Rhodium-Catalyzed Intra- and Intermolecular Alkene Hydroacylation Inaugural-Dissertation Zur Erlangung des Doktorgrades vorgelegt von Damien Régent aus Mulhouse (Frankreich) Fakultät für Chemie und Pharmazie Albert-Ludwigs-Universität Freiburg im- Breisgau 2013
Transcript
Rhodium-Catalyzed Intra- and Intermolecular
Alkene Hydroacylation
Inaugural-Dissertation
Zur Erlangung des Doktorgrades
vorgelegt von
Damien Régent
aus Mulhouse (Frankreich)
Fakultät für Chemie und Pharmazie
Albert-Ludwigs-Universität
Freiburg im- Breisgau
2013
Vorsitzender des Promotionausschusses: Prof. Dr. R. Schubert
Referent: Prof. Dr. B. Breit
Korreferent: Prof. Dr. W. Bannwarth
Tag der Promotion: 3. Dezember 2013
Fakultät für Chemie, Pharmazie und Geowissenschaften
Albert-Ludwigs-Universität, Freiburg im Breisgau
AK B. BREIT
Rhodium-Catalyzed Inter- and Intramolecular
Alkene Hydroacylation
Damien D. Régent
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
Rhodium-Catalyzed Intra- and Intermolecular Alkene Hydroacylation
Inaugural-Dissertation
Zur Erlangung des Doktorgrades
vorgelegt von
Damien Régent
aus Mulhouse (Frankreich)
Fakultät für Chemie und Pharmazie
Albert-Ludwigs-Universität
Freiburg im- Breisgau
2013
Vorsitzender des Promotionausschusses: Prof. Dr. T. Koslowski Referent: Prof. Dr. B. Breit Korreferent: Prof. Dr. W. Bannwarth
”Dans la vie, rien n’est à craindre, tout est à comprendre”
Marie Sklodowska Curie, 1867-1935 À mes parents, ma femme Karine et mes soeurs Céline et Charlène À mon grand-père, qui aurait été fier de moi
Parts of this Ph.D. Thesis were published in the following publications: N. R. Vautravers, D. D. Regent, B. Breit: Inter- and Intramolecular Hydroacylation of Alkenes emplying a Bifunctional Catalyst System
Chem. Comm. 2011, 47, 6635-6637
Acknowledgement The work on this Ph.D. thesis took place in the Institut für Organische Chemie und Biochemie at the Albert-Ludwigs-Universität between October 2009 and June 2013 under the guidance of Prof. Dr. Bernhard Breit. I would like to thank him for the opportunity to join his research group and to work on an interesting research topic. I would like to thank also Prof. Dr. Willi Bannwarth to give me his opinion on my Ph.D. manuscript, and for his presence as my second referent professor. My special thank for Prof. Dr. Bernhard Breit, Dr. Jordan Page, who took their time to read my manuscript and made plenty of suggestion and correction. For the helpful discussion and all the analyses received in time, I thank the entire analytic department: Dr. Keller, Ms. Schonhardt and Mr. Reinbold (NMR), Dr. Wörth and Mr. Warth (MS), and Mr. Tonnies (CHN). My thanks belong to all that have contributed somehow to this work. First of all to Dr. Nicolas Vautravers, who developed the concept of our bifunctional catalyst system, and gave me day after day with the smile good advices for the application of this catalytic system to the rhodium-catalyzed intra- and intermolecular alkenes hydroacylation. It was a great pleasure to work with him, I learned a lot at his side about chemistry and humanly. I am also very thankful to my research trainees: Miss Fanny Cacheux (ENSCMu, France), Mr. Fabio Lima (ENSIACET, France), Mr. Jonas Ohms and Mr. Achim Link (Albert-Ludwigs-Universität) for their enthusiastic work. I cannot forget also to thank Mr. Günter Leonhardt-Lutterbeck for his “doigts de fée” and his massive contribution to the ligand synthesis. Work in an international group, with colleagues from all over the word, was for me a very enlightening experience. So I would like to give thanks to the French team for the good shared moments: Dr. Nicolas Vautravers, Dr. Lisa Diab, Mr. Thomas Gilles, Dr. Wilfried Raimondi and last but not least Dr. Nacim Abermil. Fēi cháng gàn xìe to my Chinese colleagues: Mr. Xu Kun, Dr. Li ChangKun and Ms. Ruan Qiao for their daily careful presence and all our pleasing discussions. A big thank to our secretary Ms. Anita Weidner for her behind-the-scenes work, and her communicative kindness. A big thank-you to all my other colleagues, and specially my sub-laboratory colleagues for the warm and pleasant working atmosphere: Dr. Sonja Diezel, Mr. Nils Thieme, Mr. Daniel Wüstmann, Mr. Adrian Pritzius, Mr. Alex Haydl, Dr. Mario Stein, Dr Daniela Fuchs, Mr. Gerhard Weitzel… Finally, I wish to express my deepest gratitude to my family: mes remerciements les plus grands sont dédiés à ma famille et mes amis, en particulier à mes parents, mes soeurs Céline et Charlène, et ma femme Karine pour leur constant amour et soutien. “Soyons reconnaissants aux personnes qui nous donnent du bonheur, elles sont les charmants jardiniers par qui nos âmes sont fleuries” Marcel Proust, Les Plaisirs et les Jours, 1896
Contents
A Theoretical Section 1. Introduction ................................................................................................................................ 1
2.2.1 Principle and mechanism .................................................................................................. 15
2.2.2 Strategies to avoid decarbonylation ............................................................................... 16
2.2.2.1 Use of a stable metallacyclic complex ..................................................................... 16 2.2.2.2 Saturation of the metal complex .............................................................................. 17
2.2.2.3 Coordination saturation by P-, S-, or O-coordinating atoms for intermolecular hydroacylation ......................................................................................................... 18
2.2.2.4 Coordination saturation by P-, S-, or O-coordinating atoms for intramolecular hydroacylation ................................................................................ 24 2.2.2.5 A new strategy (2013) : enantioselective ketone hydroacylation using Noyori’s hydrogen transfer catalyst ........................................................................ 29
3. Aim of the work .................................................................................................................... 31
4. Results and Discussion .................................................................................................. 34 4.1 Ligand concept and synthesis .............................................................................................. 34
4.1.1 General approach for the construction of cyclometalated phosphine-based pincer complexes ................................................................................................................. 34
4.1.2 Use of chelation auxiliary ................................................................................................. 35
4.2.2 Optimisation of the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde .................................................................................................. 51
4.2.3 Our best reaction conditions for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde ........................................................... 60
4.2.4 Proof of concept for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde ................................................................................. 61
4.2.5 Screening of our P-N ligand library in our best reaction condition for the
rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde ........................................................................................................................ 62
4.2.6 Scope of benzaldehyde with diverse alkenes .............................................................. 64
4.2.6.1 Benzaldehyde hydroacylation of alken-1-ol and hydroxyl-protected derivatives … 64 4.2.6.2 Benzaldehyde hydroacylation of 2 or 3-cyclosubstitued alkenes ............................ 65 4.2.6.3 Benzaldehyde hydroacylation of alkenamine …....................................................... 67 4.2.6.4 Benzaldehyde hydroacylation of alkenoic acid ...................................................... 67 4.2.6.5 Benzaldehyde hydroacylation of vinyl alkenoate ……............................................ 67 4.2.6.6 Benzaldehyde hydroacylation of small bulky alkene: 3,3-dimethylbutene and trimethyl-vinyl-silane .............................................................................................. 68
4.2.7 Scope of substituted benzaldehyde with 1-octene ................................................... 68
4.2.7.1 Use of electron-rich benzaldehydes ......................................................................... 68 4.2.7.2 Use of electron-poor benzaldehydes ........................................................................ 70 4.2.7.3 Use of 2-naphthaldehyde and 3-thiophene carbaldehyde ... ................................... 71
4.2.8 Summary of published results and conclusion ........................................................... 72
4.2.9 Rhodium-catalyzed intermolecular hydroacylation of 1-octene and aliphatic aldehydes ............................................................................................................................... 73
4.2.9.1 Rhodium-catalyzed intermolecular hydroacylation of 1-octene and 1-cyclohexe-1- carbaldehyde ............................................................................................................ 73 4.2.9.2 Cyclization of “double alkene” and “double alkyne” with formaldehyde .............. 73 4.2.9.3 Rhodium-catalyzed intermolecular hydroacylation of hexanal with 1-octene ........ 74 4.2.9.4 Rhodium-catalyzed intermolecular hydroacylation of 2-phenyl-propionaldehyde and
4.2.10 Intermolecular hydroacylation of salicylaldehyde with methyl acrylate followed by an intramolecular transesterification ..................................................... 76
4.2.11 Rhodium-catalyzed intermolecular asymmetric hydroacylation of 1-octene with benzaldehyde using chiral oxazoline derivative ligands ......................................... 77
4.3.7 Proof of concept for the rhodium-catalyzed intramolecular hydroacylation of o-vinylbenzaldehyde ........................................................................................... 93
4.3.8 Screening of our P-N ligand library in our best reaction condition for the
rhodium-catalyzed intramolecular hydroacylation of o-vinylbenzaldehyde ..... 95
4.3.9 Intramolecular hydroacylation of diversified o-vinylbenzaldehydes: synthesis 96
4.3.9.1 Synthesis of 6-vinylveratraldehyde (99) .................................................................. 96 4.3.9.2 Synthesis of 3-formyl-4-vinylbenzoic acid methyl ester (103) ................................. 97 4.3.9.3 Synthesis of 3-formyl-4-phtalimido-2-vinylbenzaldehyde (108) .............................. 97 4.3.9.4 Synthesis of 4-methyl-2-vinylbenzaldehyde (110) .................................................... 98 4.3.9.5 Synthesis of 1-vinylnaphthalene-2-carbaldehyde (114) .......................................... 98 4.3.9.6 Synthesis of 4-nitro-2-vinylbenzaldehyde (118) ...................................................... 99 4.3.9.7 Synthesis of 5-chloro-2-vinylbenzaldehyde (122) .................................................. 99 4.3.9.8 Synthesis of 5-fluoro-2-vinylbenzaldehyde (124) .................................................. 100 4.3.9.9 Synthesis of 4-hydroxy-2-vinylbenzaldehyde (126) ............................................... 100 4.3.9.10 Synthesis of 2-vinylpyridine-3-carbaldehyde (129) ............................................. 101 4.3.9.11 Synthesis of 2-vinylthiophene-3-carbaldehyde (134) ............................................ 101
4.3.10 Intramolecular hydroacylation of diversified o-vinylbenzaldehydes: catalytic
6. Outlook ........................................................................................................................................ 145
B Experimental part 7. General informations .................................................................................................... 147
10.1 Rhodium-catalyzed intermolecular hydroacylation of substituted benzaldehyde and 1-octene ................................................................................................. 243
10.1.1 General procedure: intermolecular hydroacylation of substituted benzaldehydes and diverse alkenes protocol (GP1) ......................................................................... 243
10.1.2 General procedure: intermolecular hydroacylation of substituted benzaldehydes
and diverse alkenes protocol using Jun’s condition (GP2) .................................. 244
10.1.3 Synthesis of 1-phenylnona-1-one (46) ......................................................................... 245
10.2 Rhodium-catalyzed intermolecular hydroacylation of substituted
benzaldehydes and diverse alkenes ................................................................................ 246
10.2.1 Synthesis of 7-hydroxy-1-phenyl-heptan-1-one (48) ................................................ 246
10.2.2 Synthesis of 1,3-diphenyl-propan-1-one (50) .............................................................. 247
10.2.3 Synthesis of 1,4-diphenyl-butan-1-one (52) ................................................................. 248
10.2.4 Synthesis of 3-cyclohex-3-enyl-1-phenyl-propan-1-one (54) ............................... 249
10.2.5 Synthesis of 3-cyclohexyl-1-phenyl-propan-1-one (56) ......................................... 250
10.2.6 Synthesis of 7-oxo-7-phenyl heptanoic acid (58) ...................................................... 251
10.2.7 Synthesis of 4-oxo-4-phenylbutyric acid methyl ester (60) ................................... 252
10.2.8 Synthesis of 4,4-dimethyl-1-phenyl-pentan-1-one (62) .......................................... 253
10.2.9 Synthesis of 3-dimethylsilanyl-1-phenyl-propan-1-one (64) ................................. 254
10.2.10 Synthesis of 1-(3-methoxy-phenyl)-nonan-1-one (66) .......................................... 255
10.2.11 Synthesis of 1-(4-methoxy-phenyl)-nonan-1-one (68) .......................................... 256 10.2.12 Synthesis of 1-biphenyl-4-yl-nonan-1-one (70) ....................................................... 257
10.2.13 Synthesis of 1-(4-chloro-phenyl)-nonan-1-one (72) ............................................... 258
10.2.14 Synthesis of 1-naphthalen-2-yl-nonan-1-one (74) .................................................. 259
10.2.15 Synthesis of 1-thiophen-2-yl-nonan-1-one (76) ....................................................... 260
10.3 Rhodium-catalyzed hydroacylation of 1-octene with aliphatic aldehydes 261
10.3.1 Synthesis of 3-phenyl-undecan-3-one (84) ................................................................. 261
10.4 Intermolecular hydroacylation of salicylaldehyde with methyl acrylate followed by an intramolecular transesterification ................................................. 262
10.4.1Synthesis of 4-(2-hydroxy-phenyl)-4-oxo-butyric acid methyl ester (87) ........... 262
10.4.2Synthesis of 2-hydroxymethyl-phenol (89) and carbonic acid 2-hydroxy-methyl-phenyl ester methyl ester (90) ....................................................................................... 263
10.10.1Synthesis of 3,4-dihydro-2H-phenanthren-1-one (191) and 2-methyl-2,3-dihydro-cyclopent[a]naphthalene-1-one (192) ........................................................... 283
10.10.2Synthesis of 6,7-dimethoxy-3,4-dihydro-2H-naphthalene-1-one (193) and 5,6-dimethoxy-2-methyl-1-indanone (194) ........................................................................ 284
10.10.3Synthesis of 6-chloro-3,4-dihydro-2H-naphthalene-1-one (195) and (196) ….... 285
10.11 Rhodium-catalyzed hydroacylation of 5-hexenal and synthesis of medium-sized ring .................................................................................................................... 286
10.11.1 Synthesis of cyclohexanone (198) and 2-methylcylopentanone (199) ... 286
10.11.2 Synthesis of cyclododecanone (202) .............................................................. 287
10.11.3 Synthesis of 6,7,8,9,10,11-hexahydro-5-oxa-benzocyclodecen-12-one (205) ……………………………………………………………………………….. 287
11.1.2 Synthesis of “[Rh(imine P-N ligand 1)2]+Cl-” obtained with [Rh(COD)Cl]2
(206) ....................................................................................................................................... 289 11.2 The influence of the P-N ligand ....................................................................................... 291
Abbreviations Ac acetate acac acetylacetonato AcOEt ethyl acetate AcOH acetic acid Ag silver AIBN azobisisobutyronitrile AlOx aluminium oxide app. apparatus aq. aqueous Ar aryl ATH asymmetric transfer hydrogenation atm. atmosphere BDPP 2,4-bis(diphenylphosphino)-pentane BINAP 2,2’-bis(diphenylphosphino)-1,1’-binaphthyle b/l branched to linear BOC N-tert-butoxycarbonyl bpym bipyrimidine Bu butyl n-Buli n-butyllithium tBuOK potassium tret-butoxide c contration °C degree Celsius cat. catalyst cc concentrated cHex cyclohexane coe cyclooctene COD cyclooctadienyle Cp pentamethylcyclopentadienyl Cy cyclohexyl d day(s) DABCO 1,4-diazabicyclooctane DBU 1,8-diazabicyclo[5.4.0]undec-7-ene DCE 1,2-dichloroethane dcpe bis(dicyclohexylphosphino)ethane degr. degradation DFT density functional theory DIBAL-H diisobutylaluminium hydride DIPA diisopropylamine DMAP 4-dimethylaminopyridine DMF N,N’-dimethylformamide DMG Directed Metalating Group DMSO dimethyl sulfoxide dmpe 1,2-bis(dimethylphosphino)ethane DoM Directed ortho-Metalation dppe 1,2-bis(diphenylphosphino)ethane dppf 1,1'-bis(diphenylphosphino)ferrocene dppp 1,3-Bbs(diphenylphosphino)propane
Abbreviations
dr diastereoisomeric ratio EDC 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide ee enantiomeric excces eq. equivalent ESI electron spray ionisation Et ethyl Et2O diethylether Et3N triethylamine EtOH ethanol Fe iron FG functional group GC gas chromatography GP general procedure h hour(s) HCl hydrochloric acid HOMO Highest Occupied Molecular Orbital HSOMO Highest Single-Occupied Molecular Orbital HPESW Hajos-Parrish-Ender-Sauser-Wiechert HRMS high resolution mass spectrum Hz Hertz iPrOH isopropanol J nuclear spin-spin coupling (Hz) k kinetic constant L or lig. ligand LDA lithium diisopropylamide liq liquid LTMP lithium tetramethylpiperidine LUMO Lowest Unoccupied Molecular Orbital M molarity Me methyl MeOH methanol min minute(s) MOCC Metal-Organic Cooperative Catalysis Mol.Wt. molecular weight Ms mesyl MS mass spectrum MTM methylthiomethyl N normality NBS N-bromosuccinimide NCS N-chlorosuccinimide NBD norboradiene NET norepinephrine transporter NHC N-heterocyclic carbine NMP N-methyl-2-pyrrolidone NMR nuclear magnetic resonance o orto OEP octaethylporphyrin o/n overnight p para P pressure PCC pyridinium chlorochromate
Abbreviations
PE petroleum ether PEG polyethylene glycol Ph phenyl Piv pivaloyl PivCl pivaloyl chloride por porphirine ppm parts per million PPTS pyridinium p-toluenesulfonate Pr propyl p-TSA para-toluenesulfonic acid quant. quantitative R alkyl RT room temperature SM starting material SN nucleophilic substitution t tert T temperature TBME methyl tert-butyl ether TBS tert-butyldimethylsilyl TCA tricyclic antidepressant Tf triflate TFA trifluoroacetic acid THF tetrahydrofurane TLC thin-layer chromatography TMP tetramethylpiperidine TMS tetramethylsilane TMSCl trimetylsilyl chloride Tol toluol Tp triptophenyl Ts tosyl UV ultra-violet W-M Wieland-Miescher
1
1
A Theoretical Section 1. Introduction The challenge for a chemist is to solve problems about which people care.[1] Many of chemistry´s fundamental discoveries were made in the course of developing practical technologies. Catalysis and polymer science, for instance, had their origins in industry. Chemistry is the key fundamental science, which starts from a basic unit of matter – the atom – and find its quintessence in the synthesis of complex natural products, useful synthons for the development of active drugs, or new high-technology eco friendly materials. Chemistry is at the end of a century of expansion.[1] At the dawn of the last century, the chemical industry was at the start of its growth. At the instigation of the academic research, this developed efficiently over the last fifty years many essential breakthroughs on the basic concepts of the field (i.e. the chemical bond, the laws of thermodynamics, theories of kinetics) were achieved.
Now, the industry is mature. Researchers in the industry have nowadays at their disposal a large and powerful toolbox for the determination of the structure and the synthesis of almost any molecule with sophistication.[2] The continual sharpening of its tools are demonstrated by all the complex natural molecules syntheses starting from the wonderful milestone synthesis of strychnine[3] by R. B. Woodward et al. (Nobel Prize in Chemistry, 1965) to the synthesis by K. C. Nicolaou and co-workers of the maitotoxin,[4] the largest metabolite ever isolated and characterized.
“Reinvention is essential for the continuing relevance and survival of chemistry”, states G. M. Whitesides from Harvard University.[1] Chemistry must undergo fundamental change, a radical new reprogramming of our thinking, a deep calling into question of our habits. The challenges of the future are more complex, and must be increasingly interdisciplinary. Chemists will be more in harmony with the world around them.
[1] G. M. Whitesides and J. Deutch, Nature 2011, 469, 21-22. [2] a) E. J. Corey, X.-M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, New-York, 1995. b) K. C. Nicolaou, D. Vourloumis, N. Winssinger, P. S. Baran, Angew. Chemie 2000, 112, 46-126; Angew. Chem. Int. Ed. 2000, 39, 44-122. [3] R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker, K. Schenker, J. Am. Chem. Soc. 1954, 76, 4749-4751; R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker, K. Schenker, Tetrahedron 1963, 19, 242-288. [4] a) K. C. Nicolaou, M. O. Frederick, Angew. Chemie 2007, 28, 5372-5376; Angew. Chem. Int. Ed. 2007, 46, 5278–5282. b) K. C. Nicolaou, K. P. Cole, M. O. Frederick, R. J. Aversa, R. M. Denton, Angew. Chemie 2007, 46, 9031-9035; Angew. Chem. Int. Ed. 2007, 46, 8875-8879. c) K. C. Nicolaou, M. O. Frederick, A .C. B. Burtoloso, R. M. Denton, F. Rivas, K. P. Cole, R. J. Aversa, R. Gibe, T. Umezawa, T. Suzuki, J. Am. Chem. Soc. 2008, 130, 7466–7476. d) K. C. Nicolaou, R. J. Aversa, J. Jin, F. Rivas, J. Am. Chem. Soc. 2010, 132, 6855–6861. e) K. C. Nicolaou, C. F. Gelin, J. H. Seo, Z. Huang, T. Umezawa, J. Am. Chem. Soc. 2010, 132, 9900–9907. f) K C. Nicolaou, J. H. Seo, T. Nakamura, R. J. Aversa, J. Am. Chem. Soc. 2011, 133, 214–219. g) K. C. Nicolaou, T. M. Baker, T. Nakamura, J. Am. Chem. Soc. 2011, 133, 220–226.
Introduction
2
The American Commitee on Challenges for the Chemical Sciences in the 21th century published in 2003, thirteen challenges for chemists and chemical engineers.[5] Drug design (develop medecines and therapies that can cure currently untreatable diseases), energy (develop renewable and inexpensive energy, especially new ways for energy generation, storage, and transportation), and environmental chemistry (understand the complex chemistry of the earth, including land, sea, atmosphere, and biosphere so we can maintain its livability especially in how to deal with pollution and others threats to earth) are three of the most interesting challenges. Sustainable development*, [6], [7] is the leading concept of the 21th century for organic chemistry.[8] Chemists must think at the start of the synthesis of a new molecule or the inception of a new industrial process, develop how to synthesize and manufacture this new substance with a high consideration in reducing the environmental impact of its synthesis. Chemists must also minimize waste production, maximize the use of raw chemicals, decrease the energy consumption [9] and use more environmentally friendly solvents.**, [10] In 1998, P. T. Anastas and J. C. Warner published twelve principles which are widely accepted as a basis of green chemistry.[11]
THE IDEAL SYNTHESIS
AtomEfficient
Simple
100% Yield
Available Materials
Environmentally acceptable
No Wasted Reagents
One Step
Safe
Figure 1. The ideal synthesis.[1] The dream of an “ideal synthesis”. In 1996, P. Wender defined his vision of the ideal synthesis: “an ideal synthesis is generally regarded as one in which the target molecule (natural or designed) is prepared from readily available, inexpensive starting materials in one simple, safe, environmentally acceptable, and resource-effective operation that proceeds quickly and in quantitative yields”.[12]
[5] Board on Chemical Sciences and Technology (BCST), Beyond the Molecular Frontier: Challenges for Chemistry and Chemical Engineering, The National Academies Press, Washington 2003. [6] G. Bruntland, Our Common Future, Oxford University Press, New-York 1987. [7] T. Clarke, Nature 2002, 418, 812-814. [8] Report of the United Nations Conference on Environment and Development, 3-14 June 1992, Rio de Janeiro, <http://www.un.org/esa/sustdev>. [9] M. Eisen, J. O. Metzger, E. Schmidt, U. Schneidewind, Angew. Chem. 2002, 114, 402-425; Angew. Chem Int. Ed. 2002, 41, 414-436. [10] C. Capello, U. Fischer, K. Hunkelbühler, Green Chem. 2007, 9, 927-934. [11] P. T. Anastas, J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New-York 1998. [12] P. A. Wender, Chem. Rev. 1996, 96, 1-2.
* Sustainable Development has been defined as: “Meeting the needs of the present generation without compromising the ability of future generations to meet their own needs”.[6]
** Green Solvents “express the goal to minimize the environmental impact resulting from the use of solvents in chemical production”.[10]
Introduction
3
The development of new catalytic reactions and environmentally acceptable processes according to the atom ecomomic concept* developed by Trost that have an optimal selectivity (i. e. chemo-, region-, diastereo-, and enantioselectivity), attempt to reach this ideal goal.[13]
Catalysis is a key tool towards green chemistry. Catalysis is a process in which the rate of a chemical reaction is changed by a catalyst.[14] The catalyst decreases the activation energy of a reaction which results in potentially milder reaction conditions and hence energy efficiency. Consequently, according to the Boltzmann distribution, more molecular collisions have the energy needed to reach the transition state. Catalysts can enable reactions that would otherwise be blocked or slowed by a kinetic barrier. Kinetics also played a crucial role to improve the efficiency of reactions. Catalysts promote the frequency of contact of the reactants, which improves the reaction rate and consequently energy consumption. The challenge of catalyst development is to find catalytic conditions which preferantially select a product among competing isomeric products via an efficient energetic differentiation.
catalyst
catalyst
catalyst
cata
lyst
product formedfirst reagent
on to catalyst
second reagent on to catalyst
reaction occurson catalyst
catalytic cycle
Figure 3. Langmuir-Hinselwood mechanism.
[13] a) B. M. Trost, Science 1991, 254, 1471-1477; b) B. M. Trost, Acc. Chem. Ress. 2002, 35, 695-705.
[14] a) B. M. Trost, Angew. Chemie. 1995, 107, 285-307; Angew. Chem. Int. Ed. 1995, 34, 259-281. b) R. A. Scheldon, Pure Appl. Chem. 2000, 72, 1233-1246. * Atom economy means maximizing the incorporation of material from the starting molecules or reagents into the final product.
Introduction
4
Catalysis is directly inspired from the nature. The design and development of self-optimizing chemical systems (new active drugs, new biocompatible therapies) copying the development of biological systems through evolution, is a new exciting source of important discoveries.
Many living cell transformations are catalyzed by enzymes. Enzymes are able to do a wide range of catalytic biosynthetic pathways like oxydation, degradation... Additionally, enzymes are very specific as to which reactions they catalyze and the substrates that are involved in these reactions. Enzymatic reactions have many advantages. They proceed at ambient pressure and temperature, and the protection of all reactive functional groups in the substrate structure is not required. Many enzymes do not need any additional coenzymes to show full activity, or when a coenzyme like the NADH is required, it is directly recycled by the human body. Enzymes can also be used in organic synthesis to form with high efficiency new functionnalized binding with the optimal configuration. Organometallic chemistry is the Pandora’s box of the catalysis. This area of the chemistry lies at the interface between organic chemistry and inorganic chemistry, and studies the chemical compounds containing bonds between carbon and a metal. The organometallic chemistry can be split in two different families : the organolithium and –magnesium species, which is an ionic chemistry (e. g. the Grignard reagent which is an organomagnesium reagent that is used extensively in the organic laboratory as an alkylating agent) ; and the chemistry with transition metals, culminating with notable recent advances worthy of the Nobel Prize by W. S. Knowles and R. Noyori in 2001 for their work on asymmetrically catalysed hydrogenation reactions, K. B. Sharpless in 2001 for his work on asymmetrically catalysed oxidation reactions, Y. Chauvin, R. H. Grubbs and R. S. Schrock in 2005 for their development of the metathesis method in organic synthesis, R. F. Heck, E.-I. Negishi and A. Suzuki in 2010 for their development of palladium-catalyzed cross-coupling reactions.
Catalysis can be either homogeneous or heterogeneous, depending on whether a catalyst exists in the same phase as the substrate. In the heterogeneous catalysis, reactants must diffuse to the catalyst surface and adsorb onto it. After reaction, the products must desorb from the surface and diffuse away from the solid surface according to the Langmuir-Hinselwood mechanism. Heterogeneous catalysis has many applications like the Bosch-Haber process to synthesize ammonia from nitrogen and hydrogen or the monohydrogenation of alkynes via the Lindlar catalyst. In the homogeneous catalysis, the catalyst is dissolved in a solvent together with the substrates. This last method is used more frequently for fine chemical synthesis.
* NADH is nicotinamide adenine dinucleotide, that a oxidizing agent found in all living cells.
Introduction
5
Start3H2(g) + N2(g)
compressor
ironcatalyst
400 - 450 °C200 atm.
reactor
condenser
2NH3(l)storage
unreacted gases recycled
Figure 4. The Bosch-Haber process. Multifunctional solids often are catalytically active, e.g. zeolites, alumina and certain forms of graphitic carbon.
Among all catalytic transformations, this thesis will focus more specifically on the activation of C-H bonds using transition metal catalysts.
6
2. Background 2.1 C-H activation
2.1.1 C-H bonds
There are few methods for activating C-H bonds to directly produce more valuable functional groups. This lack is due to the low affinity of the C-H bond, with two main points that explain this chemical inertness:
Pauling’s scale: the electronegativity difference between carbon (χ = 2.5) and hydrogen (χ = 2.1) is very small. The C-H bond generally regarded as being non-polar.
The high bond dissociation energy of a sigma C-H bond (about 413 kJ.mol-1).
The C-H bond has no empty orbital of low energy (LUMO) or filled orbital of high energy (HOMO) that could readily participate in a chemical reaction.
2.1.2 Organometallic C-H bond activation[15] Six modes of organometallic C-H activation are described in the literature, but just the electrophilic and the oxidative additions are on the top of research interests:
The oxidative addition first reported by the group of Chatt in the Journal of the American Chemical Society in 1965.[16]
The electrophilic addition initiated by the Shilov system in 1972.[17]
The sigma-bond metathesis of unactivated alkanes first reported in 1983 with the activation of cyclohexane by Watson et al.[18]
The activation by metalloradicals essentially describes with porphyrin complexes, and discovered by Wailand in 1985.[19]
The mercat system, a photochemical system reported by Crabtree in 1987.[20]
The 1,2-addition concomitantly reported by Wolczanski and Bergman in 1988 with the use of Zr(IV) amido alkyl complexes.[21]
C-H activation may be defined as a reaction that cleaves a carbon-hydrogen bond. All reactions involve organometallic complexes and proceed by coordination of a hydrocarbon to the inner-sphere of metal, either via an intermediate “alkane or arene complex” or as a transition state leading to a metal-carbone intermediate. Important to this definition is the requirement that during the C-H cleavage event, the hydrocarbonyl species remains associated in the inner-sphere and under the influence of the metal.
[15] A. S. Goldman, K. I. Goldberg, Activation and Functionalization of C-H Bonds ACS Symposium Series 885 2004. [16] J. Chatt and J. M. Davidson, J. Chem. Soc. 1965, 843-855. [17] N. F. Gol’dschleger, V. V. Es’kova, A. E. Shilov, A. A. Shteinman, Zhurnal Fizicheskoi Khimii 1972, 46, 1353. [18] P. L. Watson, J. Am. Chem. Soc. 1983, 105, 6491-6493. [19] K. J. Del Rossi, B. B. Wailand, J. Am. Chem. Soc. 1985, 107, 7941-7944. [20] S. H. Brown, R. H. Carbtree, Chem. Comm. 1987, 970-971. [21] a) C. C. Cummings, S. M. Baxter, P. T. Wolczanski, J. Am. Chem. Soc. 1988, 110, 8731-8733. b) P. J. Walsh, F. J. Hollander, R. G. Bergman, J. Am. Chem. Soc. 1988, 110, 8729-8731.
Background
7
The first C-H activation by a transition metal was reported by Chatt et al. in 1965 with insertion of a ruthenium atom ligated to dmpe* in the C-H bond of naphthalene.[16]
PRuPHP
"Ru(Me2PCH2CH2PMe2)2" P
H
Scheme 1. Preparation of an hydrido(naphtyl) ruthenium complex. 2.1.2.1 C-H activation by oxidative addition One of the most important milestones for the development of the metal-catalyzed C-H bond activation was the homogeneous catalytic hydrogenation of olefins and acetylenes at room temperature and low pressure (≤ 1 atm.) using (PPh3)3RhCl published in 1965 by Wilkinson et al.[22] The mechanism of the catalytic cycle, which alternates between Rh(I) and Rh(III), clearly describes the addition of a molecule of hydrogen and the reductive elimination of the C-H bond.
In 1970, Green et al. reported the photochemical insertion of tungsten in a benzene C-H bond.[23]
In 1976, Shaw et al. designed new bulky tertiary di-t-butylphosphines which generate, via cyclometalation a new type of “RPCP” tridentate ligated complexes.[24] These “pincer” ligands were obtained with a wide range of simple metal halide salts including Rh, Pd, Ir, Ni, Pt,etc. and played a very important role in C-H activation.[25]
PtBu2
PtBu2
H [MCl3.3H2O]+
PtBu2
PtBu2
MH
Cl
M = Ir or Rh
Scheme 2. A new type of “RPCP” tridentate ligated complexes The first intramolecular aliphatic C-H activation was reported by Whitesides and co-workers in 1979.[26] Whitesides and co-workers succeeded to convert dineopentylbis (triethylphosphine)platinum(II) to a bis(triethylphosphine)-3,3’-dimethylplatinacyclobutane via activation of an unactivated C-H bond by intramolecular oxidative addition.
[22] J. F. Young, J. A. Osborn, F. H. Jardine, G. Wilkinson, Chem. Comm. 1965, 131-132. [23] M. L. Green, P. J. Knowles, J. Chem. Soc. D 1970, 24, 1677–1677. [24] C. J. Moulton, B. L. Shaw, J. Chem. Soc, Dalton Trans. 1976, 1020-1024. [25] a) M. Albrecht, G. van Koten, Angew. Chemie 2001, 20, 3866-3898; Angew. Chem Int. Ed. 2001, 40, 3750-3781. b) M. E. van der Boom, D. Milstein, Chem. Rev. 2003, 103, 1759-1792. c) N. Selender and K. Szabó, Chem. Rev., 2011, 111(3), 2048–2076. [26] P. Foley, G. M. Whitesides, J. Am. Chem. Soc. 1979, 101(10), 2732–2733.
* dmpe : 1,2-Bis(dimethylphosphino)ethane
Background
8
Pt(C2D5)3P
(C2D5)3P
k1
k-1
Pt(C2D5)3P
+ (C2D5)3P
k2k-2
(C2D5)3PPt
H(C2D5)3P
Pt(C2D5)3P
k3-C(CH3)4
P(C2H2)3
HH
Scheme 3. The first C-H bond activation by intramolecular oxydative addition.
In 1982, Bergman and co-workers described the first photochemical C-H activation of completely saturated hydrocarbons, namely cyclohexane and neopentane, forming the hydridoalkylmetal complex Cp*Ir(PMe3)H(C6H5).[27]
IrH
HMe3P
h / C6H12
IrMe3P
H
h / Me4C
IrMe3P
H
h / C6H6
IrMe3P
H
[(Me5C5)IrCl2]2
1) PMe3
2) LiEt3BH
Scheme 4. Photochemical C-H activation of cyclohexane and neopentane. The C-H activation is of major economic interest. C-H activation is often described for alkanes, pyridine derivatives, benzene analogues and aldehydes.
[27] A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc. 1982, 104(1), 352-354.
Background
9
One of the most exciting applications of the C-H activation is the conversion of alkanes into more useful compounds. Alkanes are major constituents of natural gas and petroleum, and this energy resource is decreasing fast. Therefore it will be necessary to improve their use. For example, methane can be converted to methanol: it is first oxidized in methyl bisulphate by sulphuric acid, catalyzed by a Pt (II) complex. The resulting methyl bisulphate is then hydrolyzed to give methanol.[28] Another relevant application is the catalytic hydroborylation of alkanes. In 1999, the group of Knochel described C-H activations of tert-butyl group in organoboranes compounds via a direct borane-hydrocarbon dehydrogenation.[29]
B H+
C H
B H
C H
B
C+
H
H
Scheme 5. The four-center mechanism of borane-hydrocarbon dehydrogenation reactions
by Köster et al.[30]
The catalytic alkane dehydrogenation in alkene should also be noticed.[31] A pincer iridium complex was used to catalyse the transfer dehydrogenation of alkanes at high temperature.
Figure 5. An efficient catalyst for cycloalkane transfer-dehydrogenation: the “pincer” complex 2,6-bis[di(t-butyl)phosphinomethyl]phenyl]IrH2.
[31]
Selective C-H bond activation reaction of pyridine is also widely described. In 2005, Saak and co-workers reported the activation of pyridine with the titanocene fragment [Cp2Ti] generated in situ by using the corresponding bis(trimethylsilyl) acetylene complex.[32] The same group reported the C-H activation of other N-heterocycles such as pyrimidine, pyrazine and triazine with the same titanocene fragment.[33] In 2008, Cho et al. described a highly selective alkenylation and arylation of pyridine N-oxides.[34] C-H bound in α-N-position was activated by palladium acetate at 100 °C.
[28] R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh, H. Fujii, Science 1998, 280, 560-564. [29] a) B. Goldfuss, P. Knochel, L. O. Bromm, K. Knapp, Angew. Chemie 2000, 112(22), 4302-4305; Angew. Chem. Int. Ed. 2000, 39(22), 4136-4139. b) H. Laazari, L. O. Bromm, F. Lhermitte, R. M. Gschwind, P. Knochel, J. Am. Chem. Soc. 1999, 121, 6940-6941. [30] a) R. Köster, W. Larbig, G. W. Rotermund, Justus Liebigs Ann. Chem. 1965, 682, 21-48; b) R. Köster, G. Benedikt, W. Fenzl, K. Reinert, Justus Liebigs Ann. Chem. 1967, 702, 197-223; c) G. J. Abruscato, T. T. Tidwell, J. Org. Chem. 1972, 37, 4151-4156. [31] F. Liu, E. B. Pak, B. Singh, C. M. Jensen, A. S. Goldman, J. Am. Chem. Soc. 1999, 121, 4086-4087. [32] I. M. Piglosiewicz, S. Kraft, R. Beckhaus, D. Haase, W. Saak, Eur. J. Inorg. Chem. 2005, 938-945 [33] S. Kraft, R. Beckhaus, D. Haase, W. Saak, Angew. Chemie 2004, 12, 1609-1614; Angew. Chem. Int. Ed. 2004, 43, 1583-1587. [34] S. H. Cho, S. J. Hwang, S. Chang, J. Am. Chem. Soc. 2008, 130, 9254-9256.
* dmpe : 1,2-Bis(dimethylphosphino)ethane
Background
10
NO -
+CO2Et+
H NO -
+ CO2Et
Pd(OAc)2 (10 mol%)Ag2CO3 (1.5 mol%)
pyridine
1,4-dioxane, 100 °C96%
NO -
+ CO2Et N CO2Et
PCl3 (1.2 eq.)
toluene25 °C, 15 min.
92%
NO -
+ H
alkenylation
arylation +NO -
+
Pd(OAc)2 (10 mol%)Ag2CO3 (2.2 eq.)
pyridine
100 °C, 16 h79%
+NO -
+
3 : 1
Scheme 6. A highly selective alkenylation and arylation of pyridine N-oxides using palladium acetate. Many catalytic functionalizations of benzene and its derivatives can also be found in the literature:[35] Pd-catalyzed coupling of arenes with olefins, Pd- and Pt-catalyzed hydroarylation of alkynes, Pd-catalyzed carboxylation of arenes and hydroxylation of arenes with O2. An interesting application is the preparation of biologically active heterocycles such as coumarins, quinolinones, and thiocoumarins.[36] The synthesis of coumarins in formic acid at room temperature has been reported by Trost and Toste.[37]
OH
+ RO
O R1
H
R = H, Me, PhR1 = H, Et
Pd(OAc)2 / HCO2H O O
R
50 - 71%
RT
Scheme 7. Palladium acetate-catalyzed synthesis of coumarins in formic acid at room temperature.
Last but not least is the C-H activation adjacent to carbonyl species. One of its applications is the hydroacylation reaction in which an aldehyde is added across an alkene or an alkyne bond to yield the corresponding ketone. More details are discussed in chapter 2.2 (p. 15).
2.1.2.2 C-H activation by electrophilic addition In 1966, A. Shilov applied the concept developed by Wilkinson et al. with the H/D exchange study between methane and heavy water by a simple platinum (II) catalyst (K2PtCl4).[38]
[35] C. Jia, T. Kitamura, Y. Fujiwara, Acc of Chem Res 2001, 34(8), 633-639. [36] C. Jia, D. Piao, T. Kitamura, Y. Fujiwara, J. Org. Chem. 2000, 65, 7516-7522. [37] B. M. Trost and F. D. Toste, J. Am. Chem. Soc. 1996, 118, 6305-6306. [38] A. P. Khrushch, L. A. Tokina, A. E. Shilov, Kinetica I Kataliz 1966, 7, 901.
Background
11
Shilov´s group extended this observation to the oxidation of alkanes to alcohols catalyzed by Pt(II)Cl2 in a aqueous solution with [Pt(IV)Cl6]
2- acting as the final oxidant.[39]
PtII CH2 R
PtIVCH2 R
H
PtIV
PtII
+ H+PtIVCH2 R
Cl- or HO-
R CH2 Cl
R CH2OH
+
oxidation
alkeneactivation
or
functionalization
Scheme 8. Oxidation of alkanes to alcohol catalyzed by Pt(II)Cl2. Roy Periana et al. published in 1998 the best optimisation of the Shilov process with high-yield (72 %) via the oxidation of methane to methyl bisulfate by a platinum(II) bipyrimidine catalyst (bpym)PtCl2 in concentrated sulfuric acid. The same system was described using Hg(II)/H2SO4 for methane oxidation.[28]
By analogy, Fujiwara et al. reported the carboxylation of alkanes and arenes under mild conditions at room temperature with carbon monoxide (1 atm) using palladium (II) acetate (10 mol%) in TFA* with potassium peroxodisulfate K2S2O8 as an oxidant.[40]
Pd(OAC)2 Pd(O2CCF3)2 [PdO2CCF3]+
Ar PdO2CCF3
Ar PdO2CCF3
OAr O2CCF3
O
TFA -CF3CO2-
Ar H-H+[O]/TFA
Pd(0)
Ar
O
OH
TFA
TFAA
CO
Scheme 9. Carboxylation of alkanes and arenes under mild conditions with carbon monoxide. Finally there is the C-H activation adjacent to carbonyl species. One of its applications is the hydroacylation reaction in which an aldehyde is added over an alkene or an alkyne bond to yield the corresponding ketone.
[39] a) A. E. Shilov, A. A. Shteinman, Coord. Chem. Rev. 1977, 24, 97-143. b) L. A. Kushch, V. V. Lavrushko, Y. S. Misharin, A. P. Moravskii, A. E. Shilov, Nouveau Journal de Chimie 1983, 7, 729. [40] a) Y. Fujiwara, K. Takaki, J. Watanabe, Y. Uchida, H. Taniguchi, Chem. Lett. 1989, 1687-1688. b) K. Nakata, Y. Yamaoka, T. Miyata, Y. Taniguchi, K. Takaki, Y. Fujiwara, J. Organomet. Chem. 1994, 473, 329-334. c) C. Jia, T. Kitamura, Y. Fujiwara, Acc. Chem. Res. 2001, 34, 633-639.
* TFA: Trifluoroacetic acid
Background
12
2.1.2.3 C-H activation by sigma-bond metathesis The sigma-bond metathesis is a concerted mechanism that uses transition metal complexes with a d0 configuration, which are very efficient catalysts as oxidative addition is excluded. The transition state of this reaction is a typical four center species.
LnM R + R´ HM C
C H
#
LnM R´ + R H
Scheme 10. The sigma-bond metathesis.
Watson first reported in 1983 that Cp*LuMe underwent exchange with in cyclohexane solvent.[18], [41]
2.1.2.4 C-H activation by metalloradicals The C-H activation systems are based upon the use of porphyrin complexes, either with iron or rhodium, and developed by Wailand.[19] Wailand[42] reported that (octaethylporphyrin)rhodium(II) dimer Rh(OEP)2 thermally reacts with alkylaromatic molecules exclusively at the alkyl C-H unit to produce organometallic products Rh(III)OEP(alkylaromatic) and Rh(III)OEPH.
In 1987, Crabtree published the functionalization of alkanes by mercury photosensitisation. This reaction can be carried out with a good isolated yield of 90-95% and on large scale. The accepted mechanism involves free hydrogen atom abstraction from the alkane by the 3P1 excited atomic state of mercury to give alkyl radicals.[20], [43]
R H + Hg* R . + H . R R H H1/2 + 1/2
Scheme 12. The Mercat system.
2.1.2.6 C-H activation by 1,2-addition Wolczanski and Bergman of the university of Berkeley independently published in 1988 that (tBu3SiNH)2ZrPh could undergo 1,2-elimination of alkanes to form an imido complex.[21]
[41] P. L. Watson. G. W. Parshall, Acc. Chem. Res. 1985, 18, 51-56. [42] a) K. J. Del Rossi, B. B. Wailand, Chem. Comm. 1986, 1653-1655; b) A. E. Sherry, B. B. Wailand, J. Am. Chem. Soc. 1990, 112, 1259-1261; c) B. B. Wailand, S. Ba, A. E. Sherry, J. Am Chem. Soc. 1991, 113, 5305-5311. [43] a) S. H. Brown, R. H. Carbtree, J. Am. Chem. Soc. 1989, 11, 2935-2946; b) S. H. Brown, R. H. Carbtree, J. Am. Chem. Soc. 1989, 11, 2946-2953; c) R. H. Carbtree, S. H. Brown, C. A. Muedas, C. Boojamra, R. R. Ferguson, Chemtech 1991, 21, 634.
Background
13
Subsequent addition of a C-H bond across the Zr=N linkage generates the phenyl species.
Zr NHtBu
tBuHN
tBuHN
-
Zr NtButBuHN
tBuHN
+
Zr NHtBu
tBuHN
tBuHN
H
Scheme 13. Methane and benzene activation via transcient (tBu3SiNH)2Zr=NSitBu3.
2.1.3 Non-organometallic C-H bond activation[15] The non-organometallic C-H activation systems can be split in two categories:
The monooxygenase enzymes (essentially cytochrome P-450 and methane monooxygenase) that enzymes incorporate one hydroxyl group into substrates in many metabolic pathways
The Fenton-type chemistry published in 1894, and describes the use of iron(II) for the catalysis of hydrogen peroxide with alkanes
2.1.3.1 C-H activation by enzymes
An approach in the C-H activation was the use of enzymes.[44] The most famous enzyme catalysis is the conversion of C-H bonds to C-O bonds using cytochrome P450. In humans, these enzymes are involved in making cholesterol, steroids, and other lipids; they also metabolize drugs. The use of modified stereoselective enzymes is one of the most promising challenges for the oxidation of unactivated C-H bonds for the construction of complex molecules with high efficiency. Prof. Dr. Manfred Reetz of the Max-Planck-Institut für Kohlenforschung in Mülheim an der Ruhr developed recently many stereoselective enzyme mutants for the selective C-H activation of many organic compounds. Reetz et al. published for example in 2012 a highly regio- and enantioselective oxidative hydroxylation of cyclohexene-1-carboxylic acid methyl ester using modified monooxygenase P450 by iterative saturation mutagenesis.[45]
CO2Me
R-selectiveP450-BM3
mutant
S-selectiveP450-BM3
mutant
CO2Me
OH
CO2Me
OH
conversion : up to 99%e.e. : 84%
conversion : 89%e.e. : 93%
Scheme 14. High regio- and enantioselective oxidative hydroxylation of cyclohexene-1-carboxylic acid methyl ester.
[15] A. S. Goldman, K. I. Goldberg, Activation and Functionalization of C-H Bonds ACS Symposium Series 885 2004. [44] R. J. Bergman, Nature 2007, 446, 391-394. [45] R. Agudo, G. D. Roiban, M. T. Reetz, Chembiochem 2012, 13(10), 1465-1473.
Background
14
Another striking example of a C-H activating enzyme is methane monooxygenase, which was recently discovered in a class of bacteria that lives at the interface of aerobic and anaerobic environments. This enzyme converts methane to methanol, although it can also oxidize several other organic compounds. The specificity of these enzymes derrives from the presence of iron ions at their active sites. Iron centers react with oxygen to make a highly reactive iron-oxygen double bond Fe=O, which adds to a C-H bond yielding a carbon radical and a complex bearing an iron-hydroxy group, followed by the formation of the C-O bond. Chemists have tried to mimic these biological systems. Several synthetic iron complexes have have been made that use oxygen, or other oxidants such as hydrogen peroxide, to convert alkanes into oxygenated products. A great example of the use of iron proteins was the biosynthesis of the prodiginine natural products undecylprodigiosin, streptorubin B and metacycloprodigiosin published in 2011 in Nature Chemistry by Steven Bruner. He used a small family of enzymes involved in the oxidation degradation of aromatic compounds.[46]
NH HN
OCH3
O
H
HN
NH N
OCH3
HN H
H
NH N
OCH3
HN
NH N
OCH3
HN
12
10
H
Metacycloprodigiosin
Streptorubin B
Undecylprodigiosin
+
RedH
McpH
RedG
McpG
Rieske-typeoxygenases
CysS
FeS
SFe
N
NNH
NH
SCys
His
His
Rieske iron-sufurcenter
Scheme 15. An exemple of C-H activation by enzymes. Synthesis of the Streptorubin B and the
Metacycloprodigiosin with the use of Rieske iron-sulfur center. 2.1.3.2 The Fenton-type chemistry
The Fenton-type chemistry could be also used for the oxidation of diverse alkanes. It is an oxidation process using iron(II) and hydrogen peroxide as catalysts. H. J. H Fenton was the first to describe the oxidation of tartaric acid by H2O2 in the presence of ferrous iron ions.[47]
This system are using for treating various industrial wastewater components including aromatic amines, a wide variety of dyes, pesticides, surfactants, and explosives.
[46] S. D. Bruner, Nature Chemistry 2011, 3, 342-343. [47] H. J. H. Fenton, J. Chem. Soc., Trans. 1894, 65, 899-911.
Background
15
1) Fe2++ H2O2 + H+ Fe3++ H2O + HO .
2) Fe3++ H2O2 Fe2++ HOO . + H+
Scheme 16. The Fenton system. Although the Fenton system looks like a simple reaction, its mechanism is very controversial, consisting of either a proposed radical and non-radical pathway. 2.2 Hydroacylation 2.2.1 Principle and mechanism[48]
The hydroacylation of alkenes has the potential to become an attractive, atom-economical
synthetic method for the transformation of an aldehyde and an unsaturated hydrocarbon into a ketone. Intra- and intermolecular variants of the reaction are known. The proposed mechanism begins with a transition metal inserting into the aldehyde C-H bond to form an acyl metal hydride 1. Then, the acylmetal alkyl complex 2 is generated by insertion of the hydride 1 into an unsaturated C-C bond. Reductive elimination of the acylmetal alkyl 2 affords ketone 3 with the regeneration of the transition metal catalyst.
Scheme 17. Proposed mechanism for the alkene hydroacylation. Decarbonylation is the most important side-reaction of hydroacylation. Since metal carbonyl complexes are so stable, the acyl metal hydride 1 (see Scheme 17) is readily decarbonylated and the resulting alkylmetal hydride 4 is reductively eliminated to yield an alkane and a metal carbonyl complex as the final products. Tsuji et al. first observed in 1965 that aldehydes were decarbonylated by Wilkinson´s catalyst at room temperature.[49]
[48] C. H Jun, E. A. Jo, J. W. Park, Eur. J. Org. Chem. 2007, 1869-1881. [49] T. Kondo, Y. Tsuji, Y. Watanabe, Tet. Lett. 1987, 28, 6229-6230 ; T. Kondo, M. Akazome, Y. Tsuji, Y. Watanabe, J. Org. Chem. 1990, 55, 1286-1291.
Background
16
[RhCl(PPh3)3] [RhCl(PPh3)2]- L
+ L
R H
O
R
O
RhPPh3
PPh3
HR
RhPPh3
COPPh3 R-H +
ClRh
PPh3
COPPh3
III
III
I
II
acyl metal hydride complex
Scheme 18. Mechanism for decarbonylation using Wilkinson´s catalyst.
Suggs later demonstrated this oxidative addition with the isolation of the hydro-acyl product of addition of an aldehyde to Wilkinson´s complex. [50]
Many strategies or methods were developed in hydroacylation chemistry to limit the undesired decarbonylation pathway. 2.2.2 Strategies to avoid decarbonylation[48]
Four methods are described to stabilize the acylmetal complex in order to avoid decarbonylation:
Use of a stable metallocyclic complex
Saturation of the metal complex by a labile alkene (ethylene, vinylsilane,etc.) or carbon monoxide at high pressure.
Coordination saturation by P-, S-, or O- coordinating atoms
- For intermolecular hydroacylation - For intramolecular hydroacylation
Use of a chelating auxiliary such as 2-amino-3-picoline
2.2.2.1 Use of a stable metallocyclic complex As five- or six-membered metallocyclic complexes are more stable than other-sized metallocyclic complexes, generating a five- or six-member acylmetallocyclic complex is one of the better ways to avoid decarbonylation.[51] The intramolecular hydroacylation of 4-pentenal to furnish cyclopentanone is one good example.[52]
[50] J. W. Suggs, J. Am. Chem. Soc. 1978, 100, 640-641. [51] C.-H. Jun, H. Lee, J.-B. Hong, B. I. Kwon, Angew. Chem. 2002, 114(12), 2250-2251; Angew. Chem. Int. Ed. 2002, 41(12), 2146-2147. [52] D. Y. Lee, I. J. Kim, C. H. Jun, Angew. Chem. 2002, 114(16), 3157-3159; Angew. Chem. Int. Ed. 2002, 41(16), 3031-3033.
Background
17
H
O L = phosphane
L3RhClO
O
Rh
H
L
L
ClL
O
Rh
L
H L
Cl Rh
O
L
LCl
L3RhCl
Scheme 19. Rhodium-catalyzed hydroacylation of 4-pentenal. This intramolecular hydroacylation process can be applied to the synthesis of cyclopentanone derivatives from 4-pentenal derivatives,[53] and by extension to the asymmetric synthesis of cyclopentanone derivatives.[54]
2.2.2.2 Saturation of the metal complex
In 1988, Milstein and co-workers stabilized the indenylrhodium (I) complex with a high pressure of ethylene.[55] Ethylene was the only alkene suitable in this reaction.
H
O
H2C CH2
(1000 psi)+(3.6 mol%)
C6D6, 100 °C
ORh
Scheme 20. Stabilisation of indenylrhodium(I) complex by ethylene saturation. Watanabe and co-workers used a high pressure of CO to stabilize the carbonylruthenium (0) complex intermediate.[56] This reaction required harsh reaction conditions (20 bars, 200 °C, 2 days), but could be used with a wide range of common olefins such as cyclohexene.
H
O
+
Ru3(CO)12
(1 mol%)
CO (20 kg.cm-2)
200 °C, 48 h
O
44%
Scheme 21. Stabilisation of carbonylruthenium (0) complex by carbon monoxide saturation.
[53] a) T. Sattelkau, P. Eilbracht, Tet. Lett. 1998, 1905-1908; b) M. Tanaka, M. Takahashi, E. Sakamoto, M. Imai, A. Matsui, M. Fujio, K. Funakoshi, K. Sakai, H. Suemune, Tetrahedron 2001, 57, 1197-1204; c) G. Kim, E. J. Lee, Tetrahedron: Asymmetry 2001, 12, 2073-2076; d) Y. Oonishi, A. Taniuchi, M. Mori, Y. Sato, Tet. Lett. 2006, 47, 5617-5621. [54] a) B. Bosnich, Acc. Chem. Res. 1998, 31, 667-674; b) G. C. Fu in Modern Rhodium-Catalyzed Reactions (Ed.: P. A. Evans), Wiley-VCH, New York, 2005, 79-91. [55] T. B. Marder, D. C. Roe, D. Milstein, Organometallics 1988, 7, 1451-1453. [56] T. Kondo, Y. Tsuji, Y. Watanabe, Tet. Lett. 1987, 28, 6229-6230 ; T. Kondo, M. Akazome, Y. Tsuji, Y. Watanabe, J. Org. Chem. 1990, 55, 1286-1291.
Background
18
In 1997, Lenges and Brookhart utilized a cobalt complex as a catalyst for the intermolecular hydroacylation of vinylsilane under mild conditions.[57]
O
H
N+ SiMe3
N
O
SiMe3
Co SiMe3Me3Si
(5 mol%)
benzene, 35 °C
82%
Me5
Scheme 22. Stabilisation of cobalt complex by vinylsilane. 2.2.2.3 Coordination saturation by P-, S-, or O-coordinating atoms for intermolecular hydroacylation In 1995, Lee and Jun used the chelating properties of phosphorus in the hydroacylation of o-(diphenylphosphanyl)benzaldehyde with 1-hexene in the presence of [(C8H14)2RhCl]2, 2-(diphenylphosphanyl)heptanophenone was obtained with a good yield (66%).[58], [59]
O
H
P
Ph
Ph+ n-C4H9
O
n-C4H9
P
Ph
Ph
[(C8H14)2RhCl]2
[Rh]
PPh
Ph
[Rh]OH
P
O
Ph
Ph
[Rh]
n-C4H9
H
P
O
Ph
Ph
[Rh]
n-C4H9
L
66%
(5 mol%)THF, 90 °C, 4 h
n-C4H9
Scheme 23. Hydroacylation of o-(diphenylphosphanyl)benzaldehyde with 1-hexene in the presence of [(C8H14)2RhCl]2.
Recently, Willis et al. realized a rhodium-catalyzed intermolecular hydroacylation between β-thioether-functionalized aldehydes and 1,3-disubstituted allenes.[60] He demonstrated the favoured position of the sulfur atom in the β-position yielding a stable five-membered metallocyclic hydride intermediate.
[57] C. P. Lenges and M. Brookhart, J. Am. Chem. Soc. 1997, 119, 3165-3166. [58] H. Lee, C. H. Jun, Bull. Korean Chem. Soc. 1995, 16, 66-68. [59] H. Lee, C. H. Jun, Bull. Korean Chem. Soc. 1995, 16, 1135-1138. [60] M. C. Willis, S. J. McNally, P. J. Beswick, Angew. Chemie 2004, 116(3), 344-347; Angew. Chem. Int. Ed. 2004, 42, 340-343.
Background
19
MeS O
H+
O
OMe
[Rh(dppe)]ClO4(10 mol%)
DCM, 60 °C, 2 h MeS O
O
O71%
Scheme 24. Hydroacylation of 3-methylsulfanyl-propionaldehyde with acrylic acid methyl ester in the presence of [Rh(dppe)]ClO4.
The first good example of a hydroacylation with an oxygen atom as a coordinating atom was the use of salicylaldehyde by Suemune et al.[61] In 1999, the first intermolecular hydroacylation of norbornene with salicylaldehyde using Wilkinson’s complex as catalyst was reported by the Miura group.[62] Unfortunately, the isolated yield of exo-hydroacylated norbornane was low (6-39%). Suemune et al. significantly optimised this rhodium-catalysed intermolecular hydroacylation of norbornenes and norbornadienes with salicylaldehydes. It was the first π-facial selective intermolecular hydroacylation.[61]
OH
H
O
+DCE, 80 °C, 1-3 d
OH O
O
OHRhCl(PPh3)3
(0.2 eq.)K3PO4
6 equiv.
+
exo endo
norbornene : full yield, exo-selectivenorboradiene : full yield, (20/1 exo:endo)
Scheme 25. Rh-catalysed π-facial selective intermolecular hydroacylation of norbornenes.
Nomura and co-workers used salicylaldehyde with 4-octyne in the presence of [Rh(COD)]2Cl2 and sodium carbonate as base and a dppf ligand under reflux to obtain quantitatively the α,β-unsaturated ketone.[62], [63]
O
H
OH
+ PrPr
[RhCl(COD)]2(1 mol%)
dppf, Na2CO3
toluene, reflux, 0.5 h
OH
O
Pr
Pr
100%
Scheme 26. Hydroacylation of 3-salicylaldehyde with 4-octyne in the presence of a rhodium catalyst.
[61] M. Tanaka, M. Imai, Y. Yamamoto, K. Tanaka, M. Shimowatari, S. Nagumo, N. Kawahara, H. Suemune, Org. Lett. 2003, 5, 1365-1367 ; M. Imai, M. Tanaka, K. Tanaka, Y. Yamamoto, N. Imai-Ogata, M. Shimowatari, S. Nagumo, N. Kawahara, H. Suemune, J. Org. Chem. 2004, 69, 1144-1150 ; K. Tanaka, M. Tanaka, H. Suemune, Tet. Lett. 2005, 46, 6053-6056. [62] K. Kokubo, K. Matsumasa, Y. Nishinaka, M. Miura, M. Nomura, Bull. Chem. Soc. Jpn. 1999, 72, 303-311. [63] K. Kokubo, K. Matsumasa, M. Miura, M. Nomura, J. Org. Chem. 1997, 62, 4564-4565.
* dppf : 1,1’-Bis(diphenylphosphino)ferrocene
Background
20
In 2007, Stemmler and Bolm[64] reported the asymmetric version of the work of Suemune et al.[61] After a screening of diverse rhodium sources, chiral phosphine ligands and additives, the best combination was obtained for the use of acetylacetonatobisethylene rhodium, potassium phosphate and a basic (S)-MonoPhos ligand.
Scheme 27. Rhodium-catalyzed asymmetric intermolecular hydroacylation reaction with salicylaldehydes
In 2010, Dong and co-workers made hydroacylations between homoallylic sulfides containing a substrate-bound directing group, and salicylaldehyde derivatives in the presence of a spiro-phosphoramidite ligand, (R)-SIPHOS-PE, and it gave α-branched ketones in >20:1 selectivity and up to 97% ee.[65]
H
OOH
R + SR"´R
[Rh(COD)Cl]2(R)-SIPHOS-PE
K3PO4, CH2Cl230 °C
OH O
R´
SR"
R
20:1 selectivity, up to 97% ee
Scheme 28. Regio- and enantioselective intermolecular hydroacylation: substrate-directed addition of salicylaldehydes to homoallylic sulfides.
Hydroacylation of salicylaldehyde was already used successfully in many other examples. Suemune and co-workers made the hydroacylation of salicylaldehyde with hexadiene at room temperature.[61] He obtained quantitatively (branched/linear = 4/1) the hydroacylated product. Finally, the same group tested the hydroacylation of salicylaldehyde with norbornene in mild conditions. They obtained the exo hydroacylated product in good yield (90%).[61]
Dong and co-workers also produced cyclopropylketones containing quaternary stereocenters, with diastereoselectivity and excellent enantiomeric excess using intramolecular Rh-catalysed hydroacylation.[66]
[64] R. T. Stemmler, C. Bolm, Adv. Synth. Catal. 2007, 349, 1185-1198. [65] M. M. Coulter, K. G. M. Kou, B. Kalligan, V. M. Dong, J. Am. Chem. Soc. 2010, 132, 16330. [66] D. H. T. Phan, K. G. M. Kou, V. M. Dong, J. Am. Chem. Soc. 2010, 132, 16354.
Background
21
H
HMe
Ar
+´Ar H
O Me Ar
H
Ar´O
5% Rh Josiphos
strain- release hydroacylation
95% ee, up to 20:1 dr
Scheme 29. Enantioselective desymmetrization of cyclopropenes by hydroacylation.
Willis et al. published an efficient alkyne hydroacylation of alpha-methylthiomethyl (MTM) ether substituted aldehydes and 1-alkynes in the presence of [Rh(dppe)]ClO4 to deliver α-O-MTM-substituted enone products. The obtained product can be hydrolyzed in the presence of silver nitrate to give the corresponding free hydroxyl compound.[67]
OMeS
R1
O
H H R2OMeS
R1
O
R2HO
R1
O
R2+
AgNO3
[Rh(dppe)ClO4](10 mol%)
DCE, 70 °C
24 examples55-91% yield
Scheme 30. O-Substituted alkyl aldehydes for rhodium-catalyzed intermolecular alkyne
hydroacylation : the utility of methylthiomethyl ethers.
The same year, the first rhodium-catalyzed branched-selective hydroacylation of alkynes was also developed by Willis et al.[68] After screening different ligands, they found the use of an ortho-isopropyl-dppe* ligand to deliver the branched-selective ligand (20:1 b/l).
[67] S. R. Parsons, J. F. Hooper, M. C. Willis, Org. Lett. 2011, 13(5), 998-1000. [68] C. Gonzalez-Rodriguez, R. J. Pawley, A. B. Chaplin, A. L. Thompson, A. S. Weller, M. C. Willis, Angew. Chemie 2011, 123(22), 5240-5244; Angew. Chemie Int. Ed. 2011, 50, 5134 –5138.
* dppe : 1,2-Bis(diphenylphosphino)ethane
Background
22
A third paper by Willis et al., very useful regarding its huge application in the synthesis of pharmaceutical drugs and agrochemical products, was published about the use of alkyne hydroacylation for the synthesis of highly substituted furans.[69] It is a tandem reaction with a rhodium-catalyzed intermolecular hydroacylation of S-chelating alkyl aldehydes with substituted propargylic alcohols to give γ-hydroxy-α-β-enones with 100% atom efficiency, followed in situ by an acid-catalyzed dehydrative cyclisation of the resulted hydroxyenones to the corresponding furans. The same procedure can be used for the preparation of various heterocycles (pyrroles, thiophenes, pyridazines).[68]
SMe O
HR1
[Rh(nbd)2][BF4](5 mol%)
dppe (5 mol%)+
DCE, 65 °C
OH
R3
R2
R1
O
R2 OH
R3
O
R2
R3
SMe
R1acid
15 examplesup to 93% yield
Scheme 32. An alkyne hydroacylation route to highly substituted furans.
In 2012, Dong and co-workers reported an intermolecular hydroacylation of allylic alcohols with salicylicaldehydes to afford β-hydroxy aryl ketones in good yields (73-94% yield) as single regioisomers.[70] The reactivity and regioselectivity were promoted by catalytic amounts of a phosphinite that forms a dynamic covalent bond with the allylic alcohol. Hydroacylation of 1,2 and 1,1-disubstituted olefins generates β-hydroxy aryl ketones bearing tertiary and quaternary centers, respectively.
Ar
O
H
[RhI]Ph2POMe
+NaOAc, DCE
R1
Ar
OOH
R1
OH
R2R2
11 examples73-94% yield
Scheme 33. β-Hydroxy ketones prepared by regioselective hydroacylation.
To avoid decarbonylation, a side reaction that limits the turnover number for catalytic hydroacylation, Dong et al. proposed the use of a chiral SIPHOS-PE ligand (see Scheme 35) for the enantioselective hydroacylation of substituted salicylicaldehydes and 1-octene with a low catalyst loading (2 mol%).[71] They proved that the use of SIPHOS-PE ligand favoured the turnover–limiting insertion by lowering the barrier for reductive elimination in the linear-selective pathway. A deuterium labeling study is also described and it showed that the branched insertion is fully reversible. They applied their method to the synthesis of eight biologically active octaketide natural products, including anticancer drug candidate cytosporone B.[71]
[69] P. Lenden, D. A. Entwistle, M. C. Willis, Angew. Chemie 2011, 123(45), 10845-10848 ; Angew. Chem. Ind. Ed. 2011, 50, 10657–10660. [70] S. K. Murphy, M. M. Coulter, V. M. Dong, Chem. Sci. 2012, 3, 355. [71] M. Von Delius, C. M. Le, V. M. Dong, J. Am. Chem. Soc. 2012, 134, 15022.
Background
23
OH
H
O
+
OH O
chiral ligand:
OHR2O
O
R1O O
R5
R3
R4
O
OP N
Ph
PhMe
Me
8 oktaketide natural productssynthesized via step- and atom- economical hydroacylation route
In 2012, the first rhodium-catalysed, linear-selective hydroacylation of alkynes was published.[72] After their work employing β-S-substituted aldehydes in a linear selective intermolecular reaction using either alkenes or alkynes, Willis et al. extended their scope with the use of electron deficient alkynes, particularly in combination with synthetically useful aryl aldehydes.[72]
SMe
H
O
R
R = hexyl, 3-thiophene or Ar(EWG-substituted)
+
[Rh(nbd)2BF4](5 mol%)
dcpe* (5 mol%)
acetone, RT, 1-2 h
SMe O
R
H
Scheme 35. Rhodium-catalysed linear-selective alkyne hydroacylation. A mechanistic study with the isolation of the vinyl intermediate that precedes reductive elimination in the alkyne intermolecular hydroacylation was published by Willis et al. in 2012.[73] This study included selective deuteration experiments that showed the irreversible insertion of the hydride, and also revealed that an interesting isomerization process is occuring between the two branched alkenyl protons that is suggested to occur via a metallacyclopropene intermediate.
[72] S.-J. Poingdestre, J. D. Goodacre, A. S. Weller, M. C. Willis, Chem. Commun. 2012, 48, 6354-6356. [73] R. Pawley, M. Huertos, G. Lloyd-Jones, A. S. Weller, M. C. Willis, Organometallics 2012, 31, 5650-5659.
* dcpe : 1,2-Bis(dicyclohexylphosphino)ethane
Background
24
2.2.2.4 Coordination saturation by P-, S-, or O-coordinating atoms for intramolecular hydroacylation Dong and co-workers worked on the intramolecular hydroacylation mediated by coordinating atoms. They worked not only with S-chelating groups, but also on analogues of salicylaldehyde with an O-chelating group. In both cases, the strategy was the same: they made seven- or eight- membered rings with the chelating atom in the α-position of the benzyl moiety.
In 2008, Dong and co-workers described the design and execution of a novel approach to form chiral lactones via C−H bond activation. The strategy featured an unprecedented enantioselective Rh-catalyzed hydroacylation of carbon−oxygen double bonds. Representative keto-aldehydes (derived from salicylaldehyde) underwent cyclization with complete regioselectivity to afford seven-membered lactones in great enantiomeric excess.[74]
O
H
OO
O
O
O
up to 99% eePh
[Rh((R)-DTBM-SEGPHOS)]BF4 (5 mol%)
CH2Cl2, RTH
Ph
Scheme 36. Hydroacylation of ether-linked substrates in the presence of [Rh(dppp)2]BF4 with a [Rh(R)-DTBM-SEGPHOS]BF4 catalyst.
In 2009, Dong and co-workers described the hydroacylation of sulphide-linked substrates. Hydroacylations were all performed using Wilkinson’s complex at room temperature in dichloromethane.[75]
S
O
H
S
O
S
O
S
O
62% 82% 65%
S
O
92%
RhCl(PPh3)3(5 - 10 mol%)
CH2Cl2, RT
Scheme 37. Hydroacylation of sulphide-linked substrates in the presence of Wilkinson’s catalyst.
[74] a) Z. Shen, P. K. Dornan, H. A. Khan, T. K. Woo, V. M. Dong, J. Am. Chem. Soc. 2009, 131, 1077-1091; b) Z. Shen, H. A. Khan, V. M. Dong, J. Am. Chem. Soc. 2008, 130, 2916. [75] M. M. Coulter, P. K. Dornan, V. M. Dong, J. Am. Chem. Soc. 2009, 131, 6932.
Background
25
In 2009, Dong and co-workers described also the first rhodium-catalyzed intramolecular olefin hydroacylation to produce medium-sized heterocyclic ketones with high regio- and enantiocontrol. Both α- and β-substituted ketones can be produced, depending on catalyst choice and substrate structure. In this stereoselective C−H bond functionalization, ethers, sulfides, and sulfoxides functional groups performed as effective directing groups. Moreover, the mechanistic studies were also detailed by Dong and co-workers.[75]
O
O
H
O
O
88%, 98% ee
[Rh((R,R')-Me-DuPhos)]BF4
(5 mol%)
CH2Cl2, RT
O
O
86%, 98% ee
S
O
89%, 97% ee
(R)-DTBM-SEGPHOS
S
O
89%, 97% ee
n = 1, 85%, 99% een = 2, 86%, 93% ee(R,R')-Me-DuPhos
n
Scheme 38. Enantioselective hydroacylation of ether-linked substrates in the presence of a Me-DuPhos, BDPP, DTBM-SEGPHOS catalysts.
S
O
H
O
O
91%, 95% ee
[Rh((R,R')-Me-DuPhos)]BF4
(5 mol%)
CH2Cl2, RT
[Rh((R,R')-Me-DuPhos)]BF4
(5 mol%)
CH2Cl2, RT
91%S
O
Scheme 39. Enantioselective hydroacylation of sulphide-linked substrates in the presence of a Me-DuPhos and BDPP catalyst.
[75] M. M. Coulter, P. K. Dornan, V. M. Dong, J. Am. Chem. Soc. 2009, 131, 6932.
Background
26
In 2010, Dong and co-workers published the first atom-economical approach to synthesize phthalides by using enantioselective ketone hydroacylation.[76] In the presence of Rh[(Duanphos)]X (X = NO3, OTf, OMs), various 2-ketobenzaldehydes undergo intramolecular hydroacylation to produce phthalides which are biologically relevant five-membered lactones found in herbs, fruits, and vegetables.
H
O
R RO
R´
[Rh(COD)Cl]2
Duanphos
AgX, toluene, 90 °CO
O
R´H
Phthalides 92-98% ee
Scheme 40. Phthalides by rhodium-catalyzed ketone hydroacylation.
In 2009, Glorius and co-workers described an intramolecular N-heterocyclic carbene (NHC)-catalyzed hydroacylation of unactivated double bonds.[77] Systematic variation of the catalyst structure revealed an N-mesitylthiazolylidene annulated with a seven-membered ring to be especially reactive. This NHC enables a unique C−C bond-forming reaction to afford substituted chroman-4-ones in moderate to excellent yields, even ones containing all-carbon quaternary centers.
O
HN S
ClO4
MeS
OMe
O 1,4-dioxane, 1 h, 120 °C O
OMe
O
Scheme 41. N-Heterocyclic carbene-catalyzed hydroacylation of unactivated double bonds.
In 2010, Glorius and co-workers described the N-heterocyclic carbene (NHC)-catalyzed hydroacylation of unactivated alkynes to provide α,β-unsaturated ketones.[78]
R
OR2
OR
O
R2N S
ClO4
MeS
5 mol%
K2CO3 (10 mol%), THF, 70 °C, 2 h
H
O
Scheme 42. N-Heterocyclic carbene-catalyzed cascade reaction involving the hydroacylation of unactivated alkynes.
[76] D. H. T. Phan, B. Kim, V. M. Dong, J. Am. Chem. Soc. 2009, 131, 15608. [77] K. Hirano, A. T. Biju, I. Piel, F. Glorius, J. Am. Chem. Soc. 2009, 131, 14190. [78] A. T. Biju, N. E. Wurz, F. Glorius, J. Am. Chem. Soc. 2010, 132, 5970-5971.
Background
27
In 2010, Glorius and co-workers demonstrated the compatibility of nucleophilic NHCs with electrophilic arynes with the conceptually new N-heterocyclic carbene catalyzed formal insertion arynes into the C-H bond of aldehydes. This NHC-catalyzed hydroacylation of arynes allows the conversion of aliphatic, α,β-unsaturated, and aromatic aldehydes into aryl ketones.[79]
N SClO4
MeS
Br
TMS
OTf
10 mol%
KF (2.0 eq)THF, 4 h, 25 °C
K2CO3 (20 mol%)
Br
OO
H+
Scheme 43. Intermolecular N-heterocyclic carbene-catalyzed hydroacylation of arynes.
In 2011, Dong and co-workers realized a nitrogen-directed ketone hydroacylation to furnish eight-membered N-containing lactones (benzoxazecinones) in high yields and high enantiomeric excesses.[80]
O
H
NO N
O
O
85% eeR
5% [Rh(L)]BF4
CH2Cl2, 8 hrsH
R
MeMe
L: (R)-3,4,5-OMe-MeOBIPHEP
Scheme 44. Nitrogen-directed ketone hydroacylation: enantioselective synthesis of benzoxazepinones and benzoxazecinones.
In 2011, Piel and Glorius reported a highly assymetric hydroacylation of unactivated olefins, resulting in the formation of twenty-one different chroman-4-one-type products in good yields and excellent enantioselectivities, and in each case building up a new all-carbon quaternary stereocenter.[81] A pre-study of the reaction mechanism of this NHC-catalyzed transformation was described based on DFT calculations.
O
HR1
X
R2
O
R1
X
R2
21 exemplesup to 99% yield
96-99% ee
O
N NN
MesBn
..DBU, 1,4-dioxane
80 °C, 20 h
Scheme 45. Highly asymmetric NHC-catalyzed hydroacylation of unactivated alkenes.
[79] A. T. Biju, F. Glorius, Angew. Chemie 2010, 122(50), 9995-9958; Angew. Chem. Ind. Ed. 2010, 49, 9761-9764. [80] A. H. Khan, K. G. M. Kou, V. M. Dong, Chem. Sci. 2011, 2, 407. [81] I. Piel, M. Steinmetz, K. Hirano, R. Fröhlich, S. Grimme, F. Glorius, Angew. Chemie 2011, 123, 5087-5091; Angew. Chem. Int. Ed. 2011, 50, 4983-4987.
Background
28
In 2012, Douglas et al. reported the rhodium-catalysed formation of 1-tetralones and 1-suberones via an intramolecular hydroacylation using a simple pyridine ligand and triphenylphosphine.[82] He obtained 3-methyl-1-tetralone in 65% yield using 2.5 mol% of catalyst, 5 mol% of triphenylphosphine and additives at 100 °C.
N NH2
R´
a : R´ = H
b : R´ = N
H
O
R
O
R
[RhCl(coe)2]2 (2.5 mol%)ligand (25 mol% a
or 100 mol% b)PPh3 (5 mol%)
aniline (1.2 equiv.)benzoic acid (10 mol%)
PhCF3, 100 °C
R = Me (65% yield a)R = Ph (48% yield a) (58% yield b)
N
H
O
N
Osame reaction
conditions
67% yield a
N
H
O
N
Osame reaction
conditions
69% yield a
Scheme 46. Cooperative Catalysis Approach to Intramolecular Hydroacylation. To avoid the undesired decarbonylation side-reaction, Saegusa et al. reported in 1990 the first Ni0/PR3-catalyzed intermolecular alkyne hydroacylation to give ketones.[83] Following this work, Ogoshi et al. investigated the mechanism of the Ni(0)/NHC hydroacylation, and they concluded that the reaction pathway proceeds through an oxo-nickelacyclic intermediate, and not through an acyl metal intermediate as described in our rhodium/P-N ligand strategy.[84]
They could apply this catalyst for the synthesis of a broad range of 1-indanone derivatives in good to excellent yields. Only the fluoro-substituted example did not work due to the deactivation of the catalyst by the oxidative addition of an Ar-Cl bond to nickel (0). They also synthesized six 1-tetralone derivatives in good yield and with very fast reaction times (1-2 h).
[82] E. V. Beletskiy, C. H. Sudheer, C. J. Douglas, J. Org. Chem 2012, 77, 5884-5893. [83] T. Tsuda, T. Kiyoi, T. Saegusa, J. Org. Chem. 1990, 55, 2554-2558. [84] Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi, S. Ogoshi, Angew. Chemie 2012, 124(43), 10970-10973; Angew. Chemie Int. Ed. 2012, 51(43), 10812-10815.
Background
29
H
OR2
n
O
nR2
R1
R1
Ni(COD)2/ItBu (5 mol%)
mesitylene, 130 °C
O O
OMe
O
Bn
O
SiEt3
O O
Cl
O
F
O
O OPhO O
OMe
O
FO
O
O O O
5h, 98% 6h, 97% 1h, 99% 2h, 99%
5h, 93%a 5h, 75% 6h, < 1% 6h, 92%
6h, 84% 24h, 54% 1h, 92% 1h, 99%
1h, 70% 2h, 99% 1h, 99% 2h, 75%
n = 1 or 2
ItBu :N
NC :
Scheme 47. Synthesis of five and six-membered benzocyclic ketones through intramolecular alkene hydroacylation catalyzed by nickel(0)/N-heterocyclic carbenes.
2.2.2.5 A new strategy (2013): enantioselective ketone hydroacylation using Noyori’s hydrogen transfer catalyst Inspired by the work of M. Krische,[85] S. K. Murphy and Vy M. Dong in 2013 reached a milestone with their enantioselective ketone hydroacylation using Noyori’s hydrogen transfer catalyst.[86]
[85] F. Shibahara, J. F. Bower, M. J. Krische, J. Am. Chem. Soc. 2008, 130, 14120-14122. [86] S. K. Murphy, Vy M. Dong, J. Am. Chem. Soc. 2013, 135, 5553-5556.
Background
30
Contrary to their rhodium-catalyzed strategy for the preparation of enantioriched lactones, the cyclization of 1,4-keto aldehydes failed to generate the corresponding γ-butyrolactones. They observed significant decarbonylation, probably due to the high flexibility of these substrates. The other classical strategies including N-heterocyclic carbenes NHCs,[87] ruthenium[88] or iridium hydrides,[89] gave poor reactivity and chemoselectivity. The use of an asymmetric hydrogen transfer catalyst with Noyori’s ATH ruthenium catalyst to form the 1,4-keto aldehyde substrate in situ by oxidation of a stable 1,4-keto alcohol gave good results at room temperature, without decarbonylation, utilizing an inexpensive transition metal.
R
O
OH
ROH
OH
RO
OH
H
O
R
OHH
O
R
O
NH2
Ru
TsNPh
Ph
Me
ClMe
Me
+"H2"
-"H2"
-"H2"
Ru-H catalyst
Noyori's ATH catalyst
R
O
OHO
R
O
5 mol % Noyori [Ru] cat./tBuONa
1.2 eq. acetone3 eq. iPrOH
EtOAc, 22 °C, 24 h10 examplesyield: 65-91%ee: 87-93%
R1
O
OH
R2R2 5 mol % Noyori [Ru] cat./tBuONa
1.2 eq. acetone
3 eq. iPrOHEtOAc, 22 °C, 24 h
O
O
R1R2
R2
10 examplesyield: 32-98%ee: 86-96%
Principle:
Scheme 48. Enantioselective ketone hydroacylation using Noyori’s hydrogen transfer catalyst.
[87] A. Chan, K. A. Scheidt, J. Am. Chem. Soc. 2006, 128, 4558. [88] S. Omura, T. Fukuyama, H. Murakami, Chem. Comm. 2009, 6741. [89] T. Suzuki, T. Yamada, K. Watanabe, T. Katoh, Bioorg. Med. Chem. Lett. 2005, 15, 258.
31
3. Aim of the work The aim of this Ph.D. thesis was to develop the concept of a new bifunctional catalyst system, to synthesize a library of new PNN pincer ligands containing an aminopicoline moiety and a phosphine function, to apply these new ligands to the formation of cyclic and acyclic ketones via an intra- or intermolecular hydroacylation respectively, and finally to complete a mechanistic study of the catalyst system. Decarbonylation is the most significant side-reaction of hydroacylation. Since metal carbonyl complexes are so stable, the acyl metal hydride intermediate complex is readily decarbonylated and the resulting alkylmetal hydride is reductively eliminated to yield an alkane and a metal carbonyl complex as the final products. Many strategies were described in the literature to stabilize the acylmetal complex in order to avoid decarbonylation.* The most efficient strategy was developed by Prof. Jun (University of Seoul) in 1994 called MOCC (Metal-Organic Cooperative Catalysis).[90], ** It combined the catalytic properties of a standard rhodium catalyst and an organic catalyst as 2-amino-3-picoline. Unfortunately, in many cases high loadings (between 20 and 100 mol%) of the aminopicoline cocatalyst are necessary to get efficient turnover.[90], [91] According to this method, we wondered whether tethering a phosphine binding site to the pyridine ring of this cocatalyst, thus generating a new bifunctional catalyst system, could be a more efficient solution.[92]
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
Figure 6. Hydroacylation of alkenes: Concept for a new bifunctional catalyst system LRh(I) and proposed transition state.
[90] Y. J. Park, J. W. Park, C. H. Jun, Acc. of Chem. Res. 2008, 41(2), 222-234. [91] C. H Jun, E. A. Jo, J. W. Park, Eur. J. Org. Chem. 2007, 1869-1881. [92] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637.
* Look at p. 16 to see more details about the different strategies to prevent the decarbonylation. ** Look at p. 36 to see more details about the MOCC (Metal-Organic Cooperative Catalysis).
Aimofthework
32
The catalyst design was based on the following reflections: the aminopicoline moiety would on the one hand, act as a reversibly bound directing group allowing for facile C-H activation while simultaneously preventing the undesired decarbonylation through formation of an o-vinylbenzaldimine.[92] Integration of the phosphine function would, on the other hand, not only form an active hydroacylation catalyst, but also enhance the binding constant of the directing group to the catalytically active rhodium center and would thus increase the effective concentration of the rhodium catalyst, further promoting the C-H activation step.[83] Finally, the role of the 3-positioned methyl group would be to induce a specific imine conformation, which will place the imino C-H bond in close proximity to the catalytically active metal center, again facilitating the oxidative addition of the C-H bond. The use of a cocatalyst would thus become unnecessary.
By modifying the length of the amino and phosphino side arms, and replacing the pyridine core unit by a standard phenyl ring, the reactivity of the pincer ligands can be significantly impacted. The interaction with the rhodium center in the cavity of the different PNN ligand can be finely tuned. For each substrate in the inter- and intramolecular hydroacylation of alkenes, the same procedure will be used:
Optimization of reaction conditions (screening of diverse neutral and cationic rhodium catalysts, temperature, scale of the reaction, ratio ligand/catalyst, catalyst loading, reaction time,etc.) with the standard PNN ligand.
Screening a library of PNN ligands.
Determining the scope of compatible substrates (electron-donating group or electron-withdrawing, aliphatic or aromatic,etc.).
N NH2
Ph2P
N NH2
Ph2P
N NH2
Ph2P
N NH2
Ph2P
Figure 7. The standard 6-[[(diphenylphosphanyl)-methyl]-3-methylpyridin-ylamine ligand.
[83] T. Tsuda, T. Kiyoi, T. Saegusa, J. Org. Chem. 1990, 55, 2554-2558. [92] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637.
Aimofthework
33
In order to clarify the mechanism of this new catalytic system, several mechanistic studies will be performed:
Isolation and characterisation of a rhodium-P-N ligand complex.
Using preformed imines in a stoechiometric reaction with an alkene.
Comparative study of the imine formation rate for the 3-substituted methyl and the 3-substituted methoxy PNN ligands.
Control experiment with a ligand lacking the amino functionality.
Deuterium-labeling study with deuterated benzaldehyde and 1-octene.
34
4. Results and Discussion 4.1 Ligand concept and synthesis 4.1.1 General approach for the construction of cyclometalated phosphine-based pincer complexes[25]
Bernard Shaw working with Brian Weeks was the first in 1973 to focus his research on the development of cyclometalated phosphine-based pincer complexes.[93] They reported that diphosphines of the types tBu2P(CH2)nP
tBu2 (n= 9, 10 or 12) with the very bulky tertbutylphosphine donor groups form large-ring complexes of types trans-[{PdCl2[
tBu2P(CH2)10PtBu2]}x] (x = 1,2 or 3) and trans-[{IrCl(CO)[tBu2P(CH2)10P
tBu2]}x].
P
P
Pt ClCl
tBu
tButBu
tBu
Figure 8. The mononuclear trans-[{PtCl2[tBu2P(CH2)10P
tBu2]}] complex.
Quickly after their publication, the idea of pincer complexes emerged as a hot topic covering all aspects of modern organometallic chemistry. Over the past decade, the cyclometalated phosphine-based pincer complexes were efficiently used for dehydrogenation, Heck olefin arylation, Suzuki biaryl coupling reactions,etc.[25]
[25] a) M. Albrecht, G. van Koten, Angew. Chemie 2001, 20, 3866-3898; Angew. Chem Int. Ed. 2001, 40, 3750-3781. b) M. E. van der Boom, D. Milstein, Chem. Rev. 2003, 103, 1759-1792. c) N. Selender and K. Szabó, Chem. Rev., 2011, 111(3), 2048–2076. [93] a) E. Miller, B. L. Shaw, J. Chem. Soc, Dalton Trans. 1974, 480-485. b) B. L. Shaw, M. F. Uttley, J. Chem. Soc, Chem. Commun. 1974, 918-919. c) C. E. Jones, B. L. Shaw, B. L. Turtle, J. Chem. Soc., Dalton Trans. 1974, 992-999. d) C. J. Moulton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1976, 1020-1024. e) H. D. Empsall, E. M. Hyde, R. Markham, W. S. McDonald, M. C. Norton, B. L. Shaw, J. Chem. Soc., Chem. Commun. 1977, 589-590. f) C. Crocker, R. J. Errington, W. S. McDonald, K. J. Odell, B. L. Shaw, J. Chem. Soc., Chem. Commun. 1977, 589-590. g) N. A. Al-Salem, H. D. Empsall, R. Markham, B. L. Shaw, B. J. Weeks, J. Chem. Soc., Dalton Trans. 1979, 1972-1982. h) N. A. Al-Salem, W. S. McDonald, R. Markham, M. C. Norton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1980, 59-63. i) C. Crocker, R. J. Errington, R. Markham, C. J. Moulton, K. J. Odell, B. L. Shaw, J. Am. Chem. Soc. 1980, 102, 4373-4379. j) R. J. Errington, B. L. Shaw, J. Organomet. Chem. 1982, 238, 319-325. k) C. Crocker, R. J. Errington, R. Markham, C. J. Moulton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 387-395. l) C. Crocker, H. D. Empsall, R. J. Errington, E. M. Hyde, W. S. McDonald, R. Markham, M. C. Norton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 1217-1225. m) J. R. Briggs, A. G. Constable, W. S. McDonald, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 1225-1230. n) R. J. Errington, F. E. McDonald, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 1829-1835.
ResultsandDiscussion:Ligandconceptandsynthesis
35
The generic structure of the pincer complexes is a typical ECE architecture in which E is a bulky donor chelating group such as an imine, a phosphorus, a sulfur or a selenium. To diversify the strategy for using the pincer ligands, the two different donors on the side arms could be different. The palladium pincer ligands have many advantages; they are thermally stable due to their tridentate coordination, and resistant in most cases toward moisture and air, they allow for storage under normal conditions and easy handling. By modifying the steric and electronic properties of the different donor groups on their side arms, the reactivity of the pincer ligands can be significantly modified.
MY
XnLm
E E
E = NR2, PR2, SR, SeR
Y = C, N, ...
R
- chiral pocket- steric contrains
- anchoring site- remote electronic modulations
- cavity for metal binding with tunable accessibility- sites for counterions or ancillary ligands
- hardness/softness- metal-binding rigidity- steric constrains of substituents- coordinating 2 e- donor or free Lewis base
Figure 9. The generic structure of a metal pincer complex.
The role of pincer ligands for the selectivity of the catalysis is crucial. Under ambient conditions, the oxidation state of palladium is largely restricted to Pd(II). Thus in the d8 square planar Pd(II) pincer complexes, only one free coordination site is available for catalysis. This implies that formation of undesired side products arising from ligand exchange processes can be avoided. However, the synthesis of pincer complexes is sometimes considered as a limitating factor for their application in catalysis. Moreover, the reductive elimination of the pincer ligand to give Pd(0) leads to a usually irreversible cleavage of the Pd-Y bond, which eventually leads to decomposition of the complex. 4.1.2 Use of a chelation auxiliary This strategy developed by Jun in 1994 is called MOCC (Metal-Organic Cooperative Catalysis). It combined the catalytic properties of a standard rhodium catalyst and an organic catalyst as 2-amino-3-picoline.[89]
N NH2
Figure 10. 2-amino-3-picoline.
ResultsandDiscussion:Ligandconceptandsynthesis
36
This strategy was used to develop our ligand, and is described in the following catalyst design part. 4.1.3 Catalyst Design In 1979, Suggs published the activation of aldehyde carbon-hydrogen bonds through oxidative addition in route to the formation of 3-methyl-2-aminopyridyl aldimines-rhodium based catalytic system.[94] Jun and co-workers developed particularly a new efficient and selective hydroacylation of terminal alkenes using aldehyde by a chelation-assisted catalytic system.[95] Their new concept, called Metal-Organic Cooperative Catalysis (MOCC) combined the well-known catalytic properties of Wilkinson’s catalyst and the 2-amino-3-picoline (organic catalyst), which enables the formation of a temporary aldimine. 2-amino-3-picoline is a temporary chelating auxiliary, which stabilizes the acyl-metal hydride species. This stabilisation allows for an efficient C-H activation while simultaneously preventing the undesired decarbonylation through formation of an o-vinylbenzaldimine.
R H
O N N
R H R'
ConventionalHydroacylation
N N
R R'R'
R R'
O
N NH2
New Catalytic Cycleof Organic Molecules
(Ph3P)3RhCl
Figure 11. Comparative scheme of the MOCC hydroacylation system using 2-amino-3-picoline with the conventional system.
The position 3 of the methyl group plays a key-role, as it makes free rotation of the imine group rather difficult. The resulting geometrical bias also facilitates the cleavage of the imino C-H bond through decreasing the distance between the catalyst and the C-H bond.
[94] J. W. Suggs, J. Am. Chem. Soc. 1979, 101, 489. [95] D. Y. Lee, I. J. Kim, C.-H. Jun, Angew. Chem. 2002, 114(12), 2250-2251; Angew. Chem. Int. Ed. 2002, 41(12), 2146-2147.
ResultsandDiscussion:Ligandconceptandsynthesis
37
Figure 12. The key-role of 3-positionned methyl group. Jun in particular described the catalytic effect of 2-amino-3-picoline in hydroacylation of an olefin with aldehydes.[90], [91] Jun’s system gave good results in the hydroacylation reaction, but the high loading of the 2-amino-3-picoline catalyst (between 40 and 100 %) is a drawback.
N N
Rh ClL
L
O
+toluene, 150 °C, 48 h
Ph
2-amino-3-picoline (100 mol%)
(Ph3P)3RhCl (5 mol%)
O
84%
trace
Scheme 49. An exemple of hydroacylation used the Jun´s sytem with 100% of 2-amino-3-picoline. After several years of investigation, Prof. Jun and Prof. Lee completed their strategy for the rhodium-catalyzed intermolecular hydroacylation, with the help of two additives, aniline and benzoic acid.[96] They succeeded in isolating the desired final ketone in a nearly quantitative yield after one hour.
[96] a) C.-H. Jun, D. Y. Lee, H. Lee, J.-B- Hong, Angew. Chemie 2000, 112(17), 3214-3216; Angew. Chem. Int. Ed. 2000, 39, 3070-3072. b) C.-H. Jun, J. H. Lee, Pure Appl. Chem. 2004, 76(3), 577-587.
ResultsandDiscussion:Ligandconceptandsynthesis
38
The aniline, which is more nucleophilic than 2-amino-3-picoline, is first reacted with benzaldehyde to give an aldimine. The transimination of the resulting aldimine with 2-amino-3-picoline, facilitated by the benzoic acid, gives a second aldimine and regeneration of the aniline. After undergoing the classical hydroacylation pathway as described before, the final ketone is obtained.
O
H+
N
H
N
H
N N N
O
NH2
H2O
NH2N
H2N
+ benzoic acid
H2O
2-amino-3-picoline (20 mol%)
98%
Scheme 50. Rhodium-catalyzed intermolecular hydroacylationwith the help of two additives: aniline and benzoic acid.
Another revelant example published by Jun et a l. is the direct one-pot synthesis of ketones from primary alcohols and terminal alkenes using a transition metal catalyst and 2-amino-3-picoline as a temporary chelating auxiliary.[97] In this method, the starting aldehyde is directly prepared in situ by oxidation through hydrogen transfer by Wilkinson´s catalyst. The stoechiometric amount of chelating auxiliary and the three days of reaction time are the main drawback of this method.
O
H
[(Ph3P)3RhCl](10 mol%)OH
N NH2
-H2O
N N
H
N N
(100 mol%)
OH2O
N NH2
-74%
Scheme 51. One-pot synthesis of ketones from primary alcohols and terminal alkenes.
Thus, our strategy was to combine the diphenylphosphine part of the Wilkinson’s catalyst together with the 2-amino-3-picoline co-catalyst in one ligand.[92] This strategy would allow for decreasing the loading of 2-amino-3-picoline. Ligand 1 was used in conjunction with a neutral rhodium catalyst [Rh(COD)Cl]2 or a cationic rhodium catalyst [Rh(COD)2]BF4, as rhodium represents the main metal used in hydroacylation.
N NH2
Ph2P
N NH2
Ph2P
N NH2
Ph2P
N NH2
Ph2P
Figure 13. The standard 6-[[(diphenylphosphanyl)-methyl]-3-methylpyridin-ylamine ligand 1. 4.1.4 Synthesis of ligands 1 and 7[92]
The 6-[[(diphenylphosphanyl)-methyl]-3-methylpyridin-ylamine ligand 1 was prepared in six steps via its 2-amino-6-methylpyridine precursor.[92]
2-Amino-6-methylpyridine a was protected quantitatively by pivaloylchloride at room temperature overnight to give the corresponding protected product b. This was then methylated in meta position through directed ortho-lithiation, followed by a quench with iodomethane to yield 74% of the dimethylated product c. The Schlosser base (mix of DIPA, n-BuLi and tBuOK) was used to selectively convert the 6-methyl group of c to give 72% of d. Caesium fluoride and hexachloroethane were used to chlorinate d yielding e with 88 % yield, stirring in concentrated HCl solution gives f (72% of yield). Finally, the diphenylphosphine group is introduced by substitution of the chlorine via a dissolving metal reduction to give 80% of ligand 1.
[92] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637.
ResultsandDiscussion:Ligandconceptandsynthesis
40
1) n-BuLi, THF, 90 min, -78 °C to 0 °C2) MeI, o/n, 0 °C to RT
a
PivCl, Et3N, CH2Cl2, o/n, 0 °C to RT
N NH2 N NH
O
b
quant.
N NH
O
c
74%
N NH
O
d
72%
Si
88% CsF, C2Cl6, MeCN, 5 h, 60 °C
N NH
O
eCl
1) Schlosser Base, THF, -78 °C2) TMSCl, THF, o/n, -78 °C to RT
Scheme 52. Synthesis of 6-[[(diphenylphosphanyl)-methyl]-3-methylpyridin-ylamine ligand 1.
Removal of the phthalimide group has shown to be not compatible with the adjacent methoxy group; decomposition of the substrate or yield inferior to 10% were observed under a wide range of standard reaction conditions (N2H4, basic or acidic conditions). Fortunately, its deprotection under Birch conditions, followed by the nucleophilic attack of the diphenylphosphide moiety allowed the formation of ligand 41 in an improved yield (27%).
Scheme 53. Synthesis of 6-[(diphenylphosphanyl)-methyl]-3-methoxy-pyridin-2-ylamine, ligand 7. The 6-[(diphenylphosphanyl)-methyl]-3-methoxy-pyridin-2-ylamine, ligand 7 was prepared in five steps starting from 3-hydroxy-6-methyl-2-nitropyridine 2.[92] First, 3-methoxy-6-methyl-2-nitropyridine 3 was synthesized by a Williamson ether synthesis of 2 with iodomethane and K2CO3.
[98] Then, 3 was reduced with NaBH4 and NiCl2·6H2O to give the amine 4. The latter was protected by phthalic anhydride to give the corresponding protected compound 5. The next step was a radical bromination of 5 with N-bromosuccinimide activated by azobisisobutyronitrile in benzene. In the final step, the ligand 7 was obtained via a Birch reduction, followed by deprotection of the amine.
[98] M. Malamas, W. Fobare, W. Solvibile, F. Lovering, J. Condon, A. Robichaud, U. S. Pat. Appl. Publ., 20060173049, 03 Aug. 2006 (ligand 7, step 1).
ResultsandDiscussion:Ligandconceptandsynthesis
42
4.1.5 Synthesis of a library of bifunctionnal ligands[92]
Although Jun’s system is an efficient method for the hydroacylation reaction, the high loading of cocatalyst (between 20 and 100 mol%) is a real drawback. We thus investigated the design and application of new bifunctional P-N ligands, combining the properties of Jun´s aminopyridine cocatalyst together with a phosphine moiety, and its further application in the rhodium-catalyzed intra- and intermolecular hydroacylation of alkenes using a 1:1 Rh/Ligand (5-10 mol%) ratio, therefore decreasing dramatically the amount of 2-amino-3-picoline. Several ligands have thus been prepared with different linkers between the pyridine core, the phosphorus atom and the amine residue.
N NH2P
43
N NH2
22
P
NP
36
NH2
NP
41
NH2
N NH2
P
11
N NH2
P
7
O
NH2
P
15
O
N NH2
31
P
N NH2
27
P
Figure 14. Library of bifunctionnal P-N ligands While common organic transformations were used to form the aforementioned ligands, two keys steps must be notably pointed out in terms of their efficiency and their scarce use in the literature. The first reaction was the generation of diphenylphosphide under Birch type conditions from triphenylphosphine and its nucleophilic substitution of a suitably halogenated substrate. This method proved to be quite effective as a last step towards those ligands.
n = 0, 1, 2 PPh3, Na (2 eq.)
NH3(liq)
X = Cl, Br
FGR
X
n = 0, 1, 2FGR
PPh2
Scheme 54. A first key-step in the bifunctionnal P-N ligands synthesis: the dissolving metal reaction.
ResultsandDiscussion:Ligandconceptandsynthesis
43
The other interesting reaction was the radical bromination of a picoline or tolyl moiety, in the presence of a phthalimide protected amine. Indeed, several standard amine protecting groups have only shown poor yields in this reaction, probably related to the presence of the free hydrogen on the nitrogen atom.
Scheme 55. A second key-step in the bifunctionnal P-N ligands synthesis : the radical bromination with N-bromosuccinimide NBS and azobisisobutyronitrile, as a radical initiator.
Ligand 11 was prepared according to literature procedures.
4.1.5.1 Synthesis of 6-[(diphenylphosphanyl)-methyl]-pyridin-2-ylamine 11[99],
[100]
The 6-[(diphenylphosphanyl)-methyl]-pyridin-2-ylamine, ligand 11 was prepared in four steps via 6-methyl-pyridin-2-ylamine a. Amine a was protected in good yield (95%) by phthalic anhydride to give the corresponding protected product 8. The next step was a radical bromination of 8 with N-bromosuccinimide activated by azobisisobutyronitrile in benzene for sixteen hours. The phthalimide protecting group was cleaved under diluted acidic conditions to yield 78% of 6-chloromethyl-pyridin-2-ylamine 10. Finally, the diphenylphosphine group was introduced by substitution of the chlorine with diphenylphosphide generated under Birch type conditions to give 70 % of ligand 11.
Scheme 56. Synthesis of 6-[(diphenylphosphanyl)-methyl]-pyridin-2-ylamine, ligand 11.
[99] X. Collin, J.-M. Robert, G. Wielgosz, G. Le Baut, C. Bobin-Dubigeon, N. Grimaud, J.-Y. Petit, Eur. J. of Med. Chem. 2001, 36(7-8), 639-649 (ligand 11, step 1). [100] G. Shyamaprosad, D. Swapan, J. Subrata, A. Avijit Kumar, Chemistry Letters 2004, 33(7), 916-917 (ligand 11, step 2, same reaction butlower yield (40%).
ResultsandDiscussion:Ligandconceptandsynthesis
44
4.1.5.2 Synthesis of 6-(diphenylphosphinomethyl)-2-methoxyaniline 15 The 5-[(diphenylphosphanyl)-methyl]-2-methoxy-phenylalanine, ligand 15 was prepared in three steps via 2-methoxy-5-methyl-phenylamine 12. 2-methoxy-5-methyl-phenylamine 12 was protected in good yield (86%) by phthalic anhydride to give the corresponding protected product 13. The next step was a radical bromination of 13 with N-bromosuccinimide activated by azobisisobutyronitrile in benzene for sixteen hours. Finally, the diphenylphosphine group is introduced by substitution of the chlorine with diphenylphosphide generated under Birch type conditions, and the phthalimide protecting group was cleaved during the following acidic work-up to perform the final ligand 15 in 27% yield.
Scheme 57. Synthesis of 6-(diphenylphosphinomethyl)-2-methoxyaniline, ligand 15. 4.1.5.3 Synthesis of 8-diphenylphosphanyl-quinolin-2-ylamine 23[101], [102]
The 8-diphenylphosphanyl-quinolin-2-ylamine, ligand 23 was prepared in six steps via 2-bromo-phenylamine 16 and 3-phenyl-acryloyl chloride 17. The α,β-unsaturated amide 18 was obtained under basic conditions from the amine 16 and the acyl chloride 17 in good yield (92%). Then, the resulting amide 18 was quickly cyclized in a hot chlorobenzene solution to the quinolinone 19 (56% yield). Phosphoryl chloride was used as a chlorinating, dehydrating agent to form the chloroquinoline 20, which was converted with 75% yield to its corresponding aminoquinoline 21. The next step was a standard BOC protection to obtain 22. The final Birch reduction, followed by a BOC deprotection by TFA at room temperature (63% for the two steps), gave the final quinoline ligand 23.
[101] F. Cottet, M. Marull, O. Lefebvre, M. Schlosser, Eur. J. Org. Chem. 2003, 1559-1568 (ligand 23, steps 1, 2 and 3). [102] Y. Cheng, T. C. Judd, M. T. Bartberger, J. Brown, K. Chen, R. T. Fremeau Jr., D. Hickman, S. A. Hitch-cock, B. Kordan, V. Li, P. Lopez, S. W. Louie, Y. Luo, K. Michelsen, T. Nixey, T. S. Powers, C. Rattan, E. A. Sickmeier, D. J. St. Jean Jr., R. C. Wahl, P. H. Wen, S. Wood, J. Med. Chem. 2011, 54(16), 5836-5857 (ligand 23, step 4, same reaction but lower yield).
ResultsandDiscussion:Ligandconceptandsynthesis
45
K2CO3, H2O, acetone, 2 h, 0 °C
92%
16
Br
NH2
O
Cl+
17
O
N
H Br
18
56%
NH
O
Br
AlCl3, chlorobenzene2 h, 125 °C
19
57%
POCl3, 2 h, 125°C
Br
N Cl
20
75%N NH2
Br
NH4OH, 16 h, 190 °C
21
78%
N NH
Br
o/n, 60 °C
22
O
O
O
O
O
O
O 1) n-BuLi, THF, 30 min, -78 °C2) PPh2Cl, o/n, -78 °C to 0 °C3) TFA, CH2Cl2, 2 h, RT
63% N NH2
P
23
Scheme 58. Synthesis of 8-diphenylphosphanyl-quinolin-2-ylamine, ligand 23.
ResultsandDiscussion:Ligandconceptandsynthesis
46
4.1.5.4 Synthesis of 6-(2-diphenylphosphanyl-ethyl)-3-methyl-pyridin-2-ylamine 27
a
PivCl, Et3N, CH2Cl2o/n, 0 °C to RT
N NH2N N
H
O
b
quant.
1) n-BuLi, THF, 90 min. -78 °C to 0 °C2) MeI, o/n, 0 °C to RT
Scheme 59. Synthesis of 6-(2-diphenylphosphanyl-ethyl)-3-methyl-pyridin-2-ylamine, ligand 27.
A similar synthetic strategy* was adopted to prepare ligand 27, which differs from ligand 31 by the presence of a 3-methyl group in the pyridine nucleus, aiming at inducing a specific imine conformation, which will locate the imino C-H bond in close proximity to the catalytically active metal center. The 6-(2-diphenylphosphanyl-ethyl)-3-methyl-pyridin-2-ylamine, ligand 27 was prepared in six steps from 2-amino-6-methylpyridine a with a overall yield of 21%. 2-Amino-6-methylpyridine a was protected quantitatively by pivaloylchloride at room temperature overnight to give the corresponding protected product b. This was then methylated in meta position through directed ortho-lithiation, followed by a quench with iodomethane to yield 74% of the dimethylated product c. The ethanoyl compound 24 was obtained by lithiation of the 6-methyl group of c, quenched with formaldehyde. Then, the chlorination of 24 with CCl4 gives 25 in moderate yield (71%). The pivaloyl protecting group was cleaved under acidic conditions to yield 70% of 2-amino-6-chloroethyl-3-methyl-pyridine 26. Finally, the diphenylphosphine group is introduced by substitution of the chlorine via a Birch reaction to give 68% of ligand 27.
* Look at p. 47 for the synthetic strategy (Scheme 60).
ResultsandDiscussion:Ligandconceptandsynthesis
47
4.1.5.5 Synthesis of 6-(2-diphenylphosphanyl-ethyl)-pyridin-2-ylamine 31[91]
A similar building block was used in the synthesis of ligand 31, as the one already developed for the synthesis of ligand 1. The hydroxyl-ethyl pyridine 28 was converted to its corresponding N-[6-(2-bromo-ethyl)-pyridin-2-yl]-2,2-dimethyl-propionamide 29 in moderate yield (72%). The final ligand 31 was obtained in two remaining steps: the pivaloyl group was cleaved under diluted acidic conditions and the diphenylphosphine group was introduced by substitution of the chlorine with diphenylphosphide generated under Birch type conditions (48% after 2 steps).
Scheme 60. Synthesis of 6-(2-diphenylphosphanyl-ethyl)-pyridin-2-ylamine, ligand 31. Aromatic amino phosphines ligand 36 and ligand 41 were synthesized from readily available starting materials, prepared according to literature procedures. 4.1.5.6 Synthesis of 2-(6-diphenylphosphinopyridin-2-yl)propan-2-amine 36[103], [104]
The 1-(6-diphenylphosphanyl-pyridin-2-yl)-1-methyl-ethylamine, ligand 36 was prepared in four steps via 2,6-dibromopyridine 32. The lithiation of 32 followed by the addition of excess acetone and subsequent quenching with ammonium chloride aqueous solution results in the formation of 2-bromo-6-(2-hydroxyisopropyl)pyridine 33 in high yield (96%). In the presence of BF3·Et2O, the tertiary alcohol 33 reacted with acetonitrile under reflux conditions to afford N-[1-(6-bromopyridin-2-yl)-1methylethyl]acetamide 34 in 30% yield, with 50% of the starting material 33 subsequently recovered. The final ligand 36 was obtained in two remaining steps: the acetyl group was cleaved under diluted acidic conditions and the diphenylphosphine group was introduced by substitution of the chlorine with diphenylphosphide generated under Birch type conditions (76% after 2 steps).
[91] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637. [103] D. Song, R. H. Morris, Organometallics 2004, 23, 4406-4413 (ligand 36, steps 1 and 2). [104] J. Hu, PCT Int. Appl., 2009131814, 29 Oct. 2009 (ligand 36, step 3, similar conditions but lower yield (81%).
Scheme 61. Synthesis of 2-(6-diphenylphosphinopyridin-2-yl)propan-2-amine, ligand 36.
4.1.5.7 Synthesis of C-(6-diphenylphosphanyl-pyridin-2-yl)-methylamine 41 The C-(6-diphenylphosphanyl-pyridin-2-yl)-methylamine, ligand 41 was prepared in four steps via 2-bromo-6-methylpyridine 37. The amino-phthalimide protected C-(6-bromo-pyridin-2-yl)-methylamine 39 was obtained in two remaining steps: a radical monobromination with NBS and AIBN, followed by an analog Gabriel synthesis with the potassium salt of phthalimide (23% after 2 steps). The phthalimide protecting group was cleaved under diluted acidic conditions to yield 81% of C-(6-bromo-pyridin-2-yl)methylamine 40. Finally, the diphenylphosphine group was introduced by substitution of the chlorine with diphenylphosphide generated under Birch type conditions to give 70% of ligand 41.
Scheme 62. Synthesis of C-(6-diphenylphosphanyl-pyridin-2-yl)-methylamine, ligand 41.
4.1.5.8 Synthesis of 6-diphenylphosphanyl-pyridin-2-yl-amine 43[105], [106]
As described in the Ph.D. manuscript of Dr. W. Seiche (Group of Prof. Dr. B. Breit, 2005), the 6- diphenylphosphanyl-pyridin-2-yl-amine, ligand 43 was obtained in one step from 6-bromo-pyridin-2-ylamine 42 via a dissolving metal reduction with triphenylphosphine.
Scheme 63. Synthesis of 6-diphenylphosphanyl-pyridin-2-yl-amine, ligand 43. A wide range of electronically and sterically diverse amino phosphine ligands could thus be easily accessed in a few steps. With those ligands in hands, we chose the model reaction between benzaldehyde and 1-octene to probe their influence on the outcome of this reaction, under optimized conditions.
[105] M. Weis, C. Waloch, W. Seiche, B. Breit, J. Am. Chem. Soc. 2006, 128(13), 4188-4189. [106] Dr. W. Seiche (PhD thesis, Breit group, 2009).
50
4.2 Scope of rhodium catalyzed intermolecular hydroacylation 4.2.1 Introduction
In 2000, Jun and co-workers published in Angewandte Chemie a highly active catalyst system for intermolecular hydroacylation.[107] The advantages of the method are the reaction time, and the good yield, the main drawbacks are the high loading of the chelating auxiliary (20 mol%) and the use of two additives in significant amount, aniline (60 mol%) and benzoic acid (6 mol%).
R H
O+ R´
R
O
R´
[Rh(PPh3)3Cl] (2 mol%)aniline (60 mol%)
2-amino-3-picoline (20 mol%)benzoic acid (6 mol%)
toluene, 130 °C, 1 h
O
O
O
O
O
Si
O F
F
FF
F
O
O
O
O
O
F
FF
O
N
O
O
98%a
83%a
99%a
84%a
95%a, b
98%a, c
95%a, c
79%a
71%a
60%a
95%a
71%a, d
5 eq.
a Yield of product after isolation. b 1.1 equivalents of vinyl trimethylsilane was used. c Reaction time was 40 min. d 10% of the aldol condensation product of 3-phenylpropionaldehyde was obtained.
Scheme 64. Jun´s method applied to the rhodium-catalyzed intermolecular hydroacylation.
To improve this method, we decided to explore a new kind of ligand combining the diphenylphosphine part of the Wilkinson’s catalyst together with the 2-amino-3-picoline co-catalyst in one ligand. We started our investigation with the optimisation of the rhodium catalyzed intermolecular hydroacylation of 1-octene with benzaldehyde.
4.2.2 Optimization of the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde We started our optimisation to mimic the reaction condition developed by Pr. Jun and co-workers using three self made P-N ligands with THF and 10 mol% of rhodium (III) trichloride trihydrate at 150 °C. In the three cases, with our quinoline P-N ligand 22, the γ-phosphino methyl P-N ligand 27, and our standard methyl P-N ligand 1, low yields of hydroacylation product was obtained after 4 days (<5 to 17%) (entries 2-3-4, Table 1). The change of solvent to an aprotic apolar solvent as toluene showed a positive impact on the reaction with the same P-N ligands. A noteworthy increase in yield (34%) was obtained with our γ-phosphino methyl P-N ligand 27 (entry 6), and to a lesser extent (17%) with our standard ligand 1 (entry 7). The methyl group in the position 3 of our PNN ligand series plays a crucial role in inducing the imine conformation and facilitating the oxidative addition of the C-H bond. To prove it by practice, no reaction was observed for the 3-unmethylated ligands 11 and 31 (entries 8-9). The use of DCE as a polar aprotic solvent had no positive influence in the reaction (entries 10-11).
H
O
+
[RhCl3.3H2O] (10 mol%)
THF, 4 d, 150 °C
Oligand (100 mol%)
PPh3 (30 mol%)
Scheme 65. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: ligand screening.
Table 1. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: ligand screening.
benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [RhCl3.3H2O] (0.022 mmol, 6.0 mg), ligand 1 (0.066 mmol, 20 mg).l Determined by 1H-NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes. According to the Table 2, we continued our study with toluene as solvent, and changed the rhodium source. Chloro (1,5-cyclooctadiene) rhodium (I) dimer [Rh(COD)Cl]2 was very often described as a neutral rhodium source in the C-H activation, for its reactivity and its great stability to air and moisture. The screening of five PNN ligands with [Rh(COD)Cl]2 was a milestone in our project. Excellent reproducible results ranging from 90% to 93% were obtained with the use of 10 mol% of neutral rhodium catalyst and our standard ligand 1 at 150 °C, validating the positive synergetic effect of aminopicoline and phosphine moieties once combined within bifunctional ligand (entry 2). Our γ-phosphino methyl P-N ligand 27 gives also a good yield (68%) (entry 1). A significant decrease in the yield was observed for the two corresponding 3-unmethylated P-N ligands 11 and 31, respectively 46% and 33% (entries 3 and 4). No reaction was observed in the case of the quinoline P-N ligand 22 (entry 5).
H
O
+
[Rh(COD)Cl]2 (10 mol%)
toluene, 4 d , 150 °C
O
1 : 1 lig/cat
Scheme 66. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: ligand screening (2).
Table 2. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: ligand screening (2).
entry
P-N ligand
[Rh] catalyst
conditions
NMR yield (%)f
1a
N NH2
Ph2P Ligand 27
[Rh(COD)Cl]2
10 mol%
Toluene (200 μL), 150 °C, 4 d
68
2b
N NH2
PPh2 Ligand 1
[Rh(COD)Cl]2
10 mol%
Toluene (200 μL), 150 °C, 4 d
90-93
3c
N NH2
PPh2 Ligand 11
[Rh(COD)Cl]2
10 mol%
Toluene (200 μL), 150 °C, 4 d
46
(continued on the next page)
ResultsandDiscussion:Intermolecularhydroacylation
54
4d
N NH2
Ph2P Ligand 31
[Rh(COD)Cl]2
10 mol%
Toluene (200 μL), 150 °C, 4 d
33
5e
N NH2
PPh2 Ligand 22
[Rh(COD)Cl]2
10 mol%
Toluene (200 μL), 150 °C, 4 d
0
a benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [Rh(COD)2]Cl2 (0.022 mmol, 10.8 mg), ligand 27 (0.044 mmol, 14.1 mg). b benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [Rh(COD)2]Cl2 (0.022 mmol, 10.8 mg), ligand 1 (0.044 mmol, 13.5 mg). c benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [Rh(COD)2]Cl2 (0.022 mmol, 10.8 mg), ligand 11 (0.044 mmol, 12.8 mg). d benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [Rh(COD)2]Cl2 (0.022 mmol, 10.8 mg), ligand 31 (0.044 mmol, 13.4 mg). e benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [Rh(COD)2]Cl2 (0.022 mmol, 10.8 mg), ligand 22 (0.044 mmol, 14.4 mg). f Determined by 1H-NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes. A screening of the influence of different rhodium precursors was initiated. Reducing the rhodium catalyst loading to 5 mol% with the same ratio ligand/catalyst with [Rh(COD)Cl]2 gave a good result 82% (entry 1, Table 3). A similar good result (81%) was obtained with the dicarbonylacetylacetonato rhodium (I) catalyst [Rh(CO)2acac] (entry 4). The same reaction conditions with chloro 1,3-cycloheptadiene rhodium (I) dimer [Rh(cycloheptadiene)Cl]2 and rhodium (I) carbonyl chloride [Rh(CO)Cl]2 was unsuccessful, respectively 27% and 31% (entries 2 and 3). We chose to continue our optimisation with [Rh(COD)Cl]2 as a rhodium catalyst for the ease of synthesis.
H
O
+
[Rh] source (10 mol%)
toluene, 24 h, 150 °C
O
1 : 1 lig/cat
N NH2
PPh2
(10 mol%)
Scheme 67. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: rhodium catalyst screening.
ResultsandDiscussion:Intermolecularhydroacylation
55
Table 3. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: rhodium catalyst screening.
entry
[Rh] catalyst
conditions
NMR yield (%)e
1a
[Rh(COD)Cl]2
5 mol%
Toluene (200 μL), 150 °C, 24 h
82
2b
[Rh(cycloheptadiene)Cl]2
5 mol%
Toluene (200 μL), 150 °C, 24 h
27
3c
[Rh(CO)Cl]2
5 mol%
Toluene (200 μL), 150 °C, 24 h
31
4d
[Rh(CO)2acac]
10 mol%
Toluene (200 μL), 150 °C, 24 h
81
a benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(COD)2]Cl2 (0.011 mmol, 5.5 mg), ligand 1 (0.022 mmol, 6.5 mg). b benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(cycloheptadiene)2]Cl2 (0.011 mmol, 5.1 mg), ligand 1 (0.022 mmol, 6.5 mg). c benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(CO)2]Cl2 (0.011 mmol, 4.3 mg), ligand 1 (0.022 mmol, 6.5 mg). d benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(CO)2acac] (0.022 mmol, 5.7 mg), ligand 1 (0.022 mmol, 6.5 mg). e Determined by 1H NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes.
The unsaturated coupling partner must be in excess in comparison to the aldehyde partner, in order to react fast with the acyl metal hydride intermediate complex, and avoid the decarbonylation pathway. With a high excess of alkene (5 eq.), a moderate yield was obtained (50%) at 100 °C in toluene with 5 mol% of [Rh(COD)Cl]2 (entry 1, Table 4). A decrease of the amount of alkene by two allows performing the best conditions with a good yield of 68% (entry 2). Under 2% of catalyst, the yield was drastically reduced, respectively to 59% with 2 equivalents (entry 3) and 9% under equimolar condition (entry 4).
Pr. Dong et al. showed in 2012 in their mechanistic study of the rhodium-phosphoramidite catalyzed alkene hydroacylation of salicylaldehyde, that the use of an alkene excess (eight-fold) increases the rate of the hydroacylation and disfavours decarbonylation.[108] An analysis by 31P-NMR of her reaction mixture proved that a relatively small amount of CO complex was formed. The major species observed was the resting state complex [Rh(COD)(ligand)]. In contrast, a predominance of the CO complex was observed in 31P NMR with a lack of alkene.
In 2007, Brookhart and co-workers concluded in their mechanistic studies of the intermolecular addition of aromatic aldehydes to olefins catalyzed by Rh(I) olefin complexes, that the hydroacylation reaction has a first-order dependence on aldehyde concentration and an inverse first-order dependence on olefin concentration when aromatic aldehydes are employed. [109]
[108] M. Delius, C. M. Le, V. M. Dong, J. Am. Chem. Soc. 2012, 134(36), 15022-32. [109] A. H. Roy, C. P. Lenges, M. Brokkhart, J. Am. Chem. Soc. 2007, 129(7), 2082-2093.
ResultsandDiscussion:Intermolecularhydroacylation
56
H
O
+
[Rh(COD)Cl]2 (5 mol%)
toluene, 24 h, 100 °C
O
1 : 1 lig/cat
1 to 5 eq.
N NH2
PPh2
(10 mol%)
Scheme 68. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: rhodium catalyst screening.
Table 4. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: rhodium catalyst screening.
(0.011 mmol, 5.5 mg), ligand 1 (0.022 mmol, 6.5 mg). c benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.44 mmol, 66 μL), [Rh(COD)2]Cl2 (0.011 mmol, 5.5 mg), ligand 1 (0.022 mmol, 6.5 mg). d benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.22 mmol, 32 μL), [Rh(COD)2]Cl2 (0.011 mmol, 5.5 mg), ligand 1 (0.022 mmol, 6.5 mg). e Determined by 1H NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes. The rhodium-to-ligand ratio is a very important factor for optimal activity of the catalytic reaction. With an excess of three equivalents of ligand in comparison with the rhodium catalyst, no reaction was observed (entry 2, Table 5). This is due to the acyl metal hydride intermediate complex saturated with the excess of ligand. No alkene can be added under the acyl metal hydride complex. We finally used for the synthesis of our scope published in Chemical Communications a ligand to rhodium ratio of 1.3:1.[92] [92] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637.
ResultsandDiscussion:Intermolecularhydroacylation
57
H
O
+[Rh(COD)Cl]2 (10 mol%)
toluene, 1 d , 150 °C
O
N NH2
PPh2
(10-30 mol%)
Scheme 69. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: ratio ligand/catalyst
Table 5. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: ratio ligand/catalyst.
(0.022 mmol, 10.8 mg), ligand 1 (0.066 mmol, 40.5 mg). c Determined by 1H NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes. Jun and co-workers described an increased reactivity in the intermolecular hydroacylation with the use of benzoic acid.[107] The role of benzoic acid might be to catalyze the condensation of the aldehyde coupling partner and the P-N ligand to generate the aldimine intermediate.[91] Unfortunately, we observed a decrease of 11% of the yield with 5% of benzoic acid (entry 2, Table 6). With 10 mol% of benzoic acid, the yield falls drastically by 25% (entry 3). Therefore, the use of benzoic acid showed no positive effect in our reaction.
H
O
+
[Rh(CO)2acac] (10 mol%)
toluene, 24 h, 150 °C
O
P-N ligand 1 (10 mol%)
benzoic acid (0-10 mol%)
Scheme 70. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: benzoic acid effect.
[107] C.-H. Jun, D.-Y. Lee, H. Lee, J.-B. Hong, Angew. Chemie 2000, 112(17), 3214-3216; Angew. Chem. Int. Ed. 2000, 39(17), 3070-3072. [92] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637.
ResultsandDiscussion:Intermolecularhydroacylation
58
Table 6. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: benzoic acid effect.
entry
P-N ligand
[Rh] catalyst
benzoic acid (mol %)
conditions
NMR yield(%)d
1a
N NH2
PPh2 Ligand 1
[Rh(CO)2acac]
10 mol%
0
THF (200 μL)
150 °C, 1 d
76
2b
N NH2
PPh2 Ligand 1
[Rh(CO)2acac]
10 mol%
5
THF (200 μL),
150 °C, 1 d
65
3c
N NH2
PPh2 Ligand 1
[Rh(CO)2acac]
10 mol%
10
THF (200 μL),
150 °C, 1 d
25
a benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(CO)2acac] (0.022 mmol, 5.7 mg), ligand 1 (0.022 mmol, 6.8 mg). b benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), benzoic acid (0.011 mmol, 1.3 mg), [Rh(CO)2acac] (0.022 mmol, 5.7 mg), ligand 1 (0.022 mmol, 6.8 mg). c benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), benzoic acid (0.022 mmol, 2.5 mg), [Rh(CO)2acac] (0.022 mmol, 5.7 mg), ligand 1 (0.022 mmol, 6.8 mg). d Determined by 1H-NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes. Finally, we studied the effect of various additives in the intermolecular hydroacylation of benzaldehyde with 1-octene. As described before, aniline and benzoic acid are used successfully in the Jun system. The first one, more nucleophilic than 2-amino-3-picoline, is first condensed with benzaldehyde to give a first aldimine, used as an active intermediate species. Then, the transimination of this resulting aldimine with 2-amino-3-picoline gives a second aldimine with the help of benzoic acid and regeneration of the aniline. In our case, the use of 1.6 equivalents of aniline and 0.6 equivalent of benzoic acid in our standard reaction conditions, showed a decrease of the yield by 14% (entry 1, Table 7). Just the addition of 0.3 equivalent of benzoic acid gives a reduction of the yield (entry 2), confirming the results described before (entries 2 and 3). Montmorillonite K10[110] is a phyllosilicate mineral constituent of volcanic ash weathering product, bentonite. This green clay catalyst contains both Brønsted and Lewis acidic sites, with the Brønsted site mainly associated with the interlamellar region and the Lewis sites mainly associated with main sites. It is used in many applications such as the deprotection of tetrahydropyranyl ethers, the Biginelli reaction, or the Fischer indole cyclisation. Montmorillonite K10 is inefficient in our reaction; just traces of product was detected by 1H NMR (entry 4). [110] N. Kaur and D. Kishore, J. Chem. Pharm. Res. 2012, 4(2), 991-1015.
ResultsandDiscussion:Intermolecularhydroacylation
59
H
O
+
[Rh(COD)Cl]2 (5 mol%)
toluene, 24 h, 100 °C
O1 : 1 lig/catadditives
Scheme 71. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: additives.
Table 7. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: additives.
(0.011 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.8 mg). c benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 11 (0.022 mmol, 6.8 mg). d benzaldehyde (0.22 mmol, 25 μL), 1-octene (1.08 mmol, 169 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 31 (0.022 mmol, 6.8 mg). e Determined by 1H NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes. To improve the yield of the Stille reaction, lithium chloride is often added to the reaction mixture.[111] This reagent stabilizes the intermediate complex formed by the oxidative addition of a catalyst and accelerates the reaction. By analogy, we observe negative effects after addition of one equivalent of lithium chloride in our hydroacylation reaction. A poor yield of 23% was obtained (entry 2, Table 8). Quaternary ammonium salts are often used as green and mild catalysts.[112] The most well-known application in the literature is the use of chiral quaternary ammonium salts derived from the cinchona alkaloid family of natural products for the asymmetric alkylations, Michael additions, Darzens reactions, etc. In our case, the additionnal use of methyl diethylbenzyl ammonium chloride gave a moderate yield of 63%, a satisfying result but with a loss of 18% (entry 3).
[111] J. K. Stille, Angew. Chemie 1986, 98(6), 504-519; Angew. Chem. Int. Ed. 1986, 25, 508-524. [112] J. Novacek and M. Waser, Eur. J. Org. Chem. 2013, 637–648.
ResultsandDiscussion:Intermolecularhydroacylation
60
H
O
+
[Rh(CO)2acac] (10 mol%)
toluene, 24 h, 150 °C
O1 : 1 lig/cat
N NH2
PPh2
(10 mol%)
additives
Scheme 72. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: additives (2). Table 8. Rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene: additives (2).
entry
additives (1 eq.)
[Rh] catalyst
conditions
NMR yield (%)d
1a
-
[Rh(CO)2acac]
10 mol%
Toluene (200 μL)
150 °C, 24 h
81
2b
LiCl
[Rh(CO)2acac]
10 mol%
Toluene (200 μL)
150 °C, 24 h
23
3c N
+Cl -
[Rh(CO)2acac]
10 mol%
Toluene (200 μL)
150 °C, 24 h
63
a benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(CO)2acac] (0.022 mmol, 5.7 mg), ligand 1 (0.022 mmol, 6.8 mg). b benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), LiCl (0.22 mmol, 1mg), [Rh(CO)2acac] (0.022 mmol, 5.7 mg), ligand 1 (0.022 mmol, 6.8 mg). c benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), methyl diethylbenzyl ammonium chloride (0.22, 4.3 mg), [Rh(CO)2acac] (0.022 mmol, 5.7 mg), ligand 1 (0.022 mmol, 6.8 mg). d Determined by 1H-NMR methods. All reactions were performed under argon in 8 mL Schlenk tubes.
4.2.3 Our best reaction conditions for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde
Scheme 73. Best reaction conditions for rhodium-catalyzed hydroacylation of benzaldehyde and 1-octene.
ResultsandDiscussion:Intermolecularhydroacylation
61
In summary of our optimisation of the reaction parameters, we can conclude:
The temperature: 150 °C, we succeed to decrease with moderate yields (50-60%) at 120 °C, but under 120 °C a significant decrease of the yield was observed.
The solvent: toluene, more polar solvents were tested unsuccessfully, low yield for THF (up to 17%) and no reaction for DCE.
The ligand: “Me” PNN ligand 1, the yield was halved for the corresponding 3-unmethylated PNN ligand 11.
The analog γ-phosphino methyl PNN ligand 27 also gave a moderate yield (68%).
The rhodium catalyst: [Rh(COD)Cl]2, similar results were reported for [Rh(CO)2acac], [Rh(CO)Cl]2, [RhCl3·3H2O], [Rh(cycloheptadiene)Cl]2 give lower yields (respectively 31%, 27% and 34%).
The time reaction: 24h.
The alkene proportion: 2.5 eq. of alkene in comparison with the aldehyde. Under equimolar conditions, no reaction was observed.
The ligand to rhodium ratio: 1.3:1, a significant excess of ligand annihilates the reaction.
The concentration: c = 1.1 mol.L-1.
The additives: No additives were tested successfully: benzoic acid, aniline, lithium chloride, quaternary ammonium salt, montmorillonite K10.
4.2.4 Proof of concept for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde Before the screening of diverse alkenes with substituted benzaldehydes, we proved the concept of our new bifunctionnal catalyst system with three control experiments:
Use of ligand L’ , lacking the amino functionality
N
PPh2
Ligand L´ (229)
Scheme 74. Ligand L’ lacking the amino functionnality. [107] C.-H. Jun, D.-Y. Lee, H. Lee, J.-B. Hong, Angew. Chemie 2000, 112(17), 3214-3216; Angew. Chem. Int. Ed. 2000, 39(17), 3070-3072.
ResultsandDiscussion:Intermolecularhydroacylation
62
N NH2
Ph2P
our system
[Rh]
[Rh(PPh3)3Cl]
Wilkinsoncatalyst
N NH2
[Rh(PPh3)3Cl]
Jun system
N
Ph2P
control systemlacking the NH2
[Rh]+ cocatalyst (20 mol%)
L L'
entry 2 entry 3 entry 4entry 1
Scheme 75. Control experiments for the intermolecular hydroacylation. Wilkinson´s catalyst on its own was completely inefficient in this transformation (entry 1, Table 9). However, upon adding 2-amino-3-picoline (20 mol%) as a cocatalyst, appreciable yield of 76% of the ketone product could be obtained as already reported by Jun et al (entry 2, Table 9). [107] In addition, a control experiment with ligand L’, lacking the amino functionality, resulted in a complete lost of activity in the reaction, thus enhancing the importance of the amino group within ligand 1 for the reaction to proceed via iminium formation and the prevention of decarbonylation (entry 4, Table 9). Table 9. Proof of concept for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde.
entry
[Rh]/ligand 1:1 (mol%)
Cocatalyst (mol%)
NMR yield (%)e
1a
10
-
0
2b
10
2-amino-3-picoline (20)
76
3c
10
-
82
4d
10
-
0
a benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(PPh3)3Cl] (0.022 mmol, 10.1 mg). b
benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(PPh3)3Cl] (0.022 mmol, 10.1 mg), 2-amino-3-picoline (0.022 mmol, 4.4 μL). d benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(COD)2]Cl2 (0.011 mmol, 5.5 mg), ligand 1 (0.022 mmol, 6.5 mg). c benzaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 85 μL), [Rh(COD)2]Cl2 (0.011 mmol, 5.5 mg), ligand L´ (0.022 mmol, 6.1 mg). e Determined by 1H-NMR methods. All reactions were performed under argon in 8 mL Schlenk tubes.
4.2.5 Screening of our P-N ligand library in our best reaction condition for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde We next turned our attention toward the ligand screening of our P-N ligand library in the intramolecular hydroacylation and chose o-vinylbenzaldehyde as a model substrate. [107] C.-H. Jun, D.-Y. Lee, H. Lee, J.-B. Hong, Angew. Chemie 2000, 112(17), 3214-3216; Angew. Chem. Int. Ed. 2000, 39(17), 3070-3072.
ResultsandDiscussion:Intermolecularhydroacylation
63
Table 10. Screening of P-N ligands for the intermolecular hydroacylation of 1-octene and benzaldehyde.
entry
P-N ligand (reference + structure)
NMR yield (%)e
1a
Ligand 1
N NH2
Ph2P
82
2b
Ligand 7
N NH2
Ph2P
O
0
3c
Ligand 11
N NH2
Ph2P
17
4d
Ligand 15
NH2
Ph2P
O
0
5e
Ligand 22
N NH2
Ph2P
0
6f
Ligand 27
N NH2
Ph2P
56
7g
Ligand 31
N NH2
Ph2P
24
8h
Ligand 36
NPh2P
NH2
12
(continued on the next page)
ResultsandDiscussion:Intermolecularhydroacylation
64
entry
P-N ligand (reference + structure)
NMR yield (%)e
9i
Ligand 41
NPh2P
NH2
33
10j
Ligand 43
N NH2Ph2P
61
All reactions are performed with benzaldehyde (0.22 mmol, 24 μL), 1-octene (0.55 mmol, 85 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), 24 h, 150 °C, neat, 8 mL Schlenk tubes. a Ligand 1 (0.022 mmol, 6.7 mg). b Ligand 7 (0.022 mmol, 7.2 mg). c Ligand 11 (0.022 mmol, 6.4 mg). d Ligand 15 (0.022 mmol, 7.2 mg). e Ligand 27 (0.022 mmol, 7.0 mg). f Ligand 31 (0.022 mmol, 6.7 mg). g Ligand 36 (0.022 mmol, 7.0 mg). h Ligand 41 (0.022 mmol, 6.4 mg). i Ligand 43 (0.022 mmol, 6.1 mg). j Determined by 1H NMR methods. We observed a significant influence of ligand backbone in the intermolecular hydroacylation of benzaldehyde and 1-octene, with a privileged balance between steric and electronic effects, found in ligand 1 (entry 1, Table 10).
4.2.6 Scope of benzaldehyde with diverse alkenes Due to the highest reactivity of the benzaldehyde (mesomeric effect), we start the scope for the rhodium-catalyzed intermolecular hydroacylation with benzaldehyde and diverse alkenes. Benzaldehyde is easily converted by air oxidation to benzoic acid. Jun showed the positive impact of benzoic acid in the hydroacylation.[107] Therefore, to prevent the impact of benzoic acid in the obtained results, benzaldehyde was freshly distilled before every catalytic reaction. THF was also freshly distilled under a system of sodium/benzophenone to obtain a moisture, oxygen, and peroxide-free solvent. [Rh(COD)Cl]2 is not highly air-sensitive, and could be weighed outside the glovebox.
4.2.6.1 Benzaldehyde hydroacylation of alken-1-ol and hydroxyl-protected derivatives To start the scope, a series of catalytic hydroacylation reactions between benzaldehyde and several alken-1-ols was performed. Three relevant points must be considered for these substrates:
The possible extra binding of the hydroxyl function to the rhodium center
The weight of the substrate: small spacer (volatility problem) and large spacer (solubility problem)
The steric hinderance close to the alkene function [107] C.-H. Jun, D.-Y. Lee, H. Lee, J.-B. Hong, Angew. Chemie 2000, 112(17), 3214-3216; Angew. Chem. Int. Ed. 2000, 39(17), 3070-3072.
ResultsandDiscussion:Intermolecularhydroacylation
65
The hydroacylation of benzaldehyde with 5-hexen-1-ol 47 succeeded in obtaining 75% of 7-hydroxy-1-phenyl-heptan1-one 48 (identified by the characteristic triplet for the formed C(2)H2 at 2.9-3.0 ppm) (entry 1, Table 11). In decreasing the spacer to two (but-3-en-1-ol) and one (propen-1-ol), we drastically decreased the yield, respectively, to 18% (entry 2) and 20% (entry 3). No reaction was observed with the bulky 2-methyl-but-3-en-2-ol (entry 4). The benzaldehyde hydroacylation with a acetate-protected 5-hexen-1-ol yielded 44% of the final ketone, and suggestes a required interaction of the hydroxyl group with the rhodium center (entry 5). Table 11. Rhodium-catalyzed hydroacylation of benzaldehyde and alken-1-ols.
The catalytic hydroacylations of benzaldehyde 44 with styrene 49 and α-methylstyrene were not conclusive. We had hoped for a gain in reactivity due to the electronic mesomeric effect of the benzene ring to the alkene function. No reaction was observed with cumene (entries 1 and 2, Table 12). Poor results were obtained for the hydroacylation of styrene (45% at 150 °C (entry 3, 50) and 10% at 100 °C (entry 4)). The polymerization of styrene (and α-methylstyrene) was in competition with the desired hydroacylation above ca. 100 °C. In both cases, the observed polymerization was due to the high stability of the benzyl radical (styrene)
ResultsandDiscussion:Intermolecularhydroacylation
66
and tertiary radical (cumene) intermediates. Moreover, the low conversions could be explained by the steric hindrance in α-position (general remark for the hydroacylation of α,α’-disubstitued-alkenes). To prove the problem of steric hindrance of styrene and cumene, the hydroacylation of benzaldehyde 44 with allyl benzene 51 was performed in a good 72% isolated yield at 150 °C (entry 5, 52). In this case, less polymerization was observed due to the less stability of the secondary radical indermediate. Finally, the negative contribution of the mesomeric donor phenyl group on the alkene was attested by the moderate isolated yield (55%) obtained for the hydroacylation of benzaldehyde 44 and 4-vinylcyclohexene 53 (entry 6, 54), and the moderate yield (44%) obtained for the 4-vinylcyclohexane 55 (entry 7, 56). Table 12. Rhodium-catalyzed hydroacylation of 2 or 3-cyclosubstituted alkenes.
(0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg).f Determined by 1H NMR methods. g Isolated yield. 4.2.6.3 Benzaldehyde hydroacylation of alkenamine No reaction was observed for the hydroacylationa of benzaldehyde and allyl amine (Scheme 76), similar to the result obtianed for the allyl alcohol (entry 3, Table 10).
H
O
+
O[Rh(COD)Cl]2 (5 mol%)ligand 1 (10 mol%)
ratio lig/cat 1:1
neat, 150 °C, 24 h2.5 eq.
NH2NH2
no reaction
Scheme 76. Rhodium-catalyzed hydroacylation of benzaldehyde with allyl amine.
a benzaldehyde (0.22 mmol, 84 μL), allyl amine (0.55 mmol, 41 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg. b Determined by 1H-NMR methods. 4.2.6.4 Benzaldehyde hydroacylation of alkenoic acid A similar result (51%) was obtained for the hydroacylationa of benzaldehyde 44 with 5-hexenoic acid 57 (Scheme 77, 58) than for the weight similar acetate-protected 5-hexen-1-ol (entry 5, Table 11).
H
O
+
O[Rh(COD)Cl]2 (5 mol%)ligand 1 (10 mol%)
ratio lig/cat 1:1
neat, 150 °C, 24 h2.5 eq.
51% yieldb
OH
O
44 57 58
OH
O
Scheme 77. Rhodium-catalyzed hydroacylation of benzaldehyde with 5-hexenoic acid.
a benzaldehyde (0.22 mmol, 84 μL), 5-hexenoic acid (0.55 mmol, 65 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg. b Isolated yield. 4.2.6.5 Benzaldehyde hydroacylation of vinyl alkenoate The hydroacylationa of benzaldehyde 44 with acrylic acid methyl ester 59 was successful in obtaining 73% isolated yield of 4-oxo-4-phenyl-butyric acid methyl ester 60 (Scheme 78).
ResultsandDiscussion:Intermolecularhydroacylation
68
H
O
+
O[Rh(COD)Cl]2 (5 mol%)ligand 1 (10 mol%)
ratio lig/cat 1:1
neat, 150 °C, 24 h
2.5 eq.
OMeOMe
73% yieldb
44 59 60
OO
Scheme 78. Rhodium-catalyzed hydroacylation of benzaldehyde with acrylic acid methyl ester.
4.2.6.6 Benzaldehyde hydroacylation of small bulky alkene (3,3-dimethyl-butene and trimethyl-vinyl-silane) No reaction was observed for the hydroacylation of benzaldehyde with 3,3-dimethyl-butene 61 and trimethyl-vinyl-silane 63. These results confirmed the negative effect of steric hinderance and elctronic inductive donor group (entries 1 and 2, Table 13, 62 and 64). Table 13. Rhodium-catalyzed hydroacylation of benzaldehyde with small bulky alkene.
entry
aldehyde
alkene
conditions
NMR yieldc (%)
1a
H
O
61
24 h, 150 °C, neat
0 (19d)
product 62
2b H
O
Si
63
24 h, 100 °C, neat
Traces
product 64
a benzaldehyde (0.22 mmol, 84 μL), 3,3-dimethyl-butene (0.55 mmol, 71 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg). b benzaldehyde (0.22 mmol, 84 μL), trimethyl-vinyl-silane (0.55 mmol, 80 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg). c Determined by 1H-NMR methods. d Jun’s conditions. 4.2.7 Scope of substituted benzaldehyde with 1-octene To extend the scope of this transformation, we describe herein a screening of 1-octene with a wide range of electronically substituted benzaldehydes. 4.2.7.1 Use of electron-rich benzaldehydes
We report the positive effect of electron-donating group substitution of the benzaldehyde coupling partner. A meta- 65 and para- 67 substituent with a strong electron-donating
ResultsandDiscussion:Intermolecularhydroacylation
69
methoxy group gave respectively 91% and 93% yield (entries 2 and 3, Table 14, 66 and 68). These two substrates were the best results obtained for the hydroacylation obtained with our bifunctional system, higher than the reference reaction between benzaldehyde and 1-octene (entry 1). A moderate yield (65%) was performed with the weakly donating group 4-biphenyl-carbaldehyde 69 (entry 4, 70). Surprisingly, a small amount (< 5%) of hydroacylated product was observed in the 1H NMR with 2-bromo-4,5-dimethoxy-benzaldehyde (entry 5) in comparison with the good results obtained with 3- and 4-anisaldehydes. This result could be explained by the steric hinderance in ortho-position with respect to the aldehyde. No reaction was noted for 3- and 4-hydroxybenzaldehydes, despite the strong donating effect of the hydroxyl group (entries 6 and 7). The weak EDG-effect of the two methyl substituted 2,4-dimethylbenzaldehyde could explained the lack of yield observed (entry 8). Table 14. Rhodium-catalyzed hydroacylation of EDG-substituted benzaldehydes with 1-octene.
In a previous report from Jun, the hydroacylation was favoured with electron-rich benzaldehydes and electron-poor alkenes.[107] Nevertheless, one example with an -substituted benzaldehyde was successfully obtained.[107] The hydroacylation of p-chlorobenzaldehyde 71 and 1-octene 45 yield 70% of 1-(4-chloro-phenyl)-nonan-1-one at the standard conditions (entry 2, Table 15, 72). The other exemples of fluoro- or bromobenzaldeydes gave no reaction at all (entry 3, 4 and 5).The hydroacylation with the most strongly deactivated nitro substituted substrate corroborated this tendency (entry 6).
Table 15. Rhodium-catalyzed hydroacylation of EWG-substituted benzaldehydes with 1-octene.
4.2.7.3 Use of 2-naphthaldehyde and 3-thiophene carbaldehyde
To finish the diversity of the aromatic aldehydes, the hydroacylation of 1-octene with 2-naphthaldehyde 73 was achieved in low yield (11%) (entry 2, Table 16). A similar poor yield (8%) was obtained with 3-thiophene carbaldehyde 75 (entry 3, 76). To be noted, an excellent yield (96%) for this thiophene substrate was obtained using Jun’s reaction conditions.
Table 16. Rhodium-catalyzed hydroacylation of 2-naphthaldehyde and 3-thiophene carbaldehyde with 1-octene
(0.022 mmol, 10.8 mg), ligand 1 (0.066 mmol, 40.5 mg). c 3-thiophene carbaldehyde (0.22 mmol, 25 mg), 1-octene (0.55 mmol, 85 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 14.1 mg). d 3-thiophene carbaldehyde (0.22 mmol, 25 mg), 1-octene (0.55 mmol, 85 μL), [Rh(PPh3)3Cl] (0.022 mmol, 20.4 mg), ligand 1 (0.022 mmol, 14.1 mg). d Determined by 1H NMR methods. 4.2.8 Summary of published results and conclusion After having established optimized reaction conditions, we extended the scope of this transformation to a wide range of electronically and structurally diverse alkenes and substituted benzaldehydes. Reasonable yields ranging from 93% to 51% were obtained for the addition of various terminal alkenes to benzaldehyde. The protocol is tolerant to a variety of functional groups on the alkene moiety including ester, hydroxyl, carboxylic acid, as well as allowing for internal alkenyl groups. Both electron donating as well as electron withdrawing substituents on the aryl benzaldehyde system are efficient reaction partners, thus highlighting the wide functional group tolerance of this catalyst system. A particularly relevant example is the hydroacylation of benzaldehyde with 6-hexen-1-ol 47. Despite of the possible coordination of the oxygen-atom to the rhodium catalytic center, a good yield of 75% was obtained. Our best result was performed with the hydroacylation of electron-donating para-methoxy substituted benzaldehyde 67 with 1-octene, with a top yield of 93%.
H
O
R1FG+FGR
O
FGRR1
FG
[Rh(COD)Cl]2 (5 mol%)ligand 1 (10 mol%)
toluene, 150°C, 24h
Scheme 79. Rhodium-catalyzed hydroacylation of substituted benzaldehydes and diverse alkenes.
O
O
O
73% yield
O
83% yield
O
57% yield
O
OH
75% yield
O
OH
51% yield
O
O
72% yield
O
70% yield
Cl
O
93% yield
MeO
68% yield
7268 70
605854
524846
O
Scheme 80. Best results obtained for the hydroacylation using our bifunctionnal system.
ResultsandDiscussion:Intermolecularhydroacylation
73
4.2.9 Rhodium-catalyzed intermolecular hydroacylation of 1-octene and aliphatic aldehydes Unfortunately, even a large diverse library of substrates and many harsh reaction conditions, only low yields were obtained for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and aliphatic aldehydes. All the PNN ligands synthesized were unsuccessful for this reaction, a massive change in the core structure must be designed for a better activation of the aldehyde C-H bond. 4.2.9.1 Rhodium-catalyzed intermolecular hydroacylation of 1-octene and 1-cyclohexene-1-carbaldehyde No reaction was observed for the hydroacylation of 1-cyclohexene-1-carbaldehyde and 1-octene (entry 1, Table 17), even with the use of benzoic acid (entry 2). Table 17. Rhodium-catalyzed hydroacylation of 2-naphthaldehyde with 1-octene.
entry
aldehyde
alkene
conditions
NMR yieldd (%)
1a
H
O
24 h, 150 °C, neat
0
2b
H
O
24 h, 150 °C, neat benzoic acid (10 mol%)
0
3c H
O
24 h, 150 °C, neat
83
a 1-cyclohexene-1-carbaldehyde (0.22 mmol, 25 μL), 1-octene (0.55 mmol, 84 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg). b 1-cyclohexene-1-carbaldehyde (0.22 mmol, 25μL), 1-octene (0.55 mmol, 84 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg), benzoic acid (0.022mmol, 2.7 mg). c benzaldehyde (0.22 mmol, 24μL), 1-octene (0.55 mmol, 84 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg). d Determined by 1H NMR methods. 4.2.9.2 Cyclization of “double alkene” or “double alkyne” with formaldehyde No reaction was observed for the double hydroacylation of formaldehyde in polymeric form with either N,N-diallyl-4-methyl-benzenesulfonamide (entry 1, Table 18), or with 2-allyl-2-ethyl-malonic acid dimethyl ester (entry 2).
ResultsandDiscussion:Intermolecularhydroacylation
74
Table 18. Cyclization of “double alkene” or “double alkyne” with formaldehyde.
entry
aldehyde
alkene
conditions
NMR yieldc (%)
1a
O
HHpolymeric form
N
Ts
24 h, 150 °C, neat
0
2b
O
HHpolymeric form
O
OO
O
24 h, 150 °C, neat
0
a formaldehyde polymeric form (0.22 mmol, 6.6 mg), N,N-diallyl-4-methyl-benzenesulfonamide (0.55 mmol, 138 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg). b formaldehyde polymeric form (0.22 mmol, 6.6 mg), alkene (0.55 mmol, 117 μL), [Rh(COD)2]Cl2 (0.022 mmol, 5.4 mg), ligand 1 (0.022 mmol, 6.7 mg). c Determined by 1H NMR methods.
4.2.9.3 Rhodium-catalyzed intermolecular hydroacylation of hexanal with 1-octene The hydroacylation of aliphatic aldehydes with aliphatic alkenes represents a great challenge. We proved in the hydroacylation of benzaldehyde and diverse alkenes, that the size of the alkyl chains plays a crucial role (Table 11). According to this observation, we selected hexanal 78 and 1-octene 45 for the two hydroacylation coupling partners. At 150 °C, a total conversion was obtained, but one alkenyl proton was observed. The structure of this product 79 results from an intermolecular aldol-condensation of aldehyde 77.
O
H +
O
O
H
H
[Rh(COD)2BF4] (10 mol%)
ligand 7 (13 mol%)
toluene (c = 1.1 M)
quant. (79)77 150 °C, 1 h45
79
78
Scheme 81. Rhodium-catalyzed intermolecular hydroacylation of hexanal with 1-octene.
4.2.9.4 Rhodium-catalyzed intermolecular hydroacylation of 2-phenyl-propionaldehyde and 1-octene According to the result obtained for the hydroacylation of hexanal and 1-octene, we ventured the hypothesis that the α-substitution of the aldehyde will avoid the isomerisation and allow the hydroacylation. 2-phenyl-propionaldehyde 80 was obtained by a regioselective hydroformylation of styrene using the protocol developed by Seiche and Breit.[113]
ResultsandDiscussion:Intermolecularhydroacylation
75
3 phosphorus ligands were tested in good yields: triphenylphosphine 81 (90% yield), tri-(o-tolyl)-phosphine 82 (70% yield) and tri-(di-t-butylphenyl)phosphinite Van Leeuwen ligand 83 (70% yield).[114]
THF, RT, o/n
[Rh(CO)2acac](0.007 mol%)
Phosphorus ligand(0.033 mol%)
O
H
CO/H2
49 80C9H10O
Mol. Wt.: 134,18C8H8
Mol. Wt.: 104,15
ligand 81: PPh3
ligand 82:
1)
2) P
C18H15PMol. Wt.: 262,29
C21H21PMol. Wt.: 304,37
3) ligand 83:
P
O
OO
C42H63O3PMol. Wt.: 646,92
3 phosphorus ligands screened:
(90% yield)
(70% yield)
(70% yield)
Scheme 82. Regioselective hydroformylation of styrene. No hydroacylated product 84 was observed in 1H NMR, starting material was recovered.
+
[Rh(COD)2BF4] (10 mol%)
ligand 7 (13 mol%)
toluene (c = 1.1 M)
no reaction80 150 °C, 1 h45
H
O O
84
Scheme 83. Rhodium-catalyzed hydroacylation of 2-phenyl-propionaldehyde and 1-octene.
[113] W. Seiche, A. Schuschkowski, B. Breit, Adv. Synth. Cat. 205, 347, 1488-1494. [114] P. C. J. Kramer, P. W. N. M. Van Leeuwen, Phosphorus(III) ligands in homogeneous catalysis: design and synthesis 2012, John Wiley & Sons.
ResultsandDiscussion:Intermolecularhydroacylation
76
4.2.10 Intermolecular hydroacylation of salicylaldehyde with methyl acrylate followed by an intramolecular transesterification As an extension of the good results obtained (63% neat, and 73% in toluene) for the catalytic hydroacylation 60 of benzaldehyde 44 and methyl acrylate 59 (Scheme 78), we tried to perform the same with salicylaldehyde. And we hypothesized, that the use of a base would allow for the transesterification, and yielded in situ an interesting 3,4-dihydro-benzo[b]oxepine-2,5-dione.
The reaction of salicylaldehyde and methylacrylate at 150 °C gave each time exclusively the decarbonylated phenol product. The same reaction under mild condition (100 °C) yielded the desired ketone 87 quantitatively. The greater reactivity of salicylaldehyde in comparison with benzaldehyde for hydroacylation is due to the extra coordination of the rhodium center by the hydroxyl group as shown by Suemune[115] in his “double chelation” assisted hydroacylation between salicyclaldehydes and by Dong in her regio- and selective intermolecular hydroacylation of salicylaldehydes to homoallylic sulphides.[116]
The same reaction performed with different bases usually used for the alkene hydroacylation (like K3PO4, Cs2CO3, Na2CO3 and K2CO3) gave quantitative yields with < 1/4 equivalent of base 2-hydroxymethyl-phenol 89 and with > 1 equivalent of base gave quantitatively the carbonate product 90 (carbonic acid 2-hydroxymethyl-phenyl ester methyl ester). In both cases, a reduction of the aldehyde was observed.
H
O
+
O
OMe
[Rh(COD)Cl]2 (10 mol%)ligand 1, ratio lig/cat 1:1
neat or toluene150 °C, 24 h
O
OMe
O
63% (neat)73% (toluene)
1) benzaldehyde + methyl acrylate
2) salicylaldehyde + methyl acrylate at 150 °C
N NH2
PPNN ligand 1
H
O
OH+
O
OMeOH
[Rh(COD)Cl]2 (10 mol%)ligand 1, ratio lig/cat 1:1
neat, 150 °C, 24 h+
O
OMe
86
44 59
60
85 59 59
(continued on the next page)
[115] I. Inui, M. Tanaka, M. Imai; K. Tanaka, H. Suemune, Chem. Pharm. Bull. 2009, 57, 1158. [116] M. M. Coulter, K. G. M. Kou, B. Galligan, Vy. M. Dong, J. Am. Chem. Soc. 2010, 132, 16330-16333.
ResultsandDiscussion:Intermolecularhydroacylation
77
H
O
OH+
O
OOH
O
O
OO
O
O
O
OH
O
O
[Rh(COD)Cl]2 (10 mol%)ligand 1, ratio lig/cat 1:1
base
neat, 100 °C, 24 h
+
+
expected expected
product 90 obtained quant. yield
H
O
OH+
O
OMe
[Rh(COD)Cl]2 (10 mol%)ligand 1, ratio lig/cat 1:1
OH
O
O
OMe
quant.
3) salicylaldehyde + methyl acrylate + a base at 100 °C
OH
OH
product 89 obtained quant. yield
neat, 100 °C, 24 h
Bases tested: K3PO4, Cs2CO3, Na2CO3, K2CO3
Results :
< ¼ eq of base : product 89¼< x <1 éq. of base: mix of product 89 and product 90> 1 eq of base: product 90
85
85
59
59 87
87 88
Scheme 84. Rhodium-catalyzed intermolecular hydroacylation of salicylaldehyde and methyl acrylate.
4.2.11 Rhodium-catalyzed intermolecular hydroacylation of 1-octene with benzaldehyde using chiral oxazoline-derived ligands We next turned our attention to the development of an eventual asymmetric version of the intermolecular hydroacylation using new oxazoline-derived ligands. Inspired by our bifunctional ligand, we replaced the phosphino moiety by a chiral oxazoline, which coordinated the rhodium center and introduced chirality with the isopropyl group.
NH2N
N
ONMeHN
N
O NN
N
O
H
N
H
S
CF3
F3C
C11H15N3OMol. Wt.: 205,26
C12H17N3OMol. Wt.: 219,28
C20H18F6N4OSMol. Wt.: 476,44
9291 93
Figure 15. Three chiral oxazoline P-N ligands.
For ligands 91 and 92, we anticipate similar activation of the aldehyde through an aldimine formation (Scheme 85, activation mode 1). For ligand 93, we replace the amino function with a thiourea. The thiourea acts as aldehyde activator through a hydrogen-bonding bridge similar to that described in the aldehyde hydrogenation by acyl guanidine published by Breit and co-workers (Scheme 83, activation mode 2).[117]
ResultsandDiscussion:Intermolecularhydroacylation
78
NH2N
N
ONN
N
O
H
N
H
S
CF3
F3C
O
H
O
H
[Rh]chirality
metal binding
aldehyde activation through hydrogen-bonding
aldehyde activation through aldimine formation
[Rh]
metal binding
chirality
activation mode 1 activation mode 2
Scheme 85. Oxazoline ligands used two different aldehyde activation modes.
H
O
+
Ooxazoline chiral ligand[Rh] catalyst
ratio lig/cat 1.3:1
150 °C, 24 hneat or toluene2.5 eq.
Scheme 86. Rhodium-catalyzed enantioselective intermolecular hydroacylation of benzaldehyde with 1-octene.
Table 19. Attempts of the rhodium-catalyzed intermolecular hydroacylation of benzaldehyde with 1-octene employ chiral N,N,N-ligands.
entry
ligand
conditions
NMR yieldj (%)
1a
NH2N
N
O
24 h, 150 °C, Toluene
[Rh(COD)2BF4]
0
2b
NH2N
N
O
24 h, 120 °C, Toluene
[Rh(COD)Cl]2
1
3c
NH2N
N
O
24 h, 150 °C, Neat [Rh(COD)2BF4]
2
(continued on the next page)
[117] a) L. Diab, T. Šmejkal, J. Geier, B. Breit, Angew. Chemie 2009, 121, 8166-8170; Angew. Chem. Int. Ed. 2009, 48, 8022-8026. b) D. Fuchs, G. Rousseau, L. Diab, U. Gellrich, B. Breit, Angew. Chemie 2012, 124, 2220-2224; Angew. Chem. Int. Ed. 2012, 51, 2178-2180.
The three oxazoline chiral ligands were tested using different reaction conditions, but no significant yield was observed. The replacement of the phosphino moiety by an oxazolidine inhibited the hydroacylation. A maximum of 7% yield was observed in 1H NMR with the use of oxazoline ligand 91, [Rh(COD)Cl]2, at 150 °C while running the reaction neat (entry 4, Table 19).
81
4.3 Indanones: rhodium-catalyzed intramolecular cyclization 4.3.1 Introduction 1-Indanones are found in many natural products or are important building blocks for the development of other biologically active compounds. One of the most well-known drugs containing an indanone backbone is Donepezil®.[118] This acetylcholinesterase inhibitor was used to avoid the dramatic decrease in cholinergic innervation in the cortex and hippocampus due to the loss of neurons in the basal forebrain.
O
N
.HCl
Figure 16. Donepezil® hydrochloride. 4.3.2 Stoechiometric reactions Because of the importance of 1-indanones, many synthetic pathways have been developed for their preparation. The basic process for the preparation of 1-indanones is the intramolecular Friedel-Crafts acylation of 3-arylpropanoic acids[119] and 3-arylpropanoyl halides,[120] or the intramolecular alkylation using 1-aryl-2-propen-1-ones.[121] These methods require the use of an equivalent of Lewis or Brönsted acid and the heating of the reaction mixture.
[118] H. Sugimoto, Pure Appl. Chem. 1999, 71, n°11, 2031-2037. [119] R. Rendy, Y. Zhang, A. McElrea, A. Gomez, D. A. Klumpp, J. Org. Chem. 2004, 69, 2340-2347 ; J. Koo, J. Am. Chem. Soc. 1953, 75, 1891-1895 ; V. Premasagar, V. A. Palaniswamy, E. J. Eisenbraun, J. Org. Chem. 1981, 46, 2974-2976 ; R. S. Budhhram, V. A. Palaniswamy, E. J. Eisenbraun , J. Org. Chem. 1986, 51, 1402-1406 ; T.-L. Ho, K. Y. Lee, C. K. Chen, J. Org. Chem. 1997, 62, 3365-3369. [120] S. Hanessian, J. Ma, Tet. Lett. 2001, 42, 8785-8788 ; H. O. House, C. B. Hudson, J. Org. Chem. 1970, 647-651 ; B. Hulin, M. Koreeda, J. Org. Chem. 1984, 207-209 ; T. Yamato, C. Hideshima, G. K. Surya Prakash, G. A. Olah, J. Org. Chem. 1991, 56, 3955-3957 ; T. F. Buckley III, H. Rapoport, J. Am. Chem. Soc. 1980, 102, 3056-3062. [121] K. Koltunov, S. Walspurger, J. Sommer, Tet Lett 2005, 46, 8391-8394 ; C. Vial, G. Bernardinelli, P. Schneider, M. Aizenberg, B. Winter, Helv. Chim. Acta 2005, 88, 3109-3117 ; A. Bhattacharya, B. Segmuller, A. Ybarra, Synthetic Commun. 1996, 26, 1775-1784 ; G. Sartori, F. Bigi, R. Maggi, G. L. Bernardi, Tet. Lett. 1993, 34, 7339-7342 ; C. Roussel, H. G. Rajoharison, L. Bizzari, L. Shaimi, J. Org. Chem. 1988, 53, 683-685 ; T. Suzuki, T. Ohwada, K. Shudo, J. Am. Chem. Soc. 1997, 119, 6774-6780 ; J. H. Burckhalter, R. C. Fuson, J. Am. Chem. Soc. 1948, 70, 4184-4186 ; Fuson , W. E. Ross, C. H. McKeever, J. Am. Chem. Soc. 1938, 60, 2935-2936.
In 2005, Yonezawa and co-workers[122] and Womack et al.[123] reported that the acetal of 2-alkylcinnamaldehydes cyclized in the presence of BF3·OEt2 and trimethyl orthoformate for three hours at room temperature to yield 1-alkoxy-2-alkyl-1H-indenes, a precursor of 1-indanone.
R
H
O
(MeO)3CH, BF3.Et2O
R
OMe45% (R = Me)48% (R = Et)
Scheme 87. Conversion of 2-alkylcinnamaldehydes to 1-alkoxy-2-alkyl-1H-indenes
via a BF3·Et2O catalytic intramolecular Friedel-Crafts reaction. In 2007, Womack et al. completed their study and found that the use of FeCl3 as a catalyst could improve the yield of 1-alkoxy-2-alkyl-1H-indenes up to 90%.[124] And then, they converted 1-alkoxy-2-alkyl-1H-indenes in 1-indanones in two steps via isomerization and hydrolysis.
Scheme 88. Preparation of 2-alkylindanones catalyzed by iron(III) trichloride. In 2008, Saito et al. realized a novel one-pot approach to the synthesis of indanones through a Sb(V)-catalyzed reaction of phenylalkynes with aldehydes.[125]
[122] T. Jobashi, T. Jobashi, A. Kawai, S. Kawai, K. Maeyama, H. Oike, Y. Yoshida, N. Yonezawa, Tetrahedron 2006, 62, 5717-5724 ; T. Jobashi, K. Maeyama, K. Noguchi, Y. Yoshida, N. Yonezawa, Bull. Chem. Soc. Jpn. 2006, 79, 627-633. [123] G. B. Womack, R. L. Snowden, R. L. Mosimann, A. A. Birkbeck, A Process for Producing Indenol Esters or Ethers. WO Pat. Appl. WO 2005/113473 A2, December 1, 2005. [124] G. B. Womack, J. G. Angeles, V. E. Fanelli, C. A. Heyer, J. Org. Chem. 2007, 72, 7046-7049. [125] A. Saito, M. Umakoshi, N. Yagyu, Y. Hanzawa, Org. Lett. 2008, 10, 1783-1785.
Scheme 89. One-pot approach to the synthesis of indanones through Sb(V)-catalyzed reaction of phenylalkynes with aldehydes.
4.3.3 Catalytic reactions A number of palladium-catalyzed annulation processes have been reported for the synthesis of indanones. Negishi et al. developped in 1996 an effective method to produce indenones and indanones through the palladium-catalyzed carbonylation of o-iodostyrene.[126]
I
5% PdCl2(PPh3)21.5-4 eq. Et3N, CO (1 atm)
MeCN/PhH, 80 °C, 6 h
O
+
NEt2
O
50% 9%
I 5% PdCl2(PPh3)2, 1.5-4 eq. Et3N, CO (40 atm)MeOH, DMF, 100 °C, 6 h
O
+
CO2Me
O
2% 74%
+
2%
O
O
Scheme 90. Synthesis of indenones through the palladium-catalyzed carbonylation
of o-iodostyrenes derivates. In 1999, Yamamoto and co-workers used the annulation of internal alkynes with o-bromobenzaldehyde palladium acetate-catalyzed, followed by isomerization of the resulting indenol to form indanone.[127]
[126] E. Negishi, S. M. Coperet, S. Ma, T. Mita, T. Sugihara, J. M. Tour, J. Am. Chem. Soc. 1996, 118, 5919 [127] V. Gevorgyan, L. G. Quan, Y. Yamamoto, Tet. Lett. 1999, 40, 4089.
Scheme 91. Synthesis of indanones through a palladium acetate-catalyzed annulation of internal alkynes by o-bromobenzaldehyde.
Gagnier and Larock used the Yamamoto’s method in 2003 to prepare successfully indanones in good yields.[128]
I+ CO (1 atm)
10% Pd(OAc)2, 2-pyridine, n-Bu4NClDMF, 100 °C, 8 h
O
100%
Scheme 92. Synthesis of indanones through the palladium acetate-catalyzed carboannulation of o-iodostyrene.
In 2004, Park et al. provided another approach to the synthesis of indanones via intramolecular Heck reaction of Baylis-Hillman adducts of 2-iodobenzaldehyde using 5 mol % of palladium acetate for the Heck coupling to obtain the indanone products.[129]
I
H
O
DABCO
Z
95% (Z = CO2Me)86% (Z = CO2Et)58% (Z = CO2Bu-t)65% (Z = COMe)
I
Z
OH
Pd(OAc)2, (o-Tol)3P, Et3N
34% (Z = CO2Me)32% (Z = CO2Et)27% (Z = CO2Bu-t)35% (Z = COMe)
O
Z
Scheme 93. Synthesis of indanones via intramolecular Heck reaction of
Baylis-Hillman adducts of 2-iodobenzaldehyde. In 2007, Odreda et al. developped the cyclization of electron-rich 2-alkyl-1-ethynylbenzene derivates catalyzed by 10 mol % of TpRuPPh3(CH3CN)2PF6 at 100 °C to obtain 60 % yield after one day.[130]
[128] S. V. Gagnier and R. C. Larock, J. Am. Chem. Soc. 2003, 125, 4804-4807 [129] J. B. Park, S. H. Ko, W. P. Hong, K. J. Lee, Bull. Korean Chem. Soc. 2004, 25, n°6, 927-930. [130] A. Odreda, S. Datta, R. S. Liu, J. Org. Chem. 2007, 72, 3289-3292.
Scheme 94. Cyclization of electron-rich 2-alkyl-1-ethynylbenzene derivatives
catalyzed by 10 mol% of TpRuPPh3(CH3CN)2PF6. Directly inspired by the work of Hallberg et al. published in 2011, [131] Xiao et al. synthesized a wide range of monosubstituted 1-indanones of potential pharmaceutical use in a one-pot fashion in moderate to excellent yields via palladium catalysis in ethylene glycol.[132] A Heck reaction first installs an enol functionality on the aromatic ring; this is followed by an ethylene glycol promoted aldol-type annulation with a neighboring carbonyl group, resulting in the formation of various 1-indanones.
RBr/Cl
O
R´+
R´´ OR´´´HO OH
[Pd]
O
R´´
OHR´
30 exemples
R
O
R´
OR´´´
R´´
[Pd]
R´´
OHR´
OO
H3O+
HO OH
Scheme 95. Direct synthesis of 1-indanones via palladium-catalysed olefination and ethylene glycol-promoted aldol-type annulations cascade.
4.3.4 Towards the hydroacylation of o-vinylbenzaldehyde In 1971, Schrock and Osborn published the preparation and the properties of some cationic complexes of Rhodium [I] and Rhodium [III].[133] Reactions of the dimeric diene complexes of rhodium [I], [Rh(diene)Cl]2, with uncharged monodentate ligands such as triphenylphosphine, lead to the cleavage of the halogen bridge and the formation of monomeric species of a type Rh[diene]PPh3Cl. Schrock and Osborn demonstrated under the appropriate conditions further reaction can occur in which the chloride ion is displaced from the metal by a neutral ligand to form a cationic, four-coordinated rhodium [I] complex.
It was observed for a wide range of counter-anions (PF6-, ClO4
-, B(C6H5)4-), ligands (tertiary
phosphine, phosphite, amine or arsine) and dienes (cyclooctadiene or norbornadiene).
[131] J. Ruan, J. A. Iggo, J. Xiao, Org. Lett. 2011, 13(2), 268-271. [132] A. Arefalk, M. Larhed, A. Hallberg, J. Org. Chem. 2005, 70, 938-942. [133] R. Schrock and J. Osborn, J. Am. Chem. Soc., 1971, 2397-2402.
Figure 17. Deplacement of chloride ion from the metal by neutral ligand to form
a cationic four-coordinated rhodium [I] complex. In 1988, Fairlie and Bosnich applied these results to the conversion of 4-pentenal to cyclopentanone.[134], [135] in 1976, Miller first reported the cyclization of 4-pentenal to cyclohexanone.[136] He obtained 72% of yield at room temperature with the Wilkinson’s catalyst in ethylene saturated solvent after eighty-eight hours. Fairlie and Bosnich have shown that many parameters have an influence onto the increase of 1-indanone over the decarbonylation product:
The substrate-catalyst interaction: the main factor to improve this interaction is saturation of the metal. For this reason, the choice of the rhodium precatalyst is crucial. The best rhodium catalysts are dimeric rhodium(I) complexes such as [Rh(COD)]2Cl2, [Rh(COD)]2(ClO4)2 or [Rh(dppe)]2(ClO4)2.
The solvent effect: The dimeric rhodium complex exists in a monomeric form in DCM and as an arene-bridging dimer in CH3NO2. The dimeric form is a coordinatively saturated 18-electron species which therefore is not expected to engage into oxidative addition. In general, DCM and toluene are key solvents for the hydroacylation.
The substrate decarbonylation: The choice of the catalyst is the most important factor
to avoid decarbonylation. But other factors do influence the decarbonylation rate, such as dilution and the turnover rate.
The Pd and Rh “black” degradation: The degradation of Pd and Rh catalyst is very
scarse in this hydroacylation reaction.
The ligand bite angle: An optimal ligand bite angle is required to optimize the steric and electronic influence on metal catalysts.
Fairlie and Bosnich obtained interesting results with analogs of 4-pentenal (88-100%), the main constraint being the steric hinderance at the 5-position. Substituents in the 5-position gave 89% for the methyl, 70% for the phenyl and 0% for the dimethyl.
[134] D. Fairlie and B. Bosnich, Organometallics 1988, 7, 936-945. [135] D. Fairlie and B. Bosnich, Organometallics 1988, 7, 946-954. [136] C. F. Lochow and R. G. Miller, J. Am. Chem. Soc. 1976, 98(5), 1281-1283.
By analogy to the cyclization of 4-pentenal derivatives, Bosnich and co-workers also tried to cyclize o-vinylbenzaldehyde.[134] He obtained 30% of the indanone, 5% of the decarbonylated compound and 65% of an unidentified product. In 2005, A. T. Morehead Jr. and co-workers identified this by-product as the o-vinylbenzaldehyde cyclized dimer with exclusive selectivity for the trans-isomer.[137]
O
RhL2+
O
H
O
H
O
RhL2+
H
O
H
RhL2+
O
RhL2+
O OH
O
O H
L2Rh+
1,2 migratoryinsertion
2,1 migratoryinsertion
(Dimerization)
Figure 18. Mechanistic rationale for the formation of the dimer. After the initial oxidative addition, the migratory 1,2-insertion will lead to the desired 1-indanone. The migratory 2,1-insertion will lead to a branched intermediate which, in the presence of excess o-vinylbenzaldehyde, yields the dimerized product. Keeping in mind the proposed mechanism, steric hinderance was introduced to the α-position of o-vinylbenzaldehyde to avoid the 2,1-insertion pathway; 1 mol % of [Rh(dppe)(NBD)]ClO4 was used at room temperature in DCM to afford the hydroacylation of 2-formyl styrenes (88% COOEt - 97% Me, Et).[137] This study was completed when the test of the asymmetric hydroacylation of 2-formyl styrenes with a Rh-BINAP catalyst found excellent selectivity, particularly with electron-withdrawing substituents.[137]
[134] D. Fairlie and B. Bosnich, Organometallics 1988, 7, 936-945. [137] K. Kundu, J. McCullagh, A. T. Morehead Jr, J. Am. Chem. Soc. 2005, 127, 16042-16043.
Scheme 96. Hydroacylation of 2-formyl styrenes with 1 mol % of [Rh(dppe)(NBD)]ClO4. Table 21. Hydroacylation of 2-formyl styrenes with 1 mol % of [Rh(dppe)(NBD)]ClO4.
entry
substrate (R=)
yield (%)
1
Me
97
2
Et
97
3
Ph
98
4
2-Naphthyl
88
5
CH2CH2OH
97
6
SiMe3
93
7
CF3
90
8
CO2Et
88
O
H
R
2 mol %[Rh(R-BINAP)]ClO4
CH2Cl2, 23 °C
O
R
Scheme 97. Asymmetric hydroacylation of 2-formyl styrenes with a Rh-BINAP catalyst.
Table 22. Asymmetric hydroacylation of 2-formyl styrenes.
entry
substrate (R=)
yield (%)
ee (%)
1
Me
97
99
2
Et
97
99
4
2-Naphthyl
88
96
5
CH2CH2OH
97
96
6
SiMe3
93
70
7
CF3
90
99
8
CO2Et
88
96
Finally, Morehead and co-workers also demonstrated the key role of the concentration of the substrate. In fact, he succeeded in obtaining 95% of 1-indanone when the substrate was added slowly by a syringe pump over eighteen hours.[137]
O
Hfast
addition
O
(95%)
slowaddition
(18 h)
O
HO
(75%)
Scheme 98. Concentration dependence of the hydroacylation of o-vinylbenzaldehyde.
4.3.5 Substrate synthesis Substrate 95 has been prepared by the Bouveault aldehyde synthesis.[138] In this one-step reaction, n-BuLi was added to substrate 94 to make the bromo-lithium exchange. Upon addition of DMF, a hemiaminal is formed, which can be hydrolyzed to the desired o-vinylbenzaldehyde 95.
[137] K. Kundu, J. McCullagh, A. T. Morehead Jr, J. Am. Chem. Soc. 2005, 127, 16042-16043. [138] M. Inoue and M. Nakada, J. Am. Chem. Soc. 2007, 129, 4164-4165.
4.3.6 Catalytic experiments Substrate 95 was tested in the rhodium catalyzed intramolecular alkene hydroacylation of o-vinylbenzaldehyde using ligand 1, cationic [Rh(COD)2]BF4 or neutral [Rh(COD)Cl]2 rhodium precursors. Solvent effect, reaction time and temperature were optimized. Our system was compared with the Jun system[90] and the use of Wilkinson’s catalyst.[139]
4.3.6.1 Hydroacylation of o-vinylbenzaldehyde (95)
O
H [Rh]/ligand+
96 97
O
95
Scheme 100. Hydroacylation of o-vinylbenzaldehyde 45.
Table 23. Hydroacylation of o-vinylbenzaldehyde (95).
entry
catalyst (mol%)
ligand (mol%)
conditions
NMR yield (%)e
1a
N NH2
P
1
(10)
[Rh(COD)Cl]2 (5)
24 h, 150 °C Neat
96 (99)
2 a
Ligand 1 (10)
[Rh(COD)Cl]2 (5)
12 h, 150 °C Neat
96 (96) 95 (4)
3 a
Ligand 1 (10)
[Rh(COD)Cl]2 (5)
2 h, 150 °C
Neat
96 (95) 95 (5)
4 a Ligand 1 (10) [Rh(COD)Cl]2 (5) 24 h, 120 °C Neat
96 (70) 95 (30)
5 a Ligand 1 (5) [Rh(COD)Cl]2 (2.5) 2 h, 150 °C Neat
96 (46) 95 (54)
6a
Ligand 1 (5)
[Rh(COD)Cl]2 (2.5)
2 h, 120 °C Neat
96 (10) 95 (90)
7b
Ligand 1 (10)
[Rh(COD)2]BF4 (10)
24 h, 150 °C Neat
96 (73) 97 (27)
(continued on the next page)
[139] J. A. Osborn and G. Wilkinson, J. Am. Chem. Soc. A: Inorganic, Physical, Theoretical 1966, 1711-173.
18c 2-amino-3-picoline (40) [Rh(PPh3)3]Cl (10) 24 h, 150 °C Toluene
-
19c 2-amino-3-picoline (40) [Rh(PPh3)3]Cl (10) 24 h, 120 °C Toluene
-
20d - [Rh(PPh3)3]Cl (10) 24 h, 150°C Toluene
96 (22) 95 (78)
21d - [Rh(PPh3)3]Cl (10) 24h, 120°C Toluene
96 (36) 95 (64)
a o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), ligand 1 (0.022 mmol, 6.7 mg), [Rh(COD)Cl]2 (0.011 mmol, 5.4 mg). b o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), ligand 1 (0.022 mmol, 6.7 mg), [Rh(COD)2]BF4 (0.022 mmol, 9.0 mg), 8 mL Schlenk tubes. c o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), 2-amino-3-picoline (0.088 mmol, 9.2 μL), [RhCl(PPh3)3] (0.022 mmol, 20.4 mg), 8 mL Schlenk tubes. d o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), [RhCl(PPh3)3] (0.022 mmol, 20.4 mg), 8 mL Schlenk tubes.e Determined by 1H NMR methods. Reactions performed in 8 mL Schlenk tubes. Neutral catalyst [Rh(COD)Cl]2 was first used to hydroacylate o-vinylbenzaldehyde. No solvent was used. Hydroacylation with 5 mol% of [Rh(COD)Cl]2 and 10 mol% of ligand at 150 °C for twenty four hours gave 99% of 1-indanone (entry 1, Table 23). The same reaction was stopped after twelve hours (entry 2) and two hours (entry 3) give respectively 96% and 95%.
The formation of the 5-membered ring is significantly favoured thermodynamically, so the formation of 1-indanone is very fast. Temperature was decreased to 120 °C for 24 h (entry 4) to obtain 70% of 1-indanones and 30% of starting material. Finally, the loading of ligand and catalyst were divided by two lowering the yield to 42% (entry 5) at 150 °C and 10% (entry 6) at 120 °C. Cationic catalyst [Rh(COD)2]BF4 was also investigated at 150 °C with 10 mol% of ligand 1 for 24 h (entry 7). Full conversion was obtained, but 27% of decarbonylated starting material was detected by 1H NMR. To avoid this decarbonylation, the temperature was decreased at 120 °C to obtain 98% of yield (entry 8). Contrary to [Rh(COD)Cl]2 (150 °C), the optimal temperature was lower for [Rh(COD)2]BF4 (120 °C). The reaction was stopped after 12 h (entry 9) and 2 h (entry 10) to obtain in both cases 98% yield. Finally, the loading of ligand and catalyst were decreased. With 5 mol%, 96% (entry 11) and 87% (entry 12) were respectively obtained at 150 °C and 120 °C. With 2.5 mol%, 84% (entry 13) and 4% (entry 14) were respectively obtained at 150 °C and 120 °C. With 2.5 mol% and less, 150 °C is required to boost the reaction. Hydroacylation of o-vinylbenzaldehyde was performed for two hours with 1 mol% of ligand 1 yielding 7% of 1-indanone (entry 15). To complete the conversion, the same reaction was performed for 6 hours (entry 16). An increase of 6 % was obtained, but the rest of the starting material was decarbonylated. Finally, the loading was divided by two (0.5 mol%) (entry 17) to obtain 67% of 1-indanone and 32% of vinyl benzene. To finish the study, our best results (entry 1) and (entry 8), were compared with the Jun’s system, and the Wilkinson’s catalyst. With the Jun’s system (2-amino-3-picoline 40 mol%, [Rh(PPh3)3]Cl 10 mol%) at 150 °C (entry 18) and 120 °C (entry 19), neither 1-indanone nor starting material were detected. Decomposition crude was observed. Using only 10 mol% of [Rh(PPh3)3]Cl gave 22% at 150 °C (entry 20) and 36% at 120 °C (entry 21). The rest is starting o-vinylbenzaldehyde in both case. 4.3.6.2 Conclusion
Good yields (entry 1) and (entry 8) were obtained with [Rh(COD)Cl]2 and [Rh(COD)2]BF4. In the formation of 1-indanone from o-vinylbenzaldehyde in comparison to Jun’s system (entry 18) and (entry 19), and Wilkinson’s catalyst (entry 20) and (entry 21). Reaction time (twenty-four to two hours) and loading of catalyst/ligand (10 to 0.5 mol%) were decreased with success. 4.3.7 Proof of concept for the rhodium-catalyzed intramolecular hydroacylation of o-vinylbenzaldehyde Before the intramolecular hydroacylation of diversified o-vinylbenzaldehydes, we proved the concept of our new bifunctionnal catalyst system with three control experiments:
Scheme 101. Ligand L’ lacking the amino functionnality.
N NH2
Ph2P
our system
[Rh]
[Rh(PPh3)3Cl]
Wilkinsoncatalyst
N NH2
[Rh(PPh3)3Cl]
Jun system
N
Ph2P
control systemlacking the NH2
[Rh]+ cocatalyst (20 mol%)
L L'
entry 2 entry 3 entry 4entry 1
Scheme 102. Control experiments for the intermolecular hydroacylation.
Wilkinson´s catalyst on its own gave poor results in this transformation (entry 1, Table 24). Jun´s system gave also only 5% of yield(entry 2, Table 24). In addition, a control experiment with ligand L’, lacking the amino functionality, resulted in a significantly less efficient catalyst in the reaction, thus enhancing the importance of the amino group within ligand 1 for the reaction to proceed via iminium formation and the prevention of decarbonylation (entry 4, Table 24). Table 24. Proof of concept for the rhodium-catalyzed intermolecular hydroacylation of 1-octene and benzaldehyde
entry
[Rh]/ligand 1:1 (mol%)
Cocatalyst (mol%)
NMR yield (%)e
1a
10
-
22
2b
10
2-amino-3-picoline (20)
5
3c
5
-
96
4d
5
-
42
a o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), [Rh(PPh3)3Cl] (0.022 mmol, 10.1 mg). b o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), [Rh(PPh3)3Cl] (0.022 mmol, 10.1 mg), 2-amino-3-picoline (0.022 mmol, 4.4 μL). c o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), [Rh(COD)2]Cl2 (0.011 mmol, 5.5 mg), ligand 1 (0.022 mmol, 6.5 mg). d o-vinylbenzaldehyde (0.22 mmol, 29.1 mg). e Determined by 1H-NMR methods. All reactions are performed under argon in 8 mL Schlenk tubes. [107] C.-H. Jun, D.-Y. Lee, H. Lee, J.-B. Hong, Angew. Chemie 2000, 112(17), 3214-3216; Angew. Chem. Int. Ed. 2000, 39(17), 3070-3072.
4.3.8 Screening of our P-N ligand library in our best reaction condition for the rhodium-catalyzed intramolecular hydroacylation of o-vinylbenzaldehyde We next turned our attention toward the ligand screening of our P-N ligand library in the intramolecular hydroacylation and chose o-vinylbenzaldehyde as a model substrate. Table 25. Ligand screening in the intramolecular hydroacylation reaction of o-vinylbenzaldehyde.
All reactions are performed with o-vinylbenzaldehyde (0.22 mmol, 29.1 mg), [Rh(COD)Cl]2 (0.011 mmol, 5.4 mg), 1 h, 150 °C, toluene (200 μL), 8 mL Schlenk tubes. a Ligand 1 (0.022 mmol, 6.7 mg). b Ligand 7 (0.022 mmol, 7.2 mg). c Ligand 11 (0.022 mmol, 6.4 mg). d Ligand 15 (0.022 mmol, 7.2 mg). e Ligand 22 (0.022 mmol, 7.4 mg). f Ligand 27 (0.022 mmol, 7.0 mg). g Ligand 31 (0.022 mmol, 6.7 mg). h Ligand 36 (0.022 mmol, 7.0 mg). i Ligand 41 (0.022 mmol, 6.4 mg). j Ligand 43 (0.022 mmol, 6.1 mg). k Determined by 1H NMR methods. The influence of the ligand backbone and its parameters is less pronounced in the intramolecular reaction than in the intermolecular process.* This could be explained by the good ability of such cyclic substrate to coordinate the rhodium metal center and bring in close proximity the reactive alkene and C-H aldehydic bond, without the need of an ancillary ligand (entry 1, Table 25). However, ligand 1 proved to be one of the best ligands in terms of activity. A decrease in the yield was observed significantly in the case of the 3-methoxy-substituted ligand 7 (entry 2), due to the slow formation of the active imine in this case comparing to the reference ligand 1.** The increase of the phosphino side chain length had negligeable impact on the yield (entries 6 and 7), in comparing with the amino moiety (entry 9). The use of the quinoline core structure P-N ligand 22 decreased the yield to 27% (entry 5). Finally, good yield (95%) was obtained with the simple ligand 43 (entry 10). 4.3.9 Intramolecular hydroacylation of diversified o-vinylbenzaldehydes: Substrate synthesis 4.3.9.1 Synthesis of 6-vinylveraltraldehyde 99 6-Vinylveratraldehyde 99 was prepared by a Stille coupling. The reaction between freshly prepared tributyl(vinyl)tin and 6-bromoveratraldehyde 98 was made in a Schlenk tube in the presence of tetrakis(triphenylphosphine)palladium (0).[140] 2,6-Di-tert-butyl-p-cresol, a weak acid, was used as an additive to performe the Stille coupling.
[140] M. Murakami, S. Kadowaki, A. Fujimoto, M. Ishibashi, T. Matsuda, Org. Lett. 2005, 7(10), 2059-2061.
* Look at p. 60 to see the ligand screening on the intermolecular hydroacylation of benzaldehyde with 1-octene. ** Look at p. 148 to see the compared imine rate formation with ligand 1 and ligand 7 in the intermolecular hydroacylation version.
Scheme 103. Synthesis of 6-vinylveraltraldehyde 99.
4.3.9.2 Synthesis of 3-formyl-4-vinylbenzoic acid methyl ester 103 The 3-formyl-4-vinylbenzoic acid methyl ester substrate 103 was obtained in three steps. The methyl group of the 4-bromo-3-methylbenzoic acid methyl ester 100 was diacetoxylated by chromium trioxide and acetic acid in the presence of cold sulfuric acid.[141]. The resulting diacetylated product 101 was then hydrolyzed at reflux in sulfuric acid followed by a diluted hydrochloric acid solution to give the corresponding aldehyde 102.[141] The final dimethoxy substituted vinyl benzaldehyde 103 was obtained by a Suzuki coupling with potassium vinyl trifluoroborate in a sealed tube.
Scheme 104. Synthesis of 3-formyl-4-vinylbenzoic acid methyl ester 103.
4.3.9.3 Synthesis of 3-formyl-4-phtalimido-2-vinylbenzaldehyde 108 The 3-formyl-4-phtalimido-2-vinylbenzaldehyde 108 was performed in four steps. At first, 3-bromo-4-methylaniline 104 was protected with a very good yield by phthalic anhydride at 190 °C for one hour to give the corresponding phthalimide 105. This was then dibrominated by N-bromosuccinimide and AIBN as a radical initiator to give the dibrominated product 106. Concentrated sulfuric acid was used to oxidize the last dibrominated compound 106 to the aldehyde 107. The final ester substituted vinyl benzaldehyde 108 was obtained by a common Stille coupling.
[141] A. D. Burrows, C. G. Frost, M. F. Mahon, C. Richardson, Angew. Chemie 2008, 120(44), 8610-8614; Angew. Chem. Int. Ed. 2008, 47, 8482-8486.
Scheme 105. Synthesis of 3-formyl-4-phtalimido-2-vinylbenzaldehyde 108.
4.3.9.4 Synthesis of 4-methyl-2-vinylbenzaldehyde 110 4-Methyl-2-vinylbenzaldehyde 110 was prepared by a Suzuki-Miyaura cross-coupling reaction. Potassium vinyl trifuloroborate was used in association with triphenylphosphine, palladium chloride and caesium carbonate as a base, to provide a good yield.
Scheme 106. Synthesis of 4-methyl-2-vinylbenzaldehyde 110.
4.3.9.5 Synthesis of 1-vinylnaphtalene-2-carbaldehyde 114 The naphthalene-substituted substrate 114 was obtained in three steps. At first, 1-bromo-2-methyl-naphthalene 111 was dibrominated by NBS to give product 112. [142] The classical method to hydrolyze the dibromo moiety with sulfuric acid could not be used for this substrate. A degradation of the naphthyl group was observed by 1H-NMR. Therefore, a biphasic system with calcium carbonate was used for the hydrolysis to yield the aldehydic product 113.[142]
[142] G. J. Domski, J. B. Edson, I. Keresztes, E. B. Lobkovsky, Chem. Commun. 2008, 6137-6139.
Finally, 1-bromonaphthalene-2-carbaldehyde underwent a Stille coupling to give the final substrate 114 in a good yield.[143]
NBS, AIBN every 3 hoursbenzene, 16 h, reflux
97%
Br Br
Br
Br
CaCO3, water 8 h, reflux
95%
Br
O
H
92%
O
H
tributyl(vinyl)tinPd(PPh3)4 (2 mol%)toluene, 16 h, 110 °C
111
114
113112
Scheme 107. Synthesis of 1-vinylnaphthalene-2-carbaldehyde 114.
4.3.9.6 Synthesis of 4-nitro-2-vinylbenzaldehyde 118 The nitro-substituted substrate 118 was obtained with the same three step route previously used in the substrate synthesis. The starting material 115 was dibrominated with NBS by a radical bromination initiated by AIBN to give the dibrominated compound 116, which was then hydrolyzed with sulfuric acid to its corresponding aldehyde 117. A final Suzuki-Miyaura cross-coupling reaction with potassium vinyl trifuloroborate gave 4-nitro-2-vinylbenzaldehyde 118.
A radical dibromination with NBS and using AIBN as an initiator to give the dibrominated compound 120, followed by hydrolysis with sulfuric acid at room temperature to obtain the corresponding aldehydic product 121, and lastly a Suzuki-Miyaura cross-coupling was used to obtain the final chlorinated substrate 122 with a good yield.
Scheme 109. Synthesis of 5-chloro-2-vinylbenzaldehyde 122. 4.3.9.8 Synthesis of 5-fluoro-2-vinylbenzaldehyde 124 5-Fluoro-2-vinylbenzaldehyde 124 was prepared by a Suzuki-Miyaura cross-coupling reaction. Potassium vinyl trifluoroborate was used in association with triphenylphosphine, palladium chloride (0) and caesium carbonate as a base, to give a good yield.
Scheme 110. Synthesis of 5-fluoro-2-vinylbenzaldehyde 124.
4.3.9.9 Synthesis of 5-hydroxy-2-vinylbenzaldehyde 126 The hydroxy-substituted substrate 126 was obtained by Stille cross-coupling with tributyl(vinyl)tin and tetrakis(triphenylphosphine)palladium.[144]
Br
O
H
tributyl(vinyl)tin, Pd(PPh3)4 (2 mol%)toluene, 16 h, 110 °C
81%
HO HOH
O
125 126
Scheme 111. Synthesis of 5-hydroxy-2-vinylbenzaldehyde 126.
[144] H. Tanaka, A. K. M. Abdul Hai, H. Ogawa, S. Torii, Synlett 1993, 11, 835-836 (For the synthesis of the starting tributylvinyltin, 99% yield).
4.3.9.10 Synthesis of 2-vinylpyridine-3-carbaldehyde 129 Directed metalating groups (DMGs) can be used to selectively deprotonate pyridine rings, much like benzene rings. DoM* on bromo- and iodopyridines, rather than lithium-halogen exchange is possible with lithium amides as long as temperatures are kept sufficiently low to prevent dehydropyridine formation via 1,2-elimination.[145] Consequently, 2-bromopyridine was converted at -78 °C into its corresponding 3-substituted carbaldehyde 128 with lithium diisopropylamide, and DMF with good regioselectivity.[146]
Then, 2-bromopyridine-3-carbaldehyde 128 was substituted with a vinyl group in the 2-position. A Stille cross-coupling provided the substrate 129.
127 128
NN 75%Br Br
H
O1) LDA THF, 4 h, -78°C2) DMF 2.5 h, -78°C to r.t
N71%
H
Otributyl(vinyl)tinPd(PPh3)4 (2 mol%)toluene, 16 h, 110 °C
129
Scheme 112. Synthesis of 2-vinylpyridine-3-carbaldehyde 129.
4.3.9.11 Synthesis of 2-vinylthiophene-3-carbaldehyde 134 The thiophene substrate 134 was obtained in three steps. At first, thiophene-3-carbaldehyde 130 was protected quantitatively by ethylene glycol and p-toluenesulfonic acid at reflux using a Dean-Stark trap to give the corresponding protected product 131. Although the sulfur atom is relatively unreactive, the flanking carbon centers at the 2- and 5-positions are highly susceptible to attack by electrophiles. The acetal substituent has not enough electronic effect to favour regioselectively the 2-position. Therefore, the lithiation with n-BuLi of the protected compound 131 followed by its quenching with DMF gave 2,3-thiophenedicarbaldehyde-3-(ethylene acetal) 132 with a moderate yield. The resulting aldehyde 132 was converted into its corresponding vinyl product 133 by Wittig olefination. Finally, the 2-vinylthiophene-3-carbaldehyde ethylene acetal 133 was deprotected with potassium hydrogen sulfate at 60 °C to yield the desired thiophene substrate 134.
[145] T. Kauffmann, R. Wirthwein, Angew. Chemie 1971, 10, 20-33; Angew. Chem. Int. Ed. 1971, 83, 21-34. [146] A. Numata, Y. Kondo, T. Sakamoto, Synthesis 1999, 2, 306-311.
OO 1) n-BuLi (2.5 M in n-hexanes) THF, 30 min., 0 °C2) DMF, THF, 3 h, RT
45 % S
H
OO
H
O132
84 %n-BuLi (2.5 M in n-hexanes)methyl triphenylphosphonium bromideTHF, 30 min., 0 °C; then o/n, RT
S
H
OO
133
54 %
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
S
H
O
134
Scheme 113. Synthesis of 2-vinylthiophene-3-carbaldehyde 134.
4.3.10 Intramolecular hydroacylation of diversified o-vinylbenzaldehydes: Catalytic experiments With a highly active catalyst system in hands, we investigated the scope of our protocol by diversifying the backbone of o-vinylbenzaldehyde. Table 26. Intramolecular hydroacylation of diversified o-vinylbenzaldehydes.
The catalyst tolerates electron donating, neutral and electron withdrawing substituents on the aromatic nucleus. A wide range of functional groups including carboxylic esters, halogens (chlorine and fluorine), nitro groups, and phenol functional groups are compatible with the reaction conditions and excellent yields were obtained. Conversely, employing 2-vinylpyridine-3-carbaldehyde did not show any reactivity, and the starting material was recovered quantitatively; the substrate may act itself as a competitive ligand for ligand 1 at the rhodium center thus preventing turnover. 4.3.11 Intramolecular hydroacylation of 2-vinylthiophen-3-carbaldehyde : cyclubutane dimer formation
Cycloaddition reaction of alkenes to give cyclobutane dimers is one of the most studied reaction in organic photochemistry. Pr. Dr. Maurizio D’Auria of the University of Roma “La Sapienza” is focusing his research of the photochemical dimerization of methyl 2-furyl- and 2-thienylacrylate in the presence of benzophenone.[147 a] He obtained with hard photochemical condition (twenty-four hours of irradiation, 500 W light) cyclobutane dimers with a maximal yield of 50%. The cyclobutane dimerization is typical of the five-membered ring aromatic heterocycles, with the reactivity of the furan being higher than the thiophene. Pr. Dr. M. D’Auria found that the cyclobutane dimerization occurs in the triplet state of the molecule and that this triplet state is obtained via energy transfer from benzophenone. For the dimerization of substituted 2-furyl- and 2-thienylacrylate (-Me, -OMe or –OH), a mixture of eight isomers could be obtained (trans and cis, head-to-head or head-to-tail). The choice of the solvent plays a key-role in the selectivity of the reaction. For example, the formation of charge-transfer complexes, favoured in polar solvent, results in a predominance of head to head dimers.[147 a] The photochemical dimerization of (E)-methyl 3-(2-thienyl)acrylate gave a mixture of two head-to-head cyclobutane derivatives in 1:1 ratio with an overall yield of 25%.
S
OMe
O 500 W lightbenzophenone
acetonitrile1 h, inert atm.
S
S
OMe
O
OMe
O
S
S
OMe
O
OMe
O
+
25%
1:1 ratio
Scheme 114. Photochemical dimerization of (E)-methyl 3-(2-thienyl)acrylate.
[147] a) M. D´Auria, L. Emanuele, G. Mauriello, R. Racioppi, J. of Photochem. and Photobio. A: Chemistry 2000, 134, 147-15.
In 1975, Ingemar Ander and co-workers studied the base-catalyzed cyclobutane formation from 3-nitro-2-vinylthiophene. [147 b]
S
NO2
ArAr
Ar
Ar ArAr
Ar
Ar
34% formaldehyde in wateracetic acidpyrrolidine
methanol3 h, 65 °C
Ar =S
NO2
cis-1,2 cis-1,3trans-1,2 trans-1,3
1 isomerhighly probable: trans-1,2
60%
Mass spectrum: Found m/e = 310 (2.2%); 282 (0.5%, loss of C2H4);155 (100%)
Scheme 115. Base-catalyzed reaction of 2-methyl-3-nitrothiophene with aqueous formaldehyde.
They supposed that decoupling of the signals centered at δ = 4.5 ppm should convert the methylene signals to an AA´BB´ spectrum for cis-1,2, trans-1,2 and cis-1,3 or an A4 spectrum for trans-1,3, since cyclobutanes are known to invert rapidly. They observed with the NMR spectrum, an unsymmetrical pattern with the methylene signals, and excluded the possibility of a trans-1,3-cyclobutane, but did not provide any interpretation.[147 b] It has been demonstrated by Dodson and Zielske that in the case of 1,2-diphenylcyclobutanes, the trans-isomer is the thermodynamically more stable isomer (less than 1% of the cis-isomer remained). [147 c] It has been found that in the 1,3-disubstituted cyclobutanes the cis isomers are thermodynamically favoured over the trans isomers. [147 d] Furthermore, Lambert and Roberts found that for a substituent on a cyclobutane ring the equatorial position is favoured over the axial position. [147 e] These facts together with the finding by Dodson and Zielske make us believe that the product obtained by Ander and co-workers is either trans-1,2-bis(3-nitro-2-thienyl)cyclobutane or cis-1,2-bis(3-nitro-2-thienyl)cyclobutane. In both cases, the mass spectral analysis showed the loss of ethylene. But the symmetric nature of the product was confirmed by its mass spectra, which showed a peak at m/e = 310 (relative abundance 2.2%) and a parent peak at m/e = 155. This behaviour is diagnostic for a head-to-head configuration of the substituents (trans-1,2).[147 a], [147 b] Doughty and co-workers showed also in their study of cyclobutanes, that the cis and the trans-1,3-cyclobutanes have similar NMR spectra, but differ in the ring methylene region. The trans isomer exhibits a narrower and more intense resonance than the cis isomer. [147 d] The preferential formation of the trans-1,2-cyclobutane can also be explained by the need of the complete superposition of the two aromatic core units (π-stacking) to initiate the reaction.
[147 a] A last criterium to confirm the cyclobutanes structures by 1H-NMR is the J-value of the A2X2 system at δ = 4-5 ppm (JAX = 7 Hz for the cis and JAX = 10 Hz for the trans).[147 a]
[147] a) M. D´Auria, L. Emanuele, G. Mauriello, R. Racioppi, J. of Photochem. and Photobio. A: Chemistry 2000, 134, 147-15. b) S. Gronowitz, I. Ander, Acta Chemica Scandinavia B 1975, 29, 513-523. c) R. M. Dodson, A. G. Zielske, J. Org. Chem. 1967, 32, 28-31. d) N. L. Allinger, L. A. Tushaus, J. Am. Chem. Soc. 1965, 30, 1935-1939; I. Lillien, R. A. Doughty, Tetrahedron 1967, 23, 3321-3326. e) J. B. Lambert, J. D. Roberts, J. Am. Chem. Soc. 1965, 87, 3884-3890.
Rhodium-catalyzed intramolecular hydroacylation of 2-vinylthiophene-3-carbaldehyde 134 gave surprisingly a thiophene cyclobutane dimer 146 instead of the previous 5,6-dihydro-cyclopenta[b]thioph-4-one 145. We obtained a thiophene cyclobutane dimer quantitatively in one hour at 150 °C.
+
[Rh(COD)2]BF4 (5 mol%)
ligand 7 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C not obtained !
S
H
O
134
S
145
O
S
H
O
146 trans-1,2-cyclobutane
S
O
H
Scheme 116. Rhodium-catalysed intramolecular hydroacylation of 2-vinylthiophene-3-carbaldehyde. The presence of only one cyclobutane dimer product, was confirmed by the 13C-NMR spectrum where we observed only seven carbons δC = 184.1 (CO), 158.4, 137.2, 128.3, 123.8 (thiophene carbons), 44.3 and 28.7 (cyclobutane carbons). The 1H-NMR spectra of compound 146 showed a singlet at δH = 9.81 ppm (aldehydic protons), a doublet at δH = 7.37 ppm and a doublet at δH = 7.17 ppm (furyl protons), three intense multiplets at δH = 4.48 ppm, δH = 2.60 ppm and δH = 2.31 ppm (cyclobutane protons). The homonuclear correlation spectroscopy (COSY) showed a clear correlation between the methylene protons. The challenge for this project is, for the first time, to calculate the HOMO, the LUMO, the LSOMO and the HSOMO of different five-membered ring aromatic heterocycles derivatives (furanes, thiophenes, imidazoles...), and afterwards benzene derivatives. We must also study the influence of the catalyst, the ligand, and the benzaldehyde substituent during the dimerization. And finally apply our method to the dimerization of substituted 2-furyl- , 2-imidazol and 2-thienylacrylate (-Me, -OMe or –OH). 4.3.12 Pharmaceutical application: Synthesis of Donepezil hydrochloride®
The 1-indanone core produced during this intramolecular process is a structural element found in many natural products and an important building block for the development of other biologically active compounds and pharmaceutical agents. In this part, we tried to prove the practicality of our method with the synthesis of Donepezil hydrochloride®, a well-known acetylcholineesterase inhibitor. This efficient drug, which features a 1-indanone core unit, is used for the treatment of Alzheimer´s type diseases, such as memory loss or other neuro-degenerative disorders. As of 2011, Donepezil hydrochloride® developed by Eisai and Pfizer was the world's best-selling Alzheimer's treatment. Several syntheses of Donepezil hydrochloride® have been reported, which are either too long or contain unacceptable operations and thus are not suitable for plant-scale operations. The best synthetic method was developed by Sugimoto et al in 1995. He reported an efficient synthesis with the condensation of commercially available 5,6-dimethoxy-1-indanone with 1-benzyl-4-piperidinecarboxaldehyde, in the presence of potassium carbonate, in aqueous methanol medium at reflux temperature, as a key step.[148]
He showed also the high and selective affinity of Donepezil for the acetylcholineesterase (IC50 = 5.7 nM), 1250 times greater than the butyrylcholineesterase. [148]
Scheme 117. Synthesis of Donepezil Hydrochloride® 152. We were able to realize a 3-step synthesis of Donepezil starting from 6-vinylveratraldehyde 99. First, 1-benzyl-4-piperidinecarboxaldehyde 150 was synthetised in two steps.[149] The relatively inexpensive ethyl isonipecotate 147 was N-alkylated with benzyl chloride 148 in toluene in the presence of triethylamine, and the resulting benzyl piperidine compound 149 was reduced to its corresponding piperidine carboxaldehyde 150 with a good yield. The 5,6-dimethoxyindanone partner 151 was performed via the rhodium-catalyzed intramolecular hydroacylation method, in one hour at 150 °C with an excellent yield (98% yield).[92] To finish the synthesis, the two coupling partners 5,6-dimethoxyindanone 145 and 1-benzyl-4-piperidinecarboxaldehyde 150 were reacted in the presence of sodium hydroxide in distilled THF at reflux temperature, to give the alkylidene product 151. [149]
The alkylidene product 151 was reduced with cobalt(II)-chloride hexahydrate and sodium borohydride to give the desired Donepezil®. Treatment with hydrochloric acid gives finally the Donepezil hydrochloride® 152 with a moderate yield of 55% after three steps. [149]
[148] H. Sugimoto, Y. Yamanishi, Y. Limura, Y. Kawakami, Current Med. Chem. 2000, 7, 303-339. [92] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637. [149] A. V. V. Srinivasrao, Y. Venkateshwarlu: An improved process for the preparation of Donepezil - WIPO Patent Application WO/2008/010235 – 2008-01-24, Torrent Pharmaceuticals Limit
107
4.4 Tetralones: rhodium-catalyzed intramolecular cyclization 4.4.1 Introduction α-Tetralones represent an important class of starting materials for the synthesis of biologically active substances. The anthracyclines (daunorubicin, doxorubicin, epirubicin, idarubicin,etc.) are one of the most important,[150], [151] because these compounds are used to treat a wide range of cancers including leukemias and lung cancers. Another outstanding example of an active α-tetralone is the tetracycline,[152] as these protein synthesis inhibitor are used against many bacterial infections like acne.
O
OO
OH
OH
OH
O
OH
HO
NH2
OH
Figure 19. Doxorubicin.
OH O OH O O
OH
NH2
OH
NHO
Figure 20. Tetracyclin. 4.4.2 Stoechiometric and catalytic reactions The classic way to synthesize 1-tetralones is the intramolecular Friedel-Crafts acylation of 4-arylbutyric acids, described by W. S. Johnson in 1949.[153] In 1984, B. Hulin and M. Koreeda described a mild method for the cyclization of 4-arylbutyric acid via its trifluoromethanesulfonic anhydride derivative, where they obtained 87% yield[154]
COOH1) SOCl22) CF3SO2OH
O
87%
Scheme 118. Cyclization of 4-aryl alkanoic acid to α-tetralone via its trifluoromethanesulfonic anhydride derivative.
[150] M. Braun, Tetrahedron 1984, 40, 4585-4591. [151] J.P. Rizzi, S.A. Kende, Tetrahedron 1984, 40, 4693-4700. [152] G. Stork, J. J. La Clair, V. G. Young Jr, X. Xie, R. E. McCarley, J. G. Verkade, J. Am. Chem. Soc. 1996, 118, 5304-5305. [153] W. S. Johnson, Org React 1949, 2, 114-177. [154] B. Hulin, M. Koreeda, J. Org. Chem. 1984, 49, 207-209.
More examples were described without resolving the strongly acidic conditions, especially when the aromatic ring is desactivated by electron withdrawing substituents.[155]
A few other routes may be found in the literature to form 1-tetralones. In every case, they are very specific, and not transposable into a general procedure. These include, for instance, the oxidation of tetrahydronaphthalenes,[156] the photolysis of 1-o-methylaryl-1,3-diketones,[157] or the Michael addition of the anion derived from an o-toluate to an α,β-unsaturated ester followed by a Dieckmann condensation.[158]
In 2012, Douglas et al. reported the rhodium-catalysed formation of 1-tetralones and 1-suberones via an intramolecular hydroacylation using a simple pyridine ligand and triphenylphosphine.[82] He obtained 3-methyl-1-tetralone with 65% yield using 2.5 mol% of catalyst, 5 mol% of triphenylphosphine and additives at 100 °C.
N NH2
R´
a : R´ = H
b : R´ = N
H
O
R
O
R
[RhCl(coe)2]2 (2.5 mol%)ligand (25 mol% a
or 100 mol% b)PPh3 (5 mol%)aniline (1.2 eq.)
benzoic acid (10 mol%)
PhCF3, 100 °C
R = Me (65% yield a)R = Ph (48% yield a) (58% yield b)
(continued on the next page)
[155] a) B. B. Snider, T. J. Kwon, J. Org. Chem. 1990, 55, 4786-4788 ; b) F. Z. Yang, K. M. Trost, W. E. Fristad, Tet. Lett. 1987, 28, 1493-1496 ; c) J. H. Rigby, A. Kotnis, J. Kramer, Tet. Lett. 1983, 24, 2939-2940 ; d) F. Johnson, E. R. Marinelli, J. Org. Chem. 1986, 51, 3911-3913 ; e) R. Beugelmans, J. Chastanet, H. Ginsburg, L. Quintero-Cortes, G. Roussi, J. Org. Chem. 1985, 50, 4933-4938 ; f) R. C. Klix, M. H. Cain, A. V. Bhatia, Tet. Lett. 1995, 36, 6413-6414 ; g) J. Thibonnet, M. Abarbri, J.-L. Parrain, A. Duchêne, Tet. Lett. 1996, 37, 7507-7510. [156] D. Zhao, D. G. Lee, Synthesis 1994, 915-916. [157] J. M. Hornback, M. L. Poundstone, B. Vadlamani, S. M. Graham, J. Gabay, S. T. Patton, J. Org. Chem. 1988, 53, 5597-5601. [158] B. Tarnchompoo, C. Thebtaranonth, Y. Thebtaranonth, Synthesis 1986, 785-786.
[82] E. V. Beletskiy, Ch. Sudheer, C. J. Douglas, J. Org. Chem. 2012, 77(14), 5884-5893.
Scheme 119. Cooperative catalysis approach to intramolecular hydroacylation.
To avoid the undesired decarbonylation side-reaction, Saegusa et al. reported in 1990 the first Ni0/PR3-catalyzed intermolecular alkyne hydroacylation to give α,β-enones.[83]
Following this work, Ogoshi et al. investigated the mechanism of the Ni(0)/NHC hydroacylation, and they concluded that the reaction pathway proceeds through an oxanickelocycle intermediate, and not through an acyl metal intermediate as described in our rhodium/P-N ligand strategy.[84] They could apply this catalyst for the synthesis of a broad range of 1-indanone derivatives in good to excellent yields. More details are described in the background (p. 30).
4.4.3 Substrate synthesis
o-Allylbenzaldehyde was obtained in two steps; in the first step, vinyl magnesium bromide reacted with 2-bromobenzylbromide 153 in the presence of copper iodide and 2,2’-bipyridine to form 1-bromo-2-allylbenzene 154. In the second step, 154 was converted via the Bouveault aldehyde synthesis into o-allylbenzaldehyde 155 in 58 % yield.[159]
[83] T. Tsuda, T. Kiyoi, T. Saegusa, J. Org. Chem. 1990, 55, 2554-2558. [84] Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi, S. Ogoshi, Angew. Chemie 2012, 124(43), 10970-10973; Angew. Chem. Int. Ed. 2012, 51(43), 10812-10815. [159] I. D. G. Watson, S. Ritter, F. D. Toste, J. Am. Chem. Soc. 2009, 131(6), 2056-2057.
1) CuI, 2,2'-bipyridine, toluene, 0 °C2) vinyl magnesium bromide, 4 h, 0 °C to RT Br
154
1) n-BuLi, THF, 30 min., -78°C2) DMF, THF, o/n, -78 °C to RT
155
O
H
58%
68%
Scheme 120. Synthesis of o-allylbenzaldehyde 155.
4.4.4 Catalytic experiments Substrate 155 was tested in the rhodium catalyzed intramolecular alkene hydroacylation of o-vinylbenzaldehyde 95 using ligand 1, cationic [Rh(COD)2]BF4 or neutral [Rh(COD)Cl]2 rhodium precursors. Solvent effect, reaction time and temperature were optimized. Our system was compared with the Jun’s system[90], [91] and Wilkinson’s catalyst.[160]
Rhodium catalyzed intramolecular hydroacylation of o-allylbenzaldehyde give two products according to the Baldwin’s rules:
The 6-endo-trig product : α-tetralone
The 5-exo-trig product : 2-methylindanone 4.4.4.1 Hydroacylation of o-allylbenzaldehyde 155
H
O
toluene (c = 1.1 M)120 °C, 24 h
O
+
O
[Rh(COD)2]BF4
155
156 157
158 159
decarbonylated products
Scheme 121. Rhodium-catalyzed intramolecular hydroacylation of o-allylbenzaldehyde 155.
[160] J. A. Osborn, G. Wilkinson, J. Am. Chem. Soc. A: Inorganic, Physical, Theoretical 1966, 1711-1732.
(0.022 mmol, 20.4 mg). d o-vinylbenzaldehyde 155 (0.22 mmol, 29.1 mg), [RhCl(PPh3)3] (0.022 mmol, 20.4 mg). e Determined by 1H NMR methods. Reactions performed in 8 mL Schlenk tubes.
The neutral catalyst precursor [Rh(COD)Cl]2 was used first to hydroacylate o-allylbenzaldehyde 155. No solvent was used. Hydroacylation (entry 1, Table 27) with 5 mol% of [Rh(COD)Cl]2 and 10 mol% of ligand 1 at 150 °C for 24 h gave a mixture of 1-tetralone 156 (44%), 2-methylindanone 157 (26%), allylstyrene 158 (21%) and isomer of allylstyrene 159 (9%). The same reaction was performed at 120 °C (entry 2) for 24 hours yielding a mixture of 1-tetralone 156 (38%), 2-methylindanone 157 (37%) and isomer of allylstyrene 159 (27%). 1-tetralone 156 is obtained with [Rh(COD)2]Cl2 but the yield was not satisfied.
Cationic catalyst [Rh(COD)2]BF4 was tested with 5 mol% of [Rh(COD)Cl]2 and 10 mol% of ligand at 150 °C for 24 hours yielding 45% of 1-tetralone 156 and 47% of the 5-exo-trig-product 157 (entry 3). The proportion of by-products 158 and 159 was decreased, but the selectivity 6-endo-trig 156 / 5-exo-trig 157 was low. Two parameters were changed to improve this selectivity 156 / 157. The temperature was decreased to 120 °C (entry 4) and 90 °C (entry 5). At 120 °C, no selectivity was obtained, but no by-products were detected. When entry 4 was performed in toluene, 1-tetralone 156 (78%) and 2-methylindanone 157 (22%) were obtained without by-products. Solvent plays a key role in the hydroacylation of o-allylbenzaldehyde 155. The loading of the ligand and the catalyst was divided by two (entry 7) which resulted in a low conversion. To improve the selectivity of the reference hydroacylation (entry 6), ligand was added (entry 8) and (entry 9) (1.3 equivalent). A 5% increase of the yield was observed (1-tetralone 156 83%, 2-methylindanone 157 17%).
To finish the study our best results (entry 6) and (entry 9) were compared with the Jun’s system, and the Wilkinson’s catalyst. With the Jun’s system (entry 10) (2-amino-3-picoline 40 mol%, [Rh(PPh3)3]Cl 10 mol%) at 120°C (entry 10), a mixture of 1-tetralone 156 (54%), 2-methylindanone 157 (16%), allylstyrene 158 (25%) and o-allylbenzaldehyde 155 (5%) was obtained. Using 40 mol % of [Rh(PPh3)3]Cl without ligand at the same temperature (entry 11) gave no products. Starting material was totally degraded.
Table 28. Hydroacylation of o-allylbenzaldehyde 155 (solvent and ratio ligand/[Rh] catalyst).
entry
ligand (mol%)
ligand (mol%) condition
conversion (%)
NMR yield (%)
1
N NH2
P
1
(13)
[Rh(COD)2]BF4 (10)
72 h, 110 °C Toluene (200 μL)
30
156 (20), 157 (10)
(continued on the next page)
* Thanks to my bachelor student: Fanny Cacheux (National Graduate School of Chemistry, Mulhouse (FRANCE), 2011, Master 1 student).
Cationic catalyst [Rh(COD)2]BF4 in dry toluene was first used to hydroacylate o-allylbenzaldehyde 155. Hydroacylation (entry 1, Table 28) with 10 mol% of [Rh(COD)2]BF4
and 13 mol% of ligand 1 at 110 °C for 72 hours gave a mixture of 1-tetralone 156 (20%) and 2-methylindanone 157 (10%). The same reaction realized at 100 °C (entry 2) for 72 h gave only 5% of conversion. Hydroacylation with 10 mol% of [Rh(COD)2]BF4 and 13 mol% of ligand 1 at 120 °C was then tested with different solvents. No conversion was observed in pyridine (entry 3) and in dichloromethane (entry 8). Reaction in acetonitrile (entry 4) gave a mixture of 1-tetralone 107 (54% yield) and 2-methylindanone 157 (35% yield). The conversion was complete with ethanol (entry 5), and a mixture of 1-tetralone 156 (63%) and 2-methylindanone 157 (37%) was obtained. The same reaction with TBME gave a mixture of 1-tetralone 156 (28% yield) and 2-methylindanone 157 (32% yield) and a conversion quite low. Finally, a very low conversion (14%) was obtained with ethyl acetate (entry 7). Another series of experiments were made in toluene using another ligand 7 in order to compare the reactivity of the ligand. Hydroacylation (entry 9) with 10 mol% of [Rh(COD) 2]BF4 and 13 mol% of ligand 7 at 120 °C in 200 L of toluene for 72 h gave any conversion. The same result was obtained with [Rh(COD)2]BF4 and 16 mol% of ligand 7 at 120 °C for 72 h (entry 10). Then, hydroacylation (entry 11) with 10 mol% of [Rh(COD) 2]BF4 and 13 mol% of ligand 7 at 120 °C neat for 72 h gave a ratio of 85% of 1-tetralone 156 and 15% of 2-methlyindanone 157 but the conversion was not complete (91%). Finally, hydroacylation (entry 12) with 10 mol% of [Rh(COD)2]BF4 and 13 mol% of ligand 7
at 120 °C in 400 L of toluene for 72 h gave a ratio of 5% of 1-tetralone 156 and 95% of 2-methlyindanone 157, but with a very low conversion. 4.4.4.2 Conclusion Good yields (entry 6) were obtained with [Rh(COD)2]BF4. In the formation of 1-tetralone 107 from o-allylbenzaldehyde 155 in comparison to Jun’s system (entry 18) and (entry 19), and Wilkinson’s catalyst (entry 20) and (entry 21).
* Thanks to my bachelor student: Fabio Lima (National Graduate School of Chemistry, Toulouse (FRANCE), 2011, Master 1 student).
Scheme 122. Substrate synthesis of substituted o-allylbenzaldehyde.
The first step in the synthesis of o-allylbenzaldehyde was a radical bromination of bromotoluene with N-bromosuccinimide activated by AIBN in benzene for sixteen hours. Then, the dibrominated compound was treated with H2SO4 to give the corresponding aldehyde. 2-Bromobenzaldehyde was then protected by ethylene glycol in benzene. The reaction mixture was heated to reflux with a Dean-Stark trap for twenty-six hours. To a solution of the corresponding 1,3-dioxolane protected product at -78 °C, was added 1.2 equivalent of n-BuLi (2.5 M in hexane).[161] The mixture was stirred for two hours at -78 °C.[161] The lithiated intermediate was not isolated; it was directly quenched in situ with MgBr2 freshly prepared from magnesium and dibromoethane.[161] After two hours, allyl bromide was added to give the o-allyl-1,3-dioxolane protected product.[161] The final step to obtain o-allylbenzaldehyde was the deprotection with KHSO4 in water/acetone at 60 °C for 4.5 hours, or with pTSA (or PPTS) in a mixture of water in THF overnight.
A library of seven diverse o-allylbenzaldehydes containig electron withdrawing and electron donating groups was tested successfully under our optimal condition for the rhodium-catalyzed selective 1-tetralone synthesis. Diversity in the core unit was taken in consideration with the hydroacylation of 2-allyl-naphthalene-1-carbaldehyde 180.
[161] K. Knobloch, M. Keller, W. Eberbach, Eur. J. of Org. Chem. 2001, 17, 3313-3332.
4.4.5.1 Synthesis of 2-allyl-4-methyl-benzaldehyde 164
Me Br
H
O O
Me Br
H
O
Li
H
O O
MgBr
H
O OH
O O
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a Dean-Stark trap
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
KHSO4, H2O/ acetone (1 : 6)4.5 h, 60 °C
Me
MeMeMe
109 160 161
162163164
98%
23%(after 3 steps)
quant.
Scheme 123. Synthesis of 2-allyl-4-methylbenzaldehyde 164.
The 2-allyl-4-methylbenzaldehyde substrate 164 was obtained from 2-bromo-4-methyl benzaldehyde 109 with a 22% overall yield after five steps according to the synthetic pathway described in Scheme 123.
4.4.5.2 Synthesis of 2-allyl-5-methoxy-benzaldehyde 170
Br
H
O O
Br
H
O
Li
H
O O
MgBr
H
O O
H
O O
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a Dean-Stark trap
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
165 166 167
168169170
quant.
29%after 3 steps
MeO MeO MeO
MeOMeOMeO quant.
Scheme 124. Synthesis of 2-allyl-5-methoxybenzaldehyde 170. The 2-allyl-5-methoxybenzaldehyde substrate 170 was obtained from 2-bromo-5-methoxy benzaldehyde 165 with a 29% overall yield after five steps according to the synthetic pathway described in Scheme 124.
4.4.5.3 Synthesis of 2-allyl-5-fluoro-benzaldehyde 175
98%
61%(after 3 steps)
Br
H
O O
Br
H
O
Li
H
O O
MgBr
H
O O
H
O O
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a Dean-Stark trap
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
KHSO4, H2O/ acetone (1 : 6)4.5 h, 60 °C
123 171 172
173174175
F F F
FFF 88%
Scheme 125. Synthesis of 2-allyl-4-fluorobenzaldehyde 175.
The 2-allyl-4-fluoro-benzaldehyde substrate 175 was obtained from 2-bromo-5-fluoro benzaldehyde 123 with a 53% overall yield after five steps according to the synthetic pathway described in Scheme 125.
4.4.5.4 Synthesis of 2-allyl-naphthalene-1-carbaldehyde 180
98% (after 3 steps)
BrNBS, AIBN every 3 hours
benzene, 16 h, reflux Br
Br
Br
Br
O
H
Br
H
Li
H
O OO O
MgBr
H
O O
H
O O
H
O
ethylene glycolpTSA, H2O (cat.)benzene, 26 hreflux with a Dean-Stark trap
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h
reflux (70 °C)KHSO4, H2O / acetone (1 : 6)
4.5 h, 60 °C
97% 95%
111 112 113
176
180179
178 177
quant.
97%
CaCO3, H2O8h, reflux
Scheme 126. Synthesis of 2-allyl-naphtalene-1-carbaldehyde 180.
The 2-allyl-naphthalene-1-carbaldehyde substrate 180 was obtained from 2-bromo-1-methyl naphthalene 111 with a 93% overall yield after five steps according to the synthetic pathway described in Scheme 126. 4.4.5.5 Synthesis of 2-allyl-4,5-dimethoxy-benzaldehyde 185
MeO Br
H
O O
MeO Br
H
O
Li
H
O O
MgBr
H
O O
H
O O
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a
Dean-Stark trap n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
MeO
MeOMeOMeO
98 181 182
183184185
MeO MeO MeO
MeOMeOMeO
76%
43% (after 3 steps)
quant.
Scheme 127. Synthesis of 2-allyl-4,5-dimethoxybenzaldehyde 185.
The 2-allyl-4,5-dimethoxy-benzaldehyde substrate 185 was obtained from 2-bromo-4,5-dimethoxybenzaldehyde 98 with a 33% overall yield after five steps according to the synthetic pathway described in Scheme 127.
4.4.5.6 Synthesis of 2-allyl-5-chloro-benzaldehyde 190
BrNBS, AIBN every 3 hours
benzene, 16 h, reflux Br
Br
Br
Br
O
H
Br
H
Li
H
O OO O
MgBr
H
O O
H
O O
H
O
ethylene glycolpTSA, H2O (cat.)benzene, 26 hreflux with a Dean-Stark trapn-BuLi (2.5 M in n-hexanes)
THF, 2 h, -78 °C.MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h
reflux (70 °C)KHSO4, H2O / acetone (1 : 6)
4.5 h, 60 °C
72% 95%
Cl
Cl
H2SO4, 1 h, RT
Cl Cl
ClClCl
Cl
119 120 121
186
190189
188 187
quant.
20%(after 3 steps)
95%
Scheme 128. Synthesis of 2-allyl-5-chloro-benzaldehyde 190. The 2-allyl-5-chloro-benzaldehyde substrate 190 was obtained from 2-bromo-5-chloro-benzaldehyde 119 with a 19% overall yield after five steps according to the synthetic pathway described in Scheme 128.
In summary, the rhodium-catalyzed intramolecular hydroacylation of o-allylbenzaldehyde derivatives was very slow (3 days of reaction time required). A good conversion (86%) and a favorable ratio (91:9) for the desired 1-tetralone 156 was obtained under neat conditions for the hydroacylation of o-allylbenzaldehyde 155. A similar favorable ratio was observed for the 4,5-dimethoxy-substituted tetralone 193 (79:21) and the 5-chloro-substituted tetralone 195 (86:14). For the naphtha tetralone 191, the reaction was slower (5 days). A full conversion under neat conditions was observed, but with a less selective ratio (66:34).
4.5.1 Introduction In 2005, K. Yoshikai et al. achieved in moderate yield (56-90%) the atom-economical synthesis of six-membered cyclic ketones through an intramolecular addition to olefins of in situ generated acyl radicals by tert-dodecanethiol and AIBN.[162] This method is resctricted to electron-deficient olefins, and requires thermal activation (refluxing chlorobenzene).
OMe
OO O
O
OMe
O
H
O
tert-dodecanethiol (0.3 eq)AIBN (0.3 eq)
toluene (0.1 M)19 h, reflux
same conditions
85%
58%
Scheme 129. Synthesis of six-membered cyclic ketones using a free radical activation.
Another efficient method for the synthesis of 6-membered-ring carbocycles is the cyclization of alkenyl β-keto esters and amides by an efficient and reusable PdCl2(MeCN)2/CuCl2/PEG-400 system.[163] A large library of substituted alkenyl substrates was successfully tested up to furnish the corresponding cyclohexanones in 98% yield. However, the cyclization of a hexenyl keto ester substrate failed
O
OMe
O
O
OMe
O
O
OMe
O
OMe
OO
O
OMe
OH
H
O
OMe
O
PdCl2,(MeCN)2, CuCl2
PEG-400, 4 h
97%
same conditions
80%
PdCl2,(MeCN)2, CuCl2
PEG-400, 24 h
<5%
Scheme 130. Palladium cyclization of alkenyl β-keto esters and amides.
[162] K. Yoshikai, T. Hayama, K. Nishimura, K.-I. Yamada, K. Tomioka, J. Org. Chem., 2005, 70, 681-683. [163] J.-H. Li, Q.-M. Zhu, Y. Liang, D. Yang, J. Org. Chem., 2005, 70, 5347-5349.
The use of the so-called Hajos-Parrish-Eder-Sauer-Wiechert (HPESW) reaction is an efficient organocatalytic process for the synthetis of the bicyclic Wieland-Miescher (W-M) and the Hajos-Parrish (H-P) ketones. [152] This method developped by J.-P. Cheng and co-workers is a gram scale method using a low catalyst loading, efficient (high enantioselectivity up to 96%), excellent yields up to 98%, and potentially green (solvent free, room temperature).[164]
O
O
O
O
OR
N
NH2
. TfOH
(10 mol%)
m-NO2C6H4CO2H (5 mol%)
neat, RT, 12 h
yield: 95%ee: 92%
W-M ketone
O
O
O
O
O
same conditions
yield: 95%ee: 96%
H-P ketone
Scheme 131. Cyclohexanone synthesis via a Hajos-Parrish-Ender-Sauser-Wiechert reaction. 4.5.2 Stoechiometric and catalytic reactions From the interaction of different strain effects (binding, torsion, Van der Walls radii), combinded with the entropy factor, it is found that five and six-membered rings are usually formed most easily. And the cyclization of the five-membered ring is significantly faster than the 6-membered one (100 times in the case of a lactonisation). In 1980, R. E. Campbell Jr. and R. G. Miller published a rhodium-catalyzed hydroacylation of 5-hexen-1-al.[165] The reaction does not provide the cyclohexanone but delivers the cyclopentanone as the sole product in low yield while employing a high catalyst loading.
O O
H
O RhCl(PPh3)3 (50 mol%)
CH2Cl2, RT
19%
Scheme 132. Rhodium-catalyzed hydroacylation of 5-hexen-1-al. Only one specific example has been described in the literature for the six-membered ring formation using a rigid carbohydrate-derived scaffold.
[164] P. Zhou, L. Zhang, S. Luo, J.-P. Cheng, J. Org. Chem., 2012, 77, 2526-2530. [165] R. E. Campbell Jr. and R. G. Miller, J. of Organomet. Chem. 1980, 186, C27-C31.
K. Gable and G. A. Benz used the Wilkinson catalyst [RhCl(PPh3)2]2 and ethylene (1 atm) for the saturation of the metal center and to avoid the decarbonylation.[166] In this special case, the cyclopentanone is disfavoured due to the ring strain that would be present in the resulting 5,5,5-tricyclic product. This method cannot be considered as a general process.
H
O
O
O
O
Me
MeH
H
HO
O
O
O
O
H
H Me
Me
H
HO
[(Ph3P)2RhCl]2 (30 mol%)
C2H4 (1 atm)CDCl3, 70 °C, 6 h
60%
Scheme 133. Cyclohexanone synthesis via a Hajos-Parrish-Ender-Sauser-Wiechert reaction. 4.5.3 Synthesis of 5-hexen-1-al A more accessible method for the synthesis of 5-hexenal 197 is the alcohol oxidation of the inexpensive, commercially available 5-hexen-1-ol 47. Three different oxidation methods were tested : manganese dioxide, Swern oxidation with oxalyl chloride, and Corey oxidation with pyridinium chlorochromate PCC.
OH H
O[ox.]
Methods: 1) MnO2 2) DMSO, (ClCO)2, NEt3 3) PCC
47 197
Scheme 134. Oxidation of 5-hexen-1-ol to 5-hexenal.
The Corey and Swern oxidation both gave excellent yields for the oxidation of 5-hexen-1-ol. The high toxicity and the carcinogenic affect of the chromium (VI) compounds encourage us to favour the Swern oxidation for a large scale reaction. 4.5.4 Catalytic experiments 4.5.4.1 Hydroacylation of 5-hexen-1-al The catalytic intramolecular hydroacylation of 5-hexenal 197 is not described in the literature, and is a great challenge especially with its asymmetric version for the synthesis of 2-substituted chiral cyclohexanones. The best conditions obtained for the analog cyclisation of the o-allylbenzaldehyde to its corresponding cyclized 1-tetralone product (cationic rhodium catalyst [Rh(COD)2BF4], 120 °C, neat, “OMe” ligand 7 was first attempted for the cyclisation of 5-hexenal. Modifying the ligand/catalyst ratio, solvent screening with the two best solvents (toluene and ethanol) described for the hydroacylation of 1-indanones and 1-tetralones, and a comparison between [Rh(COD)2]BF4 and [Rh(COD)Cl]2 were also performed here.
H
O
OO
N
OMe
NH2
P
[Rh]-cat.+
197 198 199
Scheme 135. Cyclisation of 5-hexenal. Table 31. Hydroacylation of 5-hexen-1-al.
5-hexen-1-al (0.22 mmol, 23 μL), [Rh(COD)2]BF4 (0.022 mmol, 9.0 mg), ligand 7 (0.029 mmol, 9.2 mg), ethanol (200 μL). f 5-hexen-1-al (0.22 mmol, 23 μL), [Rh(COD)2]BF4 (0.022 mmol, 9.0 mg), ligand 7 (0.035 mmol, 11.4 mg). All reactions are performed under argon in 8 mL Schlenk tubes. In a previous chapter (1-tetralone synthesis), we propose a thermodynamic control between the 5-exo-trig 2-methyl-indanone 199 (obtained mostly at 120 °C) and the 6-endo-trig 1-tetralone 198 (obtained predominantly at 150 °C). In our standard condition (entry 1, Table 31), the cyclization was performed in excellent yield (98%) and with a good endo excess (endo/exo: 82/18). When increasing the ratio ligand/catalyst (1/1 to 1.3/1 and 1.6/1), resulted in a better endo excess (endo/exo: 84/16), but with a significant decrease of the yield, 57% and 52%, respectively (entries 5 and 7). A similar inverse endo/exo ratio at 150 °C furnished in excess the 2-methylcyclopentanone, but at higher temperature, many decarbonylated and isomerized products were obtained. A full conversion was observed, but a lower yield in 2-methylcyclopentanone (entry 2). Using the neutral rhodium catalyst [Rh(COD)Cl]2 gave a lower 55% yield (33% less than the use of [Rh(COD)2BF4]) and a worth endo/exo selectivity (71/29) (entry 4). Finally, the solvent screening with toluene (entry 3) and ethanol (entry 6) gave no significant improve, respectively 53% and 72% of yield with a smaller endo excess. 4.5.4.2 Conclusion A promissing result (98% of yield, endo/exo : 82/18) was obtained (entry 1) for the cyclization of the 5-hexenal. However, a decreasing of the reaction time and the catalyst loading must be performed to complete this study. An asymetric version with the rhodium-catalyzed intramolecular hydroacylation of 6-substituted 5-hexen-1-al to synthesize chiral α-substituted cyclohexanones will be considered.
Scheme 136. Asymetric rhodium-catalyzed intramolecular hydroacylation of 6-substituted 5-hexen-1-al
4.5.6 Synthesis of medium-sized rings Medium-sized rings are motifs in natural products and regarded difficult structures to access in organic synthesis. We proved in this project, the possible use of transition-metal catalysts to achieve the formation of cyclic ketones, with the successful synthesis of cyclohexanone, obtained with 98% of yield. By extension of this result, we tried to synthesize cyclodecanone 202 from dodec-11-enal 201. Starting dodec-11-enal 201 was obtained by a quantitative one-step Swern-type oxidation of dec-9-en-1-ol 200. No hydroacylated product was observed by 1H NMR, starting material was recovered.
OH
oxalyl chloride, DMSO
-78 °C, 1 hH
O
H
O[Rh(COD)2BF4] (10 mol%) O
ligand 7 (13 mol%)
toluene (c = 1.1 M)
no reaction
quant.200
202201
201
150 °C, 1 h
Scheme 137. Rhodium-catalyzed synthesis of cyclodecanone 202. According to the strategy used by Dong and co-workers for the synthesis of medium-sized heterocyclic ketones via oxygen-assisted hydroacylation, we tried to synthesize a ten-membered heterocycle with the use of the P-N ligand 7 and [Rh(COD)2BF4].[167] The starting 2-hex-5-enyloxy-benzaldehyde 204 was obtained in two steps. Toluene-4-sulfonic acid hex-5-enyl ester 203 was performed in 64% yield by O-tosylation of hex-5-en-1-ol 47.[168] And then, salicylaldehyde 85 was converted to the O-alkylated starting 2-hex-5-enyloxy-benzaldehyde 204 (45% yield). No hydroacylated product was observed by 1H NMR, starting material was recovered.
[167] K. Mori, Eur. J. of Org. Chem. 2005, 10, 2040-2044. [168] M. M. Coulter, P. K. Dornan, Vy. M. Dong, J. Am. Chem. Soc. 2009, 131, 6932-6933.
Scheme 138. Rhodium-catalyzed synthesis of ten-membered heterocycle 205.
129
4.6 Mechanistic studies 4.6.1 Substrate-catalyst interaction We started our investigation with the initial substrate-catalyst interaction. In 1988, Michael Anderson and co-workers published crystal and molecular structures of rhodium complexes with the P-N donor ligand (C6H5)2P(CH2)2C5H4N, and its potential use in the catalytic decarbonylation of aldehydes.[169] In particular, the cationic rhodium complex [Rh(PN)2]
+ which showed good catalytic activities, high yield, high selectivity, and a remarkable robustness in diverse organic syntheses was studied. They synthesized this rhodium [Rh(PN)2]
+ complex with the BF4-
counterion in good yield upon treatment of [Rh(NBD)Cl]2 in acetone solution with AgBF4, followed by the addition of 2 equiv. of the P-N ligand in toluene. This P-N bis chelated complex with the phosphorus atoms trans to the nitrogen atoms has a square planar geometry with a moderate tetrahedral distortion due to steric hinderance provided by the phosphine groups (Figure 22).[169]
+ . In our case, the active cationic rhodium complex intermediate [Rh(PN)2]
+ 206 was formed by reacting rhodium catalysts ([Rh(COD)Cl]2 with ligand 1 for ten minutes. in deuterated chloroform directly in a special argon flushed NMR tube closed with a septum (entry 1, Table 32). A mixture of two rhodium complexes was observed in 31P NMR
doublet, 59.4 ppm, JRh,P = 168.8 Hz: [Rh(PN)(COD)]Cl 207 [169] M. P. Anderson, A. L. Casalnuovo, B. J. Johnson, B. M. Mattson, A. M. Mueting, Inorg. Chem. 1988, 27, 1649-1658. * 31P NMR spectrum of [Rh(PN)2]Cl 206 and [Rh(PN)(COD)]Cl 207 in the appendix.
ResultsandDiscussion:Mechanisticstudies
130
Table 32. Rhodium complex with P-N Ligand 1
entry
Rh source
conditions
31P NMR
1a
[Rh(COD)Cl]2
10 min., RT, CDCl3
Sonication
doublet, 59.4 ppm, JRh,P = 168.8 Hz
[Rh(PN)(COD)]Cl 207
doublet, 30.7 ppm, JRh,P = 143.96 Hz
[Rh(PN)2]Cl 206
2a
[Rh(COD)Cl]2
1 h, 40 °C, CDCl3
Sonication
doublet, 30.7 ppm, JRh,P = 143.96 Hz
[Rh(PN)2]Cl 206
a benzylidene-{6-[(diphenylphosphanyl)-methyl]-3-methyl-pyridin-2-yl}-amine (50 mg, 0.12 mmol), [Rh(COD)Cl]2 (125 mg, 0.24 mmol, 2 eq), deuterated chloroform (1 mL). To complete the reaction, the reaction mixture was heated for one hour at 40 °C (entry 2, Table 32). The unique bis-chelated PN ligand rhodium complex [Rh(PN)2]Cl was observed. The interaction of the benzylidene-{6-[(diphenylphosphanyl)-methyl]-3-methyl-pyridin-2-yl}-amine (imine-PN ligand 1) with the active rhodium center was confirmed in 1H NMR with a shift of 0.45 ppm of the iminic proton:
* 1H, 13C, 31P NMR spectra of imine-PN ligand 1 and imine-PN ligand 1 + [Rh(COD)Cl]2 in the appendix.
ResultsandDiscussion:Mechanisticstudies
131
The 15N NMR analysis indicated a double interaction of the rhodium with the two nitrogen atoms from the amino group and the pyridine moiety. Finally, the mass of the active [Rh(PN)2]
+ species was confirmed by high resolution mass spectrum (dir. pos. ESI, M = 891.2 ([Rh(PN)2]
+)). 4.6.2 The evidence of the imine as a key intermediate In order to get more insight into the mechanism of the reaction, we pre-formed the imine between ligand 1 and benzaldehyde and added a stoichiometric amount of [Rh(COD)Cl]2 under the same reaction conditions used for catalysis. Aqueous work-up released the desired ketone, supporting the existence of a crucial imine as a key intermediate.
with a pre-formed Imine and 1-Octene. In addition, a control experiment with ligand L’ 209, lacking the amino functionality, resulted in a complete loss of activity in the reaction, thus highlighting the importance of the amino group within ligand 1 for the reaction to proceed via iminium formation and the prevention of decarbonylation (Schemes 140 and 141).
N
PPh2
Ligand L´ (209)
Scheme 140. Ligand L’ 209 lacking the amino functionnality. * HR mass spectrum of the active [Rh(PN)2]
+ species in the appendix.
ResultsandDiscussion:Mechanisticstudies
132
O
H+
OLigand L´ 209 (10 mol%)[Rh(COD)Cl]2 (5 mol%)
toluene (c = 1.1 M)150 °C, 24 hno reaction
O
H
Ligand L´ 209 (10 mol%)[Rh(COD)Cl]2 (5 mol%)
toluene (c = 1.1 M)150 °C, 24 h
42%
O
44 45 46
95 96
Scheme 141. Rhodium-catalyzed inter- and intramolecular hydroacylation with the use of our P-N ligand lacking the amino functionality.
4.6.3 The influence of the P-N ligand The successful reaction of the preformed imine supports the in situ activation of the starting aldehyde as an imine via condensation with the P-N ligand. To verify this result, we studied the rate of the imine formation respectively with the similar Me P-N ligand 1 and OMe P-N ligand 7 (Scheme 142).
O
H+
toluene (c = 1.1 M)150 °C
N NH2
Me
PPh2
N N
Me
PPh2
O
H+
N NH2
OMe
PPh2
N N
MeO
PPh2
reaction time
1 d2 d3 d
yield
92%96%96%
toluene (c = 1.1 M)150 °C
reaction time
1 d2 d3 d
yield
42%72%94%
H
H
44 1
44 7
208
210
Scheme 142. Comparative study of the imine formation rate for ligand 1 and ligand 7. * 1H and 13C spectra of Me P-N ligand 1 and OMe P-N ligand 7 in the appendix.
ResultsandDiscussion:Mechanisticstudies
133
The slow formation (42% after 1 day) of the imine in the case of the OMe PN ligand 7 could explain the lower yield (24%) obtained with this ligand in the intermolecular hydroacylation of benzaldehyde and 1-octene, in comparing with the 82% of yield performed with the Me P-N ligand 1 (entry 1, Table 25). 4.6.4 The deteurium-labeling studies To gain further insight into the reaction mechanism, we prepared deuterium-labeled benzaldehyde, and we studied its reaction with 1-octene in our standard reaction conditions. The hydrolysis of benzil in deuterium oxide in the presence of potassium cyanide gives the corresponding deuterated benzaldehyde in 96% at room temperature for one hour (Scheme 143).[170]
+
N
Ph2P
NH2
O
O
O
DKCN, D2O
dioxane, RT, 30 min
O
D
O
D
+
O D
ratio 213/214 (1.4:1)
[Rh(COD)Cl]2, toluene
150 °C, 24 h
96%
82%
211 212
212 45
213
214
Scheme 143. Rhodium-catalyzed intermolecular hydroacylation with deuterium-labeled benzaldehyde and 1-octene.
The reaction of deuterated benzaldehyde and 1-octene gives a ratio α/β deuterated ketone of 2.9:1. The triplet at δH = 2.96 ppm (α-ketone methylene protons) and the quintuplet at δH = 1.73 ppm (β-ketone methylene protons) are used to calculate the ratio α/β deuterated ketone. These two charasterictic peaks integrated for 1.29 (α) and 1.75 (β) instead of a theoretical base 2H (non-deteurated case).
α/β excess = 75.12
29.12
= 2.9
[170] A. G. Griesbeck, S. Bondock, P. Cygon, J. Am. Chem. Soc. 2003, 125(30), 9016-9017.
ResultsandDiscussion:Mechanisticstudies
134
Scheme 144. Rhodium-catalyzed intermolecular hydroacylation with deuterium-labeled benzaldehyde and 1-octene (1H-NMR).
A high-resolution mass analysis of the ketone product showed a unique deuterium incorporation in the molecule. This result proves that the deuterium relocation is an intramolecular process according to the conclusion of Bosnich et al. (Scheme 144).[171], [172] Bosnich et al. showed in their study of the intramolecular hydroacylation of 4-pentenal that the resulting 2.7:1 ratio is due to the involvement of a hydrogen atom or a deuterium atom during the β-elimination, after the deuteride transfer to the olefin (Scheme 145). [171], [172]
This β-elimination is reversible, and subsequently the deuterated benzaldehyde and the formed deuterated olefin are in equilibrium. The hydrogen five-membered metallocycle and the deteurated one give respectively the β and the α-deuterated ketones (Scheme 145). [171], [172]
[171] D. P. Fairlie, B. Bosnich, Organometallics 1988, 936-945. [172] D. P. Fairlie, B. Bosnich, Organometallics 1988, 946-954.
ratio α/β = 2.9/1
α-ketone methylene
protons
β-ketone methylene
protons
ResultsandDiscussion:Mechanisticstudies
135
N
D+ + [Rh]
N
RhD
Rh
N
O D
N
Rh
N
RhH
Rh
N
O
D
N
H+
D
D
n = 5
+ [Rh]
[Rh]
+ [Rh]
2.9 1.0
+n = 5
D
+
n = 5
D
n = 5n = 5
n = 5
n = 5
n = 5
n = 5
n = 5
R R
R R R
RR
C-H activation C-H activation
reductiveelimination
reductiveelimination
hydro-metallation
hydro-metallation
hydro-metallation
hydro-metallation
D
(213) (214)
Scheme 145. Insight into the mechanism of the intermolecular hydroacylation with the use of deuterated benzaldehyde. Explanation for the final 2.9:1 ratio α/β final ketones.
Hence, the catalytic cycle would consist of imine formation, C-H activation, hydrometallation, and reductive elimination, which would furnish the corresponding ketimine leading to the desired ketone after hydrolysis. [171], [172]
[171] D. P. Fairlie, B. Bosnich, Organometallics 1988, 936-945. [172] D. P. Fairlie, B. Bosnich, Organometallics 1988, 946-954.
ResultsandDiscussion:Mechanisticstudies
136
The aminopicoline moiety of L would thus, on the one hand, act as a reversibly bound directing group allowing for facile C-H activation while simultaneously preventing the undesired decarbonylation through formation of an o-vinylbenzaldimine. Integration of the phosphine function would, on the other hand, not only form an active hydroacylation catalyst, but also enhance the binding constant of the directing group to the active rhodium center and would thus increase the effective concentration of the rhodium catalyst, further promoting the C-H activation step (Scheme 146).
4.6.5 The substrate decarbonylation In 1979, Doughty and co-workers suggested that M-P bond rupture was an important and rate-limiting step in the catalytic decarbonylation cycle.[173] M-N bonds are known to be more labile than M-P bonds in some complexes, therefore Anderson et al. make the hypothesis that the active [Rh(PN)2]
+ complex resulting from the heating of [Rh(COD)2BF4] catalyst in toluene would lead to higher catalytic activities.[169]
The [Rh(PN)2]
+ is very electron rich, and must facilitate the oxidative addition. Moreover, they showed the unactivity of the [Rh(PN)2]
+ complex for the decarbonylation of benzaldehyde. 4.6.6 The proposed mechanism In agreement with literature reports, we can summarize (Scheme 146) the four main steps of the rhodium-catalyzed inter or intramolecular hydroacylation. The first step consists of an initial condensation of the starting aldehyde with the P-N ligand to form an active imine. The second step involves oxidative addition into the aldehydic C-H bond by a rhodium (I) catalyst to form an acyl metal hydride complex (C-H activation). After a third alkene coordination step, the acyl metal hydride complex is added to a double bond (hydrometallation). These three first steps constitute endothermic processes with low activation energies, and are consequently fast and reversible. Finally, reductive elimination of the previously formed acyl metal alkyl complex gives the final desired ketone with the regeneration of the transition metal catalyst. This final reductive elimination of the metallocyclohexanone step is due to its significant kinetic barrier. Therefore, the reverse of this final C-C formation is kinetically unfavourable. A water molecule generated in situ during the process is necessary for the final cleavage of the imine. Since the acyl metal hydride complex is so stable, a second decarbonylative reaction pathway can be observed. The metallocycloalkane is decomposed via a π-allyl pathway to an alkyl metal hydride complex, and is subsequently quickly eliminated to the corresponding simple alkane and a metal carbonyl complex. This side reaction is even described for similar processes at room temperature (Scheme 147).
[173] D. H. Doughty, M. F. McGuiggan, H. Wang, L. H. Pignolet, Fundamental Research in Homogeneous Catalysis 1979, 909-919. [169] M. P. Anderson, A. L. Casalnuovo, B. J. Johnson, B. M. Mattson, A. M. Mueting, Inorg. Chem. 1988, 27, 1649-1658.
ResultsandDiscussion:Mechanisticstudies
137
R H
O+ R´
R M
O
H
M
decarbonylation
RM
H
CO
M CO + R H
acyl metal hydride complex
alkyl metal hydride complex
alkanemetal carbonyl complex
elimination
oxydative addition(C-H activation)
Scheme 146. The decarbonylative reaction pathway.
N NH2
Ph2P [Rh]
H
O
R´
R
R[Rh]
H
N
Ph2P
N
R´
R
N
Ph2P
N
[Rh]
R´
N
Ph2P
N
[Rh]
R´
R
O
H2OH2O
R
R´
Scheme 147. Proposed mecanism of the rhodium-catalyzed inter- and intramolecular hydroacylation
using our P-N Ligand 1.
138
Conclusions
The aim of this Ph.D. thesis was to develop the concept of a new bifunctional catalyst system, the synthesis of a library of new PNN pincer ligands containing an aminopicoline moiety and a phosphine function, testing their application to the formation of cyclic and acyclic ketones via an intra- or intermolecular hydroacylation, and finally a mechanistic studies of the catalyst system. 1) A new concept in bifunctional catalysis was developed by combining the diphenyl-phosphine part of the Wilkinson’s catalyst together with the 2-amino-3-picoline co-catalyst in one ligand. In cooperation with a rhodium active center, the new ligand was used to prevent the decarbonylation side-reaction, to efficient promote the C-H activation, and to avoid the need of a cocatalyst.
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
N NH2
Ph2P Rh
new bifunctional catalyst
LRh(I)
metalbinding
substratebinding
nocatalyst
N NH2
[Rh(PPh3)3Cl]
Jun´s catalyst system
high cocatalystloading (20-100 mol%)
R1 +H R2
O
R1
O
R2
N N
Ph2P RhH R2
R1
#
Figure 23. Hydroacylation of alkenes: Concept for a new bifunctional catalyst system LRh(I) and proposed transition state.
2) Ten ligands have been prepared with different linkers between the pyridine core, the phosphorus atom and the amine residue. The role of the substituent (methyl or methoxy) in the 3-position, the impact of the nitrogen of the pyridine for enhancing the binding constant to the catalytically active rhodium center, and the modulation of core structure (pyridine or quinoline) were discussed.
N NH2
P
1reference ligand
Conclusions
139
N NH2P
43
N NH2
22
P
NP
36
NH2
NP
41
NH2
N NH2
P
11
N NH2
P
7
O
NH2
P
15
O
N NH2
31
P
N NH2
27
P
Figure 24. Library of bifunctionnal P-N ligands. 3) The bifunctional catalyst system was also applied successfully to the intermolecular hydroacylation of various terminal alkenes to substituted benzaldehydes with reasonable yields ranging from 51% to 93%. The protocol tolerates a variety of functional groups on the alkene moiety including ester, hydroxyl, carboxylic acid, as well as an internal alkenyl group. Both electron donating as well as electron withdrawing substituents on the aryl benzaldehyde system are efficient reaction partners, thus highlighting the wide functional group tolerance of this catalyst system. A particularly relevant exemple is the hydroacylation of benzaldehyde with 1-hexenol. Despite of the possible coordination of the oxygen to the rhodium center, a good yield of 75% was obtained.
FGR1H
9 examplesup to 93% yield
O
+ R2FG FGR1
O
R2FGligand L (5 mol%)
toluene (c = 1.1 M)
[Rh(COD)Cl]2 (10 mol%)
150 °C, 24 h
Scheme 148. Rhodium-catalyzed intermolecular hydroacylation of diverse alkenes with substituted benzaldehydes.
4) The bifunctional catalyst system was applied successfully to the intramolecular hydroacylation of substituted o-vinylbenzaldehydes with excellent yield up to 98%. The catalyst tolerates electron donating, neutral, and electron withdrawing substituents on the
Conclusions
140
aromatic nucleus. A wide range of functional groups, including carboxylic esters, halogens (chlorine and fluorine), nitro groups, and free phenols are compatible with the reaction conditions and excellent yields are obtained.
FGRH
O
FGR
10 examplesup to 98% yield
O
ligand 1 (5 mol%)
toluene (c = 1.1 M)
[Rh(COD)2]BF4 (5 mol%)
150 °C, 1 h
Scheme 149. Rhodium-catalyzed intramolecular hydroacylation of o-vinylbenzaldehydes. To prove the praticality of our intramolecular hydroacylation of o-vinylbenzaldehydes, the synthesis of Donepezil hydrochloride®, a well-known acetylcholineesterase inhibitor, was realized. This efficient drug is used for the treatment of Alzheimer´s type diseases, which features a 1-indanone core unit and is the world's best-selling Alzheimer's treatment (Pfizer).
O
O
O NH+
Cl-
Donepezil®
Hydrochloride
1-indanonecore unit
152
Figure 25. Donepezil Hydrochloride® 152. 5) A surprising observation was the quantitative formation of thiophene cyclobutane dimer in one hour at 150 °C, instead of the previous 5,6-dihydro-cyclopenta[b]thioph-4-one. This result (quantitative yield) is particularly appreciable regarding to the maximal 50% yield obtained in the literature employing photochemical condition.
+
[Rh(COD)2]BF4 (5 mol%)
ligand 7 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C not obtained !
S
H
O
134
S
145
O
S
H
O
146 trans-1,2-cyclobutane
S
O
H
Scheme 150. Rhodium-catalysed intramolecular hydroacylation of 2-vinylthiophene-3-carbaldehyde.
Conclusions
141
6) An extension to larger ring structures was performed with the thermodynamically controlled intramolecular hydroacylation of substituted o-allylbenzaldehydes into 1-tetralone derivatives, and its aliphatic version with the conversion of 5-hexenal into cyclohexanone.
H
O
OO
N
OMe
NH2
P
[Rh]-cat.+
197 198 199
(10 mol%)
(10 mol%)
1 d, 120 °CNeat
98% combined yield198/199 82:18 (ratio)
Scheme 151. Cyclisation of 5-hexenal into cyclohexanone.
Table 33. Intramolecular hydroacylation of diversified o-allylbenzaldehydes.
H
O
FGR
[Rh(COD)2]BF4 (10 mol%)ligand 7 (10 mol%)
120 °C, 72 h
O O
+FGR FGR
O O
157156
92% (combined yield)107/108 85:15 (ratio)
O O
192191
100% (combined yield)191/192 66:34 (ratio)
neat, 3 d neat, 5 d
100% (combined yield)191/192 45:55 (ratio)
toluene, 5 d
O O
194193
91% (combined yield)193/194 79:21 (ratio)
toluene, 3 d
MeO
MeO
MeO
MeO
O O
196195
76% (combined yield)195/196 86:14 (ratio)
toluene, 3 d
Cl Cl
Conclusions
142
7) To provide further into the hydroacylation using our bifunctional catalyst system, different mechanistic studies were performed. A key P-N bis chelated complex intermediate was isolated and proved to be the active cationic species of our hydroacylation reaction, but also crucially inactive towards the decarbonylation of benzaldehyde.
We highlighted the role of our bifunctional ciral auxiliary with the successful stoechiometric reaction of 1-octene and a pre-formed imine obtained by condensation of benzaldehyde and ligand 1.
N N
PPh2
+
O
[Rh(COD)Cl]2 (100 mol%)
toluene (c = 1.1 M)150 °C, 24 h
73%208 45 46
Scheme 153. Stoechiometric rhodium-catalyzed intermolecular hydroacylation with a pre-formed imine and 1-octene.
In addition, a control experiment with ligand L’, lacking the amino functionality, resulted in a complete lost of activity in the reaction, thus enhancing the importance of the amino group on lignad L for the reaction to proceed via iminium formation and the prevention of decarbonylation.
N
PPh2
Ligand L´
Scheme 154. Ligand L’ lacking the amino functionnality.
Conclusions
143
O
H+
OLigand L´ 209 (10 mol%)[Rh(COD)Cl]2 (5 mol%)
toluene (c = 1.1 M)150 °C, 24 hno reaction
O
H
Ligand L´ 209 (10 mol%)[Rh(COD)Cl]2 (5 mol%)
toluene (c = 1.1 M)150 °C, 24 h
42%
O
44 45 46
95 96
Scheme 155. Rhodium-catalyzed inter- and intramolecular hydroacylation with the use of our P-N ligand lacking the amino functionality.
The difference between results for the intermolecular hydroacylation of benzaldehyde with 1-octene using ligand 1 (82%) and ligand 7 (36%) is explained by the slowly forming (42% after one day) of the imine in the case of the ligand 7 in comparing with the 92% obtained with the ligand 1 after one day. Finally, a deteurium-labeling study with the reaction of deuterated benzaldehyde and 1-octene, gave a ratio α/β deuterated ketone of 2.9:1. An high-resolution mass analysis of the ketone product showed a unique deuterium incorporate per molecule. This result proves that the deuterium relocation is an intramolecular process according to the conclusion of Bosnich et al.
+
O
O
O
DKCN, D2O
dioxane, RT, 30 min
O
D
O
D
+
O D
ratio 213/214 (2.9/1)
[Rh(COD)Cl]2, toluene
150 °C, 24 h
96%
82%
211 212
212 45
213
214
Ligand 1
Scheme 156. Rhodium-catalyzed intermolecular hydroacylation with deuterium-labeled benzaldehyde and 1-octene.
In agreement with literature reports, we can summarize in the following catalytic cycle (Scheme 157), the four main steps of the rhodium-catalyzed inter or intramolecular hydroacylation.
Conclusions
144
The catalytic cycle would consist of imine formation, C-H activation, hydrometallation, and reductive elimination, which would furnish the corresponding ketimine leading to the desired ketone after hydrolysis. Since the acyl metal hydride complex is so stable, a second decarbonylative reaction pathway can be observed.
N NH2
Ph2P [Rh]
H
O
R´
R
R[Rh]
H
N
Ph2P
N
R´
R
N
Ph2P
N
[Rh]
R´
N
Ph2P
N
[Rh]
R´
R
O
H2OH2O
R
R´
Scheme 157. Proposed mecanism of the inter- and intramolecular hydroacylation using our P-N Ligand 1.
145
Outlook The utility of our new bifunctional system in different applications in inter- or intramolecular hydroacylation has been demonstrated. Many investigations or applications can be still taken into consideration. 1) A more economical ruthenium catalyst system.
+R
O
H R'[Ru]-cat.
R
O
R'
H?
2) A rhodium-catalyzed enantioselective branched intermolecular hydroacylation using a chiral P-N bifunctional ligand.
+R
O
H R'
[Rh]-cat.
R
O
R'
?
branched product
chiral PN ligand
3) An efficient application of our system to aliphatic aldehydes. 4) Synthesis of 6-substituted 5-hexen-1-al, and application to an asymmetric version of the rhodium-catalyzed intramolecular hydroacylation of 5-hexen-1-al.
[Rh]-cat.
?
chiral P-N ligandHR
O
O
R
5) Application to the synthesis of larger cyclic ketones
[Rh]-cat.
?
H
O
O
n
n
6) Synthesis of 1- and 3-suberone
[Rh]-cat.
?
H
O O
[Rh]-cat.
?O
O
HH+
Outlook
146
7) Application of the synthesis of 1-tetralone derivatives via a rhodium-catalyzed hydro-acylation to the Berchemiaside B synthesis, a cytotoxic inhibitor used for the treatment of Leukemia. A proposed eleven-step synthesis* with the assembling of three moieties: a 1-tetralone obtained by rhodium-catalyzed hydroacylation, a β-D-glucopyranose sugar and a para-methoxy-cinnamoyl chloride or cinnamic acid.
OH
OH
O
OO
HO
O
O
OH
OHOH
eleven-step synthesis
* Proposed synthetical pathway for berchemiaside B, look at appendix (p. 297-298).
147
B Experimental Part 7. General information All experiments were performed at the Institute für Organische Chemie und Biochemie in Freiburg except for the high resolution mass spectra of intermediate ligands 28 and 29 at the Laboratory of Organic Chemistry in ETH Zurich. Thanks to Veronika Ehmke (ex-Diederich Group) for these two analyses. All reactions carried out at high temperatures or with oxygen and moisture sensitive materials were performed using pre-dried glassware (oil pump vacuum, 300 °C) under an atmosphere of argon (5.0, Sauerstoffwerck Friedrichshaffen (D)). Schlenk tubes, distillation appareal, cannula, NMR tubes, stirring bars were dried in oven at 90 °C prior to use. Organic solutions were concentrated under reduced pressure by rotary evaporation. All reagents are available commercially unless otherwise noted.
Membrane pump: 40 mbar Oil pump : 0.1 mbar
7.1 Solvents
Petroleum ether and cyclohexane for extraction and column chromatography were distilled on rotary evaporator prior to use. Reactions requiring water-free solvents were dried by Mrs M. Braun (using a Solvent Purification System 800), who used two successive columns.
Diethylether: Dried by refluxing over potassium-sodium alloy/benzophenone and distilled under argon.
Toluene: Dried by refluxing over sodium/benzophenone and distilled under argon.
Dichloromethane: Dried by refluxing over CaH2 and distilled under argon.
Tetrahydrofuran: Dried by refluxing over potassium and distilled under argon
immediately before use.
Methanol: Dried by distillation from magnesium.
Pyridine: Dried by distillation from CaH2 under argon.
Toluene used for catalytic hydroacylation was renewed regularly. Peroxides in THF and diethylether monitored by starch iodide test paper.
ExperimentalPart:Generalinformation
148
7.2 Chromatography
Chromatographic purification of products was accomplished using Machery-Nagel silica gel 60® (230-400 mesh). Thin-Layer Chromatography (TLC) was performed using TLC aluminium sheets with Silica gel 60 with fluorescent indicator F254 (Merck). UV-active substances were detected in UV-light with wavelength λ = 254 nm. Staining was achieved via dipping into the appropriate agent followed by heating with a heat-gun. Coloring agents for TLC:
7.3 Melting points Melting points were determined using a Dr. Tottoli apparatus (Büchi) and were not corrected.
7.4 Mass spectrometry
Each sample were dried under vaccum prior to analysis. Mass analysis were performed at the analytical departement by Mr. C. Warth and Mr. Dr. J. Wörth. Methods for Low Resolution Mass Spectrometry (LRMS):
Electron-Impact Ionization (EI): MAT 95XL, Thermo, ionization energy 70 eV, temperature 200 °C.
Chemical Ionization (CI): LCQ Advantage, Thermo, spray-voltage 4-5 kV, temperature 250-300 °C, solvent MeOH or MeCN/H2O.
temperature 250-300 °C, solvent MeOH or MeCN/H2O. Methods for High Resolution Mass Spectrometry (HRMS):
Electron-Impact Ionization (EI): MAT 95XL, Thermo, ionization energy 70 eV,
temperature 200 °C.
ExperimentalPart:Generalinformation
149
7.5 NMR spectroscopy
Nuclear magnetic resonance spectra were acquired on Varian Mercury 300 [300 MHz (1H), 121 MHz (31P), 75 MHz (13C)Bruckner Advance AMX 400 400 MHz (1H), 161 MHz (31P), 100 MHz (13C) spectrometers. All 1H NMR spectra are reported in parts per million (ppm) downfield from TMS and were measured relative to the signals at 7.26 ppm (CHCl3) and 2.50 ppm (d6-DMSO). All 13C NMR spectra were reported in ppm relative to residual CHCl3 (77.16 ppm) and d6-DMSO (39.52 ppm) and were obtained with 1H decoupling. The chemical shifts (δ) are reported in ppm for all core reasonances, and the coupling constants in Hertz (Hz). Multiplicities for 1H NMR are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, sept = septet, oct = octuplet, non = nonuplet, dec = decuplet, m = multiplet, br s = broad singlet.
7.6 Elemental analysis
Every samples were dried under vaccum prior to analysis. Elemental analysis was performed on an Elementar vario EL (Elementar Analysensystem GmbH) at the analytical departement by Mr F. Tönnies.
7.7 Hydroacylation experiments
Hydroacylation reactions were perfomed in the hot night fume hood, and protected with a front shield. 8 mL Schlenk tubes (ø = 1 cm, height = 10 cm) and stirring bars (ø = 3 mm, lenght = 8 mm) was used for optimized stirring. Schlenk tubes were taken out of the oven just before the reaction. Schlenk tubes were purged three times with argon. Reactions mixtures were filtered over Celite (Fluka) using deuterated chloroform (for neat reactions), and the used solvent for the other reactions before NMR analysis.
150
8. Synthesis of ligands 8.1 Overview
N NH2P
43
N NH2
22
P
NP
36
NH2
NP
41
NH2
N NH2
P
11
N NH2
P
7
O
NH2
P
15
O
N NH2
31
P
N NH2
27
P
N NH2
P
1
reference ligand
Figure 28. Library of bifunctionnal P-N ligands
ExperimentalPart:Synthesisofligands
151
8.2 Synthesis of ligands 8.2.1 Synthesis of 6-[(diphenylphosphanyl)-methyl]-3-methylpyridin-
A solution of trimethylacetylchloride (12.51 mL, 101.7 mmol, 1.1 eq) in CH2Cl2 (15 mL) was slowly added to an ice-cold solution of 2-amino-6-picoline a (10 g, 92.5 mmol) and NEt3 (16.11 mL, 115.6 mmol, 1.25 eq) in CH2Cl2 (200 mL). The reaction mixture was stirred at 0 °C for 15 minutes and overnight at room temperature. It was then poured into water (150 mL) and extracted with CH2Cl2 (3 x 60 mL). The organic phase was washed with dilute NaHCO3 (60 mL), dried over MgSO4 and concentrated in vacuo. The residue was recrystallized from hot pentane leaving 6-methyl-2-(pivaloylamino)-pyridine b as white crystals (17.7 g, 92.0 mmol, quant.). 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 1.33 (s, 9H, C(CH3)3), 2.45 (s, 3H, CH3), 6.88 (d, 3J5-4 = 7.5 Hz, 1H, Ar-H5), 7.58 (dd, 3J4-3 = 8.3 Hz, 3J4-5 = 7.5 Hz, 1H, Ar-H4), 7.96 (bs, 1H, NH), 8.05 (d, 3J3-4 = 8.3 Hz, 1H, H3). 13C {1H} NMR (100.612 MHz, CDCl3): δ (ppm) = 24.3 (s, 1C, C7), 27.9 (s, 3C, C10), 40.2 (s, 1C, C9), 111.3 (s, 1C, C3), 119.5 (s, 1C, C5), 139.1 (s, 1C, C4), 151.3 (s, 1C, C2), 154.7 (s, 1C, C6), 177.5 (s, 1C, C8). LRMS (Cl(NH3), C11H16N2O, Exact Mass = 192.3): m/z = 193.2 ([M+H]+, 100), 135.0 ([M-tBu]+, 5.1), 108.0([M-Piv]+, 6.2). CHN (%): calcd: C: 68.72 H: 8.39 N: 14.57 found: C: 68.76 H: 8.55 N: 14.44 Tm = 68-69 °C Rf = 0.53 (cHex/AcOEt 7:3) Analytic data are in accordance with the literature.[174]
[174] J. A. Turner, J. Org. Chem. 1983, 48, 3401-3408.
ExperimentalPart:Synthesisofligands
152
8.2.1.2 Synthesis of 3,6-dimethyl-2(pivaloylamino)-pyridine c, ligand 1 step 2
N NH
O
b
1) n-BuLi, THF, 90 min, -78 °C to 0 °C2) MeI, o/n, 0 °C to RT
N NH
O
c
2
45
67 8
910
10
103
11
74% 1
C12H18N2OMol. Wt.: 206,28
C11H16N2OMol. Wt.: 192,26
n-BuLi (2.5 M in hexanes, 34.4 mL, 85.9 mmol, 2.2 eq) was added dropwise at -78 °C to a solution of 6-methyl-2-(pivaloylamino)-pyridine b (7.5 g, 39.0 mmol) in 100 mL of THF. The resulting orange solution was stirred at this temperature for 30 minutes before being stirred at 0 °C for one additional hour. Methyl iodide (2.68 mL, 43 mmol, 1.1 eq) was then slowly added to the ice-cooled solution and the reaction mixture was allowed to warm to room temperature overnight. The mixture was then poured into water, and extracted three times with Et2O (3 x 30 mL). The combined organic layers were washed with brine, dried over MgSO4 and evaporated to dryness. The crude was purified by silica gel column chromatography (eluting with cHex/AcOEt 7:3) yielding 3,6-dimethyl-2-(pivaloylamino)-pyridine c as white crystals (5.2 g, 28.9 mmol, 74%). 1H NMR (400.13 MHz, CDCl3): δ (ppm) = 1.35 (s, 9H, C(CH3)3), 2.17 (s, 3H, CH3 (11)), 2.46 (s, 3H, CH3 (7)), 6.90 (d, 3J5-4 = 7.7 Hz, 1H, Ar-H5), 7.44 (d, 3J4-5 = 7.7 Hz, 1H, Ar-H4), 7.66 (bs, 1H, NH). 13C {1H} NMR (100.6 MHz, CDCl3): δ (ppm) = 18.2 (s, 1C, C11), 23.9 (s, 1C, C7), 28.0 (s, 3C, C10), 39.8 (s, 1C, C9), 121.8 (s, 1C, C5), 126.3 (s, 1C, C3), 140.6 (s, 1C, C4), 148.9 (s, 1C, C2), 155.0 (s, 1C, C6), 177.1 (s, 1C, C8). LRMS ((EI), C12H18N2O, Exact Mass = 206.3): m/z = 122.1([M-Piv]+, 95.4), ), 149.1 ([M-tBu]+, 72.9), 206.2 ([M]+, 100). HRMS (EI): Calcd. for C12H18N2O (M): 206.14191; Found: 206.14230 (difference -1.9 ppm). CHN (%): calcd: C: 69.87 H: 8.79 N: 13.58 found: C: 69.57 H: 8.91 N: 13.35 Tm = 116 °C Rf = 0.24 (cHex/AcOEt 7:3)
ExperimentalPart:Synthesisofligands
153
8.2.1.3 Synthesis of 3-methyl-2-(pivaloylamino)-6-(trimethylsilylmethyl)-pyridine d, ligand 1 step 3
N NH
O
c
N NH
O
d
2
45
67
89
10
10
103
11
72% Si12
12
12
1) Schlosser Base, THF, -78 °C2) TMSCl, THF, o/n, -78 °C to RT
1
C12H18N2OMol. Wt.: 206,28
C15H26N2OSiMol. Wt.: 278,47
An oven-dried, nitrogen-flushed 250 mL Schlenk flask was charged at -78 °C with KOtBu (2.6 g, 23.2 mmol), DIPA (3.19 mL, 22.6 mmol) and anhydrous THF (25 mL). n-BuLi (2.5 M, 9.4 mL, 23.5 mmol) wad slowly added and and the reaction mixture was for 30 min at -78 °C. A solution of 3,6-dimethyl-2(pivaloylamino)-pyridine c (2.06 g, 10.0 mmol) in 170 mL of THF was then slowly added and reacted for 1 hour at -78 °C. The red mixture was subsequently quenched with a solution of TMSCl (16 mmol, 2.1 mL) in 5 mL of THF before it was warmed to room temperature overnight. After hydrolysis (30 mL of water), CH2Cl2 extraction (3 x 30 mL) and drying (MgSO4), the solvent was removed in vacuo and the crude was purified by flash chromatography on silica gel (cHex/AcOEt 7:3) affording 3-methyl-2-(pivaloylamino)-6-(trimethylsilylmethyl)-pyridine d as white crystals (2.00 g, 7.22 mmol, 72%). 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 1.35 (s, 9H, C(CH3)3), 2.12 (s, 3H, CH3), 4.57 (s, 2H, CH2Cl), 7.26 (d, 3J5-4 = 7.7 Hz, 1H, Ar-H5), 7.57 (d, 3J4-5 = 7.7 Hz, 1H, Ar-H4), 7.70 (bs, 1H, NH). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = -1.5 (s, 3C, C12), 18.4 (s, 1C, C11), 28.0 (s, 3C, C10), 39.9 (s, 1C, C9), 46.6 (s, 1C, C7), 121.3 (s, 1C, C5), 129.3 (s, 1C, C3), 141.2 (s, 1C, C4), 150.0 (s, 1C, C2), 153.2 (s, 1C, C6), 177.3 (s, 1C, C8). LRMS ((EI), C15H26N2OSi, Exact Mass = 278.2): m/z = 278.3 ([M]+, 100). HRMS (EI): Calcd. for C15H26N2OSi (M): 278.18144; Found: 278.18140 (difference +0.2 ppm). CHN (%): calcd: C: 64.70 H: 9.41 N: 10.06 found: C: 64.50 H: 9.57 N: 9.85 Tm = 74 °C Rf = 0.53 (cHex/AcOEt 7:3)
ExperimentalPart:Synthesisofligands
154
8.2.1.4 Synthesis of 6-chloromethyl-3-methyl-2-(pivaloylamino)-pyridine e, ligand 1 step 4
N NH
O
d
88%
CsF, C2Cl6, CH3CN, 5 h, 60 °C
Si
N NH
O
e
2
45
67
89
10
10
103
11
Cl1
C12H17ClN2OMol. Wt.: 240,73
C15H26N2OSiMol. Wt.: 278,47
Dry CsF (1.1 g, 7.2 mmol, 4.0 eq) was added at 25 °C to a solution of 3-methyl-2-(pivaloylamino)-6-(trimethylsilylmethyl)-pyridine d (0.5 g, 1.79 mmol) and C2Cl6 (1.7 g, 7.2 mmol, 4.0 eq) in acetonitrile (30 mL). The heterogenous solution was stirred at 60 °C for 5 hours. After cooling to 25 °C, the mixture was poured into a separatory funnel containing ethyl acetate (45 mL), H2O (45 mL). The product was extracted with ethyl acetate (3 x 45 mL); then, the combined organic fractions were shaken with brine (70 mL) and dried over MgSO4. Filtration and concentration in vacuo gave pure 6-chloromethyl-3-methyl-2-(pivaloylamino)-pyridine e as a yellowish solid (0.38 g, 1.57 mmol, 88%). 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 1.35 (s, 9H, (CH3)3), 2.12 (s, 3H, CH3), 4.57 (s, 2H, CH2Cl), 7.26 (d, 3J5-4 = 7.7 Hz, 1H, Ar-H5), 7.57 (d, 3J4-5 = 7.7 Hz, 1H, Ar-H4), 7.70 (bs, 1H, NH). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 18.4 (s, 1C, C11), 28.0 (s, 3C, C10), 39.9 (s, 1C, C9), 46.6 (s, 1C, C7), 121.3 (s, 1C, C5), 129.3 (s, 1C, C3), 141.2 (s, 1C, C4), 150.0 (s, 1C, C2), 153.2 (s, 1C, C6), 177.3 (s, 1C, C8). LRMS (Cl(NH3), C11H15ClN2O, Exact Mass = 240.1): m/z = 240.1 ([M]+, 28.1), 241.1 ([M+H]+, 100), 242.1 ([M(isotope(Cl)]+, 20.09), 243.1([M(isotope(Cl)+H]+, 32.75). HRMS (EI): Calcd. for C12H17ClN2O (M): 240.10294; Found: 240.10310 (difference -0.7 ppm). CHN (%): calcd: C: 59.87 H: 7.12 N: 11.64 found: C: 60.61 H: 7.73 N: 10.91 Rf = 0.30 (cHex/AcOEt 7:3)
ExperimentalPart:Synthesisofligands
155
8.2.1.5 Synthesis of 6-chloromethyl-3-methyl-2-aminopyridine f, ligand 1 step 5 A solution of 6-chloromethyl-3-methyl-2-(pivaloylamino)-pyridine e (0.38 g, 1.58 mmol) was dissolved in concentrated HCl (5 mL) and refluxed overnight. After cooling to room temperature, the mixture was neutralized with K2CO3 and extracted with CH2Cl2 (3 x 20 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography on silica gel (cHex/AcOEt 70:30) affording 6-chloromethyl-3-methyl-2-aminopyridine f as white crystals (0.18 g, 1.15 mmol, 72%). The product has to be stored at -20 °C to avoid any polymerisation.
Na (236.2 mg, 9.84 mmol, 2.0 eq) was added over 10 min to liquid ammonia (30 mL) at -78 °C. The dark blue solution was stirred at this temperature for 20 minutes and then treated portionwise with PPh3 (1.29 g, 4.92 mmol, 1.05 eq) over 5 minutes. The dark red-orange solution was stirred for a further 2 hours at -78 °C before the addition of 6-chloromethyl-3-methyl-2-aminopyridine f (770 mg, 4.78 mmol). THF (30 mL) was then added after 40 minutes and the cooling bath removed. Ammonia was allowed to evaporate overnight and the residue was quenched with H2O (10 mL), extracted with Et2O (3 x 8 mL) and dried over Na2SO4. The solvent was evaporated in vacuo, and subsequent flash column chromatography on silica gel (CH2Cl2, then CH2Cl2/AcOEt, 10:3) afforded ligand 1, 6-[(diphenylphosphanyl)-3-methyl-pyridin-2-ylamine, as a white solid (1.2 g, 3.9 mmol, 80%). 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 2.05 (s, 3H, CH3), 3.44 (s, 2H, CH2), 4.38 (bs, 1H, NH2), 6.29 (dd, 3J5-4 = 7.4 Hz, 4J5-P = 1.5 Hz, 1H, Ar-H5), 7.06 (d, 3J4-5 = 7.4 Hz, 1H, Ar-H4), 7.31 (m, 6H, Ar-H11 and Ar-H12), 7.44 (m, 4H, Ar-H10). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 16.8 (s, 1C, C8), 38.1 (d, 1J7-P = 14.9 Hz, 1C, C7), 113.8 (d, 3J5-P = 6.7 Hz, 1C, C5), 114.3 (d, 4J4-P = 6.7 Hz, 1C, C4), 128.4 (C11, d, 3J11-P = 6.7 Hz, 4C, C11), 128.7 (s, 2C, C12), 133.1 (d, 2J10-P = 18.7 Hz, 4C, C10), 138.3 (s, 1C, C3), 138.8 (d, 1J9-P = 15.3 Hz, 2C, C9), 153.7 (d, 2J6-P = 8.0 Hz, 1C, C6), 156.6 (s, 1C, C2). 31P {1H} NMR (101.3 MHz, CDCl3): δ (ppm) = -12.1. HRMS (EI): Calcd. for C19H19N2P (M): 306.1286; Found: 306.1286 (difference -0.0 ppm). CHN (%): calcd: C: 74.49 H: 6.25 N: 9.14 found: C: 74.39 H: 6.15 N: 9.21 Tm = 121 °C Rf = 0.36 (CH2Cl2/AcOEt 19:1)
ExperimentalPart:Synthesisofligands
157
8.2.2 Synthesis of 6-[(diphenylphosphanyl)-methyl]-3-methoxy-pyridin-2-ylamine, ligand 7
8.2.2.1 Synthesis of 3-methoxy-6-methyl-2-nitro-pyridine 3, ligand 7 step 1
2
45
6
7
3 8
1N
OH
NO284%
2 3
MeI, K2CO3, DMSO, o/n, RT
N
O
NO2
C6H6N2O3Mol. Wt.: 154,12
C7H8N2O3Mol. Wt.: 168,15
MeI (4.5 g, 32.2 mmol, 1.0 eq) and K2CO3 (6.72 g, 48.6 mmol, 1.5 eq) were successfully added to a solution of 3-hydroxy-6-methyl-2-nitro-pyridine 2 (5 g, 32.2 mmol) in DMSO (50 mL) and stirred overnight at room temperature. H2O (70 mL) was added to the reaction mixture, followed by extraction with AcOEt (3 x 50 mL). The combined organic extracts were washed with brine (3 x 50 mL), dried over MgSO4 and concentrated in vacuo to yield a residue, which was recrystallised from CH2Cl2/Petroleum Ether leaving 3-methoxy-6-methyl-2-nitro-pyridine 3 as a white crystalline solid (4.5 g, 27.0 mmol, 84%). 1H NMR (400.130 MHz, CDCl3): δ = 2.53 (s, 3H, CH3), 3.94 (s, 3H, OCH3), 7.36 (dd, 3J5-4 = 8.5 Hz, 4J5-7 = 0.5 Hz, 1H, Ar-H5), 7.42 (d, 3J4-5 = 8.5 Hz, 1H, Ar-H4). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 23.0 (s, 1C, C8), 56.8, (s, 1C, C7), 123.4 (s, 1C, C4), 128.4 (s, 1C, C5), 145.7 (s, 2C, C2 and C3), 149.0 (s, 1C, C6). HRMS (EI): Calcd. for C7H8O3N2 (M): 168.0535; Found: 168.0531 (difference -2.3 ppm). Tm = 86-87 °C Analytic data are in accordance with the literature.[98]
[98] M. Malamas, W. Fobare, W. Solvibile, F. Lovering, J. Condon, A. Robichaud, U. S. Pat. Appl. Publ., 20060173049, 03 Aug. 2006.
ExperimentalPart:Synthesisofligands
158
8.2.2.2 Synthesis of 3-methoxy-6-methyl-2-aminopyridine 4, ligand 7 step 2
NaBH4 (13 g, 342.0 mmol, 4.0 eq) was added in portions over a 30 minute period to an ice-cooled solution of 3-methoxy-2-nitro-picoline 3 (14.4 g, 85.7 mmol), NiCl2·6H2O (41.0 g, 174.0 mmol, 2.0 eq) in MeOH (350 mL) and stirred at room temperature for 4 hours. The solvent was removed to dryness and the resulting black precipitate is dissolved in 3 M HCl (50 mL). Neutralisation of the reaction mixture with NH4OH, followed by extraction with AcOEt (3 x 50 mL) gave 3-methoxy-6-methyl-2-aminopyridine 4 as a brown crystalline solid (10.45 g, 75.7 mmol, 88%). 1H NMR (400.130 MHz, CDCl3): δ = 2.32 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 4.62 (br s, 2H, NH2), 6.44 (d, 3J5-4 = 8.1 Hz, 1H, Ar-H5), 6.80 (d, 3J4-5 = 8.1 Hz, 1H, Ar-H4). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 23.3 (s, 1C, C8), 55.5 (s, 1C, C7), 112.5 (s, 1C, C5), 116.2 (s, 1C, C4), 140.4 (s, 1C, C3), 147.2 (s, 1C, C2), 149.4 (s, 1C, C6). HRMS (EI): Calcd. for C7H10N2O (M): 138.07920; Found: 138.07931 (difference +0.7 ppm). Tm = 84 °C Rf = 0.38 (CH2Cl2 pure) Analytic data are in accordance with the literature.[98]
·
[98] M. Malamas, W. Fobare, W. Solvibile, F. Lovering, J. Condon, A. Robichaud, U. S. Pat. Appl. Publ., 20060173049, 03 Aug. 2006.
ExperimentalPart:Synthesisofligands
159
8.2.2.3 Synthesis of 3-methoxy-6-methyl-2-phthalimidopyridine 5, ligand 7 step 3
71%
O
O
O
, 1 h, 190 °C
4
N
O
NH2
C7H10N2OMol. Wt.: 138,17
2
45
6
7
3
8
1
5
N
O
N
O
O
C15H12N2O3Mol. Wt.: 268,27
9
9
10
10 11
11
12
12
A mixture of 3-methoxy-6-methyl-2-phthalimidopyridine 4 (1.10 g, 7.9 mmol) and phthalic anhydride (1.12 g, 7.9 mmol) were stirred and held at 190 °C for 1 hour. After cooling to room temperature, the residue was purified by silica gel chromatography (eluting with AcOEt) to yield 3-methoxy-2-phthalimido-6-picoline 5 as a white crystalline solid (1.52 g, 5.7 mmol, 71%). 1H NMR (400.130 MHz, CDCl3): δ = 2.53 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 7.24 (d, 3J5-4 = 8.6 Hz, 1H, Ar-H5), 7.30 (d, 3J4-5 = 8.6 Hz, 1H, Ar-H4), 7.76 (m, 2H, Ar-H12), 7.93 (m, 2H, Ar-H11). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 23.3 (s, 1C, C8), 56.2 (s, 1C, C7), 121.1 (s, 1C, C5), 123.9 (s, 1C, C4), 125.4 (s, 2C, C11), 132.5 (s, 2C, C12), 134.3 (s, 2C, C10), 134.6 (s, 1C, C3), 150.0 (s, 1C, C2), 150.3 (s, 1C, C6), 167.0 (s, 2C, C9). HRMS (EI): Calcd. for C15H12O3N2 (M): 268.08460; Found: 268.08469 (difference +0.3 ppm). Tm = 228 °C Rf = 0.77 (AcOEt pure)
ExperimentalPart:Synthesisofligands
160
8.2.2.4 Synthesis of 6-bromomethyl-(3-methoxy-2-phthalimido)-pyridine 6, ligand 7 step 4
51%
NBS, AIBN every 3 hoursbenzene, 16 h, reflux
5
N
O
N
O
O
C15H12N2O3Mol. Wt.: 268,27
2
45
67
3
8
1
6
N
O
N
O
O
9
9
10
10 11
11
12
12
Br
C15H11BrN2O3Mol. Wt.: 347,16
A mixture of 3-methoxy-2-phthalimido-6-picoline 5 (4.5 g, 16.8 mmol), N-bromosuccinimide (3.29 g, 18.5 mmol, 1.1 eq) and AIBN (150 mg) in anhydrous benzene (10 mL) was stirred and held at reflux for 16 hours. (AIBN (150 mg) was added every 3 hours). After cooling to room temperature, the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 6-bromomethyl-(3-methoxy-2-phthalimido)-pyridine 6 as a green solid (3.0 g, 8.6 mmol, 51%). 1H NMR (400.130 MHz, CDCl3): δ = 3.81 (s, 3H, OCH3), 4.51 (s, 2H, CH2Br), 7.31 (d, 3J5-4 = 2.4 Hz, 1H, Ar-H5), 7.47 (d, 3J4-5 = 2.4 Hz, 1H, Ar-H4), 7.79 (m, 2H, Ar-H12), 7.95 (m, 2H, Ar-H11). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 32.8 (s, 1C, C8), 56.2 (s, 1C, C7), 112.5 (s, 1C, C5), 120.6 (s, 1C, C4), 123.9 (s, 2C, C11), 130.5 (s, 2C, C12), 131.0 (s, 2C, C10), 131.6 (s, 1C, C3), 148.3 (s, 1C, C6), 155.6 (s, 1C, C2), 167.3 (s, 2C, C9). HRMS (CI): Calcd. for C16H13O3NBr (M+H): 346.0079; Found: 346.0071 (difference -2.3 ppm). Tm = 208 °C Rf = 0.68 (CH2Cl2 pure)
ExperimentalPart:Synthesisofligands
161
8.2.2.5 Synthesis of 6-[(diphenylphosphanyl)-methyl]-3-methoxy-pyridin-2-ylamine, ligand 7
Na (320 mg, 13.9 mmol, 2.0 eq) was added over 10 min to liquid ammonia (10 mL) at -78 °C. The dark blue solution was stirred at this temperature for 20 minutes and then treated portionwise with PPh3 (1.82 g, 6.96 mmol, 1.05 eq) over 5 minutes. The dark red-orange solution was stirred for a further 2 hours at -78 °C before the addition of 6-bromomethyl-(3-methoxy-2-phthalimido)benzene 6 (2.33 g, 6.76 mmol). THF (15 mL) was then added after 40 minutes and the cooling bath removed. Ammonia was allowed to evaporate overnight and the residue was quenched with H2O (10 mL), extracted with CH2Cl2 (3 x 8 mL) and dried over Na2SO4. The solvent was evaporated in vacuo and subsequent flash column chromatography on silica gel (CH2Cl2 then CH2Cl2/AcOEt, 95:5) afforded 6-[(diphenylphosphanyl)-methyl]-3-methoxy-pyridin-2-ylamine 7 as a white solid (586 mg, 1.83 mmol, 27%). 1H NMR (400.130 MHz, CDCl3): δ = 3.29 (s, 2H, CH2), 3.79 (s, 3H, OCH3), 4.61 (bs, 1H, NH2), 6.41 (d, 3J5-4 = 1.9 Hz, 1H, Ar-H5), 6.46 (d, 3J4-5 = 1.9 Hz, 1H, Ar-H4), 7.31 (m, 6H, Ar-H10 and Ar-H12), 7.40 (m, 4H, Ar-H11). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 35.4 (d, 1J7-P = 14.6 Hz, 1C, C7), 55.6 (s, 1C, C8), 110.4 (d, 5J3-P = 1.4 Hz, 1C, C3), 116.2 (d, 3J5-P = 7.1 Hz, 1C, C5), 119.3 (d, 4J4-P = 7.0 Hz, 1C, C4), 128.4 (d, 3J11-P = 6.5 Hz, 4C, C11), 128.7 (s, 2C, C12), 130.0 (d, 4J2-P = 8.6 Hz, 1C, C2), 133.1 (d, 2J10-P = 18.1 Hz, 4C, C10), 138.8 (d, 1J9-P = 15.2 Hz, 2C, C9), 145.9 (d, 2J6-P = 2.9 Hz, 1C, C6). 31P{1H} NMR (101.3 MHz, CDCl3) δ = -11.0. HRMS (EI): Calcd. for C20H20ONP (M): 321.1283; Found: 321.1281 (difference -0.6 ppm). Tm = 120 °C Rf = 0.29 (CH2Cl2/AcOEt 19:1)
ExperimentalPart:Synthesisofligands
162
8.2.3 Synthesis of 6-[(diphenylphosphanyl)-methyl]-pyridin-2-ylamine, ligand 11
8.2.3.1 Synthesis of 2-phthalimido-6-picoline 8, ligand 11 step 1
NH2N N
O
O
N 2
45
67
89 103
1
11
89
10
11
8C14H10N2O2
Mol. Wt.: 238,24C6H8N2
Mol. Wt.: 108,14
O
O
O
1 h, 190 °C
95%
a
A mixture of 2-amino-6-picoline a (3.0 g, 27.8 mmol) and phthalic anhydride (7.09 g, 27.8 mmol, 1.0 eq) were stirred and held at 190 °C for 1 hour. After cooling to room temperature, the residue was purified by silica gel chromatography (eluting with AcOEt) to yield 2-phthalimido-6-picoline 8 as a white crystalline solid (6.29 g, 26.4 mmol, 95%). 1H NMR (400.130 MHz, CDCl3): δ = 2.62 (s, 3H, CH3), 7.21 (d, 3J3-4 = 8.7 Hz, 1H, Ar-H3), 7.23 (d, 3J5-4 = 8.7 Hz, 1H, Ar-H5), 7.77 (t, 3J4-5 = 8.7 Hz, 3J4-3 = 8.7 Hz, 1H, Ar-H4), 7.79 (m, 2H, Ar-H11), 7.95 (m, 2H, Ar-H10). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 24.5 (s, 1C, C7), 119.5 (s, 1C, C5), 123.5 (s, 1C, C3), 124.0 (s, 2C, C11), 132.1 (s, 2C, C9), 134.6 (s, 2C, C10), 138.6 (s, 1C, C4), 145.5 (s, 1C, C6), 159.3 (s, 1C, C2), 167.0 (s, 2C, C8). HRMS (EI): Calcd. for C14H10O2N2 (M): 238.0742; Found: 238.0739 (difference -1.3 ppm). Tm = 192 °C Analytic data are in accordance with the literature.[99]
[99] X. Collin, J-M. Robert, G. Wielgosz, G. Le Baut, C. Bobin-Dubigeon, N. Grimaud, J.-Y. Petit, Eur. J. of Med. Chem. 2001, 36(7-8), 639-649.
ExperimentalPart:Synthesisofligands
163
8.2.3.2 Synthesis of 6-bromomethyl-2-phthalimido-pyridine 9, ligand 11 step 2
N
O
O
N
8
NBS, AIBN every 3 hoursbenzene, 16 h, reflux
62%
C14H10N2O2Mol. Wt.: 238,24
N
O
O
N 2
45
67
89 103
1
11
89
10
11
9
Br
C14H9BrN2O2Mol. Wt.: 317,14
A mixture of 2-phthalimido-6-picoline 8 (2.80 g, 11.8 mmol), N-bromosuccinimide (2.30 g, 12.9 mmol, 1.1 eq) and AIBN (70 mg) in anhydrous benzene (50 mL) was stirred and held at reflux for 16 hours. (AIBN (70 mg) was added every 3 hours). After cooling to room temperature, the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 6-bromomethyl-2-phthalimido-pyridine 9 as a white solid (2.31 g, 7.32 mmol, 62%). 1H NMR (300.070 MHz, CDCl3): δ = 4.59 (s, 2H, CH2Br), 7.35 (d, 3J5-4 = 7.8 Hz, 1H, Ar-H5), 7.54 (d, 3J3-4 = 7.8 Hz, 1H, Ar-H3), 7.80 (m, 2H, Ar-H11), 7.89 (t, 3J4-5 = 8.7 Hz, 3J4-3 = 8.7 Hz, 1H, Ar-H4), 7.97 (m, 2H, Ar-H10). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 33.0 (s, 1C, C7), 121.6 (s, 1C, C5), 123.5 (s, 1C, C3), 124.1 (s, 2C, C11), 131.9 (s, 2C, C9), 134.8 (s, 2C, C10), 139.4 (s, 1C, C4), 145.8 (s, 1C, C6), 157.4 (s, 1C, C2), 166.7 (s, 2C, C8). HRMS (EI): Calcd. for C14H9O2N2Br (M): 315.9847; Found: 315.9844 (difference -0.9 ppm). Tm = 170 °C Rf = 0.40 (CH2Cl2 pure) Analytic data are in accordance with the literature.[99]
[99] X. Collin, J-M. Robert, G. Wielgosz, G. Le Baut, C. Bobin-Dubigeon, N. Grimaud, J.-Y. Petit, Eur. J. of Med. Chem. 2001, 36(7-8), 639-649.
ExperimentalPart:Synthesisofligands
164
8.2.3.3 Synthesis of 2-amino-6-bromomethyl-pyridine 10, ligand 11 step 3
78%
HCl(aq), 2 h, 130 °C
N
O
O
N
9
Br
C14H9BrN2O2Mol. Wt.: 317,14
C6H7ClN2Mol. Wt.: 142,59
10
NH2N 2
45
67
3
1Cl
A solution of 6-bromomethyl-2-phthalimido-pyridine 9 (7.0 g, 22.1 mmol) was dissolved in a HCl solution (3M, 25 mL) and heated for 2 hours at 130 ºC. After cooling to room temperature, the mixture was neutralized with NaOH and extracted with AcOEt (3 x 25 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography on silica gel (CH2Cl2/AcOEt, 9:1) affording 2-amino-6-bromomethyl-pyridine 10 as white crystals (2.45 g, 17.2 mmol, 78%). The product has to be stored at -20 °C to avoid any polymerisation. 1H NMR (400.130 MHz, CDCl3): δ = 4.47 (s, 2H, CH2Cl), 4.53 (br s, 2H, NH2), 6.43 (d, 3J5-4
Na (236 mg, 9.84 mmol, 2.0 eq) was added over 10 min to liquid ammonia (7 mL) at -78 °C. The dark blue solution was stirred at this temperature for 20 minutes and then treated portionwise with PPh3 (1.29 g, 4.92 mmol, 1.05 eq) over 5 minutes. The dark red-orange solution was stirred for a further 2 hours at -78 °C before the addition of 2-amino-6-chloromethyl-pyridine 10 (679 mg, 4.78 mmol). THF (10 mL) was then added after 40 minutes and the cooling bath removed. Ammonia was allowed to evaporate overnight and the residue was quenched with H2O (10 mL), extracted with CH2Cl2 (3 x 10 mL) and dried over Na2SO4. The solvent was evaporated in vacuo, and subsequent flash column chromatography on silica gel (CH2Cl2 then CH2Cl2/AcOEt, 10:3) afforded 2-amino-6-(diphenyl phosphinomethyl)-pyridine 11 as a white solid (979 mg, 3.35 mmol, 70%). 1H NMR (400.130 MHz, C6D6): δ = 3.51 (s, 2H, CH2), 3.66 (br s, 2H, NH2), 5.70 (br d, 3J5-4 = 8.1 Hz, 1H, Ar-H5), 6.31 (br d, 3J3-4 = 7.5 Hz, 1H, Ar-H3), 6.87 (dd, 3J4-5 = 8.1 Hz, 3J4-3 = 7.5 Hz, 1H, Ar-H4), 7.05 (m, 6H, Ar-H9 and Ar-H11), 7.49 (m, 4H, Ar-H10). 13C{1H} NMR (100.613 MHz, C6D6): δ = 39.2 (d, 1J7-P = 16.8 Hz, 1C, C7), 105.3 (d, 5J3-P = 2.7 Hz, 1C, C3), 113.4 (d, 3J5-P = 6.7 Hz, 1C, C5), 128.5 (d, 2J10-P = 6.7 Hz, 4C, C10), 128.6 (s, 2H, C11), 133.3 (d, 2J9-P = 19.1 Hz, 4C, C9), 137.5 (s, 1C, C4), 139.9 (d, 1J8-P = 16.8 Hz, 2C, C8), 157.2 (d, 2J6-P = 8.3 Hz, 1C, C6), 158.6 (s, 1C, C2). 31P{1H} NMR (121.5 MHz, C6D6): δ = -10.6. HRMS (EI): Calcd. for C18H17N2P (M): 292.1129; Found: 292.1127 (difference -0.7 ppm). Tm = 111 °C Rf = 0.22 (CH2Cl2/AcOEt 19:1)
ExperimentalPart:Synthesisofligands
166
8.2.4 Synthesis of 6-(diphenylphosphinomethyl)-2-methoxyaniline, ligand 15
8.2.4.1 Synthesis of 4-methoxy-5-phthalimido toluene 13, ligand 15 step 1
12
NH2
O
O
O
, 1 h, 190 °C
O
86%2
45
67
3
8
1
13
O
N
O
O
9
9
10
10 11
11
12
12
C8H11NOMol. Wt.: 137,18
C16H13NO3Mol. Wt.: 267,28
A mixture of 2-methoxy-5-methylaniline 12 (9.8 g, 71.4 mmol) and phthalic anhydride (10.6 g, 71.4 mmol, 1.0 eq) were stirred and held at 190 °C for 1 hour. After cooling to room temperature, the residue was purified by silica gel chromatography (eluting with AcOEt) to yield 4-methoxy-5-phthalimido toluene 13 as a white crystalline solid (16.4 g, 61.3 mmol, 86%). 1H NMR (400.130 MHz, CDCl3): δ = 2.34 (s, 3H, CH3), 3.76 (s, 3H, OCH3), 6.95 (d, 3J4-5 = 8.5 Hz, 1H, Ar-H4), 7.06 (d, 4J1-5 = 2.5 Hz, 1H, Ar-H1), 7.23 (br d, 3J5-4 = 8.5 Hz, 1H, Ar-H5), 7.77 (m, 2H, Ar-H11), 7.94 (m, 2H, Ar-H12). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 20.5 (s, 1C, C7), 56.1 (s, 1C, C8), 112.2 (s, 1C, C4), 120.1 (s, 1C, C1), 123.8 (s, 2C, C11), 130.5 (s, 1C, C5), 131.3 (s, 1C, C2), 132.5 (s, 2C, C12), 134.2 (s, 2C, C10), 153.4 (s, 1C, C3), 167.6 (s, 2C, C9). HRMS (EI): Calcd. for C16H13O3N (M): 267.0895; Found: 267.0891 (difference -1.5 ppm). Tm = 206 °C Rf = 0.81 (AcOEt pure)
ExperimentalPart:Synthesisofligands
167
8.2.4.2 Synthesis of 6-bromomethyl-(4-methoxy-5-phthalimido) benzene 14, ligand 15 step 2
NBS, AIBN every 3 hoursbenzene, 16 h, reflux
62%
1413
O
N
O
O
2
45
67
3
8
1
O
N
O
O
9
9
10
10 11
11
12
12
Br
C16H12BrNO3Mol. Wt.: 346,18
C16H13NO3Mol. Wt.: 267,28
A mixture of 4-methoxy-5-phthalimido toluene 13 (4.5 g, 16.8 mmol), N-bromosuccinimide (3.29 g, 18.5 mmol) and AIBN (150 mg) in anhydrous benzene (10 mL) was stirred and held at reflux for 16 hours. (AIBN (150 mg) was added every 3 hours). After cooling to room temperature, the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with AcOEt) to yield 6-bromomethyl-(4-methoxy-5-phthalimido)benzene 14 as a white solid (4.6 g, 13.3 mmol, 79%). 1H NMR (400.130 MHz, CDCl3): δ = 3.81 (s, 3H, OCH3), 4.51 (s, 2H, CH2Br), 7.01 (d, 3J4-5
Na (320 mg, 13.9 mmol) was added over 10 min to liquid ammonia (10 mL) at -78 °C. The dark blue solution was stirred at this temperature for 20 minutes and then treated portionwise with PPh3 (1.82 g, 6.96 mmol) over 5 minutes. The dark red-orange solution was stirred for a further 2 hours at -78 °C before the addition of 6-bromomethyl-(4-methoxy-5-phthalimido)benzene 14 (2.33 g, 6.76 mmol). THF (15 mL) was then added after 40 minutes and the cooling bath removed. Ammonia was allowed to evaporate overnight and the residue was quenched with H2O (10 mL), extracted with CH2Cl2 (3 x 8 mL) and dried over Na2SO4. The solvent was evaporated in vacuo and subsequent flash column chromatography on silica gel (CH2Cl2 then CH2Cl2/OAcEt, 95:5) afforded 6-(diphenylphosphinomethyl)-2-methoxyaniline 15 as a white solid (586 mg, 1.83 mmol, 27%). 1H NMR (400.130 MHz, CDCl3): δ = 3.29 (s, 2H, CH2), 3.79 (s, 3H, OCH3), 6.41 (dt, 3J5-4 = 8.1 Hz, 4J5-1 = 1.9 Hz, 1H, Ar-H5), 6.46 (t, 4J1-5 = 1.9 Hz, 1H, Ar-H1), 6.60 (d, 3J4-5 = 8.1 Hz, 1H, Ar-H4), 7.31 (m, 6H, Ar-H10 and Ar-H11), 7.40 (m, 4H, Ar-H9). 13C{1H} NMR (100.613 MHz, CDCl3): δ = 35.4 (d, 1J7-P = 14.6 Hz, 1C, C7), 55.6 (s, 1C, C8), 110.4 (d, 4J4-P = 1.4 Hz, 1C, C4), 116.2 (d, 3J5-P = 7.1 Hz, 1C, C5), 119.3 (d, 4J4-P = 7.0 Hz, 1C, C1), 128.4 (d, 3J11-P = 6.5 Hz, 4C, C11), 128.7 (s, 2C, C12), 130.0 (d, 2J5-P = 8.6 Hz, 1C, C6), 133.1 (d, 3J10-P = 18.1 Hz, 4C, C10), 136.0 (s, 1C, C3), 138.8 (d, 1J9-P = 15.2 Hz, 2C, C9), 145.9 (d, 4J2-P = 2.9 Hz, 1C, C2). 31P{1H} NMR (101.3 MHz, CDCl3) δ = -11.0. HRMS (EI): Calcd. for C20H20ONP (M): 321.1283; Found: 321.1281 (difference -0.6 ppm). Tm = 122 °C Rf = 0.49 (CH2Cl2 pure)
ExperimentalPart:Synthesisofligands
169
8.2.5 Synthesis of 8-diphenylphosphanyl-quinolin-2-ylamine, ligand 23. 8.2.5.1 Synthesis of 2-bromocinnamanilide 18, ligand 23 step 1
K2CO3, H2O, acetone, 2 h, 0 °C
92%
16
Br
NH2
O
Cl+
17
O
N
H Br
18C6H6BrN
Mol. Wt.: 172,02C9H7ClO
Mol. Wt.: 166,60C15H12BrNO
Mol. Wt.: 302,17
A mixture of cinnamoyl chloride 17 (10.0 g, 60.0 mmol), 2-bromoaniline 16 (9.76 g, 60.0 mmol), and potassium carbonate (11.9 g) in water (30 mL) and acetone (25 mL) was kept for 2 hours at 0 °C before being poured into ice-water (60 mL). The precipitate formed was collected and crystallized from hot n-hexanes to afford 2-bromocinnamanilide 18 as colorless prisms (14.08 g, 46.6 mmol, 92%). The resulting 2-bromocinnamanilide 18 was directly engaged without further NMR analyses to the next step. Analytic data are in accordance with the literature.[101]
8.2.5.2 Synthesis of 8-bromo-2(1H)-quinolinone 19, ligand 22 step 2
O
N
H Br
18
56%NH
O
Br
AlCl3, chlorobenzene2 h, 125 °C
19C15H12BrNO
Mol. Wt.: 302,17C9H6BrNO
Mol. Wt.: 224,05
A solution of 2-bromocinnamanilide 18 (14.08 g, 46.6 mmol) and aluminium chloride (37 g, 279.6 mmol) in chlorobenzene (50 mL) was heated to 125 °C for 2 hours at 50 °C. It was then poured onto ice, and the resulting precipitate was filtered and crystallized from hot ethanol to obtain 8-bromo-2(1H)-quinolinone 19 (5.84 g, 26.1 mmol, 56%) as colorless needles. The resulting 8-bromo-2(1H)-quinolinone 19 was directly engaged without further NMR analyses to the next step. Tm = 196 °C Analytic data are in accordance with the literature.[101]
[101] F. Cottet, M. Marull, O. Lefebvre, M. Schlosser, Eur. J. Org. Chem. 2003, 1559-1568.
ExperimentalPart:Synthesisofligands
170
8.2.5.3 Synthesis of 8-bromo-2-chloro-quinoline 20, ligand 23 step 3
NH
O
Br
19
57%
POCl3, 2 h, 125 °C
Br
N Cl
20C9H6BrNO
Mol. Wt.: 224,05C9H5BrClN
Mol. Wt.: 242,50 Phosphorus oxychloride (7.30 mL, 78.4 mmol, 3.5 eq) and 8-bromo-(1H)-quinolinone 19 (5.0 g, 22.4 mmol) were heated to 125 °C for 2 h before being poured onto ice. The resultig precipitate was filtered and crystallized from hot methanol to afford 8-bromo-2-chloroquinoline 20 as purple prisms (4.15 g, 18.6 mmol, 57%). The resulting 8-bromo-2-chloroquinoline 20 was directly engaged without further NMR analyses to the next step. Tm = 113-114 °C Rf = 0.43 (cHex/AcOEt 9:1) Analytic data are in accordance with the literature.[101]
[101] F. Cottet, M. Marull, O. Lefebvre, M. Schlosser, Eur. J. Org. Chem. 2003, 1559-1568.
ExperimentalPart:Synthesisofligands
171
8.2.5.4 Synthesis of 2-amino-8-bromoquinoline 21, ligand 23 step 4
Br
N Cl
20
75%N NH2
Br
NH4OH, 16 h, 190 °C
21C9H5BrClN
Mol. Wt.: 242,50C9H7BrN2
Mol. Wt.: 223,07
2
456
7 3
89 10
1 11
8-bromo-2-chloroquinoline 20 (9.75 g, 40.2 mmol) was suspended in NH4OH(aq) (50 mL) and heated at 190 °C for 16 hours. After cooling to room temperature, H2O (50 mL) and AcOEt (100 mL) were added and the aqueous phase extracted with AcOEt (3 x 100 mL). The combined organic fractions were then dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography on silica gel (CH2Cl2 then cHex/AcOEt, 1/1) affording 2-amino-8-bromoquinoline 21 as a purple solid (6.70 g, 30.0 mmol, 75%). 1H NMR (400.13 MHz, CDCl3): δ = 4.98 (br s, 2H, NH2), 6.74 (d, 3J3-4 = 8.5 Hz, 1H, Ar-H3), 7.10 (t, 3J7-6 = 3J7-8 = 7.9 Hz, 1H, Ar-H7), 7.58 (dd, 3J6-7 = 7.9 Hz, , 4J6-8 = 1.4 Hz, 1H, Ar-H6), 7.85 (d, 3J4-3 = 8.5 Hz, 1H, Ar-H4), 7.88 (dd, 3J8-7 = 7.9 Hz, 4J8-6 = 1.4 Hz, 1H, Ar-H8). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 112.5 (s, 1C, C3), 121.1 (s, 1C, C9), 123.1 (s, 1C, C7), 125.0 (s, 1C, C5), 127.5 (s, 1C, C6), 133.5 (s, 1C, C8), 138.7 (s, 1C, C4), 145.1 (s, 1C, C10), 157.8 (s, 1C, C2). HRMS (EI): Calcd. for C9H7N2Br (M): 221.9793; Found: 221.9791. Tm = 158 °C Rf = 0.12 (cHex/AcOEt 8:2) Analytic data are in accordance with the literature.[102]
[102] Y. Cheng, T. C. Judd, M. T. Bartberger, J. Brown, K. Chen, R. T. Fremeau Jr., D. Hickman, S. A. Hitchcock, B. Kordan, V. Li, P. Lopez, S. W. Louie, Y. Luo, K. Michelsen, T. Nixey, T. S. Powers, C. Rattan, E. A. Sickmier, D. J. St. Jean Jr., R. C. Wahl, P. H. Wen, S. Wood, J. Med. Chem. 2011, 54(16), 5836-5857. Similar conditions, but lower yield (3.2% after steps 3 and 4).
ExperimentalPart:Synthesisofligands
172
8.2.5.5 Synthesis of 2-(N-tert-butoxycarbonyl)-8-bromoquinoline 22, ligand 23 step 5
N NH2
Br
21
78%
o/n, 60 °C
22
O
O
O
O
O
C9H7BrN2Mol. Wt.: 223,07
C14H15BrN2O2Mol. Wt.: 323,19
N NH
Br
2
456
7 3
89 10
1
O
O
11
14
131215
15
15
A mixture of 2-amino-8-bromoquinoline 21 (2 g, 9.0 mmol) and di-tert-butyldicarbonate (2.16 g, 9.9 mmol, 1.1 eq) was heated at 60 ºC overnight. The reaction was cooled, poured into saturated NH4Cl (100 mL) and extracted with AcOEt (2 x 100 mL). The combined organic layers were washed with brine (3 x 50 mL), dried over MgSO4, filtered and concentrated to give a yellow oil, which was recrystallised from hot pentane to give 2-(N-tert-butoxycarbonyl)-8-bromoquinoline 22 as a purple solid (2.26 g, 7.0 mmol, 78%).
8.2.5.6 Synthesis of 8-diphenylphosphanyl-quinolin-2-ylamine, ligand 23
N NH
Br
22
O
O
1) n-BuLi, THF, 30 min, -78 °C2) PPh2Cl, o/n, -78 °C to 0 °C3) TFA, CH2Cl2, 2 h, RT
63%
23C14H15BrN2O2
Mol. Wt.: 323,19C21H17N2P
Mol. Wt.: 328,35
N NH2
P
2
456
7 3
89 10
11
15
14
13
1313
1415
1413
14
12 12
n-BuLi (2.5 M in Hexanes, 8.2 mmol, 3.3 mL, 2.2 eq) was slowly added at -78 ºC to a solution of 2-(N-tert-butoxycarbonyl)-8-bromoquinoline 22 (1.20 g, 3.71 mmol) in THF (20 mL) and stirred at this temperature for 30 minutes. PPh2Cl (1.06 g, 4.82 mmol, 1.3 eq) was then added and the solution allowed to warm to room temperature overnight. After addition of H2O (20 mL) and extraction with CH2Cl2 (3 x 20 mL), the combined organic extracts were washed with brine (3 x 30 mL), dried over MgSO4 and concentrated in vacuo. The residue was dissolved in CH2Cl2 (10 mL) in the presence of TFA (2.5 mL) and stirred at this temperature for 2 hours. The mixture was then diluted with H2O (10 mL), neutralized with NaHCO3 and extracted with CH2Cl2 (3 x 10 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated in vacuo. Subsequent short flash column chromatography on silica gel (CH2Cl2) afforded 2-amino-8-(diphenylphoshino)quinoline 23 as a yellow solid (0.77 g, 2.34 mmol, 63%). 1H NMR (400.13 MHz, CDCl3): δ = 4.71 (br s, 2H, NH2), 6.67 (d, 3J3-4 = 8.6 Hz, 1H, Ar-H3), 6.96 (m, 1H, Ar-H7), 7.13 (t, 3J6-7 = 7.9 Hz, 1H, Ar-H6), 7.31 (m, 10H, Ar-H13, H14 and H15), 7.60 (d, 3J8-7 = 7.9 Hz, 1H, Ar-H8), 7.85 (d, 3J4-3 = 8.6 Hz, 1H, Ar-H4). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 112.0 (s, 1C, C3), 122.7 (s, 1C, C5), 123.1 (d, 3J7-P = 2.6 Hz, 1C, C7), 128.3 (s, 2C, C15), 128.4 (s, 4C, C14), 128.6 (s, 1C, C6), 134.2 (s, 1C, C9), 134.4 (s, 4C, C13), 134.5 (s, 1C, C8), 138.1 (C12), 138.2 (s, 1C, C4), 149.5 (d, 3J10-P = 18.8 Hz, 1C, C10), 156.3 (d, 4J2-P = 1.8 Hz, 1C, C2). 31P{1H} NMR (101.3 MHz, C6D6): δ = -15.2. HRMS (EI): Calcd. for C21H17N2P (M): 328.1129; Found: 328.1127. Tm = 198 °C Rf = 0.29 (CH2Cl2/AcOEt 19:1)
ExperimentalPart:Synthesisofligands
174
8.2.6 Synthesis of 6-(2-diphenylphosphanyl-ethyl)-3-methyl-pyridin-2-ylamine, ligand 27.
8.2.6.1 Synthesis of 6-ethanoyl-2-methyl-3-(pivaloylamino)-pyridine 24, ligand 27 step 3
a
PivCl, Et3N, CH2Cl2o/n, 0 °C to RT
N NH2 N NH
O
b
quant.
1) n-BuLi, THF, 90 min -78 °C to 0 °C2) MeI, o/n, 0 °C to RT
N NH
O
74%
N NH
O
24HO
83%
1) n-BuLi, THF, 30 min, 0 °C2) CH2O, o/n, RT
c
C6H8N2Mol. Wt.: 108,14
C11H16N2OMol. Wt.: 192,26
C12H18N2OMol. Wt.: 206,28
C13H20N2O2Mol. Wt.: 236,31
2
45
6
7
38
910
111 14
1312 14
14
Synthesis of 6-methyl-2-pivaloylamino-pyridine b, ligand 27 step 1 Refer to p. 161 to see more details about the synthesis.
Synthesis of 3,6-dimethyl-2(pivaloylamino)-pyridine c, ligand 27 step 2 Refer to p. 162 to see more details about the synthesis.
n-BuLi (2.5 M in hexanes, 6.7 mL, 16.8 mmol, 2.2 eq) was added dropwise at 0 °C to a solution of 3,6-dimethyl-2-(pivaloylamino)-pyridine c (1.57 g, 7.62 mmol) in THF (20 mL) and stirred at this temperature for 30 minutes. Paraformaldehyde (0.37 g, 12.2 mmol, 1.6 eq) was then slowly added to the ice-cold solution and the reaction mixture was allowed to warm to room temperature overnight. The mixture was then poured into water, and extracted three times with AcOEt (3 x 10 mL). The combined organic layers were washed with brine, dried over MgSO4 and evaporated to dryness. The crude was purified by silica gel column chromatography (eluting with CH2Cl2/AcOEt 4:1) yielding 6-ethanoyl-3-methyl-2-(pivaloylamino)-pyridine 24 as a white solid (1.51 g, 6.39 mmol, 84%). 1H NMR (300.07 MHz, CDCl3): δ = 1.29 (s, 9H, (CH3)3), 2.13 (s, 3H, CH3), 2.87, (t, 3J8-9 = 5.7 Hz, 2H, CH2(8)), 3.87 (t, 3J9-8 = 5.7 Hz, 2H, CH2(9)), 4.08 (br s, 1H, OH), 6.89 (d, 3J5-4 = 8.1 Hz, 1H, Ar-H5), 7.40 (d, 3J4-5 = 8.1 Hz, 1H, Ar-H4), 7.83 (br s, 1H, NH). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 17.5 (s, 1C, C7), 27.6 (s, 3C, C14), 38.7 (s, 1C, C8), 39.5 (s, 1C, C13), 61.7 (s, 1C, C9), 121.1 (s, 1C, C5), 125.5 (s, 1C, C3), 140.2 (s, 1C, C4), 148.7 (s, 1C, C2), 156.6 (s, 1C, C6), 176.8 (s, 1C, C12).
ExperimentalPart:Synthesisofligands
175
HRMS (EI): Calcd. for C13H20O2N2 (M): 236.1525; Found: 236.1523. Tm = 117 °C Rf = 0.12 (CH2Cl2/AcOEt 4:1) 8.2.6.2 Synthesis of 6-chloroethyl-3-methyl-2-(pivaloylamino)-pyridine 25, ligand 27 step 4
N NH
O
24HO
CCl4, PPh3,CH2Cl2, o/n, RT
71%
25C13H20N2O2
Mol. Wt.: 236,31C13H19ClN2O
Mol. Wt.: 254,76
N NH
O
Cl
2
45
6
7
38
91
10 13
1211 13
13
6-ethanoyl-3-methyl-2-(pivaloylamino)-pyridine 24 (2.63 g, 11.1 mmol) was dissolved in CH2Cl2 (30 mL) in the presence of carbon tetrachloride (2.56 g, 16.7 mmol, 1.5 eq). Triphenylphosphine (3.5 g, 13.3 mmol, 1.2 eq) was added in portions over a 30 minute period and the solution was stirred at room temperature overnight. The solvent was removed to dryness and the residue was purified by flash chromatography on silica gel (CH2Cl2) affording 6-chloroethyl-3-methyl-2-(pivaloylamino)-pyridine 25 as a colorless oil (2.0 g, 7.88 mmol, 71%). 1H NMR (400.13 MHz, CDCl3): δ = 1.34 (s, 9H, (CH3)3), 2.19 (s, 3H, CH3), 3.13 (t, 3J8-9 = 7.1 Hz, 2H, CH2(8)), 3.82 (t, 3J9-8 = 7.1 Hz, 2H, CH2(9)), 7.00 (d, 3J5-4 = 7.7 Hz, 1H, Ar-H5), 7.49 (br d, 3J4-5 = 7.7 Hz, 1H, Ar-H4), 7.81 (br s, 1H, NH). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 18.0 (s, 1C, C7), 27.7 (s, 3C, C13), 39.5 (s, 1C, C12), 40.4 (s, 1C, C8), 43.6 (s, 1C, C9), 121.7 (s, 1C, C5), 127.5 (s, 1C, C3), 140.4 (s, 1C, C4), 149.3 (s, 1C, C2), 154.2 (s, 1C, C6), 176.9 (s, 1C, C11). HRMS (EI): Calcd. for C13H19ON2Cl (M): 254.1186; Found: 25411.86.
ExperimentalPart:Synthesisofligands
176
8.2.6.3 Synthesis of 2-amino-6-chloroethyl-3-methyl-pyridine 26, ligand 27 step 5
N NH2
Cl
2
45
6
7
38
91
10N N
H
O
25Cl
26
HCl(aq), 2 h, 130 °C
70%
C13H19ClN2OMol. Wt.: 254,76
C8H11ClN2Mol. Wt.: 170,64
A solution of 6-chloroethyl-3-methyl-2-(pivaloylamino)-pyridine 25 (0.9 g, 3.54 mmol) was dissolved in a HCl solution (3M, 5 mL) and heated for 2 hours at 130 ºC. After cooling to room temperature, the mixture was neutralized with NaOH and extracted with EtOAc (3 x 5 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography on silica gel (CH2Cl2/AcOEt, 9:1) affording 2-amino-6-chloroethyl-3-methyl-pyridine 26 as a colorless oil (0.42 g, 2.48 mmol, 70%). Presence of phosphine oxide. 1H NMR (400.13 MHz, CDCl3): δ = 2.09 (s, 3H, CH3), 3.02 (t, 3J8-9 = 7.3 Hz, 2H, CH2(8)), 3.84 (t, 3J9-8 = 7.3 Hz, 2H, CH2(9)), 4.39 (br s, 2H, NH2), 6.51 (d, 3J5-4 = 7.3 Hz, 1H, Ar-H5), 7.20 (d, 3J4-5 = 7.3 Hz, 1H, Ar-H4). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 16.9 (s, 1C, C7), 40.8 (s, 1C, C8), 44.0 (s, 1C, C9), 114.1 (s, 1C, C5), 114.8 (s, 1C, C3), 138.3 (s, 1C, C4), 153.8 (s, 1C, C6), 156.9 (s, 1C, C2). HRMS (CI): Calcd. for C8H12N2Cl (M+H): 171.0689; Found: 171.0691.
ExperimentalPart:Synthesisofligands
177
8.2.6.4 Synthesis of 6-(2-diphenylphosphanyl-ethyl)-3-methyl-pyridin-2-ylamine, ligand 27
8.2.7.1 Synthesis of 6-(N-tert-butoxycarbonyl)-picoline 28, ligand 31 step 2
a
PivCl, Et3N, CH2Cl2o/n, 0 °C to RT
N NH2N N
H
O
b
quant.
28
1) n-BuLi, THF, 30 min, 0 °C2) CH2O, o/n, RT
82%
C11H16N2OMol. Wt.: 192,26
C6H8N2Mol. Wt.: 108,14
C12H18N2O2Mol. Wt.: 222,28
N NH
O
HO
2
45
67
3
8
9
110 13
1211 13
13
Synthesis of 6-methyl-2-pivaloylamino-pyridine b, ligand 31 step 1 Refer to p. 161 to see more details about the synthesis.
n-BuLi (2.5 M in hexanes, 14.3 mL, 35.6 mmol, 2.2 eq) was added dropwise at 0 °C to a solution of 6-(N-tert-butoxycarbonyl)-picoline b (3.37 g, 16.2 mmol) in THF (50 mL) and stirred at this temperature for 30 minutes. Paraformaldehyde (0.78 g, 25.9 mmol, 1.6 eq) was then slowly added to the ice-cold solution and the reaction mixture was allowed to warm to room temperature overnight. The mixture was then poured into water, and extracted three times with EtOAc (3 x 30 mL). The combined organic layers were washed with brine, dried over MgSO4 and evaporated to dryness. The crude was purified by silica gel column chromatography (eluting with CH2Cl2/AcOEt 4:1) yielding 6-(N-tert-butoxycarbonyl)-2-ethanoyl-pyridine 28 as a white solid (3.16 g, 13.3 mmol, 82%). 1H NMR (400.13 MHz, CDCl3): δ = 1.52 (s, 9H, (CH3)3), 2.91 (t, 3J7-8 = 5.7 Hz, 2H, CH2(7)), 3.98 (t, 3J8-7 = 5.7 Hz, 2H, CH2(8)), 4.14 (br s, 1H, OH), 6.80 (d, 3J5-4 = 7.6 Hz, 1H, Ar-H5), 7.53 (br s, 1H, NH), 7.58 (dd, 3J4-5 = 7.6 Hz, 3J4-3 = 8.1 Hz, 1H, Ar-H4), 7.78 (d, 3J3-4 = 8.1 Hz, 1H, Ar-H3). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 28.4 (s, 3C, C13), 38.6 (s, 1C, C7), 61.9 (s, 1C, C8), 81.1 (s, 1C, C12), 110.0 (s, 1C, C3), 118.0 (s, 1C, C5), 139.0 (s, 1C, C4), 151.3 (s, 1C, C2), 152.4 (s, 1C, C6), 159.1 (s, 1C, C1).
HRMS (EI): Calcd. for C12H19N2O3 (M+H2O+): 239.1390; Found: 239.1391
Tm = 103 °C
Rf = 0.28 (CH2Cl2/AcOEt 4:1)
ExperimentalPart:Synthesisofligands
179
8.2.7.2 Synthesis of 6-(N-tert-butoxycarbonyl)-2-bromoethyl-pyridine 29, ligand 31 step 3
N NH
O
Br
2
45
67
3
81
9 12
1110 12
12
N NH
O
28
HO
72%
CBr4, PPh3,CH2Cl2, o/n, RT
29C12H18N2O2
Mol. Wt.: 222,28C12H17BrN2O
Mol. Wt.: 285,18 6-(N-tert-butoxycarbonyl)-2-ethanoyl-pyridine 28 (1.92 g, 8.06 mmol) was dissolved in CH2Cl2 (30 mL) in the presence of carbon tetrabromide (4.01 g, 12.1 mmol, 1.5 eq). Triphenylphosphine (2.54 g, 9.70 mmol, 1.2 eq) was added in portions over a 30 minute period and the solution was stirred at room temperature overnight. The solvent was removed to dryness and the residue was purified by flash chromatography on silica gel (CH2Cl2) affording 6-(N-tert-butoxycarbonyl)-2-bromoethyl-pyridine 29 as a colorless oil (1.74 g, 5.80 mmol, 72%). 1H NMR (499.63 MHz, CDCl3): δ = 1.51 (s, 9H, (CH3)3), 3.20 (t, 3J7-8 = 7.1 Hz, 2H, CH2(7)), 3.70 (t, 3J8-7 = 7.1 Hz, 2H, CH2(8)), 6.83 (d, 3J5-4 = 7.7 Hz, 1H, Ar-H5), 7.25 (br s, 1H, NH), 7.59 (dd, 3J4-5 = 7.7 Hz, 3J4-3 = 8.2 Hz, 1H, Ar-H4), 7.79 (d, 3J3-4 = 8.2 Hz, 1H, Ar-H3). 13C{1H} NMR (125.6 MHz, CDCl3): δ = 28.4 (s, 3C, C12), 31.4 (s, 1C, C7), 40.8 (s, 1C, C8), 81.1 (s, 1C, C11), 110.4 (s, 1C, C3), 118.2 (s, 1C, C5), 138.8 (s, 1C, C4), 151.7 (s, 1C, C2), 152.4 (s, 1C, C6), 156.8 (s, 1C, C1). HRMS (EI): Calcd. for C12H17BrN2NaO2 (M+NaOH+): 323.0366; Found: 323.0366. Rf = 0.72 (CH2Cl2 pure)
ExperimentalPart:Synthesisofligands
180
8.2.7.3 Synthesis of 2-amino-6-chloroethyl-pyridine 30, ligand 31 step 4
N NH2
Cl
2
45
67
3
81
9
N NH
O
29
Br
30
HCl(aq), 2 h, 130 °C
70%
C12H17BrN2OMol. Wt.: 285,18
C7H9ClN2Mol. Wt.: 156,61
A solution of 6-(N-tert-butoxycarbonyl)-2-bromoethyl-pyridine 29 (1.3 g, 4.33 mmol) was dissolved in a HCl solution (3M, 5 mL) and heated for 2 hours at 130 ºC. After cooling to room temperature, the mixture was neutralized with NaOH and extracted with AcOEt (3 x 5 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (CH2Cl2/AcOEt 9:1) affording 2-amino-6-chloroethyl-pyridine 30 as a colorless oil (0.49 g, 3.16 mmol, 73%). 1H NMR (400.13 MHz, CDCl3): δ = 3.06 (t, 3J7-8 = 7.1 Hz, 2H, CH2(7)), 3.87 (t, 3J8-7 = 7.1 Hz, 2H, CH2(8)), 4.60 (br s, 2H, NH2), 6.41 (d, 3J5-4 = 8.3 Hz, 1H, Ar-H5), 6.57 (d, 3J3-4 = 7.4 Hz, 1H, Ar-H3), 7.40 (dd, 3J4-5 = 8.3 Hz, 3J4-3 = 7.4 Hz, 1H, Ar-H4). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 40.7 (s, 1C, C7), 43.6 (s, 1C, C8), 107.3 (s, 1C, C3), 113.8 (s, 1C, C5), 138.8 (s, 1C, C4), 155.7 (s, 1C, C2), 158.1 (s, 1C, C6). HRMS (CI): Calcd. for C7H10N2Cl (M+H): 157.0533; Found: 157.0535.
ExperimentalPart:Synthesisofligands
181
8.2.7.4 Synthesis of 2-amino-6-(diphenylphosphinoethyl)-pyridine, ligand 31
Na (420 mg, 18.3 mmol, 2.0 eq) was added over 10 minutes to liquid ammonia (30 mL) at -78 °C. The dark blue solution was stirred at this temperature for 20 minutes and then treated portionwise with PPh3 (2.39 g, 9.1 mmol, 1.05 eq) over 5 minutes. The dark red-orange solution was stirred for a further 2 hours at -78 °C before the addition of 2-amino-6-chloroethyl-pyridine 30 (1.38 g, 8.8 mmol). THF (20 mL) was then added after 40 minutes and the cooling bath removed. Ammonia was allowed to evaporate overnight and the residue was quenched with H2O (10 mL), extracted with CH2Cl2 (3 x 10 mL) and dried over Na2SO4. The solvent was evaporated in vacuo, and subsequent flash column chromatography on silica gel (CH2Cl2 then CH2Cl2/AcOEt 4:1) afforded 2-amino-6-(diphenylphosphinoethyl)-pyridine 31 as a white solid (1.75 g, 5.72 mmol, 68%). 1H NMR (400.13 MHz, C6D6): δ = 2.56 (m, 2H, CH2(7)), 2.83 (m, 2H, CH2(8)), 3.92 (br s, 2H, NH2), 5.80 (d, 3J5-4 = 7.5 Hz, 1H, Ar-H5), 6.22 (d, 3J3-4 = 7.5 Hz, 1H, Ar-H3), 6.97 (dd, 3J4-5 = 3J4-3 = 7.4 Hz, 1H, Ar-H4), 7.06 (m, 6H, Ar-H11 and Ar-H13), 7.46 (m, 4H, Ar-H12). 13C{1H} NMR (100.6 MHz, C6D6): δ = 28.7 (d, 2J7-P = 13.2 Hz, 1C, C7), 34.9 (d, 1J8-P = 18.1 Hz, 1C, C8), 105.7 (s, 1C, C3), 112.3 (s, 1C, C5), 128.6 (d, 3J12-P = 7.6 Hz, 4C, C12), 128.7 (s, 2C, C13), 133.2 (d, 1J10-P = 18.7 Hz, 2C, C10), 137.7 (s, 1C, C4), 139.7 (d, 2J11-P = 14.8 Hz, 4C, C11), 158.8 (s, 1C, C2), 160.8 (d, 3J6-P = 13.9 Hz, 1C, C6). 31P{1H} NMR (162.0 MHz, C6D6): δ = -15.2. HRMS (EI): Calcd. for C19H19N2P (M): 306.1286; Found: 306.1286. Tm = 137 °C Rf = 0.12 (CH2Cl2/AcOEt 19:1)
ExperimentalPart:Synthesisofligands
182
8.2.8 Synthesis of 6-(2-diphenylphosphanyl-ethyl)-pyridin-2-ylamine, ligand 36. 8.2.8.1 Synthesis of 2-(6-bromo-pyridin-2-yl)-propan-2-ol 33, ligand 36 step 1
96%
32
N BrBr
1) n-BuLi, THF, 15 min, -95 °C2) excess acetone, 30 min, -95 °C then o/n, RT3) NH4Cl, H2O
33
NBr
OH
C5H3Br2NMol. Wt.: 236,89
C8H10BrNOMol. Wt.: 216,08
n-Buli (1.6 M in hexanes, 34 mL, 84 mmol, 1.5 eq) was diluted with 40 mL of Et2O. The resulting solution was cooled to -95 °C. Then 2,6-dibromopyridine 32 (20.0 g in 140 mL of THF, 56.3 mmol) was added slowly with vigorous stirring. The resulting dark green suspension was stirred at -95 °C for 15 minutes, followed by the addition of 6.2 mL (1 equivalent) of acetone. The resulting mixture was further stirred at -95 °C for 30 minutes, warmed to ambient temperature, and stirred overnight to afford an orange solution. A saturated NH4Cl solution was added with vigorous stirring. The organic layer was combined with the CH2Cl2 extract of the aqueous layer, washed with brine, and dried over MgSO4. The removal of the solvents gave a brown oil of 6-(2-diphenylphosphanyl-ethyl)-pyridin-2-ylamine 33 (20.34 g, 94 mmol, 96%). The resulting 6-(2-diphenylphosphanyl-ethyl)-pyridin-2-ylamine 33 was directly engaged without further NMR analyses to the next step. Analytic data are in accordance with the literature.[103]
[103] D. Song, R. H. Morris, Organometallics 2004, 23, 4406-4413.
ExperimentalPart:Synthesisofligands
183
8.2.8.2 Synthesis of N-[1-(6-bromo-pyridin-2-yl)-1-methyl-ethyl]-acetamide 34, ligand 36 step 2
33
NBr
OH
19%
BF3.Et2O
CH3CN, 72 h, reflux
NBr
NHO
34C8H10BrNO
Mol. Wt.: 216,08C10H13BrN2O
Mol. Wt.: 257,13
2
45
67
3 8
1
8
910
11
To a solution of 2-(6-bromo-pyridin-2-yl)-propan-2-ol 33 (20.34 g, 93 mmol) in 80 mL of CH3CN was added 40 mL of BF3·Et2O. The resulting solution was refluxed for 72 hours, cooled to ambient temperature, neutralized with NaOH aqueous solution, and extracted with CH2Cl2. The CH2Cl2 extracts were combined, washed with brine, and dried over MgSO4. The solvent was then removed under vacuum and the residue was flushed through a silica gel column to afford, after evaporation, the white solid of N-[1-(6-bromo-pyridin-2-yl)-1-methyl-ethyl]-acetamide 34 in 19% yield (4.5 g, 17.5 mmol). 1H NMR (400.13 MHz, C6D6): δ = 1.74 (s, 6H, (CH3)2), 2.04 (s, 3H, CH3), 7.35 (dd, 3J5-4 = 3.2 Hz, 4J5-3 = 0.70 Hz, Ar-H5), 7.37 (dd, 3J3-4 = 3.2 Hz, 4J3-5 = 0.86 Hz, Ar-H3), 7.93 (dd, 3J4-5 = 3J4-3 = 3.2 Hz, Ar-H4). 13C{1H} NMR (100.6 MHz, C6D6): δ = 24.6 (s, 1C, C11), 27.6 (s, 2C, C8), 56.6 (s, 1C, C7), 118.5 (s, 1C, C3), 126.2 (s, 1C, C5), 139.3 (s, 1C, C4), 140.7 (s, 1C, C6), 166.4 (s, 1C, C2), 169.6 (s, 1C, C10). HRMS (EI): Calcd. for C10H14ON2Br (M): 257.02895; Found: 257.02900. Tm = 156 °C Rf = 0.17 (CH2Cl2/AcOEt 9:1) Analytic data are in accordance with the literature.[103]
[103] D. Song, R. H. Morris, Organometallics 2004, 23, 4406-4413.
ExperimentalPart:Synthesisofligands
184
8.2.8.3 Synthesis of 2-(6-bromopyridin-2-yl)propan-2-amine 35, ligand 36 step 3
NBr
NH2
2
45
67
3 8
1
8
9
NBr
NHO
34
69%
HCl(aq), 2 h, 130 °C
35C10H13BrN2O
Mol. Wt.: 257,13C8H11BrN2
Mol. Wt.: 215,09 A solution of N-(2-(6-bromopyridin-2-yl)propan-2-yl)acetamide 34 (1.1 g, 4.3 mmol) was dissolved in a HCl solution (3M, 6 mL) and heated for 2 hours at 130 ºC. After cooling to room temperature, the mixture was neutralized with NaOH and extracted with AcOEt (3 x 10 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography on silica gel (CH2Cl2/AcOEt, 9:1) affording 2-(6-bromopyridin-2-yl)propan-2-amine 35 as a colorless oil (0.63 g, 2.96 mmol, 69%). 1H NMR (400.13 MHz, CDCl3): δ = 1.50 (s, 6H, (CH3)2), 2.16 (bs, 2H, NH2), 7.31 (dd, 3J5-4 = 7.8 Hz, 4J5-3 = 0.9 Hz, 1H, Ar-H5), 7.39 (dd, 3J3-4 = 7.8 Hz, 4J3-5 = 0.9 Hz, 1H, Ar-H3), 7.48 (dd, 3J4-5 = 3J4-3 = 7.8 Hz, 1H, Ar-H4). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 30.9 (s, 2C, C8), 54.3 (s, 1C, C7), 117.4 (s, 1C, C3), 125.9 (s, 1C, C5), 139.0 (s, 1C, C4), 141.5 (s, 1C, C6), 170.0 (s, 1C, C2). HRMS (CI): Calcd. for C8H12N2Br (M+H): 215.0184; Found: 215.0183. Analytic data are in accordance with the literature.[104]
[104] J. Hu, PCT Int. Appl., 200913814, 29 Oct. 2009. Similar conditions, but lower yield (81%).
ExperimentalPart:Synthesisofligands
185
8.2.8.4 Synthesis of 2-(6-diphenylphosphinopyridin-2-yl)propan-2-amine, ligand 36
Na (352 mg, 15.3 mmol, 2.0 eq) was added over 10 minutes to liquid ammonia (8 mL) at -78 °C. The dark blue solution was stirred at this temperature for 20 minutes and then treated portionwise with PPh3 (2.0 g, 7.65 mmol, 1.05 eq) over 5 minutes. The dark red-orange solution was stirred for a further 2 hours at -78 °C before the addition of 2-(6-bromopyridin-2-yl)propan-2-amine (1.59 g, 7.43 mmol). THF (10 mL) was then added after 40 minutes and the cooling bath removed. Ammonia was allowed to evaporate overnight and the residue was quenched with H2O (10 mL), extracted with CH2Cl2 (3 x 8 mL) and dried over Na2SO4. The solvent was evaporated in vacuo, and subsequent flash column chromatography on silica gel (CH2Cl2/Acetone 2:1 then CH2Cl2/NEt3, 99:1) afforded ligand 2-(6-diphenyl phosphinopyridin-2-yl)propan-2-amine as a white solid (1.86 g, 5.79 mmol, 78%). 1H NMR (400.13 MHz, CDCl3): δ = 1.46 (s, 6H, (CH3)2), 2.15 (br s, 2H, NH2), 6.99 (ddd, 3J5-4 = 7.6 Hz, 4J5-3 = 3J5-P = 0.7 Hz, 1H, Ar-H5), 7.28 (d, 3J3-4 = 7.9 Hz, 1H, Ar-H3), 7.35 (m, 6H, Ar-H11 and Ar-H13), 7.40 (m, 4H, Ar-H12), 7.51 (ddd, 3J4-5 = 7.6 Hz, 3J4-3 = 7.9 Hz, 4J4-P = 2.4 Hz, 1H, Ar-H4). 13C{1H} NMR (100.6 MHz, CDCl3): δ = 31.0 (s, 2C, C8), 54.4 (s, 1C, C7), 116.9 (s, 1C, C3), 125.9 (d, 2J5-P = 21.6 Hz, 1C, C5), 128.5 (d, 3J12-P = 7.5 Hz, 4C, C12), 129.0 (s, 2C, C13), 134.3 (d, 1J10-P = 19.6 Hz, 2C, C10), 136.2 (d, 3J4-P = 4.2 Hz, 1C, C4), 136.9 (d, 2J11-P = 10.3 Hz, 4C, C11), 162.2 (d, 3J2-P = 4.1 Hz, 1C, C2), 168.2 (d, 1J6-P = 9.8 Hz, 1C, C6). 31P{1H} NMR (162.0 MHz, CDCl3): δ = -3.5. HRMS (EI): Calcd. for C20H21N2P (M): 320.1442; Found: 320.1444. Tm = 70 °C Rf = 0.12 (CH2Cl2/AcOEt 9:1)
ExperimentalPart:Synthesisofligands
186
8.2.9 Synthesis of C-(6-diphenylphosphanyl-pyridin-2-yl)-methylamine, ligand 41.
8.2.9.1 Synthesis of 2-bromo-6-bromomethylpyridine 38, ligand 41 step 1
NBr
NBS, AIBN every hourbenzene, 5 h, reflux
68%NBr
Br
37 38C6H5Br2N
Mol. Wt.: 250,92C6H6BrN
Mol. Wt.: 172,02 A mixture of 2-bromo-6-methylpyridine 37 (3.03 g, 17.7 mmol), N-bromosuccinimide (3.13 g, 17.7 mmol, 1.0 eq) and AIBN (50 mg) in anhydrous benzene (80 mL) was stirred and held at reflux for 5 hours. (AIBN (50 mg) was added every hour) After cooling to room temperature, the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 2-dibromomethyl-5-phthalimido-bromobenzene 38 as a white solid (2.79 g, 5.89 mmol, 68%). Tm = 138 °C Rf = 0.94 (CH2Cl2 pure) Analytic data are in accordance with the literature.[175]
[175] J. A. Drewry, S. Fletcher, H. Hassan, P. T. Gunning, Org. Biomol. Chem. 2009, 7(24), 5074-5077.
ExperimentalPart:Synthesisofligands
187
8.2.9.2 Synthesis of 2-bromo-6-phthalimidomethylpyridine 39, ligand 41 step 2
, DMF, o/n, RT
97%NBr
Br
38
NBr
N
39
OO
NK
O
O
C6H5Br2NMol. Wt.: 250,92
C14H9BrN2O2Mol. Wt.: 317,14
2
45
6
3
1
11 11
109
8
7
89
10
Potassium phthalimide (0.78 g, 4.2 mmol) was added to a solution of 2-bromo-6-bromomethyl-pyridine 38 (1.06 g, 4.2 mmol) in DMF (30 mL) and stirred at room temperature overnight. H2O (30 mL) was added to the reaction mixture, followed by extraction with AcOEt (3 x 50 mL). The combined organic extracts were washed with brine (3 x 50 mL), dried over MgSO4 and concentrated in vacuo to yield 2-bromo-6-phthalimidomethyl-pyridine 39 as a white crystalline solid (1.3 g, 4.1 mmol, 97%). 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 4.99 (s, 2H, CH2), 7.16 (br d, 3J5-4 = 7.6 Hz, 1H, Ar-H5), 7.37 (d, 3J3-4 = 7.9 Hz, 1H, Ar-H3), 7.48 (dd, 3J4-5 = 7.6 Hz, 3J4-3 = 7.9 Hz, 1H, Ar-H4), 7.76 (m, 2H, Ar-H11), 7.90 (m, 2H, Ar-H10). 13C{1H} NMR (100.626 MHz, CDCl3): δ (ppm) = 42.6 (s, 1C, C7), 120.0 (s, 2C, C10), 123.7 (s, 1C, C3), 127.1 (s, 2C, C11), 132.2 (s, 2C, C9), 134.4 (s, 1C, C5), 139.1 (s, 1C, C4), 142.1 (s, 1C, C6), 157.0 (s, 1C, C2), 168.0 (s, 2C, C8). HRMS (EI): Calcd. for C14H9O2N2Br (M): 315.9847; Found: 315.9850. Tm = 134 °C Rf = 0.41 (CH2Cl2 pure)
ExperimentalPart:Synthesisofligands
188
8.2.9.3 Synthesis of 6-aminomethyl-2-bromo-pyridine 40, ligand 41 step 3
HCl(aq), 2 h, 130 °C
81%NBr
N
39
OO
NBr
NH2
40C14H9BrN2O2
Mol. Wt.: 317,14C6H7BrN2
Mol. Wt.: 187,04
2
45
6
3
1
8
7
A solution of 2-bromo-6-phthalimidomethyl-pyridine 39 (2.7 g, 8.6 mmol) was dissolved in a HCl solution (3M, 10 mL) and heated for 2 hours at 130 ºC. After cooling to room temperature, the mixture was neutralized with NaOH and extracted with AcOEt (3 x 10 mL). The combined organic fractions were dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography on silica gel (CH2Cl2/OAcEt, 9/1) affording 6-aminomethyl-2-bromo-pyridine 40 as white crystals (1.3 g, 6.9 mmol, 81%). The product has to be stored at -20 °C to avoid any polymerisation 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 1.66 (br s, 2H, NH2), 3.92 (s, 2H, CH2), 7.24 (t, 3J5-4 = 7.7 Hz, 1H, Ar-H5), 7.32 (d, 3J3-4 = 7.7 Hz, 1H, Ar-H3), 7.48 (dd, 3J4-5 = 3J4-3 = 7.7 Hz, 1H, Ar-H4). 13C{1H} NMR (100.626 MHz, CDCl3): δ (ppm) = 47.5 (s, 1C, C7), 120.0 (s, 1C, C3), 126.2 (s, 1C, C5), 139.0 (s, 1C, C4), 141.9 (s, 1C, C6), 164.0 (s, 1C, C2). HRMS (EI): Calcd. for C6H7N2Br (M): 185.9793; Found: 185.9792.
ExperimentalPart:Synthesisofligands
189
8.2.9.4 Synthesis of C-(6-diphenylphosphanyl-pyridin-2-yl)-methylamine, ligand 41
Na (667 mg, 29.0 mmol) was added over 10 minutes to liquid ammonia (10 mL) at -78 °C. The dark blue solution was stirred at this temperature for 20 minutes and then treated portionwise with PPh3 (3.80 g, 14.5 mmol, 1.25 eq.) over 5 minutes. The dark red-orange solution was stirred for a further 2 hours at -78 °C before the addition of 6-amino-2-bromo-pyridine 42 (2.0 g, 11.6 mmol, 1.0 eq.). Toluene (40 mL) was then added after 40 minutes and the cooling bath removed. Ammonia was allowed to evaporate overnight and the residue was quenched with H2O (50 mL), extracted with CH2Cl2 (3 x 50 mL) and dried over Na2SO4. The solvent was evaporated in vacuo, and subsequent flash column chromatography on silica gel (CH2Cl2 then CH2Cl2/AcOEt 9:1) afforded 6-diphenylphosphanyl-pyridin-2-yl-amine 43 as a yellow liquide (1.95 mg, 7.0 mmol, 60%). 1H NMR (499.870 MHz, C6D6): δ = 4.13 (br s, 2H, NH2), 5.72 (td, 3J3-4 = 8.3 Hz, 4J3-5 = 5J3-P
[105] M. Weis, C. Waloch, W. Seiche, B. Breit, J. Am. Chem. Soc. 2006, 128(13), 4188-4189. [106] Dr. W. Seiche (PhD manuscript, Breit group, 2009).
ExperimentalPart:SynthesisofSubstrates
191
9. Synthesis of substrates
9.1 Rhodium-catalyzed intermolecular hydroacylation of 1-octene with aliphatic aldehydes
THF, RT, o/n
[Rh(CO)2acac](0.007 mol%)
Phosphorus ligand(0.033 mol%)
O
H
CO/H2
49 80C9H10O
Mol. Wt.: 134,18C8H8
Mol. Wt.: 104,15
ligand 81: PPh3
ligand 82:
1)
2) P
C18H15PMol. Wt.: 262,29
C21H21PMol. Wt.: 304,37
3) ligand 83:
P
O
OO
C42H63O3PMol. Wt.: 646,92
3 phosphorus ligands screened:
(90% yield)
(70% yield)
(70% yield)
1
2
3
4
7
6
56
5
To a Schlenk tube under an atmosphere of argon was added [Rh(CO)2acac] (2.5 mg, 9.7 μmol, 0.007 mol%) and a phosphorus ligand (40 mg (81), 46 mg (82), 97 mg (83), 48.4 μmol, 0.033 mol%). Then THF (1.5 mL) and after 5 minutes styrene 49 (166 μL, 1.45 mmol) were added. The reaction mixture was saturated with synthesis gas applying three cycles of careful evacuation and refilling with synthesis gas [CO/H2 (1:1)]. The solution was magnetically stirred employing a cross-type stirring bar for 20 hours [22 °C, approximatively 1 bar CO/H2 (1:1)]. After full conversion, the solution was converted and the resulting crude product 2-phenyl-propionaldehyde 80 (175.7 mg (81), 1.31 mmol, 90%) and (136.2 mg (82 and 83), 1.01 mmol, 70%).
Analytic data are in accordance with the literature.[176]
[176] D. J. Vyas, E. Larionov, C. Besnard, L. Guénée, C. Mazet, J. Am . Chem. Soc. 2013, 135(16), 6177-6183.
ExperimentalPart:SynthesisofSubstrates
192
9.2 Synthesis of o-vinylbenzaldehyde 95
Br
94
1) n-BuLi, THF, 1 h, -78 °C2) DMF, 20 min, -78 °C
75%
95
O
H1
23
45 6
7
8
9
C8H7BrMol. Wt.: 183,05
C9H8OMol. Wt.: 132,16
To a solution of o-bromostyrene 94 (2 g, 1.37 mL, 10.9 mmol) in THF (40 mL) was added n-Buli (2.5 M solution in hexane, 4.82 mL, 12.0 mmol, 1.1 eq) at -78 °C. The reaction mixture was stirred at the same temperature for 1 hour to the yellowish suspension was added DMF (1.02 mL, 13.12 mmol, 1.2 eq) dropwise. After stirring the mixture at -78 °C for addition 20 minutes, saturated aqueous NH4Cl (40 mL) was added and the mixture was extracted with Et2O (3 x 30 mL). The combined organic layer was dried over magnesium sulfate and concentrated in vacuo. The residue was purified by column chromatography (cHex/AcOEt 50:1 to 20:1) to afford o-vinylbenzaldehyde 95 as a colorless oil (1.08 g, 8.2 mmol, 75%). 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 5.51 (dd, 3J9trans-8 = 11.0, 2J9trans-9cis = 1.2 Hz, 1H, H9trans), 5.70 (dd, 3J9cis-8 = 17.3, 2J9cis-9trans = 1.2 Hz, 1H, H9cis), 7.43 (m, 1H, Ar-H3), 7.52 (dd, 3J8-9cis = 17.3, 3J8,9trans = 11.0 Hz, 1H, H8), 7.54 (m, 2H, Ar-H4 and Ar-H5), 7.82 (dd, 3J2-
Mol. Wt.: 192,21 A solution of 6-bromoveratraldehyde 98 (0.4 g, 1.63 mmol), tributyl(vinyl)tin (475 µL, 1.63 mmol), [Pd(PPh3)4] (38 mg, 0.03 mmol) and 2,6-di-tert-butyl-p-cresol (56 mg, 0.25 mmol) in toluene (3 mL) was heated at 110 °C for 16 hours under an argon atmosphere in a sealed tube. After cooling to room temperature, H2O (3 mL) was added, followed by extraction with CH2Cl2 (3 x 10 mL). The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 6-vinylveratraldehyde 99 as a pale yellow solid (0.285 g, 1.49 mmol, 91%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 3.92 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 5.48 (dd, 3J9trans-8 = 11.0 Hz, 2J9trans-9cis = 1.1 Hz, 1H, H9trans), 5.62 (dd, 3J9cis-8 = 17.3 Hz, 2J9cis-
Mol. Wt.: 146,19 A solution of 2-bromo-4-methylbenzaldehyde 109 (0.39 g, 1.9 mmol), potassium vinyltrifluoroborate (0.26 g, 1.9 mmol, 1.0 eq), PdCl2 (6.9 mg, 0.04 mmol, 2 mol%), PPh3 (30.5 mg, 0.12 mmol, 0.06 eq) and Cs2CO3 (1.9 g, 5.7 mmol, 3.0 eq) in THF/H2O (9:1) (3 mL) was heated at 85 °C for 22 hours under an argon atmosphere in a sealed tube. After cooling to room temperature, H2O (3 mL) was added, followed by extraction with CH2Cl2 (3 x 10 mL). The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 4-methyl-2-vinylbenzaldehyde 110 as a pale yellow solid (0.218 g, 1.5 mmol, 77%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.42 (s, 3H, CH3), 5.48 (dd, 3J10trans-9 = 11.0 Hz, 2J10trans-10cis = 1.3 Hz, 1H, H10trans), 5.68 (dd, 3J10cis-9 = 17.4 Hz, 2J10cis-10trans = 1.3 Hz, 1H, H10cis), 7.23 (d, 3J5-6 = 8.1 Hz, 1H, Ar-H5), 7.36 (s, 1H, Ar-H3) 7.52 (dd, 2J9-10cis = 17.4 Hz, 2J9-10trans = 11.0 Hz, 1H, H9), 7.72 (d, 3J6-5 = 8.1 Hz, 1H, Ar-H6), 10.22 (s, 1H, H8). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 21.9 (s, 1C, C7), 119.1 (s, 1C, C10), 128.1 (s, 1C, C3), 128.9 (s, 1C, C5), 130.8 (s, 1C, C1), 131.7 (s, 1C, C6), 133.7 (s, 1C, C9), 140.6 (s, 1C, C2), 144.8 (s, 1C, C4), 192.1 (s, 1C, C8). HRMS (EI): Calcd. for C10H10O (M): 146.0732; Found: 146.0731 (difference -0.7 ppm).
ExperimentalPart:SynthesisofSubstrates
202
9.3.5 Synthesis of 1-vinylnaphthalene-2-benzaldehyde 114 9.3.5.1 Synthesis of 1-bromo-2-dibromomethylnaphthalene 112
1
87
65
43
2
10
9
NBS, AIBN every 3 hoursbenzene, 16 h, reflux
97%
Br Br
Br
Br11
111 112C11H9Br
Mol. Wt.: 221,09C11H7Br3
Mol. Wt.: 378,89
A mixture of 1-bromo-2-methylnaphthalene 111 (9.4 g, 42.5 mmol), N-bromosuccinimide (22.7 g, 127.5 mmol, 3.0 eq) and AIBN (30 mg) in anhydrous benzene (100 mL) was stirred and held at reflux for 16 hours. (AIBN (30 mg) was added every 3 hours) After cooling to room temperature, the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 1-bromo-2-dibromomethylnaphthalene 112 as a pale yellow solid (15.6 g, 41.2 mmol, 97%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 7.49 (s, 1H, CHBr2), 7.62 (m, 2H, Ar-H8 and Ar-H7), 7.84 (d, 3J6-7 = 8.3 Hz, 1H, Ar-H6), 7.90 (d, 3J4-3 = 8.9 Hz, 1H, Ar-H4), 8.07 (d, 3J3-4
= 8.9 Hz, 1H, Ar-H3), 8.31 (d, 3J9-8 = 8.3 Hz, 1H, Ar-H9). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 41.4 (s, 1C, C11), 119.7 (s, 1C, C1), 126.9 (s, 1C, C3), 128.1 (s, 1C, C7), 128.4 (s, 1C, C6 or C8 or C9), 128.5 (s, 1C, C6 or C8 or C9), 128.6 (s, 1C, C6 or C8 or C9), 129.2 (s, 1C, C4), 131.4 (s, 1C, C10), 134.8 (s, 1C, C5), 138.1 (s, 1C, C2). HRMS (EI): Calcd. for C11H7Br3 (M): 375.8098; Found: 375.8099 (difference +0.2 ppm). Tm = 89 °C Rf = 0.92 (CH2Cl2 pure) Analytic data are in accordance with the literature.[181]
[181] A. S. Demir, O. Reis, Tetrahedron 2004, 60, 3803-3812.
ExperimentalPart:SynthesisofSubstrates
203
9.3.5.2 Synthesis of 1-bromonaphthalene-2-carbaldehyde 113
112
Br
Br
Br
CaCO3,water 8 h, reflux
95%
1
87
65
43
2
10
9
Br
O
H11
113C11H7Br3
Mol. Wt.: 378,89C11H7BrO
Mol. Wt.: 235,08
A mixture of 1-bromo-2-dibromomethylnaphthalene 112 (15.6 g, 41.2 mmol) and CaCO3 (9.1 g, 90.6 mmol, 2.2 eq) in H2O (350 mL) was heated at reflux for 8 hours. It was then cooled to room temperature and extracted with AcOEt (2 x 150 mL). The organic layer was washed with water (100 mL), brine (100 mL) and dried over MgSO4. The solvent was removed in vacuo and the crude compound was purified by recrystallisation from Petroleum Ether/AcOEt (95:5) giving 1-bromonaphtalene-2-carbaldehyde 113 as a white solid (9.31 g, 39.6 mmol, 95%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 7.68 (m, 2H, Ar-H8 and Ar-H7), 7.85 (d, 3J6-7 = 8.7 Hz, 1H, Ar-H6), 7.88 (m, 1H, Ar-H4), 7.93 (d, 3J3-4 = 8.7 Hz, 1H, Ar-H3), 8.50 (m, 1H, Ar-H9), 10.67 (d, J = 0.9 Hz, 1H, H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 124.2 (s, 1C, C3), 128.3 (s, 1C, C7), 128.4 (s, 1C, C6 or C8 or C9), 128.5 (s, 1C, C6 or C8 or C9), 128.6 (s, 1C, C6 or C8 or C9), 129.9 (s, 1C, C4), 131.3 (s, 1C, C10), 131.5 (s, 1C, C1), 132.3 (s, 1C, C5), 137.4 (s, 1C, C2), 193.0 (s, 1C, C11). HRMS (EI): Calcd. for C11H7OBr (M): 233.9680; Found: 233.9677 (difference -1.3 ppm). Tm = 106 °C Rf = 0.46 (CH2Cl2/AcOEt 20:1) Analytic data are in accordance with the literature.[182]
[182] J. W. Grissom, D. Klingberg, S. Meyenburg, B. L. Stallman, J. Org. Chem. 1994, 59, 7876-7888.
ExperimentalPart:SynthesisofSubstrates
204
9.3.5.3 Synthesis of 1-vinylnaphthalene-2-carbaldehyde 114
Mol. Wt.: 182,22 A solution of 1-bromonaphtalene-2-carbaldehyde 113 (2 g, 8.51 mmol), tributyl(vinyl)tin (2.7 mL, 9.36 mmol, 1.1 eq) and [Pd(PPh3)4] (203 mg, 0.18 mmol, 2 mol%) in toluene (12 mL) was heated at 110 °C for 16 hours under an argon atmosphere in a sealed tube. After cooling to room temperature, H2O (12 mL) was added, followed by extraction with CH2Cl2 (3 x 20 mL). The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with Petroleum Ether/CH2Cl2, 1:1) to yield 1-vinylnaphtalene-2-carbaldehyde 114 as white solid (1.43 g, 7.83 mmol, 92%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 5.50 (dd, 3J13trans-12 = 17.6 Hz, 2J13trans,13cis = 1.6 Hz, 1H, H13trans), 6.02 (dd, 3J13cis-12 = 11.3 Hz, 2J13cis-13trans = 1.6 Hz, 1H, H13cis), 7.39 (dd, 2J12-13cis = 17.6 Hz, 3J12-13trans = 11.3 Hz, 1H, H12), 7.61 (m, 2H, HAr, Ar-H8 and Ar-H7), 7.84 (d, 3J6-7 = 8.5 Hz, 1H, Ar-H6), 7.88 (br d, 3J4-3 = 8.3 Hz, 1H, Ar-H4), 8.00 (d, 3J3-4 = 8.5 Hz, 1H, H3), 8.20 (br d, 3J9-8 = 8.3 Hz, 1H, H9), 10.46 (d, 4J11-3 = 0.9 Hz, 1H, H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 123.0 (s, 1C, C3), 126.0 (s, 1C, C9 or C13), 126.2 (s, 1C, C9 or C13), 127.0 (s, 1C, C8), 128.2 (s, 1C, C4 or C6 or C7), 128.6 (s, 1C, C4 or C6 or C7), 128.9 (s, 1C, C4 or C6 or C7), 130.7 (s, 1C, C12), 131.5 (s, 1C, C2 or C5), 131.7 (s, 1C, C2 or C5), 135.9 (s, 1C, C10), 143.4 (s, 1C, C1), 192.8 (s, 1C, C11). HRMS (EI): Calcd. for C13H10O (M): 182.0732; Found: 182.0732. Tm = 96 °C (lit: 74 °C) Analytic data are in accordance with the literature.[183]
[183] K. Tanaka, D. Hojo, T. Shoji, Y. Hagiwara, M.Hirano, Org. Lett. 2007, 9, 2059-2063.
ExperimentalPart:SynthesisofSubstrates
205
9.3.6 Synthesis of 4-nitro-2-vinylbenzaldehyde 118 9.3.6.1 Synthesis of 1-bromo-2-dibromomethyl-5-nitrobenzene 116
115
NBS, AIBN every 3 hoursbenzene, 16 h, reflux
27%
Br Br
Br
Br1 76
5
43
2
116C7H6BrNO2
Mol. Wt.: 216,03C7H4Br3NO2
Mol. Wt.: 373,82
O2N O2N
A mixture of 2-bromo-4-nitrotoluene 115 (15.0 g, 69.4 mmol), N-bromosuccinimide (37.0 g, 208.3 mmol, 3.0 eq) and AIBN (50 mg) in anhydrous benzene (200 mL) was stirred and held at reflux for 16 hours. (AIBN (50 mg) was added every 3 hours) After cooling to room temperature, the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with toluene) to yield 1-bromo-2-dibromomethyl-5-nitrobenzene 116 as a pale yellow solid (7.1 g, 19.0 mmol, 27%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 7.05 (s, 1H, CHBr2), 8.20 (d, 3J3-4 = 8.6 Hz, 1H, Ar-H3), 8.31 (dd, 3J4-3 = 8.6 Hz, 4J4-6 = 2.2 Hz, 1H, Ar-H5), 8.40 (d, 4J6-4 = 2.2 Hz, 1H, Ar-H6). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 37.4 (s, 1C, C7), 120.1 (s, 1C, C2), 123.4 (s, 1C, C4), 128.0 (s, 1C, C6), 132.2 (s, 1C, C3), 146.7 (s, 1C, C1), 148.2 (s, 1C, C5). HRMS (EI): Calcd. for C7H4O3NBr2 (M): 291.8609; Found: 291.8608 (difference -0.3 ppm). Tm = 85 °C Rf = 0.86 (Toluene pure)
ExperimentalPart:SynthesisofSubstrates
206
9.3.6.2 Synthesis of 2-bromo-4- nitrobenzaldehyde 117
116
Br
Br
Br
H2SO4, 1 h, RT
96%
Br
O
H1 76
5
43
2
117
C7H4Br3NO2Mol. Wt.: 373,82
C7H4BrNO3Mol. Wt.: 230,02
O2N O2N
1-bromo-2-dibromomethyl-5-nitrobenzene 116 (7.1 g, 19.0 mmol) in concentrated H2SO4 (70 mL) was heated for 3 hours at 60 °C. AcOEt (70 mL) was added and the aqueous phase was extracted with AcOEt (3 x 70 mL). The combined organic layers were washed with water (70 mL) and brine (70 mL) and dried over MgSO4. After evaporation of the solvent, the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 2-bromo-4-nitrobenzaldehyde 117 as a white solid (4.2 g, 18.2 mmol, 96%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 8.08 (d, 3J3-4 = 8.6 Hz, 1H, Ar-H3), 8.27 (dd, 3J4-
A solution of 2-bromo-4-nitrobenzaldehyde 117 (0.343 g, 1.5 mmol), potassium vinyltrifluoroborate (0.20 g, 1.5 mmol, 1.0 eq), PdCl2 (5.3 mg, 0.03 mmol, 2 mol%), PPh3 (23.5 mg, 0.09 mmol, 0.06 eq) and Cs2CO3 (1.5 g, 4.5 mmol, 3.0 eq) in THF/H2O (9:1) (3 mL) was heated at 85 °C for 22 hours under an argon atmosphere in a sealed tube. After cooling to room temperature, H2O (3 mL) was added, followed by extraction with CH2Cl2 (3 x 10 mL). The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 4-nitro-2-vinylbenzaldehyde as a white solid 118 (0.193 g, 1.1 mmol, 73%). 1H NMR (300.070 MHz, CDCl3): δ (ppm) = 5.71 (d, 3J9trans-8 = 11.1 Hz, 1H, H9trans), 5.88 (d, 3J9cis-8 = 17.7 Hz, 1H, H9cis), 7.51 (dd, 3J8-9cis = 17.7 Hz, 3J8-9trans = 11.1 Hz, 1H, H8), 8.01 (d, 3J3-4 = 8.5 Hz, 1H, Ar-H3), 8.23 (dd, 3J4-3 = 8.5 Hz, J = 2.2 Hz, 1H, Ar-H5), 8.41 (d, 4J6-4 = 2.2 Hz, 1H, Ar-H6), 10.40 (s, 1H, H7). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 122.4 (s, 1C, C9), 122.5 (s, 1C, C4), 122.7 (s, 1C, C6), 131.4 (s, 1C, C1), 132.0 (s, 1C, C8), 136.5 (s, 1C, C3), 142.0 (s, 1C, C2), 150.8 (s, 1C, C5), 190.5 (s, 1C, C7). HRMS (EI): Calcd. for C9H7O3N (M): 177.0426; Found: 177.0428 (difference +1.1 ppm). Tm = 46 °C
ExperimentalPart:SynthesisofSubstrates
208
9.3.7 Synthesis of 5-chloro-2-vinylbenzaldehyde 122 9.3.7.1 Synthesis of 1-bromo-2-dibromomethyl-4-chlorobenzene 120
119 120
NBS, AIBN every 3 hoursbenzene, 16 h, reflux
72%
Br Br
Br
Br1 76
5
43
2
Cl Cl
C7H6BrClMol. Wt.: 205,48
C7H4Br3ClMol. Wt.: 363,27
A mixture of 2-bromo-5-chlorotoluene 119 (12.5 g, 60.8 mmol), N-bromosuccinimide (32.5 g, 182.5 mmol, 3.0 eq) and AIBN (40 mg) in anhydrous benzene (150 mL) was stirred and held at reflux for 16 hours. (AIBN (40 mg) was added every 3 hours) After cooling to room temperature, the solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with toluene) to yield 1-bromo-4-chloro-2-dibromomethylbenzene 120 as a pale yellow solid (15.9 g, 43.8 mmol, 72%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 6.98 (s, 1H, CHBr2), 7.15 (dd, 3J4-3 = 8.6 Hz, 4J4-
Mol. Wt.: 150,15 A solution of 2-bromo-5-fluorobenzaldehyde 123 (0.39 g, 1.9 mmol), potassium vinyltrifluoroborate (0.26 g, 1.9 mmol, 1.0 eq), PdCl2 (6.9 mg, 0.04 mmol, 2 mol%), PPh3 (30.5 mg, 0.12 mmol, 0.06 eq) and Cs2CO3 (1.9 g, 5.7 mmol, 3.0 eq) in THF/H2O (9:1) (3 mL) was heated at 85 °C for 22 hours under an argon atmosphere in a sealed tube. After cooling to room temperature, H2O (3 mL) was added, followed by extraction with CH2Cl2 (3 x 10 mL). The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 5-fluoro-2-vinylbenzaldehyde 124 as a yellow oil (0.231 g, 1.54 mmol, 81%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 5.53 (d, 3J9trans-8 = 11.0 Hz, 1H, H9trans), 5.64 (d, 3J9cis,8 = 17.4 Hz, 1H, H9cis), 7.34 (td, 3J4-3 = 8.6 Hz, 3J4-F = 3.0 Hz, 1H, H4), 7.41 (dd, 3J8-
[186] P. Mukherjee, S. J. Roy, T. K. Sarkar, Org. Lett. 2010, 12, 2472-2475.
ExperimentalPart:SynthesisofSubstrates
212
9.3.9 Synthesis of 5-hydroxy-2-vinylbenzaldehyde 126
125
O
H1 76
5
43
29
8
HOHO 81%
O
Br
H
tributyl(vinyl)tin, Pd(PPh3)4toluene, 16 h, 110 °C
126C7H5BrO2
Mol. Wt.: 201,02C9H8O2
Mol. Wt.: 148,16 A solution of 2-bromo-5-hydroxybenzaldehyde 125 (0.2 g, 1.0 mmol), tributyl(vinyl)tin (324 µL, 1.1 mmol, 1.1 eq) and [Pd(PPh3)4] (26 mg, 0.02 mmol, 2 mol%) in toluene (6 mL) was heated at 110 °C for 16 hours under an argon atmosphere in a sealed tube. After cooling to room temperature, H2O (5 mL) was added, followed by extraction with CH2Cl2 (3 x 15 mL). The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with AcOEt/CH2Cl2 1:50) to yield 5-hydroxy-2-vinylbenzaldehyde 126 as a brown liquid (0.12 g, 0.81 mmol, 81%). 1H NMR (300.130 MHz, CDCl3): δ (ppm) = 5.44 (dd, 3J9trans-8 = 11.0 Hz, 2J9trans-9cis = 1.1 Hz, 1H, H9trans), 5.55 (br s, 1H, OH), 5.60 (dd, 3J9cis-8 = 17.3 Hz, 2J9cis-9trans = 1.1 Hz, 1H, H9cis), 7.07 (dd, 3J4-3 = 8.5 Hz, 3J4-6 = 2.9 Hz, 1H, Ar-H4), 7.28 (d, 4J6-4 = 2.9 Hz, 1H, Ar-H6), 7.40 (dd, 3J8-9cis = 17.3 Hz, 3J8-9trans = 11.0 Hz, 1H, H8), 7.48 (d, 4J3-4 = 8.5 Hz, 1H, Ar-H3), 10.26 (s, 1H, H7). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 115.8 (s, 1C, C6), 118.5 (s, 1C, C9), 121.8 (s, 1C, C4), 129.2 (s, 1C, C2), 129.4 (s, 1C, C3), 132.3 (s, 1C, C8), 134.0 (s, 1C, C1), 155.8 (s, 1C, C5), 192.0 (s, 1C, C7). HRMS (EI): C9H8O2 (M): 148.0524; Found: 148.0526 (difference +1.3 ppm).
ExperimentalPart:SynthesisofSubstrates
213
9.3.10 Synthesis of 2-vinylpyridine-3-carbaldehyde 129 9.3.10.1 Synthesis of 2-bromopyridine-3-carbaldehyde 128
127 128
N1
7
6
54 3
2N 75%Br Br
H
O1) LDA THF, 4 h, -78 °C2) DMF 2.5 h, -78 °C to RT
C5H4BrNMol. Wt.: 158,00
C6H4BrNOMol. Wt.: 186,01
Freshly distilled 2-bromopyridine 127 (16.5 g, 104 mmol) was added dropwise to a solution of LDA at -78 °C (prepared by the dropwise addition of n-BuLi (2.5 M in hexanes, 115 mmol, 46 mL, 1.1 eq) to a solution of i-Pr2NH (135 mmol, 19.1 mL, 1.3 eq) in THF (250 mL) at -78 °C and stirred for 20 minutes. After 4 hours at this temperature, freshly distilled DMF (16 mL) was added dropwise and the reaction mixture was left to stir for a further 30 minutes at -78 °C before being warmed to room temperature over 2 hours. Saturated aqueous NH4Cl (100 mL) was added to the reaction mixture, followed by extraction with Et2O (3 x 50 mL). The combined organic extracts were washed with brine (2 x 50 mL), dried over MgSO4 and concentrated in vacuo to yield a red oil, which was purified by silica gel chromatography (eluting with cHex/AcOEt 9:1) to yield 2-bromopyridine-3-carbaldehyde 128 as a yellow crystalline solid (14.6 g, 78.3 mmol, 75%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 7.43 (ddd, 3J5-4 = 7.9 Hz, 3J5-6 = 4.7 Hz, 5J5-7 = 0.9 Hz, 1H, Ar-H5), 8.17 (dd, 3J4-5 = 7.9 Hz, 4J4-6 = 2.2 Hz, 1H, Ar-H4), 8.56 (dd, 3J6-5 = 4.7 Hz, 4J6-4 = 2.2 Hz, 1H, Ar-H6), 10.33 (d, 4J7-4 = 0.9 Hz, 1H, H7). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 123.6 (s, 1C, C5), 130.7 (s, 1C, C3), 138.1 (s, 1C, C4), 145.5 (s, 1C, C2), 154.6 (s, 1C, C6), 191.2 (s, 1C, C7). HRMS (EI): C6H4ONBr (M): 184.9476; Found: 184.9472 (difference -2.2 ppm). Tm = 83 °C Rf = 0.30 (cHex/AcOEt 9:1) Analytic data are in accordance with the literature.[187]
[187] A. C. Spivey, L. Shukla, J. F. Hayler, Org. Lett. 2007, 9, 891-894.
ExperimentalPart:SynthesisofSubstrates
214
9.3.10.2 Synthesis of 2-vinylpyridine-3-carbaldehyde 129
128
N1
7
6
54 3
2N 71%Br
H
O
H
O
9
8
tributyl(vinyl)tin, Pd(PPh3)4THF/H2O (9:1), 22 h, 85 °C
129C6H4BrNO
Mol. Wt.: 186,01C8H7NO
Mol. Wt.: 133,15 A solution of 2-bromopyridine-3-carbaldehyde 128 (0.278 g, 1.5 mmol), potassium vinyltri-fluoroborate (0.20 g, 1.5 mmol, 1.0 eq), PdCl2 (5.3 mg, 0.03 mmol, 2 mol%), PPh3 (23.5 mg, 0.09 mmol, 0.06 eq) and Cs2CO3 (1.5 g, 4.5 mmol, 3.0 eq) in THF/H2O (9:1) (3 mL) was heated at 85 °C for 22 hours under an argon atmosphere in a sealed tube. After cooling to room temperature, H2O (3 mL) was added, followed by extraction with CH2Cl2 (3 x 10 mL). The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2) to yield 2-vinylpyridine-3-carbaldehyde 129 as a pale yellow solid (0.143 g, 1.1 mmol, 71%). 1H NMR (300.07 MHz, CDCl3): δ (ppm) = 5.76 (dd, 3J9trans-8 = 10.8 Hz, 2J9trans-9cis = 1.8 Hz, 1H, H9trans), 6.48 (dd, 3J9cis-8 = 17.0 Hz, 2J9cis-9trans = 1.8 Hz, 1H, H9cis), 7.35 (dd, 3J5-4 = 7.8 Hz, 3J5-6 = 4.8 Hz, 1H, H5), 7.58 (dd, 3J8-9cis = 17.0 Hz, 3J8-9trans = 10.8 Hz, 1H, Ar-H8), 8.12 (dd, 3J4-5 = 7.8 Hz, 4J4-6 = 1.9 Hz, 1H, Ar-H4), 8.8 (dd, 3J6-5 = 4.8 Hz, 4J6-4 = 1.9 Hz, 1H, Ar-H6), 10.38 (s, 1H, H7). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 122.9 (s, 1C, C5), 124.1 (s, 1C, C9), 128.2 (s, 1C, C3), 131.7 (s, 1C, C8), 138.2 (s, 1C, C4), 153.6 (s, 1C, C6), 156.5 (s, 1C, C2), 191.0 (s, 1C, C7). HRMS (EI): C8H7ON (M): 133.0528; Found: 133.0528.
ExperimentalPart:SynthesisofSubstrates
215
9.3.11 Synthesis of 2-vinylthiophene-3-carbaldehyde 134 9.3.11.1 Synthesis of thiophene-3-aldehyde ethylene acetal 131
130 131
ethylene glycol, pTSA,H2Obenzene, 26 h,
reflux with a Dean-Stark Trap
94 % 1
87
6
5
4 3
2S
H
O
S
H
OO
C5H4OSMol. Wt.: 112,15
C7H8O2SMol. Wt.: 156,20
A solution of thiophene-3-aldehyde 130 (9.6 g, 86 mmol), ethylene glycol (9.6 mL, 172mmol) and p-toluenesulfonic acid monohydrate (1.6 g, 8.6 mmol) in benzene (400 mL) was refluxed with a Dean-Stark Trap for 26 hours. The reaction was cooled to the room temperature and anhydrous Na2CO3 was added. The reaction mixture was extracted with water (2 x 200mL) and the aqueous phase extracted once with benzene (200 mL). The combined organic phases were dried over MgSO4 and evaporated to give thiophene-3-aldehyde ethylene acetal 131 as a yellow oily liquid (12.6 g, 94%). The crude was used without purification in the next step. 1H NMR (300.07 MHz, CDCl3): δ (ppm) = 4.03 (m, 2H, H8 or H7), 4.11 (m, 2H, H8 or H7), 6.18 (s, 1H, H6), 7.21 (dd, 3J4-5 = 5.0 Hz, 4J4-2 = 1.1 Hz, 1H, Ar-H4), 7.34 (dd, 3J5-4 = 4.8 Hz, 4J5-2 = 2.9 Hz, 1H, Ar-H5), 7.46 (td, 4J2-5 = 3.1 Hz, 4J2-4 = 0.7 Hz, 1H, Ar-H2). Analytic data are in accordance with the literature.[188]
[188] L. J. Nurkkala, R. O. Steen, S. J. Dunne, Synthesis 2006, 1295-1300.
ExperimentalPart:SynthesisofSubstrates
216
9.3.11.2 Synthesis of 2,3-thiophenedicarbaldehyde 3-(ethylene acetal) 132
131
1
7
6
5
4 3
2
S
H
OO1) n-BuLi (2.5 M in n-hexanes) THF, 30 min, 0 °C2) DMF, THF, 3 h, RT
45 % S
H
OO
H
O9
132C7H8O2S
Mol. Wt.: 156,20C8H8O3S
Mol. Wt.: 184,21
8
To an ice-cold solution of thiophene-3-aldehyde ethylene acetal 131 (10.4 g, 66.6 mmol) in anhydrous THF (100 mL) was added dropwise a solution of n-BuLi (27 mL of 2.5 M solution in n-hexane, 66.6 mmol, 1.0 eq) with stirring. After 30 minutes, dry DMF (6.1 mL, 133.2 mmol, 2.0 eq) in anhydrous THF (15 mL) was added. The ice bath was removed, and stirring was continued for 3 hours at room temperature. The solution was quenched with water and extracted with benzene. The extract was washed with brine, dried over anhydrous MgSO4, and evaporated under reduced pressure. The yellow oily residue was distilled to give 2,3-thiophenedicarbaldehyde-3-(ethylene acetal) 132 (5.5 g, 29.9 mmol, 45%). 1H NMR (300.07 MHz, CDCl3): δ (ppm) = 4.03 (m, 2H, H8 or H7), 4.11 (m, 2H, H8 or H7), 6.18 (s, 1H, H6), 7.2-7.4 (2 x d, 2H, Ar-H4 and Ar-H5), 8.95 (s, 1H, H9). Analytic data are in accordance with the literature.[189]
[189] S. Hibino, S. Kano, N. Mochizuki, E. Sugino, J. Org. Chem.. 1984, 49, 5006-5008.
ExperimentalPart:SynthesisofSubstrates
217
9.3.11.3 Synthesis of 2-vinylthiophene-3-carbaldehyde ethylene acetal 133
84 %
n-BuLi (2.5 M in n-hexanes)methyl triphenylphosphonium bromide
THF, 30 min, 0 °C; then o/n, RT
S
H
OO
H
O1
87
6
5
4 3
2
S
H
OO
9
10
132 133C8H8O3S
Mol. Wt.: 184,21C9H10O2S
Mol. Wt.: 182,24 A solution of 2,3-thiophenedicarbaldehyde-3-(ethylene acetal) 132 (2.4 g, 12.9 mmol) in anhydrous THF (20 mL) was added dropwise to an ice-cold solution of methyl triphenylphosphonium bromide (5.5 g, 15.5 mmol, 1.2 eq) and n-BuLi (6.1 mL of 2.5 M solution in n-hexane, 15.5 mmol, 1.2 eq) in anhydrous THF (20 mL). The mixture was stirred at room temperature overnight, quenched with brine, dried over anhydrous Na2SO4, and evaporated to dryness. The residue was purified by silica gel chromatography (eluting with CH2Cl2) to yield 2-vinylthiophene-3-carbaldehyde ethylene acetal 133 as a yellow oil (1.98 g, 10.9 mmol, 84%). 1H NMR (300.07 MHz, CDCl3): δ (ppm) = 4.03 (m, 2H, H8 or H7), 4.11 (m, 2H, H8 or H7), 5.48 (d, 3J10trans-9 = 9.3 Hz, 1H, H10trans), 5.81 (d, 3J10cis,9 = 17.6 Hz, 1H, H10cis), 5.95 (s, 1H, H6), 7.11 (d, 3J5-4 = 6.2 Hz, 1H, Ar-H5), 7.37 (d, 3J4-5 = 6.2 Hz, 1H, Ar-H4), 7.44 (dd, 2J9-10cis = 17.6 Hz, 2J9-10trans = 9.3 Hz, 1H, H9). Analytic data are in accordance with the literature.[189]
[189] S. Hibino, S. Kano, N. Mochizuki, E. Sugino, J. Org. Chem.. 1984, 49, 5006-5008.
ExperimentalPart:SynthesisofSubstrates
218
9.3.11.4 Synthesis of 2-vinylthiophene-3-carbaldehyde 134
54 %
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
S
H
OO
9 1
6
5
4 3
2
S
H
7
8
O
133 134C9H10O2S
Mol. Wt.: 182,24C7H6OS
Mol. Wt.: 138,19 A mixture of 2-vinylthiophene-3-carbaldehyde ethylene acetal 133 (1.1 g, 6 mmol) and KHSO4 (0.9 g, 6.64 mmol) in water (3 mL) and acetone (18 mL) was heated at 60°C with stirring for 4.5h. After removal of acetone and addition of water, the mixture was extracted with benzene, and the extract was washed with brine, dried over MgSO4, and evaporated under reduced pressure below 60 °C to give 2-vinylthiophene-3-carbaldehyde 134. The residue was purified by silica gel chromatography (eluting with CH2Cl2) to yield 2-vinylthiophene-3-carbaldehyde 84 as a yellow oil (450 mg, 3.3 mmol, 54%). The product must be stored in the fridge and in the darkness to avoid its dimerization.
1) CuI, 2,2'-bipyridine, toluene, 0 °C2) vinyl magnesium bromide, 4 h, 0 °C to RT Br
154
68%
2
45
6
31
9
8
7
C7H6Br2Mol. Wt.: 249,93
C9H9BrMol. Wt.: 197,07
In a 250 mL, one-neck flask was placed 2-bromobenzyl bromide 153 (4.2 g, 16.8 mmol), CuI (324 mg, 1.7 mmol) and 2,2’-bipyridil (266 mg, 1.7 mmol) in dry toluene (25 mL) at 0 °C. Vinyl magnesium bromide (0.7 mol.L-1, 103 mL, 72 mmol) was then slowly added to this mixture, then allowed to warm to room temperature and stirred for an additionnal 4 hours when TLC indicated the reaction was complete. The reaction was quenched with sat. NH4Cl solution (40 mL), extracted with diethyl ether (3 x 40 mL) and the combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was then purified by flash chromatography (hexanes/AcOEt 17:3) to yield 1-bromo-2-allylbenzene 154 (2.25 g, 11.4 mmol, 68%) as a yellowish liquid. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.56 (d, 3J7-8 = 7.4 Hz, 2H, CH2), 5.10-5.19 (m, 2H, H9), 5.97-6.07 (m, 1H, H8), 7.09-7.14 (m, 1H, H5), 7.23-7.31 (m, 2H, H3 and H4), 7.59 (m, 1H, H2). 13C {1H} NMR (100.620 MHz, CDCl3): δ (ppm) = 40.9 (s, 1C, C7), 116.6 (s, 1C, C9), 124.6 (s, 1C, C1), 127.5 (s, 1C, C3 or C4), 127.9 (s, 1C, C3 or C4), 130.5 (s, 1C, C5), 132.8 (s, 1C, C2), 135.6 (s, 1C, C8), 139.4 (s, 1C, C6). Rf = 0.64 (hexanes/AcOEt 17:3) Analytic data are in accordance with the literature.[190]
[190] I. D. G. Watson, S. Ritter, F. D. Toste, J. Am. Chem. Soc. 2009, 131(6), 2056-2057.
ExperimentalPart:SynthesisofSubstrates
220
9.4.2 Synthesis of o-allylbenzaldehyde 155
Br
154
1) n-BuLi, THF, 30 min., -78 °C2) DMF, THF, o/n, -78 °C to RT
155
O
H
58%
C9H9BrMol. Wt.: 197,07
C10H10OMol. Wt.: 146,19
24
56
3
1
987 10
In a oven dried, 100 mL round bottom flask equipped with a magnetic stir bar and septum under nitrogen, was placed 1-bromo-2-allylbenzene 154 (1.6 g, 8.2 mmol) and dry THF (8 mL). The flask was placed at -78 °C then n-BuLi (2.5 M in hexanes, 4.9 mL, 12.0 mmol) was added dropwise slowly and the resulting solution was stirred for 30 minutes. Then DMF (1.62 mL, 20.0 mmol) in dry THF (3 mL) was slowly added and the reaction mixture was slowly allowed to warm to room temperature overnight. The reaction was quenched by addition of sat. NH4Cl solution (10 mL), extracted with diethyl ether (3 x 8 mL) and the combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by flash chromatography (hexanes/AcOEt 17:3) to yield 2-allylbenzaldehyde 155 (695 mg, 4.7 mmol, 58%) as a clear liquid. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.82 (d, 3J7-8 = 6.4 Hz, 2H, CH2), 4.92 (dq, 3J10cis-9 = 17.1 Hz, 2J10cis-10trans =
[190] I. D. G. Watson, S. Ritter, F. D. Toste, J. Am. Chem. Soc. 2009, 131(6), 2056-2057.
ExperimentalPart:SynthesisofSubstrates
221
9.5 Synthesis of o-allylbenzaldehyde derivatives 9.5.1 Synthesis of 2-allyl-4-methyl-benzaldehyde 164 9.5.1.1 Synthesis of 2-(2-bromo-4-methyl-phenyl)-[1,3]-dioxolane 160
Br
H
O O
Br
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a
Dean-Stark trap
109 160
98%
24
56
3
1
8 7
Me Me
9
C8H7BrOMol. Wt.: 199,04
C10H11BrO2Mol. Wt.: 243,10
10
A solution of 2-bromo-4-methyl-benzaldehyde 109 (2.0 g, 10.1 mmol), ethylene glycol (4.18 mL, 60.6 mmol, 6.0 eq) and p-toluenesulfonic acid monohydrate (192 mg, 1.01 mmol, 10 mol%) in benzene (80 mL) was refluxed with a Dean-Stark apparatus for 26 hours. The reaction was cooled to the room temperature and anhydrous Na2CO3 was added. The reaction mixture was extracted with water (20 mL) and the aqueous phase extracted once with toluene (20 mL). The combined organic phases were dried over MgSO4 and evaporated to give 2-(2-bromo-4-methyl-phenyl)-[1,3]-dioxolane 160 as a yellow oily liquid (1.97 g, 9.9 mmol, 98%). The crude was used without purification in the next step. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 2.26 (s, 3H, CH3), 4.10-4-17 (m, 2H, H7 or H8), 4.17-4.24 (m, 2H, H7 or H8), 5.99 (s, 1H, H9), 7.03 (d, 3J5-6 = 8.3 Hz, 1H, Ar-H5), 7.32 (s, 1H, Ar-H3), 7.40 (d, 3J6-5 = 8.3 Hz, 1H, Ar-H6).
ExperimentalPart:SynthesisofSubstrates
222
9.5.1.2 Synthesis of 2-(2-allyl-4-methyl-phenyl)-[1,3]-dioxolane 163
Br
H
O O
Li
H
O O
MgBr
H
O O
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
160 161
162163
23%(after 3 steps)
H
O O
24
56
3
1
8 7
10
119
C9H8BrClO2Mol. Wt.: 263,52
C12H13ClO2Mol. Wt.: 224,68
Me Me
MeMe12
13
A solution of n-butyllithium in n-hexanes (2.5 M, 11.89 mmol, 4.75 mL, 1.2 eq) was added very slowly to a solution of 2-(2-bromo-4-methyl-phenyl)-[1,3]-dioxolane 160 (2.4 g, 9.91 mmol) in dry THF (60 mL) at -78 °C. After this had stirred for 2 hours at -78 °C, a solution of MgBr2 in diethyl ether, freshly prepared from Mg (463 mg, 19.82 mmol, 2.0 eq) and dibromoethane (1 mL, 11.9 mmol, 1.2 eq), was added slowly and the reaction mixture was allowed to warm to room temperature. After 2 h, allyl bromide (1.01 mL, 11.9 mmol, 1.2 eq) was added and the solution was refluxed for 2 hours. After cooling to room temperature, the solution was treated with a 1:1 mixture of saturated NH4Cl and water and extracted with diethyl ether (3 x 20 mL). The combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Flash chromatography (SiO2: cyclohexane/ethyl acetate, 20:1, then 10:1) afforded 2-(2-allyl-4-methyl-phenyl)-[1,3]-dioxolane 163 (520 mg, 2.3 mmol, 23%) as pale yellow oil, which crystallized from diethyl ether. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 2.26 (d, 3J9-10 = 6.7 Hz, 2H, CH2), 3.80 (d, 3J8-9 = 6.2 Hz, 2H, CH2), 3.98-4.08 (m, 2H, H7 or H8), 4.08-4.18 (m, 2H, H7 or H8), 5.03 (dq, 3J11cis-10 = 17.1 Hz, 2J11cis-11trans =
9.5.1.3 Synthesis of 2-allyl-4-methyl-benzaldehyde 164
24
5
6
3
1H
O O
H
O
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
163 164
quant.
1098
7
Me Me11
C13H16O2Mol. Wt.: 204,26
C11H12OMol. Wt.: 160,21
2-(2-allyl-4-methyl-phenyl)-[1,3]-dioxolane 163 (520 mg, 2.3 mmol) and KHSO4 (1 g, 7.34 mmol, excess) was stirred at room temperature for 4.5 hours in a mixture of acetone (20 mL) and H2O (3 mL). Then, the reaction mixture was diluted with a saturated solution of NaHCO3, and extracted with AcOEt (4 x 10 mL) : The combined organic phases were dried over MgSO4. The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2/AcOEt 9:1) to obtain 2-allyl-4-methyl-benzaldehyde 164 (368 mg, 2.3 mmol, quant.) as a colorless oil. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 2.42 (s, 2H, CH3), 3.80 (d, 3J8-9 = 6.2 Hz, 2H, CH2), 5.00 (dq, 3J10cis-9 = 17.1 Hz, 2J10cis-10trans =
9.5.2 Synthesis of 2-allyl-5-methoxy-benzaldehyde 170 9.5.2.1 Synthesis of 2-(2-bromo-5-methoxy-phenyl)-[1,3]-dioxolane 166
Br
H
O O
Br
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a
Dean-Stark trap
165 166
quant.
24
5
6
3
1
8 7
9
C8H7BrOMol. Wt.: 199,04
C10H11BrO2Mol. Wt.: 243,10
10MeO MeO
A solution of 2-bromo-4-methoxy-benzaldehyde 165 (5 g, 23.4 mmol), ethylene glycol (7.83 mL, 140.4 mmol, 6.0 eq) and p-toluenesulfonic acid monohydrate (445 mg, 2.34 mmol, 10 mol%) in benzene (180 mL) was refluxed with a Dean-Stark apparatus for 26 hours. The reaction was cooled to the room temperature and anhydrous Na2CO3 was added. The reaction mixture was extracted with water (60 mL) and the aqueous phase extracted once with toluene (60 mL). The combined organic phases were dried over MgSO4 and evaporated to give 2-(2-bromo-5-methoxy-phenyl)-[1,3]-dioxolane 166 as a yellow oily liquid (5.69 g, 23.4 mmol, quant.). The crude was used without purification in the next step. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.76 (s, 3H, OCH3), 3.98-4.06 (m, 2H, H7 or H8), 4.07-4.15 (m, 2H, H7 or H8), 6.02 (s, 1H, H10), 6.76 (dd, 3J4-3 = 8.7 Hz , 4J4-6 = 3.1 Hz, 1H, H4), 7.15 (d, 4J6-4 = 3.1 Hz, 1H, H3), 7.41 (d, 3J3-4 = 8.7 Hz, 1H, H6). 13C {1H} NMR (100.620 MHz, CDCl3): δ (ppm) = 55.36 (s, 1C, C9), 65.32 (s, 2C, C7 and C8), 102.33 (s, 1C, C10), 113.00 (C2), 113.05 (C4 or C6), 116.48 (C4 or C6), 133.48 (C3), 137.39 (C1), 158.92 (C5). HRMS (EI): Calcd. for C10H12
79BrO3 (M): 258.9970; Found: 258.9968 (difference +0.7 ppm) Analytic data are in accordance with the literature.[191]
[191] C. Che, S. Li, Z. Yu, F. Li, S. Xin, L. Zhou, S. Lin, Z. Yang, ACS Comb. Sci. 2013, 15(4), 202-207.
ExperimentalPart:SynthesisofSubstrates
225
9.5.2.2 Synthesis of 2-(2-allyl-5-methoxy-phenyl)-[1,3]-dioxolane 169
Br
H
O O
Li
H
O O
MgBr
H
O O
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
166 167
168169
29%(after 3 steps)
H
O O
24
5
6
3
1
8 7
10
119
12
C10H11BrO3Mol. Wt.: 259,10
C13H16O3Mol. Wt.: 220,26
MeO MeO
MeOMeO
A solution of n-butyllithium in n-hexanes (2.5 M, 13.9 mmol, 5.6 mL, 1.2 eq) was added very slowly to a solution of 2-(2-bromo-5-methoxy-phenyl)-[1,3]-dioxolane 166 (3 g, 11.6 mmol) in dry THF (70 mL) at -78 °C. After this had stirred for 2 h at -78 °C, a solution of MgBr2 in diethyl ether, freshly prepared from Mg (543 mg, 29.7 mmol, 2.56 eq) and dibromoethane (1.2 mL, 18.6 mmol, 1.6 eq), was added slowly and the reaction mixture was allowed to warm to room temperature. After 2 hours, allyl bromide (1.2 mL, 13.9 mmol, 1.2 eq) and the solution was refluxed for 2 hours. After cooling to room temperature, the solution was treated with a 1:1 mixture of saturated NH4Cl and water and extracted with diethyl ether (3 x 20 mL). The combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Flash chromatography (SiO2: cyclohexane/ethyl acetate, 20:1, then 10:1) afforded 2-(2-allyl-5-methoxy-phenyl)-[1,3]-dioxolane 169 (750 mg, 3.4 mmol, 29%) as pale yellow oil, which crystallized from diethyl ether. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.52 (d, 3J9-10 = 6.2 Hz, 4J9-11 = 1.2 Hz, 2H, CH2), 3.78 (s, 3H, OCH3), 3.99-4.07 (m, 2H, H7 or H8), 4.08-4.16 (m, 2H, H7 or H8), 5.00-5.11 (m, 2H, H11), 5.94-6.14 (m, 1H, H10), 7.03 (s, 1H, Ar-H6), 7.07 (d, 3J4-3 = 8.1 Hz, 1H, Ar-H4), 7.47 (d, 3J3-4 = 8.1 Hz, 1H, Ar-H3).
ExperimentalPart:SynthesisofSubstrates
226
9.5.3 Synthesis of 2-allyl-5-methoxy-benzaldehyde 170 9.5.3.1 Synthesis of 2-(2-bromo-5-fluoro-phenyl)-[1,3]-dioxolane 171
Br
H
O O
Br
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a
Dean-Stark trap
123 171
F F
C7H4BrFOMol. Wt.: 203,01
C9H8BrFO2Mol. Wt.: 247,06
quant.
24
5
6
3
1
8 7
9
A solution of 2-bromo-5-fluoro-benzaldehyde 123 (5 g, 24.8 mmol), ethylene glycol (8.3 mL, 148.8 mmol, 6 eq) and p-toluenesulfonic acid monohydrate (471 mg, 2.48 mmol, 10 mol%) in benzene (200 mL) was refluxed with a Dean-Stark apparatus for 26 hours. The reaction was cooled to the room temperature and anhydrous Na2CO3 was added. The reaction mixture was extracted with water (60 mL) and the aqueous phase extracted once with toluene (60 mL). The combined organic phases were dried over MgSO4 and evaporated to give 2-(2-bromo-5-fluoro-phenyl)-[1,3]-dioxolane 171 as a yellow oily liquid (6.13 g, 24.8 mmol, quant.). The crude was used without purification in the next step. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 4.10-4-17 (m, 2H, H7 or H8), 4.17-4.24 (m, 2H, H7 or H8), 5.97 (d, 4J9-7 and 8 = 1.2 Hz, 1H, H9), 6.92 (td, 3J4-3 = 9.1 Hz, 3J4-F = 4J4-6 = 3.0 Hz, 1H, H4), 7.31 (dd, 1H, H6), 7.49 (dd, 1H, H3). 13C {1H} NMR (100.620 MHz, CDCl3): δ (ppm) = 65.48 (s, 2C, C7 and C8), 101.95 (s, 1C, C9), 115.12 (d, 2JC-F = 34.3 Hz, 1C, C6), 116.82 (d, 4JC-F = 3.3 Hz, 1C, C2), 117.70 (d, 2JC-F = 22.7 Hz, 1C, C4), 134.22 (d, 3JC-F = 7.7 Hz, 1C, C3), 139.00 (d, 3JC-F = 6.8 Hz, 1C, C1), 161.94 (d, 1JC-F = 247.3 Hz, 1C, C5). HRMS (EI): Calcd. for C9H9
9.5.3.2 Synthesis of 2-(2-allyl-5-fluoro-phenyl)-[1,3]-dioxolane 174
Br
H
O O
Li
H
O O
MgBr
H
O O
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
allyl bromide2 h, reflux (70 °C)
171 172
173174
F F
F
C9H8BrFO2Mol. Wt.: 247,06
61%(after 3 steps)
C12H13FO2Mol. Wt.: 208,23
H
O OF
24
5
6
3
1
8 7
10
119
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
12
A solution of n-butyllithium in n-hexanes (2.5 M, 12.19 mmol, 4.87 mL, 1.2 eq) was added very slowly to a solution of 2-(2-bromo-5-fluoro-phenyl)-[1,3]-dioxolane 171 (2.5 g, 10.16 mmol) in dry THF at -78 °C. After this had stirred for 2 hours at -78 °C, a solution of MgBr2 in diethyl ether, freshly prepared from Mg (475 mg, 19.51 mmol. 1.9 eq) and dibromoethane (1 mL, 11.18 mmol, 1.1 eq), was added slowly and the reaction mixture was allowed to warm to room temperature. After 2 hours, allyl bromide (1.03 mL, 12.19 mmol, 1.2 eq) was added and the solution was refluxed for 2 hours. After cooling to room temperature, the solution was treated with a 1:1 mixture of saturated NH4Cl and water and extracted with diethyl ether (3 x 30 mL). The combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Flash chromatography (SiO2: cyclohexane/ethyl acetate, 10:1, then 4:1) afforded 2-(2-allyl-5-fluoro-phenyl)-[1,3]-dioxolane 174 (1.3 g, 6.24 mmol, 61%) as pale yellow oil, which crystallized from diethyl ether. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.49 (d, 3J9-10 = 6.1 Hz, 2H, CH2), 4.00-4.08 (m, 2H, H7 or H8), 4.08-4.16 (m, 2H, H7 or H8), 5.00 (dq, 3J11cis-10 = 17.1 Hz, 2J11cis-11trans =
9.5.3.3 Synthesis of 2-allyl-5-fluoro-benzaldehyde 175
24
5
6
3
1H
O O
H
O
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
174 175
F F88%
C12H13FO2Mol. Wt.: 208,23
C10H9FOMol. Wt.: 164,18
1098
7
2-(2-allyl-5-fluoro-phenyl)-[1,3]-dioxolane 174 (1.3 g, 6.25 mmol) and KHSO4 (1.02 g, 7.5 mmol, 1.2 eq) was stirred at room temperature for 4.5 hours in a mixture of acetone (20 mL) and H2O (3 mL). Then, the reaction mixture was diluted with a saturated solution of NaHCO3, and extracted with AcOEt (4 x 10 mL). The combined organic phases were dried over MgSO4. The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2/AcOEt 90:10) to obtain 2-allyl-5-fluoro-benzaldehyde 175 (903 mg, 5.5 mmol, 88%) as a colorless oil. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.70 (d, 3J8-9 = 6.1 Hz, 2H, CH2), 4.88 (dq, 3J10cis-9 = 17.1 Hz, 2J10cis-10trans =
[84] Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi, S. Ogoshi, Angew. Chemie 2012, 124(43), 10970-10973; Angew. Chemie Int. Ed. 2012, 51(43), 10812-10815.
ExperimentalPart:SynthesisofSubstrates
229
9.5.4 Synthesis of 2-allyl-naphthalene-1-carbaldehyde 180
9.5.4.1 Synthesis of 2-(2-bromo-naphthalen-1-yl)-[1,3]-dioxolane 176
Br
H
O O
Br
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a
Dean-Stark trap
113 176
quant.
24
5
3
1
8
13
69
10
1112
C11H7BrOMol. Wt.: 235,08
C13H11BrO2Mol. Wt.: 279,13
7
A solution of 2-bromo-naphthalene-1-carbaldehyde 113 (1.10 g, 4.68 mmol), ethylene glycol (0.53 mL, 9.36 mmol, 2.0 eq) and p-toluenesulfonic acid monohydrate (91 mg, 0.47 mmol, 10 mol%) in benzene (40 mL) was refluxed with a Dean-Stark apparatus for 26 hours. The reaction was cooled to the room temperature and anhydrous Na2CO3 was added. The reaction mixture was extracted with water (40 mL) and the aqueous phase extracted once with toluene (40 mL). The combined organic phases were dried over MgSO4 and evaporated to give 2-(2-bromo-naphthalen-1-yl)-[1,3]-dioxolane 176 as a pale yellow oily liquid (1.30 g, 4.68 mmol, quant.). The crude was used without purification in the next step. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 4.12-4.32 (m, 4H, H7 and H8), 6.43 (s, 1H, H13), 7.52-7.64 (m, 2H, Ar-H), 7.69 (d, 3J = 8.6 Hz, 1H, Ar-H3), 7.83 (m, 2H, Ar-H), 8.39 (d, 3J = 8.6 Hz, 1H, Ar-H4). 13C {1H} NMR (100.620 MHz, CDCl3): δ (ppm) = 65.6 (s, 2C, C7 and C8), 103.4 (s, 1C, C13), 123.9 (s, 1C, C2 or C12), 124.1 (s, 1C, C2 or C12), 127.1 (CAr), 127.5 (CAr), 127.9 (CAr), 128.1 (s, 1C, C4 or C6), 132.1 (s, 1C, C4 or C6), 134.5 (s, 1C, C5), 134.9 (s, 1C, C1). Analytic data are in accordance with the literature.[192]
[192] J. Clayden, C. McCarthy, N. Westlund, C. S. Frampton, J. Chem. Soc., Perkin Trans. 1 2000, 1363-1378.
ExperimentalPart:SynthesisofSubstrates
230
9.5.4.2 Synthesis of 2-(2-allyl-naphthalen-1-yl)-[1,3]-dioxolane 179
Br
H
O O
Li
H
O O
MgBr
H
O O
H
O O
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
176 177
178179
98%(after 3 steps)
C13H11BrO2Mol. Wt.: 279,13
C16H16O2Mol. Wt.: 240,30
24
5
6
3
1
8
10
119
1514
13
12
7
16
A solution of n-butyllithium in n-hexanes (2.5 M, 1.33 mL, 3.33 mmol, 1.2 eq) was added very slowly to a solution of 2-(2-bromo-naphthalen-1-yl)-[1,3]-dioxolane 176 (770 mg, 2.77 mmol) in dry THF at -78 °C. After this had stirred for 2 hours at -78 °C, a solution of MgBr2 in diethyl ether, freshly prepared from Mg (97 mg, 4 mmol) and dibromoethane (0.27 mL, 3.05 mmol, 1.1 eq), was added slowly and the reaction mixture was allowed to warm to room temperature. After 2 hours, allyl bromide (0.29 mL, 3.33 mmol, 1.2 eq) was added and the solution was refluxed for 2 hours. After cooling to room temperature, the solution was treated with a 1:1 mixture of saturated NH4Cl and water and extracted with diethyl ether (3 x 15 mL). The combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Flash chromatography (SiO2: cyclohexane/ethyl acetate, 20:1, then 10:1) afforded 2-(2-allyl-naphthalen-1-yl)-[1,3]-dioxolane 179 (650 mg, 2.71 mmol, 98%) as pale yellow oil, which crystallized from diethyl ether. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 4.01 (dt, 3J9-10 = 5.5 Hz, 4J9-11 = 1.9 Hz, 2H, CH2), 4.15-4.24 (m, 4H, H7 and H8), 4.95 (dq, 3J11cis-10 = 17.3 Hz, 2J11cis-11trans =
9.5.4.3 Synthesis of 2-allyl-naphthalene-1-carbaldehyde 180
H
O O
KHSO4, H2O / acetone (1 : 5)4.5 h, 60 °C
179 180
97%
C16H16O2Mol. Wt.: 240,30
H
24
5
6
3
1 12
1311
78
9
10
O14
C14H12OMol. Wt.: 196,24
2-(2-allyl-naphthalen-1-yl)-[1,3]-dioxolane 179 (650 mg, 2.71 mmol) and KHSO4 (500 mg, 3.7 mmol, 1.35 eq) was stirred at room temperature for 4.5 hours in a mixture of acetone (10 mL) and H2O (2 mL). Then, the reaction mixture was diluted with a saturated solution of NaHCO3 (5 mL), and extracted with AcOEt (4 x 15 mL). The combined organic phases were dried over MgSO4. The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2/AcOEt 9:1) to obtain 2-allyl-naphthalene-1-carbaldehyde 180 (514 mg, 2.62 mmol, 97%) as a colorless oil. 1H-NMR (400.130 MHz, CDCl3): δ (ppm) = 4.30 (dt, 3J11-12 = 6.4 Hz, 4J11-13 = 1.7 Hz, 2H, CH2), 4.93 (dq, 3J13cis-12 = 17.1 Hz, 2J13cis-13trans =
9.5.5 Synthesis of 2-allyl-4,5-dimethoxy-benzaldehyde 185 9.5.5.1 Synthesis of 2-(2-bromo-4,5-dimethoxy-phenyl)-[1,3]-dioxolane 181
Br
H
O O
Br
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26 hreflux with a
Dean-Stark trap
98 181
76%
24
6
3
1
8 7
MeO MeO9
MeO MeO10
C9H9BrO3Mol. Wt.: 245,07
C11H13BrO4Mol. Wt.: 289,12
5 11
A solution of 2-bromo-4,5-dimethoxy-benzaldehyde 98 (3 g, 12.3 mmol), ethylene glycol (1.40 mL, 24.6 mmol, 2.0 eq) and p-toluenesulfonic acid monohydrate (242 mg, 1.25 mmol, 10 mol%) in benzene (120 mL) was refluxed with a Dean-Stark apparatus for 26 hours. The reaction was cooled to the room temperature and anhydrous Na2CO3 was added. The reaction mixture was extracted with water (40 mL) and the aqueous phase extracted once with toluene (40 mL). The combined organic phases were dried over MgSO4 and evaporated to give 2-(2-bromo-4,5-dimethoxy-phenyl)-[1,3]-dioxolane 181 as a yellow oily liquid (2.69 g, 9.3 mmol, 76%). Purification by silica gel chromatography (eluting with toluene/petroleum ether 2:1). 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.86 (s, 3H, OCH3(9) or OCH3(10)), 3.88 (s, 3H, OCH3(9) or OCH3(10)), 4.17 (m, 2H, H7 and H8), 5.98 (s, 1H, H11), 7.00 (s, 1H, H4), 7.31 (s, 1H, H6), 7.49 (s, 1H, H3). 13C {1H} NMR (100.620 MHz, CDCl3): δ (ppm) = 56.0 (s, 1C, C9 or C10), 56.2 (s, 1C, C9 or C10), 65.3 (s, 2C, C7 and C8), 102.6 (s, 1C, C11), 110.1 (s, 1C, C2), 113.3 (s, 1C, C6), 115.3 (s, 1C, C3), 128.2 (s, 1C, C1), 148.4 (s, 1C, C5), 150.1 (s, 1C, C4). Analytic data are in accordance with the literature.[191]
[191] C. Che, S. Li, Z. Yu, F. Li, S. Xin, L. Zhou, S. Lin, Z. Yang, ACS Comb. Sci. 2013, 15(4), 202-207.
ExperimentalPart:SynthesisofSubstrates
233
9.5.5.2 Synthesis of 2-(2-allyl-4,5-dimethoxy-phenyl)-[1,3]-dioxolane 184
Br
H
O O
Li
H
O O
MgBr
H
O O
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
181 182
183184
43%(after 3 steps)
H
O O
24
5
6
3
1
8 7
10
119
MeO MeO
MeOMeO12
MeO MeO
MeOMeO13
C11H13BrO4Mol. Wt.: 289,12
C14H18O4Mol. Wt.: 250,29
A solution of n-butyllithium in n-hexanes (1.6 M, 8.9 mL, 22.24 mmol, 2.0 eq) was added very slowly to a solution of 2-(2-bromo-5-chloro-phenyl)-[1,3]-dioxolane 181 (3.2 g, 11.12 mmol) in dry THF (110 mL) at -78 °C. After this had stirred for 2 hours at -78 °C, a solution of MgBr2 in diethyl ether, freshly prepared from Mg (432 mg, 17.78 mmol, 1.6 eq) and dibromoethane (1.2 mL, 13.9 mmol, 1.25 eq), was added slowly and the reaction mixture was allowed to warm to room temperature. After 2 hours, allyl bromide (1.17 mL, 13.4 mmol, 1.2 eq) was added and the solution was refluxed for 2 hours. After cooling to room temperature, the solution was treated with a 1:1 mixture of saturated NH4Cl and water and extracted with diethyl ether (3 x 30 mL). The combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Flash chromatography (SiO2: cyclohexane/ethyl acetate, 20:1, then 10:1) afforded 2-(2-allyl-5-chloro-phenyl)-[1,3]-dioxolane 184 (1.19 g, 4.78 mmol, 43%) as pale yellow oil, which crystallized from diethyl ether. The resulting 2-(2-allyl-4,5-dimethoxy-phenyl)-[1,3]-dioxolane 184 was directly engaged without further NMR analyses to the next step.
ExperimentalPart:SynthesisofSubstrates
234
9.5.5.3 Synthesis of 2-allyl-4,5-dimethoxy-benzaldehyde 185
24
5
6
3
1H
O O
H
O
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
184 185
quant.
1098
7
MeO MeO11
MeO MeO12
C14H18O4Mol. Wt.: 250,29
C12H14O3Mol. Wt.: 206,24
2-(2-allyl-4,5-dimethoxy-phenyl)-[1,3]-dioxolane 184 (2 g, 6.9 mmol) and KHSO4 (1 g, 7.34 mmol, 1.06 eq) was stirred at room temperature for 4.5 hours in a mixture of acetone (20 mL) and H2O (3 mL). Then, the reaction mixture was diluted with a saturated solution of NaHCO3, and extracted with AcOEt (4 x 10 mL). The combined organic phases were dried over MgSO4. The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2/AcOEt 9:1) to obtain 2-allyl-4,5-dimethoxy-benzaldehyde 185 (1.42 g, 6.9 mmol, quant.) as a colorless oil. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.72 (d, 3J8-9 = 6.0 ppm, 2H, CH2), 3.88 (s, 1H, OCH3), 3.91 (s, 1H, OCH3), 4.95 (dq, 3J10cis-9 = 17.1 Hz, 2J10cis-10trans =
4J10trans-9 = 1.1 Hz, 1H, H10trans), 5.91-6.07 (m, 1H, H9), 6.68 (s, 1H, Ar-H3), 7.35 (s, 1H, Ar-H6), 10.13 (s, 1H, H7). Analytic data are in accordance with the literature.[193]
[193] N. S. Narashiman, T. Mukhopadhyay, S. S. Kusurhar, Indian J. of Chem., section B : Organic Chemistry including Medicinal Chemistry 1981, 20(7), 546-548.
ExperimentalPart:SynthesisofSubstrates
235
9.5.6 Synthesis of 2-allyl-5-chloro-benzaldehyde 190
9.5.6.1 Synthesis of 2-(2-bromo-5-chloro-phenyl)-[1,3]-dioxolane 186
9
Br
H
O O
Br
H
O
ethylene glycolpTSA, H2O (cat.)
benzene, 26hreflux with a
Dean-Stark trap
121 186
Cl Clquant.
24
5
6
3
1
8 7
C7H4BrClOMol. Wt.: 219,46
C9H8BrClO2Mol. Wt.: 263,52
9
A solution of 2-bromo-5-chloro-benzaldehyde 121 (2.35 g, 10.8 mmol), ethylene glycol (1.25 mL, 21.6 mmol, 2.0 eq) and p-toluenesulfonic acid monohydrate (213 mg, 1.08 mmol, 10 mol%) in benzene (120 mL) was refluxed with a Dean-Stark apparatus for 26 hours. The reaction was cooled to the room temperature and anhydrous Na2CO3 was added. The reaction mixture was extracted with water (40 mL) and the aqueous phase extracted once with toluene (40 mL). The combined organic phases were dried over MgSO4 and evaporated to give 2-(2-bromo-5-chloro-phenyl)-[1,3]-dioxolane 186 as a yellow oily liquid (2.85 g, 10.8 mmol, quant.). The crude was used without purification in the next step. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.96-4.05 (m, 2H, H7 or H8), 4.05-4.15 (m, 2H, H9 or H10), 6.00 (s, 1H, H9), 7.16 (dd, 3J3-4 = 8.1 Hz, 4J6-4 = 2.7 Hz, 1H, 1H, Ar-H4), 7.44 (d, 3J3-4 = 8.1 Hz, 1H, Ar-H3), 7.56 (d, 4J6-4 = 2.7 Hz, 1H, Ar-H6). 13C {1H} NMR (100.620 MHz, CDCl3): δ (ppm) = 65.65 (C7 and C8), 102.13 (C9), 120.77 (C2), 128.15 (C6), 130.68 (C4), 133.78 (C5), 134.17 (C3), 138.55 (C1). HRMS (EI): Calcd. for C9H9O2ClBr (M): 262.9474; Found: 262.9471 (difference +1.3 ppm) Analytic data are in accordance with the literature.[191]
[191] C. Che, S. Li, Z. Yu, F. Li, S. Xin, L. Zhou, S. Lin, Z. Yang, ACS Comb. Sci. 2013, 15(4), 202-207.
ExperimentalPart:SynthesisofSubstrates
236
9.5.6.2 Synthesis of 2-(2-allyl-5-chloro-phenyl)-[1,3]-dioxolane 189
Br
H
O O
Li
H
O O
MgBr
H
O O
n-BuLi (2.5 M in n-hexanes)THF, 2 h, -78 °C.
MgBr2 freshly preparedTHF, 2 h, -78 °C to 0 °C
allyl bromide2 h, reflux (70 °C)
186 187
188189
Cl Cl
Cl20%(after 3 steps)
H
O OCl
24
5
6
3
1
8 7
10
119
C9H8BrClO2Mol. Wt.: 263,52
C12H13ClO2Mol. Wt.: 224,68
A solution of n-butyllithium in n-hexanes (2.5 M, 26.6 mmol, 12.7 mL, 1.2 eq) was added very slowly to a solution of 2-(2-bromo-5-chloro-phenyl)-[1,3]-dioxolane 186 (26.6 mmol, 7 g) in dry THF (240 mL) at -78 °C. After this had stirred for 2 hours at -78 °C, a solution of MgBr2 in diethyl ether, freshly prepared from Mg (1.65 g, 68.2 mmol, 2.56 eq) and dibromoethane (3.67 mL, 42.6 mmol, 1.6 eq), was added slowly and the reaction mixture was allowed to warm to room temperature. After 2 hours, allyl bromide (2.35 mL, 26.6 mmol, 1.2 eq) and the solution was refluxed for 2 hours. After cooling to room temperature, the solution was treated with a 1:1 mixture of saturated NH4Cl and water and extracted with diethyl ether (3 x 40 mL). The combined organic phases were washed with brine, dried (MgSO4), and concentrated in vacuo. Flash chromatography (SiO2: cyclohexane/ethyl acetate, 20:1, then 10:1) afforded 2-(2-allyl-5-chloro-phenyl)-[1,3]-dioxolane 189 (1.2 g, 5.34 mmol, 20%) as pale yellow oil, which crystallized from diethyl ether. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.49 (dt, 3J9-10 = 6.4 Hz, 4J9-11 = 1.6 Hz, 2H, CH2), 4.00-4.08 (m, 2H, H7 or H8), 4.08-4.16 (m, 2H, H7 or H8), 5.00 (dq, 3J11cis-10 = 17.2 Hz, 2J11cis-11trans =
9.5.6.3 Synthesis of 2-allyl-5-chloro-benzaldehyde 190
24
5
6
3
1H
O O
H
O
KHSO4, H2O / acetone (1 : 6)4.5 h, 60 °C
189 190
Cl Cl95%
1098
7
C12H13ClO2Mol. Wt.: 224,68
C10H9ClOMol. Wt.: 180,63
2-(2-allyl-5-chloro-phenyl)-[1,3]-dioxolane 189 (1.2 g, 5.34 mmol) and KHSO4 (837 mg, 6.14 mmol, 1.15 eq) was stirred at room temperature for 4.5 hours in a mixture of acetone (24 mL) and H2O (4 mL). Then, the reaction mixture was diluted with a saturated solution of NaHCO3, and extracted with AcOEt (4 x 10 mL). The combined organic phases were dried over MgSO4. The solvent was removed in vacuo and the crude product was purified by silica gel chromatography (eluting with CH2Cl2/AcOEt 20:1) to obtain 2-allyl-5-chloro-benzaldehyde 190 (915 mg, 5.07 mmol, 95%) as a colorless oil. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 3.70 (d, 3J8-9 = 6.2 Hz, 2H, CH2), 4.90 (dq, 3J10cis-9 = 17.1 Hz, 2J10cis-10trans =
[84] Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi, S. Ogoshi, Angew. Chemie 2012, 124(43), 10970-10973; Angew. Chemie Int. Ed. 2012, 51(43), 10812-10815.
To a stirring solution of oxalyl chloride (0.38 μL, 4.4 mmol, 1.1 eq) in CH2Cl2 (20 mL) at -78 °C was added dropwise a solution of DMSO (500 μL, 7.2 mmol, 1.8 eq) in CH2Cl2 (2 mL) over 15 min. After 5 minutes, a solution of 5-hexen-1-ol 47 (0.48 μL, 4.0 mmol) in CH2Cl2 (4 mL) was added dropwise over 15 minutes, and the mixture was allowed to stir for an additional 15 minutes at -78 °C. At this point, NEt3 (2.5 mL, 20 mmol) was added over 5 minutes, and the mixture was allowed to warm to room temperature. After warming to room temperature, the reaction was diluted with CH2Cl2 (10 mL). The organic layer was washed with 1 N HCl (2 x 6 mL) and brine (6 mL). The combined organic extracts were dried (MgSO4), filtered and concentrated by distilling off the dichloromethane. This residue was used in the next step without further purification. The desired 5-hexen-1-al 198 was obtained (0.47 μL, 3.9 mmol, 98%). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 1.70 (m, 2H, H3), 2.10 (q, 3J4-3 = 7.4 Hz, 2H, H4), 2.44 (m, 2H, H2), 4.99 (m, 2H, H6), 5.76 (m, 1H, H5), 9.77 (d, 3J1-2 = 1.6 Hz, 1H, H1). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 21.3 (C3), 33.1 (C4), 43.3 (C2), 115.7 (C6), 137.7 (C5), 202.5 (C1).
Tb = 128 °C Rf = 0.52 (cHex/AcOEt 5:1) Analytic data are in accordance with the literature.[194], [195]
[194] B. D. Kelly, J. M. Allen, R. E. Tundel, T. H. Lambert, Org. Lett. 2009, 11(6), 1381-1383. [195] M. Liniger, C. Neuhaus, T. Hofmann, L. Fransioli-Ignazio, M. Jordi, P. Drueckes, J. Trappe, D. Fabbro, K.-H. Altmann, ACS Med. Chem. Lett. 2011, 2(1), 22-27.
ExperimentalPart:SynthesisofSubstrates
239
9.6.2 Synthesis of medium sized rings
9.6.2.1 Synthesis of dec-9-enal 201
OH
oxalyl chloride, DMSO
-78 °C, 1 hH
O
quant.200 201
C10H20OMol. Wt.: 156,27
C10H18OMol. Wt.: 154,25
18
7
6 4
3
2
9 5
10
To a stirring solution of oxalyl chloride (2.7 mL, 31.0 mmol, 1.1 eq) in CH2Cl2 (30 mL) at -78 °C was added dropwise a solution of DMSO (3.6 mL, 50.8 mmol, 1.8 eq) in CH2Cl2 (5 mL) over 15 minutes. After 5 minutes, a solution of dec-9-en-1-ol 200 (5 mL, 28.2 mmol) in CH2Cl2 (20 mL) was added dropwise over 15 minutes, and the mixture was allowed to stir for an additional 15 minutes at -78 °C. At this point, NEt3 (12.5 mL, 100 mmol) was added over 5 minutes, and the mixture was allowed to warm to room temperature. After warming to room temperature, the reaction was diluted with CH2Cl2 (70 mL). The organic layer was washed with 1 N HCl (2 x 40 mL) and brine (40 mL). The combined organic extracts were dried (MgSO4), filtered and concentrated by distilling off the CH2Cl2. This residue was used in the next step without further purification. The desired dec-9-enal 201 was obtained (4.94 mL, 28.2 mmol, quant.). 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 1.16-1.32 (m, 8H, H4, H5, H6 and H7), 1.48-1.58 (q, 3J3-4 = 3J3-2 = 7.8 Hz, 2H, H3), 1.94 (qt, 3J8-7 = 7.6 Hz, 3J8-9 = 4J8-6 = 1.4 Hz, 2H, H8), 2.31 (td, 3J2-3 = 7.4 Hz, 3J2-1 = 2.0 Hz, 2H, H2), 4.80-4.85 (m, 1H, H10trans), 4.86-4.92 (m, 1H, H10cis), 5.65-5.74 (m, 1H, H9), 9.65 (t, 3J1-2 = 1.8 Hz, 1H, H1). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 22.1 (s, 1C, C3), 28.8 (s, 1C, C4 or C5 or C6 or C7), 28.9 (s, 1C, C4 or C5 or C6 or C7), 29.1 (s, 1C, C4 or C5 or C6 or C7), 29.2 (s, 1C, C4 or C5 or C6 or C7), 33.7 (s, 1C, C8), 43.8 (s, 1C, C3), 114.2 (s, 1C, C10), 139.1 (s, 1C, C9), 202.7 (s, 1C, C1). Rf = 0.78 (cHex/AcOEt 19:1) dexp. (dec-9-enal) = 0.88 g.mL-1
Analytic data are in accordance with the literature.[196]
[196] S. R. Park, C. Kim, D.-G. Kim, N. Thrimurtulu, H.-S. Yeom, J. Jun, S. Shin, Y. Ho Rhee, Org. Lett. 2013, 15(6), 1166-1169.
ExperimentalPart:SynthesisofSubstrates
240
9.6.2.2 Synthesis of 2-hex-5-enyloxy-benzaldehyde Synthesis of toluene-4-sulfonic acid hex-5-enyl ester 203
OH OTsCl, dry pyridine
quant.47 203
1 h, RT
C6H12OMol. Wt.: 100,16
C13H18O3SMol. Wt.: 254,35
13
4 210
9
7
6
5
8SOO
119
1012
Tosyl chloride (9 g, 48.3 mmol, 1.15 eq.) was added portionwidse to a stirred and ice-cooled solution of 5-hexen-1-ol 47 (5 mL, 41.7 mmol) in dry pyridine (25 mL). The mixture was stirred for 3 hours at 0 °C, poured into ice and diluted HCl, and extracted with hexane. The extract was washed successively with water, saturated NaHCO3 solution, and brine dried over MgSO4, and concentrated in vacuo to give (10.61 g, 41.7 mmol, quant.) of toluene-4-sulfonic acid hex-5-enyl ester 203 as a colourless oil. 1H-NMR (400.132 MHz, CDCl3): δ (ppm) = 1.41 (q, 3J4-5 = 3J4-3 = 6.0 Hz, 2H, H4), 1.61 (q, 3J3-4 = 3J3-2 = 6.0 Hz, 2H, H3), 2.01 (dt, 3J5-4 = 6.6 Hz, 2H, H5), 2.42 (s, 3H, Ar-CH3), 4.01 (t, , 3J2-3 = 6.0 Hz, 2H, H2), 4.95 (m, 2H, H7), 5.70 (m, 1H, H6), 7.36 (d, 3J10-9 = 6.0 Hz, 2H, H10), 7.36 (d, 3J9-10 = 6.0 Hz, 2H, H9). Analytic data are in accordance with the literature.[167]
[167] K. Mori, Eur. J. of Org. Chem. 2005, 10, 2040-2044.
ExperimentalPart:SynthesisofSubstrates
241
Synthesis of 2-hex-5-enyloxy-benzaldehyde 204
24 h, RTOTs +
O
H
OH
K2CO3, DMF
45%
O
H
O
203 85 204
8 13
12
11
109
76
5
43
21
C7H6O2Mol. Wt.: 122,12
C13H18O3SMol. Wt.: 254,35
C13H16O2Mol. Wt.: 204,26
To a stirred solution of salicylaldehyde 85 (1.9 mL, 17.9 mmol) in N,N-dimethylformamide (0.2 M) was added K2CO3 (3.72 g, 26.6 mmol, 1.5 eq) and toluene-4-sulfonic acid hex-5-enyl ester 163 (5g, 19.7 mmol, 1.1 eq). The mixture was stirred for 24 hours. The crude reaction mixture was then diluted with ethyl acetate and successively washed once with aqueous 1M NaOH and twice with brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated to dryness in vacuo. 2-hex-5-enyloxy-benzaldehyde 204 (1.81 g, 8.86 mmol, 45%) was isolated by flash chromatography (cHex/AcOEt 9:1, then 4:6). 1H-NMR (400.132 MHz, CDCl3): δ (ppm) = 1.55-1.66 (m, 2H, H10), 1.81-1.90 (m, 2H, H9), 2.10-2.17 (m, 2H, H11), 4.07 (t, 3J8-9 = 6.4 Hz, 2H, H8), 4.98 (dq, 3J13trans-12 = 10.3 Hz, 2J13trans-13cis =
[197] L. Garanti, A. Sala, G. Zecchi, J. Org. Chem. 1975, 40(16), 2403-2406.
ExperimentalPart:SynthesisofSubstrates
242
9.7 Mechanistic studies
9.7.1 Synthesis of benzaldehyde-α-d1 Precaution in handling potassium cyanide ! In acidic conditions, cyanide is a potent inhibitor of cellular respiration (strong poison). KCN can be detoxified with hydrogen peroxide to form KOCN.
O
O
O
DKCN, D2O
dioxane, RT, 30 min
96%
211C16H16O2
Mol. Wt.: 240,30
212C7H5DO
Mol. Wt.: 107,13
12
34
5 3
4
To a stirred solution of benzyl 211 (7.5 g, 36.0 mmol) in 1,4--dioxane (23 mL) was added deuterium oxide (18 mL, 39.2 mmol, 1.1 eq) and then potassium cyanide (2.6 g, 39.2 mmol, 1.1 eq) in five portions over 30 minutes. The mixture was stirred for another 30 minutes and then diluted with water (60 mL). The mixture was extracted with diethyl ether (3 x 50 mL) and the combined organic phases were washed with saturated solution sodium bicarbonate (2 x 50 mL) and saturated aqueous sodium chloride (2 x 50 mL). After evaporation of the dried (MgSO4) ether solution, distillation gave pure benzaldehyde-1-d 212 (3.08 mL, 29.9 mmol, 96%) as a colorless oil. 1H NMR (400.132 MHz, CDCl3): δ = 7.45-7.51 (m, 2H, Ar-H4), 7.55-7.61 (m, 1H, Ar-H5), 7.81-7.85 (m, 2H, Ar-H3). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 126.5 (d, 1C, C4), 128.9 (d, 1C, C3), 129.6 (d, 1C, C5), 136.2 (s, 1C, C2), 192.0 (s, 1C, C1). Tb = 76 °C (22 mmHg). Analytic data are in accordance with the literature.[170]
[170] A. G. Griesbeck, S. Bondock, P. Cygon, J. Am. Chem. Soc. 2003, 125, 9016-9017.
243
10. Catalytic experiments 10.1 Rhodium-Catalyzed Intermolecular Hydroacylation of
benzaldehyde and 1-octene 10.1.1 General procedure: intermolecular hydroacylation of substituted benzaldehydes and diverse alkenes protocol (GP4) The oil bath was set at the desired temperature before immersing the Schlenk tube. To a dry 8 mL Schlenk tube was added the ligand and the catalyst. Then, argon and vacuum were exchanged three times. The two hydroacylation partners (alkene and aldehyde) were then added to the Schlenk tube together and solvent when required. After the desired reaction time, the Schlenk tube was cooled to room temperature, the solvent was removed in vacuo, and the resulting oil or solid filtered through Celite. Products were purified by column chromatography or directly analyzed by 1H NMR, 13C NMR and HRMS.
O[Rh(COD)Cl]2, L
Toluene, 150 °C, 24 hR2
HR1 +
O
R2
R1
A mixture of substituted benzaldehyde (23.3 mg, 0.22 mmol), ligand L (0.011 mmol, 3.4 mg, 5 mol%), [Rh(COD)Cl]2 (0.011 mmol, 4.5 mg, 5 mol%) was dissolved in toluene (200 μL) in a 8 mL Schlenk tube and stirred for 24 hours at 120 °C. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2).
ExperimentalPart:Catalyticexperiments
244
10.1.2 General procedure: intermolecular hydroacylation of substituted benzaldehydes and diverse alkenes protocol using Jun conditions (GP5)
The oil bath was set at the desired temperature before immersing the Schlenk tube. To a dry 8 mL Schlenk tube was added the ligand and the catalyst. Then, argon and vacuum were exchanged three times. The two hydroacylation partners (alkene and aldehyde) were then added to the Schlenk tube together and solvent when required. After the desired reaction time, the Schlenk tube was cooled to room temperature, the solvent was removed in vacuo, and the resulting oil or solid filtered through Celite. Products were purified by column chromatography or directly analyzed by 1H NMR, 13C NMR and HRMS.
O [Rh(PPh3)3Cl]2-amino-3-picoline
Toluene, 150 °C, 24 hR2
HR1 +
O
R2
R1
A mixture of substituted benzaldehyde (24 μL, 0.22 mmol), 2-amino-3-picoline (0.011 mmol, 3.4 mg), [Rh(PPh3)3Cl] (0.022 mmol, 20.4 mg) was dissolved in toluene (200 μL) in a 8 mL Schlenk tube and stirred for 24 hours at 150 °C. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2).
ExperimentalPart:Catalyticexperiments
245
10.1.3 Synthesis of 1-phenylnona-1-one 46
O10
1112
1312
11
1 2
3
4
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
46
+
45
5
6
7
8
9
C8H16Mol. Wt.: 112,21
C7H6OMol. Wt.: 106,12
C15H22OMol. Wt.: 218,33
H
O
83%
Following GP4, oct-1-ene 45, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1-phenylnonan-1-one 46 (39.3 mg, 0.18 mmol, 83%) as a colourless liquid. 1H NMR (400.132 MHz, CDCl3): δ = 0.88 (t, J = 7.2 Hz, 3H, H9), 1.32 (m, 10H, H4-8), 1.74 (quint, 3J3-2 = 7.3 Hz, 2H, H3), 2.96 (t, 3J2-3 = 7.3 Hz, 2H, H2), 7.46 (m, 2H, Ar-H12), 7.56 (m, 1H, Ar-H13), 7.96 (m, 2H, Ar-H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 14.3 (s, 1C, C9), 22.8 (s, 1C, C8), 24.6 (s, 1C, C3), 29.3 (s, 1C, C4), 29.5 (s, 1C, C5 or C6), 29.6 (s, 1C, C5 or C6), 32.0 (s, 1C, C7), 38.8 (s, 1C, C2), 128.2 (s, 2C, C12), 128.7 (s, 2C, C11), 133.0 (s, 1C, C13), 137.3 (s, 1C, C10), 200.8 (s, 1C, C1). HRMS (EI): Calcd. for C15H22O (M): 218.1671; Found: 218.1671. Rf = 0.85 (CH2Cl2 pure) Analytic data are in accordance with the literature.[198]
[198] D. Wang, Z. Zhang, Org. Lett. 2003, 5, 4645-4648.
ExperimentalPart:Catalyticexperiments
246
10.2 Rhodium-Catalyzed Intermolecular Hydroacylation of substituted benzaldehydes and diverse alkenes 10.2.1 Synthesis of 7-hydroxy-1-phenyl-heptan-1-one 48
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
48
+
47
HO
O9
1011
1211
10
1 2
3
4 OH5
6
7
8
C13H18O2Mol. Wt.: 206,28
C7H6OMol. Wt.: 106,12
C6H12OMol. Wt.: 100,16
75%
Following GP4, 5-hexen-1-ol 47, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 7-hydroxy-1-phenyl-heptan-1-one 48 (34.0 mg, 0.17 mmol, 75 %) as a white solid. 1H NMR (400.132 MHz, CDCl3): δ = 1.42 (m, 4H, H4 and H5), 1.59 (m, 2H, H6), 1.76 (m, 2H, H3), 2.98 (t, 3J2-3 = 7.6 Hz, 2H, H2), 3.65 (t, 3J7-6 = 6.4 Hz, 2H, H7), 7.46 (m, 2H, Ar-H11), 7.56 (m, 1H, Ar-H12), 7.96 (m, 2H, Ar-H10).3 13C {1H} NMR (100.613 MHz, CDCl3): δ = 24.4 (s, 1C, C3), 25.7 (s, 1C, C5), 29.2 (s, 1C, C4), 32.7 (s, 1C, C6), 38.6 (s, 1C, C2), 63.1 (s, 1C, C7), 128.2 (s, 2C, C10), 128.7 (s, 2C, C11), 133.1 (s, 1C, C12), 137.2 (s, 1C, C9), 200.6 (s, 1C, C1).
HRMS (EI): Calcd. for C13H19O2 (M+H): 207.1385; Found: 207.1382 (difference -1.4 ppm). Tm = 33-34 °C Analytic data are in accordance with the literature.[199]
[199] C. B. Rao, D. C. Rao, D. C. Babu, Y. Vankateswarlu, Eur. J. Org. Chem. 2010, 2855-2859.
ExperimentalPart:Catalyticexperiments
247
10.2.2 Synthesis of 1,3-diphenyl-propan-1-one 50
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
50
+
49
O8
910
1110
9
1 2
3
4
56
76
C7H6OMol. Wt.: 106,12
C16H16OMol. Wt.: 224,30
45%C8H8
Mol. Wt.: 104,15
5
Following GP4, allyl-benzene 49, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1,4-diphenyl-butan-1-one 50 (35.5 mg, 0.16 mmol, 72%) as a colourless liquid. 1H NMR (400.132 MHz, CDCl3): δ = 3.04 (t, 3J3-2 = 7.7 Hz, H3), 3.26 (t, 3J2-3 = 7.7 Hz, H2), 7.16-7.29 (m, 5H, Ar-H5, Ar-H6 and Ar-H7), 7.39-7.42 (m, 2H, Ar-H10), 7.49-7.52 (m, 2H, Ar-H11), 7.91-7.93 (m, 2H, Ar-H12). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 30.3 (s, 1C, C3), 40.6 (s, 1C, C2), 126.3 (s, 1C, C7), 128.3 (s, 2C, C5 or C6 or C9 or C10), 128.7 (s, 2C, C5 or C6 or C9 or C10), 128.8 (s, 2C, C5 or C6 or C9 or C10), 128.8 (s, 2C, C5 or C6 or C9 or C10), 133.2 (s, 1C, C11), 137.1 (s, 1C, C8), 141.5 (s, 1C, C4), 199.3 (s, 1C, C1). Tm = 68-69 °C Rf = 0.81 (CH2Cl2 pure) Analytic data are in accordance with the literature.[200]
[200] J. Lin, J. Chen, W. Su, Adv. Synth. and Cat. 2013, 355(1), 41-46.
ExperimentalPart:Catalyticexperiments
248
10.2.3 Synthesis of 1,4-diphenyl-butan-1-one 52
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
52
+
51
O9
1011
1211
10
1 2
3
4
5
67
8
76
C9H10Mol. Wt.: 118,18
C7H6OMol. Wt.: 106,12
C16H16OMol. Wt.: 224,30
72%
Following GP4, allyl-benzene 51, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1,4-diphenyl-butan-1-one 52 (35.5 mg, 0.16 mmol, 72%) as a colourless liquid. 1H NMR (400.132 MHz, CDCl3): δ = 2.09 (quint, 3J3-2 = 3J3-4 = 7.6 Hz, 2H, H3), 2.73 (t, 3J4-3 = 7.6 Hz, 2H, H4), 2.98 (t, 3J2-3 = 7.6 Hz, 2H, H2), 7.21 (m, 3H, Ar-H6 and Ar-H8), 7.29 (tt, 3J7-6 = 7.4 Hz, 3J7-8 = 1.8 Hz, 2H, Ar-H7), 7.44 (tt, 3J10-11 = 8.0 Hz, 4J10-12 = 1.8 Hz, 2H, H10), 7.55 (tt, 3J12-11 = 7.4 Hz, 4J12-10 = 1.2 Hz, 1H, H12), 7.91 (dd, 3J11-10 = 8.0 Hz, 3J11-12 = 1.2 Hz, 1H, H11), 7.93 (q, J = 4.0 Hz, 1H, H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 25.8 (s, 1C, C3), 35.4 (s, 1C, C4), 37.9 (s, 1C, C2), 126.1 (s, 1C, C8), 128.2 (s, 2C, C11), 128.5 (s, 2C, C7) , 128.6 (s, 2C, C6 or C10), 128.7 (s, 2C, C6 or C10), 133.1 (s, 1C, C12), 137.2 (s, 1C, C9), 141.8 (s, 1C, C5), 200.3 (s, 1C, C1). HRMS (EI): Calcd. for C16H16O (M): 224.1201; Found: 224.1204 (difference +1.3 ppm). Tm = 54-55 °C Rf = 0.81 (CH2Cl2 pure) Analytic data are in accordance with the literature.[201]
[201] J. A. Murphy, A. G. J. Commeureuc, T. N. Snaldon, T. M. McGuire, T. A. Khan, K. Hisler, M. L. Dewis, R. Carling, Org. Lett. 2005, 7, 1427-1429.
ExperimentalPart:Catalyticexperiments
249
10.2.4 Synthesis of 3-cyclohex-3-enyl-1-phenyl-propan-1-one 54
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
54
+
53
O10
1112
1312
11
1 2
3
4
9
8
76
5
C8H12Mol. Wt.: 108,18
C7H6OMol. Wt.: 106,12
C15H18OMol. Wt.: 214,30
57%
Following GP4, 4-vinyl-1-cyclohexene 53, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 3-cyclohex-3-enyl-1-phenyl-propan-1-one 54 (26.8 mg, 0.13 mmol, 57%) as a colourless liquid. 1H NMR (400.132 MHz, CDCl3): δ = 1.29 (m, 1H, H9), 1.72 (m, 5H, H3, H5 and H9), 2.07 (m, 2H, H8), 2.16 (m, 1H, H4), 3.02 (t, 3J2-3 = 7.4 Hz, 2H, H2), 5.67 (m, 2H, H6 and H7), 7.46 (m, 2H, Ar-H12), 7.56 (m, 1H, Ar-H13), 7.96 (m, 2H, Ar-H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 25.3 (s, 1C, C8), 29.0 (s, 1C, C9), 31.1 (s, 1C, C3), 31.9 (s, 1C, C4), 33.5 (s, 1C, C5), 36.3 (s, 1C, C2), 126.4 (s, 1C, C6 or C7), 127.2 (s, 1C, C6 or C7), 128.2 (s, 2C, C11), 128.7 (s, 2C, C12), 133.1 (s, 1C, C13), 137.3 (s, 1C, C10), 200.8 (s, 1C, C1). HRMS (EI): Calcd. for C15H18O (M): 214.1358; Found: 214.1359 (difference +0.4 ppm). Rf = 0.85 (CH2Cl2 pure) Analytic data are in accordance with the literature.[202]
[202] Y. Lu, L. S. Liebeskind, J. Org. Chem. 2004, 69, 3554-3557.
ExperimentalPart:Catalyticexperiments
250
10.2.5 Synthesis of 3-cyclohexyl-1-phenyl-propan-1-one 56
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
56
+
55C8H14
Mol. Wt.: 110,20C7H6O
Mol. Wt.: 106,12C15H20O
Mol. Wt.: 216,32
10
9
12
3
46
7
89 5
O
5610
11
45%
Following GP4, vinyl-cyclohexane 55, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 3-cyclohexyl-1-phenyl-propan-1-one 56 (21.0 mg, 0.10 mmol, 45%) as a white powder. 1H NMR (400.132 MHz, CDCl3): δ = 0.90-1.80 (m, 13H, H3, H4, H5, H6 and H7), 2.97 (m, 2H, CH2(2)), 7.46 (tt, 3J10-9 = 7.3 Hz, 3J10-11 = 1.4 Hz, 2H, H10), 7.56 (tt, 3J11-10 = 7.3 Hz, 4J11-9 = 1.4 Hz, 1H, H11), 7.97 (m, 2H, H9). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 26.4 (s, 1C, C6), 26.7 (s, 1C, C3) , 31.9 (s, 1C, C7), 33.3 (s, 1C, C5), 36.3 (s, 1C, C2), 37.6 (s, 1C, C4), 128.2 (s, 2C, C9), 128.6 (s, 2C, C10), 132.9 (s, 1C, C11), 137.2 (s, 1C, C8), 201.0 (s, 1C, C1). HRMS (EI): Calcd. for C15H22O2 (M): 216.15142; Found: 216.15140.
Rf = 0.85 (CH2Cl2 pure)
Analytic data are in accordance with the literature.[203]
[203] T. Kuwahara, T. Fukuyama, I. Ryu, Org. Lett. 2012, 14(18), 4703-4705.
ExperimentalPart:Catalyticexperiments
251
10.2.6 Synthesis of 7-oxo-7-phenyl heptanoic acid 58
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
58
+
57
HO
O8
910
1110
9
1 2
3
4 OH5
6
7O
O
C6H10O2Mol. Wt.: 114,14
C7H6OMol. Wt.: 106,12
C13H16O3Mol. Wt.: 220,26
51%
Following GP4, 5-hexenoic acid 57, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 7-oxo-7-phenyl heptanoic acid 58 (24.7 mg, 0.11 mmol, 51%) as a colourless liquid. 1H NMR (400.132 MHz, CDCl3): δ = 1.46 (m, 2H, H4), 1.74 (m, 4H, H3 and H5), 2.39 (t, 3J6-5 = 7.4 Hz, 2H, H6), 2.99 (t, 3J2-3 = 7.4 Hz, 2H, H2), 7.46 (m, 2H, Ar-H10), 7.56 (m, 1H, Ar-H11), 7.96 (m, 2H, Ar-H9). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 24.0 (s, 1C, C3), 24.7 (s, 1C, C5), 28.9 (s, 1C, C4), 33.6 (s, 1C, C6), 38.4 (s, 1C, C2), 128.2 (s, 1C, C9), 128.7 (s, 1C, C11), 133.1 (s, 1C, C10), 137.1 (s, 1C, C8), 177.9 (s, 1C, C7), 200.3 (s, 1C, C1).
HRMS (EI): Calcd. for C13H17O3 (M+H): 221.1178; Found: 221.1175 (difference -1.4 ppm). Tm = 83-84 °C Rf = 0.73 (AcOEt pure) Analytic data are in accordance with the literature.[204]
[204] M. A. Rahim, T. Fujiwara, T. Takeda, Tetrahedron 2000, 56, 763-770.
ExperimentalPart:Catalyticexperiments
252
10.2.7 Synthesis of 4-oxo-4-phenylbutyric acid methyl ester 60
44
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
60
+O
O
59
O
O
O67
8
98
7
1 2
34
5
C4H6O2Mol. Wt.: 86,09
C7H6OMol. Wt.: 106,12
C11H12O3Mol. Wt.: 192,21
73%
Following GP4, acrylic acid methyl ester 59, benzaldehyde 44, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 4-oxo-4-phenylbutyric acid methyl ester 60 (30.8 mg, 0.16 mmol, 73%) as a colourless oil. 1H NMR (400.132 MHz, CDCl3): δ = 2.78 (t, 3J3-2 = 6.6 Hz, 2H, H3), 3.32 (t, 3J2-3 = 6.6 Hz, 2H, H2) , 3.71 (s, 3H, CH3), 7.47 (m, 2H, Ar-H8), 7.58 (m, 1H, Ar-H9), 7.99 (m, 2H, Ar-H7). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 28.2 (s, 1C, C3), 33.6 (s, 1C, C2), 52.0 (s, 1C, C5), 128.2 (s, 2C, C7), 128.8 (s, 1C, C9), 133.4 (s, 2C, C8), 136.7 (s, 1C, C6), 173.5 (s, 1C, C4), 198.2 (s, 1C, C1). HRMS (EI): Calcd. for C11H13O3 (M+H): 193.0865; Found: 193.0866 (difference +0.5 ppm). Analytic data are in accordance with the literature.[205]
[205] E.-A. Jo, C.-H. Jun, Eur. J. Org. Chem. 2006, 2504-2507.
ExperimentalPart:Catalyticexperiments
253
10.2.8 Synthesis of 4,4-dimethyl-1-phenyl-pentan-1-one 62
[Rh(PPh3)3Cl] (10 mol%)
2-amino-3-picoline (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
44
O
H
62
+
61
O
C6H12Mol. Wt.: 84,16
C7H6OMol. Wt.: 106,12
C13H18OMol. Wt.: 190,28
87
12
3
4
56
78
9 5
5
19%
Following GP5, 3,3-dimethyl-but-1-ene 61, 2-amino-3-picoline, benzaldehyde 44, ligand 1 and catalyst [Rh(PPh3)3Cl] were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 4,4-dimethyl-1-phenyl-pentan-1-one 62 (7.9 mg, 0.04 mmol, 19%) as a colourles liquid. 1H NMR (400.132 MHz, CDCl3): δ = 0.97 (s, 9H, (CH3)3), 1.65 (sext, 2H, CH2(3)), 2.94 (sext, 2H, CH2(2)), 7.46 (tt, 3J8-9 = 7.3 Hz, 3J8-7 = 1.4 Hz, 2H, H8), 7.56 (tt, 3J9-8 = 7.3 Hz, 4J9-7 = 1.4 Hz, 1H, H9), 7.97 (m, 2H, H7). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 29.3 (s, 3C, C5), 30.3 (s, 1C, C4) , 34.4 (s, 1C, C2), 38.3 (s, 1C, C3), 128.2 (s, 2C, C7), 128.7 (s, 2C, C8), 133.0 (s, 1C, C9), 137.2 (s, 1C, C6), 201.2 s, 1C, (C1). Analytic data are in accordance with the literature.[206]
[206] J. F. Hooper, R. D. Young, A. S. Weller, M. C. Willis, Chem. Eur. J. 2013, 19(9), 3125-3130.
ExperimentalPart:Catalyticexperiments
254
10.2.9 Synthesis of 3-dimethylsilanyl-1-phenyl-propan-1-one 64
87
12
3
4
56
78
9 5
5
44
[Rh(PPh3)3Cl] (10 mol%)
2-amino-3-picoline (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
64
+
63
Si
O
Si
C5H12SiMol. Wt.: 100,23
C7H6OMol. Wt.: 106,12
C12H18OSiMol. Wt.: 206,36
59%
Following GP5, trimethyl-vinyl-silane 63, benzaldehyde 44, ligand 1 and catalyst [Rh(PPh3)3Cl] were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 3-dimethylsilanyl-1-phenyl-pentan-1-one 64 (26.8 mg, 0.13 mmol, 59%) as a white powder. 1H NMR (400.132 MHz, CDCl3): δ = 0.06 (s, 9H, (CH3)3), 1.65 (sext, 2H, CH2(3)), 2.94 (sext, 2H, CH2(2)), 7.46 (tt, 3J8-9 = 7.3 Hz, 3J8-7 = 1.4 Hz, 2H, H8), 7.56 (tt, 3J9-8 = 7.3 Hz, 4J9-7 = 1.4 Hz, 1H, H9), 7.97 (m, 2H, H7). 13C {1H} NMR (100.613 MHz, CDCl3): δ = -1.7 (s, 3C, C5), 11.1 (s, 1C, C3), 33.3 (s, 1C, C2), 128.2 (s, 2C, C7), 128.7 (s, 2C, C8), 132.9 (s, 1C, C9), 136.9 (s, 1C, C6), 201.4 (s, 1C, C1). HRMS (EI): Calcd. for C12H17OSi (M-H): 205.10487; Found: 205.10470 (difference -0.8 ppm). Rf = 0.81 (CH2Cl2 pure) Analytic data are in accordance with the literature.[207]
[207] Z. Gan, Y. Wu, L. Gao, X. Sun, J. Lei, Z. Song, L. Li, Tetrahedron 2012, 68(34), 6928-6934.
ExperimentalPart:Catalyticexperiments
255
10.2.10 Synthesis of 1-(3-methoxy-phenyl)-nonan-1-one 66
Analytic data are in accordance with the literature.[208]
[208] A. R. Katritzky, H. Lang, Z. Wang, Z. Zhang, H. Song, J. Org. Chem. 1995, 60(23), 7619-7624.
ExperimentalPart:Catalyticexperiments
256
10.2.11 Synthesis of 1-(4-methoxy-phenyl)-nonan-1-one 68
O10
1112
13
1211
1 2
3
4
5
6
7
8
9
MeO
67
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
68
+
45
MeO14
C8H16Mol. Wt.: 112,21
C8H8O2Mol. Wt.: 136,15
C16H24O2Mol. Wt.: 248,36
93%
Following GP4, oct-1-ene 45, 4-methoxy-benzaldehyde 67, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1-(4-methoxy-phenyl)-nonan-1-one 68 (50.8 mg, 0.20 mmol, 93%) as a yellowish solid. 1H NMR (400.132 MHz, CDCl3): δ = 0.87 (t, 3J9-8 = 6.8 Hz, 3H, CH3), 1.31 (m, 10H, H4-8), 1.71 (quint, 3J3-2 = 3J3-4 = 7.5 Hz, 2H, H3), 2.90 (t, 3J2-3 = 7.5 Hz, 2H, H2), 3.86 (s, 3H, OCH3), 6.91 (d, 3J12-11 = 8.5 Hz, 2H, Ar-H12), 7.92 (d, 3J11-12 = 8.5 Hz, 2H, Ar-H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 14.2 (s, 1C, C9), 22.8 (s, 1C, C8) , 24.8 (s, 1C, C2), 29.3 (s, 1C, C4), 29.6 (s, 2C, C5 and C6), 31.9 (s, 1C, C7), 38.4 (s, 1C, C3), 55.5 (s, 1C, C14), 113.7 (s, 2C, C12), 130.3 (s, 1C, C10), 130.4 (s, 2C, C11), 163.4 (s, 1C, C13), 199.3 (s, 1C, C1). HRMS (EI): Calcd. for C16H25O2 (M+H): 249.1855; Found: 249.1852 (difference -1.2 ppm). Tm = 43-44 °C Rf = 0.76 (CH2Cl2 pure) Analytic data are in accordance with the literature.[209]
[209] S. Dohi, K. Moriyama, H. Togo, Tetrahedron 2012, 68(32), 6557-6564.
ExperimentalPart:Catalyticexperiments
257
10.2.12 Synthesis of 1-biphenyl-4-yl-nonan-1-one 70
O10
1112
1312
11
1 2
3
4
5
6
7
8
9
69
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
70
+
45
1415
16
1716
15
C21H26OMol. Wt.: 294,43
C13H10OMol. Wt.: 182,22
C8H16Mol. Wt.: 112,21
68%
Following GP2, oct-1-ene 45, biphenyl-4-carboxaldehyde 69, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1-biphenyl-4-yl-nonan-1-one 70 (44.0 mg, 0.15 mmol, 68%) as a white powder. 1H-NMR (400.132 MHz, CDCl3): δ = 0.89 (t, 3J9-8 = 6.8 Hz, 3H, H9), 1.34 (m, 10H, H4-8), 1.76 (quint, 3J3-2 = 3J3-4 = 7.5 Hz, 2H, H3), 2.99 (t, 3J2-3 = 7.5 Hz, 2H, H2), 7.41 (m, 1H, Ar-H17), 7.47 (m, 2H, Ar-H16), 7.63 (m, 2H, Ar-H15), 7.68 (m, 2H, Ar-H12), 8.03 (m, 2H, Ar-H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 14.2 (C9), 22.8 (C8), 24.6 (C3), 29.3 (C4 or C5 or C6), 29.5 (C4 or C5 or C6), 29.6 (C4 or C5 or C6), 32.0 (C7), 38.8 (C2), 127.3 (2C, C12 or C15), 127.4 (2C, C12 or C15), 128.3 (C17), 128.8 (2C, C16), 129.0 (2C, C16), 135.9 (C10), 140.1 (C15), 145.6 (C13), 200.3 (C1). HRMS (EI): Calcd. for C21H26O (M): 294.1984; Found: 294.1987 (difference +1.5 ppm). Tm =93 °C
ExperimentalPart:Catalyticexperiments
258
10.2.13 Synthesis of 1-(4-chloro-phenyl)-nonan-1-one 72
71
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
72
+
45
Cl
O1011
12
13
1211
1 2
3
4
5
6
7
8
9
Cl
C8H16Mol. Wt.: 112,21
C7H5ClOMol. Wt.: 140,57
C15H21ClOMol. Wt.: 252,78
70%
Following GP2, oct-1-ene 45, 4-chloro-benzaldehyde 71, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1-(4-chloro-phenyl)-nonan-1-one 72 (38.8 mg, 0.15 mmol, 70%) as a white powder.
10.2.14 Synthesis of 1-naphthalen-2-yl-nonan-1-one 74
73
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
O
H
74
+
45
O
C19H24OMol. Wt.: 268,39
C11H8OMol. Wt.: 156,18
C8H16Mol. Wt.: 112,21
9
1
2 3
4 6
7
8
510
19181716
15
1413 12 11
11%
Following GP2, oct-1-ene 45, naphtalene-2-carbaldehyde 73, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield naphthalen-2-yl-nonan-1-one 74 (6.5 mg, 0.02 mmol, 11%) as a yellow powder. 1H NMR (400.132 MHz, CDCl3): δ = 0.89 (t, 3J9-8 = 7.5 Hz, 3H, CH3), 1.22-1.47 (m, 10H, H4-8), 7.47 (quint, 3J3-2 = 3J3-4 = 7.5 Hz, 2H, H3), 3.10 (t, 3J2-3 = 7.5 Hz, 2H, H2), 7.53-7.58 (m, 1H, Ar-H14), 7.58-7.62 (m, 1H, Ar-H15), 7.92-7.93 (m, 1H, Ar-H16), 7.94-7.95 (m, 1H, Ar-H18), 7.97 (d, 3J13-14 = 7.5 Hz, 1H, Ar-H13), 8.02-8.05 (m, 1H, Ar-H19), 8.47 (m, 1H, Ar-H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 14.2 (s, 1C, C9), 22.8 (s, 1C, C8) , 24.7 (s, 1C, C3), 29.3 (s, 1C, C4, C5 or C6), 29.5 (s, 1C, C4, C5 or C6), 29.7 (s, 1C, C4, C5 or C6), 31.9 (s, 1C, C7), 38.8 (s, 1C, C2), 124.1 (s, 1C, C19), 126.8 (s, 1C, C14), 127.9 (s, 1C, C16), 128.4 (s, 1C, C15 or C18), 128.5 (s, 1C, C15 or C18), 129.6 (s, 1C, C11 or C13), 129.7 (s, 1C, C11 or C13), 132.7 (s, 1C, C17), 134.6 (s, 1C, C10), 135.6 (s, 1C, C12), 200.7 (s, 1C, C1). HRMS (EI): Calcd. for C19H24O (M): 268.18272; Found: 268:18290 (difference +0.6 ppm). Tm = 57 °C Rf = 0.89 (CH2Cl2 pure)
ExperimentalPart:Catalyticexperiments
260
10.2.15 Synthesis of 1-thiophen-2-yl-nonan-1-one 76
[Rh(COD)Cl]2 (10 mol%)
ligand 1 (10 mol%)
toluene (c = 1.1 M) 1 d, 150 °C
76
+S
O
H
45 75C13H20OS
Mol. Wt.: 224,36C5H4OS
Mol. Wt.: 112,15C8H16
Mol. Wt.: 112,21
O1011
12 13
1 2
3
4
5
6
7
8
9
S
8%
Following GP3, oct-1-ene 45, thiophene-3-carbaldehyde 75, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1-thiophen-2-yl-nonan-1-one 76 (3,93 mg, 0.02 mmol, 8%) as a yellow oil. 1H NMR (400.132 MHz, CDCl3): δ = 0.88 (t, 3J9-8 = 7.5 Hz, 3H, CH3), 1.21-1.41 (m, 10H, H4-8), 1.72 (quint, 3J3-2 = 3J3-4 = 7.5 Hz, 2H, H3), 2.86 (t, 3J2-3 = 7.5 Hz, 2H, H2), 7.31 (dd, 3J12-11 = 5.0 Hz, 4J12-13 = 2.8 Hz, 1H, Ar-H12), 7.55 (dd, 3J11-12 = 5.0 Hz, 4J11-13 = 1.2 Hz, 1H, Ar-H11), 8.03 (dd, 4J13-12 = 2.8 Hz, 4J13-11 = 1.2 Hz, 1H, Ar-H13). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 14.2 (s, 1C, C9), 22.7 (s, 1C, C8) , 24.5 (s, 1C, C2), 29.2 (s, 1C, C4), 29.5 (s, 2C, C5 and C6), 31.9 (s, 1C, C7), 40.1 (s, 1C, C3), 126.3 (s, 1C, C12), 127.1 (s, 1C, C11), 131.7 (s, 1C, C13), 142.6 (s, 1C, C10), 195.1 (s, 1C, C1). HRMS (EI): Calcd. for C13H20O (M): 224.12349; Found: 224.12320 (difference -1.3 ppm). Rf = 0.81 (CH2Cl2 pure) Analytic data are in accordance with the literature.[210]
[210] N. Turkman, L. An, M. Pomerantz, Org. Lett. 2010, 12(19), 4428-4430.
ExperimentalPart:Catalyticexperiments
261
10.3 Rhodium-catalyzed hydroacylation of 1-octene with aliphatic aldehydes
10.3.1 Synthesis of 3-phenyl-undecan-3-one 84
+
[Rh(COD)2BF4] (10 mol%)
ligand 7 (13 mol%)
toluene (c = 1.1 M)
no reaction80
150 °C, 1 h
45
H
O O
84C9H10O
Mol. Wt.: 134,18
C17H26OMol. Wt.: 246,39
A mixture of substituted 2-phenyl-propionaldehyde 80 (33.9 mg, 0.22 mmol), 1-octene 45 (80 μL, 0.55 mmol), ligand 7 (0.029 mmol, 8.0 mg), [Rh(COD)2]BF4 (0.022 mmol, 9 mg) was dissolved in toluene (200 μL) in a 8 mL Schlenk tube and stirred for 1 day at 120 °C. The crude was analyzed by 1H NMR, no product 3-phenyl-undec-3-one 84 was detected.
ExperimentalPart:Catalyticexperiments
262
10.4 Intramolecular hydroacylation of salicylaldehyde with methyl acrylate followed by an intramolecular transesterification
10.4.1 Synthesis of 4-(2-hydroxy-phenyl)-4-oxo-butyric acid methyl ester 87
12
34
5
67
8
9neat, 100 °C, 24 h
H
O
OH+
O
OMe
[Rh(COD)Cl]2 (10 mol%)ligand 1, ratio L/R 1:1
OH
O
O
OMe
quant.
85 59 87
11
10 12
C7H6O2Mol. Wt.: 122,12
C4H6O2Mol. Wt.: 86,09
C11H12O4Mol. Wt.: 208,21
A mixture of salicylaldehyde 85 (23 μL, 0.22 mmol), acrylic acid methyl ester 59 (50 μL, 0.55 mmol, 2.5 eq), ligand 1 (0.022 mmol, 6.7 mg, 10 mol%), [Rh(COD)2]BF4 (0.022 mmol, 9 mg, 10 mol%) was dissolved in toluene (200 μL) in a 8 mL Schlenk tube and stirred for 1 day at 100 °C. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 4-(2-hydroxy-phenyl)-4-oxo-butyric acid methyl ester 87 (45.8 mg, 0.22 mmol, quant.) as a pale yellow solid. 1H NMR (400.132 MHz, CDCl3): δ = 2.74 (t, 3J3-2 = 7.4 Hz, 2H, CH2(3)), 3.36 (t, 3J2-3 = 7.4 Hz, 2H, CH2(2)), 3.70 (s, 3H, CH3), 6.90 (t, J = 7.6 Hz, 1H, Ar-H10), 6.96 (d, J = 7.6 Hz, 1H, Ar-H8), 7.46 (t, J = 7.6 Hz, 1H, Ar-H9), 7.78 (d, J = 7.6 Hz, 1H, Ar-H7), 10.04 (s, 1H, OH). Analytic data are in accordance with the literature.[211]
[211] A. V. Dubrovskiy, R. C. Larock, Org. Lett. 2010, 12(14), 3117-3119.
ExperimentalPart:Catalyticexperiments
263
10.4.2 Synthesis of 2-hydroxymethyl phenol 89 and carbonic acid 2-hydroxy-methyl-phenyl ester methyl ester 90
H
O
OH+
O
OOH
O
O
OO
O
O
O
OH
O
O
[Rh(COD)Cl]2 (10 mol%)ligand 1, ratio L/R 1:1
base
+
+
expected expected
product 90 obtained quant. yield
OH
OH
product 89 obtained quant. yield
neat, 100 °C, 24 h
Bases tested: K3PO4, Cs2CO3, Na2CO3, K2CO3
Results :
< ¼ eq of base : product 89¼< x <1 éq. of base: mix of product 89 and product 90> 1 eq of base: product 90
85 59
87 88
C7H8O2Mol. Wt.: 124,14
C9H10O4Mol. Wt.: 182,17
10
13
12
3
45
6
7 8
9
16
18
17
1514
1112
19
A mixture of salicylaldehyde 164 (23 μL, 0.22 mmol), acrylic acid methyl ester 201 (50 μL, 0.55 mmol, 2.5 eq), ligand 1 (0.022 mmol, 6.7 mg, 10 mol%), [Rh(COD)Cl]2 (0.022 mmol, 6.7 mg, 10 mol%) and a base was added to a 8 mL Schlenk tube and stirred for 1 day at 100 °C. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 2-hydroxymethyl phenol 231 (27.3 mg, 0.22 mmol, quant., with < ¼ eq of base) or carbonic acid 2-hydroxy-methyl-phenyl ester methyl ester 232 (40.1 mg, 0.22 mmol, quant., with > 1 eq of base). 2-hydroxymethyl phenol 89: 1H NMR (400.132 MHz, CDCl3): δ = 2.91 (s, 1H, OH(8)), 4.78 (s, 2H, CH2), 6.78-6.92 (m, 1H, OH(9)), 7.02 (d, 1H, Ar-H2), 7.19 (d, 1H, Ar-H3 or Ar-H4), 7.24 (d, 1H, Ar-H4 or Ar-H3), 7.48 (d, 1H, Ar-H5). Analytic data are in accordance with the literature.[212]
[212] L. Huang, T. Su, W. Shan, Z. Luo, Y. Sun, F. He, X. Li, Biorganic and Medicinal Chemistry 2012, 20(19), 3038-3048.
ExperimentalPart:Catalyticexperiments
264
10.5 Rhodium-Catalyzed o-vinylBenzaldehyde Hydroacylation 10.5.1 General procedure: o-vinylbenzaldehyde derivatives hydroacylation
protocol (GP1) The oil bath was pre-heated before adding the Schlenk tube. To a dry, 8 mL Schlenk tube was added the ligand and the catalyst. Then, argon and vacuum were exchanged three times. The two hydroacylation partners (alkene or alkyne; ketone or aldehyde) were then added to the Schlenk tube together with additives (bases or acids) and solvent when required. After the desired reaction time, the Schlenk tube was cooled to room temperature, the solvent was removed in vacuo, and the resulting oil or solid filtered over Celite. Products were purified by column chromatography or directly analyzed by 1H NMR, 13C NMR and HRMS.
O O[Rh(COD)2]BF4, L
Toluene, 150 °C, 1 h. RR
H
A mixture of substituted o-vinylbenzaldehyde (29.1 mg, 0.22 mmol), ligand 1 (0.011 mmol, 3.4 mg, 5 mol%), [Rh(COD)2]BF4 (0.011 mmol, 4.5 mg, 5 mol%) was dissolved in toluene (200 μL) in a 8 mL Schlenk tube and stirred for 1 hour at 150 °C. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2).
ExperimentalPart:Catalyticexperiments
265
10.5.2 Synthesis of indan-1-one 96
O
H+
95
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
96
12
3
45 6
O7
8
9
C9H8OMol. Wt.: 132,16
C9H8OMol. Wt.: 132,16
97%
Following GP1, o-vinylbenzaldehyde 95, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 1-indanone 96 and vinylbenzene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (AcOEt pure) to yield 1-indanone 96 (28.2 mg, 0.21 mmol, 97%) as a white powder. 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.68 (m, 2H, H9), 3.14 (t, 3J8-9 = 5.7 Hz, 2H, H8), 7.36 (br t, 3J5-4 = 7.5 Hz, 1H, Ar-H5), 7.47 (br d, 3J3-2 = 7.7 Hz, 1H, Ar-H3), 7.57 (td, 3J4-5 = 7.5 Hz, 3J4-3 = 1.3 Hz, 1H, Ar-H4), 7.75 (d, 3J2-3 = 7.7Hz, 1H, Ar-H2). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 25.9 (s, 1C, C9), 36.3 (s, 1C, C8), 123.8 (s, 1C, C2), 126.8 (s, 1C, C5), 127.4 (s, 1C, C6), 134.7 (s, 1C, C4), 137.2 (s, 1C, C1), 155.2 (s, 1C, C6), 207.1 (s, 1C, C7). HRMS (EI): C9H8O (M): 132.0575; Found: 132.0577 (difference +1.5 ppm).
Tm = 38-40 °C Rf = 0.85 (AcOEt pure) Analytic data are in accordance with the literature.[213]
[213] T. Suzuki, T. Ohwada, K. Shudo, J. Am. Chem. Soc. 1997, 119, 6774-6780.
Following GP1, 6-vinylveratraldehyde 99, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 5,6-dimethoxy-1-indanone 135 and 1,2-dimethoxy-4-vinylbenzene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 5,6-dimethoxy-indan-1-one 135 (41.4 mg, 0.22 mmol, 98%) as a brawn crystals. 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.63 (m, 2H, H9), 3.01 (m, 2H, H8), 3.87 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 6.86 (s, 1H, Ar-H2), 7.14 (s, 1H, Ar-H5). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 25.6 (s, 1C, C9), 36.5 (s, 1C, C8), 56.1 (s, 1C, C10), 56.3 (s, 1C, C11), 104.2 (s, 1C, C5), 107.6 (s, 1C, C2), 130.0 (s, 1C, C1), 149.5 (s, 1C, C6), 150.4 (s, 1C, C3), 155.5 (s, 1C, C4), 205.7 (s, 1C, C7). HRMS (EI): C11H12O3 (M): 192.0786; Found: 192.0789 (difference +1.5 ppm).
Tm = 128 °C Rf = 0.68 (AcOEt pure) Analytic data are in accordance with the literature.[128]
[128] S. V. Gagnier; R. C. Larock, J. Am. Chem. Soc. 2003, 125, 4804-4807.
ExperimentalPart:Catalyticexperiments
267
10.6.2 Synthesis of 1-oxo-indan-5-carboxylic acid methyl ester 136
O
H
103
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
136
12
3
4
5 6
O7
8
9
MeO
O
MeO
O
1011 +
O
MeO
C11H10O3Mol. Wt.: 190,20
C11H10O3Mol. Wt.: 190,20
98%
Following GP1, 3-formyl-4-vinylbenzoic acid methyl ester 103, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 1-oxo-indan-5-carboxylic acid methyl ester 136 and 4-vinyl-benzoic acid methyl ester (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 1-oxo-indan-5-carboxylic acid methyl ester 136 (41.4 mg, 0.22 mmol, 98%) as a white powder. 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.76 (m, 2H, H9), 3.21 (m, 2H, H8), 3.94 (s, 3H, OCH3), 7.55 (dq, 3J5-4 = 8.0 Hz, 4J5-9 = 0.8 Hz, 1H, Ar-H5), 8.27 (dd, 3J4-5 = 8.0 Hz, 4J4-2 = 1.7 Hz, 1H, Ar-H4), 8.42 (dq, 4J2-4 = 1.7 Hz, 5J2-5 = 0.8 Hz, 1H, Ar-H2). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 26.2 (s, 1C, C9), 36.6 (s, 1C, C8), 52.5 (s, 1C, C11), 125.4 (s, 1C, C2), 127.0 (s, 1C, C5), 130.0 (s, 1C, C3), 135.5 (s, 1C, C4), 137.5 (s, 1C, C1), 159.6 (s, 1C, C6), 166.4 (s, 1C, C10), 206.0 (s, 1C, C7). HRMS (EI): C11H10O3 (M): 190.0630; Found: 190.0628 (difference -1.1 ppm). Tm = 123 °C Rf = 0.84 (AcOEt pure) Analytic data are in accordance with the literature.[214]
[214] S. Chiba, Y.-J. Xu, Y.-F. Wang, J. Am. Chem. Soc. 2009, 131, 12886-12887.
ExperimentalPart:Catalyticexperiments
268
10.6.3 Synthesis of 5-phthalimido-indan-1-one 137 .
+
108
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
N
OH
OO N OO
137
NO O
1
2
1111
1010
99
88
7
64
3
5
O
16
17
C17H11NO3Mol. Wt.: 277,27
C17H11NO3Mol. Wt.: 277,27
95%
Following GP1, 4-phtalimido-2-vinylbenzaldehyde 108, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 2-(1-oxo-indan-6-yl-isoindole-1,3-dione 137 and 2-(3-vinyl-phenyl)-isoindole-1,3-dione (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 2-(1-oxo-indan-6-yl-isoindole-1,3-dione 137 (52.1 mg, 0.21 mmol, 95%) as a white powder. 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.76 (m, 2H, H16), 3.23 (m, 2H, H17), 7.52 (br d, 3J4,3 = 8.3 Hz, 1H, Ar-H4), 7.63 (m, 1H, Ar-H6), 7.83 (m, 2H, Ar-H11), 7.89 (br d, 3J3,4 = 8.3 Hz, 1H, Ar-H3), 7.99 (m, 2H, Ar-H10). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 26.0 (C16), 36.6 (C17), 124.2 (C10), 124.5 (C6), 124.6 (C3), 125.7 (C4), 131.7 (C9), 134.9 (C11), 136.4 (C2), 137.4 (C5), 156.0 (C1), 167.0 (C8), 206.0 (C7). HRMS (EI): C17H11O3N (M): 277.0739; Found: 277.0738 (difference -0.4 ppm).
Tm = 232 °C Rf = 0.85 (AcOEt pure)
ExperimentalPart:Catalyticexperiments
269
10.6.4 Synthesis of 4-methyl-indan-1-one 138
O
H+
110 138
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
Me
12
3
4
5 6
O7
8
910
Me
C10H10OMol. Wt.: 146,19
C10H10OMol. Wt.: 146,19
97%
Following GP1, 4-methyl-2-vinylbenzaldehyde 110, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 4-methyl-indan-1-one 138 and 1-methyl-3-vinyl-benzene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 4-methyl-indan-1-one 138 (25.2 mg, 0.22 mmol, 97%) as a colourless liquid. 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.44 (s, 3H, CH3), 2.67 (m, 2H, H9), 3.09 (m, 2H, H8), 7.18 (br d, 3J3-2 = 7.8 Hz, 1H, Ar-H3), 7.27 (m, 1H, Ar-H5), 7.65 (d, 3J3-2 = 7.8 Hz, 1H, Ar-H2) 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 22.2 (s, 1C, C10), 25.8 (s, 1C, C9), 36.5 (s, 1C, C8), 123.7 (s, 1C, C1), 127.2 (s, 1C, C5), 128.7 (s, 1C, C3), 135.0 (s, 1C, C2), 145.9 (s, 1C, C4), 155.8 (s, 1C, C6), 206.7 (s, 1C, C7).
HRMS (EI): C10H10O (M): 146.0732; Found: 146.0730 (difference -1.4 ppm). Rf = 0.87 (AcOEt pure) Analytic data are in accordance with the literature.[215]
[215] D. W. Boykin, R. L. Hertzler, J. K. Delphon, E. J. Eisenbraun, J. Org. Chem. 1989, 54, 1418-1423.
ExperimentalPart:Catalyticexperiments
270
10.6.5 Synthesis of 5-hydroxy-indan-1-one 139
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
12
3
4
5 6
O7
8
9
HO
O
H+
126 139
HO HO
C9H8O2Mol. Wt.: 148,16
C9H8O2Mol. Wt.: 148,16
95%
Following GP1, 5-hydroxy-2-vinylbenzaldehyde 126, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 5-hydroxy-indan-1-one 139 and 1-hydroxy-4-vinyl-benzene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 5-hydroxy-indan-1-one 139 (31.9 mg, 0.22 mmol, 95%) as a white powder.
Analytic data are in accordance with the literature.[216]
[216] M. Phialas, P. Sammes, P. D. Kennewell, R. Westwood, J. Chem. Soc., Perkin Trans. 1, 1984, 687-695.
ExperimentalPart:Catalyticexperiments
271
10.6.6 Synthesis of 4-nitro-indan-1-one 140
O
H
118
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
140
12
3
4
5 6
O7
8
9
+
C9H7NO3Mol. Wt.: 177,16
C9H7NO3Mol. Wt.: 177,16
93%
O2N O2N O2N
Following GP1, 4-nitro-2-vinylbenzaldehyde 118, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 4-nitro-indan-1-one 140 and 1-nitro-3-vinyl-benzene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 4-nitro-indan-1-one 140 (36.2 mg, 0.20 mmol, 93%) as a yellowish solid. 1H-NMR (400.132 MHz, CDCl3): δ (ppm) = 2.83 (m, 2H, H9), 3.28 (m, 2H, H8), 7.90 (br d, 3J5-4 = 8.4 Hz, 1H, Ar-H5), 8.23 (br d, 3J4-5 = 8.4 Hz, 1H, Ar-H3), 8.35 (m, 1H, Ar-H2). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 26.0 (s, 1C, C9), 36.8 (s, 1C, C8), 122.2 (s, 1C, C2), 123.0 (s, 1C, C4), 124.9 (s, 1C, C5), 141.4 (s, 1C, C1), 152.0 (s, 1C, C3), 155.8 (s, 1C, C6), 205.0 (s, 1C, C7).
HRMS (EI): C9H7O3N (M): 177.0426; Found: 177.0427 (difference +0.5 ppm). Tm = 126 °C Rf = 0.87 (AcOEt pure) Analytic data are in accordance with the literature.[217]
[217] S. Kapur, N. C. Mathur, K. McManus, A. L. Pincock, J. A. Pincock, Canadian Journal of Chemistry 1988, 66(11), 2888-2893.
ExperimentalPart:Catalyticexperiments
272
10.6.7 Synthesis of 5-chloro-indan-1-one 141
O
H+
122 141
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
Cl Cl12
3
4
5 6
O7
8
9
Cl
C9H7ClOMol. Wt.: 166,60
C9H7ClOMol. Wt.: 166,60
91%
Following GP1, 5-chloro-2-vinylbenzaldehyde 122, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 5-chloro-indan-1-one 141 and 1-chloro-4-vinyl-benzene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 5-chloro-indan-1-one 141 (33.2 mg, 0.20 mmol, 91%) as a yellow oil. 1H-NMR (400.132 MHz, CDCl3): δ (ppm) = 2.73 (m, 2H, H9), 3.12 (m, 2H, H8), 7.42 (dq, 3J5-4 = 8.2 Hz, 4J5-9 = 0.7 Hz, 1H, Ar-H5), 7.54 (dd, 3J4-5 = 8.2 Hz, 5J4-9 = 2.1 Hz, 1H, Ar-H4), 7.71 (dq, 4J2-4 = 2.1 Hz, 5J2-5 = 0.7 Hz, 1H, Ar-H2). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 25.6 (s, 1C, C9), 36.8 (s, 1C, C8), 123.8 (s, 1C, C2), 128.0 (s, 1C, C5), 133.9 (s, 1C, C3), 134.7 (s, 1C, C4), 138.8 (s, 1C, C1), 153.2 (s, 1C, C6), 205.6 (s, 1C, C7).
HRMS (EI): C9H7OCl (M): 166.0185; Found: 166.0181 (difference -2.4 ppm). Rf = 0.90 (AcOEt pure) Analytic data are in accordance with the literature.[218]
[218] M. Slusarczyk, W. M. De Borggraeve, S. Toppet, G. J. Hoornaert, Eur. J. Org. Chem. 2007, 18, 2987-2994.
ExperimentalPart:Catalyticexperiments
273
10.6.8 Synthesis of 5-fluoro-indan-1-one 142
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
12
3
4
5 6
O7
8
9
F
O
H+
124 142
F F
C9H7FOMol. Wt.: 150,15
C9H7FOMol. Wt.: 150,15
92%
Following GP1, 5-fluoro-2-vinylbenzaldehyde 124, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 5-fluoro-indan-1-one 142 and 1-fluoro-4-vinyl-benzene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 5-fluoro-indan-1-one 142 (30.4 mg, 0.20 mmol, 92%) as a yellow solid. 1H-NMR (400.132 MHz, CDCl3): δ (ppm) = 2.73 (m, 2H, H9), 3.12 (m, 2H, H8), 7.29 (dt, 3J4-5 = 8.6 Hz, 4J4-2 = 2.6 Hz, 1H, Ar-H4), 7.38 (br d, 3J5-4 = 7.6 Hz, 1H, Ar-H5), 7.44 (dd, 4J2-4 = 8.6 Hz, 5J2-5 = 4.6 Hz, 1H, Ar-H2). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 25.4 (s, 1C, C9), 37.1 (s, 1C, C8), 109.7 (d, 2J2-F = 22.1 Hz, 1C, C2), 122.4 (d, 2J4-F = 23.8 Hz, 1C, C4), 128.2 (d, 3J5-F = 8.1 Hz, 1C, C5), 138.9 (d, 3J1-F = 7.2 Hz, 1C, C1), 150.6 (d, 4J6-F = 2.2 Hz, 1C, C6), 162.5 (d, 1J3-F = 248.1 Hz, 1C, C3), 206.0 (d, 4J7-F = 3.1 Hz, 1C, C7). 19F {1H} NMR (235.333 MHz, CDCl3): δ (ppm) = 114.8. HRMS (EI): C9H7OF (M): 150.0481; Found: 150.0480 (difference -0.7 ppm). Rf = 0.97 (AcOEt pure) Analytic data are in accordance with the literature.[216]
[216] M. Phialas, P. Sammes, P. D. Kennewell, R. Westwood, J. Chem. Soc., Perkin Trans. 1, 1984, 687-695.
ExperimentalPart:Catalyticexperiments
274
10.6.9 Synthesis of 1,2-dihydro-cyclopenta[α]naphthalene 143
+
114 143
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °CO
H1
87
65
43
2
10
9
11
12
13
O
C13H10OMol. Wt.: 182,22
C13H10OMol. Wt.: 182,22
97%
Following GP1, 1-vinylnaphtalene-2-carbaldehyde 114, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 1,2-dihydro-cyclopenta[α]naphthalene-3-one 143 and 1-vinyl-naphtalene (traces) were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield 6-nitro-indan-1-one 143 (38.8 mg, 0.22 mmol, 97%) as a yellowish solid. 1H-NMR (400.132 MHz, CDCl3): δ (ppm) = 2.84 (m, 2H, H9), 3.45 (m, 2H, H8), 7.66 (m, 2H, Ar-H8 and Ar-H7), 7.75 (d, 3J4-3 = 8.4 Hz, 1H, Ar-H4), 7.81 (d, 3J3-4 = 8.4 Hz, 1H, Ar-H3), 7.95 (br d, 3J6-7 = 7.9 Hz, 1H, Ar-H6), 8.06 (br d, 3J9-8 = 7.9 Hz, 1H, Ar-H9) 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 24.5 (s, 1C, C12), 36.3 (s, 1C, C13), 119.6 (s, 1C, C4), 124.5 (s, 1C, C9), 127.2 (s, 1C, C7), 128.6 (s, 1C, C3), 129.0 (s, 1C, C6), 129.3 (s, 1C, C8), 130.7 (s, 1C, C5), 134.8 (s, 1C, C2), 136.7 (s, 1C, C10), 156.5 (s, 1C, C1), 206.9 (s, 1C, C11)
Tm = 125 °C Rf = 0.87 (AcOEt pure) Analytic data are in accordance with the literature.[219]
[219] R. Takeuchi, H. Yasue, J. Org. Chem. 1993, 58, 5386-5392.
ExperimentalPart:Catalyticexperiments
275
10.6.10 Synthesis of 6,7-dihydro-[1]-pyridin-5-one 144
N
O
H
129 144
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C
not obtained !
N
12
3
4
56
O7
8
9
C8H7NOMol. Wt.: 133,15
C8H7NOMol. Wt.: 133,15
Following GP1, 2-vinylpyridine-3-carbaldehyde 129, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 6,7-dihydro-[1]-pyrindin-5-one 144 was not detected by 1H NMR in the crude, and the starting material was recovered quantitatively.
ExperimentalPart:Catalyticexperiments
276
10.7 Rhodium-catalyzed hydroacylation of 2-vinylthiophene-3-carbaldehyde
10.7.1 Synthesis of thiophene cyclobutane dimer 146
S
H
O
S
O
H
+
[Rh(COD)2]BF4 (5 mol%)
ligand 1 (5 mol%)
toluene (c = 1.1 M) 1 h, 150 °C not obtained !
1
65
4
3
2
S
H
O
134
S
145
O
146
7
8
87
1
6
3
2
C7H6OSMol. Wt.: 138,19
C7H6OSMol. Wt.: 138,19
C14H12O2S2Mol. Wt.: 276,37
4
5
quant. (96)
Following GP1, 2-vinylthiophene-3-carbaldehyde 134, ligand 1 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. No 5,6-dihydro-cyclopenta[b]thiophen-4-one 145 was detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield a 1,2-trans-thiophene cyclobutane dimer 146 (30.4 mg, 0.11 mmol, quant.) as a colorless oil. 1H-NMR (400.132 MHz, CDCl3): δ (ppm) 2.31 (m, 2H, H8), 2.60 (m, 2H, H8), 4.48 (m, 2H, H7), 7.17 (d, 3J5-4 = 5.4 Hz, 2H, Ar-H5), 7.37 (d, 3J4-5 = 5.4 Hz, 2H, Ar-H4), 9.81 (s, 2H, H6). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 28.7 (s, 2C, C7), 44.3 (s, 2C, C8), 123.8 (s, 2C, C5), 128.3 (s, 2C, C4), 137.2 (s, 2C, C3), 158.4 (s, 2C, C2), 184.1 (s, 2C, C6).
ExperimentalPart:Catalyticexperiments
277
10.8 Synthesis of Donepezil® hydrochloride 152
10.8.1 Synthesis of N-benzyl-4-ethoxycarbonylpiperidine 149
NH
OO
+Cl N
OO
147 148 149
Et3N, toluene, o/n, 80 °C
98%
C15H21NO2Mol. Wt.: 247,33
C7H7ClMol. Wt.: 126,58
C8H14NO2Mol. Wt.: 156,20
14
35
3
2
6 7
9
12
8
1311
1012
11
A solution of ethyl 4-piperidinecarboxylate 147 (15 g, 95.4 mmol), NEt3 (11.6 g, 114.7 mmol) and benzyl chloride 148 (12.1 g, 95.6 mmol, 1.2 eq) in toluene (100 mL) was heated at 80 °C overnight. After cooling to room temperature, H2O (100 mL) was added, followed by extraction with toluene (3 x 50 mL). The solvent was removed in vacuo and the yellow oil containing N-benzyl-4-ethoxycarbonylpiperidine 149 was used for the next step without purification (23.1 g, 93.4 mmol, 98%). 1H-NMR (300.07 MHz, CDCl3): δ (ppm) = 1.24 (t, 3J8-7 = 7.0 Hz, 3H, CH3), 1.85 (m, 4H, H3), 2.03 (td, J = 11.2 Hz, J = 2.8 Hz, 2H, H2 or H4), 2.26 (m, 1H, H4), 2.85 (m, 2H, H6 or H2), 3.49 (s, 2H, CH2(9)), 4.13 (q, 3J7-8 = 7.0 Hz, 2H, CH2(7)), 7.30 (m, 5H, Ar-H11, Ar-H12 and Ar-H13). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 14.2 (s, 1C, C8), 28.3 (s, 2C, C3), 41.2 (s, 2C, C9), 52.9 (s, 1C, C5), 60.2 (s, 2C, C2 and C4), 63.2 (s, 1C, C7), 126.9 (s, 1C, C13), 128.2 (s, 2C, C12), 129.1 (s, 2C, C11), 138.4 (s, 1C, C10), 175.3 (s, 1C, C6). Rf = 0.38 (cHex/AcOEt 9:1) Analytic data are in accordance with the literature.[220]
[220] T. Abe, T. Haga, S. Negi, Y. Morita, K. Takayanagi, K. Hamamura, Tetrahedron 2001, 57, 2701-2710.
ExperimentalPart:Catalyticexperiments
278
10.8.2 Synthesis of 1-benzyl-4-formylpiperidine 150
Diisobutylaluminum hydride (1 M in toluene, 44.7 mL, 44.7 mmol, 1.1 eq) was added dropwise at -78 °C to a stirred solution of ethyl 1-benzylpiperidine-4-carboxylate 149 (10.1 g, 40.6 mmol) in toluene (400 mL). The mixture was stirred at this temperature for 1 hour further before being quenched with MeOH (150 mL). The dry ice bath was removed and the solution stirred for 2 hours at room temperature. The mixture was then filtered through Celite, washed with MeOH (2 x 50 mL) and concentrated in vacuo. The crude product was purified by vacuum distillation (bp 93-97 °C/1 mmHg) giving 1-benzyl-4-formylpiperidine 150 as a yellow oil (7.6 g, 40.0 mmol, 91%). 1H-NMR (300.07 MHz, CDCl3): δ (ppm) = 1.69 (m, 2H, H3 or H5), 1.87 (m, 2H, H3 or H5), 2.11 (td, J = 10.9 Hz, J = 2.6 Hz, 2H, H2 or H6), 2.23 (m, 1H, H4), 2.81 (m, 2H, H2 or H6), 3.50 (s, 2H, CH2(8)), 7.28 (m, 5H, Ar-H10, Ar-H11 and Ar-H12), 9.64 (d, 3J7-4 = 1.3 Hz, 1H, H7). 13C{1H} NMR (100.6 MHz, CDCl3) δ = 25.4 (s, 2C, C3 and C5), 48.0 (s, 1C, C4), 52.5 (s, 2C, C2 and C6), 63.2 (s, 1C, C8), 127.0 (s, 1C, C12), 128.2 (s, 2C, C11), 129.0 (s, 2C, C10), 138.2 (s, 1C, C9), 204.0 (s, 1C, C7). Tb = 93-97 °C (1 mmHg). Analytic data are in accordance with the literature.[221]
10.8.3 Synthesis of 5,6-dimethoxy-indan-1-one 135 Refer to p. 285 to see more details about the synthesis.
[221] N. Niphade, A. Mali, K. Jagtap, R. C. Ojha, P. J. Vankawala; V. T. Mathad, Org. Process Res. Dev. 2008, 12, 731-735.
ExperimentalPart:Catalyticexperiments
279
10.8.4 Synthesis of 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-ylidenyl] methyl piperidine 151
N
O
150
H
+
O
O
N
O
O
O
76%
NaOH, THF, 3 h, reflux
135 151
O
C13H17NOMol. Wt.: 203,28
C11H12O3Mol. Wt.: 192,21
C24H27NO3Mol. Wt.: 377,48
1
6
5 4 32
78
12
109
11
2019
17
16
18
15
14
20
2122
21
13
5,6-dimethoxy-1-indanone 135 (2 g, 10.4 mmol) and 1-benzyl-4-formylpiperidine 150 (2.6 g, 12.7 mmol, 1.2 eq) were added to a solution of NaOH (2.6 g, 65 mmol, 6.2 eq) in THF (20 mL) and the mixture was refluxed for 3 hours. After cooling to room temperature, H2O (10 mL) was added, followed by extraction with DCM (3 x 30 mL). The solvent was removed in vacuo and the residue recrystallised from hot acetone to afford pure 1-benzyl-4[(5,6-dimethoxy-1-indanon)-2-ylidenyl] methyl piperidine 151 as a white crystalline solid (2.98 g, 7.91 mmol, 76%). 1H NMR (300.07 MHz, (CD3)2SO): δ (ppm) = 1.51 (m, 2H, H14 or H16), 1.67 (m, 2H, H14 or H16), 2.03 (br t, J = 10.9 Hz, 2H, H15 or H17), 2.34 (m, 1H, H13), 2.83 (m, 2H, H15 or H17), 3.48 (s, 2H, H10), 3.62 (s, 2H, CH2(18)), 3.81 (s, 3H, CH3(5) or CH3(6)), 3.88 (s, 3H, CH3(5) or CH3(6)), 6.45 (br d, J = 9.5 Hz, 1H, H12), 7.13 (s, 1H, Ar-H8 or Ar-H3), 7.16 (s, 1H, Ar-H8 or Ar-H3), 7.29 (m, 5H, Ar-H20, Ar-H21 and Ar-H22). 13C{1H} NMR (100.6 MHz, (CD3)2SO): δ = 28.9 (s, 1C, C10), 30.8 (s, 1C, C13), 36.4 (s, 2C, C14 and C16), 52.5 (s, 2C, C15 and C17), 55.6 (s, 1C, C5 or C6), 56.0 (s, 1C, C5 or C6), 62.5 (s, 1C, C18), 104.5 (s, 1C, C3 or C8), 108.1 (s, 1C, C3 or C8), 126.8 (s, 1C, C22), 128.1 (s, 2C, C21), 128.8 (s, 2C, C20), 130.7 (s, 2C, C2 and C11), 135.8 (s, 1C, C1), 138.4 (s, 1C, C9 or C19), 138.9 (s, 1C, C9 or C19), 144.6 (s, 1C, C12), 149.2 (s, 1C, C4), 155.2 (s, 1C, C7), 191.2 (s, 1C, C1). Analytic data are in accordance with the literature.[221]
[221] N. Niphade, A. Mali, K. Jagtap, R. C. Ojha, P. J. Vankawala; V. T. Mathad, Org. Process Res. Dev. 2008, 12, 731-735.
ExperimentalPart:Catalyticexperiments
280
10.8.5 Synthesis of Donepezil® Hydrochloride 152
N
O
O
O
1
6
5 4 32
78
12
109
11
2019
17
16
18
15
14
20
2122
21
13
N
O
O
O
73%
151 152
1) CoCl2.6H2O, THF, 10 min., RT
2) NaBH4, MeOH, 1 h, 10°C3) HCl(aq), 2 h, RT
C24H27NO3Mol. Wt.: 377,48
C24H30ClNO3Mol. Wt.: 415,95
.HCl
CoCl2·6H2O (60 mg, 0.28 mmol, 0.2 eq) was added to a solution of 1-benzyl-4[(5,6-dimethoxy-1-indanon)-2-ylidenyl] methyl piperidine 151 (490 mg, 1.30 mmol) in THF (12 mL) and stirred for 10 minutes at room temperature. The mixture was then cooled to 10 °C and MeOH (3 mL) followed by NaBH4 (100 mg, 2.6 mmol, 2.0 eq) were successively added. The solution was stirred for a further hour at this temperature before the addition of CH2Cl2 (15 mL). The mixture was then poured in H2O (25 mL) and extracted with CH2Cl2 (3 x 25 mL). The solvent was removed in vacuo and the resulting residue was dissolved in MeOH (500 μL) and AcOEt (4 mL). Hydrochloric acid (500 μL) was added to the solution and stirred for 2 hours at room temperature. The solvent was then removed in vacuo and the residue recrystallised from MeOH/TBME to afford pure Donepezil Hydrochloride® 152 as a white crystalline solid (0.395 g, 0.95 mmol, 73%).
Rf = 0.07 (AcOEt pure) Analytic data are in accordance with the literature.[221]
[221] N. Niphade, A. Mali, K. Jagtap, R. C. Ojha, P. J. Vankawala; V. T. Mathad, Org. Process Res. Dev. 2008, 12, 731-735.
ExperimentalPart:Catalyticexperiments
281
10.9 Rhodium-Catalyzed o-allylBenzaldehyde Hydroacylation 10.9.1 General procedure: o-allylbenzaldehyde derivatives hydroacylation
protocol (GP2) The oil bath was set at the desired temperature before immersing the Schlenk tube. In a dry 8 mL Schlenk tube was added the ligand and the catalyst. Then, argon and vacuum were exchanged three times. The o-allylbenzaldehyde derivative substrate was then added to the Schlenk tube together with additives (bases or acids) and solvent when required. After the desired reaction time, the Schlenk tube was cooled to room temperature, the solvent was removed in vacuo, and the resulting oil or solid filtered over Celite. Products were purified by column chromatography or directly analyzed by 1H NMR, 13C NMR and HRMS.
O [Rh(COD)2]BF4Ligand 7
Neat, 120 °C, 3 d.R
H
O
R
O
+
ThermodynamicProduct
KineticProduct
C10H10OMol. Wt.: 146,19
R
A mixture of substituted o-allylbenzaldehyde (32.2 mg, 0.22 mmol), ligand 7 (0.011 mmol, 3.4 mg, 5 mol%), [Rh(COD)2]BF4 (0.011 mmol, 4.5 mg, 5 mol%) in a 8 mL Schlenk tube and magnetically stirred for 3 days at 120 °C. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2).
ExperimentalPart:Catalyticexperiments
282
10.9.2 Synthesis of 1-tetralone 156 and 2-methylindanone 157
H
O
toluene (c = 1.1 M)120 °C, 24 h O
+
O
[Rh(COD)2]BF4
155 156 157C10H10O
Mol. Wt.: 146,19C10H10O
Mol. Wt.: 146,19C10H10O
Mol. Wt.: 146,19
ligand 7
(10 mol%)
(10 mol%)
12
34
56
7
89
10
1112 13
14151617
1819
20
Following GP2, o-allylbenzaldehyde 155, ligand 7 and catalyst ([Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 1-tetralone 156 and 2-methylindanone 157 were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (AcOEt pure) to yield 1-tetralone 156 (27.7 mg, 0.19 mmol, 85%) as a colourless liquid and 2-methylindanone 157 (4.0 mg, 0.03 mmol, 15%). 1-tetralone 156: 1H-NMR (400.132 MHz, CDCl3): δ (ppm) = 2.11-2.17 (m, 2H, Ar-H3), 2.66 (t, 3J4-3 = 6.2 Hz, 2H, Ar-H4), 2.97 (t, 3J2-3 = 6.1 Hz, 2H, Ar-H2), 7.24-7.26 (m, 1H, Ar-H8), 7.28-7.32 (m, 1H, Ar-H6), 7.44-7.49 (m, 1H, Ar-H7), 8.02-8.04 (m, 1H, Ar-H9). 13C {1H} NMR (100.613 MHz, CDCl3): δ (ppm) = 23.3 (s, 1C, C3), 29.7 (s, 1C, C4), 39.2 (s, 1C, C2), 126.6 (s, 1C, C8), 127.2 (s, 1C, C6), 128.8 (s, 1C, C9), 132.6 (s, 1C, C7), 133.4 (s, 1C, C10), 144.5 (s, 1C, C5), 198.4 (s, 1C, C1).
Tm = 113-116 (6 mmHg) Analytic data are in accordance with the literature.[222 a]
[222] a) T. Maji, A. Karmakar, O. Reiser, J. Org. Chem. 2011, 76(2), 736-739. b) G. B. Womack, J. G. Angeles, V. E. Fanelli, B. Indradas, R. L. Snowden, P. Sonnay, J. Org. Chem. 2009, 74(15), 5738-5741.
Following GP2, 2-allyl-naphthalene-1-carbaldehyde 180, ligand 7 and catalyst [Rh(COD)2]BF4 were heated neat in a Schlenk tube. 3,4-dihydro-2H-phenanthren-1-one 191 and 2-methyl-2,3-dihydro-cyclopenta[a]naphthalen-1-one 192 were detected by 1H NMR in the crude. 100 % conversion was observed and a yield (191/192, 66:34). Following GP2, 2-allyl-naphthalene-1-carbaldehyde 180, ligand 7 and catalyst [Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 3,4-dihydro-2H-phenanthren-1-one 191 and 2-methyl-2,3-dihydro-cyclopenta[a]naphthalen-1-one 192 were detected by 1H NMR in the crude. 100 % conversion was observed and a yield (191/192, 45:55).
[223] a) N. Kim, Y. Kim, W. Park, D. Sung, A. K. Gupta, C. H. Oh, Org. Lett. 2005, 7(23), 5289-5291. b) M. K. J ter Wiel, R. A. van Delden, A. Meetsma, B. L. Feringa, J. Am. Chem. Soc. 2003, 125(49), 15076-15086.
ExperimentalPart:Catalyticexperiments
284
10.10.2 Synthesis of 6,7-dimethoxy-3,4-dihydro-2H-naphthalene-1-one 193 and 5,6-dimethoxy-2-methyl-1-indanone 194
H
O
toluene (c = 1.1 M)120 °C, 72 h O
+
O
[Rh(COD)2]BF4
185 193 194
ligand 7
(10 mol%)
(10 mol%)
12
34
567
8 9
1012
1413
151617
19 20
21
MeO
MeO
MeO
MeO
MeO
MeO10
11
C12H14O3Mol. Wt.: 206,24
C12H14O3Mol. Wt.: 206,24
C12H14O3Mol. Wt.: 206,24
1823
22
Following GP2, 2-allyl-4,5-dimethoxy-benzaldehyde 185, ligand 7 and catalyst [Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 6,7-dimethoxy-3,4-dihydro-2H-naphthalene 193 and 5,6-dimethoxy-2-methyl-1-indanone 194 were detected by 1H NMR in the crude. 91 % conversion was observed and a yield (193/194, 79:21). 6,7-dimethoxy-3,4-dihydro-2H-naphthalene 193: 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.07 (q, 3J3-4 = 3J3-2 = 6.2 Hz, 2H, Ar-H3), 2.53 (t, 3J4-3 = 6.7 Hz, 2H, Ar-H4), 2.84 (t, 3J2-3 = 6.0 Hz, 2H, Ar-H2), 3.86 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 6.60 (s, 1H, Ar-H6), 7.44 (s, 1H, Ar-H9). Analytic data are in accordance with the literature.[224 a] 5,6-dimethoxy-2-methyl-1-indanone 194: 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 1.20 (d, 3J13-14 = 7.2 Hz, 3H, CH3), 2.51-2.64 (m, 2H, CH2), 3.22 (dd, J = 16.4 Hz, J = 7.2 Hz, 1H, H14), 3.82 (s, 1H, OCH3), 3.88 (s, 1H, OCH3), 6.79 (s, 1H, Ar-H17), 7.08 (s, 1H, Ar-H20). Analytic data are in accordance with the literature.[224 b]
[224] a) S. P. Chavan, S. Garai, A. K. Dutta, S. Pal, Eur. J. of Chem. 2012, 35, 6841-6845. b) E. Fillion, D. Fishlock, A. Wilsily, J. M. Goll, J. Org. Chem. 2005, 70(4), 1316-1327.
ExperimentalPart:Catalyticexperiments
285
10.10.3 Synthesis of 6-chloro-3,4-dihydro-2H-naphthalene-1-one 195 and 6-chloro-2-methyl-1-indanone 196
H
O
toluene (c = 1.1 M)120 °C, 72 h O
+
O
[Rh(COD)2]BF4
190 195 196
ligand 7
(10 mol%)
(10 mol%)
12
34
56
7
8 9
1011
1312
141516
18 19
20
Cl Cl Cl
17
C10H9ClOMol. Wt.: 180,63
C10H9ClOMol. Wt.: 180,63
C10H9ClOMol. Wt.: 180,63
Following GP2, 2-allyl-5-chloro-benzaldehyde 190, ligand 7 and catalyst [Rh(COD)2]BF4 were heated in dry toluene in a Schlenk tube. 6-chloro-3,4-dihydro-2H-naphthalene 195 and 6-chloro-2-methyl-1-indanone 196 were detected by 1H NMR in the crude. 76 % conversion was observed and a yield (195/196, 86:14). 6-chloro -3,4-dihydro-2H-naphthalene 195: 1H NMR (400.132 MHz, CDCl3): δ (ppm) = 2.04-2.23 (m, 2H, Ar-H3), 2.57-2.72 (m, 2H, Ar-H4), 2.86-3.01 (m, 2H, Ar-H2), 7.19 (d, 3J6-7 = 8.4 Hz, 1H, Ar-H6), 7.42 (dd, 3J7-6 = 8.4
Hz, 4J7-9 = 2.2 Hz, 1H, Ar-H7), 7.19 (d, 4J9-7 = 2.2 Hz, 1H, Ar-H9). Analytic data are in accordance with the literature.[225 a]
[225] a) F. I Carroll, B. E. Blough, P. Abraham, A. C. Mills, J. A. Holleman, S. A. Wolckenhauer, A. M. Decker, A. Landavazo, K. T. McElroy, H. A. Navarro, M. B. Gatch, M. J. Foster, J. Med. Chem. 2009, 52 (21), 6726-6781. b) Z.-G. Wang, L. Chen, J.-F. Zheng, W. Gao, Z. Zeng, H. Zhou, Z.-K. Zhang, P.-Q. Huang, Y. Su, Eur. J. Of. Med. Chem. 2013, 62, 632-648.
ExperimentalPart:Catalyticexperiments
286
10.11 Rhodium-catalyzed hydroacylation of 5-hexenal and synthesis of medium-sized ring
10.11.1 Synthesis of cyclohexanone 198 and 2-methylcyclopentanone 199
H
O
OO
N
OMe
NH2
P
+
197 198 199C6H10O
Mol. Wt.: 98,14C6H10O
Mol. Wt.: 98,14C6H10O
Mol. Wt.: 98,14
12
3
43
2 10
9
7
6
5
8Neat, 120 °C, 1 d
(10 mol%)
(10 mol%)[Rh(COD)BF4]
A mixture of substituted hex-5-enal 197 (21.6 mg, 0.22 mmol), ligand 7 (0.022 mmol, 7.1 mg), [Rh(COD)2]BF4 (0.022 mmol, 9 mg) was added to a 8 mL Schlenk tube and stirred for 1 day at 120 °C. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2). Cyclohexanone 198 and 2-methylpentanone 199 were detected by 1H NMR in the crude. The crude product was purified by flash chromatography (CH2Cl2 pure) to yield cyclohexanone 198 (21.2 mg, 0.21 mmol, 98%) as a colourless oily liquid.
Analytic data are in accordance with the literature.[227]
[226] P. Wang, J. Cai, J. Yang, C. Sun, L. Li, H. Hu, M. Ji, Tetrahedron Letters 2013, 54(6), 533-535. [227] A. Friszkowska, H. Toogood, M. Sakuma, J. M. Gardiner, G. M. Stephens, N. S. Scrutton, Adv. Synth. Cat. 2009, 351(17), 2976-2990.
ExperimentalPart:Catalyticexperiments
287
10.11.2 Synthesis of cyclododecanone 202
N
OMe
NH2
P
201 202
Toluene (c = 1.1 M)1 d, 120 °C
(10 mol%)
(13 mol%)[Rh(COD)BF4]O
H
C10H18OMol. Wt.: 154,25
O
C10H18OMol. Wt.: 154,25
A mixture of substituted hex-5-enal 201 (33.9 mg, 0.22 mmol), ligand 7 (0.022 mmol, 7.1 mg), [Rh(COD)2]BF4 (0.022 mmol, 9 mg) was dissolved in toluene (200 μL) in a 8 mL Schlenk tube and stirred for 1 day at 120 °C. The crude was analyzed by 1H NMR, no product 202 was detected.
10.11.3 Synthesis of 6,7,8,9,10,11-hexahydro-5-oxa-benzocyclodecen-12-one 205
N
OMe
NH2
P
204 205
Toluene (c = 1.1 M)1 d, 120 °C
(10 mol%)
(13 mol%)[Rh(COD)BF4]
C10H18OMol. Wt.: 154,25
O
H
O
C13H16O2Mol. Wt.: 204,26
O
O
A mixture of substituted 2-hex-5-enyloxy-benzaldehyde 204 (44.9 mg, 0.22 mmol), ligand 7 (0.022 mmol, 7.1 mg), [Rh(COD)2]BF4 (0.022 mmol, 9 mg) was dissolved in toluene (200 μL) in a 8 mL Schlenk tube and stirred for 1 day at 120 °C. The crude was analyzed by 1H NMR, no product 205 was detected.
A mixture of substituted benzaldehyde 44 (48 μL, 0.44 mmol), ligand 1 (147 mg 0.44 mmol, 1.0 eq) and MgSO4 (100 mg) was dissolved in toluene (2 mL) in a 8 mL Schlenk tube and stirred for 24 hours at 120 °C. The solution was concentrated to give a residue that was purified by a flash silica gel column chromatography (eluting with CH2Cl2) to yield “imine P-N ligand 1” benzylidene-{6[(diphenylphosphanyl)-methyl]-3-methylpyridin-2-yl}-amine 208. 1H NMR (400.130 MHz, CDCl3): δ (ppm) = 8.81 (s, iminic proton), 10.06 (aldehydic proton). 31P {1H} NMR (101.3 MHz, CDCl3): δ (ppm) = -9.8.
ExperimentalPart:Mechanisticstudies
289
11.1.2 Synthesis of “Rh(imine P-N ligand 1)2]+ Cl- obtained with
[Rh(COD)Cl]2
O
H+
N NH2
Me
PPh2
N N
Me
PPh2toluene (c = 1.1 M)
150 °C, 3 d H
C19H19N2OPMol. Wt.: 322,34
C7H6OMol. Wt.: 106,12
44 1 208
N N
Me
PPh2H
+
MgSO4
96%
[Rh(COD)Cl]2
N N
Me
PPh2
imine PN ligand 1
Rh
N
Ph2P
N
C2D2Cl2O2Rh2Mol. Wt.: 336,77
CDCl3, RT, 10 min.
Me
C26H23N2PMol. Wt.: 394,45
C52H46N4P2RhMol. Wt.: 891,80
206
quant.
ExperimentalPart:Mechanisticstudies
290
ExperimentalPart:Mechanisticstudies
291
11.2 The influence of the P-N ligand 11.2.1 Synthesis of “imine P-N ligand 7” benzylidene-
A mixture of substituted benzaldehyde 44 (48 μL, 0.44 mmol), ligand 7 (155 mg 0.44 mmol, 1.0 eq) and MgSO4 (100 mg) was dissolved in toluene (2 mL) in a 8 mL Schlenk tube and stirred for 24 hours at 120 °C. The solution was concentrated to give a residue that was purified by a flash silica gel column chromatography (eluting with CH2Cl2) to yield “imine P-N ligand 7” benzylidene-{6[(diphenylphosphanyl)-methyl]-3-methylpyridin-2-yl}-amine 210. 1H-NMR (400.130 MHz, CDCl3): δ (ppm) = 8.89 (s, iminic proton), 10.06 (aldehydic proton). 31P {1H} NMR (101.3 MHz, CDCl3): δ (ppm) = -10.1.
ExperimentalPart:Mechanisticstudies
292
11.3 The deuterium-labeling studies
11.3.1 Synthesis of 1-phenylnonan-1-one-α-1-d 233 and 1-phenylnonan-1-one- β-1-d 234
+
O
D
O
D
+
ratio 213/214 (1.4:1)
[Rh(COD)Cl]2, toluene
150 °C, 24 h82%
C7H5DOMol. Wt.: 107,13
C8H16Mol. Wt.: 112,21
C15H21DOMol. Wt.: 219,34
C15H21DOMol. Wt.: 219,34
212 45
213
214
PN ligand 1
1
2 53
4 86
7 9
13
1211
10
1211
O
1
2 5
34 86
7 9
13
1211
10
1211
D
Following GP2, oct-1-ene 45, benzaldehyde-1-d 212, ligand 1 and catalyst [Rh(COD)Cl]2 were heated in dry toluene in a Schlenk tube. The solution was concentrated to give a residue that was purified by silica gel column chromatography (eluting with CH2Cl2) to yield 1-phenylnonan-1-one-α-1-d 213 and 1-phenylnonan-1-one-β-1-d 214 (39.3 mg, 0.18 mmol, 82%), ratio 213/214 (1.4:1). 1H-NMR (400.132 MHz, CDCl3): δ = 0.88 (t, 3J9-8 = 7.2 Hz, 3H, H9), 1.32 (m, 10H, H4-8), 1.74 (quint, 3J3-4 = 3J3-2 = 7.3 Hz, 2H, H3), 2.96 (t, 3J2-3 = 7.3 Hz, 2H, H2), 7.46 (m, 2H, Ar-H12), 7.56 (m, 1H, Ar-H13), 7.96 (m, 2H, Ar-H11). 13C {1H} NMR (100.613 MHz, CDCl3): δ = 14.3 (s, 1C, C9), 22.8 (s, 1C, C8), 24.6 (s, 1C, C3), 29.3 (s, 1C, C4), 29.5 (s, 1C, C5 or C6), 29.6 (s, 1C, C5 or C6), 32.0 (s, 1C, C7), 38.8 (s, 1C, C2), 128.2 (s, 2C, C12), 128.7 (s, 2C, C11), 133.0 (s, 1C, C13), 137.3 (s, 1C, C10), 200.8 (s, 1C, C1).
293
12. Rhodium catalyst synthesis 12.1 Synthesis of rhodium(I) tris-(triphenylphosphine) chloride,
“Wilkinson’s catalyst” 216
215
RhCl3.3H2O P
Ethanol, reflux, 30 min.+ RhCl(PPh3)3
C18H15PMol. Wt.: 262,29
RhO3H6Cl3Mol. Wt.: 263.31
81RhC54H45ClP3
Mol. Wt.: 925.22
216
To a hot solution of triphenylphosphine 81 (6.0 g, 22.9 mmol, 6 eq) in ethanol (175 mL) was added a hot solution of rhodium trichloride trihydrate 215 (1.0 g, 3.79 mmol) in ethanol (64 mL) under argon. The solution was refluxed for an additional 30 minutes and filtered hot, and the red crystals were washed with deoxygenated anhydrous ether under argon. The crystals were dried under vacuum to give rhodium(I) tris-(triphenylphosphine) chloride 216 (2.98 g, 3.22 mmol, 85%). 31P {1H} NMR (101.3 MHz, CDCl3): δ (ppm) = 52.9 (dt, JRh-P = 189 Hz, JP-P = 37.5 Hz, Ptrans-Cl), 35.9 (dd, JRh-P = 139 Hz, JP-P = 37. Hz, Pcis-Cl). Analytic data are in accordance with the literature.[228]
[228] P. G. Gassman, D. W. Macomber, S. M. Willging, J. Am. Chem Soc. 1985, 107, 2380-2388.
ExperimentalPart:Rhodiumcatalystsynthesis
294
12.2 Synthesis of chloro-(1,5-cyclooctadiene)rhodium (I) dimer 218
215
RhCl3, 3H2OEthanol, reflux, 30 min.
+
C8H12Mol. Wt.: 108.18
O3H6Cl3RhMol. Wt.: 263.31
217C54H45ClP3Rh
Mol. Wt.: 925.22
218
Rh
+Cl-
A solution of RhCl3·3H2O 215 (5.0 g, 18.9 mmol) and 1,5-cyclooctadiene 217 (15.5 mL, 72.5 mmol, 3.8 eq) in H2O (8 mL) and ethanol (95 mL) is heated at reflux overnight. After cooling to room temperature, the crude dimer complex [Rh(COD)Cl]2 is filtered off, and recrystallized from CH2Cl2/hexane to yield 3.9 g (14.9 g, 16.10 mmol, 85%) of the pure complexe [Rh(COD)Cl]2 218. 1H-NMR (400.132 MHz, CDCl3): δ = 1.73 (m, 4H), 2,47 (m, 4H), 4.21 (s, 4H) 13C {1H} NMR (100.613 MHz, CDCl3): δ = 30.9, 78.7. Analytic data are in accordance with the literature.[229]
[229] D. R. Baghurst, D. M. P. Mingos, J. of Organomet. Chem. 1990, 384(3), C57-C60.
ExperimentalPart:Rhodiumcatalystsynthesis
295
12.3 Synthesis of tetrakis(triphenylphosphine)palladium (0) 220
Pd(II)Cl2
Mol. Wt.: 177,33
P+
C18H15PMol. Wt.: 262,29
81 220219
Pd(PPh3)4
PdC72H60P4Mol. Wt.: 1155,56
1) DMSO, 150 °C (until all the solid had dissolved)2) NH2NH2, RT (addition)
A mixture of palladium(II) chloride 219 (5.00 g, 28.2 mmol) and triphenylphosphine 81 (36.98 g, 141.0 mmol) in dimethyl sulfoxide (375 mL) was heated at 150 °C until all solid had dissolved. The heater was removed and hydrazine hydrate (5.5 mL, 113.0 mmol) was added carefully. The product separated as yellow crystals on cooling to room temperature and was filtered off under an argon atmosphere. After washing with ethanol (4 x 100 mL) followed by diethyl ether (4 x 100 mL), traces of solvent were removed under vacuum to give tetrakis(triphenylphosphine)palladium(0) 220 (30 g, 25.9 mmol, 92%). 31P {1H} NMR (101.3 MHz, CDCl3): δ (ppm) = 19.0. Analytic data are in accordance with the literature.[230]
[230] J. R. Malpass, D. A. Hemmings, A. L. Wallis, S. R. Fletcher, S. Patel, J. Chem. Soc., Perkin Trans 1 2001, 1044-105.
296
C Appendix 13. Twelve principles of green chemistry
Prevent waste: Design chemical syntheses to prevent waste, leaving no waste to treat or clean up.
Design safer chemicals and products: Design chemical products to be fully effective, yet have little or no toxicity.
Design less hazardous chemical syntheses: Design syntheses to use and generate substances with little or no toxicity to humans and the environment
Use renewable feedstocks: Use raw materials and feedstocks that are renewable rather than depleting. Renewable feedstocks are often made from agricultural products or are the wastes of other processes; depleting feedstocks are made from fossil fuels (petroleum, natural gas, or coal) or are mined.
Use catalysts, not stoechiometric reagents: Minimize waste by using catalytic reactions. Catalysts are used in small amounts and can carry out a single reaction many times. They are preferable to stoechiometric reagents, which are used in excess and work only once.
Avoid chemical derivatives: Avoid using blocking or protecting groups or any
temporary modifications if possible. Derivatives use additional reagents and generate waste.
Maximize atom economy: Design syntheses so that the final product contains the maximum proportion of the starting materials. There should be few, if any, wasted atoms.
Use safer solvents and reaction conditions: Avoid using solvents, separating agents, or other auxiliary chemicals. If these chemicals are necessary, use innocuous chemicals.
Increase energy efficiency: Run chemical reactions at abient temperature and pressure whenever possible.
Design chemicals and products to degrade after use: Design chemical products to break down to innocuous substances after use so that they do not accumulate in the environment.
Analyze in real time to prevent pollution: Include in-process real-time monitoring and control during syntheses to minimize or eliminate the formation of by-products.
Minimize the potential for accidents: Design chemicals and their forms (solid, liquid, or gas) to minimize the potential for chemical accidents including explosions, fires, and releases to the environment.
297
14. Proposed synthesis path for Berchemiaside B Application of the synthesis of 1-tetralone derivatives via a rhodium-catalyzed hydro-acylation to the Berchemiaside B synthesis, a cytotoxic inhibitor used for the treatment of Leukemia. A proposed 11-step synthesis with the assembling of three moieties: a 1-tetralone obtained by rhodium-catalyzed hydroacylation, a β-D-glucopyranose sugar and a para-methoxy-cinnamoyl chloride or cinnamic acid was further proposed.
O
O
H
O
CyNH2, MgSO4
CH2Cl2, RT
O
O
H
N
LTMP
THF, -15 °C
TMP + nBuLi
O
O
H
N
Li
O
O
H
N
CeCl2
O
O
H
N
OH
1)
H
O
2) acid hydrolysis
O
O
O
O
H
O
OPG
chiral separation orenzyme resolution
O
O
H
O
OPG
alcohol protecting group
rhodium-catalyzedhydroacylationfor 1-tetralone
synthesis
O
O
O
OPG
alcohol deprotection
O
O
O
OH
Moiety 1
CeCl3
O
OH
risk of lactonisation
side product
Scheme 158. Proposed synthesis for 1-tetralone moiety 1.
Appendix:ProposedsynthesispathforBerchemiasideB
298
O
O
R
R = -OH or -Cl
1) EDC/DMAP beta-D-glucopyranose RT, 24 h2) NH4Cl O
O
O
O OHOHOH
OH
Moiety 2+3
O
O
O
OH
Moiety 3
Moiety 2
Moiety 1
+
O
O
O
O OHOHOH
OH
Moiety 2+3
TMSCl
RT, 60 °C (5 h)
O
O
O
O O
OHOHOH
O
O
O
Moiety 1+2+3 (protected)
O
O
O
O O
OHOHOH
O
O
O
BBr3
OH
OH
O
O O
OHOHOH
O
O
OH
Moiety 1+2+3
Berchemiaside B
Scheme 159. Assembling moieties 1, 2 and 3 for the final synthesis of Berchemiaside B.
NMR spectra of 2-vinylthiophene-3-carbaldehyde 134
Appendix:NMRspectraofnewmolecules
374
NMR spectra of 5,6-dimethoxy-indan-1-one 135
Appendix:NMRspectraofnewmolecules
375
NMR spectra of 1-oxo-indan-5-carboxylic acid methyl ester 136
Appendix:NMRspectraofnewmolecules
376
NMR spectra of 5-phthalimido-indan-1-one 137
Appendix:NMRspectraofnewmolecules
377
NMR spectra of 4-methyl-indan-1-one 138
Appendix:NMRspectraofnewmolecules
378
NMR spectra of 5-hydroxy-indan-1-one 139
Appendix:NMRspectraofnewmolecules
379
NMR spectra of 5-nitro-indan-1-one 140
O2N
O
Appendix:NMRspectraofnewmolecules
380
NMR spectra of 5-chloro-indan-1-one 141
Appendix:NMRspectraofnewmolecules
381
NMR spectra of 5-fluoro-indan-1-one 142
Appendix:NMRspectraofnewmolecules
382
NMR spectra of 1,2-dihydro-cyclopenta[α]naphthalene 143
Appendix:NMRspectraofnewmolecules
383
NMR spectra of thiophene cyclobutane dimer 146
S
H
O
S
O
H
S
H
O
S
O
H
Appendix:NMRspectraofnewmolecules
384
ppm
12345678910 ppm
1
2
3
4
5
6
7
8
9
10
The 1H-NMR spectra of compound 146 showed a singlet at δH = 9.81 ppm (aldehydic protons), a doublet at δH = 7.37 ppm and a doublet at δH = 7.17 ppm (furyl protons), three intense multiplets at δH = 4.48 ppm, δH = 2.60 ppm and δH = 2.31 ppm (cyclobutane protons).
Aldehydic proton
Correlation between the furyl protons
S
H
O
S
O
H
1
65
4
3
27
8
87
1
6
3
2
4
5
Correlation between the two methylene groups H8 and the aliphatic proton H7
NMR spectra of “Rh(imine P-N ligand 1)2]+ Cl-” 206
10 9 8 7 6 5 4 3 2 1 ppm
9.174
Appendix:NMRspectraofnewmolecules
398
NMR spectra of “imine P-N ligand 1” benzylidene-{6[(diphenylphosphanyl)-methyl]-3-methylpyridin-2-yl}-amine 208
10 9 8 7 6 5 4 3 2 1 ppm
2.392
3.626
7.265
8.763
3.0
6
2.1
0
1.0
0
N N
Me
PPh2H
399
D References [1] G. M. Whitesides and J. Deutch, Nature 2011, 469, 21-22.
[2] a) E. J. Corey, X.-M. Cheng, The Logic of Chemical Synthesis, John Wiley & Sons, New- York, 1995. b) K. C. Nicolaou, D. Vourloumis, N. Winssinger, P. S. Baran, Angew. Chemie 2000, 112, 46-126; Angew. Chem. Int. Ed. 2000, 39, 44-122.
[3] R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker, K. Schenker, J. Am. Chem. Soc. 1954, 76, 4749-4751; R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker, K. Schenker, Tetrahedron 1963, 19, 242-288.
[4] a) K. C. Nicolaou, M. O. Frederick, Angew. Chemie 2007, 28, 5372-5376; Angew. Chem. Int. Ed. 2007, 46, 5278–5282. b) K. C. Nicolaou, K. P. Cole, M. O. Frederick, R. J. Aversa, R. M. Denton, Angew. Chemie 2007, 46, 9031-9035; Angew. Chem. Int. Ed. 2007, 46, 8875-8879. c) K. C. Nicolaou, M. O. Frederick, A .C. B. Burtoloso, R. M. Denton, F. Rivas, K. P. Cole, R. J. Aversa, R. Gibe, T. Umezawa, T. Suzuki, J. Am. Chem. Soc. 2008, 130, 7466–7476. d) K. C. Nicolaou, R. J. Aversa, J. Jin, F. Rivas, J. Am. Chem. Soc. 2010, 132, 6855–6861. e) K. C. Nicolaou, C. F. Gelin, J. H. Seo, Z. Huang, T. Umezawa, J. Am. Chem. Soc. 2010, 132, 9900–9907. f) K C. Nicolaou, J. H. Seo, T. Nakamura, R. J. Aversa, J. Am. Chem. Soc. 2011, 133, 214–219. g) K. C. Nicolaou, T. M. Baker, T. Nakamura, J. Am. Chem. Soc. 2011, 133, 220–226.
[5] Board on Chemical Sciences and Technology (BCST), Beyond the Molecular Frontier: Challenges for Chemistry and Chemical Engineering, The National Academies Press, Washington 2003.
[6] G. Bruntland, Our Common Future, Oxford University Press, New-York 1987.
[7] T. Clarke, Nature 2002, 418, 812-814.
[8] Report of the United Nations Conference on Environment and Development, 3-14 June 1992, Rio de Janeiro, <http://www.un.org/esa/sustdev>.
[9] M. Eisen, J. O. Metzger, E. Schmidt, U. Schneidewind, Angew. Chem. 2002, 114, 402-425; Angew. Chem Int. Ed. 2002, 41, 414-436.
[10] C. Capello, U. Fischer, K. Hunkelbühler, Green Chem. 2007, 9, 927-934.
[11] P. T. Anastas, J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New-York 1998.
[12] P. A. Wender, Chem. Rev. 1996, 96, 1-2.
[13] a) B. M. Trost, Science 1991, 254, 1471-1477; b) B. M. Trost, Acc. Chem. Ress. 2002, 35, 695-705.
[14] a) B. M. Trost, Angew. Chemie. 1995, 107, 285-307; Angew. Chem. Int. Ed. 1995, 34, 259-281. b) R. A. Scheldon, Pure Appl. Chem. 2000, 72, 1233-1246.
[15] A. S. Goldman, K. I. Goldberg, Activation and Functionalization of C-H Bonds ACS Symposium Series 885 2004.
[16] J. Chatt and J. M. Davidson, J. Chem. Soc. 1965, 843-855.
References
400
[17] N. F. Gol’dschleger, V. V. Es’kova, A. E. Shilov, A. A. Shteinman, Zhurnal Fizicheskoi Khimii 1972, 46, 1353.
[18] P. L. Watson, J. Am. Chem. Soc. 1983, 105, 6491-6493.
[19] K. J. Del Rossi, B. B. Wailand, J. Am. Chem. Soc. 1985, 107, 7941-7944.
[20] S. H. Brown, R. H. Carbtree, Chem. Comm. 1987, 970-971.
[21] a) C. C. Cummings, S. M. Baxter, P. T. Wolczanski, J. Am. Chem. Soc. 1988, 110, 8731-8733. b) P. J. Walsh, F. J. Hollander, R. G. Bergman, J. Am. Chem. Soc. 1988, 110, 8729-8731.
[22] J. F. Young, J. A. Osborn, F. H. Jardine, G. Wilkinson, Chem. Comm. 1965, 131-132.
[23] M. L. Green, P. J. Knowles, J. Chem. Soc. D 1970, 24, 1677–1677.
[24] C. J. Moulton, B. L. Shaw, J. Chem. Soc, Dalton Trans. 1976, 1020-1024.
[25] a) M. Albrecht, G. van Koten, Angew. Chemie 2001, 20, 3866-3898; Angew. Chem Int. Ed. 2001, 40, 3750-3781. b) M. E. van der Boom, D. Milstein, Chem. Rev. 2003, 103, 1759-1792. c) N. Selender and K. Szabó, Chem. Rev., 2011, 111(3), 2048–2076.
[26] P. Foley, G. M. Whitesides, J. Am. Chem. Soc. 1979, 101(10), 2732–2733.
[27] A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc. 1982, 104(1), 352-354.
[28] R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh, H. Fujii, Science 1998, 280, 560-564.
[29] a) B. Goldfuss, P. Knochel, L. O. Bromm, K. Knapp, Angew. Chemie 2000, 112(22), 4302-4305; Angew. Chem. Int. Ed. 2000, 39(22), 4136-4139. b) H. Laazari, L. O. Bromm, F. Lhermitte, R. M. Gschwind, P. Knochel, J. Am. Chem. Soc. 1999, 121, 6940-6941.
[30] a) R. Köster, W. Larbig, G. W. Rotermund, Justus Liebigs Ann. Chem. 1965, 682, 21-48; b) R. Köster, G. Benedikt, W. Fenzl, K. Reinert, Justus Liebigs Ann. Chem. 1967, 702, 197-223; c) G. J. Abruscato, T. T. Tidwell, J. Org. Chem. 1972, 37, 4151-4156.
[31] F. Liu, E. B. Pak, B. Singh, C. M. Jensen, A. S. Goldman, J. Am. Chem. Soc. 1999, 121, 4086-4087.
[32] I. M. Piglosiewicz, S. Kraft, R. Beckhaus, D. Haase, W. Saak, Eur. J. Inorg. Chem. 2005, 938-945
[33] S. Kraft, R. Beckhaus, D. Haase, W. Saak, Angew. Chemie 2004, 12, 1609-1614; Angew. Chem. Int. Ed. 2004, 43, 1583-1587.
[34] S. H. Cho, S. J. Hwang, S. Chang, J. Am. Chem. Soc. 2008, 130, 9254-9256.
[35] C. Jia, T. Kitamura, Y. Fujiwara, Acc of Chem Res 2001, 34(8), 633-639.
[36] C. Jia, D. Piao, T. Kitamura, Y. Fujiwara, J. Org. Chem. 2000, 65, 7516-7522.
[37] B. M. Trost and F. D. Toste, J. Am. Chem. Soc. 1996, 118, 6305-6306.
[38] A. P. Khrushch, L. A. Tokina, A. E. Shilov, Kinetica I Kataliz 1966, 7, 901.
References
401
[39] a) A. E. Shilov, A. A. Shteinman, Coord. Chem. Rev. 1977, 24, 97-143. b) L. A. Kushch, V. V. Lavrushko, Y. S. Misharin, A. P. Moravskii, A. E. Shilov, Nouveau Journal de Chimie 1983, 7, 729.
[40] a) Y. Fujiwara, K. Takaki, J. Watanabe, Y. Uchida, H. Taniguchi, Chem. Lett. 1989, 1687-1688. b) K. Nakata, Y. Yamaoka, T. Miyata, Y. Taniguchi, K. Takaki, Y. Fujiwara, J. Organomet. Chem. 1994, 473, 329-334. c) C. Jia, T. Kitamura, Y. Fujiwara, Acc. Chem. Res. 2001, 34, 633-639.
[41] P. L. Watson. G. W. Parshall, Acc. Chem. Res. 1985, 18, 51-56.
[42] a) K. J. Del Rossi, B. B. Wailand, Chem. Comm. 1986, 1653-1655; b) A. E. Sherry, B. B. Wailand, J. Am. Chem. Soc. 1990, 112, 1259-1261; c) B. B. Wailand, S. Ba, A. E. Sherry, J. Am Chem. Soc. 1991, 113, 5305-5311.
[43] a) S. H. Brown, R. H. Carbtree, J. Am. Chem. Soc. 1989, 11, 2935-2946; b) S. H. Brown, R. H. Carbtree, J. Am. Chem. Soc. 1989, 11, 2946-2953; c) R. H. Carbtree, S. H. Brown, C. A. Muedas, C. Boojamra, R. R. Ferguson, Chemtech 1991, 21, 634.
[44] R. J. Bergman, Nature 2007, 446, 391-394.
[45] R. Agudo, G. D. Roiban, M. T. Reetz, Chembiochem 2012, 13(10), 1465-1473.
[46] S. D. Bruner, Nature Chemistry 2011, 3, 342-343.
[47] H. J. H. Fenton, J. Chem. Soc., Trans. 1894, 65, 899-911.
[48] C. H Jun, E. A. Jo, J. W. Park, Eur. J. Org. Chem. 2007, 1869-1881.
[49] T. Kondo, Y. Tsuji, Y. Watanabe, Tet. Lett. 1987, 28, 6229-6230 ; T. Kondo, M. Akazome, Y. Tsuji, Y. Watanabe, J. Org. Chem. 1990, 55, 1286-1291.
[50] J. W. Suggs, J. Am. Chem. Soc. 1978, 100, 640-641. [51] C.-H. Jun, H. Lee, J.-B. Hong, B. I. Kwon, Angew. Chemie 2002, 114(12), 2250-2251; Angew. Chem. Int. Ed. 2002, 41(12), 2146-2147.
[52] D. Y. Lee, I. J. Kim, C. H. Jun, Angew. Chemie 2002, 114(16), 3157-3159; Angew. Chem. Int. Ed. 2002, 41(16), 3031-3033.
[53] a) T. Sattelkau, P. Eilbracht, Tet. Lett. 1998, 1905-1908; b) M. Tanaka, M. Takahashi, E. Sakamoto, M. Imai, A. Matsui, M. Fujio, K. Funakoshi, K. Sakai, H. Suemune, Tetrahedron 2001, 57, 1197-1204; c) G. Kim, E. J. Lee, Tetrahedron: Asymmetry 2001, 12, 2073-2076; d) Y. Oonishi, A. Taniuchi, M. Mori, Y. Sato, Tet. Lett. 2006, 47, 5617-5621.
[54] a) B. Bosnich, Acc. Chem. Res. 1998, 31, 667-674; b) G. C. Fu in Modern Rhodium-Catalyzed Reactions (Ed.: P. A. Evans), Wiley-VCH, New York, 2005, 79-91.
[55] T. B. Marder, D. C. Roe, D. Milstein, Organometallics 1988, 7, 1451-1453.
[56] T. Kondo, Y. Tsuji, Y. Watanabe, Tet. Lett. 1987, 28, 6229-6230 ; T. Kondo, M. Akazome, Y. Tsuji, Y. Watanabe, J. Org. Chem. 1990, 55, 1286-1291.
[57] C. P. Lenges and M. Brookhart, J. Am. Chem. Soc. 1997, 119, 3165-3166.
[58] H. Lee, C. H. Jun, Bull. Korean Chem. Soc. 1995, 16, 66-68.
References
402
[59] H. Lee, C. H. Jun, Bull. Korean Chem. Soc. 1995, 16, 1135-1138. [60] M. C. Willis, S. J. McNally, P. J. Beswick, Angew. Chemie 2004, 116(3), 344-347; Angew. Chem. Int. Ed. 2004, 42, 340-343.
[61] M. Tanaka, M. Imai, Y. Yamamoto, K. Tanaka, M. Shimowatari, S. Nagumo, N. Kawahara, H. Suemune, Org. Lett. 2003, 5, 1365-1367 ; M. Imai, M. Tanaka, K. Tanaka, Y. Yamamoto, N. Imai-Ogata, M. Shimowatari, S. Nagumo, N. Kawahara, H. Suemune, J. Org. Chem. 2004, 69, 1144-1150 ; K. Tanaka, M. Tanaka, H. Suemune, Tet. Lett. 2005, 46, 6053-6056.
[62] K. Kokubo, K. Matsumasa, Y. Nishinaka, M. Miura, M. Nomura, Bull. Chem. Soc. Jpn. 1999, 72, 303-311.
[63] K. Kokubo, K. Matsumasa, M. Miura, M. Nomura, J. Org. Chem. 1997, 62, 4564-4565.
[64] R. T. Stemmler, C. Bolm, Adv. Synth. Catal. 2007, 349, 1185-1198.
[65] M. M. Coulter, K. G. M. Kou, B. Kalligan, V. M. Dong, J. Am. Chem. Soc. 2010, 132, 16330.
[66] D. H. T. Phan, K. G. M. Kou, V. M. Dong, J. Am. Chem. Soc. 2010, 132, 16354.
[67] S. R. Parsons, J. F. Hooper, M. C. Willis, Org. Lett. 2011, 13(5), 998-1000.
[68] C. Gonzalez-Rodriguez, R. J. Pawley, A. B. Chaplin, A. L. Thompson, A. S. Weller, M. C. Willis, Angew. Chemie 2011, 123(22), 5240-5244; Angew. Chemie Int. Ed. 2011, 50, 5134 –5138.
[69] P. Lenden, D. A. Entwistle, M. C. Willis, Angew. Chemie 2011, 123(45), 10845-10848 ; Angew. Chem. Ind. Ed. 2011, 50, 10657–10660.
[70] S. K. Murphy, M. M. Coulter, V. M. Dong, Chem. Sci. 2012, 3, 355.
[71] M. Von Delius, C. M. Le, V. M. Dong, J. Am. Chem. Soc. 2012, 134, 15022.
[72] S.-J. Poingdestre, J. D. Goodacre, A. S. Weller, M. C. Willis, Chem. Commun. 2012, 48, 6354-6356.
[73] R. Pawley, M. Huertos, G. Lloyd-Jones, A. S. Weller, M. C. Willis, Organometallics 2012, 31, 5650-5659.
[74] a) Z. Shen, P. K. Dornan, H. A. Khan, T. K. Woo, V. M. Dong, J. Am. Chem. Soc. 2009, 131, 1077-1091; b) Z. Shen, H. A. Khan, V. M. Dong, J. Am. Chem. Soc. 2008, 130, 2916.
[75] M. M. Coulter, P. K. Dornan, V. M. Dong, J. Am. Chem. Soc. 2009, 131, 6932.
[76] D. H. T. Phan, B. Kim, V. M. Dong, J. Am. Chem. Soc. 2009, 131, 15608.
[77] K. Hirano, A. T. Biju, I. Piel, F. Glorius, J. Am. Chem. Soc. 2009, 131, 14190.
[78] A. T. Biju, N. E. Wurz, F. Glorius, J. Am. Chem. Soc. 2010, 132, 5970-5971.
[79] A. T. Biju, F. Glorius, Angew. Chemie 2010, 122(50), 9995-9958; Angew. Chem. Ind. Ed. 2010, 49, 9761-9764.
[80] A. H. Khan, K. G. M. Kou, V. M. Dong, Chem. Sci. 2011, 2, 407.
References
403
[81] I. Piel, M. Steinmetz, K. Hirano, R. Fröhlich, S. Grimme, F. Glorius, Angew. Chemie 2011, 123(21), 5087-5091; Angew. Chem. Int. Ed. 2011, 50, 4983-4987.
[82] E. V. Beletskiy, C. H. Sudheer, C. J. Douglas, J. Org. Chem 2012, 77(14), 5884-5893.
[83] T. Tsuda, T. Kiyoi, T. Saegusa, J. Org. Chem. 1990, 55, 2554-2558.
[84] Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi, S. Ogoshi, Angew. Chemie 2012, 124(43), 10970-10973; Angew. Chemie Int. Ed. 2012, 51(43), 10812-10815.
[85] F. Shibahara, J. F. Bower, M. J. Krische, J. Am. Chem. Soc. 2008, 130, 14120-14122.
[86] S. K. Murphy, Vy M. Dong, J. Am. Chem. Soc. 2013, 135, 5553-5556.
[87] A. Chan, K. A. Scheidt, J. Am. Chem. Soc. 2006, 128, 4558.
[88] S. Omura, T. Fukuyama, H. Murakami, Chem. Comm. 2009, 6741.
[89] T. Suzuki, T. Yamada, K. Watanabe, T. Katoh, Bioorg. Med. Chem. Lett. 2005, 15, 258.
[90] Y. J. Park, J. W. Park, C. H. Jun, Acc. of Chem. Res. 2008, 41(2), 222-234.1 [91] C. H Jun, E. A. Jo, J. W. Park, Eur. J. Org. Chem. 2007, 1869-1881.
[92] N. R. Vautravers, D. D. Regent, B. Breit, Chem. Commun. 2011, 47, 6635-6637.
[93] a) E. Miller, B. L. Shaw, J. Chem. Soc, Dalton Trans. 1974, 480-485. b) B. L. Shaw, M. F. Uttley, J. Chem. Soc, Chem. Commun. 1974, 918-919. c) C. E. Jones, B. L. Shaw, B. L. Turtle, J. Chem. Soc., Dalton Trans. 1974, 992-999. d) C. J. Moulton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1976, 1020-1024. e) H. D. Empsall, E. M. Hyde, R. Markham, W. S. McDonald, M. C. Norton, B. L. Shaw, J. Chem. Soc., Chem. Commun. 1977, 589-590. f) C. Crocker, R. J. Errington, W. S. McDonald, K. J. Odell, B. L. Shaw, J. Chem. Soc., Chem. Commun. 1977, 589-590. g) N. A. Al-Salem, H. D. Empsall, R. Markham, B. L. Shaw, B. J. Weeks, J. Chem. Soc., Dalton Trans. 1979, 1972-1982. h) N. A. Al-Salem, W. S. McDonald, R. Markham, M. C. Norton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1980, 59-63. i) C. Crocker, R. J. Errington, R. Markham, C. J. Moulton, K. J. Odell, B. L. Shaw, J. Am. Chem. Soc. 1980, 102, 4373-4379. j) R. J. Errington, B. L. Shaw, J. Organomet. Chem. 1982, 238, 319-325. k) C. Crocker, R. J. Errington, R. Markham, C. J. Moulton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 387-395. l) C. Crocker, H. D. Empsall, R. J. Errington, E. M. Hyde, W. S. McDonald, R. Markham, M. C. Norton, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 1217-1225. m) J. R. Briggs, A. G. Constable, W. S. McDonald, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 1225-1230. n) R. J. Errington, F. E. McDonald, B. L. Shaw, J. Chem. Soc., Dalton Trans. 1982, 1829-1835.
[94] D. Y. Lee, I. J. Kim, C.-H. Jun, Angew. Chemie 2002, 114(12), 2250-2251; Angew. Chem. Int. Ed. 2002, 41(12), 2146-2147.
[95] J. W. Suggs, J. Am. Chem. Soc. 1979, 101, 489.
[96] a) C.-H. Jun, D. Y. Lee, H. Lee, J.-B- Hong, Angew. Chemie 2000, 112(17), 3214-3216; Angew. Chem. Int. Ed. 2000, 39, 3070-3072. b) C.-H. Jun, J. H. Lee, Pure Appl. Chem. 2004, 76(3), 577-587.
[98] M. Malamas, W. Fobare, W. Solvibile, F. Lovering, J. Condon, A. Robichaud, U. S. Pat. Appl. Publ., 20060173049, 03 Aug. 2006 (ligand 7, step 1).
[99] X. Collin, J.-M. Robert, G. Wielgosz, G. Le Baut, C. Bobin-Dubigeon, N. Grimaud, J.-Y. Petit, Eur. J. of Med. Chem. 2001, 36(7-8), 639-649 (ligand 11, step 1).
[100] G. Shyamaprosad, D. Swapan, J. Subrata, A. Avijit Kumar, Chemistry Letters 2004, 33(7), 916-917 (ligand 11, step 2, same reaction butlower yield (40%).
[101] F. Cottet, M. Marull, O. Lefebvre, M. Schlosser, Eur. J. Org. Chem. 2003, 1559-1568 (ligand 23, steps 1, 2 and 3).
[102] Y. Cheng, T. C. Judd, M. T. Bartberger, J. Brown, K. Chen, R. T. Fremeau Jr., D. Hickman, S. A. Hitch-cock, B. Kordan, V. Li, P. Lopez, S. W. Louie, Y. Luo, K. Michelsen, T. Nixey, T. S. Powers, C. Rattan, E. A. Sickmeier, D. J. St. Jean Jr., R. C. Wahl, P. H. Wen, S. Wood, J. Med. Chem. 2011, 54(16), 5836-5857 (ligand 23, step 4, same reaction but lower yield).
[103] D. Song, R. H. Morris, Organometallics 2004, 23, 4406-4413 (ligand 36, steps 1 and 2).
[104] J. Hu, PCT Int. Appl., 2009131814, 29 Oct. 2009 (ligand 36, step 3, similar conditions but lower yield (81%).
[105] M. Weis, C. Waloch, W. Seiche, B. Breit, J. Am. Chem. Soc. 2006, 128(13), 4188-4189. [106] Dr. W. Seiche (PhD manuscript, Breit group, 2009).
[108] M. Delius, C. M. Le, V. M. Dong, J. Am. Chem. Soc. 2012, 134(36), 15022-32.
[109] A. H. Roy, C. P. Lenges, M. Brokkhart, J. Am. Chem. Soc. 2007, 129(7), 2082-2093.
[110] N. Kaur and D. Kishore, J. Chem. Pharm. Res. 2012, 4(2), 991-1015.
[111] J. K. Stille, Angew. Chemie 1986, 98(6), 504-519; Angew. Chem. Int. Ed. 1986, 25, 508-524.
[112] J. Novacek and M. Waser, Eur. J. Org. Chem. 2013, 637–648. [113] W. Seiche, A. Schuschkowski, B. Breit, Adv. Synth. Cat. 205, 347, 1488-1494.
[114] P. C. J. Kramer, P. W. N. M. Van Leeuwen, Phosphorus(III) ligands in homogeneous catalysis: design and synthesis 2012, John Wiley & Sons.
[115] I. Inui, M. Tanaka, M. Imai; K. Tanaka, H. Suemune, Chem. Pharm. Bull. 2009, 57, 1158.
[116] M. M. Coulter, K. G. M. Kou, B. Galligan, Vy. M. Dong, J. Am. Chem. Soc. 2010, 132, 16330-16333.
References
405
[117] a) L. Diab, T. Šmejkal, J. Geier, B. Breit, Angew. Chemie 2009, 121, 8166-8170; Angew. Chem. Int. Ed. 2009, 48, 8022-8026. b) D. Fuchs, G. Rousseau, L. Diab, U. Gellrich, B. Breit, Angew. Chemie 2012, 124, 2220-2224; Angew. Chem. Int. Ed. 2012, 51, 2178-2180. [118] H. Sugimoto, Pure Appl. Chem. 1999, 71, n°11, 2031-2037.
[119] R. Rendy, Y. Zhang, A. McElrea, A. Gomez, D. A. Klumpp, J. Org. Chem. 2004, 69, 2340-2347 ; J. Koo, J. Am. Chem. Soc. 1953, 75, 1891-1895 ; V. Premasagar, V. A. Palaniswamy, E. J. Eisenbraun, J. Org. Chem. 1981, 46, 2974-2976 ; R. S. Budhhram, V. A. Palaniswamy, E. J. Eisenbraun , J. Org. Chem. 1986, 51, 1402-1406 ; T.-L. Ho, K. Y. Lee, C. K. Chen, J. Org. Chem. 1997, 62, 3365-3369.
[120] S. Hanessian, J. Ma, Tet. Lett. 2001, 42, 8785-8788 ; H. O. House, C. B. Hudson, J. Org. Chem. 1970, 647-651 ; B. Hulin, M. Koreeda, J. Org. Chem. 1984, 207-209 ; T. Yamato, C. Hideshima, G. K. Surya Prakash, G. A. Olah, J. Org. Chem. 1991, 56, 3955-3957 ; T. F. Buckley III, H. Rapoport, J. Am. Chem. Soc. 1980, 102, 3056-3062.
[121] K. Koltunov, S. Walspurger, J. Sommer, Tet Lett 2005, 46, 8391-8394 ; C. Vial, G. Bernardinelli, P. Schneider, M. Aizenberg, B. Winter, Helv. Chim. Acta 2005, 88, 3109-3117 ; A. Bhattacharya, B. Segmuller, A. Ybarra, Synthetic Commun. 1996, 26, 1775-1784 ; G. Sartori, F. Bigi, R. Maggi, G. L. Bernardi, Tet. Lett. 1993, 34, 7339-7342 ; C. Roussel, H. G. Rajoharison, L. Bizzari, L. Shaimi, J. Org. Chem. 1988, 53, 683-685 ; T. Suzuki, T. Ohwada, K. Shudo, J. Am. Chem. Soc. 1997, 119, 6774-6780 ; J. H. Burckhalter, R. C. Fuson, J. Am. Chem. Soc. 1948, 70, 4184-4186 ; Fuson , W. E. Ross, C. H. McKeever, J. Am. Chem. Soc. 1938, 60, 2935-2936.
[122] T. Jobashi, T. Jobashi, A. Kawai, S. Kawai, K. Maeyama, H. Oike, Y. Yoshida, N. Yonezawa, Tetrahedron 2006, 62, 5717-5724 ; T. Jobashi, K. Maeyama, K. Noguchi, Y. Yoshida, N. Yonezawa, Bull. Chem. Soc. Jpn. 2006, 79, 627-633.
[123] G. B. Womack, R. L. Snowden, R. L. Mosimann, A. A. Birkbeck, A Process for Producing Indenol Esters or Ethers. WO Pat. Appl. WO 2005/113473 A2, December 1, 2005.
[124] G. B. Womack, J. G. Angeles, V. E. Fanelli, C. A. Heyer, J. Org. Chem. 2007, 72, 7046-7049.
[125] A. Saito, M. Umakoshi, N. Yagyu, Y. Hanzawa, Org. Lett. 2008, 10, 1783-1785.
[126] E. Negishi, S. M. Coperet, S. Ma, T. Mita, T. Sugihara, J. M. Tour, J. Am. Chem. Soc. 1996, 118, 5919.
[127] V. Gevorgyan, L. G. Quan, Y. Yamamoto, Tet. Lett. 1999, 40, 4089.
[128] S. V. Gagnier and R. C. Larock, J. Am. Chem. Soc. 2003, 125, 4804-4807.
[129] J. B. Park, S. H. Ko, W. P. Hong, K. J. Lee, Bull. Korean Chem. Soc. 2004, 25, n°6, 927-930.
[130] A. Odreda, S. Datta, R. S. Liu, J. Org. Chem. 2007, 72, 3289-3292.
[131] J. Ruan, J. A. Iggo, J. Xiao, Org. Lett. 2011, 13(2), 268-271.
[132] A. Arefalk, M. Larhed, A. Hallberg, J. Org. Chem. 2005, 70, 938-942.
[133] R. Schrock and J. Osborn, J. Am. Chem. Soc., 1971, 2397-2402.
References
406
[134] D. Fairlie and B. Bosnich, Organometallics 1988, 7, 936-945.
[135] D. Fairlie and B. Bosnich, Organometallics 1988, 7, 946-954.
[136] C. F. Lochow and R. G. Miller, J. Am. Chem. Soc. 1976, 98(5), 1281-1283.
[137] K. Kundu, J. McCullagh, A. T. Morehead Jr, J. Am. Chem. Soc. 2005, 127, 16042-16043.
[138] M. Inoue and M. Nakada, J. Am. Chem. Soc. 2007, 129, 4164-4165.
[139] J. A. Osborn and G. Wilkinson, J. Am. Chem. Soc. A: Inorganic, Physical, Theoretical 1966, 1711-173.
[140] M. Murakami, S. Kadowaki, A. Fujimoto, M. Ishibashi, T. Matsuda, Org. Lett. 2005, 7(10), 2059-2061. [141] A. D. Burrows, C. G. Frost, M. F. Mahon, C. Richardson, Angew. Chemie 2008, 120(44), 8610-8614; Angewandte Chemie 2008, 47, 8482-8486.
[142] G. J. Domski, J. B. Edson, I. Keresztes, E. B. Lobkovsky, Chem. Commun. 2008, 6137-6139.
[143] K. Tanaka, D. Hojo, T. Shoji, Y. Hagiwara, M. Hirano, Org. Lett. 2007, 9(11), 2059-2062.
[144] H. Tanaka, A. K. M. Abdul Hai, H. Ogawa, S. Torii, Synlett 1993, 11, 835-836 (For the synthesis of the starting tributylvinyltin, 99% yield).
[145] T. Kauffmann, R. Wirthwein, Angew. Chemie 1971, 10, 20-33; Angew. Chem. Int. Ed. 1971, 83, 21-34.
[146] A. Numata, Y. Kondo, T. Sakamoto, Synthesis 1999, 2, 306-311.
[147] a) M. D´Auria, L. Emanuele, G. Mauriello, R. Racioppi, J. of Photochem. and Photobio. A: Chemistry 2000, 134, 147-15. b) S. Gronowitz, I. Ander, Acta Chemica Scandinavia B 1975, 29, 513-523. c) R. M. Dodson, A. G. Zielske, J. Org. Chem. 1967, 32, 28-31. d) N. L. Allinger, L. A. Tushaus, J. Am. Chem. Soc. 1965, 30, 1935-1939; I. Lillien, R. A. Doughty, Tetrahedron 1967, 23, 3321-3326. e) J. B. Lambert, J. D. Roberts, J. Am. Chem. Soc. 1965, 87, 3884-3890.
[148] H. Sugimoto, Y. Yamanishi, Y. Limura, Y. Kawakami, Current Med. Chem. 2000, 7, 303-339.
[149] A. V. V. Srinivasrao, Y. Venkateshwarlu: An improved process for the preparation of Donepezil - WIPO Patent Application WO/2008/010235 – 2008-01-24, Torrent Pharmaceuticals Limit.
[152] G. Stork, J. J. La Clair, V. G. Young Jr, X. Xie, R. E. McCarley, J. G. Verkade, J. Am. Chem. Soc. 1996, 118, 5304-5305.
[153] W. S. Johnson, Org React 1949, 2, 114-177.
References
407
[154] B. Hulin, M. Koreeda, J. Org. Chem. 1984, 49, 207-209.
[155] a) B. B. Snider, T. J. Kwon, J. Org. Chem. 1990, 55, 4786-4788 ; b) F. Z. Yang, K. M. Trost, W. E. Fristad, Tet. Lett. 1987, 28, 1493-1496 ; c) J. H. Rigby, A. Kotnis, J. Kramer, Tet. Lett. 1983, 24, 2939-2940 ; d) F. Johnson, E. R. Marinelli, J. Org. Chem. 1986, 51, 3911-3913 ; e) R. Beugelmans, J. Chastanet, H. Ginsburg, L. Quintero-Cortes, G. Roussi, J. Org. Chem. 1985, 50, 4933-4938 ; f) R. C. Klix, M. H. Cain, A. V. Bhatia, Tet. Lett. 1995, 36, 6413-6414 ; g) J. Thibonnet, M. Abarbri, J.-L. Parrain, A. Duchêne, Tet. Lett. 1996, 37, 7507-7510.
[156] D. Zhao, D. G. Lee, Synthesis 1994, 915-916.
[157] J. M. Hornback, M. L. Poundstone, B. Vadlamani, S. M. Graham, J. Gabay, S. T. Patton, J. Org. Chem. 1988, 53, 5597-5601.
[158] B. Tarnchompoo, C. Thebtaranonth, Y. Thebtaranonth, Synthesis 1986, 785-786.
[159] I. D. G. Watson, S. Ritter, F. D. Toste, J. Am. Chem. Soc. 2009, 131(6), 2056-2057.
[160] J. A. Osborn, G. Wilkinson, J. Am. Chem. Soc. A: Inorganic, Physical, Theoretical 1966, 1711-1732.
[161] K. Knobloch, M. Keller, W. Eberbach, Eur. J. of Org. Chem. 2001, 17, 3313-3332.
[162] K. Yoshikai, T. Hayama, K. Nishimura, K.-I. Yamada, K. Tomioka, J. Org. Chem., 2005, 70, 681-683.
[163] J.-H. Li, Q.-M. Zhu, Y. Liang, D. Yang, J. Org. Chem., 2005, 70, 5347-5349.
[164] P. Zhou, L. Zhang, S. Luo, J.-P. Cheng, J. Org. Chem., 2012, 77, 2526-2530.
[165] R. E. Campbell Jr. and R. G. Miller, J. of Organomet. Chem. 1980, 186, C27-C31.
[166] K. Gable and G. A. Benz, Tet. Lett. 1991, 32, 3473.
[167] K. Mori, Eur. J. of Org. Chem. 2005, 10, 2040-2044.
[168] M. M. Coulter, P. K. Dornan, Vy. M. Dong, J. Am. Chem. Soc. 2009, 131, 6932-6933. [169] M. P. Anderson, A. L. Casalnuovo, B. J. Johnson, B. M. Mattson, A. M. Mueting, Inorg. Chem. 1988, 27, 1649-1658.
[170] A. G. Griesbeck, S. Bondock, P. Cygon, J. Am. Chem. Soc. 2003, 125(30), 9016-9017.
[171] D. P. Fairlie, B. Bosnich, Organometallics 1988, 936-945. [172] D. P. Fairlie, B. Bosnich, Organometallics 1988, 946-9.
[173] D. H. Doughty, M. F. McGuiggan, H. Wang, L. H. Pignolet, Fundamental Research in Homogeneous Catalysis 1979, 909-919.
[174] J. A. Turner, J. Org. Chem. 1983, 48, 3401-3408.
[175] J. A. Drewry, S. Fletcher, H. Hassan, P. T. Gunning, Org. Biomol. Chem. 2009, 7(24), 5074-5077.
References
408
[176] D. J. Vyas, E. Larionov, C. Besnard, L. Guénée, C. Mazet, J. Am . Chem. Soc. 2013, 135(16), 6177-6183.
[177] T. M. Goegsig, L. S. Soejberg, A. T. Lindhardt, K. M. Jensen, T. Skrydstrup, R. O. Steen, S. J. Dunne, J. Org. Chem. 2008, 73, 3403-3410.
[179] A. D. Burrows, C. G. Frost, M. F. Mahon, C. Richardson, Angew. Chemie 2008, 120(44), 8610-8614; Angew. Chem. Int. Ed. 2008, 47(44), 8482-8486.
[180] R. Shrestha, P. Mukherjee, Y. Than, Z. C. Litman, J. F. Hartwig, J. Am. Chem. Soc. 2013, 135 (23), 8480-8493.
[181] A. S. Demir, O. Reis, Tetrahedron 2004, 60, 3803-3812.
[182] J. W. Grissom, D. Klingberg, S. Meyenburg, B. L. Stallman, J. Org. Chem. 1994, 59, 7876-7888.
[183] K. Tanaka, D. Hojo, T. Shoji, Y. Hagiwara, M.Hirano, Org. Lett. 2007, 9, 2059-2063.
[184] B. Gabriele, L. Veltri, V. Maltese, R. Spina, R. Mancuso, G. Salerno, Eur. J. Org. Chem. 2011, 28, 5626-5635.
[185] D. R. Spring, S. Krishnan, H. E. Blackwell, S. Schreiber, J. Am. Chem. Soc. 2002, 124, 1354-1363
[186] P. Mukherjee, S. J. Roy, T. K. Sarkar, Org. Lett. 2010, 12, 2472-2475.
[187] A. C. Spivey, L. Shukla, J. F. Hayler, Org. Lett. 2007, 9, 891-894.
[188] L. J. Nurkkala, R. O. Steen, S. J. Dunne, Synthesis 2006, 1295-1300. [189] S. Hibino, S. Kano, N. Mochizuki, E. Sugino, J. Org. Chem.. 1984, 49, 5006-5008.
[190] I. D. G. Watson, S. Ritter, F. D. Toste, J. Am. Chem. Soc. 2009, 131(6), 2056-2057.
[191] C. Che, S. Li, Z. Yu, F. Li, S. Xin, L. Zhou, S. Lin, Z. Yang, ACS Comb. Sci. 2013, 15(4), 202-207.
[192] J. Clayden, C. McCarthy, N. Westlund, C. S. Frampton, J. Chem. Soc., Perkin Trans. 1 2000, 1363-1378.
[193] N. S. Narashiman, T. Mukhopadhyay, S. S. Kusurhar, Indian J. of Chem., section B : Organic Chemistry including Medicinal Chemistry 1981, 20(7), 546-548.
[194] B. D. Kelly, J. M. Allen, R. E. Tundel, T. H. Lambert, Org. Lett. 2009, 11(6), 1381-1383.
[195] M. Liniger, C. Neuhaus, T. Hofmann, L. Fransioli-Ignazio, M. Jordi, P. Drueckes, J. Trappe, D. Fabbro, K.-H. Altmann, ACS Med. Chem. Lett. 2011, 2(1), 22-27.
[196] S. R. Park, C. Kim, D.-G. Kim, N. Thrimurtulu, H.-S. Yeom, J. Jun, S. Shin, Y. Ho Rhee, Org. Lett. 2013, 15(6), 1166-1169.
[197] L. Garanti, A. Sala, G. Zecchi, J. Org. Chem. 1975, 40(16), 2403-2406.
References
409
[198] D. Wang, Z. Zhang, Org. Lett. 2003, 5, 4645-4648.
[199] C. B. Rao, D. C. Rao, D. C. Babu, Y. Vankateswarlu, Eur. J. Org. Chem. 2010, 2855-2859.
[200] J. Lin, J. Chen, W. Su, Adv. Synth. and Cat. 2013, 355(1), 41-46.
[201] J. A. Murphy, A. G. J. Commeureuc, T. N. Snaldon, T. M. McGuire, T. A. Khan, K. Hisler, M. L. Dewis, R. Carling, Org. Lett. 2005, 7, 1427-1429.
[202] Y. Lu, L. S. Liebeskind, J. Org. Chem. 2004, 69, 3554-3557.
[203] T. Kuwahara, T. Fukuyama, I. Ryu, Org. Lett. 2012, 14(18), 4703-4705.
[204] M. A. Rahim, T. Fujiwara, T. Takeda, Tetrahedron 2000, 56, 763-770.
[205] E.-A. Jo, C.-H. Jun, Eur. J. Org. Chem. 2006, 2504-2507.
[206] J. F. Hooper, R. D. Young, A. S. Weller, M. C. Willis, Chem. Eur. J. 2013, 19(9), 3125-3130.
[207] Z. Gan, Y. Wu, L. Gao, X. Sun, J. Lei, Z. Song, L. Li, Tetrahedron 2012, 68(34), 6928-6934.
[208] A. R. Katritzky, H. Lang, Z. Wang, Z. Zhang, H. Song, J. Org. Chem. 1995, 60(23), 7619-7624.
[209] S. Dohi, K. Moriyama, H. Togo, Tetrahedron 2012, 68(32), 6557-6564.
[210] N. Turkman, L. An, M. Pomerantz, Org. Lett. 2010, 12(19), 4428-4430.
[211] A. V. Dubrovskiy, R. C. Larock, Org. Lett. 2010, 12(14), 3117-3119.
[212] L. Huang, T. Su, W. Shan, Z. Luo, Y. Sun, F. He, X. Li, Biorganic and Medicinal Chemistry 2012, 20(19), 3038-3048.
[213] T. Suzuki, T. Ohwada, K. Shudo, J. Am. Chem. Soc. 1997, 119, 6774-6780.
[214] S. Chiba, Y.-J. Xu, Y.-F. Wang, J. Am. Chem. Soc. 2009, 131, 12886-12887.
[215] D. W. Boykin, R. L. Hertzler, J. K. Delphon, E. J. Eisenbraun, J. Org. Chem. 1989, 54, 1418-1423.
[216] M. Phialas, P. Sammes, P. D. Kennewell, R. Westwood, J. Chem. Soc., Perkin Trans. 1, 1984, 687-695.
[217] S. Kapur, N. C. Mathur, K. McManus, A. L. Pincock, J. A. Pincock, Canadian Journal of Chemistry 1988, 66(11), 2888-2893.
[218] M. Slusarczyk, W. M. De Borggraeve, S. Toppet, G. J. Hoornaert, Eur. J. Org. Chem. 2007, 18, 2987-2994.
[219] R. Takeuchi, H. Yasue, J. Org. Chem. 1993, 58, 5386-5392.
[220] T. Abe, T. Haga, S. Negi, Y. Morita, K. Takayanagi, K. Hamamura, Tetrahedron 2001, 57, 2701-2710.
References
410
[221] N. Niphade, A. Mali, K. Jagtap, R. C. Ojha, P. J. Vankawala; V. T. Mathad, Org. Process Res. Dev. 2008, 12, 731-735.
[222] a) T. Maji, A. Karmakar, O. Reiser, J. Org. Chem. 2011, 76(2), 736-739. b) G. B. Womack, J. G. Angeles, V. E. Fanelli, B. Indradas, R. L. Snowden, P. Sonnay, J. Org. Chem. 2009, 74(15), 5738-5741.
[223] a) N. Kim, Y. Kim, W. Park, D. Sung, A. K. Gupta, C. H. Oh, Org. Lett. 2005, 7(23), 5289-5291. b) M. K. J ter Wiel, R. A. van Delden, A. Meetsma, B. L. Feringa, J. Am. Chem. Soc. 2003, 125(49), 15076-15086.
[224] a) S. P. Chavan, S. Garai, A. K. Dutta, S. Pal, Eur. J. of Chem. 2012, 35, 6841-6845. b) E. Fillion, D. Fishlock, A. Wilsily, J. M. Goll, J. Org. Chem. 2005, 70(4), 1316-1327.
[225] a) F. I Carroll, B. E. Blough, P. Abraham, A. C. Mills, J. A. Holleman, S. A. Wolckenhauer, A. M. Decker, A. Landavazo, K. T. McElroy, H. A. Navarro, M. B. Gatch, M. J. Foster, J. Med. Chem. 2009, 52 (21), 6726-6781. b) Z.-G. Wang, L. Chen, J.-F. Zheng, W. Gao, Z. Zeng, H. Zhou, Z.-K. Zhang, P.-Q. Huang, Y. Su, Eur. J. Of. Med. Chem. 2013, 62, 632-648.
[226] P. Wang, J. Cai, J. Yang, C. Sun, L. Li, H. Hu, M. Ji, Tetrahedron Letters 2013, 54(6), 533-535.
[227] A. Friszkowska, H. Toogood, M. Sakuma, J. M. Gardiner, G. M. Stephens, N. S. Scrutton, Adv. Synth. Cat. 2009, 351(17), 2976-2990.
[228] P. G. Gassman, D. W. Macomber, S. M. Willging, J. Am. Chem Soc. 1985, 107, 2380-2388.
[229] D. R. Baghurst, D. M. P. Mingos, J. of Organomet. Chem. 1990, 384(3), C57-C60.
[230] J. R. Malpass, D. A. Hemmings, A. L. Wallis, S. R. Fletcher, S. Patel, J. Chem. Soc., Perkin Trans 1 2001, 1044-105.

































































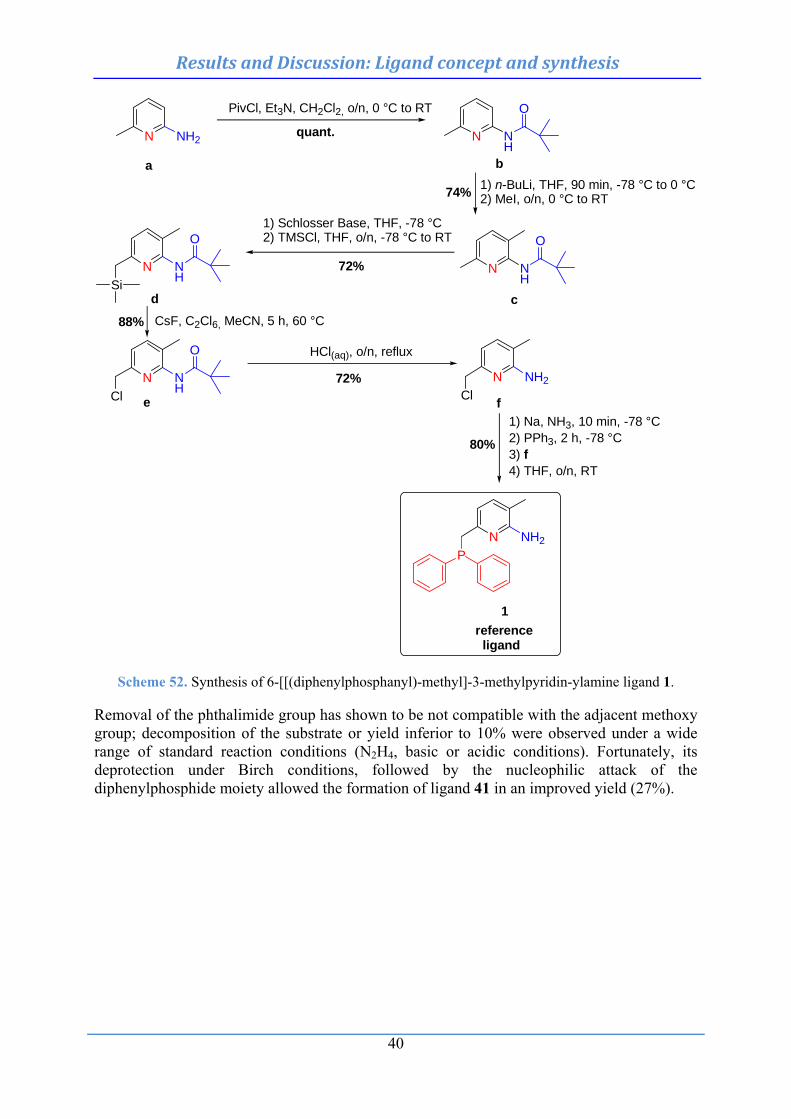





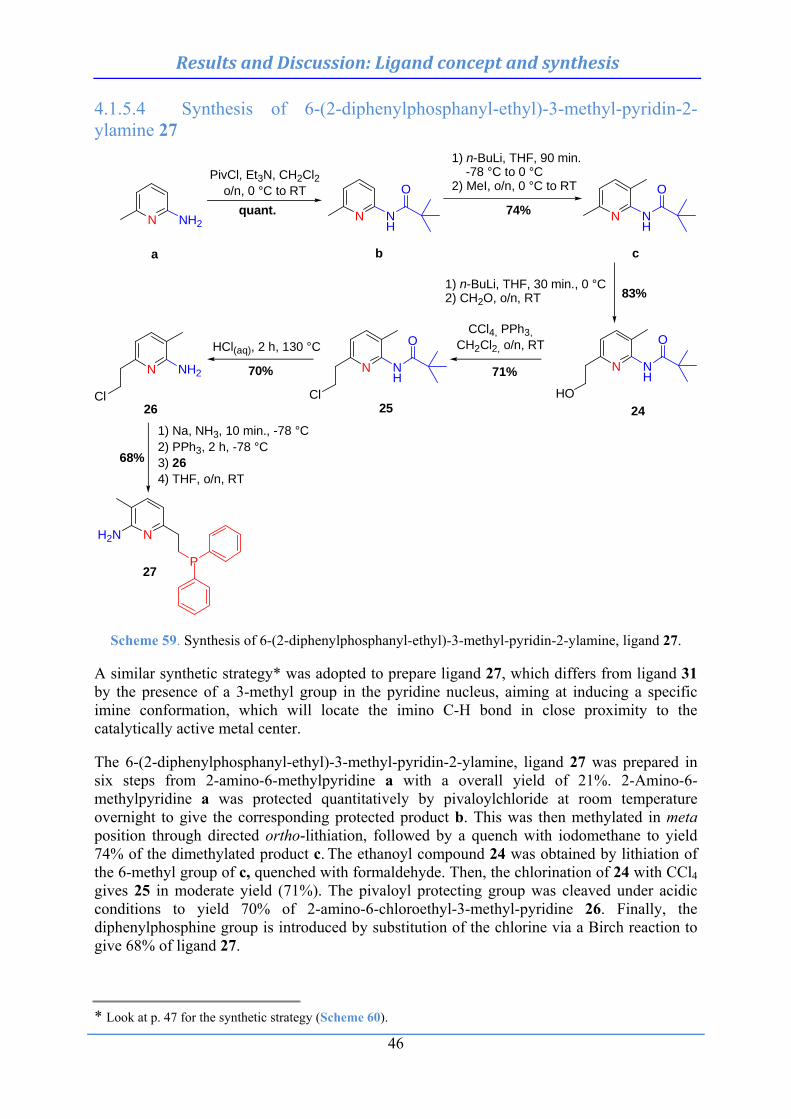




















































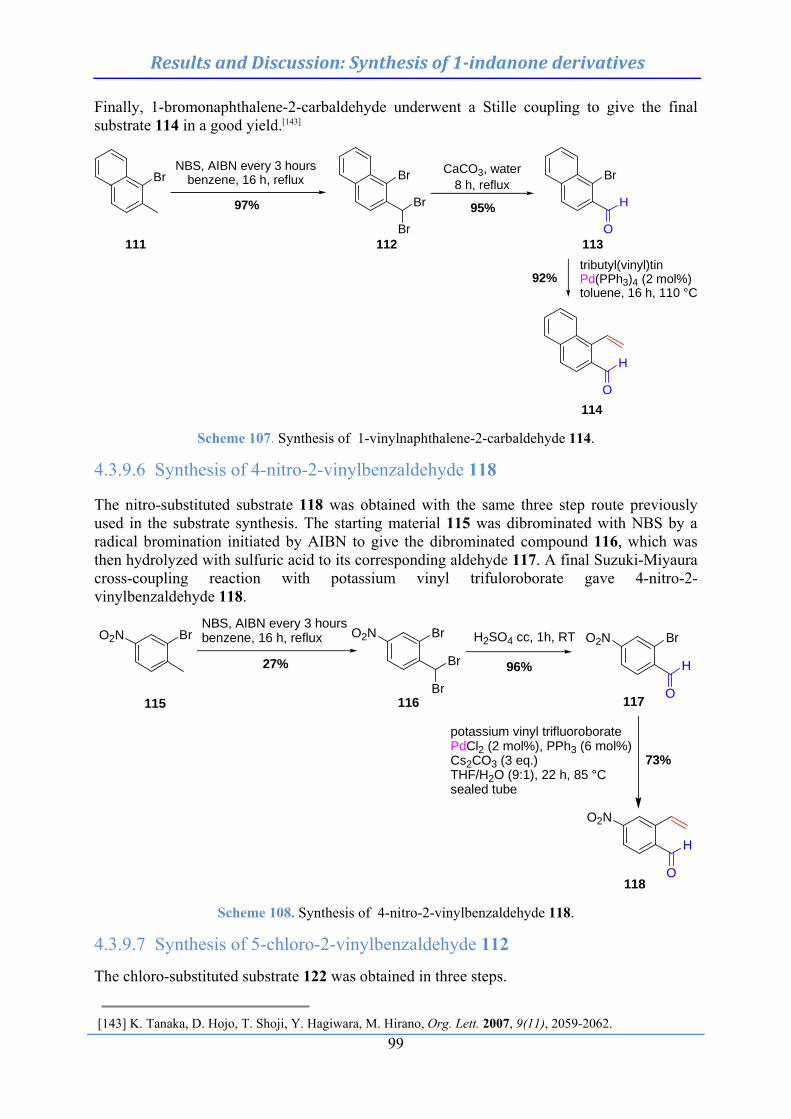























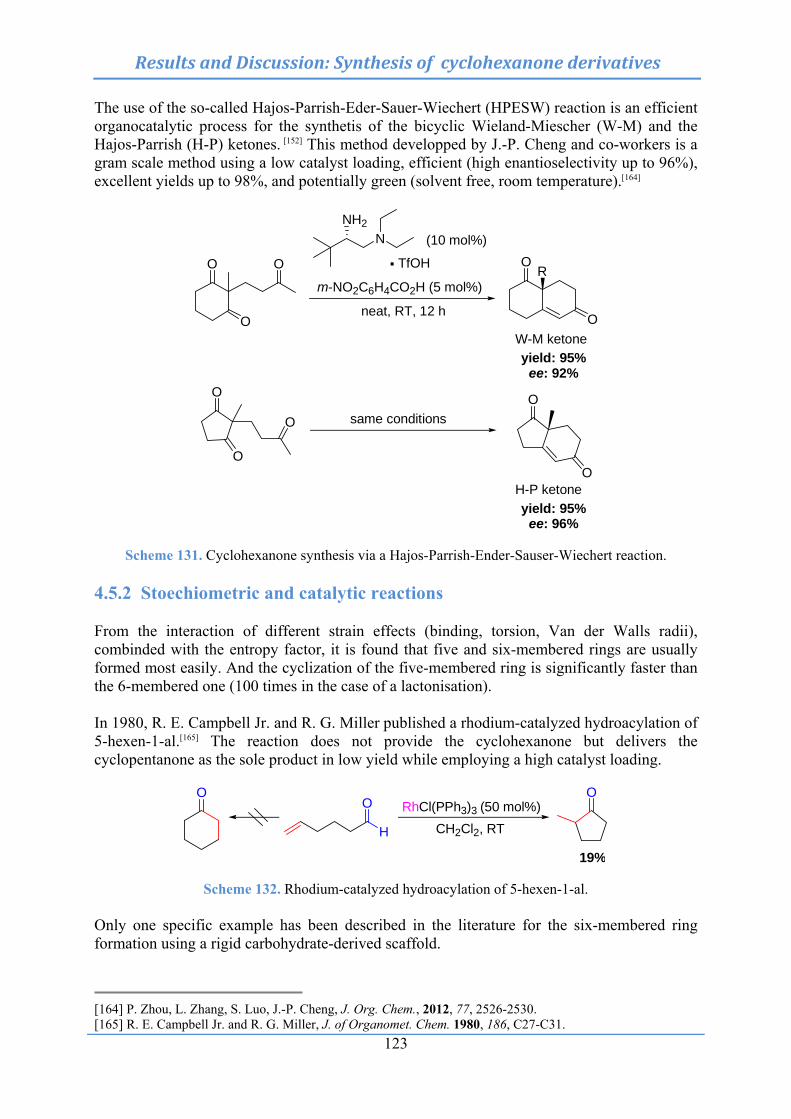




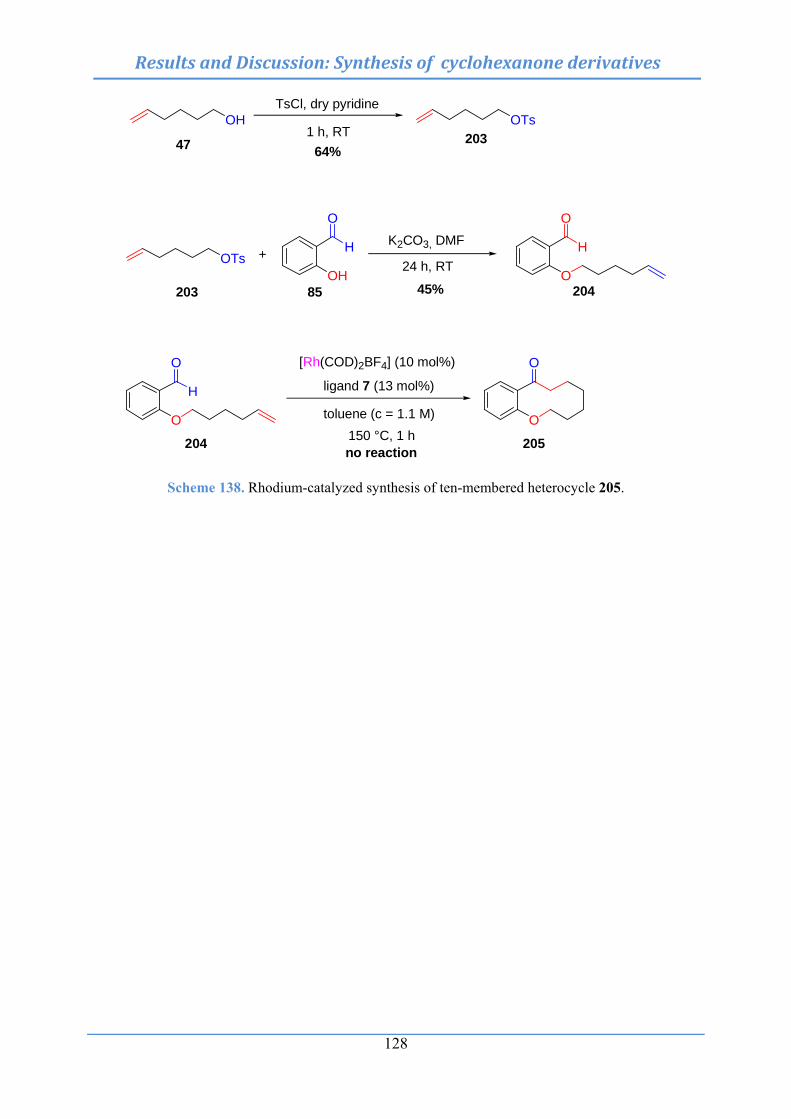










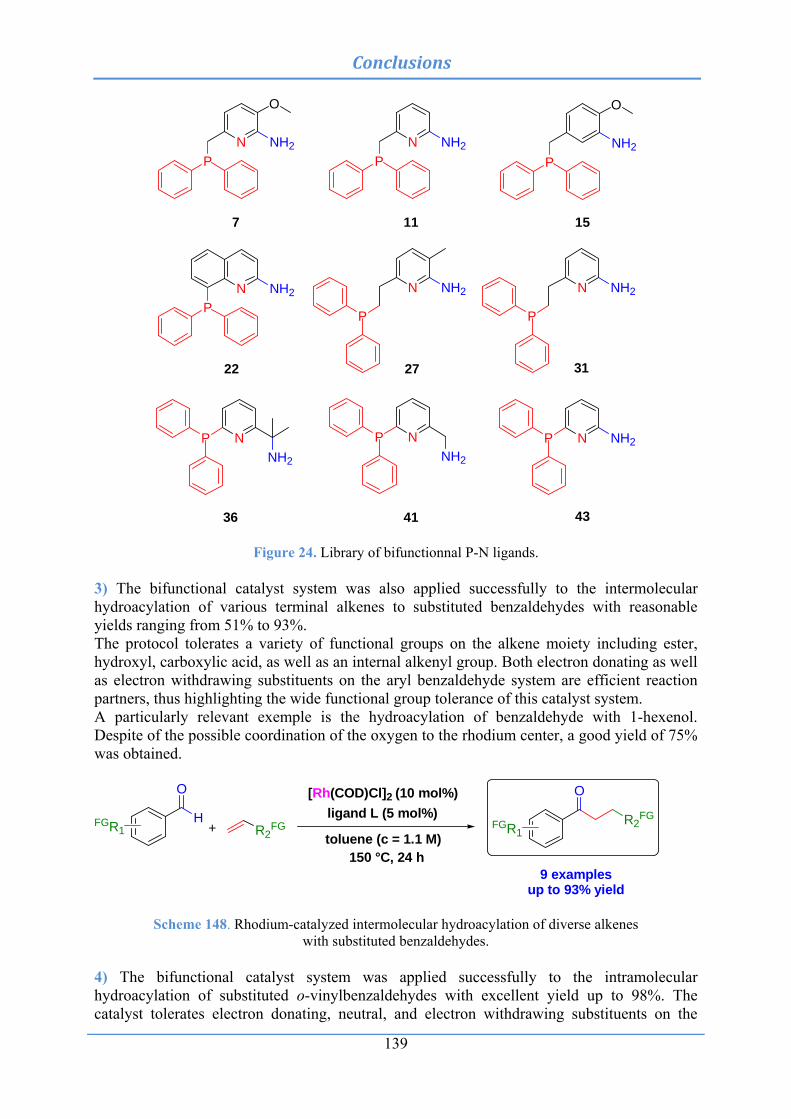
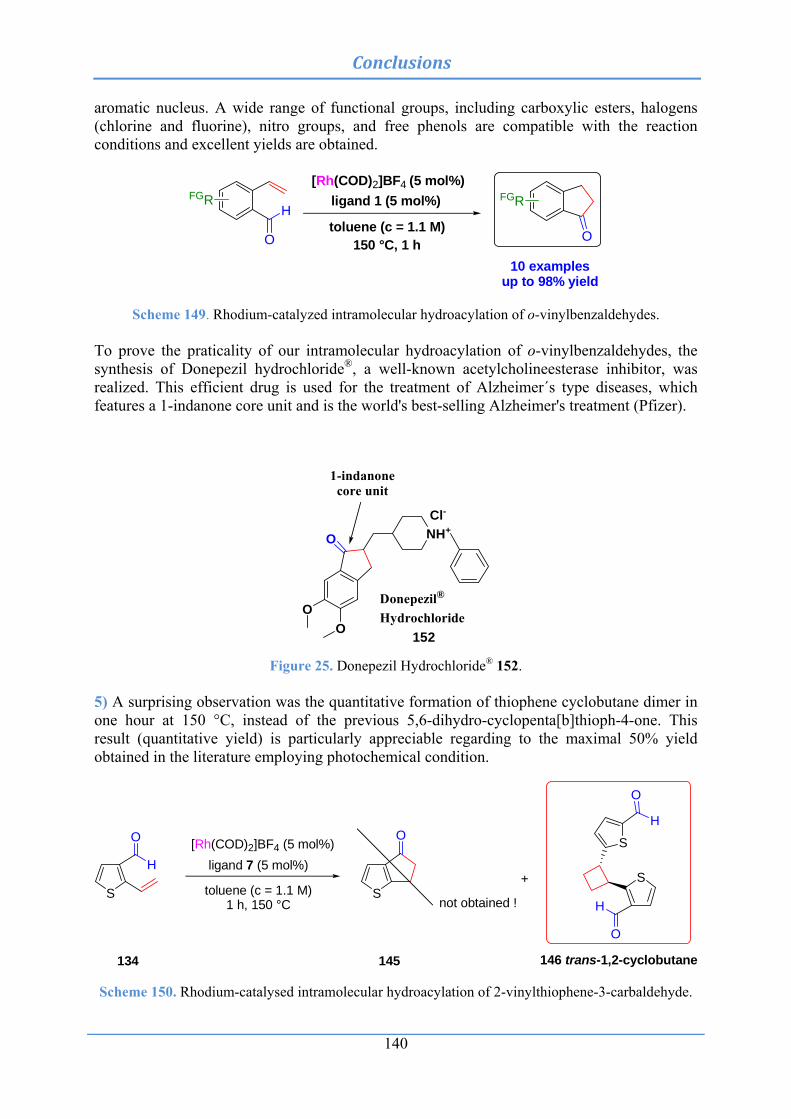






































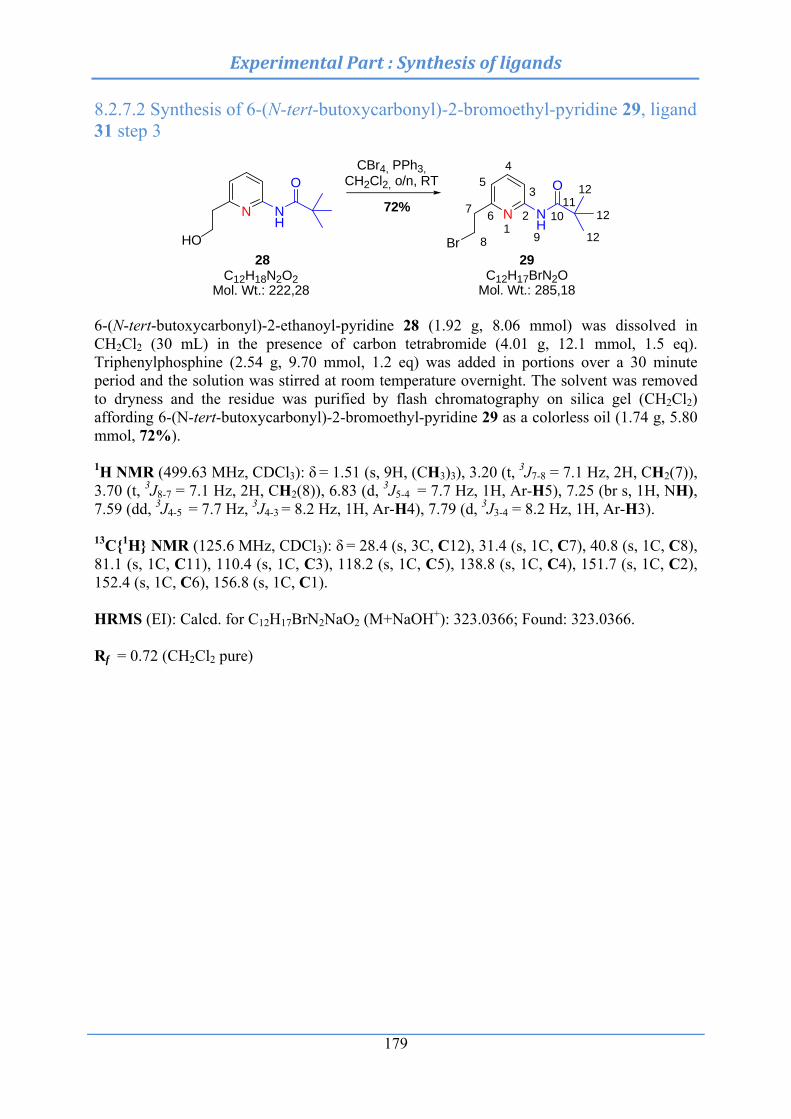


















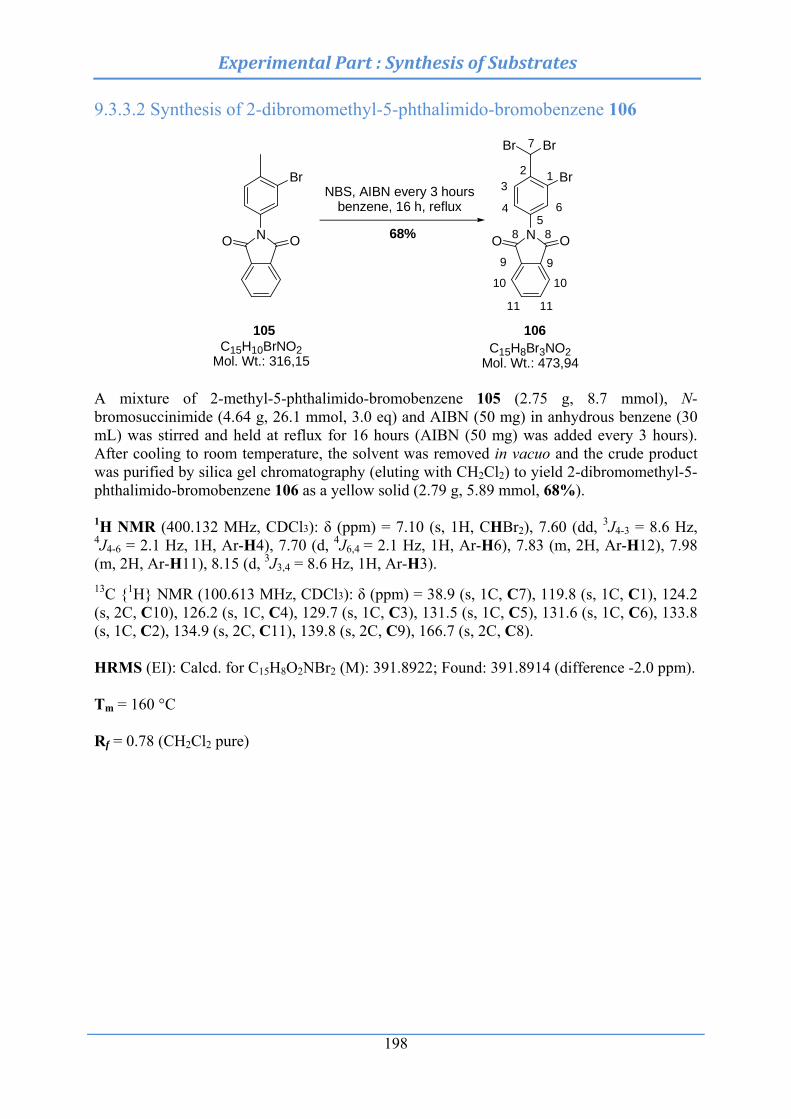
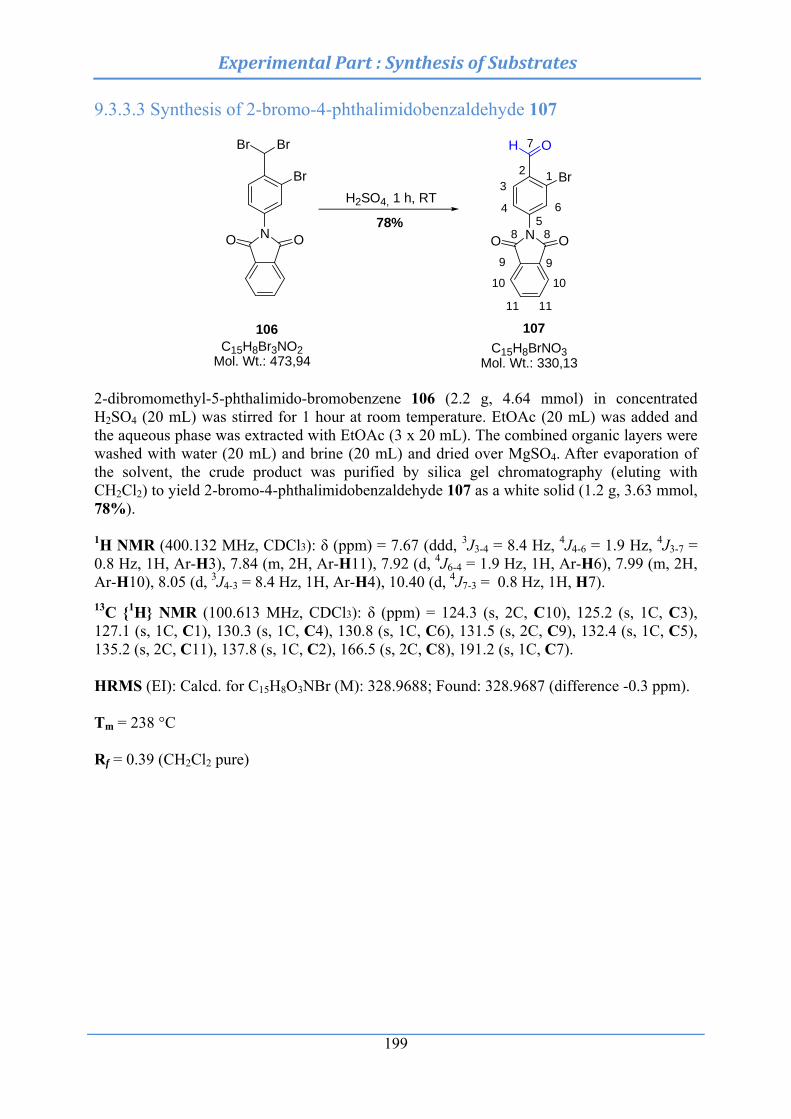




































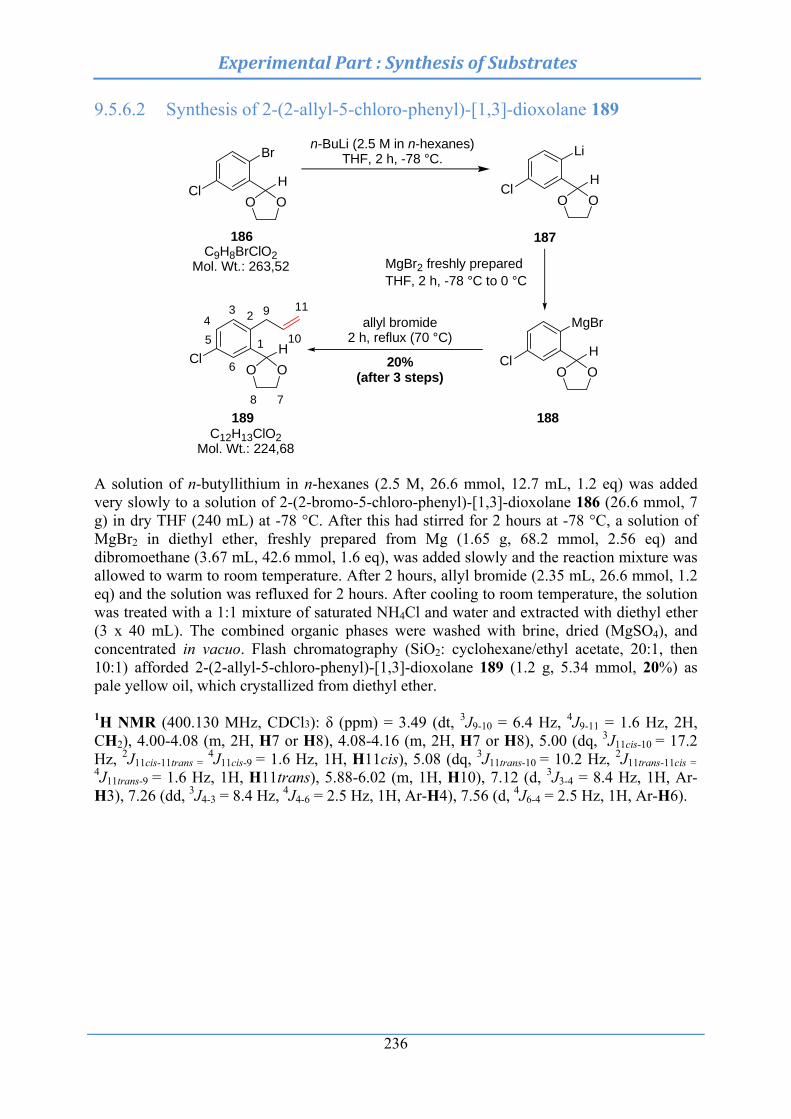

































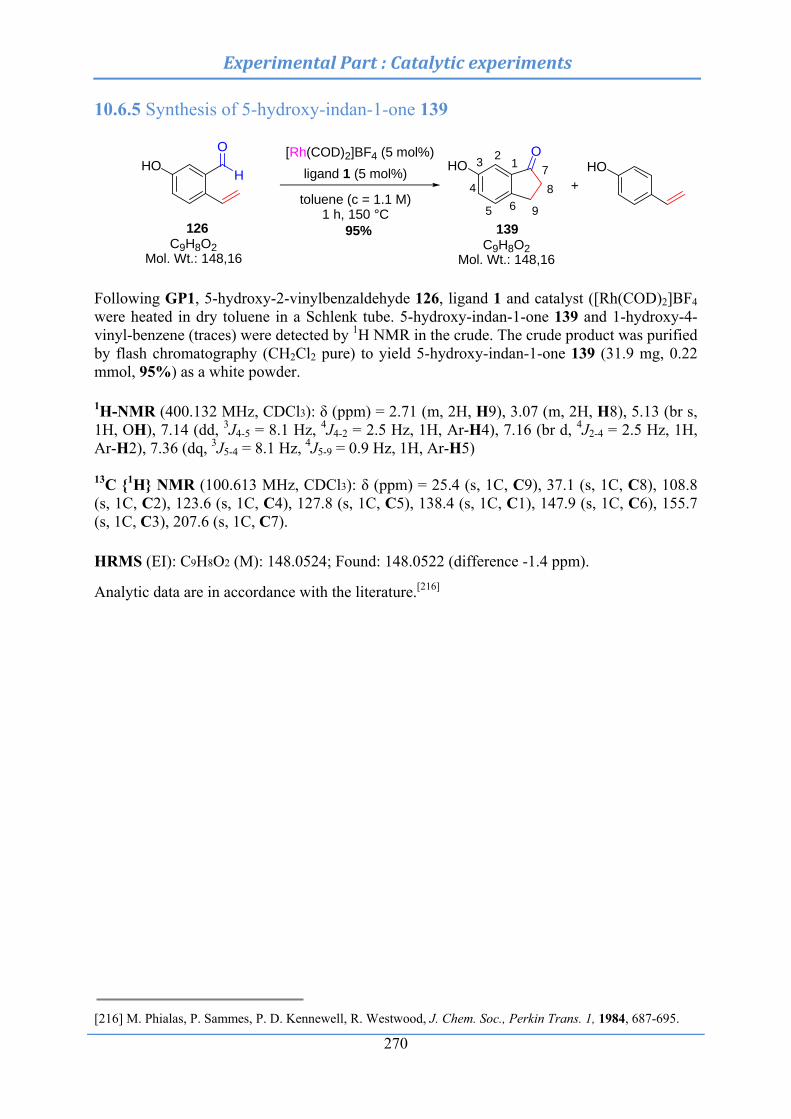






























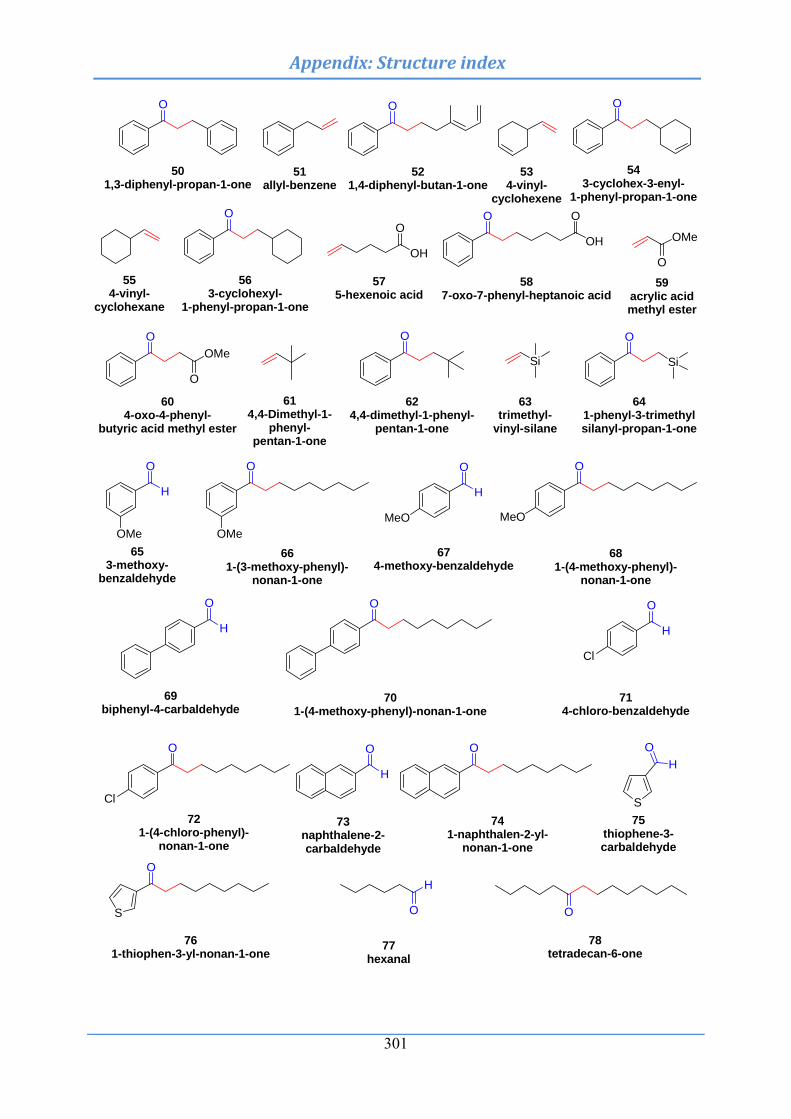









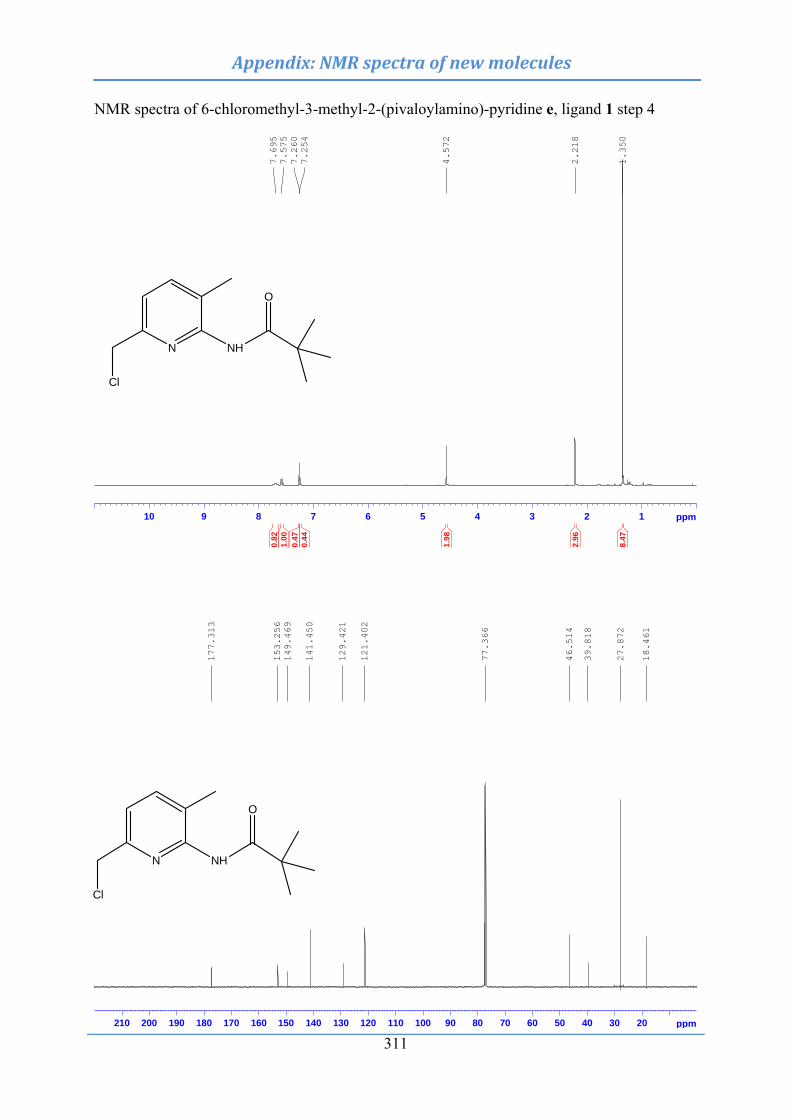


























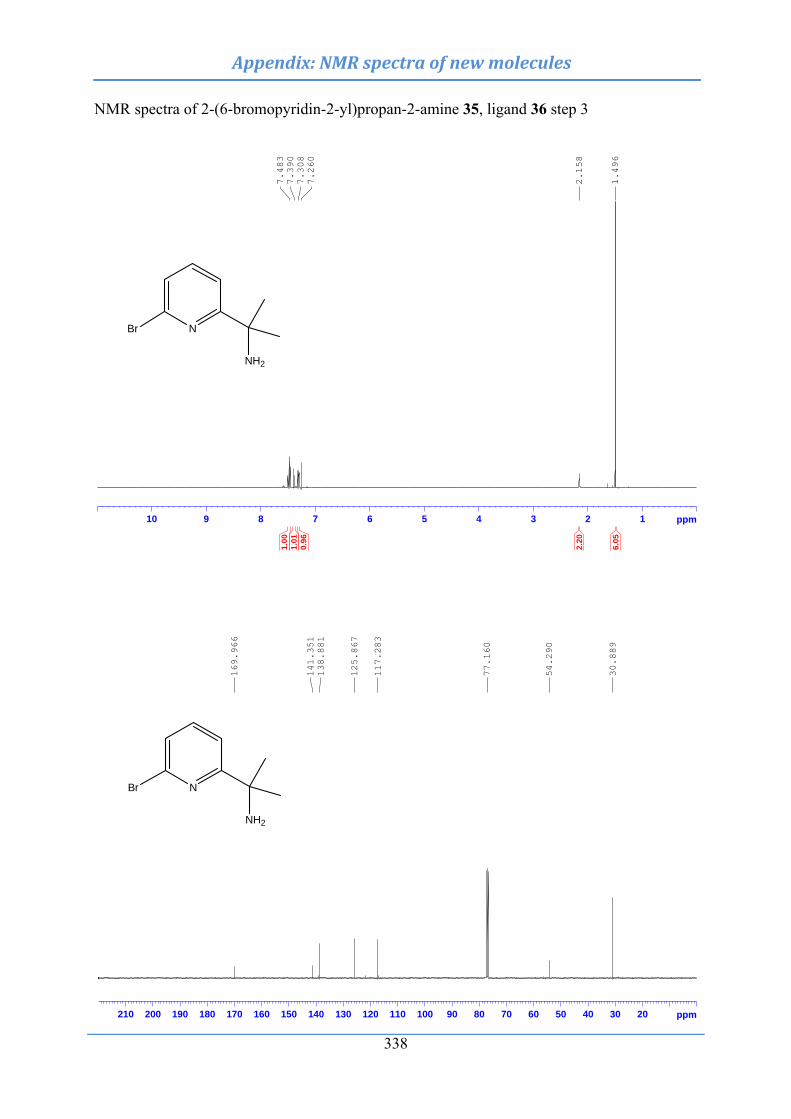














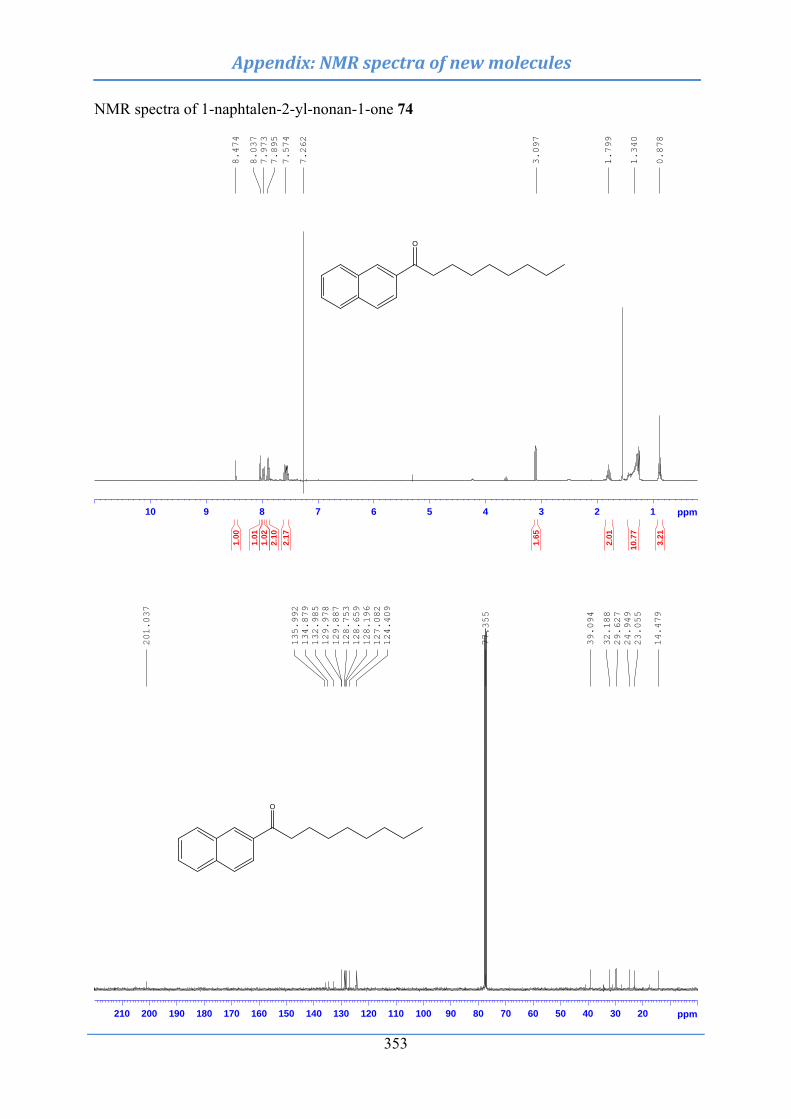











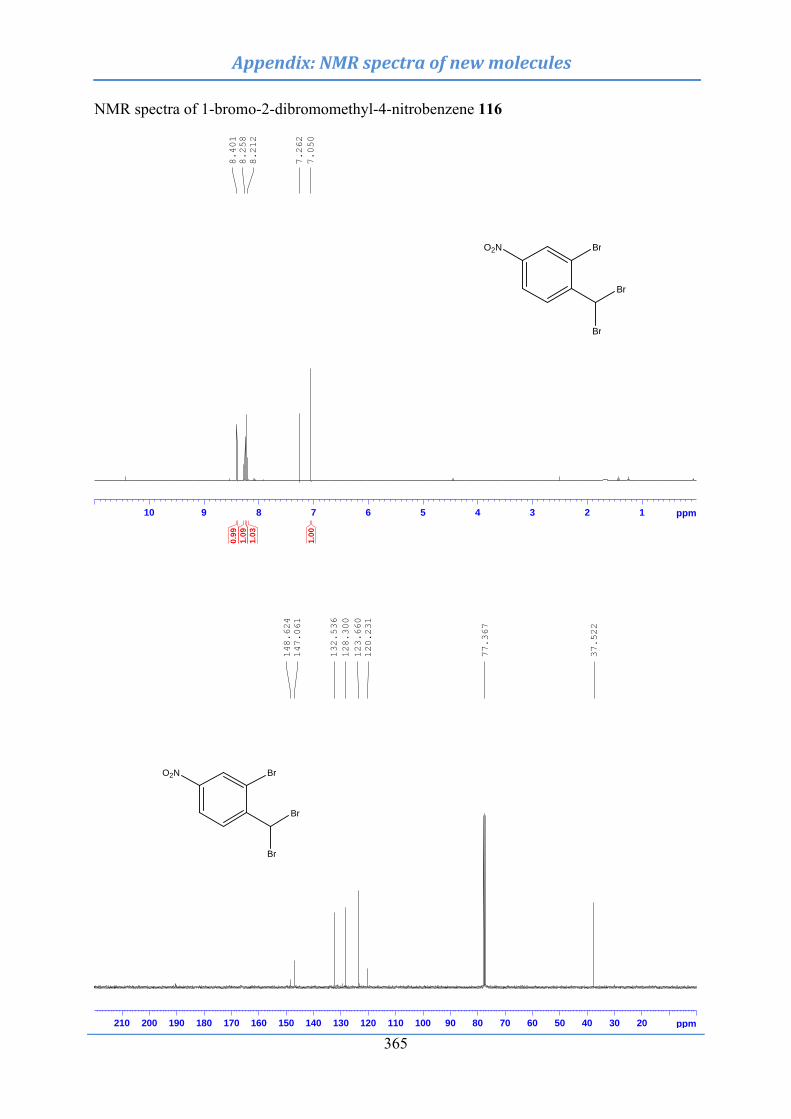










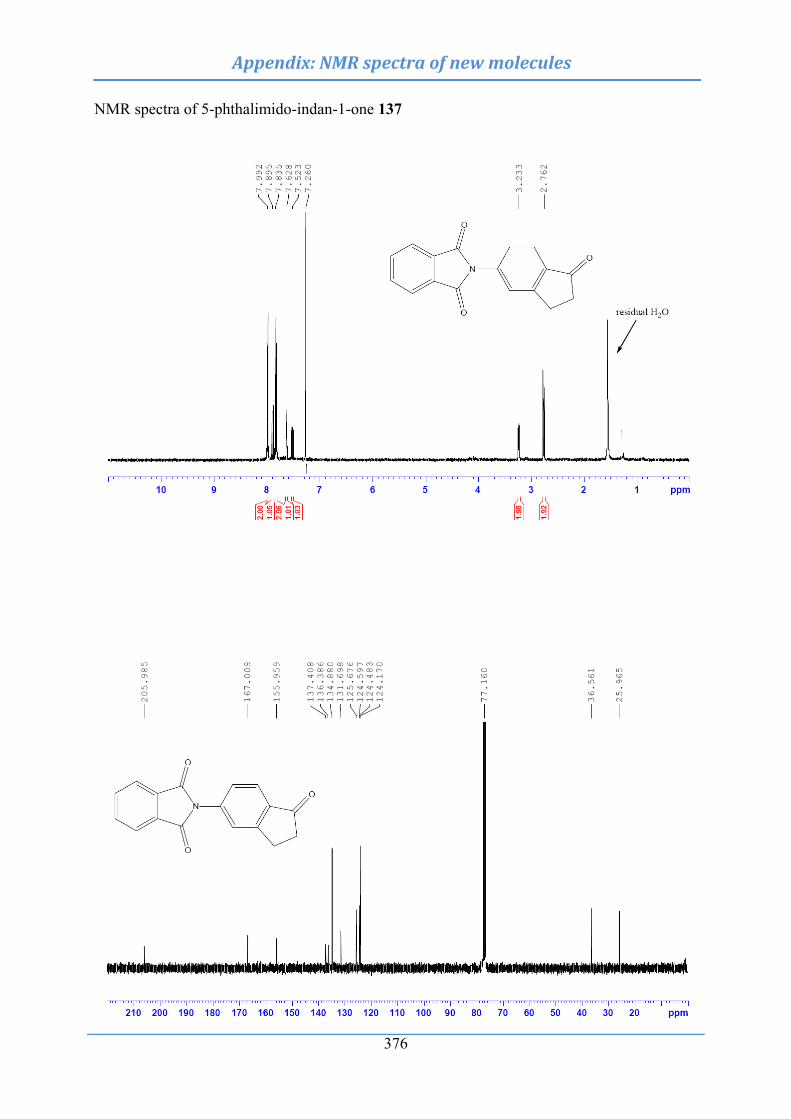

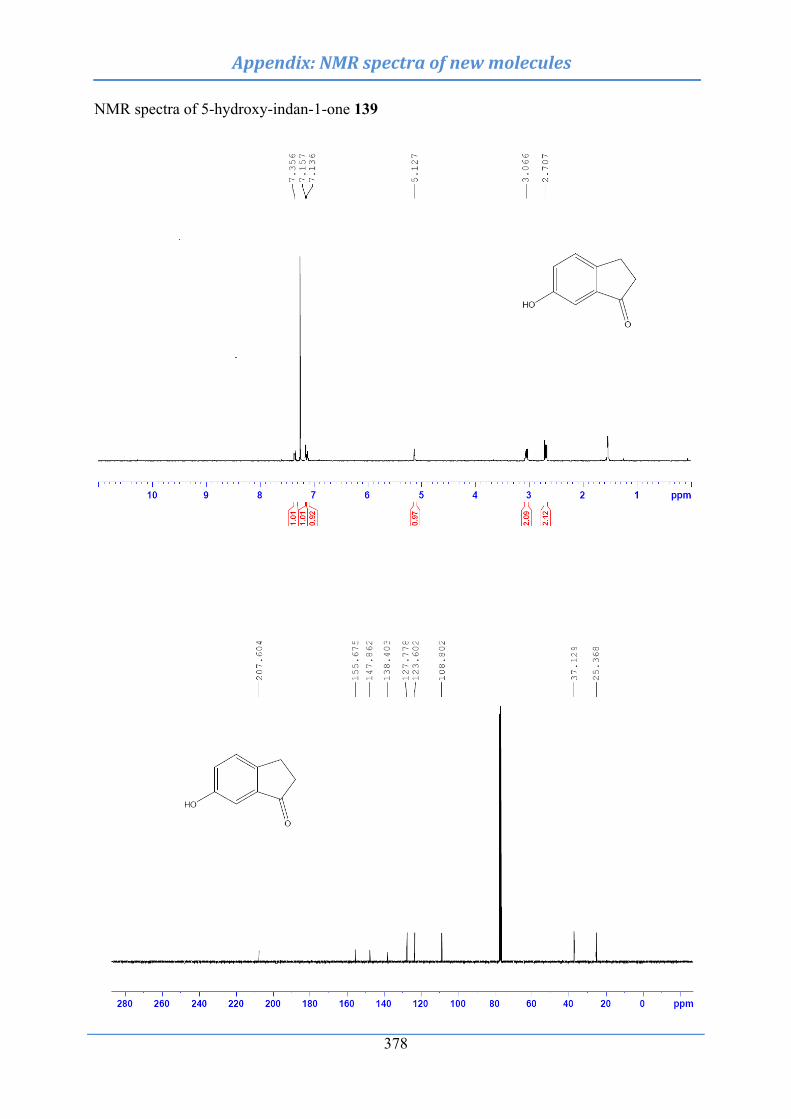


















































![Effect of Ester on Rhodium-Catalyzed Intermolecular [5 + 2 ... · S1 Effect of Ester on Rhodium-Catalyzed Intermolecular [5 + 2] Cycloaddition of 3-Acyloxy-1,4-enynes and Alkynes](https://static.documents.pub/doc/80x56/5f07e3ca7e708231d41f4414/effect-of-ester-on-rhodium-catalyzed-intermolecular-5-2-s1-effect-of-ester.jpg)
