Diabetic nephropathy (DN) is a serious complication of diabetes mellitus, and persistent inflammation incirculatory and renal tissues is an important pathophysiological basis for DN. The essence of the microin-flammatory state is the innate immune response, which is central to the occurrence and developmentof DN. Members of the inflammasome family, including both “receptors” and “regulators”, are key to theinflammatory immune response. Nucleotide binding and oligomerization domain-like receptor familypyrin domain-containing 3 (NLRP3) and other inflammasome components are able to detect endoge-nous danger signals, resulting in activation of caspase-1 as well as interleukin (IL)-1�, IL-18 and othercytokines; these events stimulate the inflammatory cascade reaction, which is crucial for DN. Hyper-glycaemia, hyperlipidaemia and hyperuricaemia can activate the NLRP3 inflammasome, which thenmediates the occurrence and development of DN through the K+ channel model, the lysosomal dam-age model and the active oxygen cluster model. In this review, we survey the involvement of the NLRP3inflammasome in various signalling pathways and highlight different aspects of their influence on DN.We also explore the important effects of the NLRP3 inflammasome on kidney function and structuralchanges that occur during DN development and progression. It is becoming more evident that NLRP3inflammasome targeting has therapeutic potential for the treatment of DN.
Diabetic nephropathy (DN), a disease that is characterized byrogressive albuminuria and a gradual decline in glomerular fil-ration rate (GFR), in association with progressive renal fibrosis orclerosis can ultimately lead to chronic renal failure [1]. Althoughany therapeutic approaches focusing on hyperglycaemia and
igh blood pressure have been implemented, many patients stilluffer from progressive and severe renal injury [2], warranting thenvestigation of other pathogenic pathways and relevant therapeu-ic strategies. Chronic hyperglycaemia, hyperlipidaemia, and otheraemodynamic and metabolic disturbances can prompt continu-us secretion of inflammatory factors, which triggers a cascade ofnflammatory reactions [3]. The innate immune response is centralo the microinflammatory state and is closely linked to the occur-ence of diabetes complications [4]. In addition, inflammasomeamily members are important components of immune inflamma-ory reactions [5].
There is a documented role for the NLRP3 inflammasome, aolyprotein complex with a relative molecular mass of approx-
mately 700000; it plays a transformation role in the process ofnflammation [6]. The NLRP3 inflammasome consists of nucleotideinding and oligomerization domain-like receptor family pyrinomain-containing 3 (NLRP3), apoptosis-associated speck-likerotein containing a caspase recruitment domain (ASC) andaspase-1 or caspase-5. NLRP3 is the core protein in the com-lex [7]. NLRP3 contains three different structural domains: (1)
pyrin domain (PYD) or C-terminal caspase-recruitment domainCARD) located at the N-terminus, which mediates protein-proteinnteractions during signal transduction; (2) a nucleotide-bindingligomerization domain (NOD)/neuronal apoptosis inhibitor pro-ein, CIITA, HET-E and TP1 (NACHT) domain located in the centref the molecule, which is vital for the activation of NLRP3 viaelf-mediated oligomerization; (3) a leucine-rich repeat (LRR)omain located at the C-terminus, which recognizes and identifiesathogenic microorganisms and other ligands through protein-rotein or protein-glycolipid interactions [8]. The function of NLRP3
n the NLPR3 inflammasome is specific ligand recognition. Whenhe ligand is recognized by the LRR in the C-terminus, the moleculesf the NLR family undergo a conformational rearrangement due tohe effects of the oligomerization of the NOD structural domain,hus exposing the effector domain and ultimately inducing theffector molecule which N-terminal has same CARD or PYD domainnd activating its biological effects [9]. NLRP3 combined withSC, recruits pro-caspase-1 to form the inflammasome and turned
nto caspase-1 by hydrolysis. Then inflammasome will promoteaspase-1 self-enzymatic hydrolysis to heterodimers which consistf the P10 and P20 subunits. The P20 subunit induces pro-IL-1�aturation and IL-1� secretion [10]. ASC, encoded by the pycard
ene, is an important 195-amino-acid protein link with upstream
f NLRP3 and downstream caspase-1. ASC is a key inflammasomedapter in the NLRP3 inflammasome complex [11] and possesseswo different structures. (1) The PYD domain located at the N-erminus is an oligomerization domain involved in protein-protein
interactions with the N-terminal PYD domain of activated NLPR3.(2) The CARD domain located at the C-terminus of ASC is involvedin recruiting caspase-1 to the inflammasome and plays a role in theactivation of caspase-1 by the effector domain [12,13].
The inflammasomes are components of the innate immune sys-tem that respond to danger signals by producing pro-inflammatorycytokines and converting the cytokines to their active forms [14].Abnormal NLRP3 inflammasome activation causes harmful inflam-mation in cryopyrin-associated periodic syndromes (CAPS), rareinherited autoinflammatory diseases characterised by recurrentfevers, leucocytosis, rashes, and arthralgias [15], as well as inautoimmune diseases, such as multiple sclerosis and lupus [16,17].The NLRP3 inflammasome has also been implicated in the patho-genesis of a number of renal conditions, including acute kidneyinjury, chronic kidney disease, DN and crystal-related nephropathy[18,19].
NLRP3 inflammasome activation occurs both in immune cells,primarily macrophages and dendritic cells, and in some intrinsickidney cells, such as renal tubular epithelium in kidney disease andautoimmunity [20].
Immune cells in the kidney, which are basically the meshworkof intrarenal mononuclear phagocytes, express all the componentsnecessary for NLRP3 complex assembly and production of IL-1� andIL-18 [19]. IL-1� and IL-18 influence adaptive immunity throughmodulation of T helper cell subsets, skewing development in favourof Th17 and Th1 cells, which are important in the developmentof autoimmunity [20]. However, the expression of inflammasomecomponents in kidney tissue cells is not clearly defined. Renal-restricted silencing of ASC attenuates proteinuria and glomerularsclerosis in mouse models of kidney disease [6,21]. However, deliv-ery of ASC siRNA via the renal artery affects both intrarenal immunecells and renal tissue cells. Thus, it is difficult to attribute theobserved protection solely to intrarenal macrophages and dendriticcells.
Murine tubular epithelial cells can secrete IL-1� and IL-18, andit is generally agreed that these cells contain all the componentsnecessary for inflammasome activation [22,23]. Mouse glomeru-lar endothelial cells, mesangial cells and podocytes do not produceIL-1� or activate caspase-1 [22], but varying degrees of evidencesupport inflammasome activation in podocytes and glomerularendothelial cells [6,21,24]. In human kidneys, renal cells includingpodocytes, mesangial cells and intercalated cells exhibit inflamma-some expression [25,26].
2. NLRP3 inflammasome’s effects and mechanisms indiabetic nephropathy
2.1. Mitogen-activated protein kinase (MAPK) signalling pathway
As an inflammatory disease, DN is influenced by secreted inflam-matory mediators, a process that is regulated by many mechanisms.One of the most important mechanisms is the MAPK signallingpathway [27]. MAPK encompasses three subfamilies [28]: the
logical
pN
tvasdJbpcehnraoii
2
ciilwpettaiItpNpIbmm
bk(p(T[pAc[nii(ptist[c
Y.-y. Qiu, L.-q. Tang / Pharmaco
38MAPK, extracellular signal-regulated kinase (ERK) and c-Jun-terminal kinase (JNK) families.
NOD2 is a member of the NLR family and has the same struc-ure as NLRP3. As NOD2 can activate the MAPK signalling pathwayia pathogen stimulation, it has been proposed that NLRP3 canlso activate MAPK signalling [29]. A study of virus-infected cellshowed that phosphorylation of ERK and a variety of chemokinesepends on ASC [30]. NOD2-mediated activation of p38MAPK and
NK is induced by recognition of the muramyl dipeptide of theacterial cell wall [31]. Activation of JNK and p38 promotes apo-tosis, while a small amount of ERK phosphorylation can protectells from damage. ERK is occasionally used as a marker for regen-ration and repair [32]. To reconcile these observations, a secondypothesis has been proposed in which the activation of MAPK sig-alling by NLRP3 is more relevant to damage than repair. NLRP3 canecognize the inflammation danger signals and induce ERK, JNK,nd p38MAPK phosphorylation; however reduced levels of NLRP3nly affect the phosphorylation of p38MAPK and JNK and do notnfluence ERK phosphorylation [29,33,34]. These observations aren support of the second hypothesis.
.1.1. P38MAPK signalling pathwayIt is reported that p38MAPK, the main function of which is to
ontrol some of the stress factors and cytokines inducing apoptosis,s associated with the development of DN under various stimulat-ng factors [35]. In vitro studies have indicated that high glucose canead to phosphorylation/activation of p38MAPK signalling path-
ay components, which can enhance mesangial cell secretion androliferation [36]. Inhibiting the p38MAPK pathway decreases thextracellular matrix (ECM) of mesangial cells (MCs), which can slowhe thickening of the glomerular basement membrane (GBM) andhus delay the process of DN [36]. In addition, high glucose can alsoctivate the NLRP3 inflammasome, The p38MAPKs are intimatelynvolved in the regulation of pro-IL-1� synthesis and in IL-1� andL-18 signalling [37,38]. The classic NLRP3 inflammasome activa-or, extracellular ATP, has been reported to trigger the activation of38MAPKs [39]. Compared with ATP, cholesterol crystals, anotherLRP3 activator, trigger a slower, more persistent activation of38MAPKs. Activation of p38MAPK is required for NLRP3-mediated
L-1� secretion in humans and mice [40]. Hence, there is reason toelieve that intracellular stress signals, such as high glucose, canediate activation of the p38MAPK pathway through the inflam-asome.P38MAPK activation and mRNA expression are controlled
y upstream agonist mitogen-activated protein kinase kinaseinase (MAPKKK) and mitogen-activated protein kinase kinaseMAPKK) [41]. MKK3/6, a dual-function kinase, activates p38MAPK.38MAPK phosphorylation is assumed to occur via a Thr-Gly-TyrTGY) motif for double phosphorylation; thus, phosphorylation ofyr and Thr in the TGY motif of p38 results in p38 protein activation42]. In cultured HMDMs, p38MAPK is activated via dual phos-horylation during the activation of the NLRP3 inflammasome byTP and cholesterol crystals, and this activation precedes or coin-ides with caspase-1 activation and the secretion of mature IL-1�40]. This is followed by p38MAPK-mediated serine and threo-ine phosphorylation of several downstream transcription factors,
ncluding cAMP response element-binding protein (CREB), activat-ng myocyte enhancer factor-2C (MEF-2C), transcription factor-1ATF-1) and ATF-2. These events regulate the transcription androduction of fibronectin (FN) as well as accumulation of the pro-ein in the basement membrane [43–45]. These mechanisms maynclude negative effects, such as renin-angiotensin-aldosterone
ystem (RAAS) activation [46], extracellular matrix (ECM) produc-ion, ROS enhancement, and growth factor-induced sensitization47]. In this process, cells become hypertrophic, with mesangialell proliferation and podocyte apoptosis [48,49]. Moreover, to
Research 114 (2016) 251–264 253
elicit positive protective effects, the podocyte cytoskeleton willbe temporarily adjusted to reduce albuminuria production [50].Additionally, p38 activation can also affect the production ofinflammatory factors and aggravate the inflammatory reaction,which, as stated above, is important for inflammasome expressionand DN. There is an obvious interaction between p38MAPK andtransforming growth factor-� (TGF-�) [51]. When activated by theinflammasome, p38MAPK usually increases the binding capacityof activation-protein-1 (AP-1) and the gene expression of TGF-�,thereby regulating p38MAPK signalling via positive feedback [52].Therefore, a vicious cycle ensues when the pathway is activatedand TGF-� is overexpressed; this is followed by the prolifera-tion of mesangial cells and accumulation of the ECM, which cancause renal glomerulosclerosis and tubulointerstitial fibrosis anddecrease the GFR. In addition, activation of p38MAPK also enhancesthe gene expression of activating transcription factor 2 (ATF2), lead-ing to significant increases in TNF-� and IL-1� [53] and resultingin the release of other inflammatory factors such as IL-6 and IL-8. These events accelerate the occurrence of glomerulonephritisand result in DN development [54]. In addition to this canoni-cal effect of NLRP3 on cytokine maturation and renal pathology,NLRP3 is thought to have non-canonical effects on various typesof renal injury [55]. Non-canonical effects of NLRP3 were observedin the promotion of TGF-�-dependent signalling in renal tubularepithelial cells [56].
Numerous studies in humans and animals such as rats andmice have also demonstrated that podocytes are one of the majorsources of glomerular IL-1� under various pathological conditionsand that activation of inflammasomes may contribute to IL-1� pro-duction [19,57]. The p38 pathways display reduced activation inASC-knockdown cells, but repeated phosphoblotting revealed thatthe levels of p-p38 in THP1 cells were too low to be measuredaccurately [30].
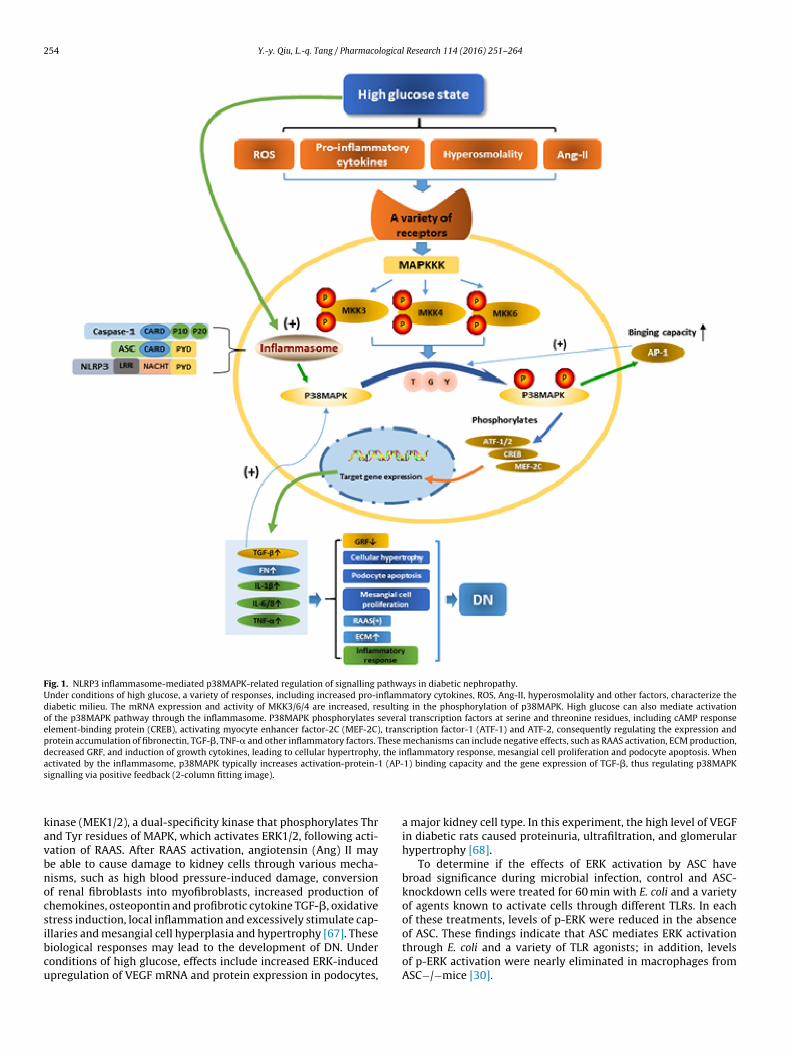
A previous study found that the p38MAPK signalling path-way mediated by the NLRP3 inflammasome in association withother pathways aggravates DN, showing that p38MAPK activa-tion increased the involvement of NF-�B signalling in renal injury[58,59]. Although documented in initial reports, it remains unclearwhether MAP kinase phosphatase-1 (MKP-1) is involved in a feed-back loop to prevent p38MAPK activation after stimulation ofthe NLRP3 inflammasome or to delay the effect [60]. Continuedresearch will reveal the mechanism of NLRP3 inflammasome-mediated p38MAPK signalling in DN and provide a new target fortreatment (Fig. 1).
2.1.2. ERK signalling pathwayUnder normal circumstances, ERK1/2 is located in the cyto-
plasm and is phosphorylated after stimulation by high glucose andmolecules such as various growth factors and hydrogen peroxide.Activated ERK1/2 then enters the nucleus and affects transcriptionfactors to promote the transcription and expression of genes par-ticipating in cell proliferation and differentiation [61]. Research hasdemonstrated that ERK plays a key role in intracellular signallingpathways, influencing the integration and transcription of genesinvolved in many cellular responses in DN [62]. Furthermore, theinflammasome stimulates ERK [33].
Previous studies of diabetes have shown that in different typesof kidney cells, including podocytes and glomerular mesangial cells,the ERK signalling pathway is activated by ROS, high blood glucoseand growth factors [63]. The NLRP3 activator regulates the produc-tion of ROS in many ways [64,65]. ERK1-mediated posttranslationalmodifications may allow the NLRP3 inflammasome to respond to
the second signal, ATP, by inducing posttranslational events thatare independent of new production of pro–IL-1� and NOD-likereceptor components [66]. In the presence of influencing factors,activated Raf-1 kinase phosphorylates and activates MAPK/ERK
Fig. 1. NLRP3 inflammasome-mediated p38MAPK-related regulation of signalling pathways in diabetic nephropathy.Under conditions of high glucose, a variety of responses, including increased pro-inflammatory cytokines, ROS, Ang-II, hyperosmolality and other factors, characterize thediabetic milieu. The mRNA expression and activity of MKK3/6/4 are increased, resulting in the phosphorylation of p38MAPK. High glucose can also mediate activationof the p38MAPK pathway through the inflammasome. P38MAPK phosphorylates several transcription factors at serine and threonine residues, including cAMP responseelement-binding protein (CREB), activating myocyte enhancer factor-2C (MEF-2C), transcription factor-1 (ATF-1) and ATF-2, consequently regulating the expression andprotein accumulation of fibronectin, TGF-�, TNF-� and other inflammatory factors. These mechanisms can include negative effects, such as RAAS activation, ECM production,decreased GRF, and induction of growth cytokines, leading to cellular hypertrophy, the inflammatory response, mesangial cell proliferation and podocyte apoptosis. Whenactivated by the inflammasome, p38MAPK typically increases activation-protein-1 (AP-1) binding capacity and the gene expression of TGF-�, thus regulating p38MAPKs
kavbnocsibcu
ignalling via positive feedback (2-column fitting image).
inase (MEK1/2), a dual-specificity kinase that phosphorylates Thrnd Tyr residues of MAPK, which activates ERK1/2, following acti-ation of RAAS. After RAAS activation, angiotensin (Ang) II maye able to cause damage to kidney cells through various mecha-isms, such as high blood pressure-induced damage, conversionf renal fibroblasts into myofibroblasts, increased production ofhemokines, osteopontin and profibrotic cytokine TGF-�, oxidativetress induction, local inflammation and excessively stimulate cap-
llaries and mesangial cell hyperplasia and hypertrophy [67]. Theseiological responses may lead to the development of DN. Underonditions of high glucose, effects include increased ERK-inducedpregulation of VEGF mRNA and protein expression in podocytes,
a major kidney cell type. In this experiment, the high level of VEGFin diabetic rats caused proteinuria, ultrafiltration, and glomerularhypertrophy [68].
To determine if the effects of ERK activation by ASC havebroad significance during microbial infection, control and ASC-knockdown cells were treated for 60 min with E. coli and a varietyof agents known to activate cells through different TLRs. In eachof these treatments, levels of p-ERK were reduced in the absence
of ASC. These findings indicate that ASC mediates ERK activationthrough E. coli and a variety of TLR agonists; in addition, levelsof p-ERK activation were nearly eliminated in macrophages fromASC−/−mice [30].
logical
pmElmi
mEleisiipietptrc
rp
2
hpTMaNpmimoJT(aaspaitscnoit
propaii
Y.-y. Qiu, L.-q. Tang / Pharmaco
Recognition of LPS by TLR4 stimulates down-stream signallingathways, such as MAPKs and NF-�B [69]. LPS treatment of humanacrophages induces the early activation of NF-�B, p38, JNK and
RK [70], and studies in the J774A.1 murine macrophage celline described the significant regulatory role of MAPK signalling
olecules, such as p38, JNK and ERK, in the expression of IL-1� andnflammasome components [33,71,72].
The ERK signalling pathway, as mediated by the NLRP3 inflam-asome, is also likely to impact other pathways to facilitate DN.
RK signalling results in phosphorylation of intracellular phospho-ipase A2 (PLA2) to promote the release of arachidonic acid andicosanoid, which can promote the changes in renal haemodynam-cs observed in DN [73,74]. In association with TGF-� signalling,ome scholars have confirmed that the ERK l/2 pathway is activatedn mesangial cells under a high glucose environment, resultingn increased TGF-�1 mRNA expression. This can affect GMCs andromote its proliferation and hypertrophy, in addition to promot-
ng the synthesis of the ECM and inhibiting ECM degradation,vents that ultimately cause glomerular sclerosis [67,75]. In addi-ion, under a high-glucose condition, the ERK pathway-induced DNrocess leads to mesangial cell proliferation and glomerular nephri-is via protein kinase C (PKC), protein tyrosine kinase (PTK), andeceptor for advanced glycation end products (RAGEs) in kidneyells [76–79] (Fig. 2).
Inflammasome-mediated ERK signalling is critical to DN. Futureesearch should explore the mechanism of ERK and identify specificroteins for targeted therapy in DN.
.1.3. JNK signalling pathwayIn type II diabetic patients and diabetic animal models, under
igh-glucose conditions, MAP3K first activates the JNK signallingathway and inflammatory cytokines, such as TNF-� [3]. TheAK1(TGF-activated kinase-1)-JNK pathway, a stress-responsiveAPK signalling pathway, is activated through lysosome rupture,
nd this activation is necessary for the complete activation of theLRP3 inflammasome through the oligomerization of the adapterrotein ASC [80]. These observations indicate that JNK activationay stimulated by the NLRP3 inflammasome function, resulting
n a cascade of responses, such as high glucose-stimulated inflam-asome activation and ASC activation of JNK or phosphorylation
f its upstream factors. The most effective MAP3K activation ofNK is thought to occur through MAPK kinase kinase 1 (MEKK1).his upstream kinase is activated by domain for versatile dockingDVD), which is located at the C-terminus of MKK4; the relevantccess domain is located at the N-terminus of P38 and JNK [81]. Inddition, the regulation and translocation of JNKs usually requirescaffold proteins for assembly and activation by kinases. JNKs phos-horylate C-Jun proteins, followed by changes in AP-1 transcriptionnd expression via post-transcriptional mechanisms after enter-ng the nucleus [82]. Notably, a connection between ASC and theranscription factor AP1 was recently established using a recon-tituted cell system that is engineered to respond to the bacterialell wall component muramyl dipeptide [83]. AP1 is one of a largeumber of transcriptional regulators that are downstream targetsf MAPKs [84]. Such behaviour can lead to insulin resistance (IR),nsulin deficiency and high blood glucose levels, clearly increasinghe development of DN (Fig. 3).
Insulin deficiency reduces the level of insulin receptor tyrosinehosphorylation and insulin receptor substrate. Tyrosine phospho-ylation is relatively reduced, and IR proteins cause mild activationf the PI3 kinase signalling pathway to regulate different metabolic
athways in renal disease. Glycogen synthase kinase signalling waslso shown to have an important relationship with JNK-related IRn the kidney [85]. Overall, these effects show that JNK plays anmportant role in the occurrence and development of DN.
Research 114 (2016) 251–264 255
2.2. Reactive oxygen species (ROS) signalling pathway
ROS-dependent activation of the NLRP3 inflammasome hasrecently been described in diabetic patients [86]. ROS generation isamong the initial activation and core events of classic DN pathogen-esis; the pathways include the polyol pathway, advanced glycationend products (AGEs) pathway, PKC pathway and hexosamine (HS)pathway [87–89]. Once activated, these pathways promote ROS for-mation in cells, and the aggravation of oxidative stress generatesa vicious cycle. ROS is an important intracellular redox signal, andthe continuous production of ROS can lead to cell damage. ROS canregulate transcription factors (NF-�B and AP-1) and the activity ofcytokines [90,91]. The important role of NLRP3 activators in ROSproduction was recently highlighted [21,92].
NLRP3 activators can alter ROS production in many ways, butthe specific underlying mechanisms are unknown. For instance,increased potassium ion outflow, resulting in decreased intracellu-lar potassium, is the most important regulatory mechanism relatedto NLRP3 activation [93], and potassium ion outflow in humangranulocytes can stimulate the generation of ROS [94]. Thus, itcan be inferred that NLRP3 activation caused by potassium ionefflux may be connected to the ROS production process. The detec-tion of NLRP3 activators, such as uric acid crystals, alum, asbestos,silica and other large particulate matter, at the cell membranemight mediate so-called “impaired phagocytosis”, which is relatedto the production of ROS [64]. ROS can activate the inflamma-some [95], and the phosphatidylinositol 3 kinase (PI3K) pathwaymay be involved in this process [96]. The involvement of TXNIPin inflammasome activation is also consistent with the reportedrequirement for ROS in activation of the NLRP3 inflammasome [24].In beta cells, TXNIP is downregulated by insulin, and TXNIP expres-sion is consistently higher in humans with type 2 diabetes [97].Furthermore, TXNIP mutations are associated with hypertriglyc-eridemia and lower plasma glucose in type 2 diabetes [98]. TXNIPis a critical regulator of NLRP3 inflammasome activation in vivo[24].
In resting cells, TXNIP/VDUP1 interacts with thioredoxin andthus does not activate NLRP3. However, upon ROS stimulation,TXNIP is released from oxidized thioredoxin and directly combineswith the LRR domain of NLRP3 to mediate assembly of the inflam-masome. Indeed, treatment of macrophages lacking TXNIP/VDUP1with ATP or uric acid crystals can weaken the activation of caspase-1 and the generation of IL-1� from pro-IL-1� [99]. In conclusion,TXNIP/VDUP1 and NLRP3 constitute a sensor of cellular stress toidentify excessive oxidative stress or danger signals and drive theinflammatory response. In acute lung injury rats, ROS can regulateNLRP3 inflammasome activation, and inhibition of ROS produc-tion suppresses NLRP3 inflammasome activation [100]. Althoughthe exact mechanism of ROS-mediated inflammasome activationis not clear, ROS are a common core component regulating NLRP3activation.
Macrophages stimulated by ATP can produce ROS, and ROS canactivate PI3K pathways, thus promoting caspase-1 activation andthe secretion of IL-1� [101]. IL-1, TNF-�, IL-6 and other cytokinesand inflammatory mediators and the generation of ROS can causedamage to the structure of the kidney tissue and accelerate theprocess of glomerular sclerosis [102]. In addition, phosphataseand tensin homolog (PTEN) inactivation by ROS leads to PI3K andERK1/2 pathway stimulation, followed by caspase-1 activation, andATP-dependent ROS generation can also induce PI3K downstreammediating factors, thereby activating the NLRP3 inflammasome[103]. ROS can also elicit NF-�B and p38MAPK signalling to activate
the NLRP3 inflammasome [59,104]. In addition, the nicotinamideadenine dinucleotide phosphate (NADPH) oxidase-dependent ROSinhibitor diphenyliodonium iodide can reduce the expression of
Fig. 2. The NLRP3 inflammasome-mediated ERK signalling pathway in diabetic nephropathy.In DN, the NLRP3 inflammasome is activated by oxidative stress, hyperglycaemia and growth factors in a variety of cell types, including podocytes and mesangial cells. Theactivated inflammasome phosphorylates and activates Raf-1, which leads to activation of MEK1/2, followed by ERK1/2 activation. ERK1/2 phosphorylates intracellular PLA2 topromote the release of arachidonic acid and eicosanoids, which can promote changes in renal haemodynamics in DN. ERK1/2-induced DN leads to mesangial cell proliferationand glomerular nephritis through PKC, PTK, RAGEs and other factors. Upregulation of TGF-�1 and VEGF mRNA and protein expression causes proteinuria, ultrafiltration, andg ed dai e and
s vascum
No
aipE[urpisrtavpididr
2
1
lomerular hypertrophy. Activation of RAAS is followed by potential Ang-II-mediatnjury, conversion of renal fibroblasts to myofibroblasts, stimulation of chemokintress. Both RAAS components and ROS can cause local inflammation and stimulateight lead to the development of DN (2-column fitting image).
LRP3 mRNA, which indicates that ROS can regulate the expressionf the NLRP3 gene [104].
In the case of diabetes, HG and high uric acid can activate ROSnd the NLRP3 inflammasome. ROS activate the expression of genesnvolved in a variety of inflammatory pathways and increase theroduction of renal inflammatory cytokines, resulting in excessiveCM accumulation and thus damage to kidney endothelial cells105]. In a state of oxidative stress, TGF-� mRNA expression ispregulated; smad signalling is activated upon TGF-� binding to itseceptor, leading to augmented expression of genes encoding ECMroteins [106]. At the same time, the smad signalling pathway can
nhibit proteases that degrade the ECM and increase the synthe-is of protease inhibitors, resulting in expansion of the mesangialegion [107]. ROS can also lead to an increase in oxygen ions byhe decoupling of nitric oxide synthase (NOS) to reduce the gener-tion of NO and cause an O2−/NO imbalance, which results in renalascular dysfunction in DN [108]. Oxygen radicals can inhibit theroliferation of vascular endothelial cells and activate NF-�B, caus-
ng vascular endothelial injury. In turn, vascular endothelial injuryecreases the synthesis and release of NO, thus decreasing bioactiv-
ty but increasing the biosynthesis and release of endothelin. Thisual-imbalance can cause strong contraction of renal blood vessels,educe renal blood flow, and cause kidney damage [109] (Fig. 4).
Enhanced activation of NF-�B p65 in DN can upregulate MCP- mRNA expression [110], a common observation in mesangial
mage to kidney cells through various mechanisms, such as pressure-induced renalosteopontin production, and profibrotic cytokine sensitization, causing oxidativelar and mesangial cell proliferation and hypertrophy. These biochemical reactions
cells and in renal tubular interstitial disease. The study foundthat activated NF-�B is translocated to the nucleus to regulatethe expression of target genes and that the release of inflam-matory mediators can cause a lasting and intense inflammatoryresponse, leading to excessive FN production and ECM accumu-lation [111]. In the DN state, the inflammasome promotes thegeneration of a considerable amount of IL-1 through NF-�B acti-vated by intracellular or extracellular receptors, with involvementof the regulatory protein TNF receptor associated factor (TRAF)family as a signal transmitter. TRAF acts on NF-�B-induced kinase(NIK). NIK does not directly phosphorylate I�B� but instead actsupstream of the I�B� kinase; however, the specific process remainsunclear. In addition, LPS-mediated priming signal-induced NLRP3mRNA expression is reduced by the NF-�B inhibitor Bay11-7082or the NADPH oxidase-dependent ROS inhibitor diphenyliodonium(DPI), indicating that NF-�B and ROS play important roles in theregulation of NLRP3 gene expression [71,104]. Studies of colitis-associated cancer-affected mice with inducible NLPR3 deficiencyhave revealed a protective role of the NLRP3 inflammasome againstcolitis and colitis-associated cancer [112,113]. This protective rolehas been attributed to NF-�B and NLRP3-inflammasome-regulatedcytokine IL-18 production, which plays a pivotal role in the repairof the ulcerated epithelium during the acute inflammatory phaseand has also been proposed to have anti-tumorigenic effects [113].
These results suggest that in the acute inflammatory phase duringthe early formation of intestinal adenomas, the NLRP3 inflam-masome may have a protective role. In contrast, if overactivated,the NLRP3 inflammasome may contribute to chronic inflamma-
Fig. 3. The NLRP3 inflammasome-mediated JNK signalling pathway in diabetic nephropathy.In T2DM patients and diabetic animal models, NLRP3 is activated in the JNK pathway primarily by environmental stress (e.g., HG) and inflammatory cytokines. Activationof the inflammasome promotes ASC-mediated phosphorylation of JNK or upstream factors, including MAP3Ks, by MKK4 via the DVD located at the C-terminus and the Dd ations teins am cy and
tttw[
id
Sgbk
omain-type docking site at the N-terminus for p38 and JNK binding. A similar situcaffold proteins for assembly of the activated kinases. JNKs phosphorylate c-Jun proechanisms after entering the nucleus. These actions will cause IR, insulin deficien
ion and, in more advanced sporadic colorectal cancer cases, leado poorer survival. In diabetic rats, significant inhibition of NF-�Bogether with decreased IL-1� and TNF-� levels were associatedith decreased TXNIP and NLRP3 expression in the diabetic kidney
114].Based on these findings, we have reason to believe that the
nflammasome may have an important role in the occurrence andevelopment of DN through the NF-�B pathway.
Expression of small ubiquitin-related modifier1 (SUMO1) andUMO2/3 increases in the DN state with a consistently high blood
lucose concentration as well as high osmotic pressure, followedy stimulation of heat shock proteins and oxidative stress in theidney. SUMO1 and SUMO2/3 act as stress proteins in the cell and
occurs with MKK7. Furthermore, JNK regulation and translocation usually requirend thereby regulate AP-1 transcription and expression through post-transcriptional
hyperglycaemia, which promote the progression of DN (2-column fitting image).
may participate in signal transduction and modifications in variouscells [115]. When SUMO2/3-induced changes in I�B� predomi-nate and significantly reduce I�B� sumoylation, these factors mayregulate the function of NF-�B by altering the balance betweensumoylated and non-sumoylated I�B� [116]. The expression ofI�B� decreases, and inflammatory NF-�B signalling is activatedin subsequent steps; activated NF-�B translocates to the nucleusto regulate the expression of downstream genes, including inter-cellular adhesion molecule 1 (ICAM-1) and TGF-�1. There is areport that inflammasome-independent NLRP3 augments TGF-
� signalling in the kidney epithelium [56]. Therefore, sustainedand enhanced inflammation and excessive FN production willpromote the proliferation of mesangial cells and accelerate the
Fig. 4. The NLRP3 inflammasome and the reactive oxygen species (ROS) signalling pathway in diabetic nephropathy.Phagocytosis of certain NLRP3 activators, such as uric acid crystals and other large particles, can cause damage at the membrane surface and result in the outflow of potassiumions, which activates the inflammasome and stimulates ROS production. The effects of high glucose and cytokines are similar. ROS are also induced through the PI3K pathway,T ough Mg ysfuna ge).
diid[
bsaosp
XNIP and genes involved in inflammasome formation. ROS can be generated thrlomerular sclerosis. ROS can lead to O2−/NO imbalance, resulting in renal vascular drea expansion and cell apoptosis. These events promote DN (2-column fitting ima
evelopment of diabetic kidney glomerular sclerosis [117]. In ann vivo study, thrombomodulin domain 1 via anti-NF-�B and NLRP3nflammasome-mediated inflammation enhanced NRF2 antioxi-ant activity and inhibition of apoptosis to ameliorate DN in mice118].
The steps of NLRP3 inflammasome activation are controlledy different mechanisms [71]: the induction of NLRP3 expres-ion at the translation level; the mechanism of NLRP3 activation
fter translation; and NLRP3 inflammasome assembly. The levelf NLRP3 expression is the limiting factor in NLRP3 inflamma-ome activation. Activation of NLRP3 first requires reaching are-excitation state, and this process is mediated by many factors.
APK pathway activation of caspase-1 and IL-1�, causing ECM accumulation andction, which causes overexpression of TGF-� and NF-�B-targeted genes, mesangial
NF-�B is an important mediator of the immune response, whichplays an important role in the formation of the preactivated-NLRP3inflammasome, and NF-�B may function upstream of ASC. Fig. 5shows the interaction between NF-�B and the inflammasome.
2.4. Inflammation mediators
The release of the pro-inflammatory cytokines IL-1� and IL-18
is controlled by the inflammasome [55]. An animal study foundthat the NLRP3 inflammasome-caspase-1-IL-1�/IL-18 axis plays animportant role in kidney disease [119]. By regulating the activa-tion of caspase-1, the NLRP3 inflammasome can promote IL-1�
Fig. 5. The NLRP3 inflammasome and the nuclear transcription factor (NF-�B) signalling pathway in diabetic nephropathy.When the inflammasome is activated by high osmotic pressure, a high concentration of glucose and ROS stimulation in vivo, a greater amount of IL-1� is produced. IL-1�interacts with receptors on the surface of the cell membrane. This interaction is followed by TRAF activation and NIK stimulation. NIK bypasses the SUMO pathway to adjustNF-�B activation. Under normal circumstances, non-active NF-�B is non-covalently bound to �B inhibitory factors (I�B�, I�B�, I�B�) in the cytoplasm and does not havea ivatedt nes arc
alioahetd
ny physiological activity. I�B� is ubiquitinated following stimulation, allowing actarget DNA, resulting in the activation of these genes. TGF-�1, ICAM-1 and other geell proliferation and glomerular sclerosis (2-column fitting image).
nd IL-18 maturation. In diabetes, the elevated levels of circu-atory and partial pro-inflammatory factors IL-1� and IL-18 cannduce sustained inflammation, causing kidney injury. Wang et al.bserved overexpression of the inflammasome components ASCnd caspase-1 in the STZ-induced DN rat model accompanied byyperuricaemia and hyperlipidaemia, in addition to elevated lev-
ls of IL-1� and IL-18 [119]. The above evidence indicates thathe NLRP3 inflammasome and inflammatory factors influence andamage the kidneys during the occurrence and development of DN.
NF-�B to translocate to the nucleus and bind to specific enhancers or promoters ofe overexpressed, which leads to aggravated inflammation, increased FN, mesangial
Reducing uric acid and blood lipid levels through the use of uricacid-lowering drugs (such as allopurinol and quercetin) may inhibitNLRP3 inflammasome activation and can prevent STZ-inducedrenal damage [119]. Other studies have found that a high uric acidlevel is an important factor in the activation of the NLRP3 inflamma-some, inducing lung injury [120]. These findings suggest that high
uric acid levels are associated with NLRP3 inflammasome activa-tion. Based on the mechanism of NLRP3 inflammasome activation, itcan be speculated the uric acid maybe through the lysosome phago-cytosis, or stimulate ROS system to activation, or Toll-like receptors
Fig. 6. NLRP3 inflammasome-mediated inflammation in diabetic nephropathy.Under the hyperuricaemic condition, the lysosome will sequester intracellular uric acid crystals and then activate the ROS system, stimulating the inflammasome, followedby secretion of a large amount of IL-1� and IL-18. However, metabolism disorders caused by high uric acid and lipid levels can stimulate TLR pathways to activate theinflammasome. Increases in IL-1� levels can increase the expression of ICAM-1, TGF-�1, VCAM-1, e-cadherin and hyaluronic acid, causing renal proximal tubule epithelialc rular hh can pi prote
(asw
atistc1gbc2tear
ell proliferation. Changes in the permeability of endothelial cells influence glomeyperplasia. Accumulation of IL-18 through activated Fas leads to cell apoptosis and
nterstitial fibrosis. IL-18 can also lead to increases in IL-1� and TNF-�, resulting in
TLRs)/scavenger receptors on macrophages identify signals, toctivate the NLRP3 inflammasome, promoting the maturation andecretion of IL-1� and IL-18 and inducing kidney inflammation,hich results in the generation and development of DN.
In experimental models, inflammatory factors are increasedlong with proteinuria, accompanied by macrophage accumula-ion [53]. Markers of inflammasome activation have been analysedn type 2 diabetic patients [26]. In albuminuric diabetic patients,erum IL-1� levels were significantly increased compared withhose in diabetic patients without albuminuria or in nondiabeticontrols, and the degree of albuminuria correlated with serum IL-�. Furthermore, NLRP3 expression was on average increased inlomeruli of albuminuric diabetic patients compared to that in dia-etic patients without albuminuria or in nondiabetic controls. Inlinical trials, inhibition of IL-1� has been shown to ameliorate type
diabetes, which suggests an important role for this proinflamma-ory cytokine in disease progression [121]. IL-1 can increase the
xpression of ICAM-1, vascular cell adhesion protein 1 (VCAM-1)nd e-cadherin [122,123]. Synthesis of prostaglandin E2 (PGE2) andelease of PLA2 are stimulated when mesangial cells are treated
aemodynamics. PLA2 and PGE2 also increase, as do mesangial cells and fibroblastroduce local inflammation and an immunopathological response, resulting in renalinuria. Such events aggravate DN (2-column fitting image).
with recombinant IL-1. Furthermore, IL-1 can induce endothelialcell permeability, alter the haemodynamics of glomeruli, influ-ence the synthesis of prostaglandins, stimulate the proliferation ofmesangial cells and fibroblasts, and induce the production of TGF-�1 [124], thereby increasing DN. Application of the IL-1 receptorantagonist anakinra in hyperglycaemic mice can not only preventbut also reverse DN. Anakinra conferred nephroprotection in bothdb/db mice and mice with STZ-induced insulinopenia and hyper-glycaemia, which mimics type 1 diabetes [26]. These results suggestthat interventions targeting this mechanism of sterile inflamma-tion may be a feasible therapeutic approach in DN. Moreover,inflammasome inhibition may have an advantage over blockinginflammatory cell recruitment [1,125], as IL-1� and IL-18 impairpodocyte and endothelial function independent of inflammatorycell recruitment [126].
The importance of IL-1 in the pathology of gout is also suggestedby promising preliminary studies in humans. Two pilot open-label
studies using inhibitors of IL-1� to treat a small number of patientswith documented acute gout attacks who could not tolerate or hadfailed standard anti-inflammatory therapies revealed a very rapid
logical
ait
nuutfmcDccdINdrto[a1fncocf
3
tbcrteptcNcrolmiisodtn
C
A
d
Y.-y. Qiu, L.-q. Tang / Pharmaco
nd efficient response to IL-1 blockade in those patients, suggest-ng that targeting IL-1 or the inflammasome could be an effectiveherapeutic alternative in gout [34,127,128].
An independent correlation has been observed between the uri-ary albumin excretion rate and IL-18 levels in the serum andrine of type 2 diabetic patients, and IL-18 levels in serum andrine were positively correlated with the degree of proteinuria inhe follow-up visit, indicating that these markers could be a riskactor for the development of DN [129]. IL-18 can also promote
onocyte and macrophage production of TNF-a, IL-1� and inter-ellular adhesion molecules, which can cause damage to the kidney.ata suggest increased local production of IL-18 in patients withoronary artery disease, similar to IL-1� and in agreement with aommon inflammasome-mediated pathway underlying their pro-uction [130]. ATP release enhances NLRP3-mediated secretion of
L-1� and IL-18 by activating the P2X purinoceptor 7 [131]. TheLRP3 inflammasome contributes to the pathogenesis of metaboliciseases as a sensor for metabolic danger and also plays a centralole in chronic inflammation, resulting in IL-1� and IL-18 matura-ion [65,132]. An experimental model has reported that knockdownf NLRP3 mitigates production of IL-1� and IL-18 against obesity133]. Although IL-18 participates in the inflammatory responsend the immune response of the renal parenchyma, the role of IL-8 in the pathogenesis of the disease remains unclear. IL-18 mayunction in the pathogenesis of DN through the following mecha-isms [102,134]: (1) promoting the production of cell inflammatoryytokines TNF-�, IFN-�, IL-1� and IL-6; (2) enhancing the functionf Fas-mediated cell apoptosis; (3) accelerating NO production andausing glomerular hyperfiltration and continuous angiectasis; (4)acilitating renal tubular atrophy and interstitial fibrosis (Fig. 6).
. Conclusion
Many distinct immunological and molecular mechanisms con-ribute to the progression of DN. The NLRP3 inflammasome is theest studied among the inflammasomes, which are multiproteinytosolic complexes that form part of the NLR family of patternecognition receptors. This review describes in detail the effects ofhe inflammasome on DN, including the association between theffects of the inflammasome and the MAPK, ROS, NF-�B signallingathways and inflammation mediators. Composed of adaptor pro-eins and signal transduction proteins, the NLRP3 inflammasomean enhance or repress pathway signalling. Indeed, the roles of theLRP3 inflammasome in human disease are becoming increasinglylear. Although much progress has been made in inflammasomeesearch, many issues need to be addressed, such as the functionf most inflammasome family members, whether there is a directigand for inflammasome activation, whether other types of inflam-
asome exist, and the mechanism by which the inflammasomes regulated. In addition, further research into drugs targeting thenflammatory pathways upstream or downstream of the inflamma-ome should be performed in the next years. Evaluation of the effectf the inflammasome in different cells at different stages of variousiseases may provide a clearer picture of how these important pro-eins are altered in disease and may promote the development ofovel approaches for treating a variety of diseases.
onflict of interest
The authors declare that they have no conflict of interest.
cknowledgements
This work was supported by the National Natural Science Foun-ation of China (No. 81073109, 81102864), the Anhui Provincial
Research 114 (2016) 251–264 261
Natural Science Foundation (China, No. 1508085MH179) and Chi-nese Traditional Medical Research of Anhui Province (China, No.2016zy08).
References
[1] F. Chow, E. Ozols, D.J. Nikolic-Paterson, R.C. Atkins, G.H. Tesch, Macrophagesin mouse type 2 diabetic nephropathy: correlation with diabetic state andprogressive renal injury, Kidney Int. 65 (2004) 116–128.
[2] M. Stolar, Glycemic control and complications in type 2 diabetes mellitus,Am. J. Med. 123 (2010) S3–S11.
[3] K.E. Wellen, G.S. Hotamisligil, Inflammation, stress, and diabetes, J. Clin.Invest. 115 (2005) 1111–1119.
[5] E. Latz, T.S. Xiao, A. Stutz, Activation and regulation of the inflammasomes,Nat. Rev. Immunol. 13 (2013) 397–411.
[6] C. Zhang, K.M. Boini, M. Xia, J.M. Abais, X. Li, Q. Liu, P.L. Li, Activation ofNod-like receptor protein 3 inflammasomes turns on podocyte injury andglomerular sclerosis in hyperhomocysteinemia, Hypertension 60 (2012)154–162.
[7] A. Halle, V. Hornung, G.C. Petzold, C.R. Stewart, B.G. Monks, T. Reinheckel,K.A. Fitzgerald, E. Latz, K.J. Moore, D.T. Golenbock, The NALP3inflammasome is involved in the innate immune response to amyloid-beta,Nat. Immunol. 9 (2008) 857–865.
[8] L.A. Carneiro, J.G. Magalhaes, I. Tattoli, D.J. Philpott, L.H. Travassos, Nod-likeproteins in inflammation and disease, J. Pathol. 214 (2008) 136–148.
[9] Inohara, Chamaillard, C. McDonald, G. Nunez, NOD-LRR proteins: role inhost-microbial interactions and inflammatory disease, Annu. Rev. Biochem.74 (2005) 355–383.
[10] J. Segovia, A. Sabbah, V. Mgbemena, S.Y. Tsai, T.H. Chang, M.T. Berton, I.R.Morris, I.C. Allen, J.P. Ting, S. Bose, TLR2/MyD88/NF-kappaB pathway,reactive oxygen species, potassium efflux activates NLRP3/ASCinflammasome during respiratory syncytial virus infection, PLoS One 7(2012) e29695.
[11] R. Stienstra, J.A. van Diepen, C.J. Tack, M.H. Zaki, F.L. van de Veerdonk, D.Perera, G.A. Neale, G.J. Hooiveld, A. Hijmans, I. Vroegrijk, S. van den Berg, J.Romijn, P.C. Rensen, L.A. Joosten, M.G. Netea, T.D. Kanneganti,Inflammasome is a central player in the induction of obesity and insulinresistance, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 15324–15329.
[12] F. Martinon, K. Burns, J. Tschopp, The inflammasome: a molecular platformtriggering activation of inflammatory caspases and processing of proIL-beta,Mol. Cell 10 (2002) 417–426.
[13] S.M. Srinivasula, J.L. Poyet, M. Razmara, P. Datta, Z. Zhang, E.S. Alnemri, ThePYRIN-CARD protein ASC is an activating adaptor for caspase-1, J. Biol.Chem. 277 (2002) 21119–21122.
[14] M. Lamkanfi, V.M. Dixit, Mechanisms and functions of inflammasomes, Cell157 (2014) 1013–1022.
[15] A. Doria, M. Zen, S. Bettio, M. Gatto, N. Bassi, L. Nalotto, A. Ghirardello, L.Iaccarino, L. Punzi, Autoinflammation and autoimmunity: bridging thedivide, Autoimmun. Rev. 12 (2012) 22–30.
[16] D. Gris, Z. Ye, H.A. Iocca, H. Wen, R.R. Craven, P. Gris, M. Huang, M.Schneider, S.D. Miller, J.P. Ting, NLRP3 plays a critical role in thedevelopment of experimental autoimmune encephalomyelitis by mediatingTh1 and Th17 responses, J. Immunol. 185 (2010) 974–981.
[17] J.M. Kahlenberg, M.J. Kaplan, The inflammasome and lupus: another innateimmune mechanism contributing to disease pathogenesis? Curr. Opin.Rheumatol. 26 (2014) 475–481.
[18] H. Wen, J.P. Ting, L.A. O’Neill, A role for the NLRP3 inflammasome inmetabolic diseases—did Warburg miss inflammation? Nat. Immunol. 13(2012) 352–357.
[19] H.J. Anders, D.A. Muruve, The inflammasomes in kidney disease, J. Am. Soc.Nephrol. 22 (2011) 1007–1018.
[20] H.L. Hutton, J.D. Ooi, S.R. Holdsworth, A.R. Kitching, The NLRP3inflammasome in kidney disease and autoimmunity, Nephrology 21 (2016)736–744.
[21] J.M. Abais, C. Zhang, M. Xia, Q. Liu, T.W. Gehr, K.M. Boini, P.L. Li, NADPHoxidase-mediated triggering of inflammasome activation in mousepodocytes and glomeruli during hyperhomocysteinemia, Antioxid. Redox.Signal. 18 (2013) 1537–1548.
[22] J. Lichtnekert, O.P. Kulkarni, S.R. Mulay, K.V. Rupanagudi, M. Ryu, R. Allam, V.Vielhauer, D. Muruve, M.T. Lindenmeyer, C.D. Cohen, H.J. Anders, Anti-GBMglomerulonephritis involves IL-1 but is independent of NLRP3/ASCinflammasome-mediated activation of caspase-1, PLoS One 6 (2011) e26778.
[23] J. Wang, Y. Wen, L.L. Lv, H. Liu, R.N. Tang, K.L. Ma, B.C. Liu, Involvement ofendoplasmic reticulum stress in angiotensin II-induced NLRP3inflammasome activation in human renal proximal tubular cells in vitro,Acta Pharmacol. Sin. 36 (2015) 821–830.
[24] R. Zhou, A. Tardivel, B. Thorens, I. Choi, J. Tschopp, Thioredoxin-interacting
protein links oxidative stress to inflammasome activation, Nat. Immunol. 11(2010) 136–140.
[25] S. Gauer, O. Sichler, N. Obermuller, Y. Holzmann, E. Kiss, E. Sobkowiak, J.Pfeilschifter, H. Geiger, H. Muhl, I.A. Hauser, IL-18 is expressed in theintercalated cell of human kidney, Kidney Int. 72 (2007) 1081–1087.
[26] K. Shahzad, F. Bock, W. Dong, H. Wang, S. Kopf, S. Kohli, M.M. Al-Dabet, S.Ranjan, J. Wolter, C. Wacker, R. Biemann, S. Stoyanov, K. Reymann, P.Soderkvist, O. Gross, V. Schwenger, S. Pahernik, P.P. Nawroth, H.J. Grone, T.Madhusudhan, B. Isermann, Nlrp3-inflammasome activation innon-myeloid-derived cells aggravates diabetic nephropathy, Kidney Int. 87(2015) 74–84.
[27] M.D. Turner, A. Chaudhry, B. Nedjai, Tumour necrosis factor receptortrafficking dysfunction opens the TRAPS door to pro-inflammatory cytokinesecretion, Biosci. Rep. 32 (2012) 105–112.
[28] J.L. Hao, Y.F. Li, R.S. Li, A novel mechanism of NALP3 inducing ischemiareperfusion injury by activating MAPK pathway in acute renal failure, Med.Hypotheses 80 (2013) 463–465.
[29] A. Terman, T. Kurz, B. Gustafsson, U.T. Brunk, Lysosomal labilization, IUBMBLife 58 (2006) 531–539.
[30] D.J. Taxman, E.A. Holley-Guthrie, M.T. Huang, C.B. Moore, D.T. Bergstralh, I.C.Allen, Y. Lei, D. Gris, J.P. Ting, The NLR adaptor ASC/PYCARD regulatesDUSP10, mitogen-activated protein kinase (MAPK), and chemokineinduction independent of the inflammasome, J. Biol. Chem. 286 (2011)19605–19616.
[31] M. Tao, P.C. Scacheri, J.M. Marinis, E.W. Harhaj, L.E. Matesic, D.W. Abbott,ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influenceinflammatory signaling pathways, Curr. Biol. 19 (2009) 1255–1263.
[32] G.L. Johnson, R. Lapadat, Mitogen-activated protein kinase pathwaysmediated by ERK, JNK, and p38 protein kinases, Science 298 (2002)1911–1912.
[33] P.C. Liao, L.K. Chao, J.C. Chou, W.C. Dong, C.N. Lin, C.Y. Lin, A. Chen, S.M. Ka,C.L. Ho, K.F. Hua, Lipopolysaccharide/adenosine triphosphate-mediatedsignal transduction in the regulation of NLRP3 protein expression andcaspase-1-mediated interleukin-1beta secretion, Inflamm. Res. 62 (2013)89–96.
[34] F. Martinon, Detection of immune danger signals by NALP3, J. Leukoc. Biol.83 (2008) 507–511.
[35] K. Ono, J. Han, The p38 signal transduction pathway: activation andfunction, Cell. Signal. 12 (2000) 1–13.
[36] R. Muller, C. Daniel, C. Hugo, K. Amann, D. Mielenz, K. Endlich, T. Braun, B.van der Veen, P. Heeringa, G. Schett, J. Zwerina, The mitogen-activatedprotein kinase p38alpha regulates tubular damage in murineanti-glomerular basement membrane nephritis, PLoS One 8 (2013) e56316.
[37] J.S. Arthur, S.C. Ley, Mitogen-activated protein kinases in innate immunity,Nat. Rev. Immunol. 13 (2013) 679–692.
[38] J.K. Lee, S.H. Kim, E.C. Lewis, T. Azam, L.L. Reznikov, C.A. Dinarello,Differences in signaling pathways by IL-1beta and IL-18, Proc. Natl. Acad.Sci. U. S. A. 101 (2004) 8815–8820.
[39] D.L. Donnelly-Roberts, M.T. Namovic, C.R. Faltynek, M.F. Jarvis,Mitogen-activated protein kinase and caspase signaling pathways arerequired for P2X7 receptor (P2X7R)-induced pore formation in humanTHP-1 cells, J. Pharmacol. Exp. Ther. 308 (2004) 1053–1061.
[40] K. Rajamaki, M.I. Mayranpaa, A. Risco, J. Tuimala, K. Nurmi, A. Cuenda, K.K.Eklund, K. Oorni, P.T. Kovanen, p38delta MAPK: a novel regulator of NLRP3inflammasome activation with increased expression in coronaryatherogenesis, Arterioscler. Thromb. Vasc. Biol. 36 (2016) 1937–1946.
[41] S.W. Kang, S.G. Adler, J. Lapage, R. Natarajan, p38 MAPK and MAPK kinase3/6 mRNA and activities are increased in early diabetic glomeruli, KidneyInt. 60 (2001) 543–552.
[42] G. Remy, A.M. Risco, F.A. Inesta-Vaquera, B. Gonzalez-Teran, G. Sabio, R.J.Davis, A. Cuenda, Differential activation of p38MAPK isoforms by MKK6 andMKK3, Cell. Signal. 22 (2010) 660–667.
[43] K. Polzer, A. Soleiman, W. Baum, R. Axmann, J. Distler, K. Redlich, A. Kilian, G.Kronke, G. Schett, J. Zwerina, Selective p38MAPK isoform expression andactivation in antineutrophil cytoplasmatic antibody-associated crescenticglomerulonephritis: role of p38MAPKalpha, Ann. Rheum. Dis. 67 (2008)602–608.
[44] A. Riad, D. Unger, J. Du, D. Westermann, Z. Mohr, M. Sobirey, M. Dorenkamp,H.P. Schultheiss, C. Tschope, Chronic inhibition of p38MAPK improvescardiac and endothelial function in experimental diabetes mellitus, Eur. J.Pharmacol. 554 (2007) 40–45.
[45] J.M. da Costa-Pessoa, R.S. Damasceno, U.F. Machado, O. Beloto-Silva, M.Oliveira-Souza, High glucose concentration stimulates NHE-1 activity indistal nephron cells: the role of the Mek/Erk1/2/p90RSK and p38MAPKsignaling pathways, Cell Physiol. Biochem. 33 (2014) 333–343.
[46] T.J. Hsieh, P. Fustier, S.L. Zhang, J.G. Filep, S.S. Tang, J.R. Ingelfinger, I.G.Fantus, P. Hamet, J.S. Chan, High glucose stimulates angiotensinogen geneexpression and cell hypertrophy via activation of the hexosaminebiosynthesis pathway in rat kidney proximal tubular cells, Endocrinology144 (2003) 4338–4349.
[47] D.J. Burt, G. Gruden, S.M. Thomas, P. Tutt, C. Dell’Anna, G.C. Viberti, L. Gnudi,P38 mitogen-activated protein kinase mediates hexosamine-inducedTGFbeta1 mRNA expression in human mesangial cells, Diabetologia 46(2003) 531–537.
[48] K. Susztak, A.C. Raff, M. Schiffer, E.P. Bottinger, Glucose-induced reactiveoxygen species cause apoptosis of podocytes and podocyte depletion at the
onset of diabetic nephropathy, Diabetes 55 (2006) 225–233.
[49] S.W. Kang, R. Natarajan, A. Shahed, C.C. Nast, J. LaPage, P. Mundel, C.Kashtan, S.G. Adler, Role of 12-lipoxygenase in the stimulation of p38mitogen-activated protein kinase and collagen alpha5(IV) in experimental
l Research 114 (2016) 251–264
diabetic nephropathy and in glucose-stimulated podocytes, J. Am. Soc.Nephrol. 14 (2003) 3178–3187.
[50] T. Dai, R. Natarajan, C.C. Nast, J. LaPage, P. Chuang, J. Sim, L. Tong, M.Chamberlin, S. Wang, S.G. Adler, Glucose and diabetes: effects on podocyteand glomerular p38MAPK, heat shock protein 25, and actin cytoskeleton,Kidney Int. 69 (2006) 806–814.
[51] S.D. Deshpande, S. Putta, M. Wang, J.Y. Lai, M. Bitzer, R.G. Nelson, L.L.Lanting, M. Kato, R. Natarajan, Transforming growth factor-beta-inducedcross talk between p53 and a microRNA in the pathogenesis of diabeticnephropathy, Diabetes 62 (2013) 3151–3162.
[52] H.L. Chen, Y.G. Wan, Q. Zhao, Y.R. Huang, X.M. Shi, X.J. Meng, J. Yao,Regulative mechanism of renal inflammatory-related p38MAPK signalingpathway in diabetic nephropathy and interventional effects of Chineseherbal medicine, Zhongguo Zhong Yao Za Zhi 38 (2013) 2268–2272.
[53] J.F. Navarro-Gonzalez, C. Mora-Fernandez, The role of inflammatorycytokines in diabetic nephropathy, J. Am. Soc. Nephrol. 19 (2008) 433–442.
[54] Q. Zhao, Y. Wan, C. Wang, Q. Wei, H. Chen, X. Meng, J. Yao, Regulatorymechanism of p38MAPK signaling pathway on renal tissue inflammation inchronic kidney disease and interventional effect of traditional Chinesemedicine, Zhongguo Zhong Yao Za Zhi 37 (2012) 1700–1704.
[55] G. Lorenz, M.N. Darisipudi, H.J. Anders, Canonical and non-canonical effectsof the NLRP3 inflammasome in kidney inflammation and fibrosis, Nephrol.Dial. Transplant. 29 (2014) 41–48.
[56] W. Wang, X. Wang, J. Chun, A. Vilaysane, S. Clark, G. French, N.A. Bracey, K.Trpkov, S. Bonni, H.J. Duff, P.L. Beck, D.A. Muruve,Inflammasome-independent NLRP3 augments TGF-beta signaling in kidneyepithelium, J. Immunol. 190 (2013) 1239–1249.
[57] Z.I. Niemir, H. Stein, G. Dworacki, P. Mundel, N. Koehl, B. Koch, F.Autschbach, K. Andrassy, E. Ritz, R. Waldherr, H.F. Otto, Podocytes are themajor source of IL-1 alpha and IL-1 beta in human glomerulonephritides,Kidney Int. 52 (1997) 393–403.
[58] T. Karrasch, K.A. Steinbrecher, B. Allard, A.S. Baldwin, C. Jobin,Wound-induced p38MAPK-dependent histone H3 phosphorylationcorrelates with increased COX-2 expression in enterocytes, J. Cell Physiol.207 (2006) 809–815.
[59] Q. He, H. You, X.M. Li, T.H. Liu, P. Wang, B.E. Wang, HMGB1 promotes thesynthesis of pro-IL-1beta and pro-IL-18 by activation of p38 MAPK andNF-kappaB through receptors for advanced glycation end-products inmacrophages, Asian Pac. J. Cancer Prev. 13 (2012) 1365–1370.
[60] D.M. Price, M.T. Wloka, C.L. Chik, A.K. Ho, Mitogen-activated protein kinasephosphatase-1 (MKP-1) preferentially dephosphorylates p42/44MAPK butnot p38MAPK in rat pinealocytes, J. Neurochem. 101 (2007) 1685–1693.
[61] S. Meloche, J. Pouyssegur, The ERK1/2 mitogen-activated protein kinasepathway as a master regulator of the G1- to S-phase transition, Oncogene 26(2007) 3227–3239.
[62] Y.S. Kanwar, J. Wada, L. Sun, P. Xie, E.I. Wallner, S. Chen, S. Chugh, F.R.Danesh, Diabetic nephropathy: mechanisms of renal disease progression,Exp. Biol. Med. 233 (2008) 4–11.
[63] N. Sakai, T. Wada, K. Furuichi, Y. Iwata, K. Yoshimoto, K. Kitagawa, S. Kokubo,M. Kobayashi, A. Hara, J. Yamahana, T. Okumura, K. Takasawa, S. Takeda, M.Yoshimura, H. Kida, H. Yokoyama, Involvement of extracellularsignal-regulated kinase and p38 in human diabetic nephropathy, Am. J.Kidney Dis. 45 (2005) 54–65.
[64] L.A. O’Neill, Immunology. How frustration leads to inflammation, Science320 (2008) 619–620.
[65] J. Tschopp, K. Schroder, NLRP3 inflammasome activation: the convergenceof multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10(2010) 210–215.
[66] M.G. Ghonime, O.R. Shamaa, S. Das, R.A. Eldomany, T. Fernandes-Alnemri,E.S. Alnemri, M.A. Gavrilin, M.D. Wewers, Inflammasome priming bylipopolysaccharide is dependent upon ERK signaling and proteasomefunction, J. Immunol. 192 (2014) 3881–3888.
[67] M. Toyoda, D. Suzuki, M. Honma, G. Uehara, T. Sakai, T. Umezono, H. Sakai,High expression of PKC-MAPK pathway mRNAs correlates with glomerularlesions in human diabetic nephropathy, Kidney Int. 66 (2004) 1107–1114.
[68] S. Hoshi, K. Nomoto, J. Kuromitsu, S. Tomari, M. Nagata, High glucoseinduced VEGF expression via PKC and ERK in glomerular podocytes,Biochem. Biophys. Res. Commun. 290 (2002) 177–184.
[69] S. Akira, K. Takeda, Toll-like receptor signalling, Nat. Rev. Immunol. 4 (2004)499–511.
[70] M.M. Budai, A. Varga, S. Milesz, J. Tozser, S. Benko, Aloe vera downregulatesLPS-induced inflammatory cytokine production and expression of NLRP3inflammasome in human macrophages, Mol. Immunol. 56 (2013) 471–479.
[71] F.G. Bauernfeind, G. Horvath, A. Stutz, E.S. Alnemri, K. MacDonald, D. Speert,T. Fernandes-Alnemri, J. Wu, B.G. Monks, K.A. Fitzgerald, V. Hornung, E. Latz,Cutting edge: NF-kappaB activating pattern recognition and cytokinereceptors license NLRP3 inflammasome activation by regulating NLRP3expression, J. Immunol. 183 (2009) 787–791.
[72] H.Y. Hsu, M.H. Wen, Lipopolysaccharide-mediated reactive oxygen speciesand signal transduction in the regulation of interleukin-1 gene expression, J.Biol. Chem. 277 (2002) 22131–22139.
[73] D.A. Long, K.L. Price, J. Herrera-Acosta, R.J. Johnson, How does angiotensin IIcause renal injury? Hypertension 43 (2004) 722–723.
[74] M. Hedl, C. Abraham, Nod2-induced autocrine interleukin-1 alters signalingby ERK and p38 to differentially regulate secretion of inflammatorycytokines, Gastroenterology 143 (2012) 1530–1543.
[75] T. Wan-Xin, C. Tian-Lei, W. Ben, W. Wei-Hua, F. Ping, Effect of mitofusin 2overexpression on the proliferation and apoptosis of high-glucose-inducedrat glomerular mesangial cells, J. Nephrol. 25 (2012) 1023–1030.
[76] S.M. Twigg, Mastering a mediator: blockade of CCN-2 shows early promisein human diabetic kidney disease, J. Cell Commun. Signal. 4 (2010) 189–196.
[77] Y. Guo, W. Yuan, L. Wang, M. Shang, Y. Peng, Parathyroidhormone-potentiated connective tissue growth factor expression in humanrenal proximal tubular cells through activating the MAPK and NF-kappaBsignalling pathways, Nephrol. Dial. Transplant. 26 (2011) 839–847.
[78] W.S. Yang, J.W. Chang, N.J. Han, S.K. Lee, S.K. Park, Spleen tyrosine kinasemediates high glucose-induced transforming growth factor-beta1up-regulation in proximal tubular epithelial cells, Exp. Cell Res. 318 (2012)1867–1876.
[79] S.C. Tang, K.N. Lai, The pathogenic role of the renal proximal tubular cell indiabetic nephropathy, Nephrol. Dial. Transplant. 27 (2012) 3049–3056.
[80] M. Okada, A. Matsuzawa, A. Yoshimura, H. Ichijo, The lysosomerupture-activated TAK1-JNK pathway regulates NLRP3 inflammasomeactivation, J. Biol. Chem. 289 (2014) 32926–32936.
[81] M. Takekawa, K. Tatebayashi, H. Saito, Conserved docking site is essential foractivation of mammalian MAP kinase kinases by specific MAP kinase kinasekinases, Mol. Cell 18 (2005) 295–306.
[83] M. Hasegawa, R. Imamura, K. Motani, T. Nishiuchi, N. Matsumoto, T.Kinoshita, T. Suda, Mechanism and repertoire of ASC-mediated geneexpression, J. Immunol. 182 (2009) 7655–7662.
[84] A. Banerjee, S. Gerondakis, Coordinating TLR-activated signaling pathwaysin cells of the immune system, Immunol. Cell Biol. 85 (2007) 420–424.
[85] R. Yang, J.M. Trevillyan, c-Jun N-terminal kinase pathways in diabetes, Int. J.Biochem. Cell Biol. 40 (2008) 2702–2706.
[86] R.E. Mirza, M.M. Fang, E.M. Weinheimer-Haus, W.J. Ennis, T.J. Koh, Sustainedinflammasome activity in macrophages impairs wound healing in type 2diabetic humans and mice, Diabetes 63 (2014) 1103–1114.
[87] J.M. Forbes, M.T. Coughlan, M.E. Cooper, Oxidative stress as a major culpritin kidney disease in diabetes, Diabetes 57 (2008) 1446–1454.
[88] T. Nishikawa, D. Edelstein, X.L. Du, S.-i. Yamagishi, T. Matsumura, Y. Kaneda,M.A. Yorek, D. Beebe, P.J. Oates, H.-P. Hammes, I. Giardino, M. Brownlee,Normalizing mitochondrial superoxide production blocks three pathways ofhyperglycaemic damage, Nature 404 (2000) 787–790.
[89] M.G. Rosca, T.G. Mustata, M.T. Kinter, A.M. Ozdemir, T.S. Kern, L.I. Szweda,M. Brownlee, V.M. Monnier, M.F. Weiss, Glycation of mitochondrial proteinsfrom diabetic rat kidney is associated with excess superoxide formation,Am. J. Physiol. Renal Physiol. 289 (2005) F420–F430.
[90] E. Ogier-Denis, S.B. Mkaddem, A. Vandewalle, NOX enzymes and toll-likereceptor signaling, Semin. Immunopathol. 30 (2008) 291–300.
[91] M. Iriti, F. Faoro, Review of innate and specific immunity in plants andanimals, Mycopathologia 164 (2007) 57–64.
[92] F. Martinon, A. Mayor, J. Tschopp, The inflammasomes: guardians of thebody, Annu. Rev. Immunol. 27 (2009) 229–265.
[93] V. Petrilli, S. Papin, C. Dostert, A. Mayor, F. Martinon, J. Tschopp, Activation ofthe NALP3 inflammasome is triggered by low intracellular potassiumconcentration, Cell. Death Differ. 14 (2007) 1583–1589.
[94] A.J. Fay, X. Qian, Y.N. Jan, L.Y. Jan, SK channels mediate NADPHoxidase-independent reactive oxygen species production and apoptosis ingranulocytes, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 17548–17553.
[95] C. Dostert, V. Petrilli, R. Van Bruggen, C. Steele, B.T. Mossman, J. Tschopp,Innate immune activation through Nalp3 inflammasome sensing of asbestosand silica, Science 320 (2008) 674–677.
[96] C.M. Cruz, A. Rinna, H.J. Forman, A.L. Ventura, P.M. Persechini, D.M. Ojcius,ATP activates a reactive oxygen species-dependent oxidative stressresponse and secretion of proinflammatory cytokines in macrophages, J.Biol. Chem. 282 (2007) 2871–2879.
[97] H. Parikh, E. Carlsson, W.A. Chutkow, L.E. Johansson, H. Storgaard, P.Poulsen, R. Saxena, C. Ladd, P.C. Schulze, M.J. Mazzini, C.B. Jensen, A. Krook,M. Bjornholm, H. Tornqvist, J.R. Zierath, M. Ridderstrale, D. Altshuler, R.T.Lee, A. Vaag, L.C. Groop, V.K. Mootha, TXNIP regulates peripheral glucosemetabolism in humans, PLoS Med. 4 (2007) e158.
[98] M.M. van Greevenbroek, V.M. Vermeulen, E.J. Feskens, C.T. Evelo, M.Kruijshoop, B. Hoebee, C.J. van der Kallen, T.W. de Bruin, Genetic variation inthioredoxin interacting protein (TXNIP) is associated withhypertriglyceridaemia and blood pressure in diabetes mellitus, Diabet. Med.24 (2007) 498–504.
[99] K.A. Fitzgerald, NLR-containing inflammasomes: central mediators of hostdefense and inflammation, Eur. J. Immunol. 40 (2010) 595–598.
100] W. Jiang, M. Li, F. He, W. Yao, Z. Bian, X. Wang, L. Zhu, Protective effects ofasiatic acid against spinal cord injury-induced acute lung injury in rats,Inflammation (2016), http://dx.doi.org/10.1007/s10753-016-0414-3.
101] T. Fernandes-Alnemri, J. Wu, J.W. Yu, P. Datta, B. Miller, W. Jankowski, S.Rosenberg, J. Zhang, E.S. Alnemri, The pyroptosome: a supramolecularassembly of ASC dimers mediating inflammatory cell death via caspase-1
activation, Cell Death Differ. 14 (2007) 1590–1604.
102] C. Mora, J.F. Navarro, Inflammation and diabetic nephropathy, Curr. Diab.Rep. 6 (2006) 463–468.
103] F. Martinon, Signaling by ROS drives inflammasome activation, Eur. J.Immunol. 40 (2010) 616–619.
Research 114 (2016) 251–264 263
[104] F. Bauernfeind, E. Bartok, A. Rieger, L. Franchi, G. Nunez, V. Hornung, Cuttingedge: reactive oxygen species inhibitors block priming, but not activation, ofthe NLRP3 inflammasome, J. Immunol. 187 (2011) 613–617.
[105] J.F. Navarro-Gonzalez, C. Mora-Fernandez, M. Muros de Fuentes, J.Garcia-Perez, Inflammatory molecules and pathways in the pathogenesis ofdiabetic nephropathy, Nat. Rev. Nephrol. 7 (2011) 327–340.
[106] Y. Li, S. Wang, Glycated albumin activates NADPH oxidase in rat mesangialcells through up-regulation of p47phox, Biochem. Biophys. Res. Commun.397 (2010) 5–11.
[107] E.A. Lee, J.Y. Seo, Z. Jiang, M.R. Yu, M.K. Kwon, H. Ha, H.B. Lee, Reactiveoxygen species mediate high glucose-induced plasminogen activatorinhibitor-1 up-regulation in mesangial cells and in diabetic kidney, KidneyInt. 67 (2005) 1762–1771.
[108] S. Prabhakar, J. Starnes, S. Shi, B. Lonis, R. Tran, Diabetic nephropathy isassociated with oxidative stress and decreased renal nitric oxideproduction, J. Am. Soc. Nephrol. 18 (2007) 2945–2952.
[109] M. Xu, D.Z. Dai, Y. Dai, Normalizing NADPH oxidase contributes toattenuating diabetic nephropathy by the dual endothelin receptorantagonist CPU0213 in rats, Am. J. Nephrol. 29 (2009) 252–256.
[110] Z. Tumur, H. Shimizu, A. Enomoto, H. Miyazaki, T. Niwa, Indoxyl sulfateupregulates expression of ICAM-1 and MCP-1 by oxidative stress-inducedNF-kappaB activation, Am. J. Nephrol. 31 (2010) 435–441.
[111] Q. Jiang, P. Liu, X. Wu, W. Liu, X. Shen, T. Lan, S. Xu, J. Peng, X. Xie, H. Huang,Berberine attenuates lipopolysaccharide-induced extracelluar matrixaccumulation and inflammation in rat mesangial cells: involvement ofNF-kappaB signaling pathway, Mol. Cell. Endocrinol. 331 (2011) 34–40.
[112] I.C. Allen, E.M. TeKippe, R.M. Woodford, J.M. Uronis, E.K. Holl, A.B. Rogers,H.H. Herfarth, C. Jobin, J.P. Ting, The NLRP3 inflammasome functions as anegative regulator of tumorigenesis during colitis-associated cancer, J. Exp.Med. 207 (2010) 1045–1056.
[113] M.H. Zaki, P. Vogel, M. Body-Malapel, M. Lamkanfi, T.D. Kanneganti, IL-18production downstream of the Nlrp3 inflammasome confers protectionagainst colorectal tumor formation, J. Immunol. 185 (2010) 4912–4920.
[114] Y.A. Samra, H.S. Said, N.M. Elsherbiny, G.I. Liou, M.M. El-Shishtawy, L.A.Eissa, Cepharanthine and Piperine ameliorate diabetic nephropathy in rats:role of NF-kappaB and NLRP3 inflammasome, Life Sci. 157 (2016) 187–199.
[115] W. Huang, L. Xu, X. Zhou, C. Gao, M. Yang, G. Chen, J. Zhu, L. Jiang, H. Gan, F.Gou, H. Feng, J. Peng, Y. Xu, High glucose induces activation of NF-kappaBinflammatory signaling through IkappaBalpha sumoylation in rat mesangialcells, Biochem. Biophys. Res. Commun. 438 (2013) 568–574.
[116] Y. Yang, F. Xia, N. Hermance, A. Mabb, S. Simonson, S. Morrissey, P. Gandhi,M. Munson, S. Miyamoto, M.A. Kelliher, A cytosolic ATM/NEMO/RIP1complex recruits TAK1 to mediate the NF-kappaB and p38mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2responses to DNA damage, Mol. Cell. Biol. 31 (2011) 2774–2786.
[117] J. Yang, Z. Zeng, T. Wu, Z. Yang, B. Liu, T. Lan, Emodin attenuates highglucose-induced TGF-beta1 and fibronectin expression in mesangial cellsthrough inhibition of NF-kappaB pathway, Exp. Cell Res. 319 (2013)3182–3189.
[118] S.M. Yang, S.M. Ka, H.L. Wu, Y.C. Yeh, C.H. Kuo, K.F. Hua, G.Y. Shi, Y.J. Hung,F.C. Hsiao, S.S. Yang, Y.S. Shieh, S.H. Lin, C.W. Wei, J.S. Lee, C.Y. Yang, A. Chen,Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice viaanti-NF-kappaB/NLRP3 inflammasome-mediated inflammation,enhancement of NRF2 antioxidant activity and inhibition of apoptosis,Diabetologia 57 (2014) 424–434.
[119] C. Wang, Y. Pan, Q.Y. Zhang, F.M. Wang, L.D. Kong, Quercetin and allopurinolameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3inflammasome activation and lipid accumulation, PLoS One 7 (2012)e38285.
[120] P. Gasse, N. Riteau, S. Charron, S. Girre, L. Fick, V. Petrilli, J. Tschopp, V.Lagente, V.F. Quesniaux, B. Ryffel, I. Couillin, Uric acid is a danger signalactivating NALP3 inflammasome in lung injury inflammation and fibrosis,Am. J. Respir. Crit. Care Med. 179 (2009) 903–913.
[121] C.M. Larsen, M. Faulenbach, A. Vaag, A. Volund, J.A. Ehses, B. Seifert, T.Mandrup-Poulsen, M.Y. Donath, Interleukin-1-receptor antagonist in type 2diabetes mellitus, N. Engl. J. Med. 356 (2007) 1517–1526.
[123] C.W. Park, J.H. Kim, J.H. Lee, Y.S. Kim, H.J. Ahn, Y.S. Shin, S.Y. Kim, E.J. Choi,Y.S. Chang, B.K. Bang, High glucose-induced intercellular adhesionmolecule-1 (ICAM-1) expression through an osmotic effect in rat mesangialcells is PKC-NF-kappa B-dependent, Diabetologia 43 (2000) 1544–1553.
[124] S. Jones, S. Jones, A.O. Phillips, Regulation of renal proximal tubularepithelial cell hyaluronan generation: implications for diabeticnephropathy, Kidney Int. 59 (2001) 1739–1749.
[125] A.S. Awad, G.R. Kinsey, K. Khutsishvili, T. Gao, W.K. Bolton, M.D. Okusa,Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renalinjury, Am. J. Physiol. Renal Physiol. 301 (2011) F1358–F1366.
[126] M. Kusuhara, K. Isoda, F. Ohsuzu, Interleukin-1 and occlusive arterialdiseases, Cardiovasc. Hematol. Agents Med. Chem. 4 (2006) 229–235.
[127] A. So, T. De Smedt, S. Revaz, J. Tschopp, A pilot study of IL-1 inhibition by
anakinra in acute gout, Arthritis Res. Ther. 9 (2007) R28.
[128] D. McGonagle, A.L. Tan, S. Shankaranarayana, J. Madden, P. Emery, M.F.McDermott, Management of treatment resistant inflammation of acute onchronic tophaceous gout with anakinra, Ann. Rheum. Dis. 66 (2007)1683–1684.
of treatment with atorvastatin or rosuvastatin, Clin. Sci. 126 (2014) 233–241.[134] A. Nakamura, K. Shikata, M. Hiramatsu, T. Nakatou, T. Kitamura, J. Wada, T.
Itoshima, H. Makino, Serum interleukin-18 levels are associated with
64 Y.-y. Qiu, L.-q. Tang / Pharmaco
[129] S. Araki, M. Haneda, D. Koya, T. Sugimoto, K. Isshiki, M. Chin-Kanasaki, T.Uzu, A. Kashiwagi, Predictive impact of elevated serum level of IL-18 forearly renal dysfunction in type 2 diabetes: an observational follow-up study,Diabetologia 50 (2007) 867–873.
[130] G.J. Martinez, S. Robertson, J. Barraclough, Q. Xia, Z. Mallat, C. Bursill, D.S.Celermajer, S. Patel, Colchicine acutely suppresses local cardiac productionof inflammatory cytokines in patients with an acute coronary syndrome, J.
Am. Heart Assoc. 4 (2015) e002128.
[131] S. Carta, F. Penco, R. Lavieri, A. Martini, C.A. Dinarello, M. Gattorno, A.Rubartelli, Cell stress increases ATP release in NLRP3inflammasome-mediated autoinflammatory diseases, resulting in cytokineimbalance, Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 2835–2840.
l Research 114 (2016) 251–264
[132] K. Schroder, R. Zhou, J. Tschopp, The NLRP3 inflammasome: a sensor formetabolic danger? Science 327 (2010) 296–300.
[133] M. Satoh, T. Tabuchi, T. Itoh, M. Nakamura, NLRP3 inflammasome activationin coronary artery disease: results from prospective and randomized study
nephropathy and atherosclerosis in Japanese patients with type 2 diabetes,Diabetes Care 28 (2005) 2890–2895.