ELSEVIER Journal of MOLECULAR S~UCT~E Journal of Molecular Structure 323 (1994) 15-28 Infrared and Raman spectra of ($-C6H6_,X,)Cr(CO)3 complexes where X = Me (n = O-6) or OMe (n = O-2). A study of metal- ligand interactions R.S. Armstrong*, M.J. Aroney*, C.M. Barnes, K.W. Nugent Department of Inorganic Chemistry, University of Sydney, Sydney, N.S. W. 2006, Australia (Received 10 November 1993) Abstract A systematic study is reported of the infrared and Raman spectra of methyl and methoxy substituted benzene chromium tricarbonyl complexes (n”-C,H,_,X,)Cr(CO)s where X = Me (n = O-6) or OMe (n = O-2). Bands have been assigned and intensities calculated for a number of vibrational modes. Trends in the wavenumbers and band intensities of the arene-metal, metal-carbonyl and carbonyl bonds are examined to gain an insight into metal-tigand interactions and intramolecular electron rearrangements with progressive substitution on the arene. The results are discussed with regard to g-, A- and ~-contributions to arene-metal bonding and of u- and 7r-components in metal- carbonyl and carbonyl bonding. The intensities are related to the polarities and polarizabilities of individual bonds within the molecules, and variations in these bond electronic properties with arene substitution are examined. 1. Introduction The nature of the bonding and of interligand electron transmission in ($-arene)M(U& com- plexes (where M is a chromium triad metal) has been contentious for many years. Whilst it is now commonly accepted that 7r-backbonding, in addition to g-bonding, occurs between the metal and the carbonyls [I], the character of the metal-arene bond is not well understood. Overlap of arene 7r-orbitals with metal valence orbitals can result in arene to metal a-donation, arene to metal x-donation, or metal to arene backdonation [2,3]; the relative contributions of these interactions are unknown. *Corresponding authors. The complexes have a “piano-stool” type geo- metry (Fig. l), with ring C-C bonds slightly longer than in the free arene [4]. Evidence exists to indicate that on coordination to the strongly electron with- drawing M(C0)3, the arene becomes z-depleted and reduced in aromaticity [5-91. With substitution on the arene, the electronic infhrencc of the substituent is felt in the carbonyl groups and, as well, in the arene ring where it is thought to play an important role in controlling the conformational disposition of M(CO)s relative to the arene [3,6]. The ($-arene)M(CO), complexes are highly dipolar [6,7,10] with the molecular dipole vector directed from the arene towards the metal tri- carbonyl moiety [l I]. This accords with the observa- tion that electron releasing substituents on the ring increase the molecular moment while electron 0022-2860/94/~7.00 @ 1994 Elsevier Science B.V. All rights reserved SSDI 0022-2860(93)07967-2
Transcript
ELSEVIER
Journal of
MOLECULAR S~UCT~E
Journal of Molecular Structure 323 (1994) 15-28
Infrared and Raman spectra of ($-C6H6_,X,)Cr(CO)3 complexes where X = Me (n = O-6) or OMe (n = O-2). A study of metal-
Department of Inorganic Chemistry, University of Sydney, Sydney, N.S. W. 2006, Australia
(Received 10 November 1993)
Abstract
A systematic study is reported of the infrared and Raman spectra of methyl and methoxy substituted benzene chromium tricarbonyl complexes (n”-C,H,_,X,)Cr(CO)s where X = Me (n = O-6) or OMe (n = O-2). Bands have been assigned and intensities calculated for a number of vibrational modes. Trends in the wavenumbers and band intensities of the arene-metal, metal-carbonyl and carbonyl bonds are examined to gain an insight into metal-tigand interactions and intramolecular electron rearrangements with progressive substitution on the arene. The results are discussed with regard to g-, A- and ~-contributions to arene-metal bonding and of u- and 7r-components in metal- carbonyl and carbonyl bonding. The intensities are related to the polarities and polarizabilities of individual bonds within the molecules, and variations in these bond electronic properties with arene substitution are examined.
1. Introduction
The nature of the bonding and of interligand
electron transmission in ($-arene)M(U& com- plexes (where M is a chromium triad metal) has been contentious for many years. Whilst it is now commonly accepted that 7r-backbonding, in addition to g-bonding, occurs between the metal and the carbonyls [I], the character of the metal-arene bond is not well understood. Overlap of arene 7r-orbitals with metal valence orbitals can result in arene to metal a-donation, arene to metal x-donation, or metal to arene backdonation [2,3]; the relative contributions of these interactions are unknown.
*Corresponding authors.
The complexes have a “piano-stool” type geo- metry (Fig. l), with ring C-C bonds slightly longer than in the free arene [4]. Evidence exists to indicate that on coordination to the strongly electron with- drawing M(C0)3, the arene becomes z-depleted and reduced in aromaticity [5-91. With substitution on the arene, the electronic infhrencc of the substituent
is felt in the carbonyl groups and, as well, in the arene ring where it is thought to play an important role in controlling the conformational disposition of M(CO)s relative to the arene [3,6].
The ($-arene)M(CO), complexes are highly dipolar [6,7,10] with the molecular dipole vector directed from the arene towards the metal tri- carbonyl moiety [l I]. This accords with the observa- tion that electron releasing substituents on the ring increase the molecular moment while electron
0022-2860/94/~7.00 @ 1994 Elsevier Science B.V. All rights reserved SSDI 0022-2860(93)07967-2
16 R.S. Armstrong et d/J. Mol. Struct. 323 (1994) IS-28
Y c- X
4 Z . . ‘X1 oc//ir\ “““t’co
cO
Fig. I. Structure of ($-arene)Cr(CO)3 complexes.
withdrawing groups reduce the moment (see Table 15, p. 1029 of Ref. 6). The prevalent opinion, based mainly on molecular orbital and XPS studies, is that the arene has a small net negative charge [12-17; others, however, have attributed to it a net positive charge [l&19]. For example, Kinomura et al. [14] have deduced that in benzene chromium tricarbonyl the benzene possesses a charge of -O.l2e, chromium +0.54e, (carbonyl) carbon +O.lSe, and oxygen -0.29e. Such calculations have also indicated that the x-orbitals of the arene make the predominant contribution to the arene-metal bond [20].
Vibrational spectroscopy has been extensively used to probe the nature of the bonding within the {~6-arene)M(CO)~ complexes (see pp. 1022- 1023 of Ref. 6 and Refs. 21-24). Such studies have tended to concentrate on the stretching frequencies of the carbonyl bands which are sensitive to changes in the donor/acceptor power of the arene ring. How- ever, the metal-arene and metal-carbonyl bands have been little studied, due to the belief that a synergic transfer of electron density from the arene to the carbonyls extensively couples these vibrations. This means that any analysis involving these bands would not be as simple or as clear cut, in terms of propounded theory, as one concerning the carbonyls alone.
In recent work, infrared and Raman spectra were determined for the ($-mesitylene)M(CO)s complexes (251, and trends in the intensities and wavenumbers of the metal-arene, metal-carbonyl and carbonyl bands interpreted in terms of arene to metal r-bonding and metal to arene 6-backbond- ing. This approach is now extended to investigating
variations in the nature of arene-metal bonding with progressive methyl or methoxy substitution of benzene chromium tricarbonyl.
2. Experimental
The (~6-arene)Cr(CO)~ complexes were purified by vacuum sublimation before use. Chloroform and tetrahydrofuran solvents (analytical grade) were degassed with dry nitrogen. Solutions were prepared in a dry nitrogen atmosphere using Schlenk techniques, and transferred to the spectro- scopic cells via a syringe. Reasons and justification for use of these solvents are given on p. 573 of Ref. 25. Pressed discs in KBr and polythene were pre- pared in the open, as the compounds are su~cien~y air stable in the solid state.
Infrared spectra were recorded using a Digilab FTS-80 Fourier transform infrared spectrometer with two optical benches, FTS lS/SO and FTS 20/ 80, configured for far-infrared and mid-infrared respectively. Far-infrared solution spectra were recorded using a 1 .Omm solution cell with poly- thene windows; mid-infrared spectra were obtained using a cell with KBr windows of 0.1 mm separation. The resultant spectra were solvent subtracted using routines contained in the software package of the spectrometer.
Raman spectra were excited using 514.52nm radiation from a Spectra-Physics Model 2025-05 Ar+ laser. This wavelength was chosen since it was sufficiently distant from the electronic absorp- tion bands to preclude any preresonance effects. The solution samples were held in a quartz cell spinning at 1600revmin-‘, and the 90” scattered radiation was focused onto the slits of a Jobin- Yvon U 1000 double monochromator equipped with a cooled RCA C31034 photomultiplier and photon counting electronics. Scanning and spec- tral acquisition and manipulation were computer controlled using a Hicom AT personal computer with ISA Enhanced Prism Software (versions 2.1 and 3.0). The spectra were plotted using a Hewlett- Packard 7475 graphics plotter.
The intensities were calculated by measuring the peak areas using the spectroscopic software packages, and also measuring the area of a standard
R.S. Armstrong et al./J. Mol. Struct. 323 (1994) IS-28 11
solvent peak for normalization purposes. The peak electron density from identical metal orbitals (dxy areas were divided by the molar concentration of the and d,t _u2). In other words, there is no conjugative compound and then by the area of the solvent peak electron flow between the arene and the carbonyls; to give a normalized area which could be compared rather the four ligands are mutually in competition directly with results from other spectra. This normal- both in their capacity as electron donors or (by a ization process was especially important in measur- different mechanism) electron acceptors. This ing the Raman intensities, as the measured peak area accords with the work of Neuse [36] who showed is affected by the optical alignment of the particular there is an impedence to electron flow from the experiment. arene to the carbonyls.
Band assignments were made after detailed inspection and collation of data available from earlier vibrational studies of a number of ($-arene) Cr(CO), complexes and of the corresponding free arenes. These data were drawn from a large range of references, most notable of which are refs. 23,24, 26-35. The solid-state infrared spectra and assign- ments of the complexes under study are given in Table 1. Solution infrared and Raman spectra and assignments were also obtained and these are avail- able on request. Tables 2 and 3 contain the band intensities and wavenumbers of the metal-arene, metal-carbonyl and carbonyl bands obtained from the solution-state infrared and Raman spectra of these complexes.
In the complexes of this study the arene energy levels have been altered by insertion of substituents on the benzenoid ring whilst maintaining a chro- mium atom metal centre. With progressive methyl substitution, the orbital energy levels of the arene inCreaSe from C&, t0 C&k6 [37-441. As a redt Of
this increase it is seen from the MO diagram (Fig. 2) that the q+i and d,Vr, dYz orbitals will converge in energy, thus increasing the n-interaction (that is, giving greater electron charge transfer from the arene to chromium). By contrast, the energy of Qk2 will increasingly diverge from that of the dxy, d,z _ ,,I orbitals, thereby lessening the extent of the metal to arene electron drift through 6-backbond- ing. Additionally, an increase would be expected in the weak arene to metal a-interaction.
3. Results and discussion
A molecular orbital scheme was proposed in ref. 25 that explained the results of a spectral study of the metal-arene (MR), metal-carbonyl (MC), and carbonyl (CO) bands of mesitylene tricarbonyl complexes of Cr, MO and W. The experimental data were analysed in terms of r-bonding between the arene @*i orbitals and the metal d,, and d,, orbitals, and S-backbondinga involving the metal d,, and dxZ_yt orbitals and the ek+* orbitals of the arene; these are the major components of the MR bond. In regard to MC0 bonding, o-donation takes place from the carbonyls to the metal, and 7r- backdonation from the metal to the carbonyl 7r*- orbitals. It was shown that both the arene and carbonyls donate electron density to the same metal orbitals (d,; and d,;) and, likewise, accept
Methyl substitution in the arene also affects the MC and CO bonding interactions. As arene to metal n-bonding is enhanced, a build-up of electron density on M occurs and this tends to inhibit the CO U- donation in the MC0 bond. With decreasing metal to arene ~-interaction, ~-backbonding in the MC0 bond becomes greater, resulting in increased occu- pancy of the carbonyl 7i-*-orbitals. The extent of occupation of the carbonyl 7r*-orbitals will, conse- quently, reflect the change in the MR S-interaction.
a This metal to arene interaction has been termed 6-backbonding [25,36] though it is sometimes referred to as arene-metal back- bonding or ~-backbonding.
The mesitylene complexes of the previous study [25] all belong to the Cj, point group, thus allowing a comparative analysis of the spectral bands of interest. That analysis, however, is complicated by variation of the metal atom polarizability with a change in M. This problem does not arise in the present work, but differences in symmetry need to be taken into account. The chromium tricarbonyl com- plexes of benzene (BCT), mesitylene (MCT) and hexamethylbenzene (HCT) have C,, symmetry; those of toluene (TCT), p-xylene (XC?‘), durene (DCT), pentamethylbenzene (PCT), anisole (ACT) and ~-dimethoxy~~ene (ZCT) are C,. This change
18 R.S. Armstrong et a/./J. Mol. Struct. 323 (1994) 15-28
Table I Band assignments and wavenumbers (cm-‘) for the (#-arene)Cr(CO)s complexesa
a CT = Cr(CO)s, B = benzene, T = toluene, X =p-xylene, M = mesityiene, D = durene, P = pentamethylbenzene, H = hexamethylbenzene, A = anisole, Z =p-dimethoxybenzene. b 6 = In-plane bend or deformation, H = out-of-plane bend or deformation. ’ Assigned as C3, for Cr(CO)s and C, for arene (C,, in brackets). Comb. = combination. MC = metal-carbonyl; R = arene; MR = metal-arene. d Uncertain assi~ment.
in symmetry presents certain difficulties in the analysis, as outlined below.
With the molecules of the C,, point group two types of vibrational mode, A, and E, are observed.
Table 2
As in ref. 25, only the symmetrical At mode of vibration has been considered, since vibrations of this type are polarized along the molecular axis [z-axis) and as such contribute to the large changes
Raman band wavenumbers (cm-‘) and relative intensities for ($-arene)Cr(CO)s complexesa
Arene Symmet~ MR MCb co
v I Y I Y I
301 334
306
1 347 364
312 359
327 359
327
{ 343 374
339 391
347
0.99 0.077
1.07 0.070 0.016
0.95 0.034
0.74 0.034
0.71 0.023 0.007
0.39 0.030
0.49
479 0.47 486 0.05
(476) (0.52)
1971 0.042 1892 0.240
1966 0.033 1888 0.240
Al E
A’
A’+A”
A’
A’+A”
Al E
A’
A’+A”
A’
A’+A”
Al E
A’
A’ -t A”
A’
A’+A”
Toluene
p-Xylene
Mesitylene
Durene
(479) (0.58) 1963 0.037 1884 0.241
1957 0.036 1882 0.231
1954 0.035 1876 0.192
480 0.67 482 0.037
479 0.62 489 0.070
477 0.41 485 0.30
(480) (0.69)
1951 0.038 1872 0.213
1947 0.038 1867 0.274
1965 0.033 1885 0.243
CsHMeS
C6Me6
Anisole 307
{ 329 353
316
{ 344 392
0.69 0.066 0.066
0.76
(478) (0.50)
475 0.14 1963 0.031 482 0.30 1883 0.237
a MR = metal-arene, MC = metal-carbonyl, CO = carbonyl, v = wavenumber (cm-‘), 2 = relative intensity. b The numbers in parentheses refer to unresolved bands, either A f + E or A’ + A”.
Table 3
R.S. Armstrong et a/./J. Mol. Struct. 323 (1994) l-T-28 21
Infrared band wavenumbers (cm-“) and relative intensities for (#-arene)Cr(CO)s complexesa
Arene Symmetry MR MCb CO
v I v I v I
Toluene
p-Xylene
Mesitylene
Durene
C&IMeJ
C6Me6
Anisole
Al
E
A’
A’+A”
A’
A’+A”
Al
E
A’
A’+A”
A’
A’+A”
Al
E
A’
A’+A”
A’
A’fA”
a MR = metal-arene, MC = metal-carbonyl, CO = carbonyl, Y =wavenumber (cm-‘), i = relative intensity. b The numbers in parentheses refer to unresolved bands, either A, + E or A’ + A”.
’ Uncertain assignment.
in molecular electronic properties known to occur treated as the point group C,. On going from the CjV along this axis with arene substitution [7]. The to C, the vibrations that were previously of A 1 sym- polarity and polarizability changes resulting from metry become A’, whilst those that were E symmetry vibrations of E symmetry are along the x- and are split into A’ and A” components. Because the y-axes, perpendicular to the molecular axis. separation between the MR vibrations of A, and E
For the complexes TCT, XCT, DCT, PCT, symmetry is not large ( < 50cm-‘) in the compounds ACT, and ZCT (C, point group), splitting of the belonging to the C3, point group, there is a definite MC and CO E modes is not observed (except as probability of mixing between the A’ components induced by the solvent in solution-state spectra). derived from the Al and E modes upon reduction This indicates that there is little perturbation of the in the symmetry to the C, point group. Conse- CJV symmetry of the Cr(CO), group on changing the quently, it is not possible to completely identify the symmetry of the arene, thus allowing comparison of intensities of the bands corresponding to the CSV Al the CO and MC bands over the range of compounds modes with changes along the z-axis only. This has studied. However, substitution on the arene directly to be considered in any comparative analysis of the affects the MR vibrations, which must therefore be compounds of the C3, and C, point groups.
302 5.23 475 5.11 333 2.08 479 2.23
307 { 345
312
{ 365’
328 ( 359
327
{
338 ( 389
345 ( 393
307 { 329
315 { 390
5.19 0.42
(477) (7.96)
4.08
1.44 (477) (6.90)
2.12 0.54
(480) (6.13)
1.24 478 4.30
492 0.90
0.87 476 0.97
0.87 483 4.41
0.60 481 3.68
0.50 489 0.73
6.02
0.38 (476) (5.67)
5.82 473 4.76
0.35 481 2.83
1975 1891 1906
1970 1884 1901
1966 1880 1897
1962 1875 1892
1957 1867 1885
1953 1863 1880
1949 1856 1874
1970 1882 1900
1967 1875 1895
521
1049
460
917
488
956
479
950
510
996
485
939
474
912
492 1020
442
912
22 R.S. Armstrong et al.lJ. Mol. Strucf. 323 (1994) 15-28
Arehe
CT
Complex M(W,
Fig. 2. MO representation for ($-arene)Cr(C0)3 complexes.
R.S. Armstrong et al./J. Mol. Struct. 323 (1994) 15-28 23
3.1. Vibrat~5nal band waven~mbe~s
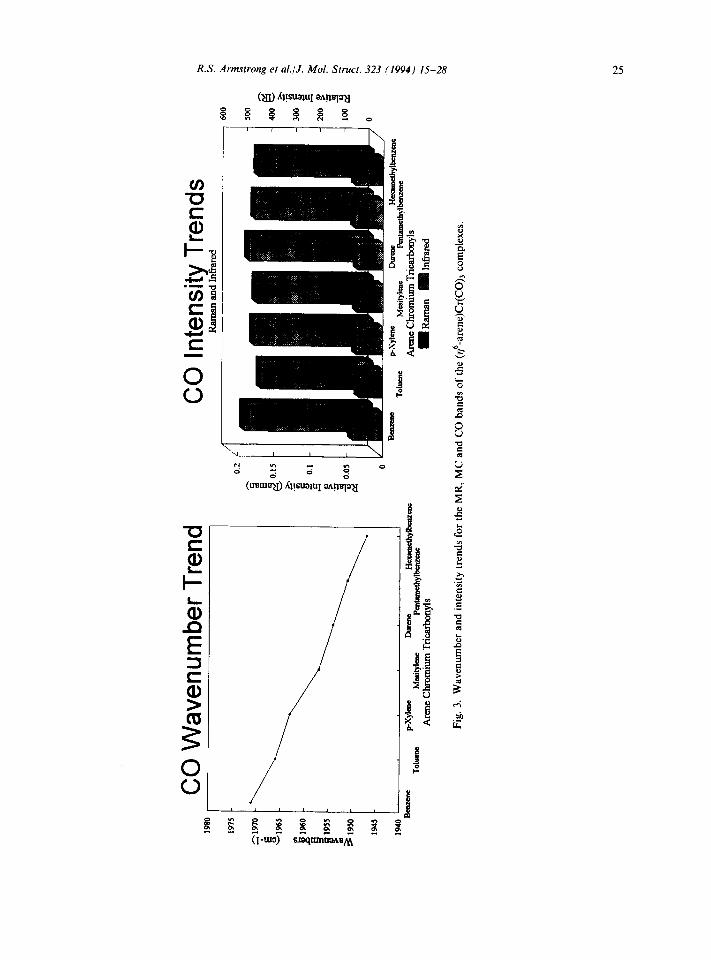
Examination of the MR, MC and CO bands (Tables 2 and 3) shows a regular trend in the wave- numbers of v(MR) and v(C0); v(MC) shows little variation. This need not have been the case since symmetry differences between the complexes could have led to severe irregularities in the sequence of band wavenumbers of consecutive compounds or even reversals of trend within the series. The regu- larity of this trend, however, indicates that the vibrational wavenumbers for these complexes are little affected by such changes in the symmetry, but rather reflect the change in the electronic properties of the compounds.
The wavenumber of a stretching vibration can often be used to provide an estimate of the strength of that particular bond, provided that the vibration is sufficiently isolated from others of similar sym- metry so as to prevent coupling. Tbe MR bond stretch is of lowest energy and more than 150cm-i from v(MC); thus coupling between the two should not be important over the range of compounds studied. Inspection of the MR vibrations for these complexes (Tables 2 and 3) shows an increase in the MR band wavenumber on going from benzene to hexamethylbenzene. This is contrary to what would be expected from reduced mass arguments, and is indicative of an increase in the bond strength of the same order. From the MO diagram we know that increasing the number of methyl substituents on the benzene ring results in an increase in the MR P interaction and a decrease in the S-interaction. The r-bond is regarded as stronger than the &bond (because of the dousers of the latter) and so an observed increase in the MR bond strength is not unexpected. Taking the MR bond as a two body oscillator between the arene and the Cr(CO), moiety, the calculated force constants (Table 4) also indicate an increase in the strength of the MR bond as the number of ring substituents becomes greater.
Since there is a lessening in the MR S-interaction with successive benzene substitution, an increased amount of electron density is available for MC0 ~-backbonding, causing a strengthening of the MC n-bond and a weakening of the CO bond. A decrease is observed in the CO stretching
wavenumber with methyl substitution, correspond- ing to the predicted weakening of that bond. It might also be expected that the MC band wave- number should increase. However, this remains relatively unchanged over the series, regardless of the increase in the MC0 Gnteraction. It is therefore suggested that the increase is offset by a decrease in the MC0 o-interaction (which as seen earlier is a consequence of increased MR r-bonding).
Wavenumber trends in the spectra of the methoxy substituted BCTs studied resemble those of the methyl substituted compounds, in accordance with a mesomeric donor capability of the methoxy group.
3.2. Band intensities of the C,, complexes (BCT, MCT, HCT)
With respect to the MR bond, in each case studied the MR band is the lowest energy Ai stretching vibration and is isolated from other absorptions in the spectrum. Both the infrared and Raman band intensities’ decrease with an
Table 4 Calculated force constants for the MR bond in (~6-arene) Cr(CO)s complexesa
a Based on a diatomic system between the arene and the Cr(CO)s moiety. b In mdyn/A (IO”2mdyn/A = lO’Nm_‘).
’ Strictly, the infrared intensities are a measure of the chunge in dipole moment p with change in vibrational state along a normal coordinate Q, i.e. (dp/dQ)a, measnred at the eq~bri~ position. If it is assumed that the normal coordinate is purely associated with the stretching of a given bond, and that the charge separation across the bond is lit& a&ted by the ~~ational state, then the infrared intensity can be regarded approximately to be a measure of the ground state bond moment. The Raman intensity can be similarly related to the bond polarizability.
MR
Wav
enum
ber
Tre
nd
2.5
f
MC
(O)
Wav
enum
ber
Tre
nd
700
. _
tscn
zene
pxykne
Dur
‘alc
H
em&
ylbm
zme
Tol
ucnc
M
esit
ylen
e P~U
ll?l
eUly
lbR
v~~
Arc
nc C
lirom
ium
Tri
carb
onyl
s
MR
lnt
ensi
t T
rend
s R
aman
and
ILed
,
6
MC
(O)
Inte
nsity
T
rend
s F
&m
an
and
Infr
ared
9
Buu
ene
PxYl
= D
uren
e H.ZTUlllechylbaaene
TO
ltb
2.W
M
esit
ylm
e P&
ME
tily
lbC
IlZ~
Are
ne C
hrom
ium
Tri
cabo
nyls
WF
23m
an
Infr
ared
*.. Y
* -
Y
-. -
_ -
- ,
m
-^
- -.
<
. .-
- r-
...,-
sm
.-
.__
*,
~I_
_
CO
Wav
enum
ber
Tre
nd
‘Mm
19
75
,“19
70
6 I
-196
5 -
1 1960
- P
195s
-
5 3 1950
1945
1940 tle
nzen
e D
urm
c T
OlW
PX
YlQ=
M
dylm
c H
arpI
vlhy
lbcn
zsns
P
alta
mah
ylbc
nzm
c A
rene
Chr
omiu
m T
riea
rtao
y~s
Are
ne
Chr
omiu
m
Tri
carb
onyl
s
CO
Int
ensi
ty T
rend
s R
aman
aodI
nfkr
ed
Fig.
3.
W
aven
umbe
r an
d in
tens
ity
tren
ds
for
the
MR
, M
C
and
CO
ba
nds
of
the
(q6-
aren
e)C
r(C
O)j
co
mpl
exes
.
600
26 R.S. Armstrong et al./J. Mol. Struct. 323 (1994) 15-28
increase in the electron donating power of the ring (Tables 2 and 3). This can be explained in terms of decreasing &-backbonding and increasing ~-bonding within the MR bond on going from benzene to mesitylene to hexamethylbenzene (as seen in the MO diagram). The S-bond is more diffuse and hence more polarizable than the x-bond, and as such is expected to contribute a larger component to the Raman band intensity. Thus any change in the degree of &bonding will have a greater effect on the Raman band intensity than a variation in n-bonding. The decrease observed in the MR Raman band intensity with methyl substitution is consistent with a decrease in the extent of the metal to arene 6-backbonding.
The infrared intensity of the MR bond is an indicator of charge separation between the arene and the Cr(CO)s moiety. The infrared intensity of this band is observed to decrease with progressive methyl substitution, indicating a diminishing MR bond dipole. This can be easily understood on the following grounds. The coordinated benzene in (#-C6H6)Cr(CO)s is electrically negative [12- 161, or at least less positive [l&19], in comparison with the chromium atom centre, so that the MR bond dipole is directed from chromium to benzene. The introduction of a methyl substituent will lead to a net electron transfer from the arene to the metal, causing the charge on the arene to become more positive and that on the chromium to become more negative (through increasing arene to metal 7r- donation and decreasing ~-backbonding), so that the MR bond dipoIe is lowered. Supporting evidence comes from comparison of the charge distributions found for BCT and XCT complexes in Refs. 12, 13 and 17.
bonyls). The experimental trend would be explained if (folIowing methylation) the balance of these effects is such that more of the electron density build-up remains on Cr than is relocated on the carbon atom of each CO. Thus the effect of each methyl insertion would be to increase the electron density on the Cr and to decrease the Cr- C bond dipole. This is true so long as the chromium in these complexes is more electrically positive than the carbon of CO, a fact borne out by much evidence [12-191. In this regard, important evidence is provided by a recent study of the EXAFS spectra of methyl substituted BCTs [45]. The K-edge absorption of these spectra displays a characteristic feature which shifts towards lower energy with increased methylation of the arene (relative shifts lie between 0.5 and 1.0 eV). The energy decrease is clearly indicative of an increase in the electron density on the chromium atom.
Both the infrared and Raman carbonyl band intensities are found to be fairly insensitive to changes in the number of ring substituents (Fig. 3). As seen earlier, a consequence of arene methyla~on is electron density shift to the carbon of CO and this should, in turn, result in greater electron density being localized on the oxygen. The CO infrared band intensities are large, relating to the highly dipo- lar nature of the COs as indicated by atomic charges on C and 0 [12-15,18,19]. By contrast, the carbonyl Raman band intensities are low since the high effec- tive nuclear charge of oxygen reduces the mobi~ty of electrons in its vicinity.
3.3. Band intensities of the C, complexes (TCT, XCT, DCT, PCT, ACT and ZCT)
In the case of MC bonding, methyl substitution Unlike the discussion of the wavenumbers, leads to increased 7r- and decreased cr-interactions, where the symmetry of the arene does not appear resulting in a more polarizable M-CO bond. to be overly important, the trends observed in the Experimental evidence for this comes from the intensities of the MR, MC and CO bands are not increasing total Raman intensity of the MC band regular with respect to the differences between con- as seen in Table 2. Also observed with methyl sub- secutive compounds. The direction of the trends stitution is a lowering of the infrared intensities seen for the C, complexes mirror those of the C,, (Table 3), suggesting decreasing bond polarity. compounds, although there is no straight line fit for Methylation of benzene leads to electron charge the series of complexes as a whole (Fig. 3). In other displacement towards Cr (increased 7r-, decreased words, whilst there is a general decrease in the MR S- interactions with arene) and, as well, away from band intensity with increasing methyl substitution Cr (lesser o-, greater 7r-interactions with car- of the benzene ring, there is no constant variation
R.S. Armstrong ef al./J. Mol. Srruct. 323 (19943 IS-28 27
in that intensity with each successive addition. The intensity trends in the MR, MC and CO bands of the less symmetrically methyl substituted benzenes support the conclusions drawn above for the CsV complexes.
The results for the methoxy substituted com- plexes, though limited in scope, are generally similar to those of the methyl substituted complexes (Tables 2 and 3), again inferring that the methoxy group in these systems functions primarily as an electron donor, in concu~ence with PES studies which show that the methoxy substituted complexes have very similar ionization potentials in comparison with the methyl substituted complexes [41] whilst there are large potential differences between the free arenes [37,39].
3.4. Dipole m~rnen~s
Although many studies have been reported on the electric dipole moments of ($-arene)M(CO)j complexes [46], vector dissection of the molecular moments into component group dipoles is fraught with uncertainty. Strohmeier and Hellmann [47] found that the MR and MC0 group dipole moments are both directed towards the metal. However, Carroll and McGlynn [18] deduced a CrCO moment directed away from Cr and a Cr- arene dipole directed towards the metal (although they entertain the possibility of a reverse Cr-arene vector direction). The results of such analyses appear to be very much dependent on the assump- tions made. Atomic charge distributions deter- mined by subsequent workers [12-191 generally agree that the metal in these complexes possesses a substantial positive charge, the carbonyls a nega- tive charge centred on the oxygen atoms, and the arene ring either a small positive or a small negative charge.
Whilst the infrared intensity data presented herein are not directly relatable to bond moment direction, trends in such intensities with arene methyl substitution do indicate that the moments of MR and of MC0 are directed away from the central metal atom, in agreement with the atomic charge distributions from XPS [14], CND0/2 [ 15,17,19] and self-consistent charge and con- figuration (SCCC) [ 12,13,16] studies. It follows
that the group dipole moment of M(CO)s, the predominant components of which are the large CO dipoles, most substantially outweighs p(MR).
It is concluded also from this work that, with each insertion of a donor substituent on the arene, an incremental charge displa~ment takes place towards M(C0)3 resulting in a small decrease in p(MR) and in p(MC), and a small increase in p(CO). The carbonyl group, unlike MR and M- CO, is highly dipolar so that small fluctuations of electron density on C or 0 atoms cause only minor variations in b(CO) and in the carbonyl infrared band intensities. The overall effect is a gradual enhancement of the molecular dipole moment with an increase in the number of electron donor substituents on the arene, a rationalization in accord with experimental observations [6,7].
4. Ack~w~~gemen~
The authors acknowledge with gratitude helpful advice from Dr. A. Masters, Mr. T. Maschmeyer and Mr. M.S. Davies of the University of Sydney, and from Professor R.J.H. Clark and Dr. S. Best of University College London.
5. References
[II
VI
[31
141
151
161
[71
F.A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley-Inters~en~, New York, 1988, pp. 58-62. G.E. Coates, M.L.H. Green and K. Wade, Organome- tallic Compounds, 3rd edn., Vol. 2, Chapman and Hall, London, 1968, Chapter 5, pp. 165-189. E.L. Muetterties, J.R. Bleeke and E.J. Wucherer, Chem. Rev., 82 (1982) 499. N.S. Chiu, L. Schafer and R. Seip, J. Organomet. Chem., 101 (1975) 331. J.A. Connor, J.A. Martinho-Simoes, H.A. Skinner and M.T. Zafarani Moattar, J. Organomet. Chem., 179 (1979) 331. R. Davis and L.A.P. Kane-Maguire, in G. Wilkinson, F.G.A. Stone and E. Abel (Eds.), Comprehensive Orga- nometalIic Chemistry, Vol. 3, Permagon, Oxford, 1982, Chapters 26.2, 27.2, 28.2. M.J. Aroney, M.K. Cooper, R.K. Pierens, S.J. Pratten and SW. Filipczuk, J. Organomet. Chem., 295 (1985) 333.
28 R.S. Armstrong et al./J. Mol. Struct. 323 (1994) 1.5-28
[8] M.J. Aroney, R.M. Clarkson, R.J. Klepetko, [27] R.E. Humphrey, Spectrochim. Acta, 17 (1961) 93. A.F. Masters and R.K. Pierens, J. Organomet. Chem., [28] G. Davidson and E.M. Riley, J. Organomet. Chem., 19 393 (1990) 371. (1969) 101.
[9] B.P. Byers and M.B. Hall, Organometallics, 6 (1987) 2319.
[29] D.M. Adams and A. Squire, J. Chem. Sot. A, (1970) 814.
[lo] M.J. Aroney, M.K. Cooper, P.A. Englert and R.K. Pierens, J. Mol. Struct., 77 (1981) 99.
[ 1 I] M.J. Aroney, M.K. Cooper, R.K. Pierens, S.J. Pratten and SW. Filipczuk, J. Organomet. Chem., 307 (1986) 191.
[30] R. Cataliotti, A. Poletti and A. Santucci, J. Mol. struct., 5 (1970) 215.
1311 G. Davidson and E.M. Riley, Spectrochim. Acta, Part A, 27 (1971) 1649.
[12] J.A. Connor, L.M.R. Derrick and I.H. Hillier, J. Chem. Sot., Faraday Trans. 2, 70 (1974) 941.
[13] M.F. Guest, I.H. Hillier, B.R. Higginson and D.R. Lloyd, Mof. Phys., 29 (1975) 113.
[14] F. Kinomura, T. Tamura, I. Watanabe, Y. Yokoyama and S. Ikeda, Bull. Chem. Sot. Jpn., 49 (1976) 3544.
[15] N.J. Fitzpatrick, J.-M. Savariault and J.-F.R. Labarre, J. Organomet. Chem., 127 (1977) 325.























![[insu-00759832, v1] Methoxy-serratenes in a soil under ...](https://static.documents.pub/doc/80x56/61dcf8ca6842e053ca61a6b5/insu-00759832-v1-methoxy-serratenes-in-a-soil-under-.jpg)






