Page 1
1
The combination of the R263K and T66I resistance 1
substitutions in HIV-1 integrase is incompatible with 2
high level viral replication and the development of high-3
level drug resistance 4
Jiaming LIANG1,2, Thibault MESPLÈDE1, Maureen OLIVEIRA1, Kaitlin ANSTETT1,3, 5
and Mark A. WAINBERG1,2,3. 6
1McGill University AIDS Centre, Lady Davis Institute for Medical Research, Jewish 7
General Hospital, Montreal, Quebec, Canada. 8
2Division of Experimental Medicine, Faculty of Medicine, McGill University, Montréal, 9
Québec, Canada. 10
3Department of Microbiology and Immunology, Faculty of Medicine, McGill University, 11
Montréal, Québec, Canada. 12
Correspondence to: Mark A. Wainberg, McGill AIDS Centre, 3755 Ch. Côte-Ste-13
Catherine, Montréal, QC H3T1E2, Canada. E-mail: [email protected] 14
Running head: Incompatibility of R263K and T66I 15
Abstract: 235 16
Importance: 131 17
Text: 2531 18
JVI Accepted Manuscript Posted Online 26 August 2015J. Virol. doi:10.1128/JVI.01881-15Copyright © 2015, American Society for Microbiology. All Rights Reserved.
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 2
2
Key Words 19
HIV-1, integrase, drug resistance, integrase inhibitors, R263K, dolutegravir, raltegravir, 20
elvitegravir 21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 3
3
ABSTRACT 37
Background: The R263K substitution in integrase has been selected in tissue culture 38
with dolutegravir (DTG) and been reported in several treatment-experienced individuals 39
receiving DTG as part of salvage therapy. R263K seems to be incompatible with the 40
presence of common resistance mutations associated with raltegravir (RAL), a different 41
integrase strand transfer inhibitor (INSTI). T66I is a substitution that is common in 42
individuals who have developed resistance against a different INSTI termed elvitegravir 43
(EVG), but it is not known whether these two mutations might be compatible in the 44
context of resistance against DTG or what impact the combination of these substitutions 45
might have on resistance against INSTIs. E138K is a common secondary substitution 46
observed with various primary resistance substitutions in RAL- and EVG- treated 47
individuals. 48
Methods: Viral infectivity, replicative capacity, and resistance against INSTIs were 49
measured in cell-based assays. Strand-transfer and 3’processing activities were measured 50
biochemically. 51
Results: The combination of R263K and T66I decreased HIV-1 infectivity, replicative 52
capacity, and strand-transfer activity. The addition of the E138K substitution partially 53
compensated for these deficits and resulted in high levels of resistance against EVG but 54
not against DTG or RAL. 55
Conclusions: These findings suggest that the presence of T66I will not compromise the 56
activity of DTG and may also help to prevent the additional generation of R263K. Our 57
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 4
4
observations support the use of DTG in second-line therapy for individuals who 58
experience treatment failure with EVG due to the T66I substitution. 59
Importance 60
The integrase strand transfer inhibitors (INSTIs) elvitegravir and dolutegravir are newly 61
developed inhibitors against human immunodeficiency virus-1 (HIV-1). HIV drug-62
resistant mutations in integrase that can arise in individuals treated with elvitegravir 63
commonly include the T66I substitution whereas R263K is a signature resistant 64
substitution against dolutegravir. In order to determine how different combinations of 65
resistance integrase mutations can influence the outcome of therapy, we report here the 66
effects of the T66I, E138K, and R263K substitutions, alone and in combination, on viral 67
replicative capacity and resistance to integrase inhibitors. Our results show that the 68
addition of R263K to the T66I substitution diminishes viral replicative capacity and 69
strand-transfer activity while not compromising susceptibility to dolutegravir. This 70
supports the use of dolutegravir in second-line therapy for patients failing elvitegravir 71
who harbor the T66I resistance substitution. 72
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 5
5
Introduction 73
Recent strategies to treat HIV-1 infection involve the use of integrase strand-transfer 74
inhibitors (INSTIs) that are the most potent antiretroviral drugs (ARVs) to date and 75
include raltegravir (RAL), elvitegravir (EVG), and dolutegravir (DTG) (1). Despite this, 76
the emergence of drug-resistance mutations in integrase (IN) represents a concern for the 77
future use of these drugs, and various resistance mutations against RAL and EVG, that 78
are associated with treatment failure, have been characterized (2). A high degree of cross-79
resistance also exists between RAL and EVG, since the major resistance substitutions for 80
RAL are located at positions G140, Y143, Q148, and N155, while those for EVG are at 81
positions T66, E92, G140, S147, Q148, and N155 (1, 3). Although resistance in initial 82
therapy has not yet been reported for DTG, patients can fail DTG if they were previously 83
treated with RAL or EVG and possess relevant mutations for those drugs (4-6). 84
In contrast, a R263K substitution was selected in tissue culture with DTG and this 85
substitution has been reported in several treatment-experienced, INSTI-naïve individuals 86
who were not fully suppressed when receiving DTG-based therapy (7). We showed that 87
R263K alone or in combination with other secondary mutations confers low-level 88
resistance to DTG and that viruses containing R263K possess significantly reduced viral 89
replication capacity (8-10). 90
It is also notable that R263K has been shown to emerge secondary to the T66I 91
substitution during tissue culture selections with EVG (11). Here, we have examined the 92
effect of combining the T66I and R263K substitutions on HIV-1 viral replicative capacity 93
and levels of resistance against various INSTIs and have also studied this in the context 94
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 6
6
of the secondary E138K mutation that commonly arises during the emergence of 95
clinically relevant resistance for both RAL and EVG. 96
Material and methods 97
Cells and reagents 98
TZM-bl and 293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM). 99
PM-1 cells were cultured in Roswell Park Memorial Institute medium (RPMI). Both 100
DMEM and RPMI were supplemented with 10% fetal bovine serum (FBS), 2 mM L-101
glutamine, 50 U/ml penicillin, and 50 µg/ml streptomycin. Cord-blood was obtained from 102
the Department of Obstetrics, Jewish General Hospital, Montréal, Canada. Primary 103
human cord-blood mononuclear cells (CBMCs) were isolated from cord-blood using 104
Ficoll Hypaque (GE Health Care Life Sciences), and the CBMCs were stimulated with 105
phytohemagglutinin. CBMCs were grown in RPMI. Cells were maintained at 37ºC under 106
5% CO2. RAL, EVG, and DTG were provided by Merck&Co., Inc., Gilead Sciences, and 107
ViiV Healthcare Inc respectively. 108
Generation of Replication-Competent Genetically Homogenous Virus 109
pNL4-3IN(T66I), pNL4-3IN(R263K), pNL4-3IN(T66I/R263K), and pNL4-3IN(T66I/E138K/R263K) were 110
produced using site-directed mutagenesis. The production of the pNL4-3IN(R263K) and 111
pNL4-3IN(E138K/R263K) plasmids has been reported previously (10). The primers used for 112
T66I mutagenesis were: sense: 5’-113
CCAGGAATATGGCAGCTAGATTGTATACATTTAGAAGGAAAAGTT-3’ and 114
antisense: 5’-AACTTTTCCTTCTAAATGTATACAATCTAGCTGCCATATTCCTGG-115
3’. All plasmids were verified by sequencing. To produce replication-competent 116
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 7
7
genetically homogenous viruses, 12.5 µg of pNL4-3IN(WT), pNL4-3IN(T66I), pNL4-3IN(R263K), 117
pNL4-3IN(T66I/R263K), or pNL4-3IN(T66I/E138K/R263K) plasmid were used to transfect 293T cells 118
using Lipofectamine 2000 (Invitrogen). Fresh medium was added at 4 h post transfection. 119
After 48 h, culture fluids were harvested and passed through a 0.45-µm filter. 120
Quantification of viruses was performed using p24 and RT assays as described previously 121
(12). 122
Tissue culture selections with RAL or EVG 123
CBMCs were infected with NL4.3IN(WT), NL4.3IN(T66I), or NL4.3IN(E138K/R263K) viruses and 124
then grown in the presence of increasing concentration of RAL or EVG. Viral replication 125
in culture was monitored by RT assay and aliquots of culture fluids were collected 126
weekly. Viral RNA was extracted from the aliquots using a RNA extraction kit (Qiagen) 127
and amplified by RT-PCR (Life Technologies) as previously described (13). The PCR 128
products were then sequenced to detect emergence of drug-resistance mutations. 129
HIV-1 infectivity and replicative capacity 130
HIV-1 infectivity was measured by short-term TZM-bl assay. Briefly, 30,000 TZM-bl 131
cells/well were infected with serially diluted viruses in a 96-well flat-bottom plate. Cells 132
were lysed at 48 h after infection and luciferase levels were measured to directly monitor 133
short-term infectivity. Fold decreases in infectivity were represented as the relative EC50, 134
which is the amount of virus (previously quantified using RT assay) needed for TZM-bl 135
cells to produce half of the maximal level of luciferase in an infection. HIV-1 replicative 136
capacity was measured as counts per minute (cpm) in PM-1 cells following HIV-1 137
infection over 21 days. Both assays have previously been described (14). 138
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 8
8
Susceptibility to antiretroviral compounds 139
Susceptibilities of virus to ARVs were measured by addition of serially diluted DTG, 140
RAL, or EVG to TZM-bl cells prior to infection using the viruses described above. 141
Luciferase levels were measured after 48 h of incubation, similar to the protocol for the 142
infectivity assay described above, and IC50 values were determined. 143
Generation of plasmids for integrase protein expression and 144
purification 145
Expression plasmids pET-15b coding for soluble integrases that were either wild-type 146
(WT) or containing the R263K or E138K/R263K substitutions were generated using site-147
directed mutagenesis as previously described (10). The T66I, T66I/R263K, and 148
T66I/E138K/R263K combinations of mutations were produced using the primers 149
described above. The pET-15b plasmids were then used to express recombinant proteins 150
in BL21 (DE3) bacterial cells. The protocol for protein expression and purification of his-151
tagged integrase has been described (9). 152
Cell-free strand-transfer assay 153
Integrase strand-transfer activities of WT integrase enzyme and integrase proteins 154
containing the T66I, R263K, T66I/R263K, E138K/R263K, or T66I/E138K/R263K 155
substitutions were measured as previously described (15). Briefly, 300 nM of processed 156
LTR-DNA duplexes were coated onto Costar 96-well DNA-binding plates (Corning) by 157
overnight incubation at 4oC. The plates were washed once with blocking buffer (20 mM 158
Tris pH 7.5, 150 mM NaCl, 0.25% BSA) and then incubated with the same buffer for 30 159
min at 37oC or overnight at 4oC. Immediately before the strand-transfer assay, plates 160
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 9
9
were washed once with PBS pH 7.4 and Assay Buffer (50 mM MOPS pH 6.8, 0.15% 161
CHAPS, 50 mM NaCl, 30 mM MnCl2, 50 µg/mL BSA). 400 nM of purified integrase 162
proteins were resuspended in assay buffer with 5 mM DTT and added to the microplates 163
for a 30 min incubation at room temperature. Serially-diluted biotinylated-target DNA (0 164
- 60 nM) was then added to each well for 1 h at 37oC. The plates were then washed twice 165
with wash buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.05% Tween 20, 2 mg/mL BSA). 166
Streptavidin-Eu solution (50 µM DTPA, 0.025 µg/mL Eu-labeled streptavidin) diluted in 167
wash buffer was added for 30 min at room temperature. Finally, the plates were washed 168
twice with wash buffer and 80 µl Wallac enhancement solution (PerkinElmer) was added. 169
Time-resolved fluorescence was read using a FlUOstar Optima multilabel plate reader 170
(BMC Labtech). 171
3’ processing assay 172
The 3’ processing activities of WT integrase enzyme or enzymes containing the T66I, 173
R263K, T66I/R263K, E138K/R263K, T66I/E138K/R263K substitutions were measured 174
as described (16). The 3’ processing assay was similar to the strand-transfer assay. Serial 175
dilutions of unprocessed-LTR DNA duplex with 3’-biotinylation were used to coat the 176
plates at concentrations between 0 to 40 nM. After addition of purified integrase proteins, 177
the plates were incubated for 2 h to allow 3’ processing to occur. 178
Data analysis 179
Each experiment was performed at least twice using three or four replicate samples. Data 180
analysis was performed using GraphPad Prism 5.0. 181
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 10
10
Results 182
Emerging substitutions in NL4.3IN(WT), NL4.3IN(T66I), and NL4.3IN(E138K/R263K) 183
under RAL or EVG drug pressure 184
To confirm previous findings and verify the possibility of T66I/E138K/R263K triple 185
substitutions, selection studies were performed using CBMCs infected with NL4.3IN(WT), 186
NL4.3IN(R263K), and NL4.3IN(E138K/R263K) under increasing concentrations of RAL or EVG 187
(Table 1). Together with several substitutions, the T66I substitution emerged from the 188
NL4.3IN(WT) or NL4.3IN(R263K) under RAL or EVG pressure, respectively, and from 189
NL4.3IN(E138K/R263K) with both drugs. In contrast, the T66I substitution was not detected 190
when RAL selection experiments were initiated with a virus containing the R263K 191
substitution nor did emerged from the WT virus under EVG pressure. 192
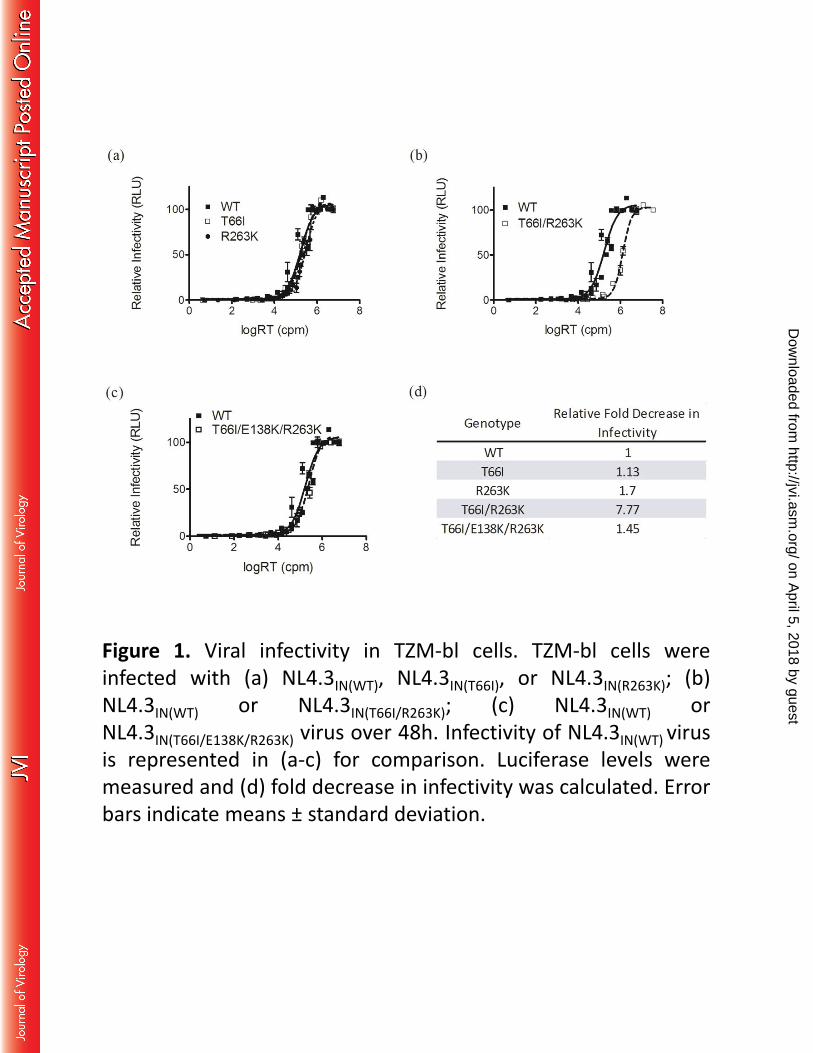
Combining the T66I and R263K substitutions impairs viral infectivity 193
To determine the effects of the T66I, E138K and R263K substitutions on viral infectivity, 194
TZM-bl cells were infected with NL4.3IN(WT), NL4.3IN(T66I), NL4.3IN(R263K), 195
NL4.3IN(T66I/R263K), or NL4.3IN(T66I/E138K/R263K) virus (Figure 1). The NL4.3IN(T66I), 196
NL4.3IN(R263K), and NL4.3IN(T66I/E138K/R263K) viruses showed only slight impairments in 197
infectivity relative to WT (Figure 1a and c), whereas NL4.3IN(T66I/R263K) displayed a 198
significant defect in infectivity (Figure 1b). Relative infectivity was decreased by 8-fold 199
by the T66I/R263K combination of substitutions. The addition of E138K to T66I/R263K 200
partially restored infectiousness (1.45-fold decrease in infectivity relative to WT) (Figure 201
1d). 202
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 11
11
The T66I/R263K combination of substitutions impairs viral replicative 203
capacity 204
A single cycle of infection does not always capture replicative defects. Therefore, we 205
also assessed the long term replicative capacity of the different viruses that contained 206
T66I using PM-1 cells. Infections were carried out with NL4.3IN(WT), NL4.3IN(T66I), 207
NL4.3IN(R263K), NL4.3IN(T66I/R263K), or NL4.3IN(T66I/E138K/R263K) over 21 days (Figure 2). RT 208
activity was measured in culture fluids at days 3, 7, 14, and 21. Similar to the results of 209
the TZM-bl infectivity assay, we found that the T66I substitution alone had little effect 210
on viral replicative capacity (Figure 2a) and R263K decreased viral replication to a 211
similar extent as previously reported (12). In contrast, the NL4.3IN(T66I/R263K) virus showed 212
a major defect in replicative capacity (Figure 2c). Although the T66I/R263K containing-213
virus yielded similar levels of RT activity at day 3 in comparison to the other viruses 214
tested, replication gradually decreased over the subsequent 18 days while the other 215
viruses attained higher levels of replication at or after day 7. In particular, the 216
NL4.3IN(T66I/E138K/R263K) virus showed partially restored replicative capacity in comparison 217
to the NL4.3IN(T66I /R263K) virus. 218
Strand-transfer activities of recombinant integrase containing the T66I, 219
E138K, and/or R263K substitutions 220
To determine whether the deficits in replicative capacity observed with mutated viruses 221
in PM1 cells were caused by changes in integrase activity, cell-free biochemical strand-222
transfer assays were performed using purified recombinant integrases containing the T66I, 223
E138K, and/or R263K substitutions. Maximal enzyme activity (Vmax) and the amount of 224
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 12
12
target LTR-DNA used to reach half Vmax (1/2MaxDNA) were calculated for each of the 225
recombinant integrase enzymes. The relative Vmax was measured for each recombinant 226
integrase and maximal strand-transfer activity of WT integrase was arbitrarily set at 100% 227
(Table 2). The results show that the presence of the T66I substitution increased 228
1/2MaxDNA by 2.4-fold while decreasing Vmax to 67% of WT. Similarly, the R263K 229
substitution increased 1/2MaxDNA by 2-fold, and the E138K/R263K substitutions in 230
tandem decreased Vmax to 38.7% of WT. For T66I/R263K substitutions, 1/2MaxDNA was 231
increased by 2.3-fold and Vmax decreased to 13% of WT. The three T66I/E138K/R263K 232
substitutions together resulted in a slightly decreased 1/2MaxDNA (1.2-fold) but Vmax was 233
only 17% of WT. 234
3’ processing activity of recombinant integrase enzymes containing the 235
T66I, E13K8, and/or R263K substitutions 236
3’ processing is a rate-limiting step in HIV-1 integration and a 3’ processing assay was 237
performed to determine the effects of the T66I, E138K, and R263K substitutions on 238
enzyme activity. The results show that no significant differences were observed among 239
the various recombinant integrase enzymes that were tested (Table 3). 240
The T66I/R263K combination of substitutions confers significant 241
resistance to EVG but remains susceptible to DTG 242
The T66I substitution in integrase has been previously reported to confer major resistance 243
to EVG while increasing HIV-1 susceptibility to DTG. Previously, we showed that 244
R263K, alone or in combination with several secondary mutations, conferred moderate-245
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 13
13
level resistance to EVG and low-level resistance against DTG (8-10). Now, we conducted 246
resistance assays using TZM-bl cells to determine the effects of the T66I substitution 247
when combined with R263K and/or with the E138K secondary substitution on resistance 248
to each of DTG, RAL, and EVG (Table 4). Compared to the IC50 of WT, and in 249
agreement with previous studies (17), the T66I substitution alone increased susceptibility 250
to DTG by 1,000-fold, conferred low-level resistance against RAL (2.4-fold), and higher 251
level resistance against EVG (10-fold). Viruses containing the T66I/R263K combination 252
of substitutions were susceptible to DTG (0.089-fold), slightly resistant to RAL (1.6-fold), 253
and more resistant to EVG (22-fold). The T66I/E138K/R263K virus was susceptible to 254
DTG (0.027-fold), slightly resistant to RAL (2.5-fold), and highly resistant to EVG (164-255
fold). 256
Discussion 257
T66I was originally described as a change in integrase selected in tissue culture under 258
EVG pressure. It was later shown to be common in the genomes of viruses isolated from 259
individuals failing EVG treatment (18). Other substitutions that are associated with 260
treatment failure under EVG-based therapy include E92Q, G140S/A, S147G, 261
Q148H/R/K, and N155H (18), of which the latter also emerged from the WT virus under 262
EVG pressure in the current study (Table 1). T66I can also be found in viruses from 263
individuals who have experienced treatment failure with RAL, though more rarely (19). 264
This may be due to the high versus low levels of resistance conferred by this substitution 265
against EVG and RAL, respectively (17), an observation that we have confirmed here 266
(Table 4). In addition, we have confirmed that T66I does not confer resistance against 267
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 14
14
DTG but significantly increases HIV-1 susceptibility to this drug. Structural models 268
derived from the crystal structure of the prototype foamy virus integrase protein suggest 269
that the T66I substitution might increase susceptibility to DTG by disrupting an 270
electrostatic interaction between T66 and N155 (15). No single integrase substitution 271
besides R263K has ever been shown to confer significant levels of resistance against 272
DTG (17, 20), helping to explain the prevalence of R263K in some treatment-273
experienced, INSTI-naïve individuals who experienced DTG-based treatment failure (7). 274
We have shown previously that the R263K substitution is also associated with decreases 275
in viral DNA integration and viral replication capacity (8), suggesting that the 276
development of resistance both in tissue culture and in vivo involves a balance between 277
levels of resistance and replicative capacity. The emergence of the T66I/R263K 278
combination of substitutions in tissue culture selection with EVG has been documented 279
(11, 21) and we show here that this combination severely impaired both integrase strand-280
transfer activity and HIV-1 replicative capacity (Table 2 and Figure 2, respectively). In 281
contrast, the T66I/R263K combination of substitutions has not been observed in the 282
presence of RAL (19, 22), suggesting that the low levels of resistance against RAL that 283
are associated with this combination are not sufficient to compensate for deficits in 284
replication capacity, that are related to decreased strand-transfer integrase activity but not 285
3’-processing activity (Tables 2 and 3). 286
The positive effect of E138K on strand-transfer activity seems to be due to an 287
improvement in DNA binding activity, as shown by decreases in 1/2MaxDNA values when 288
this substitution was present (Table 2). In contrast, E138K had little effect on maximal 289
strand-transfer activity. These findings correlate with the E138K-associated partial 290
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 15
15
compensation of defects in infectivity and replicative capacity that were observed with 291
the T66I/R263K combination of mutations (Figures 1 and 2). The addition of the E138K 292
substitution to the T66I/R263K combination also increased levels of resistance against 293
RAL and EVG by 1.5- and 7.5-fold, respectively (Table 4). 294
Importantly, the addition of R263K to T66I did not confer resistance against DTG, 295
although it may have moderated the increase in susceptibility to this drug that was 296
associated with T66I alone (Table 4). Furthermore, the T66I/R263K combination of 297
substitutions severely impaired viral replicative capacity (Figure 2). This suggests that 298
patients who experience EVG-based treatment failure with an emergent T66I substitution 299
can be successfully treated with DTG and may not be able to develop the R263K 300
substitution in combination with T66I. Given the high prevalence of the latter substitution 301
in individuals who have failed EVG (18), our results provide additional justification for 302
the use of DTG in second-line therapy after development of T66I. 303
In the current study, we also tested the ability of E138K, a secondary substitution that has 304
been observed together with R263K in tissue culture, to act together with the 305
T66I/R263K combination of mutations to modulate strand-transfer activity and 306
replicative capacity (Table 2 and Figures 1 and 2). However, viruses that contain the 307
T66I/E138K/R263K combination of substitutions remained highly susceptible to DTG 308
(Table 4). 309
Funding 310
This work was supported by the Canadian Institutes for Health Research (CIHR). 311
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 16
16
Acknowledgments 312
JL performed experiments, analysed the data and wrote the initial manuscript. TM 313
designed and performed experiments, analyzed the data and corrected the manuscript. 314
MO and KA performed experiments. MAW supervised the project and revised the 315
manuscript. All authors read and approved the final version of the paper. 316
Conflicts of interest 317
The authors declare they have no conflict of interest. 318
Legends for illustrations 319
Figure 1. Viral infectivity in TZM-bl cells. TZM-bl cells were infected with (a) 320
NL4.3IN(WT), NL4.3IN(T66I), or NL4.3IN(R263K); (b) NL4.3IN(WT) or NL4.3IN(T66I/R263K); (c) 321
NL4.3IN(WT) or NL4.3IN(T66I/E138K/R263K) virus over 48h. Infectivity of NL4.3IN(WT) virus is 322
represented in (a-c) for comparison. Luciferase levels were measured and (d) fold 323
decrease in infectivity was calculated. Error bars indicate means ± standard deviation. 324
Figure 2. Viral replicative capacity in PM-1 cells. PM-1 cells were infected with 325
NL4.3IN(WT), or (a) NL4.3IN(T66I), (b) NL4.3IN(R263K), (c) NL4.3IN(T66I/R263K), and (d) 326
NL4.3IN(T66I/E138K/R263K) viruses over 21 days. Replicative capacity of the above-327
mentioned viruses was normalized to reverse transcriptase (RT) levels of the NL4.3IN(WT) 328
virus at day 7. Supernatants were collected at days 3, 7, 14, and 21 at which time RT 329
levels were measured as counts per minute (cpm). Error bars indicate means ± standard 330
deviation. 331
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 17
17
Table 1. New substitutions emerging from NL4.3IN(WT), NL4.3IN(R263K), and 332
NL4.3IN(E138K/R263K) infections of CBMCs under raltegravir (RAL) or elvitegravir (EVG) 333
drug pressure at week 30. 334
Table 2. Strand-transfer activity of recombinant subtype B integrase containing the T66I, 335
E138K, and/or R263K substitutions. 336
Table 3. 3’ processing activity of recombinant subtype B integrase containing the T66I, 337
E138K, and/or R263K substitutions. 338
Table 4. Susceptibilities of NL4.3IN(WT), NL4.3IN(T66I), NL4.3IN(R263K), NL4.3IN(T66I/R263K), 339
and NL4.3IN(T66I/E138K/R263K) viruses to dolutegravir (DTG), raltegravir (RAL), and 340
elvitegravir (EVG) as represented by IC50 and fold-change (FC) relative to NL4.3IN(WT) 341
virus. 342
References 343
1. Mesplede T, Wainberg MA. 2013. Integrase Strand Transfer Inhibitors in HIV Therapy. 344
Infect Dis Ther 2:83-93. 345
2. Mesplede T, Wainberg MA. 2014. Is resistance to dolutegravir possible when this drug 346
is used in first-line therapy? Viruses 6:3377-3385. 347
3. Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. 2011. HIV-1 integrase inhibitor 348
resistance and its clinical implications. J. Infect. Dis. 203:1204-1214. 349
4. Eron JJ, Clotet B, Durant J, Katlama C, Kumar P, Lazzarin A, Poizot-Martin I, Richmond 350
G, Soriano V, Ait-Khaled M, Fujiwara T, Huang J, Min S, Vavro C, Yeo J. 2013. Safety and 351
efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV 352
type 1 infection: 24-week results of the VIKING Study. J. Infect. Dis. 207:740-748. 353
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 18
18
5. Castagna A, Maggiolo F, Penco G, Wright D, Mills A, Grossberg R, Molina JM, Chas J, 354
Durant J, Moreno S, Doroana M, Ait-Khaled M, Huang J, Min S, Song I, Vavro C, Nichols 355
G, Yeo JM. 2014. Dolutegravir in antiretroviral-experienced patients with raltegravir- 356
and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J. 357
Infect. Dis. 210:354-362. 358
6. Akil B, Blick G, Hagins DP, Ramgopal MN, Richmond GJ, Samuel RM, Givens N, Vavro C, 359
Song IH, Wynne B, Ait-Khaled M. 2015. Dolutegravir versus placebo in subjects 360
harbouring HIV-1 with integrase inhibitor resistance associated substitutions: 48-week 361
results from VIKING-4, a randomized study. Antivir. Ther. 20:343-348. 362
7. Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, 363
Richmond G, Buendia CB, Fourie J, Ramgopal M, Hagins D, Felizarta F, Madruga J, 364
Reuter T, Newman T, Small CB, Lombaard J, Grinsztejn B, Dorey D, Underwood M, 365
Griffith S, Min S. 2013. Dolutegravir versus raltegravir in antiretroviral-experienced, 366
integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-367
blind, non-inferiority SAILING study. Lancet 382:700-708. 368
8. Mesplede T, Quashie PK, Osman N, Han Y, Singhroy DN, Lie Y, Petropoulos CJ, Huang 369
W, Wainberg MA. 2013. Viral fitness cost prevents HIV-1 from evading dolutegravir drug 370
pressure. Retrovirology 10:22. 371
9. Wares M, Mesplede T, Quashie PK, Osman N, Han Y, Wainberg MA. 2014. The M50I 372
polymorphic substitution in association with the R263K mutation in HIV-1 subtype B 373
integrase increases drug resistance but does not restore viral replicative fitness. 374
Retrovirology 11:7. 375
10. Mesplede T, Osman N, Wares M, Quashie PK, Hassounah S, Anstett K, Han Y, Singhroy 376
DN, Wainberg MA. 2014. Addition of E138K to R263K in HIV integrase increases 377
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 19
19
resistance to dolutegravir, but fails to restore activity of the HIV integrase enzyme and 378
viral replication capacity. J. Antimicrob. Chemother. 69:2733-2740. 379
11. Lataillade M, Chiarella J, Kozal MJ. 2007. Natural polymorphism of the HIV-1 integrase 380
gene and mutations associated with integrase inhibitor resistance. Antivir. Ther. 12: 381
563-570. 382
12. Singhroy DN, Wainberg MA, Mesplede T. 2015. Combination of the R263K and M184I/V 383
resistance substitutions against dolutegravir and lamivudine decreases HIV replicative 384
capacity. Antimicrob. Agents Chemother. 59:2882-2885. 385
13. Oliveira M, Mesplede T, Quashie PK, Moisi D, Wainberg MA. 2014. Resistance 386
mutations against dolutegravir in HIV integrase impair the emergence of resistance 387
against reverse transcriptase inhibitors. AIDS 28:813-819. 388
14. Cutillas V, Mesplede T, Anstett K, Hassounah S, Wainberg MA. 2015. The R262K 389
substitution combined with H51Y in HIV-1 subtype B integrase confers low-level 390
resistance against dolutegravir. Antimicrob. Agents Chemother. 59:310-316. 391
15. Quashie PK, Mesplede T, Han YS, Veres T, Osman N, Hassounah S, Sloan RD, Xu HT, 392
Wainberg MA. 2013. Biochemical analysis of the role of G118R-linked dolutegravir drug 393
resistance substitutions in HIV-1 integrase. Antimicrob. Agents Chemother. 57:6223-394
6235. 395
16. Han YS, Quashie P, Mesplede T, Xu H, Mekhssian K, Fenwick C, Wainberg MA. 2012. A 396
high-throughput assay for HIV-1 integrase 3'-processing activity using time-resolved 397
fluorescence. J. Virol. Methods 184:34-40. 398
17. Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, Foster SA, 399
Hazen RJ, Miki S, Suyama-Kagitani A, Kawauchi-Miki S, Taishi T, Kawasuji T, Johns BA, 400
Underwood MR, Garvey EP, Sato A, Fujiwara T. 2011. In Vitro antiretroviral properties 401
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 20
20
of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob. Agents 402
Chemother. 55:813-821. 403
18. White K, Kulkarni R, Miller MD. 2015. Analysis of early resistance development at the 404
first failure timepoint in elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil 405
fumarate-treated patients. J. Antimicrob. Chemother. 70: 2632-2638. 406
19. Hurt CB, Sebastian J, Hicks CB, Eron JJ. 2014. Resistance to HIV integrase strand transfer 407
inhibitors among clinical specimens in the United States, 2009-2012. Clin. Infect. Dis. 408
58:423-431. 409
20. Quashie PK, Mesplede T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, 410
Wainberg MA. 2012. Characterization of the R263K mutation in HIV-1 integrase that 411
confers low-level resistance to the second-generation integrase strand transfer inhibitor 412
dolutegravir. J. Virol. 86:2696-2705. 413
21. Margot NA, Hluhanich RM, Jones GS, Andreatta KN, Tsiang M, McColl DJ, White KL, 414
Miller MD. 2012. In vitro resistance selections using elvitegravir, raltegravir, and two 415
metabolites of elvitegravir M1 and M4. Antiviral Res. 93:288-296. 416
22. Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, Lazzarin A, Clotet 417
B, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Zhao J, Chen J, 418
Ryan DM, Rhodes RR, Killar JA, Gilde LR, Strohmaier KM, Meibohm AR, Miller MD, 419
Hazuda DJ, Nessly ML, DiNubile MJ, Isaacs RD, Teppler H, Nguyen BY. 2008. Subgroup 420
and resistance analyses of raltegravir for resistant HIV-1 infection. N. Engl. J. Med. 421
359:355-365. 422
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 21
WT R263K E138K/R263K
RAL 0.05-2.5 T66I, T97A, G163R -H51N, T66I, T97A,
S119R, Y143H
EVG 1 N155H, R263K M50I, T66IM50I, T66I, S119R,
S147G
Virus
Drug Concentration (μM)
Table 1. New substitutions emerging from NL4.3IN(WT), NL4.3IN(R263K), and NL4.3IN(E138K/R263K)
infections of CBMCs under raltegravir (RAL) or elvitegravir (EVG) drug pressure at week 30.
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 22
Genotype Relative Vmax (%) 95% CI of Relative Vmax (%) 1/2MaxDNA (nM) 95% CI (nM)
WT 100 91.6 to 108.5 10.15 7.5 to 12.8
T66I 67.3 56.4 to 78.2 24.4 15.5 to 33.2
R263K 89.1 74 to 104.3 20.1 12.1 to 28.2
T66I/R263K 13.0 10.4 to 15.6 23.2 12.9 to 33.6
E138K/R263K 38.7 32 to 45.5 5.15 2.0 to 8.3
T66I/E138K/R263K 17.1 13.9 to 20.2 8.4 3.5 to 13.3
Table 2. Strand-transfer activity of recombinant subtype B integrase enzymes containing the
T66I, E138K, and/or R263K substitutions.
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 23
Genotype Relative Vmax (%) 95% CI of Relative Vmax (%) 1/2MaxDNA (nM) 95% CI (nM)WT 100 81.9 to 118.1 1.3 0.63 to 1.9T66I 70.85 55.8 to 85.9 0.95 0.33 to 1.58R263K 69.2 55.7 to 82.6 0.99 0.41 to 1.6T66I/R263K 106.7 77 to 135.7 1.7 0.53 to 2.9E138K/R263K 126.2 98.1 to 154.4 1.83 0.85 to 2.8T66I/E138K/R263K 97.6 76.2 to 114.9 1.26 0.55 to 1.97
Table 3. 3’ processing activity of recombinant subtype B integrase containing the T66I, E138K, and/or R263K substitutions.
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 24
Genotype IC50 (nM) 95% CI (nM) FC IC50 (nM) 95% CI (nM) FC IC50 (nM) 95% CI (nM) FC
WT 0.3 0.2 to 0.5 1 1.3 0.3 to 5.3 1 6.4 2.5 to 16.6 1
T66I 0.0004 0.0002 to 0.0007 0.001 3.2 1.3 to 7.8 2.4 61 42.6 to 87.6 10
R263K 1.5 1.2 to 1.7 4.8 1.8 0.8 to 4.2 1.3 28 16.3 to 47 4
T66I/R263K 0.03 0.02 to 0.04 0.09 2.2 0.5 to 8.9 1.6 141 95 to 209.3 22
T66I/E138K/R263K 0.008 0.004 to 0.02 0.03 3.4 0.9 to 12.6 2.5 1,054 881.6 to 1261 164
DTG RAL EVG
Table 4. Susceptibility of NL4.3IN(WT), NL4.3IN(T66I), NL4.3IN(R263K), NL4.3IN(T66I/R263K), and NL4.3IN(T66I/E138K/R263K) viruses to dolutegravir (DTG),
raltegravir (RAL), and elvitegravir (EVG) represented by IC50 and fold change (FC) relative to NL4.3IN(WT) virus.
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 25
Figure 1. Viral infectivity in TZM-bl cells. TZM-bl cells were infected with (a) NL4.3IN(WT), NL4.3IN(T66I), or NL4.3IN(R263K); (b) NL4.3IN(WT) or NL4.3IN(T66I/R263K); (c) NL4.3IN(WT) or NL4.3IN(T66I/E138K/R263K) virus over 48h. Infectivity of NL4.3IN(WT) virus is represented in (a-c) for comparison. Luciferase levels were measured and (d) fold decrease in infectivity was calculated. Error bars indicate means ± standard deviation.
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from
Page 26
on April 5, 2018 by guest
http://jvi.asm.org/
Dow
nloaded from