34
TAMIZAJE NEONATAL
| Date post: | 21-Dec-2015 |
| Category: |
Documents |
| Upload: | oscar-josue-rios-zea |
| View: | 59 times |
| Download: | 1 times |

TAMIZAJE NEONATAL

DEFINICIÓN
Estudio preventivo que debe practicarse a todo RN.
Descubrir y tratar oportunamente enfermedades graves e irreversibles que no se pueden detectar al nacimiento,
ni siquiera con una revisión médica muy cuidadosa.

IMPORTANCIA DEL TAMIZAJE NEONATAL
Si las enfermedades son diagnosticadas y tratadas durante el primer mes de vida se evitan lesiones
neurológicas irreversibles.
Por ser enfermedades genéticas, tienen alto riesgo de repetición en la familia y el diagnostico adecuado
permite el asesoramiento genético familiar.
La inversión comparada con los costos de las enfermedades es mínima.
Es obligatorio en muchos países.

MATERIALES A USAR
Guantes.
Lanceta o aguja nº 26 .
Alcohol al 70%
Algodón, gasas.
Papel filtro (SM905) específico (tarjeta de Guthrie

TOMA DE MUESTRAS
MUESTRAS se
obtienen de:
Talón
Cordón umbilical
Dorso de la mano

TIPOS DE TAMIZAJE
Existen dos tipos de tamizaje:
• Varían de un país a otro.• SIMPLE O BÁSICO: detecta de 4 a 5
enfermedades o alteraciones metabólicas.• AMPLIADO: diagnostica de 20 a 60
enfermedades o alteraciones metabólicas

TAMISAJE BÁSICO
ENTRE LAS ENFERMEDADES MÁS
FRECUENTES:
HIPOTIROIDISMO CONGÉNITO
(SE MIDE LA TSH NEONATAL )
FENILCETONURIA
GALACTOSEMIA
HIPERPLASIA SUPRARRENAL
CONGÉNITA

HIPOTIROIDISMO CONGÉNITO

HIPOTIROIDISMO CONGÉNITO
Se produce por el desarrollo anormal o por la ausencia de la glándula tiroides del bebé.
Esta glándula es la encargada de producir una hormona, la T4, indispensable para el desarrollo del cerebro y para el crecimiento, es fácil de tratar si se diagnostica precozmente.
Se estudia midiendo los niveles de la Hormona TSH neonatal.
Su tratamiento es sencillo y consiste en reponer la hormona tiroidea faltante.
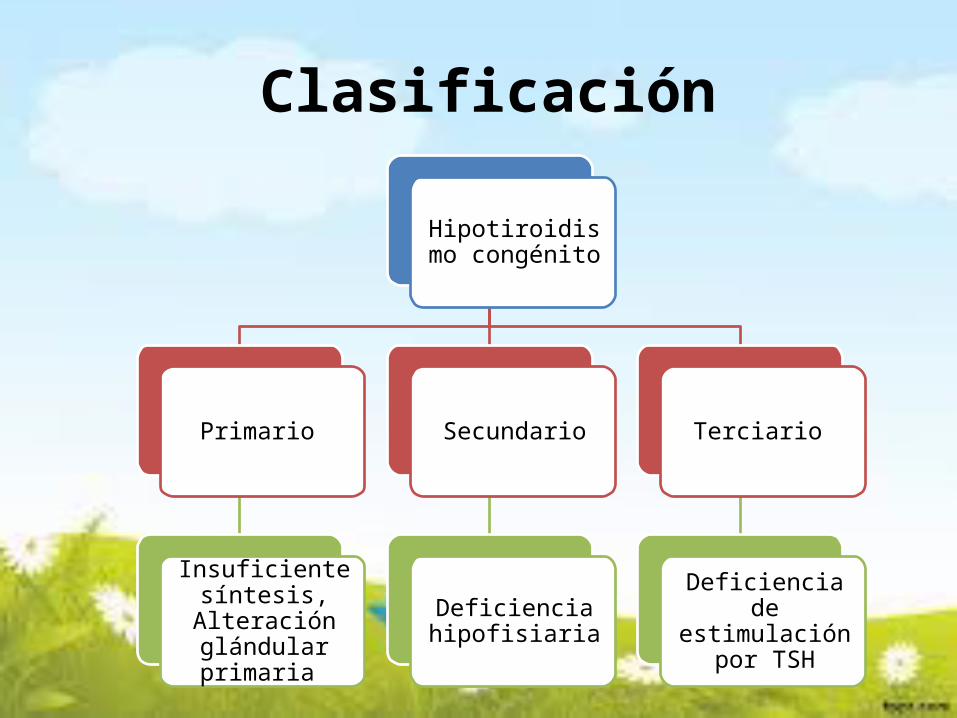
Clasificación
Hipotiroidismo congénito
Primario
Insuficiente síntesis, Alteración glándular primaria
Secundario
Deficiencia hipofisiaria
Terciario
Deficiencia de estimulación por
TSH

Richard E. Behrman et al., Nelson textbook of pediatrics. (Philadelphia, PA: Saunders, 2007).
CLASIFICACIÓN
Hipotiroidismo
primario
• Permanente: Disgenesia tiroidea• Agenesia : Ausencia de tejido funcional • Hipoplasia: Desarrollo deficiente
Dishormonogéne
sis:
• Tejido tiroideo habitual, Defecto parcial o total del proceso bioquímico en síntesis o secreción hormonal
Transitorio:
• Pasó tras placentario: Fármacos antitiroideos, Bloqueadores del receptor de TSH, Deficiencia de yodo.

SÍNTOMAS Y SIGNOS DE HIPOTIROIDISMO CONGÉNITO
•Gestación > 42 semanas
•Peso de Nacimiento > 4 kg
•Ictericia prolongada > 3 d
•Hiperbilirrubinemia no conjugada
•Edema •Hipotermia •Dificultad en la alimentación
•Fontanela posterior > 5 mm
•Hernia umbilical •Distensión abdominal •Distres respiratorio

SÍNTOMAS Y SIGNOS DE HIPOTIROIDISMO CONGÉNITO
Cianosis periférica y
livedo reticularis
Piel áspera y seca
Constipación
Letargia e hipoactividadLlanto ronco
Macroglosia
Mixedema generalizado

DIAGNÓSTICO DE SOSPECHA
DIAGNÓSTICO CONFIRMADO

TRATAMIENTO

FENILCETONURIA

FENILCETONURIA
causado por la carencia de la enzima fenilalanina hidroxilasa (cromosoma 12 gen PAH) lo que se traduce en la incapacidad de metabolizar el aminoácido tirosina a partir de FENILALANINA en el hígado, que se encuentra en las proteínas de los alimentos.


CUADRO CLÍNICOiniciándose con una
elevación en el plasma de la fenilalanina hasta
un nivel 30 veces superior al normal
excreción de ácido fenilpirúvico por la
orina.
piel clara, muy suave aterciopelada y muy
sensible.
Crisis convulsivas Olor a ratón Pelo claro y ojos azules
El desempeño escolar se puede deteriorar
levemente por retardo mental
osteopenia
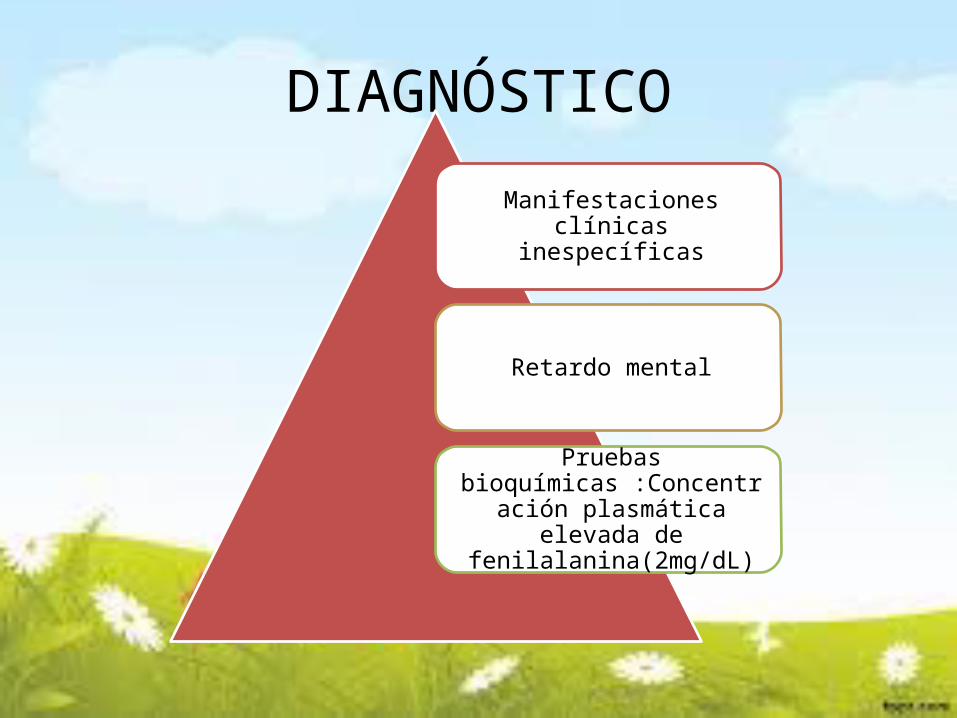
DIAGNÓSTICO
Manifestaciones clínicas inespecíficas
Retardo mental
Pruebas bioquímicas :Concentración
plasmática elevada de fenilalanina(2mg/dL)

TRATAMIENTO

GALACTOSEMIA

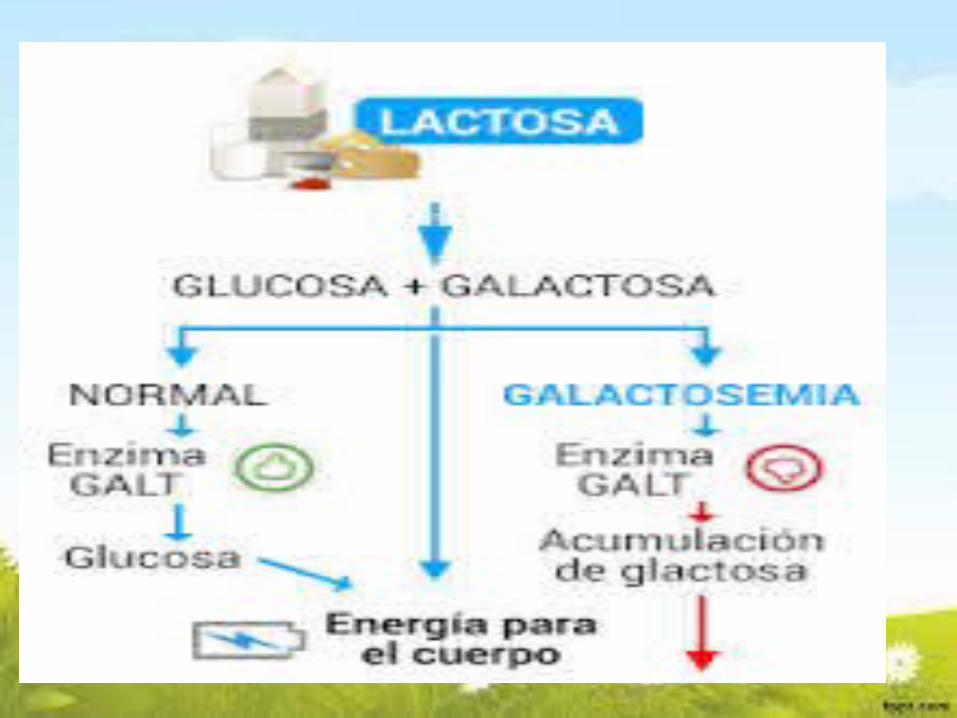
GALACTOSEMIAEs la incapacidad del organismo para utilizar el azúcar de la
leche.
LA GALACTOSA, por ende no la puede convertir EN GLUCOSA.
El depósito de galactosa en diversos órganos, lleva a retardo, cataratas e insuficiencia hepática,mala absorción tubular
renal y lesiones en el sistema nervioso central.
Puede presentarse ictericia hacia la segunda semana de vida.
El tratamiento es una dieta excentade lácteos.

CAUSAS
Deficiencia de galactosa-1-fosfatouridil transferasa (galactosemia clásica, la forma más común y la
más grave)
Deficiencia de galactosa cinasa
Deficiencia de galactosa-6-fosfato
epimerasa

SÍNTOMAS
Convulsiones Irritabilidad Letargo
Alimentación deficiente (el bebé se niega a tomar fórmula que contenga leche)
Poco aumento de peso
Coloración amarillenta de la piel y de la
esclerótica (ictericia)
Vómitos
27
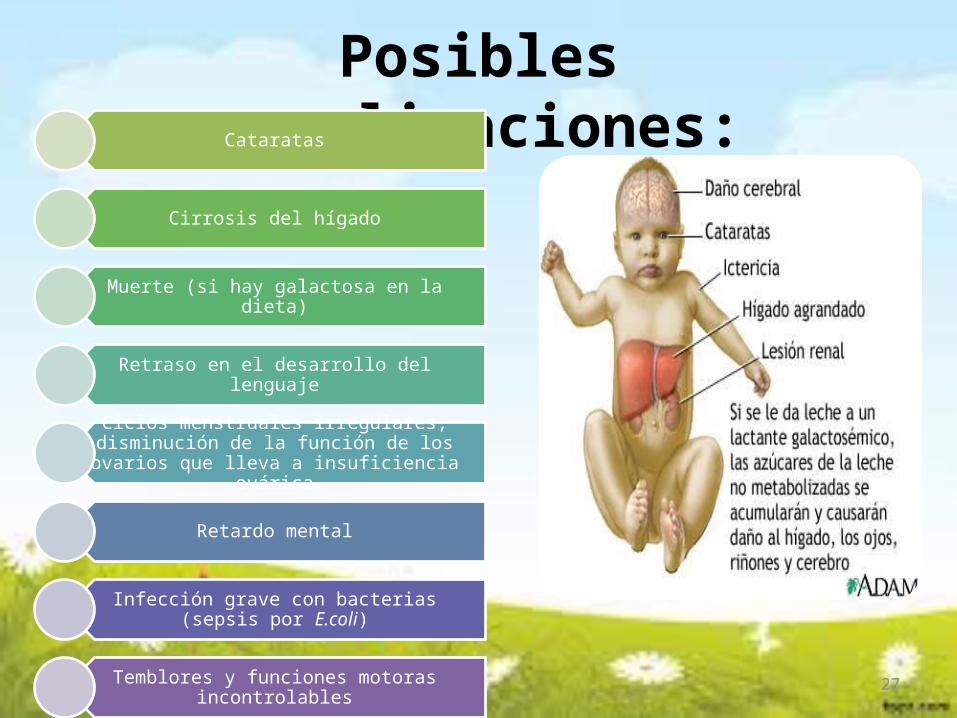
Posibles complicaciones:Cataratas
Cirrosis del hígado
Muerte (si hay galactosa en la dieta)
Retraso en el desarrollo del lenguaje
Ciclos menstruales irregulares, disminución de la función de los ovarios que lleva a insuficiencia
ovárica
Retardo mental
Infección grave con bacterias (sepsis por E.coli)
Temblores y funciones motoras incontrolables

28
Tratamiento:
• Las personas que padezcan esta afección deben evitar de por vida todos los tipos de leche, los productos que contengan leche (incluyendo la leche en polvo) y otros alimentos que contengan galactosa. Es esencial leer las etiquetas de los alimentos y ser un consumidor informado.
(3)

HIPERPLASIA ADRENAL
CONGÉNITA
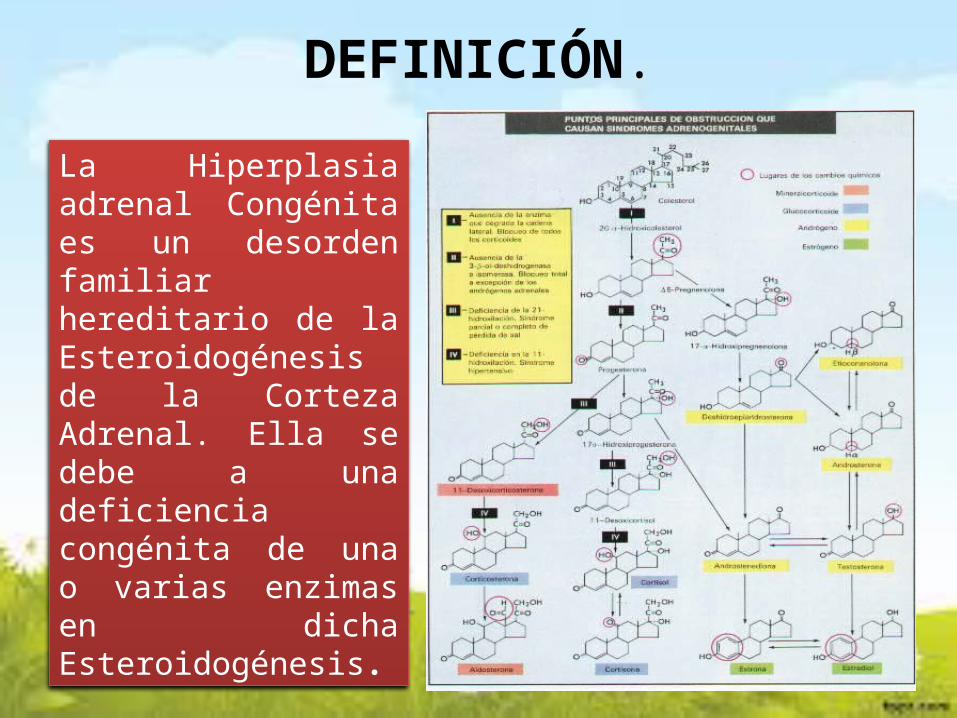
DEFINICIÓN.
La Hiperplasia adrenal Congénita es un desorden familiar hereditario de la Esteroidogénesis de la Corteza Adrenal. Ella se debe a una deficiencia congénita de una o varias enzimas en dicha Esteroidogénesis.

PATOGENIA.
La causa más frecuente de la Hiperplasia Suprarrenal
Congénita es la deficiencia congénita de la 21 -
Hidroxilasa, la cual es la resposable de la
transformación de 17 - Hidroxi Progesterona a 11 - Desoxicortisol, suprimiendo en este estado la producción
de Cortisol.

ESTEROIDOGÉNESIS SUPRARRENAL
95 %
DEHIDROEPIDANDROSTERONA

DIAGNÓSTICO
17-OH Progesterona
Test de ACTH
Actividad de Renina
Plasmática
Electrolitos plasmáticos
1 10 100 1000
basal 17-OHP, ng/mL
1000
100
10
1
Stimulated
17-OHP
ng/mL

TRATAMIENTOTRATAMIENTO SUSTITUTIVO CON GLUCOCORTICOIDES• Hidrocortisona a 15mg/m2/día• Neonatos 5 mg/día dividido en tres dosis ( 25 mg/m2/día)• Adolescentes mayores y adultos pueden ser tratados con
prednisona (5- 7,5 mg/día ) o dexametasona (0,25-0,5 mg/día
TRATAMIENTO SUSTITUTIVO CON MINERALOCORTICOIDES• 9-a-fluorhidrocortisona, dosis de 0,05-0,2 mg/día dividido en
dos o tres dosis.• Suplementos de cloruro de sodio (1-2 g por día)

















