Quelques développements de l’analyse protéomique en spectrométrie de masse en 2005-2006 Rédigé par : Wikayatou ATTANDA; Céline BASCUÑANA; Francis BAZABAS ; Christelle BENOIT ; Meryem BRIHOUM ; Thomas CORTESE ; Lila DELBOS ; Magalie GRACIA ;Gaëlle HOARAU ; Sarah JEMMAL ; Fabrice LE POGAM ; Christel LOGEZ ; Roberto MARTIN ; Adeline POIRIER ; Abdelaziz ZEMMAR. Professeur : François COUDERC
Transcript
Quelques développements de l’analyse protéomique en spectrométrie de masse en
2005-2006
Rédigé par :
Wikayatou ATTANDA; Céline BASCUÑANA; Francis BAZABAS ; Christelle BENOIT ; Meryem BRIHOUM ; Thomas CORTESE ; Lila DELBOS ; Magalie GRACIA ;Gaëlle HOARAU ; Sarah JEMMAL ; Fabrice LE POGAM ; Christel
LOGEZ ; Roberto MARTIN ; Adeline POIRIER ; Abdelaziz ZEMMAR.
Professeur : François COUDERC
Sommaire I. Introduction ........................................................................................................................ 3 II. Techniques de séparation utilisables en spectrométrie de masse....................................... 4
II.1. Techniques basées sur l’électrophorèse: .................................................................... 4 II.2. Techniques basées sur la chromatographie ................................................................ 5
III. Développement de l’ECD et l’ETD ............................................................................... 7 IV. Quantification protéique en spectrométrie de masse.................................................... 11
IV.1. 2D-DIGE (Fluorescence In Gel Electrophoresis) ................................................ 12 IV.2. Marquage isotopique in vitro : Isotope-Coded Affinity Tag (ICAT).................. 14 IV.3. Marquage isotopique in vitro : Isotope Tags for Relative and Absolute Quantification ( ITRAQ)...................................................................................................... 15 IV.4. Stable Isotope Labeling with Amino acids in Cell culture (SILAC) ................... 17 IV.5. Stable Isotopic Standards and Capture by Anti-peptides Antibodies (SISCAPA) .. .............................................................................................................................. 19
V. Les modifications post-traductionnelles : Etudes des N et O-glycosylation.................... 20
V.1. Les N-glycosylations................................................................................................ 21 V.2. Les O-glycosylations................................................................................................ 22
VI. Détection de modifications post–traductionnelles : les phosphorylations ................... 24 VII. Les modifications post-traductionnelles : acétylation et méthylation.......................... 27 VIII. Les applications médicales de la spectrométrie de masse : SELDI-TOF et Imagerie . 31
VIII.1. Le SELDI-TOF : Surface Enhanced Laser Desorption Ionisation Time of Flight.............................................................................................................................. 31
VIII.2. L’imagerie par spectrométrie de masse (Imaging Mass Spectrometry)............... 32 Bibliographie............................................................................................................................ 35
I. Introduction
Parmi les méthodes d'analyses physico-chimiques, la spectrométrie de masse occupe
une place importante grâce à ses performances (limite de détection, sensibilité et sélectivité)
et aux diverses techniques d'ionisation et de détection, qui permettent les investigations les
plus variées. Ainsi, son champ d'application s'est accru depuis les années cinquante, de la
chimie et biochimie au milieu médical et pharmaceutique, de l'étude de l'environnement à
l'agro-alimentaire.
De récentes avancées dans la séparation des protéines (électrophorèse,
chromatographie…) aussi bien que dans l’analyse en spectrométrie de masse ont permis
l’essor de la protéomique. La protéomique regroupe les méthodes qui permettent l’étude de
l’ensemble des protéines de l’organisme : en identifiant des protéines extraites d’une culture
cellulaire ou d’un tissu, leur localisation dans un compartiment cellulaire et leurs
modifications post-traductionnelles (phosphorylation, acétylation, N et O-glycosylations…),
on peut déterminer la fonction d’une protéine dans un processus biologique comme la
signalisation cellulaire.
Le développement de nouvelles méthodes en spectrométrie de masse permet
désormais de réaliser des analyses quantitatives (ICAT,iTRAQ, SILAC et SISCAPA). Dans le
domaine biologie clinique, ces approches permettent de rechercher, de détecter et de
quantifier la présence de certains marqueurs protéiques caractéristiques d’une pathologie.
De plus, la complexité des échantillons à analyser et à quantifier en protéomique
quantitative ont suscité le développement de nouvelles approches plus sensibles et beaucoup
plus résolutives, par exemple la méthode DIGE. Parallèlement les avancées techniques
permettent désormais l’amélioration de l’ionisation par ECD ou ETD.
Ce travail est écrit par des étudiants de M2 professionnel de biotechnologies en quête
de meilleures connaissances sur les techniques de séparation associées à la spectrométrie de
masse afin d’analyser les mélanges protéiques, le manuscrit se voudrait être ditactique.
Cette revue présente les dernières avancées techniques en protéomique à travers une
sélection d’articles publiés en 2006, que nous espérons être les plus pertinents.
II. Techniques de séparation utilisables en spectrométrie de masse
Actuellement, en protéomique, deux grandes familles de méthodes sont utilisées pour
séparer les protéines avant analyse par spectrométrie de masse : les techniques de
chromatographies et celles basées sur l’électrophorèse, ces techniques étant utilisées sur une
ou plusieurs dimensions afin d’augmenter la résolution finale de la séparation
chromatographique ou électrophorétique .
II.1. Techniques basées sur l’électrophorèse:
Le méthode la plus utilisée pour l’analyse protéomique est la 2D-PAGE, combinant
une première séparation des protéines en fonction de leur point isoélectrique par
isoélectrofocalisation (IEF) suivie d’une deuxième séparation en fonction de leurs masses
apparentes par SDS PAGE. Elle est en général suivie d’une caractérisation des protéines par
MALDI TOF.
Cette technique a une bonne résolution (5000 protéines séparées sur gel), mais son manque de
reproductibilité entre les gels, limite l’analyse en protéomique.
De nombreuses améliorations ont été proposées afin de palier à ce problème, comme la
technique DAGE1, ayant pour objectif d’améliorer la reproductibilité en éliminant les
différences locales de forces ioniques et pH se produisant au cours de la séparation,
notamment au cours de la première dimension. L’IEF est réalisée sur 2 strips IPG
(Immobiline pH Gradient) séparés par une membrane de dialyse. (cf. Figure. II-1).
Figure II-1 : montage DAGE avec
séparation des 2 échantillons par une
membrane de dialyse afin d’équilibrer
l’environnement ionique des échantillons.
L’inconvénient de cette innovation est que l’amélioration de la reproductibilité n’a lieu qu’au
cours de l’IEF.
La technique DIGE permet de surmonter ces problèmes : 2 échantillons, marqués par 2
cyanines excitables à des longueurs d’ondes différentes, sont mélangés puis soumis à une
séparation 2D-PAGE classique. L’expression différentielle des protéines est déterminée
ensuite grâce au ratio des intensité des fluorophores utilisés.
Malgré toutes ces innovations, ces méthodes de 2D-PAGE souffrent d’une gamme dynamique
limitée, rendant ainsi difficile l’analyse des protéines de faibles abondances, mais aussi d’un
manque de résolution pour les protéines acides ou basiques, sans parler des protéines
membranaires rarement observées.
II.2. Techniques basées sur la chromatographie
Toutes les méthodes de chromatographies (gel filtration, échange d’ions, …) sont utilisables
en analyse protéomique, mais afin d’augmenter le pouvoir de séparation et la résolution
finale, ces méthodes sont employées en général sur 2 dimensions, avec le plus souvent une
chromatographie d’échange d’ions (IEC) en premier où les protéines seront séparées en
fonction de leur charge, suivie d’une chromatographie en phase inverse où elles sont éluées en
fonction de leur degré d’hydrophobicité.
Ces méthodes sont en général couplées à l’ESI-MS.
Figure II-2: Exemple de montage 2D LC avec une colonne échangeuse d’anion en 1e dimension, et une RP
LC en 2e dimension.
La technique du chromatofocalisation est une variante de l’IEC. Elle permet de séparer les
protéines en fonction de leur pI grâce à l’établissement d’un gradient de pH dans la colonne
au cours de l’élution. L’avantage de ces techniques de séparations est le peu de matériel
biologique requis (40 µg vs 200 µg en 2D-PAGE), et surtout une meilleure gamme
dynamique puisque l’on peut détecter, en fonction de la technique, utilisée beaucoup plus de
protéines sur une plus grande gamme de pI et d’hydrophobicité qu’en 2D-PAGE, d’où
l’intérêt croissant de cette technique en protéomique2.
Figure II-3 : Comparaison du nombre de protéines identifiées issues de la gaine de myéline de souris par
2D PAGE ou MDLC
Cependant, malgré cette capacité, le temps de d’optimisation et d’analyse est beaucoup plus
important qu’en 2D-PAGE, ce qui peut limiter son utilisation dans une approche à haut débit.
De plus, malgré la bonne résolution de ces 2 techniques, celle-ci peut s’avérer insuffisante
afin de séparer et distinguer 2 protéines différentes ou 2 isoformes.
En effet, un spot de gel bidimensionnel peut être composé d’un mélange de protéines
différentes, ou d’isoformes, ayant les mêmes propriétés de migration. De même, un pic sur un
chromatogramme peut être composé, malgré les 2 dimensions, d’un mélange de
protéines/peptides différents ayant les mêmes propriétés d’élution, ce qui complique
fortement l’identification ultérieure des protéines3 ,4.
Figure II-4 Exemple de spectres MALDI obtenus au cours de l’analyse d’une fraction issue de la
2e dimension RP en 2D LC. Un pic, même de faible intensité peut être composé d’un mélange de
protéines.
Une technique alternative à ces deux grandes familles de méthode est l’électrophorèse
capillaire.
Cette approche est peu utilisée en analyse protéomique, notamment du fait qu’elle n’est pas
facilement couplable à une analyse en MS à cause d’un débit de liquide lent et d’une interface
capillaire / source d’ionisation à optimiser.
Notamment, on observe souvent une perte de résolution dans les spectres entre une analyse
avec un détecteur UV classique et celle effectuée avec un spectromètre de masse à cause de
ces problèmes d’interface entre les deux méthodes5 :
Figure. II-5 : Electrophorégrammes obtenus en utilisant un détecteur UV classique A ou un spectromètre
de masse dans le cadre d’une analyse de fractions I et II de métallotionéines. Une perte de résolution est
observée entre les 2 techniques.
A cela s’ajoute de nombreuses phases d’optimisation ( tampon, pH de travail…) afin
d’améliorer la séparation des protéines.
Cependant, c’est une technique de séparation très résolutive, avec un temps d’analyse court et
nécessitant peu de matériel biologique.
Au final, du fait de la nature complexe du protéome et de ses constituants (des milliers de
protéines, isoformes, abondance relative des protéines…), ces 2 méthodes rencontrent toutes
les mêmes difficultés à séparer deux protéines aux propriétés physico-chimiques proches au
sein d’un même spot ou pic, ou des protéines de faibles abondances, ou des protéines
membranaires.
Les trois techniques sont donc complémentaires, et le choix de l’une ou de l’autre s’effectuera
en fonction de la complexité du protéome à analyser, mais aussi de la robustesse analytique de
la technique.
III. Développement de l’ECD et l’ETD
Pour la détermination de la structure primaire des protéines, deux techniques de
spectrométrie de masse sont particulièrement intéressantes : l’Electron Capture Dissociation
(ECD) et l’Electron Transfert Dissociation (ETD). L’ECD6 est une technique permettant de
faire de la MS/MS (séquençage peptidique). Son principe repose sur l’irradiation des ions,
peptides multiprotonés, par un faisceau d’électrons de faible énergie (<0.2eV). Les électrons
sont capturés par les ions, qui à leur tour se fragmentent. L’ECD utilisable avec une
instrumentation sophistiquée, le FT-ICR (Fourier transform ion cyclotron resonance mass
Spectrometry), permet de produire un faisceau d’électrons de faible énergie et aux ions
radicalaires formés de se fragmenter dans un espace bien défini. De plus l’analyseur FT-ICR
fournit une très haute résolution m/z. Les avantages de l’ECD sont :
i) une fragmentation presque totale du peptide (dépend de sa taille),
ii) le clivage site spécifique au niveau des liaisons amides, entraîne la formation de
fragments c et z. le plus souvent mais aussi a. et y,
iii) on dispose d’une technique qui clive les ponts disulfures (dans les autres méthodes
on clive ces liaisons par traitement chimique),
iv) conserve les modifications post-traductionnelles après la fragmentation,
v) engendre peu de perte de petites molécules (NH3, CO2, H2O…).
Ses inconvénients sont que lors de la capture électronique, les peptides ionisés perdent une
charge positive.
Pour éviter la neutralisation des ions et permettre leur détection en spectrométrie de
masse, il faut que les ions parents soient au minimum doublement chargés. La fragmentation
se fait généralement sur des peptides d’une vingtaine d’acides aminés. Le rendement de la
fragmentation sur des peptides de plus grande taille est plus faible.
Equation d’une fragmentation induite par ECD : (M+2H)2++ e- c+ + z+.
Figure III-1 : Exemple d’un ECD FT-ICR MS d’un peptide (Substance P [M + 2H]2+)
Obtention des principaux fragments c+ et z+. ainsi que d’un produit a+.
L’ETD7 est une technique similaire à l’ECD. La fragmentation repose sur la réaction
ion/ion qui se produit entre le peptide multiprotoné et un anion. Cet anion se trouve séquestré
dans un piège à ions. Quand les deux ions se trouvent à une distance suffisante, il se produit le
transfert d’un électron de l’anion vers le peptide chargé positivement, ce qui induit sa
fragmentation. La fragmentation obtenue par ETD dépendra :
i) du temps de réaction entre les deux ions dans le piège,
ii) de la structure de l’anion qui va transmettre son électron.
L’ETD peut être limité par les réactions séquentielles des produits8. Ces réactions
séquentielles engendrent la neutralisation des produits. Le signal produit par de multiples
clivages est alors très difficile a interpréter. Une méthode pour inhiber la fragmentation
séquentielle de l’ETD dans une trape d’ions quadrupolaire est d’accélérer simultanément les
premières générations d’ion produits pendant la réaction ion/ion et de ce fait inhiber les
futures réactions. Cette méthode est appelé : « parallel ion parking ». Cela a pour
conséquence d’engendrer un gain de 50% dans la production relative des produits de première
génération et permet la conversion de plus de 90% de l’ion parent en produit de première
génération.
Le séquençage peptidique par ETD se fait généralement avec des spectromètres de
masse de type Triple quadrupole/Ion Trap. Le grand handicap de l’analyseur Ion trap utilisé
est sa résolution. Les dernières avancées en ETD couplent un spectromètre de masse Triple
quadrupole/Ion Trap avec un analyseur TOF (qTOF). Au niveau de l’interprétation des
spectres, la technique d’ETD est très similaire à l’ECD : on obtient un clivage spécifique
(ions de type c et z) et on conserve les modifications post-traductionnelles après le processus
de fragmentation. Comme précédemment, il faut que les ions parents soient au minimum
doublement chargés pour être observés et le rendement de la fragmentation sur des peptides
de plus grande taille est généralement faible.
Equation de la réaction d’ETD : (M+3H)3++ R- . (M+2H)2+ + R-H. c+ + z+
(M+3H)2+.+ R- c+ + z+.
Figure III-2 : Exemple d’une fragmentation induite par ETD en qTOF :
Sur ce spectre on voit bien qu’on obtient une fragmentation totale du peptide et on
produit majoritairement des ions c et z. Cependant on remarque l’apparition de deux types
d’ions z. On ne voit pas beaucoup d’ions de type z+.(en rouge). Les auteurs indiquent que les
ions radicaux sont susceptibles de réagir avec les molécules libres qui se forment lors de la
fragmentation (NH3, C(NH2)2...). On obtient alors des ions de type z+ d’un poids moléculaire
plus élevée ( en bleu).
+32Da
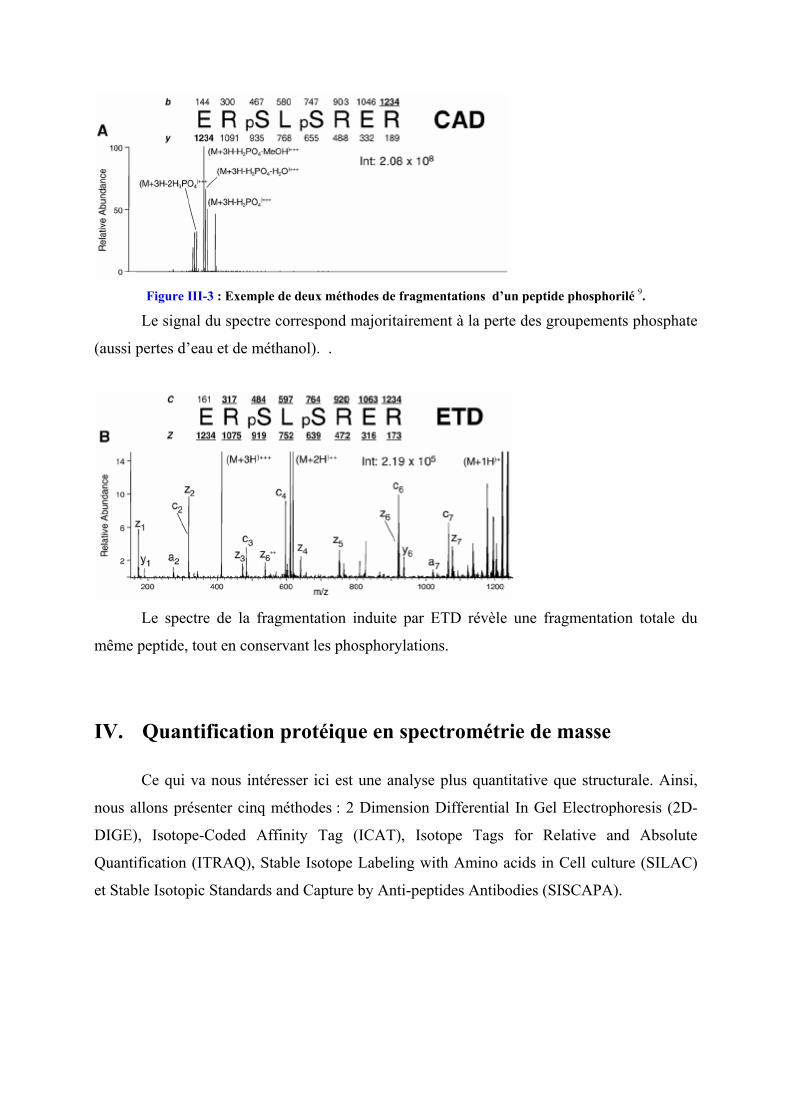
Figure III-3 : Exemple de deux méthodes de fragmentations d’un peptide phosphorilé 9.
Le signal du spectre correspond majoritairement à la perte des groupements phosphate
(aussi pertes d’eau et de méthanol). .
Le spectre de la fragmentation induite par ETD révèle une fragmentation totale du
même peptide, tout en conservant les phosphorylations.
IV. Quantification protéique en spectrométrie de masse
Ce qui va nous intéresser ici est une analyse plus quantitative que structurale. Ainsi,
nous allons présenter cinq méthodes : 2 Dimension Differential In Gel Electrophoresis (2D-
DIGE), Isotope-Coded Affinity Tag (ICAT), Isotope Tags for Relative and Absolute
Quantification (ITRAQ), Stable Isotope Labeling with Amino acids in Cell culture (SILAC)
et Stable Isotopic Standards and Capture by Anti-peptides Antibodies (SISCAPA).
IV.1. 2D-DIGE (Fluorescence In Gel Electrophoresis)
La complexité des échantillons à analyser et à quantifier en protéomique quantitative
ont suscités le développement de nouvelles approches plus sensibles et beaucoup plus
résolutives.
Parmi celles-ci la DIGE (Differential Gel Electrophoresis) 10, est une des avancées de la
protéomique quantitative très utilisé en protéomique du fait de sa fiabilité de quantification
dans les tissus complexes
Le principe de cette technique réside en la comparaison de deux profils
électrophorétiques 2D, par l’utilisation de marqueurs fluorescents spécifiques servant à
marquer les protéines pour chacun des échantillons. Le marquage est de type covalent par une
réaction spécifique, sur les groupement ε amines des chaînes latérales des lysines par des
fluorophores cyanines ( Cy3,Cy5 et Cy2).
La procédure expérimentale de la DIGE se divise en sept étapes. Les deux
échantillons protéiques à analyser sont marqués par les cyanines, l’un Cy3 et l’autre Cy5 ainsi
qu’un standard interne avec le Cy2.
Après le marquage, les trois échantillons sont déposés sur un même gel 2D ou les
protéines seront séparées dans une dimension en fonction de leur point isoélectrique et ensuite
dans une deuxième dimension en fonction de leur taille. Notons que les deux échantillons co-
migrent sur le même gel. L’analyse se fait dans un premier temps par excitation de
fluorescence du gel aux longueurs d’ondes d’excitation des marqueurs Cy3, Cy5 et Cy2. La
Figure IV-1 Propriétés structurales et chimiques des Cyanines Cya 3 ε Cy3= 150000 M-1.cm-1 λAbs:550 nm λEm:570 nm Couleur: rouge Cya 5 ε Cy5= 250000 M-1.cm-1 λAbs:640 nm λEm:670 nm Couleur: bleu Sensibilité à 125 pg, marqueurs compatibles avec la sprcctrometrie de masse
superposition des deux colorations révèlera l’expression protéique globale de chaque
échantillons. L’analyse quantitative se fait par imagerie, elle est effectuée généralement à
l’aide de logiciel spécifique (Decyder™ software (Amersham), elle consistera dans un premier
temps en la mesure de la différence de quantité de protéines de chaque spots présents sur le
gel et enfin chaque spot sera excisé, digéré et analysé par LC-MALDI-TOF MS/MS pour
identifier la protéine quantifiée correspondantes.
Cette technique a fait ses preuves dans le secteur des neurosciences en
particulier11,12,13,14, par exemple, les effets de la cocaïne sur l’expression du neuroproteome a
été étudié.
Elle offre une meilleure fiabilité que les méthodes quantitatives par comparaison de
gel 2D. Elle est aussi sensible que la coloration à l’argent, elle a l’avantage d’utiliser un
marquage compatible à la MS et de réduire le nombre de gel 2D à effectuer. Grâce aux
Figure IV-2. 2D-DIGE pour une analyse proteomique différentielle de la cristalline. Les échantillons sont marqués par les colorants fluorescents (Cy2,3 et 5) et mélangés pour une iso-electrofocalisation. L’analyse par imagerie de fluorescence est effectuée par DeCyder™ Amersham utilisant comme normalisation le standard interne marqué Cy2. Le spot protéique présentant une différence significative d’expression entre les deux profils sera analysé automatiquement par MALDI-TOF-MS. Dans un premier temps une quantification relative sera établi grâce à l’intensité des peptides marqués. L’identification de la protéine sera faite en MS/MS. La séquence peptidique sera comparée par Mass finger printing (MASCOT) avec des bases de données protéiques. Une analyse par MALDI-TOF-TOF de l’ion m/z 1667.8 est utile pour la vérification de la protéine et nous renseigne sur la nature et le site de modification post–traductionnelle associée aux différents échantillons.
standard interne (Cy2), les protéines n’étant pas présentes ou très faiblement représentées
(1ng) dans un des deux échantillons, peuvent être quantifiées15.
La procédure expérimentale prévoit un marquage minimal d’une à deux lysines par
protéine évitant de ce fait l’ajout de masse excessif de fluorophores et la précipitation des
protéines due à l’hydrophobicité des échantillons. L’efficacité du marquage sur les lysines à
permis de quantifier aussi les protéines faiblement représentées et d’éviter les problèmes de
diffusion des spots dans le gel16. Et enfin, des milliers de protéines peuvent être analysés
simultanément, ce qui est un avantage majeur.
Cependant, l’inconvénient de cette technique est qu’elle requière des colorants très
coûteux et un équipement spécifique : des scanners de fluorescences cyanines, un système
robotisé d’excision de précision couplé à un logiciel spécifique breveté lui aussi.
IV.2. Marquage isotopique in vitro : Isotope-Coded Affinity Tag
(ICAT)
Le marquage isotopique par l’ICAT est une méthode classique d’analyse quantitative de
mélanges complexes de protéines in vitro. Les protéines ou les peptides sont d’abord marqués
et sont ensuite quantifiés en MALDI-TOF. Le réactif ICAT se greffe sur les groupements
thiols des résidus cystéines, c’est une alkylation par la fonction iodoacétamide. Ce réactif est
constitué de trois parties (Figure IV-3) i) l’iodoacétamide : se lie aux SH- des cystéines
ii ) le groupement Ethylène glycol qui existe sous deux formes stables
(forme lourde : 9x13C et la forme légère : 9 x 12C)
iii) la biotine permet d’isoler les peptides marqués de part son affinité
à la streptavidine
Figure IV-3 : Structure du réactif ICAT 13C constitué de trois parties : le groupement réactif iodoacétamide : se lie aux SH- des cystéines, le groupement Ethylène glycol qui existe sous deux formes stables (forme lourde : 9x13C et la forme légère : 9 x 12C) et le tag biotine qui permet d’isoler les peptides marqués de part son affinité à la streptavidine
Le réactif ICAT est greffé sur les résidus Cystéines des protéines de deux échantillons
différents : échantillon 1 (lourd), échantillon 2 (léger) ; après mélange des deux échantillons,
les protéines sont digérées par la trypsine. Afin de simplifier le mélange, les peptides sont
séparés sur colonne d’avidine. Le tag biotine est coupé en milieu acide et les peptides sont
quantifiés par MS et sont identifiés par MS/MS.
Froment C et al17 utilisent l’ICAT pour quantifier les variations des différentes isoformes des
sous-unités du protéasome 20S dans deux types de cellules (cellules d’érythrocytes et cellules
cancéreuses U937) (Figure IV-4).
IV.3. Marquage isotopique in vitro : Isotope Tags for Relative and
Absolute Quantification ( ITRAQ)
Le marquage isotopique iTRAQ permet d’identifier et de quantifier des protéines in vitro dans
quatre échantillons différents par MS/MS. Les peptides sont marqués sur leur groupement
amine par les réactifs iTRAQ qui sont constitués de trois parties (Figure IV-5)
i ) le reporteur chargé avec des masses allant de 114 à 117
ii) la balance neutre avec des masses allant de 28 à 31
iii) le groupement réactif peptidique (PRG).
m/z Figure IV-4: Analyse en MALDI-MS des peptides marqués par le réactif
ICAT issus de la digestion de la sous-unité α du protéasome des érythrocytes
après purification sur colonne d’avidine et clivage du tag biotine. Un zoom
est réalisé pour un des peptides marqués avec un ratio expérimental 13C
/12C de 0.72 alors que le ratio théorique est de 1
La combinaison entre le reporteur et la balance doit faire 145 ; l’identification en MS/MS se
fera grâce au reporteur qui est marqué. Ce reporteur confère à la méthode sa spécificité. Dans
cette méthode, les protéines sont réduites, alkylées et digérées par la trypsine ; après le
marquage de chaque échantillon par un des quatre réactifs iTRAQ, les 4 échantillons sont
mélangés en volume égal. Les peptides sont séparés par chromatographie échangeuse d’ions
puis analysés en MS/MS.
Cong et al.18 ont établi le « profiling » des protéines de cellules fibroblastiques dans 4 états
avancés de sénescence différents par la technique iTRAQ. Après la fragmentation MS/MS,
un ion précurseur est sélectionné à m/z = 979 pour la quantification. Les ions à m/z 114, 115,
116 et 117 sont mesurés, leur abondance relative est proportionnelle à la quantité relative de
chacune des protéines (Figure IV-6). Plus on arrive à un état avancé de sénescence, plus la
protéine est stable. 240 protéines ont été identifiées et quantifiées simultanément par cette
technique.
Abdi et al19 utilisent l’iTRAQ pour détecter des biomarqueurs de pathologies
neurodégénératives telles que la maladie d’Alzheimer, la maladie de Parkinson et la démence
« Lewy body DLB ». Grâce à cette technique, ils ont pu mesurer simultanément les
changements dans le protéome du liquide céphalorachidien obtenu chez trois types de patients
comparé à un individu sain. La confirmation des résultats obtenus par Western blot montre 95
% de spécificité de la méthode.
Figure IV-5 : Structure du réactif ITRAQ constitué de trois parties : •groupement reporter : il diffère entre les quatre formes du réactif utilisé par sa masse (m=114, 115, 116 et 117) •groupement « balance »: sorte de contre poids afin qu’un peptide marqué par les réactifs iTRAQ ait la même masse d’étiquette ; sa masse varie de 28 à 31 m/z. •groupement fonctionnel réactif peptide (NHS ester), amine spécifique, qui lie de manière covalente le réactif iTRAQ isobare aux amines primaires libres du peptide, soit les fonctions amines primaires des chaînes latérales de lysine et le groupe N-Terminal d'un peptide.
IV.4. Stable Isotope Labeling with Amino acids in Cell culture (SILAC)
Cette méthode a été utilisée par Everly P.A. et al.20 afin d’identifier un marqueur
protéique caractéristique du degré de gravité du cancer de la prostate (cellules à fort ou faible
potentiel métastasique). Pour cela, ils vont utiliser des conditions de culture différentes pour
chacune des souches cellulaires ; pour les cellules à faible potentiel métastasique (PC3M) des
conditions normales de culture cellulaire (milieu avec un acide aminé « léger ») vont être
réalisées. Les cellules à fort potentiel métastasique (PC3M-LN4), seront cultivées dans un
milieu de culture contenant un acide aminé « lourd » une Lysine 613C. Le marquage par un
isotope lourd permet une co-élution lors de la séparation chromatographique (Mann M,
200621)
(YR) (RS) (YQ) (H2O2)
Figure IV-6 : Analyse comparative de l’identification et de la quantification de : (1)Fibroblastes
réplicatifs YR tag 114 , (2) Fifroblastes en senescence réplicative RS tag 115 (3) Fibroblastes
quiescents YQ tag 116 (4) Fibroblastes en sénescence provoquée par le stress oxidatif H2O2 tag
117 de cellules de W138 utilisant la vimentine comme exemple.
A = spectre MS/MS correspondant au peptide ETNLDSLPLVDTHS
Les protéines seront digérées par la trypsine pour générer des peptides qui seront nano-ESI-
MS ou nano-ESI-MS2. Ainsi les protéines issues du milieu de culture avec la lysine marquée
auront un incrément de masse de 6 unités de masse atomique (uma) pour chaque lysine
incorporée. Sur le spectre MS, en faisant ensuite le rapport de l’intensité correspondant au pic
d’un peptide marqué au 13C et celle du pic du même peptide non marqué, une quantification
relative est obtenue.
Si le rapport est supérieur à 1, cela signifie que le peptide et donc la protéine sera sur-exprimé
dans les cellules PC3M-LN4 ; si le rapport est inférieur à 1, la protéine correspondante sera
alors surexprimée dans les cellules PC3M.
Figure IV-7 : Schéma général de la méthode SILAC
2 lots de cellules sont testés : PC3M et PC3M-LN4. Les 2 lots
sont mélangés avec un ratio de 1 pour 1 puis séparés par SDS-
PAGE. Afin d’étudier que quelques protéines, le gel est
fragmenté en plusieurs sections qui seront analysées après
excision et digestion par la trypsine en MS pour la quantification
et en MS2 pour l’identification.
Figure IV-8 : Chromatogramme de peptides
après une nanoLC-ESI-MS.
On visualise les fragments qui coéluent. Le
fragment [M+2H]2+ plus lourd (m/z=772.4) est
le standard marqué par une lysine ayant 6 13C et
le peptide à quantifier est non marqué
(m/z=769.4). Le rapport des intensités relatives
entre les 2 fragments permet de retrouver la
concentration relative en protéine d’intérêt dans
le mélange complexe initial. Ici le rapport est
de 0.25, la concentration en protéine dans
l’échantillon est donc 4 fois supérieure à celle
du standard.
IV.5. Stable Isotopic Standards and Capture by Anti-peptides
Antibodies (SISCAPA)
La méthode de quantification de protéines par SISCAPA a été introduite par Anderson
NL, et al, 200422 et est basée sur le même principe que le SILAC. Le marquage du standard
interne est réalisé par assimilation d’un acide aminé modifié dans la protéine. En général les
peptides marqués sont composés de Lysine contenant des 13C. L’assimilation de ces Lysines 13C se déroule pendant l’expression dans un milieu de culture modifié (sans lysine 12C). Ceci
implique que la protéine soit connue du manipulateur pour permettre sa production (Leigh
Anderson and Christie L. Hunter, 200623)
La digestion trypsique du standard interne génère les mêmes peptides que la protéine
non marquée. Ainsi, on observera une co-élution de ces peptides par chromatographie
nanoLC. Le dosage est donc possible sur un spectre MS (ou sur le chromatogramme) en
comparant l’intensité du pic du peptide marqué (dans le spectre ; l’aire du pic pour le
chromatogramme) avec l’intensité du pic du peptide non marqué. Le but de cette méthode est
de quantifier par Single Monitoring Reaction (SRM) les protéines dans les mélanges
complexes (du type plasma sanguin).
Des difficultés de reproductibilité et de sensibilité rendent souvent compliquées ces
analyses. Ainsi, le mélange complexe à étudier sera enrichi avant la digestion trypsique. Pour
cela, on isole du mélange la protéine d’intérêt par colonne d’affinité avec un anticorps
spécifique. Cependant, cette approche est compliquée car il nécessite un anticorps très
spécifique de la protéine à quantifier. C’est pourquoi pour améliorer l’analyse, des déplétions
du mélange seront souvent effectuées pour enlever les protéines abondantes du mélange.
Figure IV-9 : Spectre MS-MS d’un
peptide d’intérêt.
Pour l’ion y7 est choisi comme fragment
spécifique pour une analyse quantitative
du peptide en Multiple Reaction
Monitoring.
Figure IV-10 : Analyse en nanoLC/MS de plasma sanguin.
Effet de la déplétion de l’échantillon par une étape de purification sur colonne d’affinité. Cette étape a été
réalisée avec des anticorps dirigés contre une protéine abondante. En A (chromatogramme), le pic de l’albumine
parasite le signal et masque les autres protéines. En B l’albumine a été enlevée du mélange ce qui nous permet
d’avoir un meilleur rapport signal sur bruit ainsi qu’une reproductibilité accrue.
Une quantification relative est calculée à partir du spectre MS, connaissant la
concentration du standard interne, il est possible de remonter à la concentration de la protéine
d’intérêt (quantification absolue) contrairement à la méthode précédente (SILAC).
Cette méthode nécessite donc une connaissance préalable de notre protéine à étudier afin de
pouvoir l’isoler de mélanges complexes. A ce jour, elle est encore peu utilisée pour la
quantification protéique car elle est longue à mettre en place et chère. Ainsi, des améliorations
doivent être développées afin de généraliser cette technique.
V. Les modifications post-traductionnelles : Etudes des N et O-glycosylation
Les N- et O-glycanes à la surface des cellules sont des récepteurs des lectines qui
modulent les événements d’adhésion et de signalisation associés à la reconnaissance cellule-
cellule. La détermination de leur structure est une étape essentielle pour établir leur rôle dans
certaines maladies. Les stratégies de spectrométrie de masse ont été optimisées pour le
séquençage de mélanges complexes constitués de N et O-glycanes. Le but est d’identifier des
marqueurs de masse de la présence des N et O-glycosylations au niveau des peptides24.
V.1. Les N-glycosylations
Les N-glycosylations des protéines sont des modifications post-traductionnelles très
communes chez les eucaryotes. La technique classique d’étude des liaisons N-glycosydiques
chez les eucaryotes est de digérer les peptides par la peptide-N-glycosidase F (PNGase F), qui
clive la liaison N-glycosidique entre l’asparagine et le N-acétylglucosamine, puis de faire une
perméthylation, et enfin de faire une analyse des molécules obtenues par MALDI-TOF.
Cependant la PNGase F est spécifique des eucaryotes et ne peut pas être utilisée chez les
bactéries car les liaisons peptide-carbohydrate sont différentes et les enzymes capable de
libérer la partie carbohydrate sont rarement utilisables . C’est pourquoi il a été développé une
alternative à la digestion par la PNGase F : la digestion par la Pronase E. Cette enzyme
protéolytique est non spécifique et provient de la bactérie Streptomyces griseus. Elle digère
l’ensemble des liaisons peptidique et peut donc être utilisée pour l’étude des glycoprotéines.
La technique d’étude est de soumettre tous les échantillons à la digestion à la pronase E puis à
une perméthylation des polysaccharides ainsi obtenus et enfin à une analyse en spectrométrie
de masse. La perméthylation permet d’avoir des informations sur la structure des molécules
étudiées et augmente la sensibilité de l’analyse en MS. Si l’on compare les spectres obtenus
après digestion à la PNGase F et à la Pronase E on constate que l’on a une différence de
masse de 111 Da pour un même résidu (Figure V-1)25.
En théorie, après perméthylation, on s’attendait à une différence de 128 Da entre les deux
digestions sur le même résidu. De plus, en MS/MS, on obtient une différence de masse de 143
Da alors qu’on s’attendait à une différence de 160 Da (résidu asparagine). Cela signifie que le
traitement à la pronase E induit un réarrangement au niveau du résidu asparagine et il a été
démontré que la différence de 17 Da correspond à la perte ou au gain d’ammoniaque (NH3) du
FigureV-1
résidu asparagine. Le gain de 111 Da en MALDI-TOF ou la perte de 143 Da en MS/MS,
peuvent donc être utilisés comme marqueurs pour la présence de N-glycosylation (Figure V-
2)26.
V.2. Les O-glycosylations
Les O-glycosylations sont des modifications post-traductionnelles ubiquitaires. Elles
sont présentes au niveau des résidus tyrosine et sérine.
La méthode d’étude des sites de glycosylation consiste à séparer les différents sucres des
protéines par électrophorèse bidimensionnelle (2-DE) puis à réaliser une β-élimination
alcaline des sucres, en rajoutant un nucléophile (ammoniaque, NH4OH). La différence de
masse permet alors de détecter les glycoprotéines par MS et fournit ainsi un tag stable qui
En comparant des spectres avant et après traitement à l’ammoniaque, on observe une
différence de masse de 1Da due à la substitution d’un hydroxyle par un groupe amine
(Figure V-3).
OH- Ammoniaque
Figure V-2. (a) Traitement à la PNGase F et à la pronase E et différence de masse attendue. (b) réarrangement de l’asparagine après traitement à la pronase E.
(a) (b)
Une autre approche est d’utiliser du 2-aminoethanethiol (AET) à la place de
l’ammoniaque. Ceci va donc générer une β-méthylaminoéthylcystéine à la place de la
thréonine et on observera donc au niveau du spectre une différence de masse de 59 Da
correspondant au résidu thréonine (Figue V-4)28.
Ainsi, en fonction des études que l’on souhaite réaliser, c'est-à-dire la détermination de
N ou O-glycosylation sur les glycoprotéines, il existe différentes méthodes29,30 (digestion à la
pronase E, pérmethylation, β-élimination, etc…). Ces méthodes ont le même but : obtenir un
marqueur de masse caractéristique des N ou O-glycosylations et qui reste stable durant les
analyses de spectrométrie de masse.
On a pu retrouver ces techniques dans de nombreuses études, notamment :
- l’étude des O-glycosylations de la caséine bovine31,
- une étude des N-glycosylations des glycoprotéines32,
- l’étude des N-glycosylation de l’aminopeptidase N de Manduca Sexta33,
- l’étude des N-glycosylations de l’α-mannosidase lysosomale bovine34.
Figure V-4. The insets highlight the regions of the spectrum containing the y5 and b8 ions which localize the glycosylation site to T152. X designates the modified threonine residue.
( M )2+
( M)2+
-59Da-59Da
Figure V-3 Identification of glycosylation sites by β-elimination with NH4OH. (A) MS of the peptic digest of nonglycosylated k-casein from spot 1 after treatment with NH4OH showing the unaltered doubly charged ion at m/z = 445.8 derived from the peptide 159AVESTVATL167. (B) MS of the peptic digest of diglycosylated k-casein from spot 3 after treatment with NH4OH showing the shift in m/z to 445.3 caused by b-elimination of the glycan and addition of ammonia. X designates the modified threonine residue.
VI. Détection de modifications post–traductionnelles : les phosphorylations
Dans les systèmes biologiques, de nombreuses protéines présentent de multiples sites
de phosphorylation. Le profil de phosphorylation des protéines peut changer en réponse à un
stimulus ou une activité cellulaire donnée. L’identification des sites de phosphorylation, qui
peuvent se trouver sur une Sérine, une Thréonine ou une Tyrosine au niveau de la séquence
primaire d’une protéine permettrait de déterminer leur signification fonctionnelle dans un
processus donné, ou en réponse à un signal intra- ou extra-cellulaire.
La spectrométrie de masse est indispensable pour étudier ces sites de phosphorylation.
La méthode consiste à calculer l’écart du rapport m/z entre deux ions correspondant au même
peptide sur un spectre MS puisqu’une phosphorylation correspond à une augmentation du
rapport m/z de 80. Cependant la détermination du site exact de phosphorylation, est parfois
difficile en MS/MS35.
En 2006, l’équipe de Sara Rinalducci et.al. a publié une étude sur la phosphorylation
de la protéine CP43 du PhotoSystème II de la feuille d’épinard, qui constitue un bon exemple
de détermination d’un site de phosphorylation.
Dans la figure VI-1 on présente le spectre ESI-Q-TOF MS/MS d’un peptide issus de la
digestion trypsique de CP43.L’ion b3 à m/z 366 (encadré en vert) correspond à la somme des
masses des résidus des acides aminés Sérine, Proline et Thréonine plus la masse d’un
groupement phosphate et celle d’un H+. On trouve également l’ion b3 phosphorylé ayant
perdu un acide phosphorique (H3PO4) à m/z 268 (encadré en rouge) et l’ion b3 phosphorylé
ayant perdu un acide phosphorique et une molécule d’eau à m/z 250. D’autre part, l’ion b2 à
m/z 185.08 (encadré en bleu) correspond à la somme des résidus sérine, proline et d’un H+.
L’ion b3 est phosphorylé mais l’ion b2 ne l’est pas donc la phosphorylation porte sur la
thréonine 3. Les auteurs ont ainsi put déterminer que ce peptide correspondant à la protéine
CP43 est phosphorylé sur la thréonine 3.
b3p m/z = 87+ 101+97+80+1 m/z = 366
b2 : m/z = 185.08 = 97+87+1
b3 m/z =87+101+97+80+1-98 m/z = 268
Figure VI-1 : spectre ESI-Q-TOF MS/MS du peptide dont la séquence est présenté sur la figure.
Par contre, dans certains cas en MS/MS, cette méthode ne permet pas de déterminer le
site exact de phosphorylation. En effet, la perte d’un acide phosphorique sur un peptide
phosphorylé correspond à perte de 18 unités de masse (+80-98). Ceci peut également
correspondre à la perte d’une molécule d’eau. Comment savoir alors à quoi correspond cette
diminution de 18 du rapport m/z ? La figure VI-2, correspondant au spectre MALDI MS/MS
du peptide N-terminal de la protéine CP43, illustre bien ce problème.
L’ion b3 à m/z de 255 (encadré en bleu) correspond à la somme des masses des trois
premiers résidus plus un H+ avec une diminution du m/z de 18. Cependant, en l’absence d’un
pic correspondant au m/z 353 (=273+80), la diminution du rapport m/z de 18 correspond soit
à la perte d’un acide phosphorique soit à la perte d’une molécule d’eau. On retrouve le même
problème avec l’ion b5, donc on ne peut pas savoir si ce peptide est phosphorylé sur la
thréonine 3 ou 5.
Cette méthode a également été utilisée dans les études de Surti et al.36 et de Borchers et al.37.
K. Chien et al.38 ont utilisé une méthode : la β-élimination et addition de Michaël pour
résoudre les problèmes d’identification des sites de phosphorylation.
b30 m/z = 114+57+101+1-18 m/z = 255
b50 : m/z = 486+1-18 m/z = 469
b3 m/z = 114+57+101+1 m/z = 273.12
b5 m/z = 486+1-18 m/z = 469
Figure VI-2 : spectre MALDI MS/MS du peptide dont la séquence est présenté sur la figure.
-98 R-CH2-S-CH2-CH2-NH2 R-CH2-O-H2PO3 R=CH2
NaOH NH2(CH2)2-SH
+77
La figure VI-3 illustre l’intérêt de cette méthode, par un exemple d’application sur un
phosphopeptide présentant deux sites de phosphorylation possibles. On peut voir dans le cadre
A, présentant le spectre MS/MS du peptide non phosphorylé, qu’on a une différence de m/z
de 87 correspondant au résidu sérine.
Dans le cadre B, les peptides sont soumis à la β-élimination et seuls les ions
phosphorylés présentent une différence de masse 69 (=87+80-98) car il y a perte d’acide
phosphorique. Dans le cadre C, les peptides sont soumis à l’addition de Michaël et seuls les
ions qui étaient phosphorylés ont subit la β-élimination et présentent une différence de masse
de 146 (=69+77).
Ainsi cette méthode permet de différencier la perte d’eau de la perte d’un acide
phosphorique puisque l’addition de Michaël se produit sur les résidus qui étaient
phosphorylés et qui ont subit la β-élimination. De plus, cette méthode permet de déterminer le
site exact de phosphorylation. Dans l’exemple suivant, la sérine 2 est phosphorylée tandis que
la sérine 6 ne l’est pas.
69
146
87 87
87
87
β-élimination
addition de Michaël
Figure VI-3 : spectre MALDI-TOF MS/MS du peptide dont la séquence est présentée sur la figure. A : peptide non phosphorylé ; B : peptide phosphorylé soumis à la β-élimination ; C : peptide ayant subis la β-élimination soumis à l’addition de Michaël.
VII. Les modifications post-traductionnelles : acétylation et méthylation
Le principe de la localisation des acétylations et méthylations repose sur l’analyse
d’ions issus de la fragmentation de peptides en ESI/MS avec un analyseur à haute résolution.
L’analyse des ions observés sur les spectres par des banques de données comme SwissProt
permet l’identification des modifications. Un incrément de masse de 14 Da est révélateur
d’une méthylation (CH3) sur les résidus Arg et Lys ; + 42 Da indique une acétylation
(CH3CO) à la place de l’hydrogène de la chaîne latérale de la Lys et de l’Arg.
Jiang et al.39ont mis au point une stratégie permettant d’identifier acétylations et
phosphorylations au sein de protéines HMGA1 (high-mobility group A1), appartenant à une
famille de facteurs de transcription. Les auteurs ont traité des cellules cancéreuses humaines
PC-3 avec ou sans inhibiteur des déacétylases, puis ils ont extrait les protéines d’intérêt par
chromatographie en phase inverse (colonne C4). Après digestion trypsique, les peptides sont
analysés en LC/ESI-MS.
Grâce à l’incrément de masse des peptides acétylés (+ 42 Da) et phosphorylés (+ 80
Da) et à la MS/MS, les auteurs ont pu déterminer la position de ces modifications. (Figure
VII-1).
Figure VII-1 : Acetylation of Lys14 in HMGA1a peptide SSQPALSKAcQEK (residues 7-17). Shown is the product ion spectrum of the [M + 2H]2+ ion (m/z 622.9) of the tryptic peptide obtained from the major HMGA1a fraction isolated from sodium butyrate-treated PC-3 cells. The y and b ions are calculated on the basis of the Lys14-acetylated peptide segment. To better view low-abundance fragment ions, some spectral regions are amplified by 2- and 5-fold as indicated by x2 and x5, respectively.
170
Les auteurs ont observé une différence de masse entre l’ion y4 et l’ion y3 et entre l’ion
b7 et b8 de 170 Da ce qui est révélateur d’une acétylation sur une lysine. Un incrément de
masse identique est observé (+42 Da) lorsque le peptide est acétylé ou triméthylé.
La différence de masse observée entre une acétylation et une triméthylation est très
faible puisque leurs incréments de masse sont respectivement 42.0105 Da (C2H2O)et 42.0468
Da (C3H6). Cette différence de 36 ppm ((42.0468-42.0105)/42) ne peut pas être mesurable
avec des analyseurs conventionnels. On peut alors utiliser le MALDI-Réflectron-TOF qui
comporte au niveau de l’analyseur un réflectron permettant de corriger le temps de vol d’ions
de même m/z mais de vitesses différentes comme le montre la figue VII-2.
Spectromètre de masse à analyseur TOF sans réflectron :
Spectromètre de masse à analyseur TOF avec réflectron :
Figure VII-2 : Représentation schématique du rôle d’un réflectron dans un analyseur de spectromètre
de masse
On a une meilleure focalisation en énergie cinétique et donc une meilleure résolution au
niveau des pics observés. La mesure se fait avec une exactitude de 5 ppm ce qui permet de
différencier un résidu acétylé d’un résidu triméthylé.
m/zm/z
Mauvaise résolution
+3000 V
-3000 V
Dét
ecte
ur
Source -3000 V Réflectron +130 VL1
L2
D
d
+3000 V
-3000 V
Dét
ecte
ur
Source -3000 V Réflectron +130 VL1
L2
D
d m/z
Meilleure résolution
m/zm/z
Meilleure résolution
Source Tube de vol
Détecteur
20 kV 0 kV -20 kV
Ions de m/z identique mais v >v
Source Tube de vol
Détecteur
20 kV 0 kV -20 kV
Ions de m/z identique mais v >v
Par cette méthode, Zhang et al.40 en 2004 ont pu caractériser le groupement porté par les
lysines des peptides obtenus après digestion trypsique des histones H3 de poulet. En effet, en
attribuant une acétylation à une lysine du peptide étudié, ils obtiennent une différence entre la
masse mesurée et la masse calculée de 35 ppm alors que l’exactitude de l’appareil est de 5
ppm. Ceci leur permet de déterminer que la lysine étudiée porte une triméthylation (exactitude
de la mesure de 1,5 ppm).
Par cette technique, on peut même déterminer les sites de modification d’un peptide
triméthylé et acétylé. En effet, en 2004, l’équipe de Zhang a synthétisé deux peptides ayant la
même masse monoisotopique et se différenciant uniquement par la position des groupements
acétyl et triméthyl portés par les lysines du peptide. Les spectres ESI-MS/MS de ces deux
peptides sont représentés sur la figure VII-3.
Une autre technique utilisée pour distinguer l’acétylation
d’une triméthylation est l’analyse, par ESI-CID-MS/MS, des ions
immonium formés après réarrangement des molécules ionisées.
Ainsi une lysine acétylée se réarrangera pour former les ions
m/z=84 et m/z=126 alors qu’une lysine triméthylée produira
seulement un ion de m/z= 84 comme le montrent les réactions ci-
contre. De plus, sur le spectre ESI-MS/MS d’un peptide comportant
une lysine triméthylée, on aura présence de l’ion (MH+-59),
caractéristique d’une perte de N(CH3)3 par l’élimination
d’Hofmann :
m=59
NH
CH
CH2
CH2
CH2
NCH3CH3
H3C+
C HH
NH
CH
CH2
CH2
CH2
CHH+
N(CH3)3
Réarrangementélimination d’Hofmann
NH
CH CH2
CH2CH2
CH2
H+
+
m/z = 143 m/z = 84m=59m=59
NH
CH
CH2
CH2
CH2
NCH3CH3
H3C++
C HH
NH
CH
CH2
CH2
CH2
CH
NH
CH
CH2
CH2
CH2
CHH+
N(CH3)3
Réarrangementélimination d’Hofmann
NH
CH CH2
CH2CH2
CH2
NH
CH CH2
CH2CH2
CH2
H+
+
m/z = 143 m/z = 84
Figure VII-3 : Spectres ESI-MS/MS de deux peptides ayant la même masse monoisotopique et ne différant
que par la position des lysines triméthylées et acétylées dans ces peptides.
Sur le spectre du peptide A caractérisé par la présence d’une lysine acétylée en N-
terminal, on peut noter la présence de l’ion m/z=126 et voir de nombreux pics correspondant
aux ions y-59 caractéristiques de la perte du groupement triméthyle à partir de l’ion y4 jusqu’à
l’ion y8. A l’inverse, on observe un ion b2-59 sur le spectre du peptide B montrant une perte
de N(CH3)3 par la lysine triméthylée en N-terminal. Par ces différentes spécificités, les auteurs
ont réussi à déterminer si une lysine triméthylée était positionnée en N-terminal (ions b-59) ou
en C-terminal (ions y-59). L’analyse ESI-MS/MS est donc un outil très puissant pour la
détermination des sites de modification. Cette méthode est notamment utilisée pour la
caractérisation des acétylations et méthylations des histones régulant l’expression génique.
Spectre ESI-MS/MS de l’ion parent [M+H]+ dont m/z=985.56
Kac S T G G Kme3A P Ry1y6 y5 y4 y3 y2y7y8
b2 b8
Kac S T G G Kme3A P Ry1y6 y5 y4 y3 y2y7y8
b2 b8
y1y6 y5 y4 y3 y2y7y8 y1y6 y5 y4 y3 y2y7y8
b2 b8
• Peptide A :
Spectre ESI-MS/MS de l’ion parent [M+H]+ dont m/z=985.56
Kme3 S T G G KacA P Ry1y6 y5 y4 y3 y2y7y8
b2
• Peptide B :
Spectre ESI-MS/MS de l’ion parent [M+H]+ dont m/z=985.56
Kme3 S T G G KacA P Ry1y6 y5 y4 y3 y2y7y8
b2
Kme3 S T G G KacA P Ry1y6 y5 y4 y3 y2y7y8
b2
y1y6 y5 y4 y3 y2y7y8 y1y6 y5 y4 y3 y2y7y8
b2
• Peptide B :
VIII. Les applications médicales de la spectrométrie de masse : SELDI-TOF et Imagerie
VIII.1. Le SELDI-TOF : Surface Enhanced Laser Desorption Ionisation
Time of Flight
La difficulté d’appliquer la spectrométrie de masse à l’étude de pathologies réside
essentiellement dans la complexité des échantillons à analyser (prélèvements
bronchoalvéolaires, sanguins, extraits cytosoliques de tumeurs…). En effet, ces échantillons
comportent une multitude de protéines qui rend difficile la détection des peptides et protéines
sous-représentés (minoritaires) ou de faible poids moléculaire.
La technique du SELDI-TOF repose sur le même principe que le MALDI-TOF (Figure VIII-
1) à une étape près qui permet justement de s’affranchir de ce problème : la séparation des
protéines selon leurs propriétés chimiques ou biologiques. Les protéines d'un échantillon sont
retenues de façon sélective sur différentes surfaces chimiques (hydrophobe (H4, H50) ;
ions métalliques chélatés (IMAC)) ou biologiques (anticorps, récepteur, ligand, acides
nucléiques…).
Figure VIII-1 : Système ProteinChip LDI-Qq-TOF-MS (SELDI-TOF) 41
Cette fixation sélective combinée à une modulation de la stringence des lavages
permet d’éliminer les protéines non fixées ou fixées de façon non spécifique et donne ainsi la
possibilité de détecter et d’analyser des protéines minoritaires ou plus petites, masquées par
des protéines plus grosses et plus abondantes. Le SELDI-TOF présente l’avantage de pouvoir
détecter des protéines de masse moléculaire inférieure à 20 kDa généralement sous-
représentées ou absentes avec la technique d’électrophorèse bidimensionnelle42.
Cette technique est utilisée pour comparer les profils protéiques («étude de profiling »)
d’échantillons susceptibles de présenter des différences (sain / malade ou traité) pour mettre
en évidence des biomarqueurs spécifiques. Les pics discriminants susceptibles de constituer
des biomarqueurs potentiels sont soumis à une analyse statistique qui permet de vérifier
l’indépendance et la significativité des pics identifiés43. D’autre part, via les couplages
covalents avec les surfaces biologiques, il est possible d’analyser les variations de quantité
d’une protéine dans différentes conditions, les variations de masse suite à des modifications
post-traductionnelles et mettre en évidence des interactions protéine/protéine.
Cette approche protéomique a par exemple, permis d’identifier deux biomarqueurs
permettant d’établir un pronostic simple dans le cancer du sein : la chaîne légère de la ferritine
et l’ubiquitine respectivement détectées avec les surfaces SAX2 et IMAC 30 Cu44. De la
même manière, des marqueurs potentiels de l’irritation des poumons par la fumée de cigarette
ont pu être mis en évidence43.
VIII.2. L’imagerie par spectrométrie de masse (Imaging Mass
Spectrometry)
Actuellement, il est possible de travailler directement sur de fines sections de tissus
pour faire de l’imagerie cellulaire en donnant une carte de la répartition protéique (Figure
VIII-2).
Figure VIII-2 : La technique de MALDI-TOF appliquée
directement sur une section tissulaire, deux modes
d’acquisition des données :
- « profiling » (à gauche) : la matrice est déposée
manuellement en différents points de la section de tissu, on
obtient alors des spectre de masse ;
- « imaging » (à droite) : la matrice peut être déposée
automatiquement avec un spotter, on obtient dans ce cas des
images 44 .
Le principe consiste à appliquer la technique de MALDI-TOF directement sur une fine
section de tissu puis deux modes d’acquisition des données sont possibles. On peut obtenir
des spectres de masse représentant l’abondance des ions des protéines (intensité des pics) liée
à l’abondance relative des protéines présentes en différents points d’une fine section de
tissu (« profiling »).
D’autre part, on peut obtenir une image représentant pour chaque ion (m/z donné) à la
fois son abondance mais également sa distribution spatiale sur l’ensemble de la section de
tissu (« imaging »). Ce nouvel outil analytique a été introduit en 1997 par Caprioli et son
équipe45.
L’avantage de l’imagerie est l’accès à des informations quant à l’expression des
protéines même dans des régions tissulaires très petites.
Ce type de technique permet d’identifier des biomarqueurs en comparant des tissus
malades avec des tissus sains mais aussi de suivre des processus biologiques en comparant
des tissus sains avec des tissus traités46. Par exemple, un biomarqueur de la néphrotoxicité de
la gentamicine chez le rat, la transthyrétine, a pu être mis en évidence au niveau du cortex
rénal46.
Dans ces 2 types de techniques (SELDI-TOF et Imaging Mass spectrometry),
l’identification des protéines d’intérêt est possible en réduisant la complexité des échantillons.
En effet, les échantillons protéiques, issus de microextractions de régions tissulaires (Imaging
Mass Spectrometry) ou de prélèvements biologiques, peuvent être fractionnés par HPLC,
nanoLC avant de séparer les protéines sur gel d’électrophorèse. Une digestion protéolytique
(trypsique le plus souvent) est ensuite réalisée en vue d’une analyse par spectrométrie de
masse en tandem des peptides de digestion. La dernière étape qui va permettre de déterminer
les protéines d’intérêt consiste à confronter avec une banque de données, les séquences des
peptides précédemment obtenues.
Conclusion :
Cette étude nous a permis de parcourir les différentes utilisation de la spectrométrie de
masse associée aux modes de séparations tels les chromatographies et l’électrophorèse, pour
l’analyse des protéines. De plus en plus les méthodes utilisées sont standardisées et rendent
cette technique utilisable par le plus grand nombre. Dans le domaine des modifications post
traductionnelles beaucoup de progrès ont été réalisés quoique les méthode nécessitent encore
toute la dextérité des préparateurs d’échantillons. De nouveaux domaine d’utilisation de la
MS sur les protéines émergent petit à petit, tel l’imagerie cellulaire qui permet de localiser la
distribution des protéines dans un tissu.
Bibliographie 1 Danos, O., Svinartchouk, F. ; 2006 ; Electrohoresis ; 27 ; 3475-3479. 2 Pepaj, M., Wilson, S.R, Novotna, K., Lundanes, E. ; 2006 ; Journal of chromatography A ; 1120 ; 132-141. 3 Vanrobaeys, F., Van Coster, R., Dhondt, G., Devreese, B., Van Beeumen, J. ; 2005 ; Journal of Proteome Research ; 4 ; 2283-2293. 4 Linke, T., Catharine Ross, A., Harrison E.H. ; 2006 ; Journal of Chromatography A ; 1123 ; 160-169. 5 Balbina, A., Barbosa, J., Sanz-Nebot, V. ; 2006 ; Electrophoresis ; 27 ; 3661-3670. 6 Cooper H.J., Kansson K.H., Marshall A.G. ; 2005 ; Mass Spectrom. Rev. ; 24 ; 201-222. 7 Xia Y., Chrisman P.A., Erickson D.E., Liu J., Liang X., Londry F.A., Yang M.J., McLuckey S.A. ; 2006 ; Anal. Chem. ; 78 ; 4146-4154. 8 Chrisman P.A., Pitteri S.J., McLuckey S.A. ; 2005 ; Anal. Chem. ; 77 ; 3411-3414. 9 Mikesh L.M., Ueberheide B., Chi A., Coon J.J., Syka J.E.P., Shabanowitz J., Hunt D.F. ; 2006 ; Biochim. Biophys. Acta.
10 Unlu M., Morgan M.E., J.S. Minden ; 1997 ; Electrophoresis ; 18. 11 Prabakaran, S., Swatton, J.E., Ryan, M.M., Huffaker, S.J., Huang, J.T., Griffin, J.L., Wayland, M., Freeman, T., Dudbridge, F., Lilley, K.S., Karp, N.A., Hester, S., Tkachev, D., Mimmack, M.L., Yolken, R.H., Webster, M.J., Torrey, E.F. and Bahn, S. ; 2004. 12 Swatton J.E., Prabakaran S., Karp N.A., Lilley K.S., Bahn S. ; 2004 ; Mol. Psychiatry ; 9. 13 Beckner M.E., Chen X., An J., Day B.W., Pollack I.F. ; 2005 ; Lab. Invest. ; 85. 14 Sitek B., Apostolov O., Stuhler K., Pfeiffer K., Meyer H.E., Eggert A., Schramm A. ; 2005 ; Mol. Cell Proteom. ; 4. 15 Shaw J., Rowlinson R., Nickson J., Stone T., Sweet A., Williams K. and Tonge R.; 2003; Proteomics. 16 Tannu, N.S., Scott E.H.; 2006; Electrophoresis. 17 Froment C., Uttenweiler-Joseph S., Bousquet-Dubouch M-P., Matondo M., Borges J-P., Esmenjaud C., Lacroix C., Monsarrat B. and Burlet-Schiltz O.; 2005; Proteomics, 5, 2351–2363. 18 Cong Y., Fan E., Wang E.; 2006; Mechanisms of Ageing and Development 127 332–343. 19 Abdi F., Quinn J. F., Jankovic J., McIntosh M., Leverenz J.B., Peskind E., Nixon R., Nutt J., Chung K., Zabetian C., Samii A., Lin M., Hattan S., Pan C., Wang Y., Jin J., Zhu D., Li G. J., Liu Y., Waichunas D., Montine T.J., Zhang J.; 2006 ; Journal of Alzheimer's Disease 9 293 – 348.
21 Mann M. ; 2006 ; Nat. Rev. Mol. Cell. Biol. ; 7(12) ; 952-958. 22 Anderson N.L., Anderson N.G., Haines L.R., Hardie D.B., Olafson R.W. and Pearson T.W. ; 2004 ; J. Proteome Res. ; 3(2) ; 235-44. 23 Anderson L. and Hunter C.L. ; 2006 ; Mol. Cell. Proteomics ; 5(4) ; 573-587.
24 Haslam S.M., North S. J ; 2004 ; Current opinion in Structural Biology. 16 : 584-591. 25 Liu X., McNally D. J., Nothaft H., Szymanski C. M., Brisson J.R., Li J.; 2006 ; Anal. Chem. 78 : 6081-6087. 26 Liu X., McNally D. J., Nothaft H., Szymanski C. M., Brisson J.R., Li J. ; 2006 ; Anal. Chem. 78 : 6081-6087. 27 Holland J. W., Deeth H. C., Alewood P. F.; 2005 ; Proteomics 5 : 990-1002. 28 Holland J. W., Deeth H. C. Alewood P. F. ; 2005 ; Proteomics 5 : 990-1002. 29 Lattova E., Kapkova P., Krokhin O., Perreault H. ;2006 ; Anal. Chem.78 : 2977-2984. 30 Peavy A. HJ, Hedrick J, Lebrilla L ; 2003 ; Anal Chem. 75 :5628-5637. 31 Holland J. W., Deeth H. C. , Alewood P. F. ; 2005 ; Proteomics 5 : 990-1002. 32Peavy TR, Hedrick JL, Lebrilla ; 2003 ; Anal Chem. 75 :5628-5637. 33 Stephens E., Sugars J., Maslen S. L., Williams D. H., Packman L. C. and Ellar D. J. ; 2004 ; Biochem. 271: 4241–4258. 34 Faid V., Evjen G., Tollersru O., Michalski J., Morelle W.; 2006 ; 5:440-461. 35 Rinalducci S., Larsen M. R., Mohammed S. Zolla L.; 2006; J. Proteome Res.; vol. 5; 973-982. 36 Surti T. S., Huang L., Jan Y. N., Jan L. Y., Cooper E. C.; 2005; PNAS ; vol. 102 ; 17828–17833. 37 Borchers C. H., Thapar R., Petrotchenko E. V., Torres M. P., Speir J. P., Easterling M., Dominski Z., Marzluff W. F.; 2006; PNAS; vol. 103; 3094–3099. 38 Chien K-Y., Chang Y-S., Yu J-S, Fan L-W., Lee C-W., Chi L-M.; 2006; Biochem. Biophys. Res. Commun.; vol.348; 47-55. 39 Jiang X., Wang Y., Biochemistry 2006, 45, 7194-7201 40 Zhang K., Yau P., Chandrasekhar B., New R., Kondrat R., Imai B., Bradbury M., Proteomics ; 2004 ; 4, 1-10 41 Reid G., Siang Gan B., She Y.M., Ens W., Weinberger S., Howard J. C. ; 2002 ; Appl Environ Microbiol. ; 68 ; 977–980. 42 Merkel, D., Rist W., Seither P., Weith A., Lenter M. C.; 2005; Proteomics, 5 ; 2972–2980. 43 Ricolleau G., Charbonnel C., Lodé L., Loussouarn D., Joalland M-P., Bogumil R., Jourdain S., Minvielle S., Campone M., Déporte-Fety R., Campion L., Jézéquel P. ; 2006 ; Proteomics ;, 6 ; 1963–1975. 44 Chaurand P., Schwartz S. A., Reyzer M. L., Caprioli R. M.; 2005 ;Toxicol Pathol; 33 ; 92–10. 45 Hsieh Y., Chen J., Korfmacher W. A.; 2006; J Pharmacol Toxicol Meth. 46 Meistermann H., Norris J. L., Aerni H.-R., Cornett D. S., Friedlein A., Erskine A. R., Augustin A., De Vera Mudry M. C., Ruepp S., Suter L., Langen H., Caprioli R. M., Ducret A.; 2006 ; Mol Cell Proteomics; 5 ; 1876–1886.