SOCIETA’ DI SCIENZE FARMACOLOGICHE APPLICATE SOCIETY FOR APPLIED PHARMACOLOGICAL SCIENCES numero 30 Poste Italiane s.p.a. - Spedizione in Abbonamento Postale - 70% - DCB PRATO SSFAoggi Notiziario di Medicina Farmaceutica Bimestrale della Società di Scienze Farmacologiche Applicate Aprile 2012 I master su Ricerca e Sviluppo dei farmaci La riflessione di oggi riguarda i master che sono disponibili in Italia. Per prima cosa, va ricordato che, anche se SSFA parla spesso dei master di Roma-Cattolica e di Milano- Bicocca, esistono anche altre offerte formative, attive in altre Università italiane. Ad esem- pio le Università di Milano-Statale, Roma-Sapienza, Pisa, Firenze, Catania e forse anche altre offrono la possibilità di seguire un master in Ricerca e Sviluppo dei farmaci. Un primo Università ha il diritto di definire il programma delle lezioni da svolgere. Ma sarebbe auspi- cabile che gli studenti avessero l’opportunità di ricevere un “curriculum di studi base” uni- forme, al quale poi aggiungere temi a discrezione di ogni direttore del corso. Per sondare la fattibilità di questa proposta, SSFA organizzerà un dibattito, con la collaborazione di SIF, nel corso della sessione pomeridiana del V corso di aggiornamento sui farmaci, in programma il prossimo 4 ottobre a Roma. Ma, veniamo ora ai due master di Roma-Cattolica e di Milano-Bicocca, che hanno un si- gnificativo supporto SSFA, e che partecipano al progetto Europeo di armonizzazione chia- mato “PharmaTrain”. Superata la fase iniziale di definizione del curriculum degli studi, negli ultimi mesi i 20 master Europei che aderiscono all’iniziativa sono stati oggetto di una visita di “ Quality Assurance”: una vera ispezione, svolta da tre valutatori, di cui parliamo nella pagina due. Come sempre succede, i tre valutatori hanno identificato alcune aree che necessitano di un miglioramento. I relativi suggerimenti sono stati inviati ai direttori dei due master, ed una seconda ispezione sarà pianificata a fine anno, per la verifica finale. Dopo questa seconda ispezione, i due master riceveranno il titolo di “centro di eccellenza” ed il valore del titolo conseguito avrà valenza Europea: un bel risultato! Fra i temi che i valutatori hanno sollevato, sia a Roma che a Milano, due meritano una riflessione: l’uso di test per la verifica dell’apprendimento, e l’uso della lingua inglese. Inte- ressante è il dibattito che si sta svolgendo fra gli studenti, in particolare quelli di Roma. Non tutti concordano sull’adozione di test di verifica (che peraltro a Milano si sono sempre fatti): la posizione di chi scrive è che un test di verifica debba essere fatto, o dopo ogni singola lezione, oppure ad intervalli regolari, oppure con una sintesi di entrambi i metodi. Ci sembra doveroso che l’Università, prima di concedere il titolo di “master”, ponga in es- sere una verifica di quanto ogni studente ha appreso. Non farlo potrebbe voler dire, per assurdo, che ha lo stesso valore il titolo di uno studente che ha seguito e preso appunti a tutte le lezioni, e quello di altro studente che ha sempre dormito sul banco! Il secondo tema riguarda l’inglese: PharmaTrain insiste perché i corsi siano svolti in inglese. I docenti (sia a Roma che a Milano) hanno sempre contestato questo punto di vista, facendo pre- sente che se il docente e tutti gli studenti sono italiani, fare lezioni in inglese è una forzatu- ra. Ed invece…proprio gli studenti sembrano apprezzare molto questa proposta. La cosa ci piace molto, perché siamo convinti che l’inglese vada coltivato giornalmente, anche acquisendo il lessico tipico del mondo dei farmaci. Alla luce di tali commenti, i master di Roma e di Milano certamente valuteranno con maggior attenzione l’utilizzo dell’inglese, che potrebbe rappresentare il primo passo verso classi internazionali di studenti. Domenico Criscuolo Sommario: I master in Italia 1 I master: visite QA 2 Il nuovo decreto CRO 3 Malattia di Alzheimer 4 Addio a Renato Dulbecco 4 10 Anni Oss.Sp.Cliniche 5 Guido Rasi at EMA 6 Tbc a Mumbai 7 PRO ed ePRO 8 Cellule staminali 9 New England J. of Medicine 10 Ipertensione 11 Notizie dall’Italia 12 Notizie dal Mondo 14 Opinioni a confronto 16 News on clinical trials 23 Nuovi Soci 24 I trapianti in Italia 22 ADR 21 IFAPP World 20 Tesi del Master 20 L’aborto clandestino 19 Seminari AB 19 Milano 4 maggio LA DEPRESSIONE IN GRAVIDANZA In collaborazione con SINPF Torino 17 - 18 maggio XXI CONGRESSO GIQAR La Qualità: la realtà di oggi e la sfida del domani Padova 24 - 25 maggio IV CONGRESSO BIAS Advanced methods in clinical trials: surrogate endpoints and adaptive designs Milano 25 giugno AGGIORNAMENTO SULLA MALATTIA DI PARKINSON In collaborazione con SINPF V CORSO DI AGGIORNAMENTO SUI FARMACI Roma, 4 ottobre obiettivo che SSFA intende proporre è l’armonizzazione di questi corsi: è chiaro che ogni
Transcript
SOCIETA’ DI SCIENZE FARMACOLOGICHE APPLICATE
SOCIETY FOR APPLIED PHARMACOLOGICAL SCIENCES
numero 30
Poste Italiane s.p.a. - Spedizione in Abbonamento Postale - 70% - DCB PRATO
SSFAoggi Notiziario di Medicina Farmaceutica
Bimestrale della Società di Scienze Farmacologiche Applicate Aprile 2012
I master su Ricerca e Sviluppo dei farmaci
La riflessione di oggi riguarda i master che sono disponibili in Italia. Per prima cosa, va ricordato che, anche se SSFA parla spesso dei master di Roma-Cattolica e di Milano-Bicocca, esistono anche altre offerte formative, attive in altre Università italiane. Ad esem-pio le Università di Milano-Statale, Roma-Sapienza, Pisa, Firenze, Catania e forse anche altre offrono la possibilità di seguire un master in Ricerca e Sviluppo dei farmaci. Un primo
Università ha il diritto di definire il programma delle lezioni da svolgere. Ma sarebbe auspi-cabile che gli studenti avessero l’opportunità di ricevere un “curriculum di studi base” uni-forme, al quale poi aggiungere temi a discrezione di ogni direttore del corso. Per sondare la fattibilità di questa proposta, SSFA organizzerà un dibattito, con la collaborazione di SIF, nel corso della sessione pomeridiana del V corso di aggiornamento sui farmaci, in programma il prossimo 4 ottobre a Roma. Ma, veniamo ora ai due master di Roma-Cattolica e di Milano-Bicocca, che hanno un si-gnificativo supporto SSFA, e che partecipano al progetto Europeo di armonizzazione chia-mato “PharmaTrain”. Superata la fase iniziale di definizione del curriculum degli studi, negli ultimi mesi i 20 master Europei che aderiscono all’iniziativa sono stati oggetto di una visita di “ Quality Assurance”: una vera ispezione, svolta da tre valutatori, di cui parliamo nella pagina due. Come sempre succede, i tre valutatori hanno identificato alcune aree che necessitano di un miglioramento. I relativi suggerimenti sono stati inviati ai direttori dei due master, ed una seconda ispezione sarà pianificata a fine anno, per la verifica finale. Dopo questa seconda ispezione, i due master riceveranno il titolo di “centro di eccellenza” ed il valore del titolo conseguito avrà valenza Europea: un bel risultato! Fra i temi che i valutatori hanno sollevato, sia a Roma che a Milano, due meritano una riflessione: l’uso di test per la verifica dell’apprendimento, e l’uso della lingua inglese. Inte-ressante è il dibattito che si sta svolgendo fra gli studenti, in particolare quelli di Roma. Non tutti concordano sull’adozione di test di verifica (che peraltro a Milano si sono sempre fatti): la posizione di chi scrive è che un test di verifica debba essere fatto, o dopo ogni singola lezione, oppure ad intervalli regolari, oppure con una sintesi di entrambi i metodi. Ci sembra doveroso che l’Università, prima di concedere il titolo di “master”, ponga in es-sere una verifica di quanto ogni studente ha appreso. Non farlo potrebbe voler dire, per assurdo, che ha lo stesso valore il titolo di uno studente che ha seguito e preso appunti a tutte le lezioni, e quello di altro studente che ha sempre dormito sul banco! Il secondo tema riguarda l’inglese: PharmaTrain insiste perché i corsi siano svolti in inglese. I docenti (sia a Roma che a Milano) hanno sempre contestato questo punto di vista, facendo pre-sente che se il docente e tutti gli studenti sono italiani, fare lezioni in inglese è una forzatu-ra. Ed invece…proprio gli studenti sembrano apprezzare molto questa proposta. La cosa ci piace molto, perché siamo convinti che l’inglese vada coltivato giornalmente, anche acquisendo il lessico tipico del mondo dei farmaci. Alla luce di tali commenti, i master di Roma e di Milano certamente valuteranno con maggior attenzione l’utilizzo dell’inglese, che potrebbe rappresentare il primo passo verso classi internazionali di studenti.
Domenico Criscuolo
Sommario:
I master in Italia 1
I master: visite QA 2
Il nuovo decreto CRO 3
Malattia di Alzheimer 4
Addio a Renato Dulbecco 4
10 Anni Oss.Sp.Cliniche 5
Guido Rasi at EMA 6
Tbc a Mumbai 7
PRO ed ePRO 8
Cellule staminali 9
New England J. of Medicine 10
Ipertensione 11
Notizie dall’Italia 12
Notizie dal Mondo 14
Opinioni a confronto 16
News on clinical trials 23
Nuovi Soci 24
I trapianti in Italia 22
ADR 21
IFAPP World 20
Tesi del Master 20
L’aborto clandestino 19
Seminari AB 19
Milano 4 maggio
LA DEPRESSIONE IN GRAVIDANZA
In collaborazione con
SINPF
Torino 17 - 18 maggio
XXI CONGRESSO GIQAR
La Qualità: la realtà di oggi e
la sfida del domani
Padova 24 - 25 maggio
IV CONGRESSO BIAS
Advanced methods in clinical trials:
surrogate endpoints and adaptive designs
Milano 25 giugno
AGGIORNAMENTO SULLA MALATTIA
DI PARKINSON
In collaborazione con SINPF
V CORSO DI AGGIORNAMENTO
SUI FARMACI Roma, 4 ottobre
obiettivo che SSFA intende proporre è l’armonizzazione di questi corsi: è chiaro che ogni
Anno VI numero 30 Pagina 2
Il master in Ricerca e Sviluppo Preclinico e Clinico dei Farmaci di Milano Bicocca partecipa al progetto Europeo IMI “PharmaTrain”, di cui vi ho già parlato. Nell’ambito del progetto, che prevede l’armonizzazione dei programmi dei corsi Europei, al fine di garantire che tutti gli specialisti Europei abbiano ricevuto lo stesso livello di formazione, è anche prevista un’ispezione di “Quality Assu-rance” svolta da tre esaminatori. Per il master di Milano gli esaminatori prescelti sono: Dominique Dubois (esperto in QA – Belgio), Thomas Szucs (esperto in master – Svizzera), Domenico Criscuolo (referente IFAPP – Italia). La data della visita era stata concordata molti mesi fa, e si è svolta regolarmente lo scorso 26 gennaio: tutta la mattina è
stata dedicata all’analisi dei documenti del master (SOP, valutazioni, elenco lezioni ed altro). Il pomeriggio è stato dedicato alla visita delle aule, ed alla riunione di fine ispezione. Il master era rappresentato dal Direttore, prof. Vittorio
Locatelli, aiutato da Antonio Tor-sello ed Elena Bresciani. SSFA era presente con Luciano Fuccel-la (docente e membro del comita-to scientifico) ed Anna Piccolboni (docente e QA del master). I risultati della visita sono stati molto positivi, come risulta dal verbale preparato dai tre esami-natori: come da procedura, ci sarà una seconda ispezione a fine anno. Dopo questa seconda ispezione, il master di Milano avrà
il titolo di “Centro di Eccellenza” nell’ambito del progetto Pharmatrain: un riconoscimento Europeo che sarà di grande aiuto a tutti gli studenti.
Domenico Criscuolo
I valutatori Pharmatrain visitano il Master Milano Bicocca
Lo scorso 24 febbraio, i valutatori desi-gnati dal Direttivo del Progetto IMI-PharmaTrain - il dr. Peter Stonier, mem-bro di British Associaton of Pharmaceuti-cal Physicians (UK) esperto di farmaco-vigilanza; il dr. Sandor Kerpel-Fronius, professore di farmacologia e farmacote-rapia alla Semmelweis University di Bu-dapest (Ungheria) e Domenico Criscuo-lo, referente IFAPP (Italia) - hanno effet-tuato una “site visit” presso la sede del Master dell’Università Cattolica di Roma. La visita aveva lo scopo di valutare la
documentazione a sostegno del Master stesso e le attività programmate e realiz-zate, nonché di acquisire una impressio-ne diretta degli ambienti in cui si svolge il Master e delle relative attrezzature didat-tiche. All’incontro hanno partecipato, oltre ai valutatori, il Direttore del Corso, prof. Pierluigi Navarra, Francesco De Tomasi del Consiglio Direttivo, Mercede Brunetti di RTC, come responsabile di Qualità e la dr.ssa Lucia Lisi, della Se-greteria Scientifica. La visita si è svolta in un sereno clima di collaborazione;
numerosi sono stati gli approfon-dimenti richiesti dai valutatori, che hanno sempre ricevuto ri-sposte adeguate a giustificare alcune differenze che il Master di Roma presenta rispetto allo schema generale impostato dal Progetto IMI. Di particolare rilievo è stata la discussione circa l’attività di formazione ‘sul campo’ fornita agli studenti trami-
te gli stages, un’esperienza – a detta dei colleghi stranieri - molto importante, e suscettibile di rappresentare un modello didattico esportabile anche ad altre real-tà. Alla usuale riunione al termine dell’incontro, i valutatori hanno fornito una prima sintesi delle loro impressioni. Un resoconto ufficiale è atteso entro un mese dalla visita: il documento definirà la situazione attuale rispetto al modello europeo e le eventuali azioni migliorative da implementare. Non sono mancati i complimenti per il lavoro svolto finora e per i risultati raggiunti in termini di eleva-to numero di domande di partecipazione, di alte percentuali di frequenza e di va-rietà ed alta qualificazione dei docenti provenienti dalle diverse aree di interes-se del Corso: Accademia, Istituzioni Pubbliche, Industrie Farmaceutiche, CRO. E’ stata già prevista una seconda visita, da svolgersi nel prossimo mese di ottobre, al fine di verificare il processo di adeguamento del Master di Roma agli standard europei: di questo vi aggiorne-remo puntualmente.
Francesco De Tomasi
I Responsabili del Master di Roma hanno accolto i Valutatori Europei
Ultime notizie dal master di Milano
Lo scorso 17 febbraio è scaduto il termine per la presentazione delle domande di iscrizione alla quarta edizione del master, che si svolgerà nel 2012. La grande notizia è che alla segreteria sono pervenute ben 100 domande di iscrizione, un vero record! Nei prossimi giorni avranno luogo le selezioni dei trenta studenti che avranno diritto all’iscrizione: la selezione sarà basata sui titoli, sulla conoscenza della lingua inglese, e su un colloquio con i candidati.
Anno VI numero 30 Pagina 3
E' stato pubblicato sulla GU del 14 gen-naio scorso il nuovo decreto sui requisiti minimi delle CRO, che sostituisce il DM del 31 marzo 2008. Il nuovo decreto è entrato in vigore lo scorso 29 gennaio 2012. Come certamente ricorderete, SSFA aveva sollecitato più volte AIFA con incontri e seminari a rivedere alcuni articoli del vecchio decreto; inoltre il Consiglio di Stato ha accolto il ricorso straordinario presentato dal Sig. Vittorio Carboni al Presidente della Repubblica per l'annullamento, previa sospensiva, del medesimo decreto. Esaminiamo ora quali sono stati i principali cambiamenti: - Articolo 1: le CRO non sono più soltan-to le organizzazioni private ma anche qualsiasi altro organismo cui il promoto-re della sperimentazione abbia affidato una parte delle proprie competenze, quindi anche le strutture pubbliche che svolgano attività di monitoraggio, auditing o data management e statisti-ca. Questo è un aspetto importante in quanto annulla possibili disparità di trattamento tra CRO private e pubbli-che. - Articolo 2: sono state introdotte le definizioni di monitor esperto e di audi-tor esperto che mancavano nel prece-dente decreto. - Articolo 3: è stato specificato che soltanto il personale che svolge attività di carattere tecnico-scientifico e di as-sicurazione controllo di qualità dovrà fare almeno 30 ore (e non più 10 giorni) di aggiornamento annuale nelle temati-che relative alle funzioni ad esso attribui-te. Il personale amministrativo, finanzia-rio, delle risorse umane e dei servizi generali è esonerato da tale aggiorna-mento. - Articolo 4: i 10 giorni di formazione teorica effettuata nei 12 mesi precedenti l'inizio dell'attività di monitoraggio sono stati ridotti a 40 ore; i 30 giorni di attività di monitoraggio in affiancamento a moni-tor esperti sono stati ridotti a 20 giorni, mentre i 6 mesi di attività nei 12 mesi precedenti l'inizio dell'attività autonoma di monitor sono stati ridotti a 4 mesi. Quest'ultimo comma 1 lettera d dell'arti-colo 4 è stato rivisto anche per quanto riguarda in alternativa gli ulteriori giorni
di attività di monitoraggio in affianca-mento che passano da 60 a 40 giorni. E' stato poi completamente modificato il comma 2, oggetto del ricorso straordina-rio che aveva portato all'annullamento del precedente decreto: mi riferisco al carattere retroattivo del precedente de-creto che è stato riconosciuto inammissi-bile dal Consiglio di Stato in quanto “la legge non dispone che per l’avvenire: essa non ha effetto retroattivo”. Sono stati infine aggiunti nel comma 3 dell'articolo 4 i punti 5 e 6 che fanno riferimento al monitor esperto che an-nualmente esegue almeno 15 giorni di visite di monitoraggio e all' "interruzione giustificata" dell'attività che non è di o-stacolo alla ripresa della stessa e non comporta la perdita della qualifica. Si tratta di una modifica importante in quanto prevede che "l'interruzione giusti-
ficata" cioè dovuta a cause di forza mag-giore, come una malattia, un incidente o l'assenza per maternità, non comporta la perdita della qualifica di monitor o di monitor esperto. - Articolo 5: i 10 giorni di formazione teorica nei 12 mesi precedenti l'inizio delle attività di auditing sono stati ridotti a 60 ore; i 30 giorni di attività di auditing in affiancamento ad auditor esperti sono stati ridotti a 20 giorni mentre i 6 mesi di attività nei 12 mesi precedenti l'inizio dell'attività autonoma di auditor sono stati ridotti a 4 mesi. Infine, il comma 2 dell'articolo 5 è stato completamente modificato come il comma 2 dell'articolo 4 che era stato appunto l'oggetto del ricorso straordinario, e che aveva porta-to all'annullamento del precedente de-
creto. Sono stati poi aggiunti gli articoli 5, 6 e 7 che fanno riferimento all'auditor esperto ed all' "interruzione giustificata" dell'attività, che non è di ostacolo alla ripresa della stessa e non comporta la perdita della qualifica. - Articolo 6: nessuna modifica. - Articolo 7: viene inserito il comma 8 dove si afferma che "le CRO, le aziende farmaceutiche ed i promotori di speri-mentazioni cliniche devono rilasciare ai dipendenti e/o ai collaboratori aventi diritto la documentazione attestante le attività eseguite inerenti al presente de-creto": anche questo è un punto molto importante, che aveva sollevato parec-chie critiche dopo la pubblicazione del primo decreto, in quanto molti dipendenti e collaboratori esterni avevano trovato difficoltà nel farsi rilasciare gli attestati delle attività svolte in precedenza.
- E' stato infine rimosso il precedente Articolo 9 riguardo "gli effetti sull'au-torizzazione all'immissione in com-mercio", che rappresenta sicuramen-te un importante successo per le aziende farmaceutiche ed i promotori più in generale. Nel complesso, vi sono stati migliora-menti rispetto al precedente decreto soprattutto nella direzione di favorire l’ingresso dei giovani nel mondo del lavoro, poiché si sono ridotti i costi per la formazione e in tal modo molte CRO potranno inserire più facilmente
neolaureati nelle proprie strutture. Restano comunque alcuni aspetti poco chiari: per esempio rimane la disparità di formazione tra i monitor/auditor delle CRO e quelli delle aziende farmaceuti-che, in quanto non esiste alcun obbligo di formazione per chi opera in quest’ultime. Come spesso accade quando si cerca di correggere qualcosa che è nato male, si fa fatica. A mio parere - ma parlo a titolo personale – sarebbe stato meglio non scrivere neppure il primo decreto CRO: si sarebbero evitati molti problemi sorti successivamente. Ma ormai è troppo tardi.
Marco Romano
Il nuovo decreto sui requisiti minimi delle CRO
Anno VI numero 30 Pagina 4
Nella prestigiosa sede dell’auditorio Ro-che (che ringraziamo per l’ospitalità), si è svolto il primo seminario di aggiorna-mento in collaborazione fra SSFA e SINPF (Società Italiana di Neuropsico-farmacologia). La SINPF condivide con SSFA, SIF e SITOX la sede di viale A-bruzzi, quindi ci è sembrato naturale proporre al suo Presidente, prof. Gio-vanni Biggio (Cagliari), un seminario scientifico in collaborazione: la proposta è stata subito accettata con entusiasmo. Il formato è stato il solito: seminario po-meridiano con quattro relazioni molto focalizzate sull’obiettivo: quello di offrire un aggiornamento sulla Malattia di Al-zheimer (MA), dal punto di vista della ricerca di base ed applicata. Dopo l’introduzione, in cui il sottoscritto ha presentato ai soci SINPF la SSFA, ed il prof. Giovanni Biggio ha presentato la sua società (circa 1000 soci, distribuiti in tutta Italia ed attivi sia in ricerca di base che ricerca applicata), la prof.ssa Moni-ca Di Luca (Milano) ha fatto una iniziale panoramica sulla patologia, ricordando che in Europa, anche a causa dell’in-vecchiamento della popolazione, le pa-tologie del SNC stanno aumentando ad un ritmo di oltre il 5% ogni anno. In parti-colare, la MA è oggi oggetto di molte
ricerche, orientate alla scoperta delle basi biologiche della malattia. Un appro-fondimento sulle linee di ricerca è stato offerto dal secondo relatore, il prof. San-dro Sorbi (Firenze). Dopo la pausa caffè, siamo entrati nel vivo della ricerca appli-cata: il prof Giuseppe Magnani (Milano) ci ha aggiornato sui più recenti orienta-menti in tema di diagnosi e terapia, men-tre la collega Grazia Dell’Agnello (Eli Lilly) ci ha raccontato le sue esperienze sugli studi clinici svolti con una molecola dell’azienda per la quale lavora. Si è trattato di un bel seminario, focaliz-zato su un tema scientifico e molto spe-cifico: oltre 80 partecipanti hanno confer-
mato il valore dell’aggiornamento propo-sto, come è stato anche documentato dall’analisi dei questionari sul gradimen-to dell’evento. Tutti si sono espressi con voti compresi fra buono ed ottimo. Nel commentare i risultati di questo se-
è detto molto soddisfatto, a nome suo personale e dei soci SINPF presenti: per cui siamo già al lavoro per programmare altri due seminari, che si svolgeranno prima della pausa estiva.
Domenico Criscuolo
La malattia di Alzheimer
Addio a Renato Dulbecco il Nobel che ha cambiato la lotta ai tumori
Il pioniere delle ricerche sulla genetica del cancro, ma anche lo scienziato gentiluomo, schierato in prima fila nelle battaglie a favore della ricerca sulle cellule staminali e per reintrodurre l'evoluzionismo nei libri scolastici. È Rena-to Dulbecco, premio Nobel della medicina nel 1975, scomparso alla soglia
a qualche mese fa le sue condizioni di salute erano buone ma nell'ultimo periodo aveva accusato alcuni problemi circolatori. Nato a Catanzaro il 22
febbraio 1914, Dulbecco si avvicina alla scienza spinto dalla passione per la fisica e arriva alla medicina dopo avere "assaporato" anche chimica e matematica. A 16 anni si iscrive alla facoltà di Medicina dell'università di Torino e segue i corsi dell'anatomista Giuseppe Levi insieme a Rita Levi Montalcini e Salvador Luria. Si laurea con lode nel 1934. Durante la seconda guerra mondiale è ufficiale medico sul fronte francese e poi su quello russo dove, nel 1942, rischia di morire. Rientrato in Italia, nel dopoguerra torna a Torino. Nel 1947 la grande decisione di trasferirsi negli Stati Uniti per raggiungere Luria, che lavorava lì già dal 1940. Un viaggio che cominciò con una sorpresa: «senza saperlo, ci ritrovammo sulla stessa nave», raccontava mezzo secolo più tardi ancora divertito, ripensando all'incontro inatteso con Rita Levi Montalcini. Dulbecco approda nel California Institute of Technology (CalTech), dove ha una cattedra e comincia a occuparsi di tumori. Nel 1960 fa la scoperta che nel 1975 lo porterà al Nobel: os-serva che i tumori sono indotti da una famiglia di virus che in seguito chiamerà oncogeni. Nel 1972 lascia gli USA per Londra, come vicedirettore dell' Imperial Cancer Research Fund. Dopo il Nobel, condiviso con David Baltimore e Howard Temin, ritorna all'Istituto Salk per studiare i meccanismi genetici responsabili di alcuni tumori, in primo luogo quello del seno. Il suo rientro in Italia, nel 1987, coincide con l'avvio del Progetto internazionale Genoma Umano, del quale Dulbecco diventa coordinatore del ramo italiano. Un'esperienza che si arena nel 1995 per mancanza di fondi e che lo riporta negli Stati Uniti.
Prof.ssa Monica Di Luca
Prof. Giovanni Biggio
dei 98 anni. Dulbecco è morto in California dove viveva con sua moglie. Fino
minario, anche il prof. Giovanni Biggio si
Anno VI numero 30 Pagina 5
Nella cornice dell’ultramoderna sede del Ministero della Salute, AIFA ha celebra-to i 10 anni dell’Osservatorio della Speri-mentazione Clinica. Si è trattato di un incontro “operativo” dove, a parte l’espressione della soddisfazione per lo sviluppo ed i risultati ottenuti, gli organiz-zatori hanno voluto cogliere l’occasione per fare il punto della situazione genera-le della ricerca, con particolare attenzio-ne allo sviluppo delle attività correlate all’Osservatorio. Quest’ impostazione è stata molto gradita da un pubblico di esperti dell’industria farmaceutica, delle CRO e delle Istituzioni che sono accorsi numerosi. Nel suo atteso intervento di apertura il prof. Pani ha sottolineato co-me AIFA consideri di importanza strate-gica aumentare il numero di studi di fasi precoci in Italia, in considerazione dell’alto valore aggiunto di queste attività ed in un contesto che vede la forte con-correnza di Paesi emergenti (la cosid-detta area BRIC). Sempre al centro dell’attenzione di AIFA il problema di un’adeguata valutazione dell’innovatività dei trattamenti. Infine, un moderno e trasparente sistema di valutazione del conflitto di interessi pone la nostra agen-zia all’avanguardia in Europa sotto que-sto punto di vista. Il dott. Tomino ha poi
esaminato i punti forti e le criticità del sistema-ricerca in Italia: tra i primi indub-biamente la competenza clinica, mentre tra le criticità la complessità amministra-tiva e le reti di eccellenza non ancora stabilizzate. Il dott. Galluccio ha ripercor-so le tappe dello sviluppo dello strumen-to osservatorio e tracciato le linee di sviluppo dello stesso, su questa linea seguito dall’intervento della dr.ssa Aita. Dagli interventi emerge un percorso con-tinuo di miglioramento con l’obiettivo di implementare una serie di funzioni che facciano dell’Osservatorio uno strumen-to attraverso il quale gestire le procedu-re di autorizzazione e di aggiornamento di uno studio. Questo anche alla luce di prossime normative europee sia in mate-ria di studi clinici, con un unico punto di accesso europeo per la domanda di autorizzazione della sperimentazione clinica, che in materia di farmacovigilan-za con la nuova normativa prossima ad entrare in vigore. Terminata la sessione più specificamente dedicata alle presen-tazioni AIFA, è seguita una sessione di presentazione di altri attori cruciali per la ricerca, iniziando dall’Istituto Superiore di Sanità, con un interessante intervento della dr.ssa Meneguz, che sottolinea il ruolo dell’Istituto come controllo e guida
delle sperimentazioni in fase precoce nel nostro Paese, puntualizzando anche gli alti standard europei ed italiani in termini di tutela dei volontari nella fase “first in man”. Il dott. Agostini di Farmindustria, nel portare il saluto dell’organizzazione, ha sottolineato come una solida collabo-razione tra Istituzioni e aziende, nel ri-spetto dei diversi ruoli e competenze, sia la base per una competitività del sistema Italia nel contesto della ricerca clinica internazionale. Le esperienze della re-gione Lombardia, del GIMEMA ed il pun-to di vista di AICRO completano l’insieme delle presentazioni che sono state seguite da un dibattito vivace. Semplificazione e razionalizzazione del-la normativa; rapidità di risposta delle amministrazioni soprattutto per quel che riguarda le Autorità Competenti ed un equilibrio tra le esigenze di tutela dei pazienti e la necessità di guadagnare competitività sono state le istanze più spesso emerse nel corso della discus-sione. Solo il termine del tempo asse-gnato alla sala interrompe un dibattito vivace, indice dell’interesse dei parteci-panti.
Salvatore Bianco
I 10 anni dell’Osservatorio delle Sperimentazioni Cliniche
Anno VI numero 30 Pagina 6
Some sly smiles flickered across the lips of the veterans of European pharmaceu-tical affairs last year, when it started to look as if an Italian was going to be put in charge of the European Medicines Agency. Those with long memories nudged one another knowingly, in com-plicit acknowledgement that the last Ital-ian in such a distinguished role wound up in prison after conviction of massive and systematic corruption. Guido Rasi, who took over as executive director of the EMA last November, has many chal-lenges ahead of him. And one of them is escaping from under the shadow cast by Duilio Poggiolini. As they like to say in serialized romantic fiction, new (or in this case, young!) readers start here. Poggi-olini was, like Rasi, head of the Italian medicines agency. Like Rasi, he was a distinguished physician and researcher. Like Rasi, he was a professor of microbi-ology. And like Rasi, he was promoted to European stardom, as chairman of the EU's top body for drug authorizations (which, back in 1991, before the EMA existed, was the Committee for Proprie-tary Medicinal Products — now reborn as the CHMP within the EMA).Unlike Rasi, Poggiolini was given a heavy jail sentence in 2000 for abusing his posi-tion and taking bribes in return for fa-vourable decisions on pricing and au-thorization. Poggiolini's misdeeds, un-covered during a "mani pulite" — clean hands — investigation into an influential Masonic lodge, were on an epic scale. He had amassed cash, jewellery, gold and works of art estimated at more than $150 million, secreted not just in Swiss bank accounts but even hidden in the sofas in his house - including gold rou-bles from the reign of Czar Nicolas II and South African Krugerrands. It took investigators twelve hours just to cata-logue the spoils secreted around the house. As if that were not enough of a record, Poggiolini was subsequently charged with delinquent management of Italy's blood transfusion services, and responsibility for thousands of cases of hepatitis C and HIV from contaminated blood. Unlike Poggiolini, Rasi already
has a profile for "mani pulite". Rasi was appointed head of the Italian health agency in 2008 as a new broom after his predecessor, Nello Martini, was un-seated in the course of an investigation into trafficking of influence. Rasi had been an advisor to the Italian agency for four years by then, but he had a reputa-tion as an honest dealer — vitally impor-tant as Martini was facing trial on charges related to agency failures that allegedly put patient safety at risk. Under its new command, the agency recovered some of the credibility it had lost over yet another incident calling into question the probity of Italian officials. Rasi oversaw a restructuring of the agency designed to make it more efficient and improve drug registration times. Only three years later, Rasi finds himself in a similar role at European level. The reputation of the once-glistening European Medicines Agency had become heavily tarnished during 2011 by accusations of conflict of interest and mismanagement. Thomas Lonngren, its executive director, pro-voked sharp criticism when he quit his job to walk straight into consultancy work in the pharmaceutical industry. At the same time, charges were flying wildly of agency incompetence and even complicity in leaving unchallenged, for years, Servier's Mediator, by then linked to hundreds of deaths in France. And the European Parliament refused to sign off on the agency's accounts because of concerns over insufficient financial and personnel controls. It was onto this un-promising stage that Rasi stepped last November, after the agency had drifted for nearly a year without an executive director. Small wonder that malicious chuckles ran round Brussels along the lines of "and after all that, they could only get an Italian!" Rasi's declaration of interests is unequivocal. He says he has no financial interest in any pharmaceuti-cal company, owns no product patents, and that he has no personal gain from any grant or other funding from a phar-maceutical company to any organization he is employed by. So mani as pulite as they could be. But Poggiolini is only one
of the shadows over the EMA. Now there are other battles of a more techni-cal nature for the London-based agency and its new boss. Rasi has shown some light thinking about squaring some of the circles he is confronted with, and doing so with what are widely recognized as insufficient resources for a growing workload and ever more-sensitive decision-making. He has publicly admitted there are prob-lems, and has publicly promised "not to hide from them". One dilemma arises from implementation of new EU pharma-covigilance rules due to come into effect in July, for which large amounts of drug information from companies is supposed to be entered in advance into the agency's EudraVigilance safety data-base. Rasi has spoken of this as his "biggest headache", because of the ad-ministrative implications in handling complex information on more than half a million products. Pragmatically, the EMA reached a compromise agreement in February with companies to smooth the process, after industry warnings that the requirements were unworkable. Rasi himself will lead from the front in tackling the problem, heading a task force set up specifically to oversee the implementa-tion of the legislation. On the thorny is-sue of biosimilars, Rasi has shown simi-lar agility. Europe's generic medicines industry is now hopeful of his backing for changes that could cut the cost of devel-oping biosimilars by harmonising Euro-pean and US regulation and avoiding the need for duplicative approval proce-dures. He is planning to create a joint US- EU biosimilar conference to simplify the work while maintaining standards. He has also promised guidelines on the harmonization of regulatory procedures for biomolecules, to increase the use of biosimilars. He has also indicated ambi-tions for the EMA to accept a broader responsibility to patients and industry, breaking a taboo that limits its role to merely showing a red or green light to marketing authorization applications. "I
(Continua a pagina 7)
The Italian's Job — Guido Rasi's EMA Challenge da Pharmaceutical Executive
Anno VI numero 30 Pagina 7
see the next phase of the agency's de-velopment to be focused on re-thinking how medicines contribute more broadly to public health, how access to medi-cines can be assured in a world of in-creasing scarcity, and a growing focus on safety," he said on his first day in office. His vision is of an agency that can contribute to ensuring access to medi-cines by taking some account of what happens to products — on pricing and reimbursement — after authorization. He has spoken of making the agency's work so credible and scientifically authorita-tive that its thinking is not challenged. He has also indicated concerns over combating falsified medicines, the chal-
lenges of new approaches to medicines development, the lack of harmonization of clinical trials rules across member states, and the need to adapt to the new demands of assessing personalized medicines on the basis of pharmacoge-netic criteria. He is alert to what he sees as the "disparity" between the risk/benefit analysis the EMA does and the emerging disciplines of health technol-ogy assessment, and he is conscious of the still-to-be-faced challenges of regu-lating medicines derived from nanotech-nology or using new routes and methods of administration. His battles will be many – but at least he comes to it with a spotless record and evident energy. So maybe Rasi should not be compared so much with Poggiolini as with some of his
compatriots currently making the running in Europe — such as Mario Draghi, who is competently piloting the European Central Bank through the turbulent wa-ters of the euro crisis, or Mario Monti, the former European Commissioner who is now trying to salvage Italy from the near-disaster of Berlusconi's misrule. And if Rasi ever gets round to writing his book, it will suffer a less ignominious fate than Poggiolini's 1989 "Practical guide for applications for marketing au-thorizations on medicinal specialities in the EEC", which is long since — and unsurprisingly — pulped.
(Continua da pagina 6)
L’India è la nazione con il numero di casi di tubercolosi più alto al mondo, e conta circa 1000 morti al giorno. E nelle ultime settimane è arrivato a Mumbai un foco-laio del tipo TDR-TB, inattaccabile dai farmaci noti, isolato nei fluidi corporei di dodici pazienti. L’acronimo TDR sta per “totally drug resistan”, ovvero totalmente resistente ai farmaci e dunque non solo ai due medicinali di prima linea (rifampicina e isoniazide), ma anche quelli di seconda linea e quindi total-mente incurabile. Attualmente, come spiega Zarir Udwadia dello Hinduja Na-tional Hospital and Medical Research Centre di Mumbai, dei dodici pazienti confermati tre sono già morti. Un pazien-te ha già contagiato la propria figlia e si calcola che la velocità di contagio sia di circa dieci-venti persone all’anno. Già la tubercolosi estensivamente resistente ai farmaci (XDR-TB) è un problema signifi-cativo, soprattutto in paesi con bassi standard sanitari, anche perché nel caso del batterio tubercolare la resistenza ai farmaci usati in prima istanza è molto più grave, poiché in questi casi si ricorre a farmaci di seconda linea, molto tossici, molto costosi, poco efficaci e in alcuni casi introvabili. E’ chiaro dunque che la resistenza totale rappresenta una mi-
naccia notevole ed è un po’ come torna-re all’era pre-antibiotici. La resistenza totale era stata rilevata in passato solo in alcuni malati di prigioni siberiane e successivamente, come spiega Paul Nunn, coordinatore del dipartimento STOP TBC dell’OMS, erano comparsi due casi totalmente resistenti in Italia nel 2007 e quindici in Iran nel 2009. Il batte-rio che causa questa patologia, Myco-bacterium tuberculosis, venne identifica-to e descritto nel 1882 da Robert Koch. Detta anche mal sottile, la malattia ri-chiama un’atmosfera quasi ottocentesca e la letteratura pullula di eroi o eroine che morirono per questa malattia (basta pensare alla Boheme per esempio). In realtà si tratta di una patologia antichis-sima: resti scheletrici mostrano che uo-mini preistorici avevano la tubercolosi già nel 4000 AC, e tracce di decadimen-to dovuto alla TB sono state ritrovate nella spina dorsale di alcu-ne mummie del 3000-2400 AC. Ma l’aspetto inquietan-te del problema è la possi-bilità che si riaffacci l’impossibilità di curare una malattia che rimane ancora oggi molto diffusa. Secondo le stime due miliardi di per-
sone, cioè un terzo della popolazione mondiale, sono stati esposti all’agente patogeno della tubercolosi, mentre an-nualmente 8 milioni di persone si amma-lano e 2 milioni muoiono in tutto il mon-do. L’inesistenza di un antibiotico risolu-tivo avrebbe risvolti drammatici, soprat-tutto in un paese come l’India, dove lo stato del sistema sanitario e le piaghe sociali aggraverebbero il problema. Uno studio condotto a Mumbai ha mostrato recentemente come solo 5 medici priva-ti, su un campione di 106, sono stati in grado di prescrivere la terapia corretta a un ipotetico paziente affetto da MDR-TB (ovvero da tubercolosi multi resistente), con il rischio oltretutto di amplificare ulteriormente la resistenza dei batteri. Solo l’1 per cento dei pazienti con MDR-TB ha infatti accesso diretto al tratta-mento pubblico e la maggior parte dei malati si rivolge ai medici del settore privato che, nel paese asiatico, è privo di una regolamentazione che ne garanti-sca l’affidabilità e la professionalità.
A cura di Domenico Criscuolo
A Mumbai una nuova forma di tubercolosi resistente a tutti i farmaci
IMPORTANTE! Uno studio reso noto dall'OMS ha evidenziato nuovi livelli record di tubercolosi farmaco-resistente. Sono stati segnalati da 80 Paesi, per il peri-odo 2007-2010, quasi il 30% di tutti i nuovi casi dia-gnosticati, afferma un articolo pubblicato sull'ultimo Bollettino OMS. Alti tassi di resistenza ai farmaci sono stati riportati in Bielorussia, Estonia, Federazione Rus-sa e Tagikistan, con punte di casi di MDR-TB in oltre il 65% dei pazienti precedentemente trattati per la TB.
Anno VI numero 30 Pagina 8
Il comitato BIAS ha inaugurato le attività del 2012 lo scorso 16 gennaio, con una giornata di approfondimento sul tema dei PRO (Patient Reported Outcomes), terreno di continuo confronto tra i vari operatori del mondo della ricerca clinica. Gli interventi hanno abbracciato diverse aree nell’intento di dare voce ai vari pun-ti di vista. Uno scambio paritetico tra relatori e partecipanti per porre il proprio sguardo da prospettive nuove ed inu-suali. Capire domande, richieste, requisi-ti e problematiche di coloro con cui spesso si collabora ma con cui si ha raramente l’occasione di sedersi con calma per un confronto costruttivo. Il convegno è stato ospitato nella sede SAS di Milano. Il prof. Niero (Verona) ha dato il via ai lavori introducendo il tema della giornata per poi affrontare gli a-spetti quali/quantitativi della creazione di un questionario. Le relazioni sono prose-guite con L. Bertù (Università dell'Insu-bria), M. Torciani (LifeBee), V. Arnera (PHT Corporation), S.Piccoli (Arithmos), E. Carzana (Chiesi) e G. Reggiardo (Medi Service). La discussione ha tocca-to aspetti tecnici, operativi, metodologici e regolatori, riscuotendo un alto gradi-mento presso i partecipanti, accorsi in
buon numero anche in que-sta occasio-ne. Comporre un’agenda che sia capa-ce di toccare i molteplici aspetti di un tema così complesso ha, tra gli altri, anche il pregio di ren-dere sempre più visibile la nostra asso-ciazione e farla conoscere a chi in passato non ha avuto modo di interagirvi. Il comitato BIAS desidera ringraziare i relatori per la disponibilità dimostrata e per la loro chiarezza espositiva, SSFA per il consu-eto ed impagabile supporto amministrativo, SAS per l’ospitalità e la collaborazione e tutti i partecipanti per l’interesse dimostrato. Il prossimo ap-puntamento è il IV Congresso BIAS che si terrà presso l’Università di Padova il 24 e 25 maggio. Come da tradizione, il
convegno si svolgerà su due piani paral-leli. La sezione di statistica verterà sul tema degli “Advanced methods in clini-cal trials: surrogate endpoints and adap-tive designs”, mentre i colleghi data manager avranno l’occasione di confron-tarsi sul tema “Data Quality in Clinical Trials”. Arrivederci a Padova!
Marco Costantini
PRO ed ePRO negli studi clinici
Anno VI numero 30 Pagina 9
Cellule staminali ottenute dalla polpa dentale (CSPD) sono in grado di pro-muovere la rigenerazione funzionale del midollo spinale (MS) di ratto lesionato sperimentalmente. Un gruppo di ricerca-tori della Facoltà di Medicina dell’Università di Nagoya (Giappone) ha dimostrato che cellule staminali, isolate dalla polpa dentale e trapiantate intrale-sionalmente, sono in grado di stimolare, in ratti con MS completamente troncato trasversalmente, la rigenerazione dura-tura dei nervi del MS. Queste cellule, oltre a promuovere la rigenerazione de-gli assoni sezionati inibendo direttamen-te gli inibitori della crescita assonale, prevengono i danni indotti dall’apoptosi di neuroni, astrociti ed oligodendrociti e si differenziano in oligodendrociti maturi, rimpiazzando le cellule perdute. L’inizio del processo rigenerativo è rilevabile già durante la fase acuta della lesione e permette, a ratti con il MS lesionato, di coordinare i movimenti delle articolazioni degli arti posteriori e di camminare, sen-za supporto fisico, già entro la quinta settimana. Risultati sperimentali hanno dimostrato che il trapianto di cellule sta-minali può promuovere vari gradi di re-cupero funzionale del MS lesionato, ma questi tipi di cellule hanno mostrato scar-sa capacità di sopravvivenza e/o di diffe-renziamento in vitro. Dalla polpa dentale di soggetti umani adulti e da denti da latte caduti spontaneamente si possono ottenere facilmente cellule staminali mul-tipotenti, altamente proliferative, clono-geniche e capaci di differenziarsi in una varietà di tipi cellulari, dalle quali si pos-sono isolare popolazioni che esprimono marker di cellule staminali mesenchimali e neuroectodermali. Come le cellule adulte del MS, anche queste cellule, in ben definite condizioni di coltivazione in vitro, possono essere indirizzate a diffe-renziarsi in osteoblasti, condrociti, adipo-citi, cellule endoteliali e neuroni funzio-nalmente attivi. Per valutare il potenziale neurorigenerativo delle cellule staminali ottenute dalla polpa dentale di denti a-dulti (CSPDA) e di denti da latte (CSPDL) nella rigenerazione assonale in fase precoce della lesione trasversale
del MS, è stata fatta una serie di esperi-menti nei quali queste cellule sono state trapiantate intralesionalmente nel MS di ratto. Analisi di citometria a flusso e di immunoistochimica hanno indicato che questi due tipi di cellule staminali espri-mono marker di cellule staminali mesen-chimali, ma non marker di cellule endoteliali/ematopoietiche. Inoltre, la maggioranza delle cellule staminali dei due tipi ha co-espresso anche vari marker di linee cellulari neurali. Analisi PCR hanno confermato che ambedue i tipi cellulari esprimono livelli di GDNF (glial cell line-neurotrophic factor), BDNF (brain-derived neurotrophic factor) e CTNF (ciliary neurotrophic factor) da tre a cinque volte maggiori di quelli espressi da fibroblasti del midollo osseo derivati dalla cute. E’ rilevante che, quando CSPDA e CSPDL sono state trapiantate nella lesione midollare di ratti il cui MS era stato completamente troncato tra-sversalmente, questi animali hanno mo-strato un recupero dell’attività locomoto-ria maggiore di quella ottenuta in animali trapiantati con cellule stromali di midollo osseo adulto o con fibroblasti. Ancor più incoraggiante è l’osservazione che un recupero più completo è stato ottenuto quando il trapianto è effettuato immedia-tamente dopo l’operazione chirurgica, durante la fase acuta della lesione al MS. Cinque settimane dopo l’induzione della lesione nervosa, i ratti trapiantati con CSPDL erano in grado di coordinare i movimenti delle tre articolazioni delle zampe posteriori e di camminare senza supporto fisico. Sebbene il grado di re-cupero fosse simile nei ratti trapiantati con CSPDA o con CSPDL, solo su que-sti ultimi l’osservazione è stata protratta per vedere come le cellule trapiantate avevano promosso la rigenerazione del MS. Analisi immunoistochimiche hanno dimostrato che, otto settimane dopo l’induzione della lesione e del trapianto di CSPDL, la preservazione di assoni neurofilamenti-positivi nel MS dei ratti trapiantati era significativamente mag-giore che in quelli di controllo. Nei ratti trapiantati, ma non in quelli di controllo, gli assoni del tratto corticospinale e quel-
li serotonergici si sono estesi caudal-mente fino a 3 mm dalla lesione. Inoltre, bottoni sinaptici di alcuni di questi assoni erano visibili, giustapposti ai neuroni nella radice caudale, e ciò fa supporre che gli assoni rigenerati abbiano ristabili-to nuove connessioni neuronali. Succes-sive analisi hanno dimostrato che le CSPDL sono anche in grado di inibire direttamente molti segnali inibitori della crescita assonale generati dagli oligo-dendrociti e dagli astrociti che si trovano nel tessuto cicatriziale della glia. In un gruppo separato di esperimenti si è di-mostrato che le CSPDL promuovono, nella zona lesionata trasversalmente del MS, la rigenerazione di strutture mielini-che. Ad otto settimane dal trapianto, circa il 30% di queste cellule erano an-cora vive nel MS e in gran parte espri-mevano due marker tipici degli oligoden-drociti maturi, ma non più marker di cel-lule staminali o di astrociti, indicando che esse si erano differenziate lungo la linea degli oligodendrociti nel MS lesionato. Le CSPDL sono capaci di una straordi-naria attività neurorigenerativa a seguito di lesione del MS: 1) inibiscono l’apoptosi indotta dalla lesione del MS per preservare le fibre nervose e le membrane mieliniche, 2) rigenerano gli assoni troncati grazie all’inibizione di molteplici segnali dell’ inibizione assona-le e 3) rimpiazzano gli oligodendrociti danneggiati grazie al loro differenzia-mento in oligodendrociti maturi. Poichè non sono noti, attualmente, altri tipi di cellule staminali capaci di svolgere le due ultime attività neurorigenerative e-splicate dalle CSPDL, queste cellule costituiscono un’eccellente e pratica risorsa cellulare per il trattamento delle lesioni del MS. Il modello animale di lesione, ottenuto tagliando trasversal-mente il MS ha permesso di studiare con precisione la capacità delle CSPD di rigenerare gli assoni troncati: tuttavia le lesioni del MS umano, a seguito di trau-mi e di gravi lesioni del rachide, si pre-sentano con quadri clinici differenti dal modello sperimentale.
Domenico Barone
Cellule staminali nella rigenerazione del midollo spinale Oggi parliamo di ...
Anno VI numero 30 Pagina 10
In 2012, the New England Journal of Medicine celebrates 200 years of pub-lishing practice-changing medical ad-vances. Throughout 2012, NEJM will mark the anniversary with events, con-tent, and applications that celebrate all those who contribute to and read the Journal, especially recognizing the vital connection between research and prac-tice. The events begin in January with the launch of NEJM’s 200th anniversary website, and continue throughout the year with the publication of articles in-vited for the anniversary, the release of a documentary film that narrates three groundbreaking findings that trans-formed practice, and many more special events. “I am grateful to the generations of researchers, authors, reviewers, and
editors upon whose shoulders we stand,” said editor in chief Jeffrey Dra-zen, M.D. “As I reflect on the 200th anni-versary of the New England Journal of Medicine, I’m proud of the historic and lasting values of this organization, and the reputation we have earned as a source of trusted and reliable informa-tion. In our third century we will continue to deliver the tools physicians need to practice better medicine so we can im-prove the lives of patients.” The anniver-
sary reinforces NEJM’s dedication to improving patient care through its mis-sion to bring the best information at the intersection of biomedical science and clinical practice to physicians and other health care professionals. NEJM is the most widely read and respected medical journal in the world and is more than a publication — it is a community, from researcher to clinician reader, that works together to bring discovery to practice every day. The 200th anniversary of NEJM celebrates all who contribute to this mission. To mark the anniversary, the NEJM an-niversary website features
An interactive historic timeline of medical advances from the last 200 years. This rich timeline links
the history of modern medicine to select content in the NEJM Ar-chive. The timeline is also avail-able as an iPad app.
An invitation to readers to submit an anniversary message, a story about how NEJM has affected them, or a video explaining why NEJM matters or what NEJM means to them.
An opportunity to vote each month for the most important article in a
specific time period, until the end of the year when readers will decide the most important article in NEJM history.
A Historical Image Challenge, with a new image each week selected from the NEJM Archive.
sary website, a new 30-minute film will examine the past 200 years of medical progress, focusing on anesthesiology and surgery, cancer, and HIV. The film connects medical groundbreaking re-search — much of it published in NEJM — to modern medical practice through a series of evocative, interwoven vi-gnettes. NEJM will also host a 200th symposium on June 22 at Harvard Medi-cal School. This event will feature note-worthy experts covering a number of important medical topics, with the intent of exploring how medicine has inter-faced with society to improve people’s lives over the past two centuries. After the event, the symposium will be avail-able for viewing on the anniversary web-site. “NEJM’s role in bringing together a global community of researchers and clinicians to improve the health care of people around the world is worth cele-brating,” said NEJM Publisher Tom Eas-ley. “Creating a platform for our 200th anniversary that brings all those connec-tions in the community to life is a rare and inspiring privilege, and we look for-ward to hearing from our readers.” NEJM history NEJM has published reports from the frontiers of medical science and practice since its earliest days. NEJM docu-mented the first public demonstration of ether anesthesia in 1846, the first full description of a spinal disk rupture in
treatment of early childhood leukemia in
new digital ways to connect with practic-ing physicians and clinicians. Videos in Clinical Medicine, peer-reviewed educa-tional videos introduced in 2006, aim to
(Continua a pagina 11)
The New England Journal of Medicine Celebrates 200th Anniversary in 2012
Coming later this year on the anniver-
1934, and the first successes in the
1948. NEJM also continues to develop
Anno VI numero 30 Pagina 11
Per abbassare la pressione arteriosa non occorre aumentare la dose o il nu-mero di medicine da prendere: in molti casi è sufficiente assumerle al momento giusto, cioè la sera prima di andare a dormire. In tal modo si possono tenere meglio sotto controllo i valori di massima e minima, ma soprattutto si può ridurre notevolmente il rischio di infarto e ictus. Il dato più recente, pubblicato poche settimane fa sul Journal of the American Society of Nephrology, si riferisce a oltre 660 persone con insufficienza renale cronica, studiate dal gruppo di R. C. Hermida dei Laboratori di Bioingegneria e Cronobiologia dell'Università di Vigo, in Spagna: a parità di condizioni iniziali, quelli a cui è stato raccomandato di prendere almeno una delle pillole pre-scritte la sera invece che alla mattina hanno avuto nei cinque anni successivi un numero di eventi cardiovascolari gra-vi inferiore di quasi il 70% rispetto agli altri. Non solo infarti e ictus, ma anche decessi, attacchi di angina o interventi resisi necessari per ristabilire il flusso di sangue nelle coronarie, scompenso car-diaco od occlusione delle arterie che vascolarizzano gli arti inferiori o la retina. L'effetto sulla salute è dipeso dal miglio-re controllo della pressione, misurata la sera: per ogni calo di 5 mmHg della pressione sistolica, si è osservata in media una riduzione del 14% del rischio cardiovascolare. Si sa che la pressione arteriosa segue un ritmo circadiano, con un calo durante il riposo notturno e un picco prima del risveglio. «Per questo è ragionevole pensare che il farmaco pre-so con la prima colazione cominci a per-dere effetto proprio nelle prime ore del mattino seguente, quando la pressione è fisiologicamente più alta e maggiore è
infatti anche il rischio di infarti» commen-ta il cardiologo milanese Carlo Schwei-ger. Ma Francesco Portaluppi, professo-re di medicina interna all'Università di Ferrara, che con l'esperto spagnolo col-labora da molti anni, va anche oltre, pre-cisando: «Se questo vale per tutti, è tanto più vero per quella categoria di pazienti che in gergo chiamiamo "non dippers", quelli cioè nei quali non si assi-ste al calo fisiologico della pressione durante la notte». A questa categoria appartengono di solito i diabetici o i ma-lati renali cronici, ma il fenomeno può insorgere con il tempo anche in chi sof-fre solo di ipertensione essenziale. «Anzi, — dice Portaluppi — ci sono per-sone in cui i valori salgono durante il sonno più che di giorno, quando posso-no addirittura sembrare normali». Que-sta ipertensione nascosta non è meno pericolosa: uno studio, pubblicato su Lancet e condotto in sei diversi Paesi su quasi 7.500 persone per poco meno di dieci anni, ha dimostrato che le più im-portanti conseguenze in termini di morta-lità si hanno proprio in relazione ai valori registrati durante la notte. Da molto tem-po si parla dell'"ipertensione da camice bianco", l' aumento transitorio dei valori di pressione che si registrano negli am-bulatori medici in persone particolarmen-te emotive, ma che si normalizzano al ritorno a casa. Ci sono però anche "falsi sani": la loro pressione misurata durante il giorno sembra nei limiti, ma i suoi rialzi notturni mettono a rischio i vasi, il cuore e più in generale la salute. «Talvolta il loro rischio cardiovascolare può essere superiore a quello di un iperteso i cui valori scendono normalmente durante il sonno» aggiunge Portaluppi «Non si può capire un film da un fotogramma, e nem-
meno da due o tre. È l'andamento dei valori pressori durante il giorno e soprat-tutto durante la notte, quando l'organi-smo è meno pronto a difendersi, a deter-minare i possibili danni ai diversi orga-ni». Per questo il National Institute for Clinical Excellence (NICE) britannico, nelle sue ultime linee guida pubblicate l'estate scorsa, ha rivoluzionato i criteri di diagnosi dell’ipertensione. Secondo le sue indicazioni, non bastano più due o tre riscontri occasionali per etichettare una persona come ipertesa: quando si trovano valori alti, bisogna sottoporla al monitoraggio continuo della pressione arteriosa per 24 ore. Ed i costi? L'analisi dell'ente britannico, sempre molto atten-to a questi aspetti, ha valutato che l'au-mento di spesa legato a questo appro-fondimento diagnostico possa essere compensato da una migliore appropria-tezza delle cure. Il semplice provvedi-mento di prendere i medicinali la sera invece che la mattina può evitare l'au-mento di dosi (e di eventuali effetti colla-terali) o l'aggiunta di un secondo farma-co quando la monoterapia non risulta sufficiente per controllare la pressione. «L'effetto positivo della somministrazio-ne serale della terapia anti-ipertensiva si manifesta anche nella popolazione ge-nerale, non solo nelle persone in cui la pressione non scende durante la notte» conclude Giuseppe Remuzzi. Ma non è uguale per tutti i medicinali: il vantaggio più eclatante si ottiene con i farmaci ACE inibitori e sartani; è meno evidente con i calcio antagonisti, mentre è assen-te con betabloccanti e diuretici.
A cura di Domenico Criscuolo
La pressione alta si cura meglio con una pillola alla sera
provide effective clinical instruction. In-teractive Medical Cases, introduced in 2009, give users the opportunity to test their diagnostic skills. And the NEJM Archive, launched in 2010, puts the en-tire collection of published articles at physicians’ fingertips. To mark this year in NEJM’s history, NEJM has commis-
sioned a series of anniversary articles that will be published biweekly in 2012. These review articles and essays au-thored by experts in their fields explore the history of medicine, disease, and therapeutics. For more information on the NEJM’s 200th anniversary, please visit http://www.nejm200.nejm.org.
The New England Journal of Medicine is the world’s leading medical journal and Web site. Founded in 1812, and owned by the Massachusetts Medical Society, NEJM publishes peer-reviewed research and interactive clinical content for physi-cians, educators and the global medical community.
Anno VI numero 30 Pagina 12
Tumori, allo IEO di Milano centro avanzato di radioterapia
È stato inaugurato all'Istituto Europeo di Oncologia (IEO) di Milano il nuovo centro di radioterapia avanzata ARC (Advanced Ra-diotherapy Center), che si pone come eccellenza per il trattamento radioterapico dei tumori, tra i primi 10 centri nel mondo, ed accoglierà fino a 4.500 pazienti all'anno. «Il nuovo centro mette a disposizione della sanità il gold standard delle terapie» ha spie-gato in conferenza stampa Roberto Orecchia, direttore della Divisione di radioterapia «rappresentato dai sei acceleratori lineari di cui è dotato l'ARC tra cui un Cyberknife, un bisturi virtuale, il sistema TomoTherapy, il sistema Trilogy, e Vero sono l'avanguardia tecnologica e sono impiegati per trattare i tumori della mammella e della prostata, ed anche i tumori del pancreas, del fegato, del polmone e del tratto testa-collo; sono nuove indicazioni della radioterapia che faranno salire la domanda di cura». L'esperto ha anche ricordato che il centro dispone di un'unità di brachiterapia e di tre acceleratori portatili della Eliot, la Electron intraoperative therapy. «È una giornata storica per lo IEO» ha dichiarato Umberto Veronesi, direttore scientifico «in cui si rafforza un principio fondante dello IEO, che è curare con trattamenti minimi efficaci, secondo l'ipotesi del minimal effect treatment (MET). Oggi la radioterapia risponde a questo obiettivo e le cure oncologiche stanno diventando sempre meno invasive e meno invalidanti». Il vantaggio è dato sia dalla capacità dei sistemi di individuare e colpire lesioni di pochi millimetri e di farlo con un'intensità tale da richiedere meno sedute rispetto alla radioterapia tradizionale: si parla di protocolli di cura di due o tre settimane al massimo. «Oggi» prosegue Veronesi «il paziente può guarire senza mutilazioni e con meno sofferenza, e questo significa che le persone avranno meno paura del tumore e andranno più spesso a sottoporsi a esami di controllo». L'investimento complessivo per allesti-re l'ARC è stato di circa 25 milioni di euro: «Diciassette milioni sono serviti per i macchinari e otto per costruire la struttura» ha spiegato Carlo Ciani, amministratore delegato IEO «un atto coraggioso da parte dei soci dell'Istituto in controtendenza rispetto a quanto accade oggi in sanità, dove ovunque viene data priorità ai tagli».
NOTIZIE DALL’ITALIA
Nuovo regolamento AIFA, più poteri al CdA
Disco verde dalla conferenza Stato-Regioni al regolamento AIFA, emanato dai ministeri Salute ed Economia in ottemperanza alla Finanziaria 2011, che assegna più poteri al CdA e ridisegna il funzionamento delle commissioni tecnico-scientifiche e prezzi. Tra le novità, la possibilità per il CdA dell'agenzia di adottare delibere che mettano mano all'assetto organizzativo e strutturale, su proposta del DG e da validare dai due Ministeri. Via al programma di autosostentamento, con l'introduzione di un diritto annuale di mille euro per ogni procedura di autorizzazione all'immissione in commercio (AIC) in corso di validità a copertura dei costi di gestione informatici e della banca dati, con la possibilità di sconto del 25% per enti pubblici e PMI. Prevista anche una serie di servizi che l'agenzia potrà offrire a pagamento (i corrispettivi saranno fissati con delibera del CdA e approvati dai Ministeri e do-vranno comunque essere competitivi rispetto al tariffario dell'EMA e delle agenzie degli altri paesi UE) tra cui scientific advice, formazione e ECM, analisi e ricerche, studi di settore, attività editoriali. Per quanto riguarda le commissioni tecnico-scientifica e prezzi, i componenti saranno dieci: oltre a quelli di diritto - DG e presidente dell'Agenzia - ce ne sono tre nominati dal ministero della Salute, uno da quello dell'economia e quattro dalla Conferenza Stato-Regioni. Arrivata anche la conferma di Luca Pa-ni nella carica di direttore generale e la nomina di Silvio Garattini nel Consiglio di amministrazione. «Due nomine di elevato profi-lo» scrive in una nota il ministro della Salute, Renato Balduzzi, «in relazione ai sempre maggiori e delicati compiti dell'agenzia».
HPV in Italia, lontana la copertura ottimale
Non è ancora ottimale la copertura del vaccino anti HPV in Italia ed è ancora troppo ampia la variabilità dell'offerta di immunizza-zione nel nostro Paese. Ad evidenziarlo, uno studio a cura del reparto di epidemiologia di malattie infettive dell’ Istituto Superiore di Sanità, in collaborazione con il gruppo sanità pubblica del coordinamento interregionale della prevenzione, che fa il punto sulla vaccinazione anti HPV in Italia, riportando i dati di copertura vaccinale aggiornati al 30 giugno 2011. All'interno della ricerca sono rappresentate le ragazze del target primario di immunizzazione (ovvero quello chiamate a vaccinarsi nel corso del 12° anno di età e dunque nate nel 1997, 1998, 1999); e le ragazze del target secondario, per le regioni che hanno esteso l'offerta gratuita a ragazze più grandi. La copertura media con tre dosi di vaccino della coorte 1997, invitata nel 2008, è del 65%. Le coperture delle coorti invitate successivamente (1998 e 1999) sembrano essere in linea con le coperture rilevate per la coorte precedente. Tutta-via, non è stato rilevato l'incremento atteso per il protrarsi delle attività vaccinali. E' dunque ancora lontano l'obiettivo sancito dall'intesa Stato-Regioni che fissa la copertura al 95% entro 5 anni dall'avvio della vaccinazione. Dall'analisi dei dati raccolti e-merge, inoltre, un'ampia variabilità tra i dati di copertura vaccinale regionali: tra le regioni più virtuose spiccano Toscana (80,7%), Basilicata (80,7%) e Sardegna (81,8%); tra i fanalini di coda la provincia autonoma di Trento (54%), la Campania (50,1%) e la Calabria (59,4%). Per individuare cause e conseguenze della disomogenea copertura vaccinale italiana e per studiare strategie di miglioramento, nel 2011 l'Istituto Superiore di Sanità ha avviato il “Progetto Valore”, il cui stato di avanzamento può essere seguito su EpiCentro.
Anno VI numero 30 Pagina 13
Decreto liberalizzazioni, tempi più brevi per i generici Tempi più brevi per l'immissione in commercio dei farmaci generici. Sarebbe questo uno degli effetti del Decreto sulle Liberaliz-zazioni, ancora in bozza. All'articolo 27 infatti sarebbe prevista una riduzione delle procedure espletate da AIFA. «Fatta salva la tutela della proprietà industriale e commerciale» si legge nella norma «l'Agenzia, nel rilasciare l'autorizzazione all'immissione in commercio dei farmaci generici (...), non ha il potere di accertare l'esistenza di protezioni brevettuali o industriali». Secondo quanto spiegano alcuni esperti del settore, le aziende produttrici potranno commercializzare il farmaco generico immediatamente alla scadenza del brevetto, senza attendere gli accertamenti da parte di AIFA, con l'effetto di accorciare i tempi per l'immissione in commercio.
NOTIZIE DALL’ITALIA
Farmaceutiche in fuga da Milano
Per le grandi multinazionali, anche farmaceutiche, la provincia di Milano non è più un territorio su cui convenga investire. Tra le Big Pharma che stanno abbandonando il mercato milanese l'ultimo caso è quello della francese Sanofi che sta chiudendo gli uffici e il centro ricerche con il licenziamento di 469 persone di cui 351 informatori scientifici della rete nazionale, 61 impiegati e 57 ricercatori. Sempre a Milano la svizzera Novartis ha venduto il proprio stabilimento, l'inglese GlaxoSmithkline ha ceduto la propria fabbrica di Baranzate e l'anglosvedese AstraZeneca ha abbandonato il sito produttivo di Caponago. Tra le cause, i defi-cit infrastrutturali e gestionali del nostro Paese e la crescente concorrenza di Paesi come Cina ed India.
AIFA approva un codice sul conflitto di interessi
Membri delle commissioni prezzi e tecnico-scientifica, personale coinvolto nelle attività istituzionali, dipendenti o consulenti, ma anche vertici e CDA. Sono solo alcuni dei soggetti operanti in AIFA che da ora in avanti saranno obbligati a dichiarare eventuali conflitti d'interesse con l'industria farmaceutica, pena l'immediata esclusione dalle attività dell'Agenzia. A stabilirlo il nuovo codice di regolamentazione del conflitto di interesse approvato dal CDA, che passa in rassegna le varie casistiche e fissa per ogni tipo-logia di conflitto le mansioni che si possono svolgere. Al livello più alto, con l'esclusione dalle attività, c'è naturalmente il posses-so di azioni o un rapporto di lavoro o consulenza con un'impresa farmaceutica. È prevista comunque la figura del testimone e-sperto, senza diritto di voto, ma il cui parere può essere ascoltato. Per chi rientra in un livello intermedio di rischio il veto vale solo per quelle attività dove il conflitto può esplicitarsi. Tra le regole del codice, l'obbligo di aggiornare almeno una volta all'anno la dichiarazione, pena l'esclusione dalla banca dati di AIFA, e sanzioni in caso di violazioni, che possono arrivare anche alla so-spensione dell'incarico. Tenuti a fare la comunicazione anche i parenti stretti del personale coinvolto.
AIFA attiva un registro per giovani “assessor” L'Agenzia Italiana del Farmaco ha istituito lo scorso 18 febbraio la banca dati YEAS (Young European Assessors), esperti in va-lutazione dei dati che riguardano i farmaci. «Si tratta di un'importante opportunità per giovani» spiega una nota di AIFA «con ade-guate competenze tecnico-scientifiche di collaborare con AIFA alla valutazione dei dati sui medicinali per uso umano. A oggi, dopo soli 10 giorni dalla sua istituzione, sono già 31 gli assessors che hanno effettuato l'iscrizione. AIFA, ove non possa far fron-te con il personale in servizio alle specifiche esigenze tecniche in materia, selezionerà gli esperti tra quelli iscritti alla Banca Da-ti». Tutti i giovani, preferibilmente al di sotto dei 35 anni, in possesso dei requisiti richiesti, fra cui ottima padronanza della lingua inglese, laurea in medicina e chirurgia, farmacia e altre aree scientifiche, adeguata qualifica e significativa esperienza professio-nale nella progettazione, conduzione, coordinamento e valutazione di trial clinici a scopo registrativo, potranno iscriversi alla ban-ca dati seguendo le istruzioni riportate sul sito dell'agenzia. La validità della banca dati sarà di tre anni e le richieste di collabora-zione potranno essere presentate durante tutto il periodo. Gli assessors dovranno attenersi scrupolosamente alle regole sul con-flitto di interessi adottata da AIFA che ha realizzato, prima fra le agenzie regolatorie europee, un regolamento ispirato ai principi stabiliti da EMA, specificando nel dettaglio i criteri per la definizione dei conflitti nel rispetto dei principi fondamentali di apparte-nenza, trasparenza e responsabilità.
Chiesi: +16,6 % l'utile nel 2011, ma crescita contenuta per il 2012
Chiesi Farmaceutici chiude in crescita il 2011 con un fatturato di 1,06 miliardi di euro (+4,2% rispetto al 2010) ed un utile netto di 112,9 milioni (+16,6%). A sostenere i ricavi della farmaceutica di Parma, la maggiore internazionalizzazione delle attività, in par-ticolare l'aumento delle vendite in Inghilterra, Austria, Grecia, Olanda, Turchia e Pakistan, che hanno compensato il calo in Spa-gna, Stati Uniti e Italia (-0,2%). Per il 2012 crescita modesta per il fatturato, atteso a 1,09 miliardi (+3,3%), in calo i profitti a 104,9 milioni (-7%). A pesare sulle previsioni alcuni fattori negativi come il taglio dei prezzi dei farmaci in Francia, Spagna, Turchia e la perdita dell’ antiaggregante piastrinico Aggrastat (tirofiban) in USA. Resta però la sicurezza dell'antiasma Foster (beclometasone/formoterolo), che garantirà ricavi di 290 milioni di euro.
Anno VI numero 30 Pagina 14
OMS: malaria in calo grazie a prevenzione e diagnosi
A partire dal 2000 l'incidenza della malaria è calata del 17% e la sua mortalità del 26% a fronte di 216 milioni di casi registrati, di cui 174 milioni nella sola Africa. Questi alcuni dei dati contenuti nel World Malaria Report 2011, pubblicato dall'OMS a dicembre. Lo studio sintetizza i dati raccolti in 106 paesi in cui la malattia è endemica. In generale, una riduzione dei casi di malaria del 50% è stata registrata tra il 2000 e il 2010 in 43 dei 99 paesi con trasmissione attiva. Dei circa 655mila decessi per malaria nel 2010, il 91% è avvenuto in Africa e l'86% ha visto coinvolti bambini al di sotto dei 5 anni di età. La lotta alla malaria è stata age-volata dall'aumento del capitale del fondo internazionale destinato alla lotta contro questa malattia, che nel 2011 si è assestato sui due milioni di dollari. Grazie all'aumento del capitale, la distribuzione gratuita di zanzariere trattate chimicamente non è più rivolta esclusivamente alle donne incinte e ai bambini, ma a tutta la popolazione. Così, nell'Africa sub-sahariana la percentuale di chi ha a casa questo tipo di protezione è passata dal 3% del 2000 al 50% nel 2011, di cui il 96% la utilizza quotidianamente. An-che l'impiego del test di diagnosi rapida è in aumento: nel 2005 lo utilizzava il 67% della popolazione sub-sahariana, nel 2010 la percentuale si è assestata al 76%. «Una maggiore attenzione alla prevenzione» si legge nel report «conduce non solamente ad una riduzione dei casi di malattia, ma anche ad un notevole risparmio nelle cure. Nel 2015, l'utilizzo corretto e diffuso di dispositi-vi quali le zanzariere trattate chimicamente nei paesi monitorati dall'OMS potrebbe condurre a un risparmio dai 31 ai 48 milioni di dollari».
NOTIZIE DAL MONDO
Stati Uniti d’America: con i generici risparmiati 931 miliardi in 10 anni
La Generic Pharmaceutical Association (GPhA) ha incaricato l'IMS Health e l'IMS Institute for Health Care Informatics di condur-re un'analisi sul risparmio indotto dai generici in un periodo di 10 anni (2001-2010). I risultati dell'analisi dell'IMS sono sorprendenti. Per la decade 2001-2010 l'uso dei generici al posto dei loro corrispettivi farmaci commerciali hanno fatto risparmiare al sistema nazionale sanitario americano $931 milioni di dollari. Solo nel 2010, l'uso dei ge-nerici approvati dall'FDA ha prodotto un risparmio di $158 milioni, una media di $3 milioni a settimana. Su questi risultati dell'IMS, la GPhA ha tratto le seguenti osservazioni:
I risparmi derivanti dai farmaci generici entrati sul mercato a partire dal 2001 hanno continuato a crescere con un tasso esponenziale, raggiungendo più di $360 milioni alla fine del 2010. I prodotti generici per i sistemi nervoso e cardiovascolare da soli hanno rappresentato il 62% del risparmio. Pur essendo molto più presenti sul mercato (circa 7 volte), i farmaci generici rappresentano ancora una quota inferiore della spesa farmaceutica rispetto ai prodotti commerciali. Negli ultimi 10 anni, accordi transattivi sui brevetti hanno fatto risparmiare miliardi di dollari con svariati farmaci generici immessi sul mercato prima della scadenza dei brevetti dei corrispettivi farmaci branded.
GPhA ritiene che lo studio sia predittivo di ancor più ampi risparmi che si potrebbero ottenere nei prossimi anni attraverso l'attua-zione delle seguenti iniziative:
Aumentare i finanziamenti all'Office of Generic Drugs della FDA per garantire tempestive revisioni e approvazione di nuovi generici. Garantire che l'FDA sviluppi processi di approvazione per i biogenerici praticabili e liberi da ostacoli che potrebbero servi-re solo a ritardare la disponibilità di farmaci biologici sicuri, efficaci e a basso costo. Fornire l'accesso a generici convenienti continuando a consentire ai produttori di generici di sostenere le contestazioni sui brevetti, immettendo sul mercato farmaci generici prima possibile generando così ulteriori risparmi per i consumatori ame-ricani.
Leucemia infantile, rischio doppio nei pressi di centrali nucleari
Dalla Francia notizie preoccupanti in merito al rischio di ammalarsi di leucemia per bambini che vivono nei pressi di centrali nu-cleari. Secondo uno studio pubblicato nell'edizione on line dell'International Journal of Cancer la probabilità di ammalarsi, infatti, è doppia rispetto alla media. L'analisi è stata realizzata tra il 2002 e il 2007 dal centro di ricerche in epidemiologia e salute delle popolazioni (Inserm) in collaborazione con l'Istituto di radioprotezione e di sicurezza nucleare (Irsn). Nel corso di questi cinque anni, gli studiosi hanno registrato 14 casi di leucemia acuta nei bambini con età inferiore ai 15 anni. Tutti vivevano in un raggio di almeno 5 km intorno alle 19 centrali nucleari distribuite sul territorio nazionale. Si tratta di circa il doppio dei casi (7,4) registrati nello stesso periodo in bambini residenti lontano dalle centrali. Il gruppo di ricercatori, diretto da Jacqueline Clavel, dovrà ora stabilire quali sono i reali fattori che determinano tale incremento. Le radiazioni emesse dagli stabilimenti nucleari sono infatti deboli e non bastano, fanno notare gli esperti, a spiegare da sole perché i bambini si ammalano di più se vivono a pochi chilome-tri da una centrale nucleare, mentre il numero di malati crolla al di là dei 5 chilometri. Per ora, quindi, spiega Clavel, non si è arri-vati al punto di 'chiedere alle famiglie di trasferirsi, almeno fino a che non si stabiliscono le cause precise. Int J Cancer, 2012 Jan 5. [Epub ahead of print]
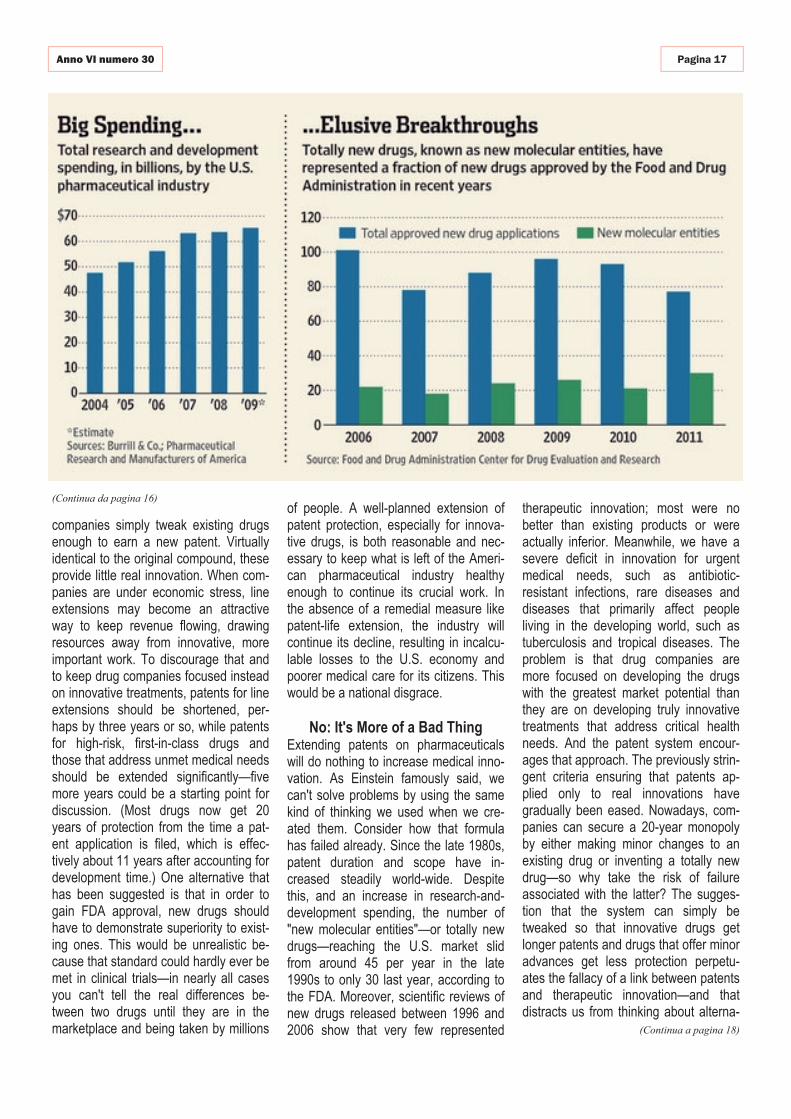
Obama envisions beating Alzheimer by 2025
The Obama administration wants the U.S. to figure out how to effectively treat and even prevent Alzheimer's disease by 2025, arguably a medical equivalent of President John F. Kennedy's challenge to America to send a man to the moon by the end of the 1960s. Of course, we eventually sent a number of men to the moon. But the question remains: Can we, in 13 years, figure out how to successfully treat and better manage patients with Alzheimer's, which robs millions of patients of their memories and iden-tities? Is a cure possible within that time frame? The goal is certainly ambitious, but the details on how to get there are still being filled in. The Associated Press reports that the government convened a two-day meeting of experts on Jan. 17 and 18 to advise the government on how to craft the fine points of its plan, which is designed to address both the medical and social challenges of Alzheimer's. These range from timely diagnosis and effective treatment to supporting family members as they care for Alz-heimer's patients. Getting there could be difficult. Right now, as the AP notes, existing treatments ease some symptoms of de-mentia, but only temporarily. And companies are struggling to find treatments that work. Pfizer, for example, recently wrote off its $725 million development pact with Medivation after its drug Dimebon failed to generate significant improvement for Alzheimer's patients in Phase III trials. AstraZeneca, by contrast, is plowing ahead with its Targacept partnership with an experimental Alz-heimer's drug advancing into midstage trials. Some of the more promising advances have been in the super-early preclinical stage and offer indirect promise. Recent research has found that exercise might help mitigate the risk of getting the disease. An-other discovery involved using nanoparticles to penetrate the blood-brain barrier in fruit flies' brains, with a future goal of using the same technology to treat Alzheimer's patients. But it is a long way to go.
Anno VI numero 30 Pagina 15
NOTIZIE DAL MONDO
AstraZeneca cutting 2.200 R&D jobs
Hit with sliding profits, a weak late-stage pipeline and a troubling track record on clinical trials, AstraZeneca has decided to trigger another round of layoffs, with 2,200 R&D staffers losing their jobs in this round. Part of a broader reorganization that will eliminate 7,300 jobs. AstraZeneca says that it is cutting way back on neuroscience, reducing the number of researchers it has in the field to a mere 40 or 50 in Boston and Cambridge, U.K. as it creates a new "virtual" group which will collaborate with academic and industry partners around the world. AstraZeneca now plans to shutter R&D facilities in Soedertaelje in Sweden and Montreal in Canada. AstraZeneca is joining a major exodus out of brain disorders, a tough field where scientists face extraordinarily high risks in the clinic and an imperfect understanding of many of the diseases. GlaxoSmithKline and Sanofi led the exit with Merck cutting back as well. Alzheimer's, Parkinson's and other conditions affect huge patient populations, raising the prospect that any pharmaceutical advances would swiftly be rewarded with blockbuster returns. Increasingly, though, the fear of high-profile clinical failures has won out over the search for big new drugs in the field, triggering deep worries among patient advocacy groups. As-traZeneca currently has two late-stage programs underway for experimental neuroscience drugs, both partnered with biotechs. There are another 9 in mid- and early-stage trials. Some of its closest ties have been forged with Nektar Therapeutics --a $1.5 billion 2009 pact centered on NKTR-118--and Targacept, which is partnered on TC-5214, a depression drug that recently failed the first two of four late-stage studies. Targacept already lost one big partner, GlaxoSmithKline, when it decided to dump its work in the field. The developer, which struck a $1.2 billion deal with AstraZeneca, can't be feeling good about this new move by Astra-Zeneca. "We've made an active choice to stay in neuroscience though we will work very differently to share cost, risk and reward with partners in this especially challenging but important field of medical research," says Martin Mackay, the company's R&D chief. "The creation of a virtual neuroscience iMed will make us more agile scientifically and financially - we will be able to col-laborate flexibly with the best scientific expertise, wherever it exists in the world."
Farmaci, in Spagna si prescriverà solo il nome della molecola
Nel tentativo di risparmiare risorse del budget statale, il Governo socialista spagnolo ha appena approvato una nuova normativa secondo la quale i medici potranno prescrivere unicamente il nome generico del farmaco e non più quello di fantasia, il cosiddet-to brand. Inoltre, le farmacie spagnole dovranno dispensare il farmaco più economico tra tutti quelli disponibili per quella determi-nata molecola e formulazione. La decisione dovrebbe servire a far risparmiare circa 2,4 miliardi di euro ogni anno. Questa misura fa parte di un pacchetto di iniziative che hanno lo scopo di ridurre il deficit statale e contrastare la gravissima crisi finanziaria che attanaglia il paese iberico, schiacciato da una disoccupazione oltre il 20% e un deficit pubblico molto elevato. Il Primo Ministro José Luis Rodríguez Zapatero, ha riferito al Parlamento spagnolo che queste misure contribuiranno a ridurre il costo dei farmaci a carico dello Stato. Lo scorso anno il budget statale per la spesa farmaceutica per la prima volta è risultato inferiore all'anno precedente, grazie a misure volte a favorire la prescrizione dei farmaci generici. Anche quest'anno la spesa per i farmaci dovreb-be ridursi di un altro 10%. Farmaindustria, l'associazione che raggruppa le aziende farmaceutiche spagnole, in un comunicato ha fatto sapere che queste decisioni rappresentano un durissimo colpo al settore, già penalizzato da precedenti decisioni, che avrà inevitabilmente forti ripercussioni occupazionali. Nell'ultimo anno, tra occupati diretti e indotto, l'occupazione nel settore farma-ceutico si è già ridotta di circa il 20%.
Anno VI numero 30 Pagina 16
OPINIONI A CONFRONTO Should Patents on Pharmaceuticals Be Extended to Encourage Innovation?
Pharmaceuticals have improved and ex-tended the lives of millions of people. But the many advances over the past couple of decades haven't come without contro-versy, much of it centering on the mas-sive profits the industry makes on block-buster drugs. The drug makers say those profits fund the research that produces breakthrough treatments. They warn that with patents expiring on several big-money drugs, their ability to develop new drugs will be severely hampered. Longer-lasting patents, they say, would protect the profits that they need to keep innova-tive products moving through the pipeline. Critics question that assumption. There's no proof, they say, of a link between pat-ent life and innovation. In their view, drug companies focus on developing the most marketable drugs instead of the most urgently needed medications. So extend-ing patents would serve mainly to boost drug companies' profits, not to encourage the innovation needed to address the world's unmet medical needs.
Yes: Innovation Demands It The American pharmaceutical industry is seriously ill. And extended patent protec-tion is just the medicine the drug compa-nies need. Pharmaceutical companies have long been demonized by many poli-ticians as heartless people that place profit ahead of people's well-being. But that perception couldn't be more wrong. The profits these companies make on blockbuster medications support the re-search that produces such break-throughs. And the scientists working in the labs are committed to finding useful new medicines. Unfortunately, there are far fewer of those scientists at work than there were 10 years ago, and their com-panies are in trouble. What's the prob-lem? A confluence of events in recent years has made drug discovery more difficult, expensive and time consuming. Most important, it has become less profit-able, largely because longer development times mean companies have less time left under patents to exclusively market their discoveries. Now, the industry faces
a financial crisis because of the recent or imminent expiration of the patents on many of its most profitable drugs. Without extended patent protection for new dis-coveries, the industry won't be able to fund the current level of research. And the consequences are profound: de-creased innovation, fewer new drugs and more job losses. Next time you hear about a drug making billions of dollars for its maker, consider this: currently, bring-ing one new drug to market takes roughly 14 years, at a cost of about $1.3 billion. For every drug that makes it to market, more than 50 other research programs fail. After all that, only two of every 10 newly approved drugs will be profitable. Those profits must fund not only all the research programs that failed, but also all the drugs that are launched but lose money. When the industry was producing a steady stream of blockbuster drugs, as it did beginning in the 1990s (for exam-ple, all the AIDS drugs), the math worked in its favor. But in recent years the num-bers have turned against the drug indus-try, for several reasons. For one, the Food and Drug Administration has be-come more risk-averse in the wake of the 2004 Vioxx debacle. Drug makers are now required to conduct more studies with many more subjects. That adds to costs and stretches out development times. And every year spent in clinical trials equals one year of lost patent cov-erage. In 1968, when development time was much shorter than today, most drugs had an effective patent life of about 17 years. Now companies usually have only about 11 years of market exclusivity for their drugs. And this number is expected to continue dropping as development times grow even longer—approaching a point where the costs and risks of devel-opment outweigh the rewards and re-search will stop. Many of the diseases addressed in the 1990s were simply eas-ier to tackle. Since then, despite in-creased research spending, fewer break-through drugs have been discovered. Difficult conditions such as cancer, Alz-heimer's, Parkinson's and obesity remain
problematic. Amid all these challenges, the drug industry is losing its financial cushion as patents from the 1990s ex-pire. Since 2006, brand drugs have lost an estimated $60 billion in sales because of patent expirations; by 2015, this figure is projected to rise to $160 billion. This is the so-called patent cliff. It shouldn't be surprising, then, that the industry is show-ing signs of stress. The share prices of the major drug makers have fallen sharply in the past decade, and weak-ened companies have succumbed to mergers and acquisitions, causing the elimination of 300,000 jobs during this time. Extension of patent life for the most innovative drugs would, at the very least, postpone the rush toward the patent cliff, providing drug companies with extra time to discover the next cycle of new, innova-tive therapies. With U.S.-based drug companies scaling back their research, there will be fewer discoveries to fill the gap and keep new treatments coming to market. Academic researchers are very good at studying the basic biology of a disease, but this is just the very beginning of the discovery process. The lion's share of the work—progressing from basic biol-ogy to an actual drug—requires the ex-pertise and resources that academic and government labs simply don't have. Of course, longer patents would mean that important drugs would remain relatively expensive for a longer time. But the ex-pense of new drugs is preferable to not having them at all. The fact that drug companies thrived in the past without patent protection is irrelevant. Companies didn't face the regulatory and competitive environment of today. For example, ge-neric competition was minimal until the 1980s. Remember that manufacturers of generic drugs contribute nothing to inno-vation. Yet they take up to 90% of sales away from the comparable brand-name drugs whose makers risked the time and money to bring breakthrough treatments to market. There are some drugs that deserve less patent protection. These are the so-called line extensions—where
(Continua a pagina 17)
Anno VI numero 30 Pagina 17
companies simply tweak existing drugs enough to earn a new patent. Virtually identical to the original compound, these provide little real innovation. When com-panies are under economic stress, line extensions may become an attractive way to keep revenue flowing, drawing resources away from innovative, more important work. To discourage that and to keep drug companies focused instead on innovative treatments, patents for line extensions should be shortened, per-haps by three years or so, while patents for high-risk, first-in-class drugs and those that address unmet medical needs should be extended significantly—five more years could be a starting point for discussion. (Most drugs now get 20 years of protection from the time a pat-ent application is filed, which is effec-tively about 11 years after accounting for development time.) One alternative that has been suggested is that in order to gain FDA approval, new drugs should have to demonstrate superiority to exist-ing ones. This would be unrealistic be-cause that standard could hardly ever be met in clinical trials—in nearly all cases you can't tell the real differences be-tween two drugs until they are in the marketplace and being taken by millions
of people. A well-planned extension of patent protection, especially for innova-tive drugs, is both reasonable and nec-essary to keep what is left of the Ameri-can pharmaceutical industry healthy enough to continue its crucial work. In the absence of a remedial measure like patent-life extension, the industry will continue its decline, resulting in incalcu-lable losses to the U.S. economy and poorer medical care for its citizens. This would be a national disgrace.
No: It's More of a Bad Thing Extending patents on pharmaceuticals will do nothing to increase medical inno-vation. As Einstein famously said, we can't solve problems by using the same kind of thinking we used when we cre-ated them. Consider how that formula has failed already. Since the late 1980s, patent duration and scope have in-creased steadily world-wide. Despite this, and an increase in research-and-development spending, the number of "new molecular entities"—or totally new drugs—reaching the U.S. market slid from around 45 per year in the late 1990s to only 30 last year, according to the FDA. Moreover, scientific reviews of new drugs released between 1996 and 2006 show that very few represented
therapeutic innovation; most were no better than existing products or were actually inferior. Meanwhile, we have a severe deficit in innovation for urgent medical needs, such as antibiotic-resistant infections, rare diseases and diseases that primarily affect people living in the developing world, such as tuberculosis and tropical diseases. The problem is that drug companies are more focused on developing the drugs with the greatest market potential than they are on developing truly innovative treatments that address critical health needs. And the patent system encour-ages that approach. The previously strin-gent criteria ensuring that patents ap-plied only to real innovations have gradually been eased. Nowadays, com-panies can secure a 20-year monopoly by either making minor changes to an existing drug or inventing a totally new drug—so why take the risk of failure associated with the latter? The sugges-tion that the system can simply be tweaked so that innovative drugs get longer patents and drugs that offer minor advances get less protection perpetu-ates the fallacy of a link between patents and therapeutic innovation—and that distracts us from thinking about alterna-
(Continua da pagina 16)
(Continua a pagina 18)
Anno VI numero 30 Pagina 18
tive policy tools to promote real health innovation. It's perfectly possible to achieve a major medical breakthrough with a product that isn't patented, while the fact of obtaining a patent doesn't say anything about a compound's actual medical value. Moreover, the patent office isn't equipped to judge therapeutic benefit. A real boost to innovation would be if the FDA required new medicines to show a therapeutic benefit over existing treatments before giving market ap-proval—a judgment the agency is per-fectly capable of making, but somehow doesn't now. Without such a prerequi-site, companies will continue to focus on pharmaceuticals with the high-est market potential, rather than innovating to address medical need. It's important to remember the purpose of the patent system—to benefit soci-ety through innovation. It makes no sense to grant pat-ent monopolies unless they produce the kind of innovation that will benefit society. Unfor-tunately, the increasingly high prices that companies charge for drugs that are under patent blunt the societal benefit of those medications, because many peo-ple can't afford the treatment they need. Extending patent protection would only worsen that situation. The usual justifica-tion for patent monopolies and high medicine prices is that these are needed to recoup the high cost of research and development. While developing a new drug undoubtedly is expensive, it's clear that pharmaceutical companies easily recoup their investment and much more. Annual sales of blockbuster drugs range in the multiple billions of dollars, and the pharmaceutical industry has been among the most profitable industrial sec-tors for many years—despite a long-recognized innovation crisis. Consider-ing that each of the top 20 best-selling drugs has annual sales of more than $4 billion, it's easy to see why patent exten-sions are highly attractive for pharma-ceutical companies. But while patent monopolies are powerful tools to maxi-mize return on investment, they fail to
provide effective incentives to encour-age innovation that addresses priority health needs. Extending patents in this broken innovation model will merely in-crease sales of those same market-friendly drugs. The fact that drug compa-nies are now faced with significant de-clines in revenue because of the expira-tion of patents on blockbuster drugs is no reason to extend patent life. Patents are a social contract, meant to reward drug companies for innovations that benefit society. For the drugs in ques-tion, that contract has been more than fulfilled. So while extending patents now would indeed provide some financial
relief to drug makers, there's no reason to believe that the result would be an increase in innovation. The suggestion that patents should be extended be-cause drug development times have been stretched by tougher FDA require-ments also rings hollow. The FDA is fulfilling its primary mandate by requiring more rigorous studies to document the safety of new medicines in the wake of cases like the Vioxx disaster. The price of that consumer safety should not be the expectation that pharmaceutical companies should be compensated by even bigger profits for meeting the agency's standards and delivering medi-cines with proven safety and efficacy. History has repeatedly shown that inno-vation can thrive without patents. Just look at penicillin or the polio vaccine. When asked who owned the patent for the polio vaccine, Jonas Salk answered, "There is no patent. Could you patent the sun?" Another example of innova-
tion without patents can be found in Switzerland, one of the global leaders in the pharmaceutical industry. In the early 1900s, the Swiss chemical companies Ciba, Geigy and Sandoz (all precursors of today's Novartis) began to produce pharmaceuticals such as aspirin, devel-oped by the German company Bayer. Subsequently, Switzerland quickly devel-oped one of the most innovative and successful pharmaceutical industries in the world, all while rejecting the idea of putting patents on chemical inventions. It was only in 1977 that Switzerland gave in to international pressure and intro-duced patents on medicines, long after it
had become highly successful with innovative pharmaceutical research and development. So, if patent extension isn't the answer, what is? I believe we should institute a regulatory environment that prioritizes health innovation instead of market opportunities, by mak-ing approval of new drugs con-tingent on therapeutic ad-vances that address unmet health needs. In parallel, we should mobilize public and private resources to finance
research and development independ-ently of patents, so that we can stop relying on pharmaceutical sales as the primary source of funding for research. International experts are exploring such alternatives on behalf of the World Health Organization, while recent experi-ence with not-for-profit drug develop-ment is showing that it can be done more cost-effectively than so far as-sumed. A pharmaceutical business model based on these premises would ensure that research on critical health needs is prioritized, and that medicines resulting from this research are afford-able. Twenty-first century science and technology have the potential to tackle many important unmet health needs. It would be a tragedy if we miss this unique opportunity.
A cura di Domenico Criscuolo
Anno VI numero 30 Pagina 19
I seminari SSFA sono sempre all’avanguardia!
Nello scorso numero di SSFAoggi vi abbiamo parlato del seminario “ La ricerca sugli antibiotici”, organizzato dal GdL Medicina Farmaceutica Milano in collaborazione con il prof. Francesco Scaglione dell’Università di Milano. Il seminario ha avuto molto suc-cesso, ed ha trattato di un problema di rilevanza mondiale, come risulta dalla notizia di questi giorni riportata in basso.
Partecipate ai Seminari SSFA! Vi offrono il panorama dell’attualità del mondo farmaceutico!
Trasformare le infezioni in malattie rare, è questo il nuovo approccio nella lotta ai batteri resistenti proposto dalla società americana di malattie infettive (IDSA = Infectious Disease Society of America). Nella proposta presentata alla Food and Drug Administration e al Congresso USA gli infettivologi americani chiedono di rivedere alcuni tipi di antibiotici e classifi-carli come farmaci orfani per le malattie rare, in modo da rendere più semplice l'approvazione per le aziende farmaceuti-che. L'uso scorretto dei farmaci ed altri fattori hanno favorito l'evoluzione di batte-ri multi-resistenti per cui ci sono poche
opzioni terapeutiche ma che causano migliaia di morti. Molte aziende hanno, però, smesso di fare ricerca in questa che è un'area poco remunerativa, la-sciando pochi farmaci contro questi ceppi come quelli MRSA ed il Clostridium diffici-le. Attualmente solo due aziende hanno in fase di sviluppo antibiotici, mentre nel 1990 erano 20. Nel 1983 c'era un proble-ma simile per i farmaci orfani per le ma-lattie rare. Per questo negli USA si offriro-no alle aziende crediti d'imposta, diritti di commercializzazione e altri incentivi: l’iniziativa ha avuto successo, ed infatti negli ultimi anni sono arrivati sul mercato
ben 135 farmaci orfani. «Un programma simile si potrebbe realizzare per gli anti-biotici» spiega Robert Guidos, vicepresi-dente dell'IDSA. «La nostra proposta crea un nuovo meccanismo per cui le aziende che vogliono sviluppare nuovi antibiotici possono arruolare meno pa-zienti per gli studi clinici e ottenere una risposta da FDA in tempi più rapidi. Per ottenere l'approvazione, le aziende dovranno avere come obiettivo antibiotici per particolari ceppi di malattie che hanno poche opzioni terapeutiche».
USA, proposta per stimolare ricerca su nuovi antibiotici
L’aborto senza le adeguate condizioni di sicurezza è una delle principali cause di mortalità tra le partorienti: una ogni sette muore perché l’interruzione della gravi-danza non è stata condotta da persona-le medico preparato e in condizioni igie-niche adeguate. E’ uno dei dati contenuti nel lungo articolo pubblicato da Lancet, che ha esaminato i dati raccolti dall’Organizzazione Mondiale della Sani-tà, in tutto il mondo, su un arco di tredici anni, dal 1995 al 2008. Nonostante la difficoltà di avere dati certi specialmente per quei paesi dove la pratica è illegale, dalla ricerca emergono con chiarezza alcuni punti. Per l’OMS, il numero com-plessivo di interruzioni di gravidanza, a livello mondiale, tra il 2003 e il 2008, si è stabilizzato, attestandosi attorno ai 28 casi ogni mille donne tra i 15 ed i 44 anni di età, con un importante calo ri-spetto al dato di partenza, quello del 1995, di 35 IVG ogni mille donne, ma anche con l’arresto del trend in diminu-zione. Anche all’interno di singole regio-ni del mondo, comunque, ci sono diffe-
renze importanti, da correlare con il con-testo sociale ed economico: in Europa occidentale (al 2008) il tasso di aborto era di 19 casi ogni mille donne, in Euro-pa orientale, di 34. Quelle che sono mol-to significative, però, sono le differenze tra le varie aree del mondo in termini di protezione della salute delle donne: il numero degli aborti classificati come “insicuri” è aumentato dal 44 al 49 per cento. Il 97 per cento delle interruzioni di gravidanza in Africa rientra in questa categoria, e il 95 per cento di quelle ef-fettuate in America Latina. In Asia que-sta percentuale scende al 40 per cento, in Oceania al 15 e in Europa al 9 per cento. Complessivamente, l’86 per cento delle interruzioni di gravidanza ha avuto luogo nei paesi del sud del mondo. In cifre assolute, nel 2008, in tutto il pia-neta 47mila donne sono morte per aborti insicuri e 8 milioni e mezzo hanno avuto gravi conseguenze sulla propria salute. I ricercatori di Lancet sottolineano poi che “il tasso di aborto è più basso nei paesi
con leggi più permissive” e che “leggi più restrittive sull’aborto non sono correlate con un abbassamento del tasso di inter-ruzione di gravidanza”. “E’ esattamente lì dove è illegale che l’aborto deve diven-tare più sicuro, tutte queste morti e que-ste complicazioni si possono evitare facilmente”. Ciò che fa la differenza, e che secondo gli autori potrebbe essere la chiave per avvicinarsi agli obiettivi del millennio in termini salute materna, sono “misure per ridurre l’incidenza delle gra-vidanze non volute e degli aborti insicuri, compresi investimenti nei servizi di piani-ficazione familiare e assistenza per a-borti in sicurezza”. L’editore di Lancet, in un editoriale, ha così commentato i dati: “Sono numeri profondamente preoccupanti. I progressi compiuti negli anni novanta vengono erosi. Condannare, stigmatizzare e cri-minalizzare l’aborto sono strategie cru-deli e fallimentari”.
A cura di Domenico Criscuolo
Lancet: “L’aborto clandestino è una delle principali cause di morte per le donne”
Cari Soci, aprite una finestra sul mondo. Il nuovo numero di IFAPP world è sul nostro sito. Troverete notizie che pro-vengono dai colleghi di tutto il mondo! E le anticipazioni del prossi-mo Congresso IFAPP,
Barcellona
14-16 novembre 2012
Anno VI numero 30 Pagina 20
Il profilo di sicurezza di un farmaco viene inizialmente determinato durante la fase sperimentale, cioè da indagini precliniche sugli animali ed in vitro, e durante gli stu-di clinici di fase I, II, III. I dati sulla sicu-rezza del farmaco dopo la commercializ-zazione provengono principalmente da studi post-registrativi e segnalazioni spontanee di reazione avversa da farma-co (ADR- adverse drug reaction), che attualmente rappresenta la fonte più im-portante di informazione sulla sicurezza di un farmaco ed il principale strumento di farmacovigilanza. La reazione avversa è una reazione nociva e involontaria che si verifica a dosi normalmente somministra-te ai soggetti umani. Le principali norme che regolano la farmacovigilanza in Italia derivano dal recepimento di direttive e linee guida comunitarie. In riferimento a
D.leg. 219-2006 (direttiva 2001/183/CE modificata) e D.leg. 12-12-2003 (nuovo modello di segnalazioni di reazione av-versa a farmaci e vaccini). Gli Stati Mem-bri garantiscono che tutte le segnalazioni di presunti gravi effetti collaterali verifica-tisi sul territorio siano messe immediata-mente a disposizione del titolare dell’autorizzazione all’immissione in com-mercio e comunque entro 15 giorni di calendario alla data della loro comunica-zione. Quando è necessaria un’ azione urgente per tutelare la salute pubblica, AIFA può sospendere la AIC di un medi-cinale, a condizione che ne informi EMA, la Commissione Europea e gli altri Stati Membri della Unione Europea entro il primo giorno lavorativo successivo. Il responsa-bile del servizio di farma-
covigilanza deve essere persona diversa dal responsabile del servizio scientifico previsto dall’articolo 14 del decreto leg.30-dic-1992 n.541 e deve essere posto in condizione di usufruire di tutti i dati di tale servizio.
Master Università Cattolica - Roma : una tesi.
Le attività di farmacovigilanza nell’industria farmaceutica e la gestione delle segnalazioni spontanee di reazione avversa
Vahid Komari Tabrizi è nato a Tabriz (Iran) nel 1979. Dopo gli studi scientifici arriva in Italia dove si iscrive alla facoltà di far-macia dell’Università di Parma nel 2003; termina i suoi studi nel 2009 nell’Ateneo di Sassari conseguendo la laurea magistrale in farmacia. Nel 2011 ottiene il diploma di Master di II livello in sviluppo preclinico e clinico del farmaco: aspetti tecnico-scientifici, regolatori ed etici, presso l’Università Cattolica del Sacro Cuore (Roma), nella facoltà di medicina e chirurgia. At-tualmente vive in Sardegna, dove lavora in una farmacia territo-riale nella provincia di Olbia-Tempio.
Anno VI numero 30 Pagina 21
LIFE-THREATENING DRUG-ASSOCIATED HYPERKALEMIA: A RET-ROSPECTIVE STUDY FROM LABORATORY SIGNALS. Noize P, Bagheri H, Durrieu G, et al. Pharmacoepidemiol Drug Saf, pubblicato on line il 16 marzo 2011
SCOPO L'iperkaliemia potenzialmente fatale può essere indotta dai farmaci ed è prevenibile nei pazienti a rischio. Questo studio è stato disegnato per descrivere casi gravi di iperkaliemia da farmaci.
METODI Soggetti adulti con concentrazioni seriche di potassio oltre 6,5 mmol/L, rilevate al momento del ricovero o durante la degenza ospedaliera nei reparti di nefrologia, cardiologia, geriatria, pronto soccorso o unità di terapia intensiva, sono stati identifi-cati dai laboratori biologici di ospedali e cliniche nella regione del Midi-Pirenei (Francia sud-occidentale). Sono stati esclusi i pa-zienti dializzati con nefropatia allo stadio terminale. L'iperkaliemia era definita farmaco-associata se al momento dell'insorgenza il paziente era in terapia con almeno un farmaco noto per la capacità di aumentare le concentrazioni seriche di potassio (farmaci assunti in regime extraospedaliero per quanto riguarda la iperkaliemia rilevata al ricovero; farmaci assunti in regime extraospeda-liero e proseguiti in ospedale o farmaci introdotti al ricovero per quanto riguarda la iperkaliemia rilevata durante la degenza in ospedale).
RISULTATI Di 168 casi di iperkaliemia, 102 (60,7%) sono stati classificati come farmaco-associati. Riguardavano pazienti anziani (età media 76,1 anni) spesso con ipertensione arteriosa e/o patologie cardiache (88,2%). Fattori di rischio, soprattutto l'insuffi-cienza cardiaca acuta, erano presenti in quasi tutti i casi (98,0%). I farmaci maggiormente coinvolti erano ACE-inibitori (47,1%), spironolattone (41,2%), sartani (23,5%) e integratori di potassio (23,5%).
CONCLUSIONI I dati di laboratorio hanno consentito un'esauriente identificazione dei casi di iperkaliemia. La frequenza di questo evento avverso farmaco-correlato e le caratteristiche dei casi esaminati suggeriscono che il trattamento con farmaci noti per au-mentare le concentrazioni seriche di potassio possa essere inappropriato, specialmente in merito a eventuali associazioni con altri farmaci o alle indicazioni d'uso, e ad alto rischio per i pazienti predisposti.
MULTIPLE DRUG EXPOSURE AS A RISK FACTOR FOR THE SERI-OUSNESS OF ADVERSE DRUG REACTIONS. Macedo AF, Alves C, Craveiro N, Marques FB J Nurs Manag 2011; 19:395-9
SCOPO Lo scopo del presente studio era validare l'ipotesi che l'esposizione a più farmaci possa essere un fattore di rischio indi-pendente per reazioni avverse da farmaci (ADR) gravi.
CONTESTO Le ADR sono una causa importante di malattia iatrogena, e sono per lo più prevenibili. L'esposizione a più farmaci, l'età anziana e il sesso femminile sono stati identificati come importanti fattori di rischio per una maggiore incidenza di ADR.
METODO Sono state studiate le segnalazioni di ADR ricevute dall'unità centrale regionale di Farmacovigilanza del Portogallo, tra gennaio 2001 e dicembre 2009.
RISULTATI Circa metà (47,4%) dei report di reazioni avverse sono stati considerati gravi, di cui il 66,7% riportava esposizione a più farmaci (media 3,07±2,2; massimo 13). Dopo aggiustamento per sesso, l'esposizione simultanea a tre o più farmaci risultava significativamente associata a un aumentato rischio di ADR gravi (odds ratio [OR] 1,23; IC 95% 1,02-1,51).
CONCLUSIONI Questi risultati supportano l'ipotesi che l'esposizione a più farmaci sia un fattore di rischio indipendente per le ADR gravi. Questi dati sono importanti sia nella valutazione del profilo rischio/beneficio dei farmaci che nel controllo della sicurez-za dei pazienti.
IMPLICAZIONI PER GLI INFERMIERI È necessario un nuovo livello di coinvolgimento del personale infermieristico sia nell'identi-ficazione delle ADR che nella prevenzione degli effetti gravi, soprattutto nei pazienti ad alto rischio. Odds ratio di reazioni avverse gravi da farmaci (ADR), secondo l'esposizione
Esposizione a più farmaci OR non aggiustato (IC 95%) OR aggiustato per sesso (IC 95%)
n >=2 1.07 (0.86-1.33) 1.17 (0.86-1.33)
n >=3 1.22 (0.99-1.50) 1.23 (1.02-1.51)
n >=4 1.30 (1.05-1.63) 1.30 (1.04-1.62)
A cura di Raimondo Russo
Anno VI numero 30 Pagina 22
I trapianti in Italia : due pareri a confronto
Trapianti: nel 2011 trend positivo delle attività …….. È stato positivo l'andamento complessivo delle attività di donazione e trapianto, nel 2011, registrando una crescita dello 0,6% nel numero complessivo dei donatori e un calo del 7% dei pazienti in lista di attesa. I dati emergono dalla relazione del centro nazio-nale trapianti, presentato al ministero della salute. A livello regionale il tasso di donazione più alto si è registrato in Toscana, ma è cresciuto in Veneto, nelle Marche e in Liguria, mentre è peggiorato in Piemonte ed Emilia. Significativi anche la diminuzione del 3,2% delle opposizioni e l'aumento del numero dei trapianti di rene da vivente, pari al 13%, e delle donazioni di cornee, dove l'Ita-lia, che ha avuto 7246 donatori nel 2011 (+8%), risulta essere il primo paese europeo. Migliorata anche la lista di attesa, con 706 pazienti in meno rispetto al 2010. Tra i pazienti iscritti in lista, 6.594 sono in attesa di un trapianto di rene, con un tempo medio di attesa pari a 3 anni, mille sono in attesa per un trapianto di fegato con un tempo medio di attesa pari a 2 anni circa, 733 per un trapianto di cuore, con un tempo medio di attesa pari a 2 anni e mezzo, e 238 sono in attesa di un trapianto di pancreas, con un tempo medio di attesa pari a 3 anni e mezzo. Secondo il ministro Renato Balduzzi, intervenuto alla presentazione, si tratta di dati «molto positivi» segno che la «rete funziona anche in un sistema articolato». E ha concluso: «L'Italia, con 21,7 donatori per milio-ne, è terza tra i grandi Paese europei, dopo la Spagna (29,2) e la Francia (22,8), ed il dato italiano è superiore del 35% alla me-dia europea (16,9). …..I dati sui trapianti non sono positivi, anzi! I dati del Ministero sui trapianti in Italia rivelano spreco di risorse e standard insufficienti: sono queste le riflessioni di Ignazio Mari-no, voce fuori dal coro di lodi che ha accompagnato la pubblicazione della relazione 2011 su donazioni e trapianti in Italia. Nel divulgare i dati il Ministero parla di un «quadro molto positivo per il nostro paese». Perché lei dissente? A leggere quelle stesse cifre, io dico invece che il sistema nazionale trapianti ha urgente bisogno di una riorganizzazione. Tra i 110 centri di trapianto esistenti in Italia, quelli che non rispettano i parametri fissati dal CNT (Centro Nazionale Trapianti) sono perfino troppi. Dieci anni fa, quando ero nel CNT, fissammo gli standard minimi che i centri trapianti territoriali avrebbero dovuto soddisfare, sulla base delle evidenze provenienti dalla letteratura internazionale. Per esempio, una struttura specializzata nel trapianto del cuore dovrebbe eseguire almeno 25 interventi l'anno, cioè due al mese. Stesso discorso per il trapianto del fegato, mentre per il rene servono 30 interventi in un anno. E i dati 2011 cosa dicono? Prendiamo Roma: risultano in attività cinque centri per il trapianto del fegato ma nessuno arriva a 25 interventi all'anno. Sa cosa vuol dire? Che il SSN spende per tenere in funzione cinque team chirurgici, cinque team di anestesia, cinque team di anatomia patologica e via di seguito. Invece ne basterebbero molti di meno: nella Capitale cinque centri eseguono in un anno poco meno di un centinaio di interventi, a Torino una sola struttura ne fa 137. Insomma si spreca denaro pubblico! Proprio quando si chiedono ai cittadini sacrifici sotto forma di nuovi ticket per finanziare la Sanità. Faccio miei due concetti e-spressi dal presidente Monti nel suo primo intervento alla Camera come capo del governo: benchmarking e spending review. Ce n'è bisogno anche nel SSN. Stessa analisi che ha recentemente investito i punti nascita. Esatto. Perché a parte i soldi, è anche un problema di qualità delle cure. C'è un articolo del New England Journal of Medicine, dicembre 1999, che dimostra senza appello l'esistenza di una relazione tra un basso numero di interventi ad alta complessità e un più elevato rischio d'errore. Quindi? Quindi il CNT non solo dovrebbe emanare le regole, ma dovrebbe anche applicarle. Se ne occuperà anche la Commissione par-lamentare d'inchiesta sul SSN di cui sono presidente. Abbiamo già messo in agenda una ricognizione, acquisiremo dati ed effet-tueremo audizioni, vogliamo capire come stanno le cose.
A cura di Domenico Criscuolo
Anno VI numero 30 Pagina 23
CANCER CytRx initiated an open-label Phase Ib safety and dose-escalation trial with INNO-206. The trial will in-clude up to 24 patients with advanced solid tumors who have failed standard therapies. INNO-206 is a tumor-targeting pro-drug of the commonly prescribed chemotherapeutic doxorubicin, and was designed to improve efficacy and reduce adverse events through controlled release and preferential targeting of tu-mors. Doxorubicin, which is currently the only FDA-approved drug on the market for soft tissue sarcoma, is a standard chemo-therapeutic treatment for a variety of other cancers. Agennix completed the enrolment of patients in the talactoferrin FORTIS-M Phase III registration trial. The trial is evaluating ta-lactoferrin plus best supportive care compared to placebo plus best supportive care in patients with non-small-cell lung cancer whose disease has progressed following two or more prior treatment regimens. The study enrolled 742 patients at over 160 sites globally. Talactoferrin is an oral biologic therapy with immunomodulatory and antibacterial properties that is being studied for the treatment of cancer and severe sepsis. Polaris enrolled the first patient in a Phase II trial of ADI-PEG 20 (pegylated arginine deaminase), the company’s enzyme-based treatment for malignant mesothelioma. This trial, called ADAM (arginine deaminase and mesothelioma), will evaluate the treat-ment efficacy of ADI-PEG 20 as a single agent compared to the best supportive care. The primary endpoint of the study is pro-gression-free survival. GenSpera reported that the first patient was dosed in an ongoing Phase I safety study evaluating G-202 in patients whose can-cers have progressed after treatment with other chemotherapies. The G-202 Phase I study is designed to enrol up to 30 patients. The primary endpoints of the open-label, dose-escalation study are to determine safety, tolerability, and pharmacokinetics of the drug, although the design allows the collection of efficacy data as well. The trial also has the provision to enrol up to 18 additional patients in an expanded Phase Ib cohort at the maximum tolerated dose of the drug. HYPERPLASIA Nymox Pharmaceuticals reported new positive results from a long-term outcome study of NX-1207 for benign prostatic hyper-plasia. The study evaluated symptomatic change and treatment status of patients involved in the company’s NX02-0012 and NX02-0013 programs. Phase I-II U.S. studies of NX-1207 were initially undertaken in 2003. The new data indicate that a signifi-cant percentage of patient given a single treatment of NX-1207 have now shown sustained improvement in their symptoms with-out other treatments for over 7.5 years. KIDNEY DISEASE FibroGen initiated a Phase IIb studies of FG-4592, an oral HIF (hypoxia inducible factor) prolyl hydroxylase inhibitor, for treat-ment of anemia in chronic kidney disease. In the study, 50 patients receive FG-4592 or epoetin alfa for 19 weeks and are moni-tored for a total of 23 weeks. Four to six cohorts of non responsive patients with stable baseline epoetin alfa of 25 to 115 IU/kg/dose three times weekly (N=64 to N=96) are planned. For hypo-responsive patients who require >115 IU/kg/dose epoetin alfa three times weekly, the Phase IIb is a multicentre active and placebo-controlled study (N=48). Screening begun in Q4 2010. Cer-tain patients from the Phase IIa program will be permitted to continue treatment in the Phase IIb study. Use of iv iron and red blood cell transfusion are not allowed per protocol except as rescue therapy. FG-4592 was tolerated in all patients to date. Concert Pharmaceuticals initiated a Phase I single ascending-dose study of CTP-499. CTP-499, which exploits Concert’s DCE Platform TM (deuterated chemical entity platform: improvement of the metabolic stability of a compound by selective incorporation of deuterium), is a potential first-in-class treatment for chronic kidney disease. CTP-499 was derived from an active metabolite of a drug approved for a different indication. It possesses a pleiotropic mechanism of action with anti-inflammatory, anti-oxidant, and anti-fibrotic properties that are different from the current standard of care. It is initially being developed for the treatment of type 2 diabetic nephropathy.
A cura di Domenico Barone
NEWS ON CLINICAL TRIALS
SSFA oggi Stampa: MEDIA PRINT, Livorno Registrazione del Tribunale di Milano, N. 319 del 14/05/2007 “Poste Italiane s.p.a. - Spedizione in Abbonamento Postale - 70% - DCB PRATO” Numero progressivo 30 Periodicità: bimestrale
Anno VI numero 30 Pagina 24
WWW.SSFA.IT
CONSIGLIO DIRETTIVO Presidente: Gianni De Crescenzo VVice—presidente: Marco Romano SSegretario: Luigi Godi TTesoriere: Anna Piccolboni Consiglieri: Rossana Benetti, Salvatore Bianco, Marco Corsi, Domenico Criscuolo, Gioacchino D’Alò, Giovanni Fiori, GiovanBattista Leproux Direttore Responsabile: Domenico Criscuolo CComitato editoriale: Giovanni Abramo, Domenico Criscuolo, Gianni De Crescenzo, Francesco De Tomasi, Luciano M. Fuccella, Marco Romano Segreteria editoriale: Sabrina Lucioni SSegreteria Organizzativa: Viale Abruzzi 32—20131 MILANO Tel. 02-29536444 Fax. 02-89058506 E-mail [email protected]