histo-aspartic
protease (HAP) (Berry et al., 1999) cysteine proteases (falcipain-1,2,3) (Banerjee et al.,
2002), a metalloprotease (falcilysin) (Pérez et al., 2013) and dipeptidyl aminopeptidase 1
(DPAP1) (Klemba et al., 2004). Two aspartic proteases, plasmepsin (PM) I and II, appear to
initiate the degradative process by cleaving a conserved hinge region in the α chain of
native hemoglobin (Gluzman et al., 1994). Several proteases are then capable of acting on
denatured or fragmented globin. These include two cysteine proteases called falcipain 2 and
3 (Banerjee et al., 2003; Sijwali et al., 2001) and two plasmepsin paralogs, histoaspartic
protease (HAP) and PM IV (Berry et al., 1999; Karolina Ersmark, Bertil Samuelsson,
2006). HAP and PM IV are approximately 60% identical to PM I and II at the amino acid
level (Coombs et al., 2001). HAP, briefly known as PM III, is an active enzyme despite
having an unusual active site with a histidine in place of one of the two standard catalytic
aspartates. Further downstream in the pathway a metalloprotease, falcilysin, acts on 15–20
amino acid globin fragments to generate small peptides (Murata & Goldberg, 2003). The
general pathway is outlined in Figure 3.66 Initial cleavage between aPhe33 and aLeu34 in
the hinge region of the domain responsible for holding the native hemoglobin tetramer
together, unravels the protein exposing it to further cleavage. Subsequent degradation into
smaller peptides can be accomplished by both plasmepsins and falcipains (Figure 2.4).
These peptides may be exported out of the FV for terminal degradation to amino acids in
the parasite cytosol (Kolakovich et al., 1997).
2.14 Drug Targets in Plamsodium falciparum
The global effort to defeat malaria has expanded greatly in the last few years. In this
process, a number of novel strategies to obstruct malarial growth have emerged. Among the
most appealing strategies is an attempt to produce a long-lasting vaccine which would help
eradicate the disease (Alonso et al., 2004). The vaccine developed by Alonso et al., appears
promising, but has so far only given limited protection (Alonso et al., 2005). Until there is a
vaccine on the market offering full protection against malaria, we must consider therapeutic

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 33
drugs for preventive chemotherapy and treatment of acute malaria. However, a problem
with present drugs is growing resistance. The limited number of malaria drugs and their
extensive use has led to increasing resistance among malaria strains. Hence, drugs with
novel mechanisms of action are desperately needed to combat the disease. In the wake of
the full genome sequencing of P. falciparum, numerous new drug targets have been
proposed.
Figure 2.4 : General Pathway for hemoglobin metabolism in P. falciparum
These targets are quite diverse and include enzymes from the respiratory chain in the
parasite mitochondria (Biagini et al., 2006; Mi-Ichi et al., 2005), several transport proteins
(Beitz, 2005; Reguera et al., 2005), enzymes in the fatty acid synthetic pathway (R.T. et al.,
2006), a number of proteases (Coppi et al., 2006; Ersmark K1, Nervall M, Gutiérrez-de-
Terán H, Hamelink E, Janka LK, Clemente JC, Dunn BM, Gogoll A, Samuelsson B, Qvist
J, 2006; Micale et al., 2006), Dihydrofolate reductase (DHFR) (Graffner-Nordberg et al.,
2000), Protein Kinase 5 (PfPK5) (Graeser et al., 1996) and DNA replication and regulation
(Yanow et al., 2006).
The proteases from the malaria parasite, named plasmepsins (Plm), have previously been
validated as potential drug targets (Goldberg et al., 1991; Liu et al., 2005; Omara-Opyene et
al., 2004). Falcipain-2 is best studied P. falciparum cysteine proteases and represents major
part of the total cysteine protease activity in the food vacuole (Hogg et al., 2006).
Ersmark et al.,(2003) also reported that, the doubling time for the malaria parasite was
longer for the plasmepsin/falcipain knock-out (plasmepsin I, plasmepsin IV and falcipain-2)
than for either plasmepsin or falcipain knock-outs alone (Ersmark et al., 2003).

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 34
Furthermore, the aspartic protease inhibitor pepstatin A was highly potent against falcipain
knock-out. Thus plasmepsin inhibitors could potentially be part of an effective drug cocktail
in combination with falcipain inhibitors. However, either knockout of falcipain-2 alone or
knockout of individual vacuole plasmepsin has no lethal effect on P. falciparum 3D7 in
vitro.
The studies suggested that, falcipain-2 and plasmepsin II can act cooperatively to mediate
the hydrolysis of hemoglobin and take part in the degradation of the cytoskeleton of
erythrocyte (Dhawan et al., 2003; Le Bonniec et al., 1999). The antiparasitic effects on P.
falciparum in culture and animal models of Plasmodium berghei infection will be
synergized by blockade of both falcipains and plasmepsins with potent inhibitors (Liu et al.,
2006). The dual inhibitors targeting falcipains and plasmepsins may lead to the occurrence
of new antimalarial therapeutics that are unaffected by the existing mechanisms of drug
resistance.
Several scaffolds from AIDS drug discovery projects have been reused to aid malaria drug
development (Ersmark et al., 2003; Luksch et al., 2008). The list of already reported targets
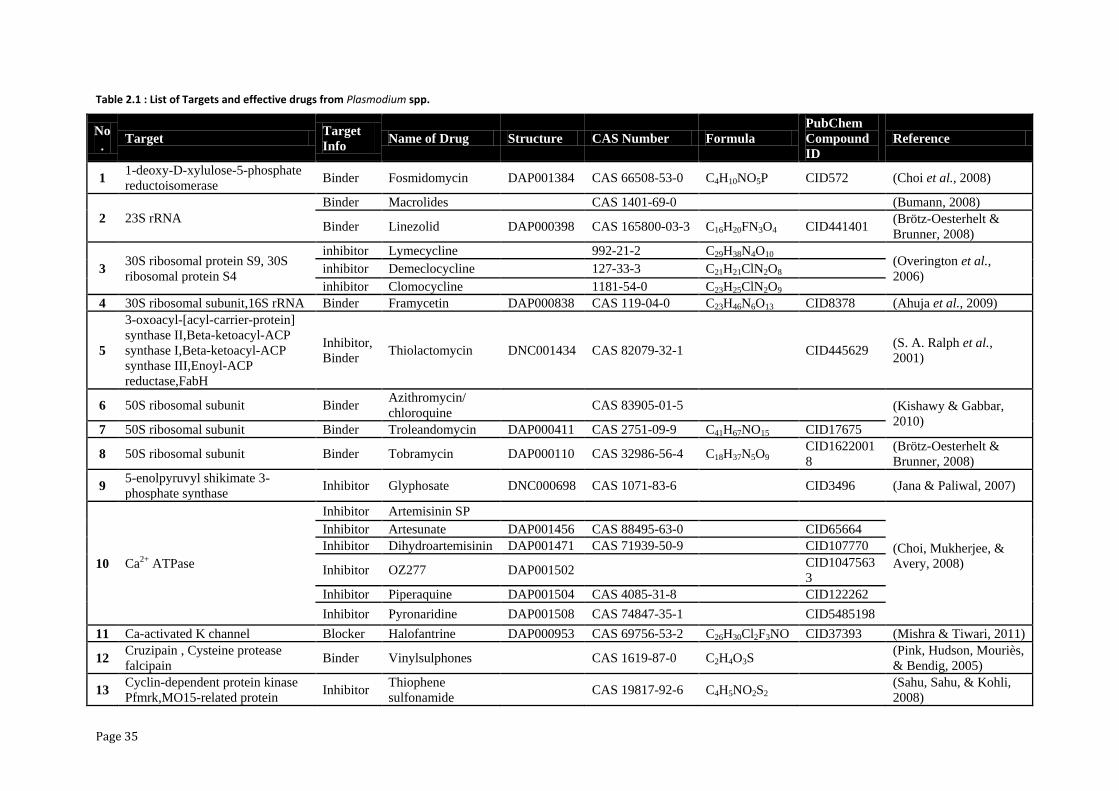
and it relevant drug or inhibitors are presented in Table 2.1.
Page 35
Table 2.1 : List of Targets and effective drugs from Plasmodium spp.
No
. Target
Target
Info Name of Drug Structure CAS Number Formula
PubChem
Compound
ID
Reference
1 1-deoxy-D-xylulose-5-phosphate
reductoisomerase Binder Fosmidomycin DAP001384 CAS 66508-53-0 C4H10NO5P CID572 (Choi et al., 2008)
2 23S rRNA
Binder Macrolides CAS 1401-69-0
(Bumann, 2008)
Binder Linezolid DAP000398 CAS 165800-03-3 C16H20FN3O4 CID441401 (Brötz-Oesterhelt &
Brunner, 2008)
3 30S ribosomal protein S9, 30S
ribosomal protein S4
inhibitor Lymecycline 992-21-2 C29H38N4O10 (Overington et al.,
2006) inhibitor Demeclocycline 127-33-3 C21H21ClN2O8
inhibitor Clomocycline 1181-54-0 C23H25ClN2O9 4 30S ribosomal subunit,16S rRNA Binder Framycetin DAP000838 CAS 119-04-0 C23H46N6O13 CID8378 (Ahuja et al., 2009)
5
3-oxoacyl-[acyl-carrier-protein]
synthase II,Beta-ketoacyl-ACP
synthase I,Beta-ketoacyl-ACP
synthase III,Enoyl-ACP
reductase,FabH
Inhibitor,
Binder Thiolactomycin DNC001434 CAS 82079-32-1
CID445629
(S. A. Ralph et al.,
2001)
6 50S ribosomal subunit Binder Azithromycin/
chloroquine CAS 83905-01-5 (Kishawy & Gabbar,
2010) 7 50S ribosomal subunit Binder Troleandomycin DAP000411 CAS 2751-09-9 C41H67NO15 CID17675
8 50S ribosomal subunit Binder Tobramycin DAP000110 CAS 32986-56-4 C18H37N5O9 CID1622001
8
(Brötz-Oesterhelt &
Brunner, 2008)
9 5-enolpyruvyl shikimate 3-
phosphate synthase Inhibitor Glyphosate DNC000698 CAS 1071-83-6
CID3496 (Jana & Paliwal, 2007)
10 Ca2+ ATPase
Inhibitor Artemisinin SP
(Choi, Mukherjee, &
Avery, 2008)
Inhibitor Artesunate DAP001456 CAS 88495-63-0
CID65664
Inhibitor Dihydroartemisinin DAP001471 CAS 71939-50-9 CID107770
Inhibitor OZ277 DAP001502
CID1047563
3
Inhibitor Piperaquine DAP001504 CAS 4085-31-8 CID122262
Inhibitor Pyronaridine DAP001508 CAS 74847-35-1
CID5485198
11 Ca-activated K channel Blocker Halofantrine DAP000953 CAS 69756-53-2 C26H30Cl2F3NO CID37393 (Mishra & Tiwari, 2011)
12 Cruzipain , Cysteine protease
falcipain Binder Vinylsulphones CAS 1619-87-0 C2H4O3S
(Pink, Hudson, Mouriès,
& Bendig, 2005)
13 Cyclin-dependent protein kinase
Pfmrk,MO15-related protein Inhibitor
Thiophene
sulfonamide CAS 19817-92-6 C4H5NO2S2
(Sahu, Sahu, & Kohli,
2008)
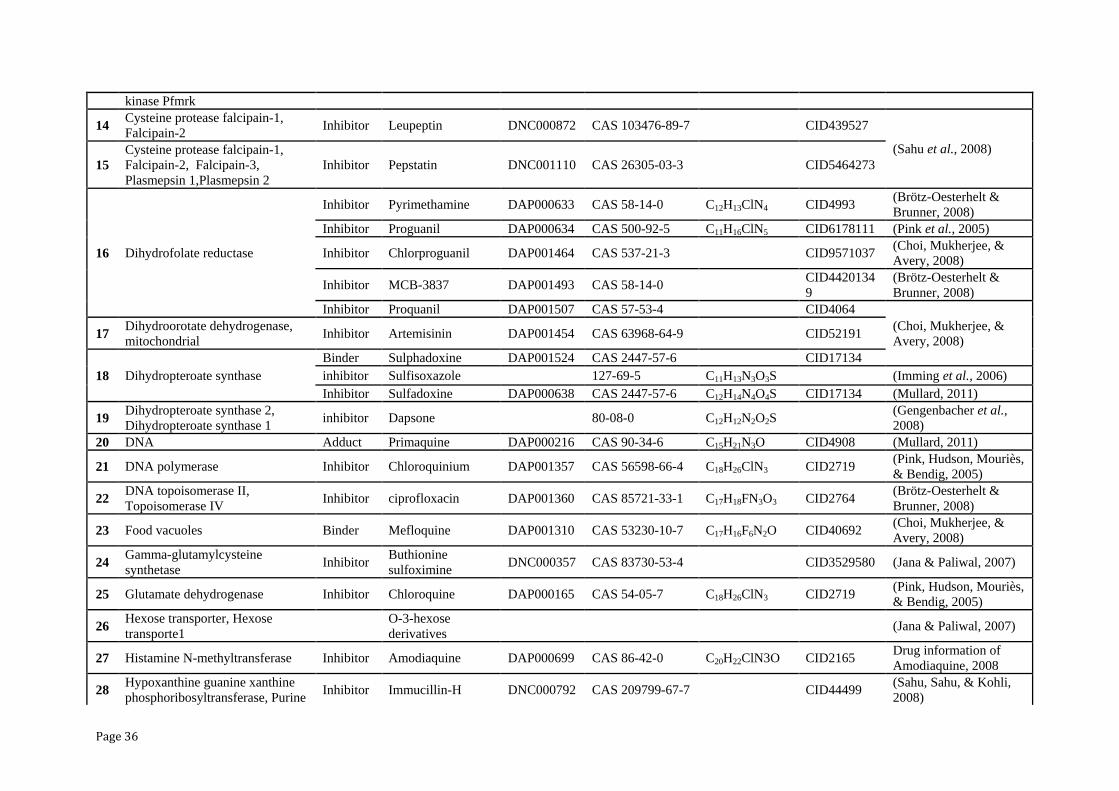
Page 36
kinase Pfmrk
14 Cysteine protease falcipain-1,
Falcipain-2 Inhibitor Leupeptin DNC000872 CAS 103476-89-7
CID439527
(Sahu et al., 2008)
15
Cysteine protease falcipain-1,
Falcipain-2, Falcipain-3,
Plasmepsin 1,Plasmepsin 2
Inhibitor Pepstatin DNC001110 CAS 26305-03-3 CID5464273
16 Dihydrofolate reductase
Inhibitor Pyrimethamine DAP000633 CAS 58-14-0 C12H13ClN4 CID4993 (Brötz-Oesterhelt &
Brunner, 2008)
Inhibitor Proguanil DAP000634 CAS 500-92-5 C11H16ClN5 CID6178111 (Pink et al., 2005)
Inhibitor Chlorproguanil DAP001464 CAS 537-21-3
CID9571037 (Choi, Mukherjee, &
Avery, 2008)
Inhibitor MCB-3837 DAP001493 CAS 58-14-0 CID4420134
9
(Brötz-Oesterhelt &
Brunner, 2008)
Inhibitor Proquanil DAP001507 CAS 57-53-4
CID4064
(Choi, Mukherjee, &
Avery, 2008) 17
Dihydroorotate dehydrogenase,
mitochondrial Inhibitor Artemisinin DAP001454 CAS 63968-64-9 CID52191
18 Dihydropteroate synthase
Binder Sulphadoxine DAP001524 CAS 2447-57-6
CID17134
inhibitor Sulfisoxazole 127-69-5 C11H13N3O3S (Imming et al., 2006)
Inhibitor Sulfadoxine DAP000638 CAS 2447-57-6 C12H14N4O4S CID17134 (Mullard, 2011)
19 Dihydropteroate synthase 2,
Dihydropteroate synthase 1 inhibitor Dapsone 80-08-0 C12H12N2O2S
(Gengenbacher et al.,
2008)
20 DNA Adduct Primaquine DAP000216 CAS 90-34-6 C15H21N3O CID4908 (Mullard, 2011)
21 DNA polymerase Inhibitor Chloroquinium DAP001357 CAS 56598-66-4 C18H26ClN3 CID2719 (Pink, Hudson, Mouriès,
& Bendig, 2005)
22 DNA topoisomerase II,
Topoisomerase IV Inhibitor ciprofloxacin DAP001360 CAS 85721-33-1 C17H18FN3O3 CID2764
(Brötz-Oesterhelt &
Brunner, 2008)
23 Food vacuoles Binder Mefloquine DAP001310 CAS 53230-10-7 C17H16F6N2O CID40692 (Choi, Mukherjee, &
Avery, 2008)
24 Gamma-glutamylcysteine
synthetase Inhibitor
Buthionine
sulfoximine DNC000357 CAS 83730-53-4
CID3529580 (Jana & Paliwal, 2007)
25 Glutamate dehydrogenase Inhibitor Chloroquine DAP000165 CAS 54-05-7 C18H26ClN3 CID2719 (Pink, Hudson, Mouriès,
& Bendig, 2005)
26 Hexose transporter, Hexose
transporte1
O-3-hexose
derivatives
(Jana & Paliwal, 2007)
27 Histamine N-methyltransferase Inhibitor Amodiaquine DAP000699 CAS 86-42-0 C20H22ClN3O CID2165 Drug information of
Amodiaquine, 2008
28 Hypoxanthine guanine xanthine
phosphoribosyltransferase, Purine Inhibitor Immucillin-H DNC000792 CAS 209799-67-7
CID44499
(Sahu, Sahu, & Kohli,
2008)
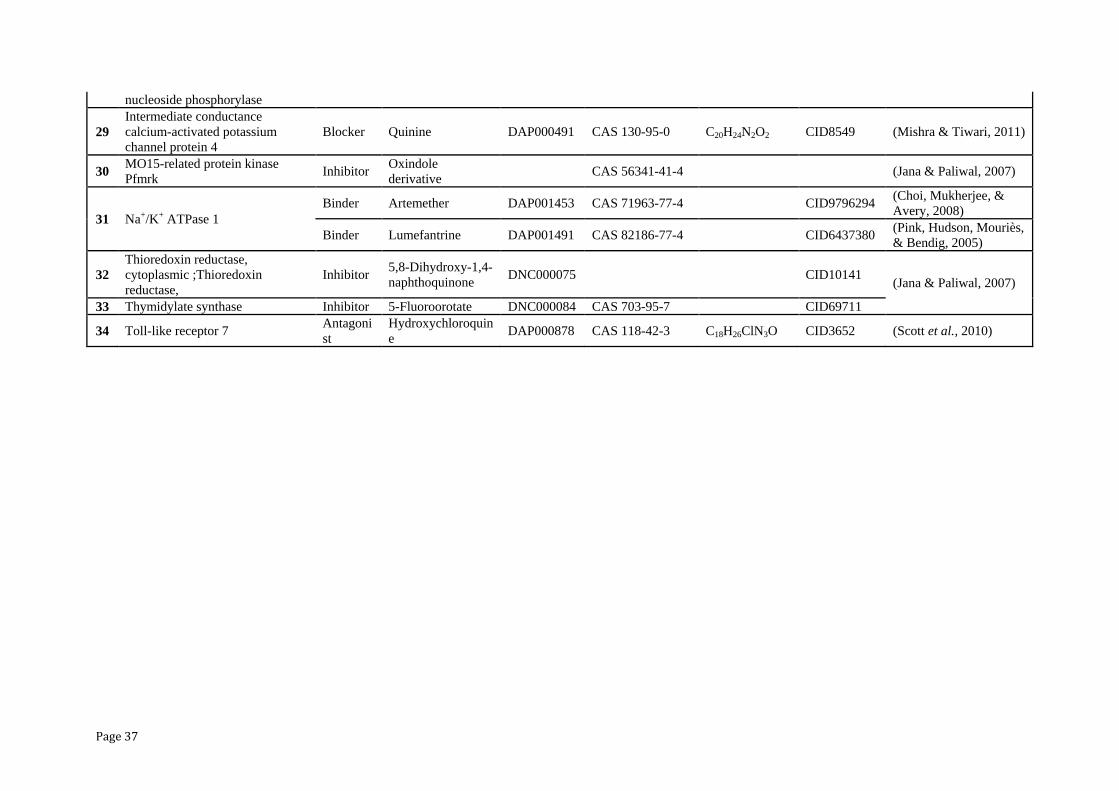
Page 37
nucleoside phosphorylase
29
Intermediate conductance
calcium-activated potassium
channel protein 4
Blocker Quinine DAP000491 CAS 130-95-0 C20H24N2O2 CID8549 (Mishra & Tiwari, 2011)
30 MO15-related protein kinase
Pfmrk Inhibitor
Oxindole
derivative CAS 56341-41-4
(Jana & Paliwal, 2007)
31 Na+/K+ ATPase 1
Binder Artemether DAP001453 CAS 71963-77-4 CID9796294 (Choi, Mukherjee, &
Avery, 2008)
Binder Lumefantrine DAP001491 CAS 82186-77-4
CID6437380 (Pink, Hudson, Mouriès,
& Bendig, 2005)
32
Thioredoxin reductase,
cytoplasmic ;Thioredoxin
reductase,
Inhibitor 5,8-Dihydroxy-1,4-
naphthoquinone DNC000075 CID10141
(Jana & Paliwal, 2007)
33 Thymidylate synthase Inhibitor 5-Fluoroorotate DNC000084 CAS 703-95-7
CID69711
34 Toll-like receptor 7 Antagoni
st
Hydroxychloroquin
e DAP000878 CAS 118-42-3 C18H26ClN3O CID3652 (Scott et al., 2010)

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 38
2.15 Plasmepsin in Plasmodium falciparum
The genome sequencing of P. falciparum has led to the identification of ten different genes
encoding plasmepsins (Coombs et al., 2001). There are ten aspartic proteases or plasmepsin
reported in the genome of P. falciparum and four of them (Plm I, Plm II, HAP and Plm IV)
have been localized in the food vacuole and shown to be involved in Hb degradation and as
such constitute possible drug targets. (Liu et al., 2005; Omara-Opyene et al., 2004). The
degradative process, taking place in an acidic food vacuole of P. falciparum, provides a
source of amino acids and helps maintain intracellular osmolarity during rapid parasite
growth (Hogg et al., 2006). Plasmepsin V, IX and X are expressed concurrently with Plm I–
IV, but are not transported to the food vacuole. The remaining plasmepsins (Plm VI, VII,
VIII) are not expressed during the intraerythrocytic stage (Banerjee et al., 2002). The
plasmepsins are aspartic proteases with pepsin like fold (Figure 2.5).
Figure 2.5 : Crystal structure of Plasmepsin 1 from Plasmodium falciparum; Protein chains are colored from the N-terminal to C-terminal using rainbow color gradient
These enzymes are composed of two domains with the active site situated in a deep cleft in
between and a loop that covers the active site. In contrast, retroviral proteases, for example
HIV-1 protease, consist of two identical subunits with two equivalent flaps covering the
active site (Figure 2.6) (Brik & Wong, 2003).
Upon substrate binding to this class of enzymes, the flap (or flaps in Aspartic protease)
closes around a substrate part containing the scissile bond, thereby forming a solvent-
shielded environment inside the enzyme. Between the flap region and the substrate, a

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 39
structurally conserved water molecule is situated in Aspartic protease (Brik & Wong, 2003)
which is lacking in Plm II. The apparent lack of symmetry in Plm II can be deceiving, as
very potent symmetrical inhibitors against it have been designed (Ersmark et al., 2004).
Each domain in plasmepsins contributes an aspartic acid residue to the active site, except for
the histo-aspartic protease (HAP, corresponding to Plm III), in which one of the catalytic
aspartates ismutated to a histidine residue (Banerjee et al., 2002; Berry et al., 1999; Sinisa
Bjelic & Aqvist, 2004). The peptide bond of the substrate is cleaved in Plm I–IV by a
catalytic aspartate that acts as a general base by activating a water molecule through
abstraction of a proton. The resulting hydroxide ion attacks the peptide bond forming a
transient tetrahedral intermediate. It has also been shown that the formation of a tetrahedral
intermediate can occur via a concerted path during which the hydroxide ion is not
completely formed (Sinisa Bjelic & Aqvist, 2004).
Figure 2.6 : Aspartic Protease with active sites and inhibitor
2.16 X-Ray Structure of Plasmepsin
Among the plasmepsins, Plm II has been the most extensively characterized, with a number
of X-ray structures reported (Asojo et al., 2002, 2003; Prade et al., 2005; Silva et al., 1996).
These were co-crystallized with several different hydroxyethylamine/statin-based inhibitors
as well as an achiral inhibitor (Asojo et al., 2002, 2003; Prade et al., 2005). The structures
of un-complexed plasmepsin (Asojo et al., 2003) and proplasmepsin have also been
determined. Several structures pending publication have further been deposited in the PDB
(1M43, 1ME6, 1XDH, 1XE5, 1XE6, 1W6H, 1W6I and 1J8J). Most of them are complexed
with pepstatin A or statin-based inhibitors. Plasmepsin IV has also captured the attention as
a drug target, when it was found that deletion of Plm I and Plm IV genes reduced malaria
parasite growth rate (Liu et al., 2005). The reports show that poly-inhibition of plasmepsins,

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 40
and other proteases such as falcipains, may be desirable in potential antimalarials to ensure
clinical efficacy (Liu et al., 2006; Omara-Opyene et al., 2004).
2.17 Perspectives of virtual screening for Drug discovery
Virtual screening is a computational technique used in drug discovery to search libraries of
small molecules in order to identify those structures which are most likely to bind to a drug
target, typically a protein receptor or enzyme (Rester, 2008; Rollinger et al., 2008). Virtual
screening has been defined as the "automatically evaluating very large libraries of
compounds" using computer programs (Walters et al., 1998). As this definition suggests,
Virtual screening has largely been a numbers game focusing on how the enormous chemical
space of over millions of conceivable compounds (Ma et al., 2011) can be filtered to a
manageable number that can be synthesized, purchased, and tested. Although searching the
entire chemical universe may be a theoretically interesting problem, more practical VS
scenarios focus on designing and optimizing targeted combinatorial libraries and enriching
libraries of available compounds from in-house compound repositories or vendor offerings.
As the accuracy of the method has increased, virtual screening has become an integral part
of the drug discovery process (McInnes, 2007). There are two broad categories of screening
techniques: ligand-based and structure-based (Rester, 2008).
2.18 Molecular docking tools utilized for antimalarial drug discovery
virtual screening
The Virtual screening is widely used to predict the binding of a large database of ligands to
a particular target, with the goal of identifying the most promising compounds from the
database for further study (Kitchen et al., 2004; Kolb et al., 2009). Millions of compounds
can be tested in very less time depending upon the available computational power for the
software. Popular programs for docking include AutoDock (Morris et al., 1998), Vina (Trott
& Olson, 2010), Dock (Ewing & Kuntz, 1997), FlexX (Rarey et al., 1996), Glide (Halgren
et al., 2004; Zhou et al., 2001) and Gold (Verdonk et al., 2003).
Recently, AutoDock Vina was introduced as a member of AutoDock suite in 2010
developed by Dr. Oleg Trott, from Molecular Graphics Lab at The Scripps Research
Institute (Trott & Olson, 2010). It uses a sophisticated gradient optimization method in its
local optimization procedure. The calculation of the gradient effectively gives the

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 41
optimization algorithm a ―sense of direction‖ from a single evaluation. By using
multithreading, Vina can further speed up the execution by taking advantage of multiple
CPUs or CPU cores. Only three-dimensional structures of molecules (with polar hydrogen
only, as AutoDock Vina still uses United Atom model) and a box definition of a search
space is required. Partial charges, solvation parameters or pre-calculated interaction energy
grids are not necessary for the simulation.
The wide ranges of software were reported to study the antimalarial drug discovery.
In the studies involved in the identification of Plm IV and the macrocyclic inhibitors 18 –
21 GOLD 1.2 was employed (Jones et al., 1997). Both the Goldscore and Chemscore
functions of GOLD 2.2 also utilized in parallel docking runs. To capture the flexible nature
of the plasmepsins against multiple protein models PDB ID:1LF2 and 1LF3 for Plm II
(Asojo et al., 2002); 1LS5 (Nezami et al., 2003) and a homology model for Plm IV.
In year 2005, Erskmark et al., used Autodock for Molecular Dynamics Simulations and
Free Energy Calculations for binding of seven synthesized di-amide compounds for
Plasmepsin II target (Ersmark et al., 2005).
Warner et al., used Glide software package for Falcipain inhibitors of Plasmodium
falciparum (Warner et al., 2006). The DOCK was utilized for FK506 immunophilin BCL6,
oncogene in B-cell lymphomas (Zhao et al., 2006). Gutierre-De-Teran et al., performed
docking using GOLD version 2.2 allowing full flexibility for structure based approach for
20 independent runs of using the default genetic algorithm (GA) to calculate Goldscore
(Gutierrez-De-Teran et al., 2006).
The open source Autodock 3.2 was used by Kasam et al., in WISDOM based design of new
plasmepsin inhibitor (Kasam et al., 2007). S Bjelic et al., used two empirical scoring
functions to predict the inhibitor binding affinity and ranking (Chemscore and Goldscore)
initially to select the most potent stereoisomer (S Bjelic et al., 2007)
FlexX was utilized for Plasmepsin II and IV inhibitors (Luksch et al., 2008) and malaria
Anthrax edema factor (D. Chen et al., 2008). Along with that, Structure and mechanism of a
transmission blocking vaccine candidate protein Pfs25 from P. falciparum: a molecular
modeling and docking study.

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 42
Friedman and Caflisch used SEED algorithm based software for Plasmepsin (Friedman &
Caflisch, 2009). Saied et al., used FLEXX for docking and QSAR based study for
identification of New benzimidazole derivatives as antiplasmodial agents and plasmepsin
inhibitors (Saied et al., 2012). Nag et al., utilized Autodock 4.0 and MGL Tool package for
screening of heterocyclic compound libraries to identify novel inhibitors for PfRIO-2 kinase
through docking (Nag et al., 2013).
Agrawal et al., had utilized the open source Autodock Vina 4.2 for their work for finding
inhibitory effect of Thiazides on Plasmepsins (Agarwal et al., 2014). The autodock Vina
was used by Sarfar Alam for molecular docking studies to validate the LibDock score in his
work (Sarfaraz Alam, 2014). While, Jin et al., used autodock for identification of dietary
flavonoids fisetin and myricetin for inhibition study of falcipain-2 and plasmepsin II of
Plasmodium falciparum (Jin et al., 2014).
2.19 Pharmacophore in Antimalarial drug Discovery
The concept of pharmacophore was first introduced in 1909 by Ehrlich, who defined the
pharmacophore as ‗a molecular framework that carries (phoros) the essential features
responsible for a drug‘s (pharmacon) biological activity‘ (Ehrlich, 1909). After a century‘s
development, the basic pharmacophore concept still remains unchanged, but its intentional
meaning and application range have been expanded considerably. According to the very
recent definition by IUPAC (Camille Georges Wermuth et al., 1998), a pharmacophore
model is ‗an ensemble of steric and electronic features that is necessary to ensure the
optimal supramolecular interactions with a specific biological target and to trigger (or
block) its biological response‘. Apart from this official definition, some other similar
definitions, as well as remarks, have been described in the literature (Dror et al., 2004;
Güner, 2002; Camille G Wermuth, 2006). The overall development and history of the
pharmacophore concept through the past century has been reviewed by Gund and Wermuth
(Güner, 2002; Camille G Wermuth, 2006).
A pharmacophore model can be established either in a ligand-based manner, by superposing
a set of active molecules and extracting common chemical features that are essential for
their bioactivity, or in a structure-based manner, by probing possible interaction points
between the macromolecular target and ligands. Pharmacophore approaches have been used
extensively in virtual screening, de novo design and other applications such as lead

Integrative Biology and Systems Biology exploration for anti-malarial Drug Discovery
Vishal A Mevada Page 43
optimization and multi-target drug design. Many successful stories of pharmacophore
approaches in facilitating drug discovery have been reported in recent years (Kubinyi, 2006;
Mustata et al., 2009).
Brunner et al., identified a pharmacophore for potent antimalarial activity for Medicinal
compounds. The predicted compounds were inhibiting the growth of erythrocytic P.
falciparum drug-sensitive and drug-resistant strains at nanomolar concentrations,
comparable to the reference antimalarials chloroquine and artesunate (Brunner et al., 2012).
In year 2013, Wadood done effort to design new structure-based pharmacophore model to
design new and potent P. falciparum dihydrooratate dehydrogenase (PfDHODH) inhibitors
(Wadood & Ulhaq, 2013).










![Life Sciences...76 3 Contribution of Natural Products to Drug Discovery in Tropical Diseases mosquito [2]. Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malariae,andPlasmodium](https://static.documents.pub/doc/80x56/6049cbda4f3447749747f712/life-sciences-76-3-contribution-of-natural-products-to-drug-discovery-in-tropical.jpg)






