Similarity and Differences in the Lactobacillus acidophilus GroupIdentified by Polyphasic Analysis and Comparative Genomics�†
Bernard Berger,* R. David Pridmore, Caroline Barretto, Francoise Delmas-Julien,Kerstin Schreiber, Fabrizio Arigoni, and Harald Brussow*
Nestle Research Center, CH-1000 Lausanne 26, Vers-chez-les-Blanc, Switzerland
Received 31 August 2006/Accepted 21 November 2006
A set of lactobacilli were investigated by polyphasic analysis. Multilocus sequence analysis, DNA typing,microarray analysis, and in silico whole-genome alignments provided a remarkably consistent pattern ofsimilarity within the Lactobacillus acidophilus complex. On microarray analysis, 17 and 5% of the genes fromLactobacillus johnsonii strain NCC533 represented variable and strain-specific genes, respectively, when testedagainst four independent isolates of L. johnsonii. When projected on the NCC533 genome map, about 10 largeclusters of variable genes were identified, and they were enriched around the terminus of replication. A quarterof the variable genes and two-thirds of the strain-specific genes were associated with mobile DNA. Signaturesfor horizontal gene transfer and modular evolution were found in prophages and in DNA from the exopoly-saccharide biosynthesis cluster. On microarray hybridizations, Lactobacillus gasseri strains showed a shift tosignificantly lower fluorescence intensities than the L. johnsonii test strains, and only genes encoding veryconserved cellular functions from L. acidophilus hybridized to the L. johnsonii array. In-silico comparativegenomics showed extensive protein sequence similarity and genome synteny of L. johnsonii with L. gasseri, L.acidophilus, and Lactobacillus delbrueckii; moderate synteny with Lactobacillus casei; and scattered X-typesharing of protein sequence identity with the other sequenced lactobacilli. The observation of a stepwisedecrease in similarity between the members of the L. acidophilus group suggests a strong element of verticalevolution in a natural phylogenetic group. Modern whole-genome-based techniques are thus a useful adjunctto the clarification of taxonomical relationships in problematic bacterial groups.
Lactobacilli belong to the phylum Firmicutes (low-G�C-content gram-positive bacteria), class Bacilli, order Lactobacil-lales. Lactobacilli have great importance for humans asstarter bacteria in food and feed fermentation (e.g., Lactoba-cillus delbrueckii in yogurt and Lactobacillus plantarum in sau-erkraut and silage fermentation) and as commensals in thevagina (e.g., Lactobacillus jensenii) or the gut (e.g., Lactobacil-lus gasseri) (50). While lactobacilli play a certain role in endo-carditis (45) and are occasionally found in clinical samples (6),no professional pathogens are known in the genus Lactobacil-lus. Consequently, lactobacilli enjoy a “generally regarded assafe” status (4). In addition, lactobacilli have attracted theattention of the medical community as vaccine carriers and asprobiotic, i.e., health-promoting, bacteria (42). Immunostimu-latory activity has been demonstrated in animal experiments,and antidiarrhea effects were shown in carefully conductedclinical trials (Lactobacillus rhamnosus and Lactobacillus para-casei) (46). However, the genetic basis for the probiotic prop-erties is not well defined—for example, it is not known to whatextent the different reported probiotic effects of lactobacilli arestrain-, species-, or genus-specific properties (48).
Notably, the ecological and phenotypic diversity of lactoba-
cilli is mirrored in a taxonomical diversity. Currently, 100 spe-cies are described in the genus Lactobacillus. Taxonomists re-alized as early as 15 years ago that the genus Lactobacilluscould be subdivided into groups (for a recent review, see ref-erence 15). Initially, three groups were proposed. Amongthem, the L. delbrueckii group was later renamed the Lacto-bacillus acidophilus group, and the Lactobacillus casei/Pedio-coccus group was split into further subgroups and a new genus.The overall phylogenetic structure of the rRNA tree in thegenus Lactobacillus is quite complicated, leading to the sug-gestion of eight groups (15).
Until about 1970, new Lactobacillus isolates from mucosalsurfaces were identified as L. acidophilus. Then, six homologygroups were distinguished among them by DNA-DNA hybrid-ization (23), leading to the definition of the species L. acidoph-ilus, Lactobacillus amylovorus, Lactobacillus crispatus, Lactoba-cillus gallinarum, L. gasseri, and Lactobacillus johnsonii (21).Within the L. acidophilus/L. delbrueckii group, there is sub-stantial genetic diversity, as mirrored by an overall genomeG�C content ranging from 33% (L. gasseri) to 51% (L.delbrueckii). This span of G�C values is about twice as large asthat normally accepted for well-defined bacterial genera (47),raising questions as to whether we are dealing with a naturalgroup of bacteria. Taxonomists know very well that the genusLactobacillus contains many deep branches and that its rela-tionship to the genera Paralactobacillus and Pediococcus is notbased on the phylogenetic data. However, a phylogenetic ge-nus concept does not yet exist in bacterial taxonomy.
For some genomics researchers with an interest in bacterialphylogeny, it is a perennially vexatious question whether bac-
* Corresponding author. Mailing address: Nestle Research Center,CH-1000 Lausanne 26, Vers-chez-les-Blanc, Switzerland. Phone for B.Berger: 41 21 785 8955. Phone for H. Brussow: 41 21 785 8676. Fax: 4121 785 8544. E-mail for B. Berger: [email protected] for H. Brussow: [email protected].
† Supplemental material for this article may be found at http://jb.asm.org/.
teria have species (19). Taxonomists have a more pragmaticapproach to the problem and define the bacterial species as thebasic unit of bacterial taxonomy and operationally as a groupof strains sharing 70% or greater DNA-DNA relatedness un-der standardized hybridization conditions. Phenotypic andchemotaxonomic features should agree with this definition.The use of many genotypic, phenotypic, and phylogenetic datain defining bacterial species became known as polyphasic tax-onomy, which has been the consensus approach to bacterialsystematics for 35 years (54). Meanwhile, 16S rRNA sequenc-ing and its cataloguing have revolutionized bacterial systemat-ics. To this technique have been added still other DNA-basedapproaches, like rapid DNA typing methods, multilocus se-quence analysis (MLSA) of housekeeping genes, and lately,sequence analyses of complete genomes. This technicalprogress led the ad hoc committee for the reevaluation of thespecies definition in bacteriology to the following species def-inition. “A species is a category that circumscribes a (prefera-bly) genomically coherent group of individual isolates/strainssharing a high degree of similarity in (many) independentfeatures, comparatively tested under highly standardized con-ditions” (49). This committee identified the sequencing ofhousekeeping genes, DNA profiling, and DNA arrays as meth-ods with great promise in bacterial systematics. While the firsttwo methods became standard tools of the bacterial taxono-mist, DNA arrays are still not routinely used in bacterial tax-onomy. Lactobacilli have been intensively investigated bypolyphasic taxonomy (54), DNA typing (59), and rRNA anal-ysis (9, 15) and lately also by the comparison of completegenome sequences (9, 34). Only one study with L. plantarum,using microarray technology, addressed the intraspecies diver-sity in lactobacilli (36). We conducted a microarray analysis inthe L. acidophilus complex for intra- and interspecies diversity,which we complemented with multilocus sequence analysis,sugar fermentation analysis, DNA typing, and comparativegenomics. A remarkably consistent pattern emerged from thiscomparative analysis, underlining the fact that we are dealingin the L. acidophilus complex with a natural group, despite thehighly variable genomic G�C contents.
MATERIALS AND METHODS
Strains. The following bacteria were used for phenotypic and molecular typ-ing, and a subset of them was used for comparative genomic hybridization(CGH) on microarrays. Lactobacillus delbrueckii subsp. bulgaricus strain ATCC11842, L. plantarum strain WCFS1, Lactobacillus salivarius subsp. salivarius strainUCC118, L. acidophilus strains ATCC 11975 and NCFM, L. gasseri strainsATCC 33323 (the type strain) and NCC2547, and L. johnsonii strains NCC533(41), NCC2767, NCC2822 (both isolates from dog feces), ATCC 33200 (the typestrain), and ATCC 11506. The two fecal isolates were derived from healthy dogsin France, and the species assignment was initially based on 16S rRNA data. TheL. johnsonii strains were selected from a collection of 15 strains based on theirextensive differences from NCC533 and each other in insertion element (IS)mapping (less than 4 insertion sites were detected out of 14 in NCC533 [unpub-lished data]). All lactobacilli were grown anaerobically in MRS broth (DifcoLaboratories, Detroit, Mich.) at 37°C. They were characterized for their rangesof fermentable carbohydrates by using the API50-CHL kit (bioMerieux, Lyon,France).
Microarray description, labeling, and hybridization. The NCC533 microar-rays covered 96% of the open reading frames (ORFs) from the complete ge-nome. PCR amplicons of 127 to 800 bp corresponding to 1,756 ORFs werespotted in duplicate onto slides (Eurogentec). Amplicons from the luciferasegene (pSP-luc�; Promega) were also spotted as spiking controls. Genomic DNAwas prepared as previously described (16). An additional purification step on
QIAGEN Genomic-tip 20/G proved to be essential to obtain the reproducibilityneeded for our normalization method (see below). Five nanograms of pSP-luc�plasmid was added to 1 �g of genomic DNA and labeled with FluoroLink Cy5-or Cy3-dUTP (Amersham) using the DNA High Prime Kit (Roche AppliedScience) according to the supplier’s instructions. Reactions were stopped after5 h by heating the sample to 65°C for 10 min. The combined reactions (Cy5 andCy3) were purified with the MinElute Reaction Cleanup Kit (QIAGEN) andeluted in 10 �l 10 mM Tris-HCl, pH 8.0. The labeled DNA and 1.5 �l of 1 mg/mlMB grade fish sperm DNA in DIG Easy Hyb Buffer (Roche Applied Science)were denatured by heating them for 2 min at 95°C and were applied to themicroarrays under a coverslip. Hybridization was performed overnight at 42°C.The slides were washed for 5 min at 42°C with 2� SSC (Invitrogen) (1� SSC is0.15 M NaCl plus 0.015 M sodium citrate), 0.1% sodium dodecyl sulfate and thenat room temperature with the same solution. Additional washes for 1 min atroom temperature with 0.2� SSC and then 0.1� SSC were conducted twice. Theslides were immediately dried by centrifugation.
Microarray data acquisition and treatment. Fluorescence scanning was per-formed on a ScanArray 4000 confocal laser scanner (Perkin-Elmer). Signalintensities for each spot were determined using Imagene software (Biodiscov-ery). Data analysis was performed with scripts written in Python (http://www.python.org). The local background value was subtracted from the intensity ofeach spot. Each spot was tagged and eliminated from the analysis if its imagesshowed clear alteration (scratches, leaking, printing, dust, etc.) or if its signalstrength was lower than twice the standard deviation of the local backgroundwhen hybridized with NCC533 genomic DNA. To preserve the signal intensitydifferences obtained when strains from different species were hybridized, weintroduced into the labeling reactions a spiking control (pSP-luc�) and used itssignal ratio to correct for differences in the labeling-reaction efficiencies. Thesignal ratio (Cy5-labeled unknown strain DNA versus Cy3-labeled NCC533DNA) of each spot was normalized using this spiking signal ratio as a reference(ratio � 1).
CGH was performed in triplicate, with duplicate spots on the slide. Thedistribution of the log2-transformed signal ratios was analyzed for each hybrid-ization reaction separately. The mean of a normal distribution fitting the mainpeak was calculated. For each group of replicates, the median of these meansprovided the final position of the main peak for the corresponding unknownstrain. The log2 signal ratio of each spot was then modified in order to shift themean of the main peak of each hybridization reaction to this value. Finally, foreach amplicon, we obtained the mean R of the ratios and the correspondingstandard deviation. The distribution of these values is depicted in the inset of Fig. 2.
CGH score and gene presence determination. For each NCC533 amplicon, theBlastN result (2) against the ATCC 33323T genome was compared to its CGHresult. When the BlastN score was plotted against the CGH ratio multiplied bythe amplicon length in base pairs, a good correlation (coefficient � 0.87) wasobserved (data not shown). This allowed the transformation of the CGH ratiosinto BlastN-like scores, which are more intuitive for biologists. This CGH scorewas calculated by the following empirically determined equation: with L beingthe amplicon length and R the mean CGH ratio, CGH score � (1.8327 · R · L) �[(0.0006) · (R · L)2]. For low values of R · L, a better correlation was given by thefollowing two equations: for 187 � R · L � 90, CGH score � �346.13 � (5.1979 ·R · L) � [(0.0087) · (R · L)2]; for R · L � 90, CGH score � 40. Similar to the bitscore of a BLAST analysis (http://www.ncbi.nlm.nih.gov/BLAST), this CGHscore reflects the level of conservation between the two genomes, taking intoaccount the size of the amplicon sequence. This approach was also validated by usingthe sequence data from the eps-rfb clusters of three test strains (see below). TheCGH scores are color coded in Fig. 2. Clustering of the CGH results was performedusing the unweighted-pair group method with average linkages (UPGMA) algo-rithm and cosine correlation (Spotfire, Somerville, MA).
A gene corresponding to an amplicon was considered to be present in theunknown genome when the CGH score of R � standard deviation was higherthan 200. Using this threshold, which is commonly used with bit scores of BLASTanalysis, all the CGH results of NCC533 were qualified as present, despite a fewlow CGH scores.
PCR and sequencing. The DNA sequencing of the eps regions from L. johnso-nii strains ATCC 33200T, ATCC 11506, and NCC2767 was initiated from theflanking conserved regions and followed by filling the intervening gaps. This wasachieved by alternating inverted PCR (35), to generate new DNA sequence data,and long-range PCR using the Expand long-template PCR system (Roche Ap-plied Science), with primers designed from the new inverted PCR sequences,until the intervening gap was bridged. The amplicons were used as templates forDNA sequence determination from both strands. For the annotation of the epsregions, sequence similarity analyses were performed with the gapped BLASTalgorithm (E value � 10�11) by using local copies of a nonredundant protein
database and the Cluster of Orthologous Genes (COG) database from theNational Center for Biotechnology Information. Protein two-dimensional struc-tures were predicted with the EMBOSS software suite (43).
Multilocus sequence analysis methodology. The different gene sequences wereamplified by PCR using the primers specified in Table 1, and the PCR productswere sent out for sequencing of both strands (Fasteris SA, Geneva, Switzerland).Design of the primers was performed using Primaclade (22). Alternatively, thesequences were retrieved from the published genomes (see references below).Raw sequence data were transferred into BioNumerics (Applied Maths, Sint-Martens-Latem, Belgium), where consensus sequences were determined usingtwo reads for each gene (one read for the 16S rRNA genes). A similarity matrixand phylogenetic trees were created based on the maximum-parsimony andneighbor-joining methods. The reliability of the groups was evaluated by boot-strap with 500 resamplings.
DNA typing. Enterobacterial repetitive intergenic consensus sequence (ERIC)and repetitive extragenic palindromic element (REP) PCRs were performedusing the primers specified in Table 1. Amplification reactions were alwaysperformed in 50 �l of a solution containing 0.2 mM each of the two opposingprimers, 2.5 �M of each deoxynucleoside triphosphate, 1.5 mM MgCl2, 5 �l of10� amplification buffer, 2.5 U of Platinium Taq polymerase (Invitrogen), and 1�l of 10-fold-diluted bacterial DNA. All amplifications were performed in thesame DNA thermocycler (GeneAmp PCR System 9700; Applied Biosystems).For REP-PCR, the PCR profile consisted of 35 cycles at 94°C for 30 s, 40°C for30 s, and 72°C for 4 min and, finally, 1 cycle at 72°C for 6 min. For ERIC-PCR,the PCR profile consisted of 35 cycles at 94°C for 30 s, 48°C for 30 s, and 72°Cfor 4 min and, finally, 1 cycle at 72°C for 6 min. The PCR products were loadedon 1% agarose gels. Images of the gels were transferred into BioNumerics.Phylogenetic trees were created using UPGMA and the Dice similarity coeffi-cient.
API 50-CHL analysis. Data were transferred into BioNumerics. A tree wascreated by cluster analysis using UPGMA and a simple matching similaritycoefficient.
MUMmer analysis. Genome comparisons were performed with the MUMmer3 package (1, 30). Only completed genome sequences were used for the align-ments: L. johnsonii strain NCC533 (NC_005362), L. gasseri strain ATCC 33323(NC_008530); L. acidophilus strain NCFM (NC_006814), L. delbrueckii subsp.bulgaricus strain ATCC 11842 (NC_008054), L. plantarum strain WCFS1(NC_004567), Lactobacillus sakei subsp. sakei strain 23K (NC_007576), L. sali-varius subsp. salivarius strain UCC118 (NC_007929), L. casei strain ATCC 334(NC_008526), Lactobacillus brevis strain ATCC 367 (NC_008497), Pediococcuspentosaceus strain ATCC 25745 (NC_008525), Oenococcus oeni strain PSU-1(NC_008528), and Leuconostoc mesenteroides strain ATCC 8293 (NC_008531).All of the analyses were performed with default parameters, except that theminimum length of a maximal exact match was set to 20. The results weredisplayed with Mummerplot script with default parameters.
Nucleotide sequence accession numbers. The sequences of the eps regionsfrom L. johnsonii strains ATCC 33200T, ATCC 11506, and NCC2767 weredeposited in GenBank under accession numbers EF138833, EF138834, andEF138835, respectively.
RESULTS AND DISCUSSION
Polyphasic analysis in the L. acidophilus complex. (i) 16SrRNA sequence. Sequencing of 16S rRNA genes from theinvestigated lactobacilli confirmed the clustering of the L.delbrueckii/L. acidophilus complex, which was clearly separatedfrom L. plantarum, L. sakei, and L. salivarius (Fig. 1A). The last
FIG. 1. Polyphasic analysis of the indicated Lactobacillus isolates by MLSA (A to E), DNA typing (F and G), fermentation capacity (H), andclustering of the microarray analysis (I). In each tree, the strain is identified at the right end of the branch by a strain and an abridged genus/speciesidentifier. La, L. acidophilus; Ld, L. delbrueckii subsp. bulgaricus; Lg, L. gasseri; Lj, L. johnsonii; Lp, L. plantarum; Lsk, L. sakei subsp. sakei; Lsl,L. salivarius subsp. salivarius. The numbers at the nodes give the bootstrap probabilities. The scale above the MLSA gives the percentage of basepair sequence identity.
VOL. 189, 2007 SIMILARITY AND DIFFERENCES IN THE L. ACIDOPHILUS GROUP 1313
three species belonged to three distinct clusters in the moreextensive 16S rRNA tree of Canchaya et al. (9). On our 16SrRNA tree, L. gasseri and L. johnsonii could be distinguisheddue to five nucleotide differences in the V1 variable region(26).
(ii) Multilocus sequence analysis. For lactic acid bacteria,the sequence analysis of genes encoding the RNA polymerase� subunit (rpoA) and the phenylalanyl-tRNA synthase (pheS)were useful taxonomic criteria (37, 38). Indeed on the rpoAtree (Fig. 1B), the investigated L. johnsonii strains formed acluster distinct from L. gasseri. As in the 16S rRNA tree, theirnearest neighbors were L. acidophilus, L. delbrueckii, L. plan-tarum/L. sakei, and then L. salivarius, in that order. On thepheS tree (Fig. 1C), L. johnsonii strains split into two sub-groups, but both were clearly separated from the two L. gasseristrains. The nearest neighbor was again L. acidophilus, while L.delbrueckii took a more distant position on the pheS tree thanon the rpoA trees. On the chaperonin groEL tree (Fig. 1D), thesequence from one L. gasseri strain could not be clearly sepa-rated from the L. johnsonii cluster, while for the other lacto-bacilli, the pattern observed for the rpoA sequences was repro-duced. In contrast, the tuf gene, encoding the elongation factorTu, again clearly separated L. johnsonii from L. gasseri in a treeanalysis and defined an L. acidophilus/L. delbrueckii group(Fig. 1E) (60).
(iii) DNA typing. Next, we applied ERIC-PCR to the strainsfrom the L. acidophilus complex. In this analysis, the se-quenced L. johnsonii strain NCC533 and the dog feces isolateNCC2822 were closely related. Two other L. johnsonii strains,namely, the type strain, NCC1680, and the other dog fecesisolate, NCC2767, were nearly identical (Fig. 1F). In a treeanalysis, the L. johnsonii strains could be clearly separatedfrom L. gasseri and L. acidophilus. In REP-PCR, L. johnsoniiNCC1680 and NCC2767 yielded identical patterns, while theremaining three L. johnsonii strains gave a distinct but re-
lated pattern. The two L. gasseri strains gave a distinctpattern (Fig. 1G).
(iv) Fermentation phenotype. The sugar fermentation pat-tern clustered the different species of the L. acidophilus com-plex together and excluded L. plantarum and L. salivarius, butno species differentiation could be obtained for this metabolicphenotype in the L. acidophilus complex (Fig. 1H). This obser-vation agrees with the analysis by taxonomists, who observed anabsence of correlation between phylogenetic placement and met-abolic properties in lactobacilli.
L. johnsonii intraspecies differences as revealed by microar-ray analysis. (i) Overall view. Next, we explored the degree ofgenetic diversity within a single species of the L. acidophiluscomplex. For that purpose, we asked what number of ORFsfrom the sequenced L. johnsonii strain NCC533 failed to hy-bridize with the four L. johnsonii strains investigated for themultilocus sequences, DNA typing, and sugar fermentationphenotype and what their distribution was. Overall, DNA fromthe test L. johnsonii strains failed to efficiently hybridize with8% to 17% of the ORFs from the reference L. johnsonii strain,NCC533. When projected on the genome map of NCC533,these CGH results showed some clustering of conserved andNCC533-specific ORFs (Fig. 2). The region around the originof replication represented the largest genome segment of rel-ative gene conservation (denoted I-a and I-b in Fig. 2), fol-lowed by two shorter regions of conservation (II and III in Fig.2). In contrast, the region around the terminus of replicationwas a major area of genetic diversity (IV in Fig. 2). The sym-metrical orientation of this diversity region around the termi-nus is remarkable, since the terminus itself was asymmetricallyplaced with respect to the origin of replication (1, 41) (Fig. 2).Two further regions of high gene diversity were identified (Vand VI in Fig. 2). In L. plantarum, the opposite was observed,as the so-called “lifestyle adaptation island,” representing thelargest region of diversity, was close to the replication origin(36).
A priori, one expects two types of genomic diversity betweenstrains belonging to the same species. One type comes fromselfish mobile DNA that invaded or left the genome, whichdoes not necessarily add to the fitness of the strain (a “mo-bilome”). The second type (“diversity regions”) may under-lie the ecological adaptation of the investigated strains andcould represent laterally acquired DNA or remnants of an-cestral DNA that were not lost during genome reductionthat occurred in the species. According to the gene annota-tion, suggestive evidence for both types of diversity wasobtained (Fig. 2).
(ii) Mobilome. Mobile DNA is suggested when a DNAsegment fulfils a combination of the following criteria: as-sociation with a recombinase gene, presence of recombination
FIG. 2. Genomic diversity in the Lactobacillus acidophilus group as seen from the viewpoint of L. johnsonii strain NCC533. (Left) CGH data.Each horizontal row corresponds to an amplicon on the array, and the genes are vertically ordered according to their positions on the NCC533genome. The columns represent the analyzed strains, and the strains are identified by their code numbers. The color code corresponding to theCGH score (BlastN-like score) is given at the bottom right of the figure; the gradient goes from black to yellow to depict the presence, divergence,or absence of a gene sequence. Some relevant gene and genetic-element positions are shown on the left side along the genome. ori, origin ofreplication; ter, terminus of replication. (Right inset) Signal ratio distribution of the CGH data. The reference is L. johnsonii strain NCC533. Ratiosare expressed in a log2 scale. See the text for details.
TABLE 1. Primers used for PCR in DNA typing and MLSA of theinvestigated Lactobacillus strains
sites, gene annotations compatible with known mobile DNA(phages, plasmids, transposons, IS, etc.), and restricted distri-bution between strains (10). Three large DNA segments, whichare essentially restricted to NCC533, clearly represent mobileDNA: two prophages, Lj965 and Lj928, and a 6-kb DNA ele-ment (Fig. 2, annotated as a, b, and c), were all previouslydescribed (58, 58a). The third element (LJ01749 to LJ1755)was apparently integrated via a Campbell-like mechanism(showing an integrase and attL and attR recombination sites)but lacks further phage links. It contained a copy of the celldivision gene ftsK and thus resembled three “potentially au-tonomous units” described in L. acidophilus (1). The attL andattR recombination sites of the two prophages exactly flankedthe region of genetic diversity. In all three cases, PCRs withprimers located to the left and right of the attachment sitesdemonstrated an unoccupied attB site in the test strains (datanot shown). In some strains, stretches of hybridizing prophagegenes matched individual modules of the prophages (Fig. 2,lane ATCC 33200T in Lj928), an observation which agrees withthe standard model of modular phage evolution (7). This ob-servation suggests the presence of related modules in proph-ages occupying distinct genomic sites in the L. johnsonii teststrains. Four further integrases were observed in the NCC533genome; only one was associated with a cluster of divergentgenes. This cluster was annotated as an arsenic resistance cas-sette (Fig. 2, bracket d). Two fragments of an insertion elementflanked a small cluster of divergent genes that lacked a bioin-formatic annotation (Fig. 2, e), and several IS flanked or in-terrupted diversity regions, e.g., the cluster of genes involved inthe synthesis of the bacteriocin lactacin F (Fig. 2, f) and theexopolysaccharide (eps) biosynthesis gene cluster (Fig. 2, g).Cumulatively, the 110 genes of the mobilome accounted for28% (93 genes) of the 335 NCC533 variable genes and 58% (56genes) of the 96 NCC533-specific genes.
(iii) Diversity regions. Within the remaining variable regionsof the CGH map, three categories of annotations dominated.They were (i) genes associated with bacterium-environmentinteraction, (ii) metabolic genes, and (iii) genes encoding un-known functions. In the first category were mucin-binding pro-teins, the eps cluster, a putative fimbrial-biosynthesis regulon,the lactacin F gene cluster, and other cell wall-anchored pro-teins, including a putative immunoglobulin A protease (Fig. 2shows the locations and extents of the diversity regions). Theeps cluster was associated with the dTDP-rhamnose biosynthe-sis operon (rfb) and represented the largest genome segment inthis diversity group showing substantial genetic variability.Genes from this variability category were previously discussedas candidate probiotic genes (41), suggesting strain-specificrather than species-specific probiotic properties. In the secondcategory were genes involved in peptide metabolism and ac-quisition of sugars from unusual polysaccharides, phospho-transferase genes, a lactose operon, a transketolase genegroup, and a pentose utilization cluster.
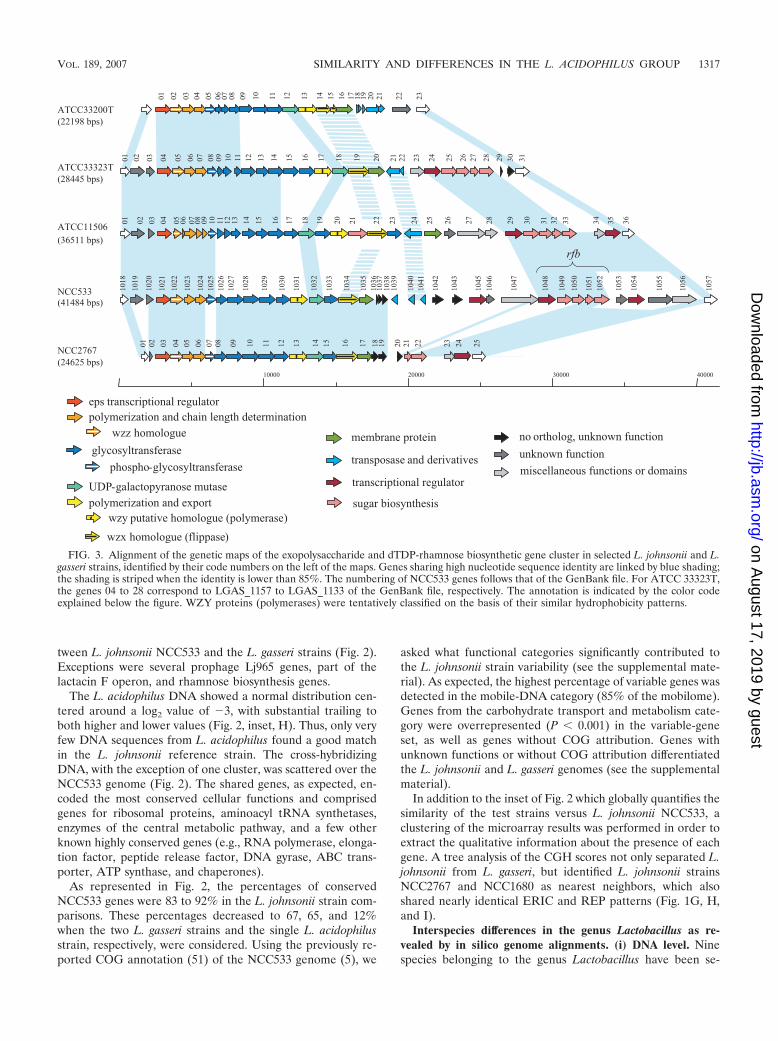
(iv) Genetic diversity in the eps cluster. In the first categoryof diversity regions, the eps cluster is of particular interest, asit is expected that non-sequence-related DNAs fulfilling com-parable functions are found at the same locus in the otherstrains (31, 56). Therefore, we sequenced the eps-rfb regions inthree L. johnsonii test strains and compared them to the eps-rfbcluster from the reference strain NCC533 and the L. gasseri
type strain, ATCC 33323T (Fig. 3). Regions of sequence iden-tity alternated with regions of sequence diversity in a morecomplex patchwork pattern than had previously been reported(24). Only at their 5� ends were the variable eps core genesclearly embedded between conserved regions. Moreover, the3� halves of the clusters (including rfb) differed strikingly inlength. Even if the eps genes did not always share DNA se-quence identity, the gene order was well conserved. The gly-cosyltransferase genes, involved in repeating-unit assembly andtherefore responsible for differences in the EPS composition atthe cell surface, formed the major component of the variableeps core. This region showed a clear modular organization, asalready described for other lactic acid bacteria (57). Pairs ofstrains shared distinct parts of the eps operon. Notably, themost similar eps operons were found in NCC533 andNCC2767, which otherwise showed the least conserved genecontents in the microarray analysis (see below). The L. johnsoniiand L. gasseri comparison showed more sharing of genes thansome intraspecies L. johnsonii comparisons, suggesting thatthis locus differentiated after speciation. The high base devia-tion index shown by the genes LJ1027 to LJ1047 in NCC533 (5)argues for their recent acquisition and supports this interpre-tation. Interestingly, the eps region is separated from the rfbregion by one or several insertion sequences or their remnants,as has been frequently observed in the 3� regions of eps oper-ons (39, 57). However, no data clearly point to an involvementof these mobile elements in the acquisition of these diversityregions. Taken together, all these observations suggest a com-plicated evolutionary history for the eps cluster, which might bea hot spot of recombination that creates variability under thepressure of positive selective forces.
Intra- versus interspecies similarities in the L. acidophiluscomplex by microarray analysis. A simplified visualization ofthe CGH results is presented in the inset of Fig. 2. Raw CGHdata come in couples of intensity values associated with eachprobe on the NCC533 array. These values are expressed asmean ratios of the results from the test to the reference strain,normalized so that a ratio of 1 expresses the perfect conserva-tion of the gene. Due to technical variability, the control hy-bridization with the reference genome versus itself results in anormal distribution when expressed on a log2 scale. It is asharp peak centered around zero, as depicted in Fig. 2, inset,A. When the four test L. johnsonii strains were hybridizedagainst the reference strain NCC533, we observed a majorpeak at the same position, suggesting very similar DNA se-quences (Fig. 2, inset, B to E). In addition, all four L. johnsoniitest strains showed a reduction in peak height and a leftwardspread in the distribution of the hybridization ratios, indicatingdecreasing DNA sequence identity between the test DNA and13% to 19% of the ORFs from NCC533.
The two L. gasseri strains, in contrast, showed a markedleftward shift for the bulk of the DNA and substantial trailingin the display of the fluorescence ratios (Fig. 2, inset, F and G).Obviously, the majority of the reference DNA only imperfectlymatched the test DNA. In addition to this shift to �1 on thelog2 scale, the main peak showed a broadening correspondingto a higher variability of DNA sequence identity. When theCGH results were mapped on the NCC533 genome, we ob-served that the genetic diversity represented by L. johnsoniistrains was a subgroup of the genetic diversity detected be-
tween L. johnsonii NCC533 and the L. gasseri strains (Fig. 2).Exceptions were several prophage Lj965 genes, part of thelactacin F operon, and rhamnose biosynthesis genes.
The L. acidophilus DNA showed a normal distribution cen-tered around a log2 value of �3, with substantial trailing toboth higher and lower values (Fig. 2, inset, H). Thus, only veryfew DNA sequences from L. acidophilus found a good matchin the L. johnsonii reference strain. The cross-hybridizingDNA, with the exception of one cluster, was scattered over theNCC533 genome (Fig. 2). The shared genes, as expected, en-coded the most conserved cellular functions and comprisedgenes for ribosomal proteins, aminoacyl tRNA synthetases,enzymes of the central metabolic pathway, and a few otherknown highly conserved genes (e.g., RNA polymerase, elonga-tion factor, peptide release factor, DNA gyrase, ABC trans-porter, ATP synthase, and chaperones).
As represented in Fig. 2, the percentages of conservedNCC533 genes were 83 to 92% in the L. johnsonii strain com-parisons. These percentages decreased to 67, 65, and 12%when the two L. gasseri strains and the single L. acidophilusstrain, respectively, were considered. Using the previously re-ported COG annotation (51) of the NCC533 genome (5), we
asked what functional categories significantly contributed tothe L. johnsonii strain variability (see the supplemental mate-rial). As expected, the highest percentage of variable genes wasdetected in the mobile-DNA category (85% of the mobilome).Genes from the carbohydrate transport and metabolism cate-gory were overrepresented (P � 0.001) in the variable-geneset, as well as genes without COG attribution. Genes withunknown functions or without COG attribution differentiatedthe L. johnsonii and L. gasseri genomes (see the supplementalmaterial).
In addition to the inset of Fig. 2 which globally quantifies thesimilarity of the test strains versus L. johnsonii NCC533, aclustering of the microarray results was performed in order toextract the qualitative information about the presence of eachgene. A tree analysis of the CGH scores not only separated L.johnsonii from L. gasseri, but identified L. johnsonii strainsNCC2767 and NCC1680 as nearest neighbors, which alsoshared nearly identical ERIC and REP patterns (Fig. 1G, H,and I).
Interspecies differences in the genus Lactobacillus as re-vealed by in silico genome alignments. (i) DNA level. Ninespecies belonging to the genus Lactobacillus have been se-
FIG. 3. Alignment of the genetic maps of the exopolysaccharide and dTDP-rhamnose biosynthetic gene cluster in selected L. johnsonii and L.gasseri strains, identified by their code numbers on the left of the maps. Genes sharing high nucleotide sequence identity are linked by blue shading;the shading is striped when the identity is lower than 85%. The numbering of NCC533 genes follows that of the GenBank file. For ATCC 33323T,the genes 04 to 28 correspond to LGAS_1157 to LGAS_1133 of the GenBank file, respectively. The annotation is indicated by the color codeexplained below the figure. WZY proteins (polymerases) were tentatively classified on the basis of their similar hydrophobicity patterns.
VOL. 189, 2007 SIMILARITY AND DIFFERENCES IN THE L. ACIDOPHILUS GROUP 1317
FIG. 4. DNA and protein sequence similarities between completely sequenced lactobacilli (identified on the y axis) as revealed by in silicogenome alignments with L. johnsonii NCC533, which was used as the sequenced reference strain (MUMmer analysis). (Left) Alignments obtainedwith NUCmer script, highlighting the conserved regions at the DNA level. The dots represent the positions of conserved DNA sequences on thegenomes. (Right) Alignments obtained with PROmer script, highlighting the conserved regions at the protein level. The dots represent thepositions of conserved protein sequences on the genomes. Identities in direct or reverse orientation are indicated in blue and red, respectively. Notethat the sequenced L. acidophilus strain does not correspond to the strain used in the CGH analysis.
quenced: L. plantarum, L. johnsonii, L. delbrueckii, L. acidoph-ilus, L. sakei, L. salivarius, and, very recently, L. casei, L. gas-seri, and L. brevis (1, 11, 13, 25, 41, 55). When the differentgenomes from the sequenced lactobacilli were plotted againstthe DNA sequence of L. johnsonii strain NCC533 (Fig. 4, left),L. gasseri showed the closest alignment. Only two large inver-sions located on either side of the terminus of replicationinterrupt the straight alignment of the two species genomes,demonstrating close synteny. Only a few segments of the L.gasseri genome did not find a match in the L. johnsonii se-quence when the MUMmer program was applied. L. acidoph-ilus showed the next-best alignment at the DNA sequencelevel. Here again, a clear diagonal line along the entire genomelength was detectable. However, the extent of the alignmentwas substantially weaker than the L. gasseri/L. johnsonii align-ment. L. delbrueckii still shared with L. johnsonii some DNAsequence identity, resulting in a faint diagonal line. L. planta-rum, L. sakei, and L. salivarius shared DNA sequence identitywith L. johnsonii mainly over one repetitive DNA segment(rRNA at 0.56 Mb).
(ii) Protein level. L. gasseri and L. acidophilus share proteinsequence identity with L. johnsonii along the entire genomelength when analyzed by MUMmer (Fig. 4, right). The numberand length of the alignment gaps were greater in the L. aci-dophilus than in the L. gasseri comparison. In contrast tothe marked loss of sequence identity in the L. johnsonii/L.delbrueckii DNA comparison, both species still showed astraight line in the protein MUMmer display (Fig. 4, right),with the exception of one major inversion (0.4 to 0.6 Mb) andone poorly aligned segment (1.0 to 1.2 Mb). The conservedsynteny of these four lactobacilli was previously observed byBlastP comparisons (24, 55). When lactobacilli outside the L.acidophilus/L. delbrueckii group were used for alignment withL. johnsonii, protein sequence identities were restricted tosmall genome segments, but despite all dispersion of thealigned regions, an X-shaped alignment pattern could still beclearly detected (especially pronounced with L. sakei), suggest-ing multiple inversions around the terminus of replication.
Evolutionary and taxonomical implications. The sequencedL. johnsonii strain differed from the test L. johnsonii strains inup to 17% of its gene content and displayed 5% strain-specificgenes. Comparable and sometimes even higher percentages ofvariable-gene content were reported for many sequenced bac-terial strains (17, 20, 32, 33, 40, 44), including L. plantarum(36). For example, the sequencing of eight Streptococcus aga-lactiae strains demonstrated a core genome shared by all iso-lates, accounting for 80% of any single genome, and a dispens-able genome part consisting of partially shared and strain-specific genes. Extrapolation of the data suggested that the S.agalactiae “pangenome” is vast and that the sum of the variablegenes within the confines of a bacterial species will rapidlyexceed the number of conserved genes (53). This hypothesisraises important questions with respect to the origin of thesevariable genes, since only a third of the variable genes andtwo-thirds of the strain-specific genes in L. johnsonii wereidentified as mobile DNA. Microbiologists are thus confrontedwith the problem of the roles of horizontally versus verticallyinherited genes in the evolution of bacterial genomes (8).Some microbiologists have rejected the idea of a phylogenetictree for bacteria (3, 18), while more recent large-scale genome
comparisons have stressed the overwhelming dominance ofvertical over horizontal gene transfers (27–29) (for a recentreview, see reference 14).
What do these considerations mean for a natural system ofbacteria and a taxonomy based on it? Our analyses within theacidophilus group of the genus Lactobacillus show a clear-cutconcordance between the different techniques. The 16S rRNAanalysis can provide an overview of the phylogenetic relation-ships between the investigated lactobacilli that was largely con-firmed by the other techniques applied. In multilocus sequenceanalysis of housekeeping genes, only three of the four genesallowed a clear separation of L. johnsonii from L. gasseri (rpoAand pheS, as suggested by Naser et al. [38], and tuf, as sug-gested by Chavagnat et al. [12], but not groEL, contrary to thereport by Teng et al. [52]). The two DNA-typing methodsconfirmed the close relationships within strains of the speciesL. johnsonii and allowed intraspecies strain differentiation,confirming earlier reports (59). As already reported (15), themetabolic phenotypes of lactobacilli have only poor taxonom-ical power in the genus Lactobacillus.
What is the contribution of newer whole-genome-based typ-ing methods to the taxonomical discussion? The microarrayanalysis clearly classified all investigated L. johnsonii strainsinto a single close-knit group of genomes, which was clearlyseparated from L. gasseri. There was no continuous transitionfrom L. johnsonii into L. gasseri—the investigated genomesformed two clearly separated clusters defining them as genomi-cally coherent groups, which agrees with the current taxonomicbacterial-species definition. Although this conclusion is at themoment based on a relatively small number of investigatedstrains, the chosen strains likely represent a sufficient variety,as testified by their extensive IS-mapping differences (see Ma-terials and Methods). Therefore, the gap separating L. johnso-nii from L. gasseri might get smaller, but will remain, whenmore strains add more breadth to each species. The microarrayanalysis revealed, furthermore, that L. gasseri was more closelyrelated to L. johnsonii than L. acidophilus, demonstrating agradual loss of similarity in the group. As the microarray anal-ysis is based on DNA-DNA hybridization, more distantly re-lated lactobacilli could not be evaluated with this technology,since these strains lacked sufficient DNA sequence similaritywith the reference strain, as confirmed by the MUMmer anal-ysis of the whole-genome sequences. Since the whole-genomein silico analysis can be extended to the protein sequence level,one can also explore the similarity of the reference strain toeven more distantly related bacteria. This analysis identified L.delbrueckii as the most closely related bacterium to the L.acidophilus complex, confirming previous taxonomical classifi-cations (54). The most closely related lactobacilli from theperspective of L. johnsonii were L. casei and L. sakei, whichagrees with a previously reported tree constructed on the basisof concatenated ribosomal protein sequences (34) and the 16SrRNA tree (9), respectively. At the next lower level of similar-ity was L. salivarius, and even more distantly related are L.brevis and L. plantarum. In the last case, the larger genome sizeof L. plantarum might dilute the similarity visually, suggestinga lower degree of similarity to L. johnsonii. This stepwise de-crease in similarity in the genome alignments still approxi-mately reflects the trends of the 16S rRNA tree.
The stepwise-decreasing degrees of similarity observed in
VOL. 189, 2007 SIMILARITY AND DIFFERENCES IN THE L. ACIDOPHILUS GROUP 1319
the L. delbrueckii/L. acidophilus group are a hallmark of Dar-winian evolution. If strong elements of vertical evolution areobserved in such a problematic taxonomical group as lactoba-cilli, there is hope that in many bacterial groups, whole-genome-based analyses will lead to a taxonomy that alsoreflects the phylogenetic relationships of the investigated bacteria,at least to a first approximation. It is currently not clearwhether the observed protein sequence relationships among allthe sequenced lactobacilli are a genomics argument for thecoherence of the genus Lactobacillus. A critical test for agenomics-supported Lactobacillus genus concept will be thedemonstration that bacteria classified outside of the genusLactobacillus share less genomic relatedness with L. johnsoniithan L. brevis, currently the most distant L. johnsonii relativeamong the sequenced lactobacilli. In a PROmer analysis, L.johnsonii was as closely related to L. brevis as to Pediococcus,while Oenococcus and Leuconostoc share practically no syntenywith L. johnsonii (Fig. 4, right, and Fig. 5). Interestingly, treesbased on ribosomal protein (34) and 16S rRNA sequences (15)place Pediococcus close to L. brevis. The congruency betweenthe “new” whole-genome comparisons and “the old methods”used for studying intra- and interspecific variation is gratifying.With the growing bacterial-genome database, whole-genome-based comparisons will become an increasingly popular aid insettling controversial taxonomical issues at both the speciesand the genus levels.
ACKNOWLEDGMENTS
We thank Todd Klaenhammer for providing us the unfinished L.gasseri genome sequence before its publication, Maria Karmirantzoufor her work on the annotation of the L. johnsonii NCC533 genomesequence, Jean-Marc Aeschlimann for helpful discussions on statistics,Deborah Moine and Juan Olivera for help with MLSA, and EneaRezzonico for editing the references.
REFERENCES
1. Altermann, E., W. M. Russell, M. A. Azcarate-Peril, R. Barrangou, B. L.Buck, O. McAuliffe, N. Souther, A. Dobson, T. Duong, M. Callanan, S. Lick,
A. Hamrick, R. Cano, and T. R. Klaenhammer. 2005. Complete genomesequence of the probiotic lactic acid bacterium Lactobacillus acidophilusNCFM. Proc. Natl. Acad. Sci. USA 102:3906–3912.
2. Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990.Basic local alignment search tool. J. Mol. Biol. 215:403–410.
3. Bapteste, E., Y. Boucher, J. Leigh, and W. F. Doolittle. 2004. Phylogeneticreconstruction and lateral gene transfer. Trends Microbiol. 12:406–411.
4. Bernardeau, M., M. Guguen, and J. P. Vernoux. 2006. Beneficial lactobacilliin food and feed: long-term use, biodiversity and proposals for specific andrealistic safety assessments. FEMS Microbiol. Rev. 30:487–513.
5. Boekhorst, J., R. J. Siezen, M. C. Zwahlen, D. Vilanova, R. D. Pridmore, A.Mercenier, M. Kleerebezem, W. M. de Vos, H. Brussow, and F. Desiere.2004. The complete genomes of Lactobacillus plantarum and Lactobacillusjohnsonii reveal extensive differences in chromosome organization and genecontent. Microbiology 150:3601–3611.
6. Brook, I., and E. H. Frazier. 1993. Significant recovery of nonsporulatinganaerobic rods from clinical specimens. Clin. Infect. Dis. 16:476–480.
7. Brussow, H., and F. Desiere. 2001. Comparative phage genomics and theevolution of Siphoviridae: insights from dairy phages. Mol. Microbiol. 39:213–222.
8. Bushman, F. 2002. Lateral DNA transfer mechanisms and consequences.Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
9. Canchaya, C., M. J. Claesson, G. F. Fitzgerald, D. van Sinderen, and P. W.O’Toole. 2006. Diversity of the genus Lactobacillus revealed by comparativegenomics of five species. Microbiology 152:3185–3196.
10. Canchaya, C., C. Proux, G. Fournous, A. Bruttin, and H. Brussow. 2003.Prophage genomics. Microbiol. Mol. Biol. Rev. 67:238–276.
11. Chaillou, S., M. C. Champomier-Verges, M. Cornet, A. M. Crutz-Le Coq,A. M. Dudez, V. Martin, S. Beaufils, E. Darbon-Rongere, R. Bossy, V. Loux,and M. Zagorec. 2005. The complete genome sequence of the meat-bornelactic acid bacterium Lactobacillus sakei 23K. Nat. Biotechnol. 23:1527–1533.
12. Chavagnat, F., M. Haueter, J. Jimeno, and M. G. Casey. 2002. Comparisonof partial tuf gene sequences for the identification of lactobacilli. FEMSMicrobiol. Lett. 217:177–183.
13. Claesson, M. J., Y. Li, S. Leahy, C. Canchaya, J. P. van Pijkeren, A. M.Cerdeno-Tarraga, J. Parkhill, S. Flynn, G. C. O’Sullivan, J. K. Collins, D.Higgins, F. Shanahan, G. F. Fitzgerald, D. van Sinderen, and P. W. O’Toole.2006. Multireplicon genome architecture of Lactobacillus salivarius. Proc.Natl. Acad. Sci. USA 103:6718–6723.
14. Dagan, T., and W. Martin. 2006. The tree of one percent. Genome Biol.7:118.
15. Dellaglio, F., and G. E. Felis. 2005. Taxonomy of lactobacillus and bifidobac-teria, p. 25–49. In G. W. Tannock (ed.), Probiotics and prebiotics: scientificaspects. Caister Academic Press, Wymondham, Norfolk, United Kingdom.
16. Delley, M., B. Mollet, and H. Hottinger. 1990. DNA probe for Lactobacillusdelbruecki. Appl. Environ. Microbiol. 56:1967–1970.
17. Dobrindt, U., F. Agerer, K. Michaelis, A. Janka, C. Buchrieser, M. Samuel-son, C. Svanborg, G. Gottschalk, H. Karch, and J. Hacker. 2003. Analysis ofgenome plasticity in pathogenic and commensal Escherichia coli isolates byuse of DNA arrays. J. Bacteriol. 185:1831–1840.
18. Doolittle, W. F. 1999. Phylogenetic classification and the universal tree.Science 284:2124–2129.
19. Doolittle, W. F., and R. T. Papke. 2006. Genomics and the bacterial speciesproblem. Genome Biol. 7:116.
20. Fitzgerald, J. R., D. E. Sturdevant, S. M. Mackie, S. R. Gill, and J. M.Musser. 2001. Evolutionary genomics of Staphylococcus aureus: insights intothe origin of methicillin-resistant strains and the toxic shock syndrome epi-demic. Proc. Natl. Acad. Sci. USA 98:8821–8826.
21. Fujisawa, T., Y. Benno, T. Yaeshima, and T. Mitsuoka. 1992. Taxonomicstudy of the Lactobacillus acidophilus group, with recognition of Lactobacil-lus gallinarum sp. nov. and Lactobacillus johnsonii sp. nov. and synonymy ofLactobacillus acidophilus group A3 (Johnson et al. 1980) with the type strainof Lactobacillus amylovorus (Nakamura 1981). Int. J. Syst. Bacteriol. 42:487–491.
22. Gadberry, M. D., S. T. Malcomber, A. N. Doust, and E. A. Kellogg. 2005.Primaclade—a flexible tool to find conserved PCR primers across multiplespecies. Bioinformatics 21:1263–1264.
23. Johnson, J. L., C. F. Phelps, C. S. Cummins, J. London, and F. Gasser. 1980.Taxonomy of the Lactobacillus acidophilus group. Int. J. Syst. Bacteriol.30:53–68.
24. Klaenhammer, T. R., R. Barrangou, B. L. Buck, M. A. Azcarate-Peril, and E.Altermann. 2005. Genomic features of lactic acid bacteria affecting biopro-cessing and health. FEMS Microbiol. Rev. 29:393–409.
25. Kleerebezem, M., J. Boekhorst, R. van Kranenburg, D. Molenaar, O. P.Kuipers, R. Leer, R. Tarchini, S. A. Peters, H. M. Sandbrink, M. W. Fiers,W. Stiekema, R. M. Lankhorst, P. A. Bron, S. M. Hoffer, M. N. Groot, R.Kerkhoven, M. de Vries, B. Ursing, W. M. de Vos, and R. J. Siezen. 2003.Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl.Acad. Sci. USA 100:1990–1995.
26. Kullen, M. J., R. B. Sanozky-Dawes, D. C. Crowell, and T. R. Klaenhammer.2000. Use of the DNA sequence of variable regions of the 16S rRNA gene
FIG. 5. Protein sequence similarities (PROmer) of Pediococcuspentosaceus, Oenococcus oeni, and Leuconostoc mesenteroides with L.johnsonii NCC533 as the sequenced reference strain.
for rapid and accurate identification of bacteria in the Lactobacillus acidoph-ilus complex. J. Appl. Microbiol. 89:511–516.
27. Kunin, V., D. Ahren, L. Goldovsky, P. Janssen, and C. A. Ouzounis. 2005.Measuring genome conservation across taxa: divided strains and united king-doms. Nucleic Acids Res. 33:616–621.
28. Kunin, V., L. Goldovsky, N. Darzentas, and C. A. Ouzounis. 2005. The netof life: reconstructing the microbial phylogenetic network. Genome Res.15:954–959.
29. Kurland, C. G., B. Canback, and O. G. Berg. 2003. Horizontal gene transfer:a critical view. Proc. Natl. Acad. Sci. USA 100:9658–9662.
30. Kurtz, S., A. Phillippy, A. L. Delcher, M. Smoot, M. Shumway, C. Antonescu,and S. L. Salzberg. 2004. Versatile and open software for comparing largegenomes. Genome Biol. 5:R12.
31. Lamothe, G. T., L. Jolly, B. Mollet, and F. Stingele. 2002. Genetic andbiochemical characterization of exopolysaccharide biosynthesis by Lactoba-cillus delbrueckii subsp. bulgaricus. Arch. Microbiol. 178:218–228.
32. Le Gall, T., P. Darlu, P. Escobar-Paramo, B. Picard, and E. Denamur. 2005.Selection-driven transcriptome polymorphism in Escherichia coli/Shigellaspecies. Genome Res. 15:260–268.
33. Leonard, E. E., L. S. Tompkins, S. Falkow, and I. Nachamkin. 2004. Com-parison of Campylobacter jejuni isolates implicated in Guillain-Barre syn-drome and strains that cause enteritis by a DNA microarray. Infect. Immun.72:1199–1203.
34. Makarova, K., A. Slesarev, Y. Wolf, A. Sorokin, B. Mirkin, E. Koonin, A.Pavlov, N. Pavlova, V. Karamychev, N. Polouchine, V. Shakhova, I. Grigoriev, Y.Lou, D. Rohksar, S. Lucas, K. Huang, D. M. Goodstein, T. Hawkins, V.Plengvidhya, D. Welker, J. Hughes, Y. Goh, A. Benson, K. Baldwin, J. H. Lee,I. Diaz-Muniz, B. Dosti, V. Smeianov, W. Wechter, R. Barabote, G. Lorca, E.Altermann, R. Barrangou, B. Ganesan, Y. Xie, H. Rawsthorne, D. Tamir, C.Parker, F. Breidt, J. Broadbent, R. Hutkins, D. O’sullivan, J. Steele, G.Unlu, M. Saier, T. Klaenhammer, P. Richardson, S. Kozyavkin, B. Weimer,and D. Mills. 2006. Comparative genomics of the lactic acid bacteria. Proc.Natl. Acad. Sci. USA 103:15611–15616.
35. Martin, V. J. J., and W. W. Mohn. 1999. An alternative inverse PCR (IPCR)method to amplify DNA sequences flanking Tn5 transposon insertions. J.Microbiol. Methods 35:163–166.
36. Molenaar, D., F. Bringel, F. H. Schuren, W. M. de Vos, R. J. Siezen, and M.Kleerebezem. 2005. Exploring Lactobacillus plantarum genome diversity byusing microarrays. J. Bacteriol. 187:6119–6127.
37. Naser, S. M., K. E. Hagen, M. Vancanneyt, I. Cleenwerck, J. Swings, andT. A. Tompkins. 2006. Lactobacillus suntoryeus Cachat and Priest 2005 is alater synonym of Lactobacillus helveticus (Orla-Jensen 1919) Bergey et al.1925 (Approved Lists 1980). Int. J. Syst. Evol. Microbiol. 56:355–360.
38. Naser, S. M., F. L. Thompson, B. Hoste, D. Gevers, P. Dawyndt, M.Vancanneyt, and J. Swings. 2005. Application of multilocus sequence anal-ysis (MLSA) for rapid identification of Enterococcus species based on rpoAand pheS genes. Microbiology 151:2141–2150.
39. Pluvinet, A., F. Charron-Bourgoin, C. Morel, and B. Decaris. 2004. Poly-morphism of eps loci in Streptococcus thermophilus: sequence replacement byputative horizontal transfer in S-thermophilus IP6757. Int. Dairy J. 14:627–634.
40. Porwollik, S., R. M. Y. Wong, and M. McClelland. 2002. Evolutionarygenomics of Salmonella: gene acquisitions revealed by microarray analysis.Proc. Natl. Acad. Sci. USA 99:8956–8961.
41. Pridmore, R. D., B. Berger, F. Desiere, D. Vilanova, C. Barretto, A. C. Pittet,M. C. Zwahlen, M. Rouvet, E. Altermann, R. Barrangou, B. Mollet, A.Mercenier, T. Klaenhammer, F. Arigoni, and M. A. Schell. 2004. The ge-nome sequence of the probiotic intestinal bacterium Lactobacillus johnsoniiNCC 533. Proc. Natl. Acad. Sci. USA 101:2512–2517.
42. Reid, G., J. Jass, M. T. Sebulsky, and J. K. McCormick. 2003. Potential usesof probiotics in clinical practice. Clin. Microbiol. Rev. 16:658–672.
43. Rice, P., I. Longden, and A. Bleasby. 2000. EMBOSS: the European molec-ular biology open software suite. Trends Genet. 16:276–277.
44. Salama, N., K. Guillemin, T. K. McDaniel, G. Sherlock, L. Tompkins, and S.Falkow. 2000. A whole-genome microarray reveals genetic diversity amongHelicobacter pylori strains. Proc. Natl. Acad. Sci. USA 97:14668–14673.
45. Salvana, E. M., and M. Frank. 2006. Lactobacillus endocarditis: case reportand review of cases reported since 1992. J. Infect. 53:e5–e10.
46. Sarker, S. A., S. Sultana, G. J. Fuchs, N. H. Alam, T. Azim, H. Brussow, andL. Hammarstrom. 2005. Lactobacillus paracasei strain ST11 has no effect onrotavirus but ameliorates the outcome of nonrotavirus diarrhea in childrenfrom Bangladesh. Pediatrics 116:e221–e228.
47. Schleifer, K. H., and W. Ludwig. 1995. Phylogeny of the genus Lactobacillusand related genera. Syst. Appl. Microbiol. 18:461–467.
48. Servin, A. L. 2004. Antagonistic activities of lactobacilli and bifidobacteriaagainst microbial pathogens. FEMS Microbiol. Rev. 28:405–440.
49. Stackebrandt, E., W. Frederiksen, G. M. Garrity, P. A. Grimont, P.Kampfer, M. C. Maiden, X. Nesme, R. Rossello-Mora, J. Swings, H. G.Truper, L. Vauterin, A. C. Ward, and W. B. Whitman. 2002. Report of thead hoc committee for the re-evaluation of the species definition in bacteri-ology. Int. J. Syst. Evol. Microbiol. 52:1043–1047.
50. Tannock, G. W. 2004. A special fondness for lactobacilli. Appl. Environ.Microbiol. 70:3189–3194.
51. Tatusov, R. L., D. A. Natale, I. V. Garkavtsev, T. A. Tatusova, U. T. Shanka-varam, B. S. Rao, B. Kiryutin, M. Y. Galperin, N. D. Fedorova, and E. V.Koonin. 2001. The COG database: new developments in phylogenetic clas-sification of proteins from complete genomes. Nucleic Acids Res. 29:22–28.
52. Teng, L. J., P. R. Hsueh, Y. H. Wang, H. M. Lin, K. T. Luh, and S. W. Ho.2001. Determination of Enterococcus faecalis groESL full-length sequenceand application for species identification. J. Clin. Microbiol. 39:3326–3331.
53. Tettelin, H., V. Masignani, M. J. Cieslewicz, C. Donati, D. Medini, N. L.Ward, S. V. Angiuoli, J. Crabtree, A. L. Jones, A. S. Durkin, R. T. Deboy,T. M. Davidsen, M. Mora, M. Scarselli, I. Ros, J. D. Peterson, C. R. Hauser,J. P. Sundaram, W. C. Nelson, R. Madupu, L. M. Brinkac, R. J. Dodson,M. J. Rosovitz, S. A. Sullivan, S. C. Daugherty, D. H. Haft, J. Selengut, M. L.Gwinn, L. Zhou, N. Zafar, H. Khouri, D. Radune, G. Dimitrov, K. Watkins,K. J. O’Connor, S. Smith, T. R. Utterback, O. White, C. E. Rubens, G.Grandi, L. C. Madoff, D. L. Kasper, J. L. Telford, M. R. Wessels, R.Rappuoli, and C. M. Fraser. 2005. Genome analysis of multiple pathogenicisolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. USA 102:13950–13955.
54. Vandamme, P., B. Pot, M. Gillis, P. de Vos, K. Kersters, and J. Swings. 1996.Polyphasic taxonomy, a consensus approach to bacterial systematics. Micro-biol. Rev. 60:407–438.
55. van de Guchte, M., S. Penaud, C. Grimaldi, V. Barbe, K. Bryson, P. Nicolas,C. Robert, S. Oztas, S. Mangenot, A. Couloux, V. Loux, R. Dervyn, R. Bossy,A. Bolotin, J. M. Batto, T. Walunas, J. F. Gibrat, P. Bessieres, J. Weissen-bach, S. D. Ehrlich, and E. Maguin. 2006. The complete genome sequenceof Lactobacillus bulgaricus reveals extensive and ongoing reductive evolution.Proc. Natl. Acad. Sci. USA 103:9274–9279.
56. van Kranenburg, R., I. C. Boels, M. Kleerebezem, and W. M. de Vos. 1999.Genetics and engineering of microbial exopolysaccharides for food: ap-proaches for the production of existing and novel polysaccharides. Curr.Opin. Biotechnol. 10:498–504.
57. van Kranenburg, R., H. R. Vos, I. I. van Swam, M. Kleerebezem, and W. M.de Vos. 1999. Functional analysis of glycosyltransferase genes from Lacto-coccus lactis and other gram-positive cocci: complementation, expression,and diversity. J. Bacteriol. 181:6347–6353.
58. Ventura, M., C. Canchaya, R. D. Pridmore, and H. Brussow. 2004. Theprophages of Lactobacillus johnsonii NCC 533: comparative genomics andtranscription analysis. Virology 320:229–242.
58a.Ventura, M., C. Canchaya, R. D. Pridmore, B. Berger, and H. Brussow. 2003.Integration and distribution of Lactobacillus johnsonii prophases. J. Bacte-riol. 185:4603–4608.
59. Ventura, M., and R. Zink. 2002. Specific identification and molecular typinganalysis of Lactobacillus johnsonii by using PCR-based methods and pulsed-field gel electrophoresis. FEMS Microbiol. Lett. 217:141–154.
60. Ventura, M., and R. Zink. 2003. Comparative sequence analysis of the tufand recA genes and restriction fragment length polymorphism of the internaltranscribed spacer region sequences supply additional tools for discriminat-ing Bifidobacterium lactis from Bifidobacterium animalis. Appl. Environ. Mi-crobiol. 69:7517–7522.
VOL. 189, 2007 SIMILARITY AND DIFFERENCES IN THE L. ACIDOPHILUS GROUP 1321