JOURNAL OF COMPUTATIONAL BIOLOGY Volume 7, Numbers 3/4, 2000 Mary Ann Liebert, Inc. Pp. 537–558 The NOESY Jigsaw: Automated Protein Secondary Structure and Main-Chain Assignment from Sparse, Unassigned NMR Data CHRIS BAILEY-KELLOGG, 1 ALIK WIDGE, 1 JOHN J. KELLEY, 1;2 MARCELO J. BERARDI, 2 JOHN H. BUSHWELLER, 3 and BRUCE RANDALL DONALD 1;4 ABSTRACT High-throughput, data-directed computational protocols for Structural Genomics (or Pro- teomics) are required in order to evaluate the protein products of genes for structure and function at rates comparable to current gene-sequencing technology. This paper presents the JIGSAW algorithm, a novel high-throughput, automated approach to protein structure characterization with nuclear magnetic resonance (NMR). JIGSAW applies graph algorithms and probabilistic reasoning techniques, enforcing rst-principles consistency rules in order to overcome a 5–10% signal-to-noise ratio. It consists of two main components: (1) graph- based secondary structure pattern identication in unassigned heteronuclear NMR data, and (2) assignment of spectral peaks by probabilistic alignment of identied secondary structure elements against the primary sequence. Deferring assignment eliminates the bottleneck faced by traditional approaches, which begin by correlating peaks among dozens of experiments. JIGSAW utilizes only four experiments, none of which requires 13 C-labeled protein, thus dramatically reducing both the amount and expense of wet lab molecular biology and the total spectrometer time. Results for three test proteins demonstrate that JIGSAW correctly identies 79–100% of -helical and 46–65% of -sheet NOE connectivities and correctly aligns 33–100% of secondary structure elements. JIGSAW is very fast, running in minutes on a Pentium-class Linux workstation. This approach yields quick and reasonably accurate (as opposed to the traditional slow and extremely accurate) structure calculations. It could be useful for quick structural assays to speed data to the biologist early in an investigation and could in principle be applied in an automation-like fashion to a large fraction of the proteome. Key words: nuclear magnetic resonance spectroscopy, automated resonance assignment, structural genomics / proteomics, protein secondary structure, graph algorithms, probabilistic reasoning. 1 Dartmouth Computer Science Department, Hanover, NH 03755. 2 Dartmouth Chemistry Department, Hanover, NH 03755. 3 Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, VA 22906. 4 Dartmouth Computer Science Department, Hanover, NH 03755. 537
Transcript
JOURNAL OF COMPUTATIONAL BIOLOGYVolume 7 Numbers 34 2000Mary Ann Liebert IncPp 537ndash558
The NOESY Jigsaw Automated Protein SecondaryStructure and Main-Chain Assignment from
Sparse Unassigned NMR Data
CHRIS BAILEY-KELLOGG1 ALIK WIDGE1 JOHN J KELLEY12
MARCELO J BERARDI2 JOHN H BUSHWELLER3
and BRUCE RANDALL DONALD14
ABSTRACT
High-throughput data-directed computational protocols for Structural Genomics (or Pro-teomics) are required in order to evaluate the protein products of genes for structure andfunction at rates comparable to current gene-sequencing technology This paper presentsthe JIGSAW algorithm a novel high-throughput automated approach to protein structurecharacterization with nuclear magnetic resonance (NMR) JIGSAW applies graph algorithmsand probabilistic reasoning techniques enforcing rst-principles consistency rules in orderto overcome a 5ndash10 signal-to-noise ratio It consists of two main components (1) graph-based secondary structure pattern identi cation in unassigned heteronuclear NMR data and(2) assignment of spectral peaks by probabilistic alignment of identi ed secondary structureelements against the primary sequence Deferring assignment eliminates the bottleneck facedby traditional approaches which begin by correlating peaks among dozens of experimentsJIGSAW utilizes only four experiments none of which requires 13C-labeled protein thusdramatically reducing both the amount and expense of wet lab molecular biology and thetotal spectrometer time Results for three test proteins demonstrate that JIGSAW correctlyidenti es 79ndash100 of -helical and 46ndash65 of -sheet NOE connectivities and correctlyaligns 33ndash100 of secondary structure elements JIGSAW is very fast running in minuteson a Pentium-class Linux workstation This approach yields quick and reasonably accurate(as opposed to the traditional slow and extremely accurate) structure calculations It couldbe useful for quick structural assays to speed data to the biologist early in an investigationand could in principle be applied in an automation-like fashion to a large fraction of theproteome
Key words nuclear magnetic resonance spectroscopy automated resonance assignment structuralgenomics proteomics protein secondary structure graph algorithms probabilistic reasoning
1Dartmouth Computer Science Department Hanover NH 037552Dartmouth Chemistry Department Hanover NH 037553Molecular Physiology and Biological Physics University of Virginia Charlottesville VA 229064Dartmouth Computer Science Department Hanover NH 03755
537
538 BAILEY-KELLOGG ET AL
1 INTRODUCTION
Modern automated techniques are revolutionizing many aspects of biology for example sup-porting extremely fast gene sequencing and massively parallel gene expression testing (eg Chen
et al (1999) Hartuv et al (1999) Karp et al (1999)) Protein structure determination however remainsa long hard and expensive task The use of high-throughput structural genomics is required in order toapply modern techniques such as computer-aided drug design on a much larger scale In particular a keybottleneck in structure determination by nuclear magnetic resonance (NMR) is the resonance assignmentproblemmdashthe mapping of spectral peaks to tuples of interacting atoms in a protein For example spectralpeaks in a 3D nuclear Overhauser enhancement spectroscopy (NOESY) experiment establish distance re-straints on a proteinrsquos structure by identifying pairs of protons interacting through space Assignment isalso directly useful in techniques such as structure-activity relation (SAR) by NMR (Shuker et al 1996Hajduk et al 1997) and chemical shift mapping (Chen et al 1993) which compare NMR spectra for anisolated protein and a protein-ligand or proteinndashprotein complex
JIGSAW is a novel algorithm for automated main-chain assignment and secondary structure determina-tion It has been successfully applied to experimental spectra for three different proteins Human Glutare-doxin (Sun et al 1997) Core Binding Factor-Beta (Huang et al 1998) and Vaccinia Glutaredoxin-1(Kelley and Bushweller 1998) In order to enable high-throughput data collection JIGSAW utilizes onlyfour NMR experiments heteronuclear single quantum coherence spectroscopy (HSQC) HNndashHreg -correlationspectroscopy (HNHA) 80 ms total correlation spectroscopy (TOCSY) and NOESY This set of experi-ments requires only days of spectrometer time rather than the months required for the traditional set ofdozens of experiments Furthermore JIGSAW requires a protein to be only 15N-labeled a much cheaperand easier process than 13C labeling From a computational standpoint JIGSAW adopts a minimalistapproach demonstrating the large amount of information available in a few key spectra
Given the set of four spectra listed above JIGSAW identi es spectral peaks belonging to secondarystructure elements and assigns them to the corresponding residues in the proteinrsquos primary sequence Incontrast to theoretical and statistical approaches for secondary structure (eg Dealeage et al (1987) andCuff et al (1998)) and global fold (eg CASP (1999)) JIGSAW works in a data-driven manner Thecontinued necessity of experimental approaches is illustrated by the fact that one of our test proteins CBF-macr has a unique fold so that homology-based structure determination would not be applicable In contrastto secondary structure predictors JIGSAW provides not only an indication of secondary structure butalso tertiary macr-sheet connectivity Finally as noted above the spectral assignment produced by JIGSAWis itself an important product One use of assigned NMR data in addition to structure determination isthe analysis of protein structural dynamics from nuclear spin relaxation (eg Palmer et al (1996) Palmer(1997) Kay (1998)) Assignment is necessary to determine the residues implicated in the dynamics dataAnother important use of NMR assignments previously mentioned is SAR by NMR one of the mostimportant recent breakthroughs in experimental methods for high-throughput drug activity screening Evenif a crystal structure is already known these studies perform NMR experiments in order to analyze chemicalshift changes and determine ligand binding modes JIGSAW offers a high-throughput mechanism for therequired assignment process
In order to identify and assign spectral peaks belonging to secondary structure JIGSAW relies on two keyinsights graph-based secondary structure pattern discovery and assignment by alignment Atoms in regularsecondary structure interact in prototypical patterns experimentally observable in a NOESY spectrumTraditional NMR techniques determine residue sequentiality from a set of through-bond experiments andthen use NOE connectivities to test the secondary structure type of the residues JIGSAW on the other handstarts by looking for these patterns and uses their existence as evidence of residue sequentiality JIGSAWapplies a set of rst-principles constraints on valid groups of NOE interactions to manage the large searchspace of possible secondary structure patterns Subsequently JIGSAW assigns spectral peaks by aligningidenti ed residue sequences to the proteinrsquos primary sequence To do this JIGSAW uses side-chain peaksidenti ed in a TOCSY spectrum to estimate probable amino acid types for the residue sequence It ndssuch a sequence in the proteinrsquos primary sequence and assigns the spectral data accordingly
In its philosophy of starting with NOESY connectivities JIGSAW is in the same spirit as the partiallyautomated Main-Chain Directed (MCD) approach of Wand and coworkers (eg Stefano and Wand (1987)Englander and Wand (1987) and Nelson et al (1991)) MCD was developed for homonuclear spectraand was applied to experimental data for only one small protein human Ubiquitin (Stefano and Wand1987) JIGSAW on the other hand is fully automated and has been successfully applied to experimentalheteronuclear spectra for three different larger proteins (for example CBF-macr is nearly twice the size of
THE NOESY JIGSAW 539
Ubiquitin) JIGSAW takes the steps necessary to deal with the signi cant amount of degeneracy in spectrafor large proteins it also provides a formal graph-theoretic framework for understanding and analyzingthe algorithm Finally JIGSAW utilizes a novel TOCSY-based method for aligning residue sequences tothe primary sequence
The JIGSAW and MCD approaches differ greatly from other (automated and partially automated) as-signment protocols used today in the NMR community Most modern approaches rely on a large suiteof 13C-labeled triple resonance NMR spectra (eg HNCA HNCACB HN(CO)CACB hellip) either to es-tablish sequential connectivities by through-bond experiments (eg AUTOASSIGN (Zimmerman et al1997) and PASTA (Leutner et al 1998)) or to match chemical shift patterns (eg Lukin et al (1997) andCroft (1997)) As previously discussed JIGSAW requires only four spectra making it much more suit-able for high-throughput studies Many automated assignment packages boot-strap the assignment processFor example NOAH (Mumenthaler and Braun 1997 Mumenthaler et al 1997) uses assignments fromthrough-bond spectra to assign the NOESY GARANT (Bartels et al 1997) correlates observed peaksacross multiple spectra with peaks predicted by a sophisticated model Partially-computed structures canbe used to re ne peak predictions (eg Hare and Wagner (1999) Mumenthaler et al (1997) and Pearlman(1999))
The 13C labeling of a protein required by most automated assignment approaches is quite expensivemaking these approaches unsuitable for large-scale structural studies In return these protocols yield agreat deal of information (eg extensive side chain interactions) In contrast JIGSAW is much cheaper andfaster but does not obtain as much information Thus JIGSAW is especially suitable for quick structuralassays to speed data to the biologist early in an investigation and could in principle be applied in anautomation-like fashion to a large fraction of the proteome Furthermore the JIGSAW approach could alsoboth help and bene t from current work on large proteins and sparse NOE sets For example protocolsdeveloped for the analysis of large proteins use complete predeuteration to alleviate spectral crowding andto sharpen resolution in NOESY spectra (Grzesiek et al 1995 Venters 1995 Gardener et al 1997)These protocols yield only HNndashHN interactions and perhaps sparse HNndash1H interactions yet have provenuseful in structural studies even though they do not yield the extensive amount of information used bymost 13C-based approaches Synergies between such protocols and JIGSAW work in both directions Onone hand JIGSAW also uses HNndashHN and sparse HNndash1H interactions to perform its assignment andthus the protocols could be combined for studies of larger proteins On the other hand JIGSAW couldpotentially compute complete three-dimensional structures even with its limited set of spectra by leveragingthe techniques developed to determine global folds from sparse NOEs (eg Ayers et al (1999) Xu et al(2000) Standley et al (1999))
Solving the NMR jigsaw puzzle raises a number of interesting algorithmic pattern-matching and com-binatorial issues This paper presents an analysis of the problem algorithms to solve it and experimentalresults Section 2 reviews the information content available in the NMR spectra used by JIGSAW Section 3presents the graph-based formalism and algorithm for nding secondary structure elements in NOESY spec-tra Section 4 discusses the alignment process Sections 33 and 41 provide results on experimental datafrom three different proteins
2 NMR DATA
NMR spectra capture interactions between atoms as peaks in R2 or R3 where the axes indicate resonancefrequencies (chemical shifts) of atoms In the 15N spectra used by JIGSAW peaks correspond to an 15Natom an HN atom and possibly another 1H atom of particular resonance frequencies JIGSAW takes asinput in addition to a protein primary sequence lists of peak maxima and intensities correlated acrossspectra1
21 NMR spectra
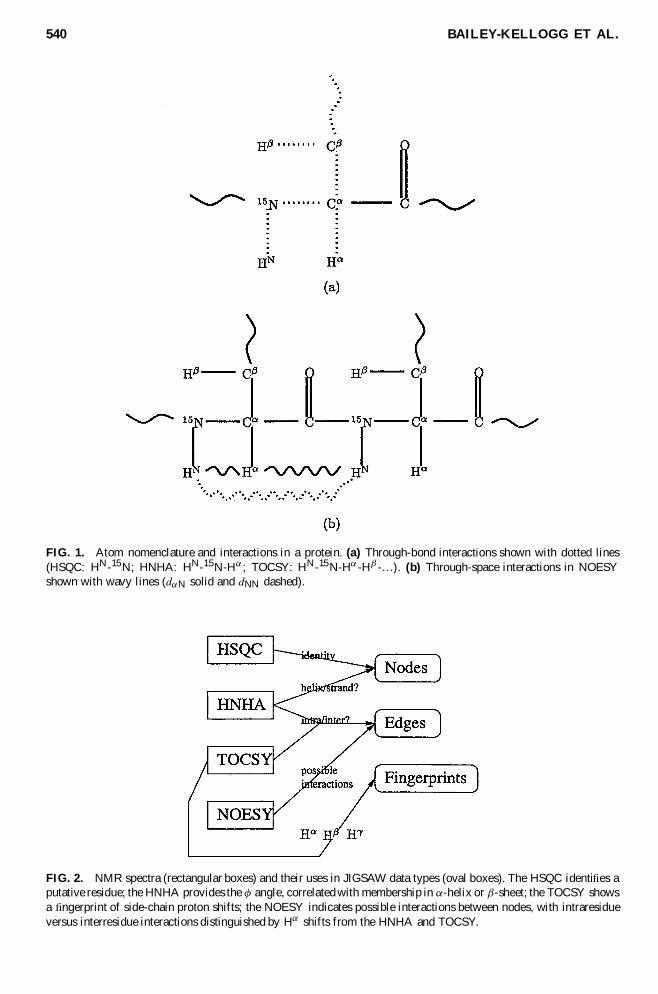
Figure 1 illustrates the experiments utilized by the JIGSAW algorithm and Figure 2 shows how theinformation content is encoded in data structures used by JIGSAW
1Automated peak picking is an interesting and well-studied signal processing problem (eg AUTOPSY (Koradiet al 1998))
540 BAILEY-KELLOGG ET AL
FIG 1 Atom nomenclature and interactions in a protein (a) Through-bond interactions shown with dotted lines(HSQC HN-15N HNHA HN-15N-Hreg TOCSY HN-15N-Hreg -Hmacr -hellip) (b) Through-space interactions in NOESYshown with wavy lines (dregN solid and dNN dashed)
FIG 2 NMR spectra (rectangular boxes) and their uses in JIGSAW data types (oval boxes) The HSQC identi es aputative residue the HNHA provides the Aacute angle correlated with membership in reg-helix or macr-sheet the TOCSY showsa ngerprint of side-chain proton shifts the NOESY indicates possible interactions between nodes with intraresidueversus interresidue interactions distinguished by Hreg shifts from the HNHA and TOCSY
THE NOESY JIGSAW 541
HSQC An HSQC spectrum (Cavanagh et al 1996 411ndash447) identi es unique pairs of through-bondcorrelated HN and 15N atoms Every residue has a unique such HNndash15N pair on the protein backbonethe coordinates for the pair are shared by all interactions within that residue and serve to referenceinteractions across all spectra2 Thus the HSQC serves to identify nodes (putative residues) for JIGSAW
HNHA An HNHA spectrum (Cavanagh et al 1996 524ndash528) captures interacting intraresidue through-bond HNndash15NndashHreg peak intensities estimate the J coupling constant 3JHNHreg which is correlated withthe Aacute bond angle of a residue Since this angle is characteristically different for reg-helices and macr-sheetsJIGSAW uses it as an estimator of the secondary structure type
TOCSY A TOCSY spectrum (Gronenborn et al 1989) includes through-bond interactions with 1Hatoms on a residuersquos side chain the 80 ms TOCSY in particular reaches many atoms on a residuersquosside chain Since the chemical shifts of 1H atoms for different amino acid types are characteristicallydifferent JIGSAW uses the shifts of a TOCSY as a ngerprint of the amino acid type
NOESY The 3D 15N-edited NOESY experiment (Gronenborn et al 1989) correlates an amide proton HN
and its 15N with a second proton that interacts through space at a distance less than 6 Aring via the NuclearOverhauser Effect (NOE) In the terminology of Wuumlthrich (1986) a dNN interaction represents an HNndashHNpair while a dregN interaction represents an HregndashHNpair (see Figure 1(b)) these can be distinguishedby the characteristically different chemical shifts of Hreg and HN atoms JIGSAW uses the NOE peaksto form edges between nodes for potentially interacting residues
22 NMR data structures
Using the information content of NMR spectra discussed in the preceding subsection JIGSAW buildstwo data structures an interaction graph connecting residue nodes and a set of ngerprints for each node
The rst data structure the NOESY interaction graph is an abstraction of a NOESY spectrum thatindicates potential residue interactions that could explain the peaks in a spectrum Each 3D interresidueNOE peak has the HN and 15N coordinates of one residue and the 1H coordinate of the Hreg or HN protonof another residue The HSQC indicates which is the rst residue by its unique HN and 15N coordinatesThe TOCSY and HNHA indicate residues whose Hreg or HN has the given 1H coordinate Unfortunatelyprojection onto the 1H dimension yields a large amount of spectral overlapmdash many protons have the samechemical shift within a tolerance For example there are 10ndash20 possible explanations for each peak in theNOESY spectrum of CBF-macr (see Section 33) yielding a 5ndash10 signal-to-noise ratio This spectral overlapis the major source of complexity in the JIGSAW approach The NOESY interaction graph captures thecomplete set of possible explanations for the peaks the JIGSAW search algorithm then determines thecorrect ones
De nition 1 (NOESY Interaction Graph) A NOESY interaction graph G 5 V E is a labeleddirected multigraph with vertices V corresponding to residues and edges E raquo V pound V such that e 5v1 v2 2 E iff there is a NOESY interaction between a proton of v1 and a proton of v2 Vertices andedges are labeled as follows
Secondary structure type label s V freg macr frac12g pound [0 1] indicates whether a residue is believed to bein an reg-helix a macr-sheet or other (random-coil) conformation and the level of con dence in that belief
Interaction type t E fdNN dregNg indicates a dregN or dNN interactionMatch score m E R1 is the 1H frequency difference between the observed peak and the shift of the
correlated Hreg or HNAtom distance d E R1 computed from the NOE peak intensity estimates the proximity of the
correlated atoms
A high match score suggests that a given edge rather than one of its competitors is the correct oneIn practice the NOESY interaction graph includes only edges for which the match score is below somethreshold (eg 005 ppm) Different atom distances are expected for atom pairs in different conformations(eg a pair of HN atoms in an reg-helix is expected to be quite close)
2Some side chains such as Gln have their own HNndash15N pairs as well These can be removed in preprocessing ordetected and handled specially
542 BAILEY-KELLOGG ET AL
This data structure provides a more abstract view of the NOESY information than typical atom-basedrepresentations (Wuumlthrich 1986 Stefano and Wand 1987) and is more amenable to search and analysis
The second data structure the TOCSY ngerprint collects all proton chemical shifts associated with agiven node
De nition 2 (Fingerprint) A ngerprint is a set of 1H chemical shifts correlated with a given residue(HN-15N pair)3
Section 4 uses ngerprints as indications of probable amino acid type in order to nd where in theprimary sequence to align a sequence of nodes belonging to a secondary structure element
In order to nd the correct secondary structure of a protein from the highly ambiguous NOESY inter-action graph JIGSAW employs a multistage search algorithm that enforces a set of consistency rules inpotential groups of edges The following subsections detail these consistency rules and the JIGSAW graphsearch algorithm
31 NOESY interaction graph constraints
Figure 3 shows some prototypical NOE interactions in (a) an reg-helix and (b) an anti-parallel macr-sheet(after Wuumlthrich 1986)4 Due to the way a helix is twisted the HN of one residue is close to the HN residueof the next and the Hreg of one residue is close to the HN of the residue one complete turn up the helixSince a macr-sheet is more stretched out only the HregndashHN sequential interactions are experimentally visible inthe NOESY but a rich pattern of cross-strand interactions are possible Figure 4 represents these patternsin NOESY interaction graphs and enumerates the interaction graph constraints imposed on these graphsby the geometry of helices and sheets5
De nition 3 (Consistency with Interaction Graph Constraints) A subgraph G0 of a NOESY interactiongraph G is consistent with the interaction graph constraints if there exists an ordering of the verticesV G0 into sequences such that every edge e 2 EG0 satis es one of the forms listed in Figure 4
While a NOESY interaction graph from experimental data contains many false edges (and some missingedges as well) the interaction graph constraints strongly limit how the correct edges t together As anexample consider the pattern in Figure 3(a) The large amount of noise in a NOESY interaction graphimplies that a vertex will have many (around 10mdashsee Section 2) dNN edges to vertices that could followit sequentially in an reg-helix However based on a simple joint probability model (and con rmed by thestatistics of Table 5 discussed below) an incorrect dNN edge is less likely also to have its symmetriccounterpart Similarly the probability of stringing together an incorrect sequence of four vertices andconnecting them with an additional dregN edge from the rst to the last is even less and the probabilitythat multiple such sequences adjoin each other is even less Intuitively while correct edges consistentlyreinforce each other incorrect edges tend to be randomly distributed and thus mutually inconsistent Thisinsight is repeatedly utilized in the JIGSAW algorithm
32 NOESY interaction graph search
The goal of the JIGSAW NOESY graph search is to nd a subgraph of a given interaction graphthat encodes the secondary structure of the protein Such a graph will have interactions indicative of thecorresponding secondary structure elements and thus will satisfy the interaction graph constraints
3The main-chain 15N chemical shift can also be included in the ngerprint4Parallel macr-sheets have similar interactions we illustrate JIGSAWrsquos approach by concentrating on anti-parallel
macr-sheets5Note that since 12Creg is not NMR-active dregN interactions are asymmetric
THE NOESY JIGSAW 543
FIG 3 NOESY dregN (solid) and dNN (dotted) interactions in (a) reg-helices and (b) macr-sheets
De nition 4 (Secondary Structure Graph) A secondary structure graph Gcurren is a subgraph of a NOESYinteraction graph G that is consistent with the interaction graph constraints (De nition 3)
Since a globally consistent graph consists of multiple locally consistent subgraphs each of constantsize JIGSAW does not have to solve a large subgraph isomorphism problem to obtain the entire secondarystructure
Figure 5 illustrates the key steps of the JIGSAW graph search algorithm Given an interaction graphJIGSAW identi es small fragment subgraphs (ldquojigsaw piecesrdquo) satisfying the interaction graph constraintsmerges them into reg-helices and pairs of adjacent macr-strands and collects the sequences into entire secondarystructure representations In practice there are many incorrect fragments among the correct ones but asdiscussed at the end of the previous section and supported in the results section mutual inconsistenciesgenerally keep them from merging into larger graphs A nal step is to rank the best solved jigsaws Thefollowing subsections detail these steps
321 Identify fragments The rst step of JIGSAW is to nd small consistent subgraphs of an inter-action graph JIGSAW searches for fragment instances of a set of fragment patterns evident in canonicalinteraction graphs (Figure 4)
De nition 5 (Fragment Pattern) A fragment pattern is a set of constraints on the connectivitiesinteraction types match scores and atom distances for a set of edges along with the secondary structuretype labels for the vertices
De nition 6 (Fragment) A fragment is a subgraph of an interaction graph satisfying the constraintsof a particular fragment pattern (Denition 5)
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
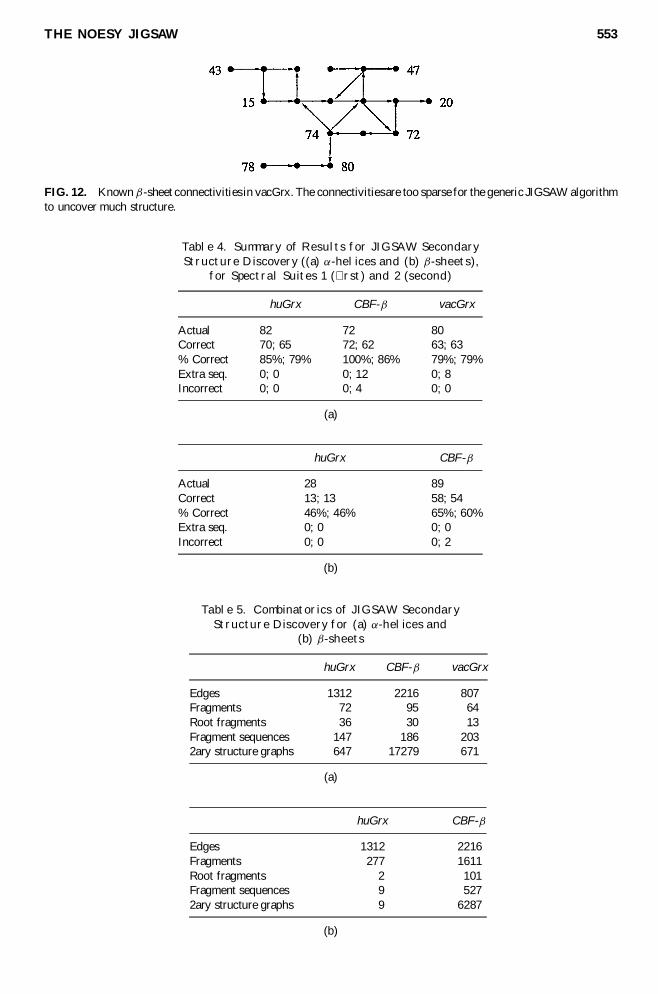
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
538 BAILEY-KELLOGG ET AL
1 INTRODUCTION
Modern automated techniques are revolutionizing many aspects of biology for example sup-porting extremely fast gene sequencing and massively parallel gene expression testing (eg Chen
et al (1999) Hartuv et al (1999) Karp et al (1999)) Protein structure determination however remainsa long hard and expensive task The use of high-throughput structural genomics is required in order toapply modern techniques such as computer-aided drug design on a much larger scale In particular a keybottleneck in structure determination by nuclear magnetic resonance (NMR) is the resonance assignmentproblemmdashthe mapping of spectral peaks to tuples of interacting atoms in a protein For example spectralpeaks in a 3D nuclear Overhauser enhancement spectroscopy (NOESY) experiment establish distance re-straints on a proteinrsquos structure by identifying pairs of protons interacting through space Assignment isalso directly useful in techniques such as structure-activity relation (SAR) by NMR (Shuker et al 1996Hajduk et al 1997) and chemical shift mapping (Chen et al 1993) which compare NMR spectra for anisolated protein and a protein-ligand or proteinndashprotein complex
JIGSAW is a novel algorithm for automated main-chain assignment and secondary structure determina-tion It has been successfully applied to experimental spectra for three different proteins Human Glutare-doxin (Sun et al 1997) Core Binding Factor-Beta (Huang et al 1998) and Vaccinia Glutaredoxin-1(Kelley and Bushweller 1998) In order to enable high-throughput data collection JIGSAW utilizes onlyfour NMR experiments heteronuclear single quantum coherence spectroscopy (HSQC) HNndashHreg -correlationspectroscopy (HNHA) 80 ms total correlation spectroscopy (TOCSY) and NOESY This set of experi-ments requires only days of spectrometer time rather than the months required for the traditional set ofdozens of experiments Furthermore JIGSAW requires a protein to be only 15N-labeled a much cheaperand easier process than 13C labeling From a computational standpoint JIGSAW adopts a minimalistapproach demonstrating the large amount of information available in a few key spectra
Given the set of four spectra listed above JIGSAW identi es spectral peaks belonging to secondarystructure elements and assigns them to the corresponding residues in the proteinrsquos primary sequence Incontrast to theoretical and statistical approaches for secondary structure (eg Dealeage et al (1987) andCuff et al (1998)) and global fold (eg CASP (1999)) JIGSAW works in a data-driven manner Thecontinued necessity of experimental approaches is illustrated by the fact that one of our test proteins CBF-macr has a unique fold so that homology-based structure determination would not be applicable In contrastto secondary structure predictors JIGSAW provides not only an indication of secondary structure butalso tertiary macr-sheet connectivity Finally as noted above the spectral assignment produced by JIGSAWis itself an important product One use of assigned NMR data in addition to structure determination isthe analysis of protein structural dynamics from nuclear spin relaxation (eg Palmer et al (1996) Palmer(1997) Kay (1998)) Assignment is necessary to determine the residues implicated in the dynamics dataAnother important use of NMR assignments previously mentioned is SAR by NMR one of the mostimportant recent breakthroughs in experimental methods for high-throughput drug activity screening Evenif a crystal structure is already known these studies perform NMR experiments in order to analyze chemicalshift changes and determine ligand binding modes JIGSAW offers a high-throughput mechanism for therequired assignment process
In order to identify and assign spectral peaks belonging to secondary structure JIGSAW relies on two keyinsights graph-based secondary structure pattern discovery and assignment by alignment Atoms in regularsecondary structure interact in prototypical patterns experimentally observable in a NOESY spectrumTraditional NMR techniques determine residue sequentiality from a set of through-bond experiments andthen use NOE connectivities to test the secondary structure type of the residues JIGSAW on the other handstarts by looking for these patterns and uses their existence as evidence of residue sequentiality JIGSAWapplies a set of rst-principles constraints on valid groups of NOE interactions to manage the large searchspace of possible secondary structure patterns Subsequently JIGSAW assigns spectral peaks by aligningidenti ed residue sequences to the proteinrsquos primary sequence To do this JIGSAW uses side-chain peaksidenti ed in a TOCSY spectrum to estimate probable amino acid types for the residue sequence It ndssuch a sequence in the proteinrsquos primary sequence and assigns the spectral data accordingly
In its philosophy of starting with NOESY connectivities JIGSAW is in the same spirit as the partiallyautomated Main-Chain Directed (MCD) approach of Wand and coworkers (eg Stefano and Wand (1987)Englander and Wand (1987) and Nelson et al (1991)) MCD was developed for homonuclear spectraand was applied to experimental data for only one small protein human Ubiquitin (Stefano and Wand1987) JIGSAW on the other hand is fully automated and has been successfully applied to experimentalheteronuclear spectra for three different larger proteins (for example CBF-macr is nearly twice the size of
THE NOESY JIGSAW 539
Ubiquitin) JIGSAW takes the steps necessary to deal with the signi cant amount of degeneracy in spectrafor large proteins it also provides a formal graph-theoretic framework for understanding and analyzingthe algorithm Finally JIGSAW utilizes a novel TOCSY-based method for aligning residue sequences tothe primary sequence
The JIGSAW and MCD approaches differ greatly from other (automated and partially automated) as-signment protocols used today in the NMR community Most modern approaches rely on a large suiteof 13C-labeled triple resonance NMR spectra (eg HNCA HNCACB HN(CO)CACB hellip) either to es-tablish sequential connectivities by through-bond experiments (eg AUTOASSIGN (Zimmerman et al1997) and PASTA (Leutner et al 1998)) or to match chemical shift patterns (eg Lukin et al (1997) andCroft (1997)) As previously discussed JIGSAW requires only four spectra making it much more suit-able for high-throughput studies Many automated assignment packages boot-strap the assignment processFor example NOAH (Mumenthaler and Braun 1997 Mumenthaler et al 1997) uses assignments fromthrough-bond spectra to assign the NOESY GARANT (Bartels et al 1997) correlates observed peaksacross multiple spectra with peaks predicted by a sophisticated model Partially-computed structures canbe used to re ne peak predictions (eg Hare and Wagner (1999) Mumenthaler et al (1997) and Pearlman(1999))
The 13C labeling of a protein required by most automated assignment approaches is quite expensivemaking these approaches unsuitable for large-scale structural studies In return these protocols yield agreat deal of information (eg extensive side chain interactions) In contrast JIGSAW is much cheaper andfaster but does not obtain as much information Thus JIGSAW is especially suitable for quick structuralassays to speed data to the biologist early in an investigation and could in principle be applied in anautomation-like fashion to a large fraction of the proteome Furthermore the JIGSAW approach could alsoboth help and bene t from current work on large proteins and sparse NOE sets For example protocolsdeveloped for the analysis of large proteins use complete predeuteration to alleviate spectral crowding andto sharpen resolution in NOESY spectra (Grzesiek et al 1995 Venters 1995 Gardener et al 1997)These protocols yield only HNndashHN interactions and perhaps sparse HNndash1H interactions yet have provenuseful in structural studies even though they do not yield the extensive amount of information used bymost 13C-based approaches Synergies between such protocols and JIGSAW work in both directions Onone hand JIGSAW also uses HNndashHN and sparse HNndash1H interactions to perform its assignment andthus the protocols could be combined for studies of larger proteins On the other hand JIGSAW couldpotentially compute complete three-dimensional structures even with its limited set of spectra by leveragingthe techniques developed to determine global folds from sparse NOEs (eg Ayers et al (1999) Xu et al(2000) Standley et al (1999))
Solving the NMR jigsaw puzzle raises a number of interesting algorithmic pattern-matching and com-binatorial issues This paper presents an analysis of the problem algorithms to solve it and experimentalresults Section 2 reviews the information content available in the NMR spectra used by JIGSAW Section 3presents the graph-based formalism and algorithm for nding secondary structure elements in NOESY spec-tra Section 4 discusses the alignment process Sections 33 and 41 provide results on experimental datafrom three different proteins
2 NMR DATA
NMR spectra capture interactions between atoms as peaks in R2 or R3 where the axes indicate resonancefrequencies (chemical shifts) of atoms In the 15N spectra used by JIGSAW peaks correspond to an 15Natom an HN atom and possibly another 1H atom of particular resonance frequencies JIGSAW takes asinput in addition to a protein primary sequence lists of peak maxima and intensities correlated acrossspectra1
21 NMR spectra
Figure 1 illustrates the experiments utilized by the JIGSAW algorithm and Figure 2 shows how theinformation content is encoded in data structures used by JIGSAW
1Automated peak picking is an interesting and well-studied signal processing problem (eg AUTOPSY (Koradiet al 1998))
540 BAILEY-KELLOGG ET AL
FIG 1 Atom nomenclature and interactions in a protein (a) Through-bond interactions shown with dotted lines(HSQC HN-15N HNHA HN-15N-Hreg TOCSY HN-15N-Hreg -Hmacr -hellip) (b) Through-space interactions in NOESYshown with wavy lines (dregN solid and dNN dashed)
FIG 2 NMR spectra (rectangular boxes) and their uses in JIGSAW data types (oval boxes) The HSQC identi es aputative residue the HNHA provides the Aacute angle correlated with membership in reg-helix or macr-sheet the TOCSY showsa ngerprint of side-chain proton shifts the NOESY indicates possible interactions between nodes with intraresidueversus interresidue interactions distinguished by Hreg shifts from the HNHA and TOCSY
THE NOESY JIGSAW 541
HSQC An HSQC spectrum (Cavanagh et al 1996 411ndash447) identi es unique pairs of through-bondcorrelated HN and 15N atoms Every residue has a unique such HNndash15N pair on the protein backbonethe coordinates for the pair are shared by all interactions within that residue and serve to referenceinteractions across all spectra2 Thus the HSQC serves to identify nodes (putative residues) for JIGSAW
HNHA An HNHA spectrum (Cavanagh et al 1996 524ndash528) captures interacting intraresidue through-bond HNndash15NndashHreg peak intensities estimate the J coupling constant 3JHNHreg which is correlated withthe Aacute bond angle of a residue Since this angle is characteristically different for reg-helices and macr-sheetsJIGSAW uses it as an estimator of the secondary structure type
TOCSY A TOCSY spectrum (Gronenborn et al 1989) includes through-bond interactions with 1Hatoms on a residuersquos side chain the 80 ms TOCSY in particular reaches many atoms on a residuersquosside chain Since the chemical shifts of 1H atoms for different amino acid types are characteristicallydifferent JIGSAW uses the shifts of a TOCSY as a ngerprint of the amino acid type
NOESY The 3D 15N-edited NOESY experiment (Gronenborn et al 1989) correlates an amide proton HN
and its 15N with a second proton that interacts through space at a distance less than 6 Aring via the NuclearOverhauser Effect (NOE) In the terminology of Wuumlthrich (1986) a dNN interaction represents an HNndashHNpair while a dregN interaction represents an HregndashHNpair (see Figure 1(b)) these can be distinguishedby the characteristically different chemical shifts of Hreg and HN atoms JIGSAW uses the NOE peaksto form edges between nodes for potentially interacting residues
22 NMR data structures
Using the information content of NMR spectra discussed in the preceding subsection JIGSAW buildstwo data structures an interaction graph connecting residue nodes and a set of ngerprints for each node
The rst data structure the NOESY interaction graph is an abstraction of a NOESY spectrum thatindicates potential residue interactions that could explain the peaks in a spectrum Each 3D interresidueNOE peak has the HN and 15N coordinates of one residue and the 1H coordinate of the Hreg or HN protonof another residue The HSQC indicates which is the rst residue by its unique HN and 15N coordinatesThe TOCSY and HNHA indicate residues whose Hreg or HN has the given 1H coordinate Unfortunatelyprojection onto the 1H dimension yields a large amount of spectral overlapmdash many protons have the samechemical shift within a tolerance For example there are 10ndash20 possible explanations for each peak in theNOESY spectrum of CBF-macr (see Section 33) yielding a 5ndash10 signal-to-noise ratio This spectral overlapis the major source of complexity in the JIGSAW approach The NOESY interaction graph captures thecomplete set of possible explanations for the peaks the JIGSAW search algorithm then determines thecorrect ones
De nition 1 (NOESY Interaction Graph) A NOESY interaction graph G 5 V E is a labeleddirected multigraph with vertices V corresponding to residues and edges E raquo V pound V such that e 5v1 v2 2 E iff there is a NOESY interaction between a proton of v1 and a proton of v2 Vertices andedges are labeled as follows
Secondary structure type label s V freg macr frac12g pound [0 1] indicates whether a residue is believed to bein an reg-helix a macr-sheet or other (random-coil) conformation and the level of con dence in that belief
Interaction type t E fdNN dregNg indicates a dregN or dNN interactionMatch score m E R1 is the 1H frequency difference between the observed peak and the shift of the
correlated Hreg or HNAtom distance d E R1 computed from the NOE peak intensity estimates the proximity of the
correlated atoms
A high match score suggests that a given edge rather than one of its competitors is the correct oneIn practice the NOESY interaction graph includes only edges for which the match score is below somethreshold (eg 005 ppm) Different atom distances are expected for atom pairs in different conformations(eg a pair of HN atoms in an reg-helix is expected to be quite close)
2Some side chains such as Gln have their own HNndash15N pairs as well These can be removed in preprocessing ordetected and handled specially
542 BAILEY-KELLOGG ET AL
This data structure provides a more abstract view of the NOESY information than typical atom-basedrepresentations (Wuumlthrich 1986 Stefano and Wand 1987) and is more amenable to search and analysis
The second data structure the TOCSY ngerprint collects all proton chemical shifts associated with agiven node
De nition 2 (Fingerprint) A ngerprint is a set of 1H chemical shifts correlated with a given residue(HN-15N pair)3
Section 4 uses ngerprints as indications of probable amino acid type in order to nd where in theprimary sequence to align a sequence of nodes belonging to a secondary structure element
In order to nd the correct secondary structure of a protein from the highly ambiguous NOESY inter-action graph JIGSAW employs a multistage search algorithm that enforces a set of consistency rules inpotential groups of edges The following subsections detail these consistency rules and the JIGSAW graphsearch algorithm
31 NOESY interaction graph constraints
Figure 3 shows some prototypical NOE interactions in (a) an reg-helix and (b) an anti-parallel macr-sheet(after Wuumlthrich 1986)4 Due to the way a helix is twisted the HN of one residue is close to the HN residueof the next and the Hreg of one residue is close to the HN of the residue one complete turn up the helixSince a macr-sheet is more stretched out only the HregndashHN sequential interactions are experimentally visible inthe NOESY but a rich pattern of cross-strand interactions are possible Figure 4 represents these patternsin NOESY interaction graphs and enumerates the interaction graph constraints imposed on these graphsby the geometry of helices and sheets5
De nition 3 (Consistency with Interaction Graph Constraints) A subgraph G0 of a NOESY interactiongraph G is consistent with the interaction graph constraints if there exists an ordering of the verticesV G0 into sequences such that every edge e 2 EG0 satis es one of the forms listed in Figure 4
While a NOESY interaction graph from experimental data contains many false edges (and some missingedges as well) the interaction graph constraints strongly limit how the correct edges t together As anexample consider the pattern in Figure 3(a) The large amount of noise in a NOESY interaction graphimplies that a vertex will have many (around 10mdashsee Section 2) dNN edges to vertices that could followit sequentially in an reg-helix However based on a simple joint probability model (and con rmed by thestatistics of Table 5 discussed below) an incorrect dNN edge is less likely also to have its symmetriccounterpart Similarly the probability of stringing together an incorrect sequence of four vertices andconnecting them with an additional dregN edge from the rst to the last is even less and the probabilitythat multiple such sequences adjoin each other is even less Intuitively while correct edges consistentlyreinforce each other incorrect edges tend to be randomly distributed and thus mutually inconsistent Thisinsight is repeatedly utilized in the JIGSAW algorithm
32 NOESY interaction graph search
The goal of the JIGSAW NOESY graph search is to nd a subgraph of a given interaction graphthat encodes the secondary structure of the protein Such a graph will have interactions indicative of thecorresponding secondary structure elements and thus will satisfy the interaction graph constraints
3The main-chain 15N chemical shift can also be included in the ngerprint4Parallel macr-sheets have similar interactions we illustrate JIGSAWrsquos approach by concentrating on anti-parallel
macr-sheets5Note that since 12Creg is not NMR-active dregN interactions are asymmetric
THE NOESY JIGSAW 543
FIG 3 NOESY dregN (solid) and dNN (dotted) interactions in (a) reg-helices and (b) macr-sheets
De nition 4 (Secondary Structure Graph) A secondary structure graph Gcurren is a subgraph of a NOESYinteraction graph G that is consistent with the interaction graph constraints (De nition 3)
Since a globally consistent graph consists of multiple locally consistent subgraphs each of constantsize JIGSAW does not have to solve a large subgraph isomorphism problem to obtain the entire secondarystructure
Figure 5 illustrates the key steps of the JIGSAW graph search algorithm Given an interaction graphJIGSAW identi es small fragment subgraphs (ldquojigsaw piecesrdquo) satisfying the interaction graph constraintsmerges them into reg-helices and pairs of adjacent macr-strands and collects the sequences into entire secondarystructure representations In practice there are many incorrect fragments among the correct ones but asdiscussed at the end of the previous section and supported in the results section mutual inconsistenciesgenerally keep them from merging into larger graphs A nal step is to rank the best solved jigsaws Thefollowing subsections detail these steps
321 Identify fragments The rst step of JIGSAW is to nd small consistent subgraphs of an inter-action graph JIGSAW searches for fragment instances of a set of fragment patterns evident in canonicalinteraction graphs (Figure 4)
De nition 5 (Fragment Pattern) A fragment pattern is a set of constraints on the connectivitiesinteraction types match scores and atom distances for a set of edges along with the secondary structuretype labels for the vertices
De nition 6 (Fragment) A fragment is a subgraph of an interaction graph satisfying the constraintsof a particular fragment pattern (Denition 5)
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 539
Ubiquitin) JIGSAW takes the steps necessary to deal with the signi cant amount of degeneracy in spectrafor large proteins it also provides a formal graph-theoretic framework for understanding and analyzingthe algorithm Finally JIGSAW utilizes a novel TOCSY-based method for aligning residue sequences tothe primary sequence
The JIGSAW and MCD approaches differ greatly from other (automated and partially automated) as-signment protocols used today in the NMR community Most modern approaches rely on a large suiteof 13C-labeled triple resonance NMR spectra (eg HNCA HNCACB HN(CO)CACB hellip) either to es-tablish sequential connectivities by through-bond experiments (eg AUTOASSIGN (Zimmerman et al1997) and PASTA (Leutner et al 1998)) or to match chemical shift patterns (eg Lukin et al (1997) andCroft (1997)) As previously discussed JIGSAW requires only four spectra making it much more suit-able for high-throughput studies Many automated assignment packages boot-strap the assignment processFor example NOAH (Mumenthaler and Braun 1997 Mumenthaler et al 1997) uses assignments fromthrough-bond spectra to assign the NOESY GARANT (Bartels et al 1997) correlates observed peaksacross multiple spectra with peaks predicted by a sophisticated model Partially-computed structures canbe used to re ne peak predictions (eg Hare and Wagner (1999) Mumenthaler et al (1997) and Pearlman(1999))
The 13C labeling of a protein required by most automated assignment approaches is quite expensivemaking these approaches unsuitable for large-scale structural studies In return these protocols yield agreat deal of information (eg extensive side chain interactions) In contrast JIGSAW is much cheaper andfaster but does not obtain as much information Thus JIGSAW is especially suitable for quick structuralassays to speed data to the biologist early in an investigation and could in principle be applied in anautomation-like fashion to a large fraction of the proteome Furthermore the JIGSAW approach could alsoboth help and bene t from current work on large proteins and sparse NOE sets For example protocolsdeveloped for the analysis of large proteins use complete predeuteration to alleviate spectral crowding andto sharpen resolution in NOESY spectra (Grzesiek et al 1995 Venters 1995 Gardener et al 1997)These protocols yield only HNndashHN interactions and perhaps sparse HNndash1H interactions yet have provenuseful in structural studies even though they do not yield the extensive amount of information used bymost 13C-based approaches Synergies between such protocols and JIGSAW work in both directions Onone hand JIGSAW also uses HNndashHN and sparse HNndash1H interactions to perform its assignment andthus the protocols could be combined for studies of larger proteins On the other hand JIGSAW couldpotentially compute complete three-dimensional structures even with its limited set of spectra by leveragingthe techniques developed to determine global folds from sparse NOEs (eg Ayers et al (1999) Xu et al(2000) Standley et al (1999))
Solving the NMR jigsaw puzzle raises a number of interesting algorithmic pattern-matching and com-binatorial issues This paper presents an analysis of the problem algorithms to solve it and experimentalresults Section 2 reviews the information content available in the NMR spectra used by JIGSAW Section 3presents the graph-based formalism and algorithm for nding secondary structure elements in NOESY spec-tra Section 4 discusses the alignment process Sections 33 and 41 provide results on experimental datafrom three different proteins
2 NMR DATA
NMR spectra capture interactions between atoms as peaks in R2 or R3 where the axes indicate resonancefrequencies (chemical shifts) of atoms In the 15N spectra used by JIGSAW peaks correspond to an 15Natom an HN atom and possibly another 1H atom of particular resonance frequencies JIGSAW takes asinput in addition to a protein primary sequence lists of peak maxima and intensities correlated acrossspectra1
21 NMR spectra
Figure 1 illustrates the experiments utilized by the JIGSAW algorithm and Figure 2 shows how theinformation content is encoded in data structures used by JIGSAW
1Automated peak picking is an interesting and well-studied signal processing problem (eg AUTOPSY (Koradiet al 1998))
540 BAILEY-KELLOGG ET AL
FIG 1 Atom nomenclature and interactions in a protein (a) Through-bond interactions shown with dotted lines(HSQC HN-15N HNHA HN-15N-Hreg TOCSY HN-15N-Hreg -Hmacr -hellip) (b) Through-space interactions in NOESYshown with wavy lines (dregN solid and dNN dashed)
FIG 2 NMR spectra (rectangular boxes) and their uses in JIGSAW data types (oval boxes) The HSQC identi es aputative residue the HNHA provides the Aacute angle correlated with membership in reg-helix or macr-sheet the TOCSY showsa ngerprint of side-chain proton shifts the NOESY indicates possible interactions between nodes with intraresidueversus interresidue interactions distinguished by Hreg shifts from the HNHA and TOCSY
THE NOESY JIGSAW 541
HSQC An HSQC spectrum (Cavanagh et al 1996 411ndash447) identi es unique pairs of through-bondcorrelated HN and 15N atoms Every residue has a unique such HNndash15N pair on the protein backbonethe coordinates for the pair are shared by all interactions within that residue and serve to referenceinteractions across all spectra2 Thus the HSQC serves to identify nodes (putative residues) for JIGSAW
HNHA An HNHA spectrum (Cavanagh et al 1996 524ndash528) captures interacting intraresidue through-bond HNndash15NndashHreg peak intensities estimate the J coupling constant 3JHNHreg which is correlated withthe Aacute bond angle of a residue Since this angle is characteristically different for reg-helices and macr-sheetsJIGSAW uses it as an estimator of the secondary structure type
TOCSY A TOCSY spectrum (Gronenborn et al 1989) includes through-bond interactions with 1Hatoms on a residuersquos side chain the 80 ms TOCSY in particular reaches many atoms on a residuersquosside chain Since the chemical shifts of 1H atoms for different amino acid types are characteristicallydifferent JIGSAW uses the shifts of a TOCSY as a ngerprint of the amino acid type
NOESY The 3D 15N-edited NOESY experiment (Gronenborn et al 1989) correlates an amide proton HN
and its 15N with a second proton that interacts through space at a distance less than 6 Aring via the NuclearOverhauser Effect (NOE) In the terminology of Wuumlthrich (1986) a dNN interaction represents an HNndashHNpair while a dregN interaction represents an HregndashHNpair (see Figure 1(b)) these can be distinguishedby the characteristically different chemical shifts of Hreg and HN atoms JIGSAW uses the NOE peaksto form edges between nodes for potentially interacting residues
22 NMR data structures
Using the information content of NMR spectra discussed in the preceding subsection JIGSAW buildstwo data structures an interaction graph connecting residue nodes and a set of ngerprints for each node
The rst data structure the NOESY interaction graph is an abstraction of a NOESY spectrum thatindicates potential residue interactions that could explain the peaks in a spectrum Each 3D interresidueNOE peak has the HN and 15N coordinates of one residue and the 1H coordinate of the Hreg or HN protonof another residue The HSQC indicates which is the rst residue by its unique HN and 15N coordinatesThe TOCSY and HNHA indicate residues whose Hreg or HN has the given 1H coordinate Unfortunatelyprojection onto the 1H dimension yields a large amount of spectral overlapmdash many protons have the samechemical shift within a tolerance For example there are 10ndash20 possible explanations for each peak in theNOESY spectrum of CBF-macr (see Section 33) yielding a 5ndash10 signal-to-noise ratio This spectral overlapis the major source of complexity in the JIGSAW approach The NOESY interaction graph captures thecomplete set of possible explanations for the peaks the JIGSAW search algorithm then determines thecorrect ones
De nition 1 (NOESY Interaction Graph) A NOESY interaction graph G 5 V E is a labeleddirected multigraph with vertices V corresponding to residues and edges E raquo V pound V such that e 5v1 v2 2 E iff there is a NOESY interaction between a proton of v1 and a proton of v2 Vertices andedges are labeled as follows
Secondary structure type label s V freg macr frac12g pound [0 1] indicates whether a residue is believed to bein an reg-helix a macr-sheet or other (random-coil) conformation and the level of con dence in that belief
Interaction type t E fdNN dregNg indicates a dregN or dNN interactionMatch score m E R1 is the 1H frequency difference between the observed peak and the shift of the
correlated Hreg or HNAtom distance d E R1 computed from the NOE peak intensity estimates the proximity of the
correlated atoms
A high match score suggests that a given edge rather than one of its competitors is the correct oneIn practice the NOESY interaction graph includes only edges for which the match score is below somethreshold (eg 005 ppm) Different atom distances are expected for atom pairs in different conformations(eg a pair of HN atoms in an reg-helix is expected to be quite close)
2Some side chains such as Gln have their own HNndash15N pairs as well These can be removed in preprocessing ordetected and handled specially
542 BAILEY-KELLOGG ET AL
This data structure provides a more abstract view of the NOESY information than typical atom-basedrepresentations (Wuumlthrich 1986 Stefano and Wand 1987) and is more amenable to search and analysis
The second data structure the TOCSY ngerprint collects all proton chemical shifts associated with agiven node
De nition 2 (Fingerprint) A ngerprint is a set of 1H chemical shifts correlated with a given residue(HN-15N pair)3
Section 4 uses ngerprints as indications of probable amino acid type in order to nd where in theprimary sequence to align a sequence of nodes belonging to a secondary structure element
In order to nd the correct secondary structure of a protein from the highly ambiguous NOESY inter-action graph JIGSAW employs a multistage search algorithm that enforces a set of consistency rules inpotential groups of edges The following subsections detail these consistency rules and the JIGSAW graphsearch algorithm
31 NOESY interaction graph constraints
Figure 3 shows some prototypical NOE interactions in (a) an reg-helix and (b) an anti-parallel macr-sheet(after Wuumlthrich 1986)4 Due to the way a helix is twisted the HN of one residue is close to the HN residueof the next and the Hreg of one residue is close to the HN of the residue one complete turn up the helixSince a macr-sheet is more stretched out only the HregndashHN sequential interactions are experimentally visible inthe NOESY but a rich pattern of cross-strand interactions are possible Figure 4 represents these patternsin NOESY interaction graphs and enumerates the interaction graph constraints imposed on these graphsby the geometry of helices and sheets5
De nition 3 (Consistency with Interaction Graph Constraints) A subgraph G0 of a NOESY interactiongraph G is consistent with the interaction graph constraints if there exists an ordering of the verticesV G0 into sequences such that every edge e 2 EG0 satis es one of the forms listed in Figure 4
While a NOESY interaction graph from experimental data contains many false edges (and some missingedges as well) the interaction graph constraints strongly limit how the correct edges t together As anexample consider the pattern in Figure 3(a) The large amount of noise in a NOESY interaction graphimplies that a vertex will have many (around 10mdashsee Section 2) dNN edges to vertices that could followit sequentially in an reg-helix However based on a simple joint probability model (and con rmed by thestatistics of Table 5 discussed below) an incorrect dNN edge is less likely also to have its symmetriccounterpart Similarly the probability of stringing together an incorrect sequence of four vertices andconnecting them with an additional dregN edge from the rst to the last is even less and the probabilitythat multiple such sequences adjoin each other is even less Intuitively while correct edges consistentlyreinforce each other incorrect edges tend to be randomly distributed and thus mutually inconsistent Thisinsight is repeatedly utilized in the JIGSAW algorithm
32 NOESY interaction graph search
The goal of the JIGSAW NOESY graph search is to nd a subgraph of a given interaction graphthat encodes the secondary structure of the protein Such a graph will have interactions indicative of thecorresponding secondary structure elements and thus will satisfy the interaction graph constraints
3The main-chain 15N chemical shift can also be included in the ngerprint4Parallel macr-sheets have similar interactions we illustrate JIGSAWrsquos approach by concentrating on anti-parallel
macr-sheets5Note that since 12Creg is not NMR-active dregN interactions are asymmetric
THE NOESY JIGSAW 543
FIG 3 NOESY dregN (solid) and dNN (dotted) interactions in (a) reg-helices and (b) macr-sheets
De nition 4 (Secondary Structure Graph) A secondary structure graph Gcurren is a subgraph of a NOESYinteraction graph G that is consistent with the interaction graph constraints (De nition 3)
Since a globally consistent graph consists of multiple locally consistent subgraphs each of constantsize JIGSAW does not have to solve a large subgraph isomorphism problem to obtain the entire secondarystructure
Figure 5 illustrates the key steps of the JIGSAW graph search algorithm Given an interaction graphJIGSAW identi es small fragment subgraphs (ldquojigsaw piecesrdquo) satisfying the interaction graph constraintsmerges them into reg-helices and pairs of adjacent macr-strands and collects the sequences into entire secondarystructure representations In practice there are many incorrect fragments among the correct ones but asdiscussed at the end of the previous section and supported in the results section mutual inconsistenciesgenerally keep them from merging into larger graphs A nal step is to rank the best solved jigsaws Thefollowing subsections detail these steps
321 Identify fragments The rst step of JIGSAW is to nd small consistent subgraphs of an inter-action graph JIGSAW searches for fragment instances of a set of fragment patterns evident in canonicalinteraction graphs (Figure 4)
De nition 5 (Fragment Pattern) A fragment pattern is a set of constraints on the connectivitiesinteraction types match scores and atom distances for a set of edges along with the secondary structuretype labels for the vertices
De nition 6 (Fragment) A fragment is a subgraph of an interaction graph satisfying the constraintsof a particular fragment pattern (Denition 5)
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
540 BAILEY-KELLOGG ET AL
FIG 1 Atom nomenclature and interactions in a protein (a) Through-bond interactions shown with dotted lines(HSQC HN-15N HNHA HN-15N-Hreg TOCSY HN-15N-Hreg -Hmacr -hellip) (b) Through-space interactions in NOESYshown with wavy lines (dregN solid and dNN dashed)
FIG 2 NMR spectra (rectangular boxes) and their uses in JIGSAW data types (oval boxes) The HSQC identi es aputative residue the HNHA provides the Aacute angle correlated with membership in reg-helix or macr-sheet the TOCSY showsa ngerprint of side-chain proton shifts the NOESY indicates possible interactions between nodes with intraresidueversus interresidue interactions distinguished by Hreg shifts from the HNHA and TOCSY
THE NOESY JIGSAW 541
HSQC An HSQC spectrum (Cavanagh et al 1996 411ndash447) identi es unique pairs of through-bondcorrelated HN and 15N atoms Every residue has a unique such HNndash15N pair on the protein backbonethe coordinates for the pair are shared by all interactions within that residue and serve to referenceinteractions across all spectra2 Thus the HSQC serves to identify nodes (putative residues) for JIGSAW
HNHA An HNHA spectrum (Cavanagh et al 1996 524ndash528) captures interacting intraresidue through-bond HNndash15NndashHreg peak intensities estimate the J coupling constant 3JHNHreg which is correlated withthe Aacute bond angle of a residue Since this angle is characteristically different for reg-helices and macr-sheetsJIGSAW uses it as an estimator of the secondary structure type
TOCSY A TOCSY spectrum (Gronenborn et al 1989) includes through-bond interactions with 1Hatoms on a residuersquos side chain the 80 ms TOCSY in particular reaches many atoms on a residuersquosside chain Since the chemical shifts of 1H atoms for different amino acid types are characteristicallydifferent JIGSAW uses the shifts of a TOCSY as a ngerprint of the amino acid type
NOESY The 3D 15N-edited NOESY experiment (Gronenborn et al 1989) correlates an amide proton HN
and its 15N with a second proton that interacts through space at a distance less than 6 Aring via the NuclearOverhauser Effect (NOE) In the terminology of Wuumlthrich (1986) a dNN interaction represents an HNndashHNpair while a dregN interaction represents an HregndashHNpair (see Figure 1(b)) these can be distinguishedby the characteristically different chemical shifts of Hreg and HN atoms JIGSAW uses the NOE peaksto form edges between nodes for potentially interacting residues
22 NMR data structures
Using the information content of NMR spectra discussed in the preceding subsection JIGSAW buildstwo data structures an interaction graph connecting residue nodes and a set of ngerprints for each node
The rst data structure the NOESY interaction graph is an abstraction of a NOESY spectrum thatindicates potential residue interactions that could explain the peaks in a spectrum Each 3D interresidueNOE peak has the HN and 15N coordinates of one residue and the 1H coordinate of the Hreg or HN protonof another residue The HSQC indicates which is the rst residue by its unique HN and 15N coordinatesThe TOCSY and HNHA indicate residues whose Hreg or HN has the given 1H coordinate Unfortunatelyprojection onto the 1H dimension yields a large amount of spectral overlapmdash many protons have the samechemical shift within a tolerance For example there are 10ndash20 possible explanations for each peak in theNOESY spectrum of CBF-macr (see Section 33) yielding a 5ndash10 signal-to-noise ratio This spectral overlapis the major source of complexity in the JIGSAW approach The NOESY interaction graph captures thecomplete set of possible explanations for the peaks the JIGSAW search algorithm then determines thecorrect ones
De nition 1 (NOESY Interaction Graph) A NOESY interaction graph G 5 V E is a labeleddirected multigraph with vertices V corresponding to residues and edges E raquo V pound V such that e 5v1 v2 2 E iff there is a NOESY interaction between a proton of v1 and a proton of v2 Vertices andedges are labeled as follows
Secondary structure type label s V freg macr frac12g pound [0 1] indicates whether a residue is believed to bein an reg-helix a macr-sheet or other (random-coil) conformation and the level of con dence in that belief
Interaction type t E fdNN dregNg indicates a dregN or dNN interactionMatch score m E R1 is the 1H frequency difference between the observed peak and the shift of the
correlated Hreg or HNAtom distance d E R1 computed from the NOE peak intensity estimates the proximity of the
correlated atoms
A high match score suggests that a given edge rather than one of its competitors is the correct oneIn practice the NOESY interaction graph includes only edges for which the match score is below somethreshold (eg 005 ppm) Different atom distances are expected for atom pairs in different conformations(eg a pair of HN atoms in an reg-helix is expected to be quite close)
2Some side chains such as Gln have their own HNndash15N pairs as well These can be removed in preprocessing ordetected and handled specially
542 BAILEY-KELLOGG ET AL
This data structure provides a more abstract view of the NOESY information than typical atom-basedrepresentations (Wuumlthrich 1986 Stefano and Wand 1987) and is more amenable to search and analysis
The second data structure the TOCSY ngerprint collects all proton chemical shifts associated with agiven node
De nition 2 (Fingerprint) A ngerprint is a set of 1H chemical shifts correlated with a given residue(HN-15N pair)3
Section 4 uses ngerprints as indications of probable amino acid type in order to nd where in theprimary sequence to align a sequence of nodes belonging to a secondary structure element
In order to nd the correct secondary structure of a protein from the highly ambiguous NOESY inter-action graph JIGSAW employs a multistage search algorithm that enforces a set of consistency rules inpotential groups of edges The following subsections detail these consistency rules and the JIGSAW graphsearch algorithm
31 NOESY interaction graph constraints
Figure 3 shows some prototypical NOE interactions in (a) an reg-helix and (b) an anti-parallel macr-sheet(after Wuumlthrich 1986)4 Due to the way a helix is twisted the HN of one residue is close to the HN residueof the next and the Hreg of one residue is close to the HN of the residue one complete turn up the helixSince a macr-sheet is more stretched out only the HregndashHN sequential interactions are experimentally visible inthe NOESY but a rich pattern of cross-strand interactions are possible Figure 4 represents these patternsin NOESY interaction graphs and enumerates the interaction graph constraints imposed on these graphsby the geometry of helices and sheets5
De nition 3 (Consistency with Interaction Graph Constraints) A subgraph G0 of a NOESY interactiongraph G is consistent with the interaction graph constraints if there exists an ordering of the verticesV G0 into sequences such that every edge e 2 EG0 satis es one of the forms listed in Figure 4
While a NOESY interaction graph from experimental data contains many false edges (and some missingedges as well) the interaction graph constraints strongly limit how the correct edges t together As anexample consider the pattern in Figure 3(a) The large amount of noise in a NOESY interaction graphimplies that a vertex will have many (around 10mdashsee Section 2) dNN edges to vertices that could followit sequentially in an reg-helix However based on a simple joint probability model (and con rmed by thestatistics of Table 5 discussed below) an incorrect dNN edge is less likely also to have its symmetriccounterpart Similarly the probability of stringing together an incorrect sequence of four vertices andconnecting them with an additional dregN edge from the rst to the last is even less and the probabilitythat multiple such sequences adjoin each other is even less Intuitively while correct edges consistentlyreinforce each other incorrect edges tend to be randomly distributed and thus mutually inconsistent Thisinsight is repeatedly utilized in the JIGSAW algorithm
32 NOESY interaction graph search
The goal of the JIGSAW NOESY graph search is to nd a subgraph of a given interaction graphthat encodes the secondary structure of the protein Such a graph will have interactions indicative of thecorresponding secondary structure elements and thus will satisfy the interaction graph constraints
3The main-chain 15N chemical shift can also be included in the ngerprint4Parallel macr-sheets have similar interactions we illustrate JIGSAWrsquos approach by concentrating on anti-parallel
macr-sheets5Note that since 12Creg is not NMR-active dregN interactions are asymmetric
THE NOESY JIGSAW 543
FIG 3 NOESY dregN (solid) and dNN (dotted) interactions in (a) reg-helices and (b) macr-sheets
De nition 4 (Secondary Structure Graph) A secondary structure graph Gcurren is a subgraph of a NOESYinteraction graph G that is consistent with the interaction graph constraints (De nition 3)
Since a globally consistent graph consists of multiple locally consistent subgraphs each of constantsize JIGSAW does not have to solve a large subgraph isomorphism problem to obtain the entire secondarystructure
Figure 5 illustrates the key steps of the JIGSAW graph search algorithm Given an interaction graphJIGSAW identi es small fragment subgraphs (ldquojigsaw piecesrdquo) satisfying the interaction graph constraintsmerges them into reg-helices and pairs of adjacent macr-strands and collects the sequences into entire secondarystructure representations In practice there are many incorrect fragments among the correct ones but asdiscussed at the end of the previous section and supported in the results section mutual inconsistenciesgenerally keep them from merging into larger graphs A nal step is to rank the best solved jigsaws Thefollowing subsections detail these steps
321 Identify fragments The rst step of JIGSAW is to nd small consistent subgraphs of an inter-action graph JIGSAW searches for fragment instances of a set of fragment patterns evident in canonicalinteraction graphs (Figure 4)
De nition 5 (Fragment Pattern) A fragment pattern is a set of constraints on the connectivitiesinteraction types match scores and atom distances for a set of edges along with the secondary structuretype labels for the vertices
De nition 6 (Fragment) A fragment is a subgraph of an interaction graph satisfying the constraintsof a particular fragment pattern (Denition 5)
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 541
HSQC An HSQC spectrum (Cavanagh et al 1996 411ndash447) identi es unique pairs of through-bondcorrelated HN and 15N atoms Every residue has a unique such HNndash15N pair on the protein backbonethe coordinates for the pair are shared by all interactions within that residue and serve to referenceinteractions across all spectra2 Thus the HSQC serves to identify nodes (putative residues) for JIGSAW
HNHA An HNHA spectrum (Cavanagh et al 1996 524ndash528) captures interacting intraresidue through-bond HNndash15NndashHreg peak intensities estimate the J coupling constant 3JHNHreg which is correlated withthe Aacute bond angle of a residue Since this angle is characteristically different for reg-helices and macr-sheetsJIGSAW uses it as an estimator of the secondary structure type
TOCSY A TOCSY spectrum (Gronenborn et al 1989) includes through-bond interactions with 1Hatoms on a residuersquos side chain the 80 ms TOCSY in particular reaches many atoms on a residuersquosside chain Since the chemical shifts of 1H atoms for different amino acid types are characteristicallydifferent JIGSAW uses the shifts of a TOCSY as a ngerprint of the amino acid type
NOESY The 3D 15N-edited NOESY experiment (Gronenborn et al 1989) correlates an amide proton HN
and its 15N with a second proton that interacts through space at a distance less than 6 Aring via the NuclearOverhauser Effect (NOE) In the terminology of Wuumlthrich (1986) a dNN interaction represents an HNndashHNpair while a dregN interaction represents an HregndashHNpair (see Figure 1(b)) these can be distinguishedby the characteristically different chemical shifts of Hreg and HN atoms JIGSAW uses the NOE peaksto form edges between nodes for potentially interacting residues
22 NMR data structures
Using the information content of NMR spectra discussed in the preceding subsection JIGSAW buildstwo data structures an interaction graph connecting residue nodes and a set of ngerprints for each node
The rst data structure the NOESY interaction graph is an abstraction of a NOESY spectrum thatindicates potential residue interactions that could explain the peaks in a spectrum Each 3D interresidueNOE peak has the HN and 15N coordinates of one residue and the 1H coordinate of the Hreg or HN protonof another residue The HSQC indicates which is the rst residue by its unique HN and 15N coordinatesThe TOCSY and HNHA indicate residues whose Hreg or HN has the given 1H coordinate Unfortunatelyprojection onto the 1H dimension yields a large amount of spectral overlapmdash many protons have the samechemical shift within a tolerance For example there are 10ndash20 possible explanations for each peak in theNOESY spectrum of CBF-macr (see Section 33) yielding a 5ndash10 signal-to-noise ratio This spectral overlapis the major source of complexity in the JIGSAW approach The NOESY interaction graph captures thecomplete set of possible explanations for the peaks the JIGSAW search algorithm then determines thecorrect ones
De nition 1 (NOESY Interaction Graph) A NOESY interaction graph G 5 V E is a labeleddirected multigraph with vertices V corresponding to residues and edges E raquo V pound V such that e 5v1 v2 2 E iff there is a NOESY interaction between a proton of v1 and a proton of v2 Vertices andedges are labeled as follows
Secondary structure type label s V freg macr frac12g pound [0 1] indicates whether a residue is believed to bein an reg-helix a macr-sheet or other (random-coil) conformation and the level of con dence in that belief
Interaction type t E fdNN dregNg indicates a dregN or dNN interactionMatch score m E R1 is the 1H frequency difference between the observed peak and the shift of the
correlated Hreg or HNAtom distance d E R1 computed from the NOE peak intensity estimates the proximity of the
correlated atoms
A high match score suggests that a given edge rather than one of its competitors is the correct oneIn practice the NOESY interaction graph includes only edges for which the match score is below somethreshold (eg 005 ppm) Different atom distances are expected for atom pairs in different conformations(eg a pair of HN atoms in an reg-helix is expected to be quite close)
2Some side chains such as Gln have their own HNndash15N pairs as well These can be removed in preprocessing ordetected and handled specially
542 BAILEY-KELLOGG ET AL
This data structure provides a more abstract view of the NOESY information than typical atom-basedrepresentations (Wuumlthrich 1986 Stefano and Wand 1987) and is more amenable to search and analysis
The second data structure the TOCSY ngerprint collects all proton chemical shifts associated with agiven node
De nition 2 (Fingerprint) A ngerprint is a set of 1H chemical shifts correlated with a given residue(HN-15N pair)3
Section 4 uses ngerprints as indications of probable amino acid type in order to nd where in theprimary sequence to align a sequence of nodes belonging to a secondary structure element
In order to nd the correct secondary structure of a protein from the highly ambiguous NOESY inter-action graph JIGSAW employs a multistage search algorithm that enforces a set of consistency rules inpotential groups of edges The following subsections detail these consistency rules and the JIGSAW graphsearch algorithm
31 NOESY interaction graph constraints
Figure 3 shows some prototypical NOE interactions in (a) an reg-helix and (b) an anti-parallel macr-sheet(after Wuumlthrich 1986)4 Due to the way a helix is twisted the HN of one residue is close to the HN residueof the next and the Hreg of one residue is close to the HN of the residue one complete turn up the helixSince a macr-sheet is more stretched out only the HregndashHN sequential interactions are experimentally visible inthe NOESY but a rich pattern of cross-strand interactions are possible Figure 4 represents these patternsin NOESY interaction graphs and enumerates the interaction graph constraints imposed on these graphsby the geometry of helices and sheets5
De nition 3 (Consistency with Interaction Graph Constraints) A subgraph G0 of a NOESY interactiongraph G is consistent with the interaction graph constraints if there exists an ordering of the verticesV G0 into sequences such that every edge e 2 EG0 satis es one of the forms listed in Figure 4
While a NOESY interaction graph from experimental data contains many false edges (and some missingedges as well) the interaction graph constraints strongly limit how the correct edges t together As anexample consider the pattern in Figure 3(a) The large amount of noise in a NOESY interaction graphimplies that a vertex will have many (around 10mdashsee Section 2) dNN edges to vertices that could followit sequentially in an reg-helix However based on a simple joint probability model (and con rmed by thestatistics of Table 5 discussed below) an incorrect dNN edge is less likely also to have its symmetriccounterpart Similarly the probability of stringing together an incorrect sequence of four vertices andconnecting them with an additional dregN edge from the rst to the last is even less and the probabilitythat multiple such sequences adjoin each other is even less Intuitively while correct edges consistentlyreinforce each other incorrect edges tend to be randomly distributed and thus mutually inconsistent Thisinsight is repeatedly utilized in the JIGSAW algorithm
32 NOESY interaction graph search
The goal of the JIGSAW NOESY graph search is to nd a subgraph of a given interaction graphthat encodes the secondary structure of the protein Such a graph will have interactions indicative of thecorresponding secondary structure elements and thus will satisfy the interaction graph constraints
3The main-chain 15N chemical shift can also be included in the ngerprint4Parallel macr-sheets have similar interactions we illustrate JIGSAWrsquos approach by concentrating on anti-parallel
macr-sheets5Note that since 12Creg is not NMR-active dregN interactions are asymmetric
THE NOESY JIGSAW 543
FIG 3 NOESY dregN (solid) and dNN (dotted) interactions in (a) reg-helices and (b) macr-sheets
De nition 4 (Secondary Structure Graph) A secondary structure graph Gcurren is a subgraph of a NOESYinteraction graph G that is consistent with the interaction graph constraints (De nition 3)
Since a globally consistent graph consists of multiple locally consistent subgraphs each of constantsize JIGSAW does not have to solve a large subgraph isomorphism problem to obtain the entire secondarystructure
Figure 5 illustrates the key steps of the JIGSAW graph search algorithm Given an interaction graphJIGSAW identi es small fragment subgraphs (ldquojigsaw piecesrdquo) satisfying the interaction graph constraintsmerges them into reg-helices and pairs of adjacent macr-strands and collects the sequences into entire secondarystructure representations In practice there are many incorrect fragments among the correct ones but asdiscussed at the end of the previous section and supported in the results section mutual inconsistenciesgenerally keep them from merging into larger graphs A nal step is to rank the best solved jigsaws Thefollowing subsections detail these steps
321 Identify fragments The rst step of JIGSAW is to nd small consistent subgraphs of an inter-action graph JIGSAW searches for fragment instances of a set of fragment patterns evident in canonicalinteraction graphs (Figure 4)
De nition 5 (Fragment Pattern) A fragment pattern is a set of constraints on the connectivitiesinteraction types match scores and atom distances for a set of edges along with the secondary structuretype labels for the vertices
De nition 6 (Fragment) A fragment is a subgraph of an interaction graph satisfying the constraintsof a particular fragment pattern (Denition 5)
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
542 BAILEY-KELLOGG ET AL
This data structure provides a more abstract view of the NOESY information than typical atom-basedrepresentations (Wuumlthrich 1986 Stefano and Wand 1987) and is more amenable to search and analysis
The second data structure the TOCSY ngerprint collects all proton chemical shifts associated with agiven node
De nition 2 (Fingerprint) A ngerprint is a set of 1H chemical shifts correlated with a given residue(HN-15N pair)3
Section 4 uses ngerprints as indications of probable amino acid type in order to nd where in theprimary sequence to align a sequence of nodes belonging to a secondary structure element
In order to nd the correct secondary structure of a protein from the highly ambiguous NOESY inter-action graph JIGSAW employs a multistage search algorithm that enforces a set of consistency rules inpotential groups of edges The following subsections detail these consistency rules and the JIGSAW graphsearch algorithm
31 NOESY interaction graph constraints
Figure 3 shows some prototypical NOE interactions in (a) an reg-helix and (b) an anti-parallel macr-sheet(after Wuumlthrich 1986)4 Due to the way a helix is twisted the HN of one residue is close to the HN residueof the next and the Hreg of one residue is close to the HN of the residue one complete turn up the helixSince a macr-sheet is more stretched out only the HregndashHN sequential interactions are experimentally visible inthe NOESY but a rich pattern of cross-strand interactions are possible Figure 4 represents these patternsin NOESY interaction graphs and enumerates the interaction graph constraints imposed on these graphsby the geometry of helices and sheets5
De nition 3 (Consistency with Interaction Graph Constraints) A subgraph G0 of a NOESY interactiongraph G is consistent with the interaction graph constraints if there exists an ordering of the verticesV G0 into sequences such that every edge e 2 EG0 satis es one of the forms listed in Figure 4
While a NOESY interaction graph from experimental data contains many false edges (and some missingedges as well) the interaction graph constraints strongly limit how the correct edges t together As anexample consider the pattern in Figure 3(a) The large amount of noise in a NOESY interaction graphimplies that a vertex will have many (around 10mdashsee Section 2) dNN edges to vertices that could followit sequentially in an reg-helix However based on a simple joint probability model (and con rmed by thestatistics of Table 5 discussed below) an incorrect dNN edge is less likely also to have its symmetriccounterpart Similarly the probability of stringing together an incorrect sequence of four vertices andconnecting them with an additional dregN edge from the rst to the last is even less and the probabilitythat multiple such sequences adjoin each other is even less Intuitively while correct edges consistentlyreinforce each other incorrect edges tend to be randomly distributed and thus mutually inconsistent Thisinsight is repeatedly utilized in the JIGSAW algorithm
32 NOESY interaction graph search
The goal of the JIGSAW NOESY graph search is to nd a subgraph of a given interaction graphthat encodes the secondary structure of the protein Such a graph will have interactions indicative of thecorresponding secondary structure elements and thus will satisfy the interaction graph constraints
3The main-chain 15N chemical shift can also be included in the ngerprint4Parallel macr-sheets have similar interactions we illustrate JIGSAWrsquos approach by concentrating on anti-parallel
macr-sheets5Note that since 12Creg is not NMR-active dregN interactions are asymmetric
THE NOESY JIGSAW 543
FIG 3 NOESY dregN (solid) and dNN (dotted) interactions in (a) reg-helices and (b) macr-sheets
De nition 4 (Secondary Structure Graph) A secondary structure graph Gcurren is a subgraph of a NOESYinteraction graph G that is consistent with the interaction graph constraints (De nition 3)
Since a globally consistent graph consists of multiple locally consistent subgraphs each of constantsize JIGSAW does not have to solve a large subgraph isomorphism problem to obtain the entire secondarystructure
Figure 5 illustrates the key steps of the JIGSAW graph search algorithm Given an interaction graphJIGSAW identi es small fragment subgraphs (ldquojigsaw piecesrdquo) satisfying the interaction graph constraintsmerges them into reg-helices and pairs of adjacent macr-strands and collects the sequences into entire secondarystructure representations In practice there are many incorrect fragments among the correct ones but asdiscussed at the end of the previous section and supported in the results section mutual inconsistenciesgenerally keep them from merging into larger graphs A nal step is to rank the best solved jigsaws Thefollowing subsections detail these steps
321 Identify fragments The rst step of JIGSAW is to nd small consistent subgraphs of an inter-action graph JIGSAW searches for fragment instances of a set of fragment patterns evident in canonicalinteraction graphs (Figure 4)
De nition 5 (Fragment Pattern) A fragment pattern is a set of constraints on the connectivitiesinteraction types match scores and atom distances for a set of edges along with the secondary structuretype labels for the vertices
De nition 6 (Fragment) A fragment is a subgraph of an interaction graph satisfying the constraintsof a particular fragment pattern (Denition 5)
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 543
FIG 3 NOESY dregN (solid) and dNN (dotted) interactions in (a) reg-helices and (b) macr-sheets
De nition 4 (Secondary Structure Graph) A secondary structure graph Gcurren is a subgraph of a NOESYinteraction graph G that is consistent with the interaction graph constraints (De nition 3)
Since a globally consistent graph consists of multiple locally consistent subgraphs each of constantsize JIGSAW does not have to solve a large subgraph isomorphism problem to obtain the entire secondarystructure
Figure 5 illustrates the key steps of the JIGSAW graph search algorithm Given an interaction graphJIGSAW identi es small fragment subgraphs (ldquojigsaw piecesrdquo) satisfying the interaction graph constraintsmerges them into reg-helices and pairs of adjacent macr-strands and collects the sequences into entire secondarystructure representations In practice there are many incorrect fragments among the correct ones but asdiscussed at the end of the previous section and supported in the results section mutual inconsistenciesgenerally keep them from merging into larger graphs A nal step is to rank the best solved jigsaws Thefollowing subsections detail these steps
321 Identify fragments The rst step of JIGSAW is to nd small consistent subgraphs of an inter-action graph JIGSAW searches for fragment instances of a set of fragment patterns evident in canonicalinteraction graphs (Figure 4)
De nition 5 (Fragment Pattern) A fragment pattern is a set of constraints on the connectivitiesinteraction types match scores and atom distances for a set of edges along with the secondary structuretype labels for the vertices
De nition 6 (Fragment) A fragment is a subgraph of an interaction graph satisfying the constraintsof a particular fragment pattern (Denition 5)
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
544 BAILEY-KELLOGG ET AL
FIG 4 Interaction graphs (dregN edges solid and dNN dotted) and constraints for (a) reg-helices and (b) macr-sheets This gure shows perfect patterns Interaction graphs in experimental NMR data contain signi cant noise manifested assome missing and many extra graph edges
Figure 6 illustrates the connectivities of some such fragment patterns Each pattern instance groups asmall set of edges (representing NOE peaks) that are mutually consistent Fragment patterns also allowthe possibility of missing edges in experimental data The directions of the missing edges are howeverdetermined by those of the other edges For example in Figure 6(b) patterns 3 and 4 are similar to patterns 1and 2 respectively the direction of the missing vertical edge can be inferred from the correspondence
Fragments are identi ed by a straightforward graph search search from each node forming paths ofedges that remain consistent with the pattern Table 1 provides pseudocode for this search The algorithmassumes that the connectivities of a fragment pattern are ordered so that the rst p1 edges each connectto exactly one new node and the remaining edges up to p total each connect only already-visited nodesSince each pattern speci es a connected subgraph such an ordering is guaranteed to exist Arguments F
and T to the algorithm specify the indices of the from- and to-nodes for an edge respectively For examplethe search for pattern 1 in Figure 6(a) would start from the leftmost node nd all edges from that nodeforward to the second node nd all edges from that node forward to the third node (so that the secondnode found the to-node of the rst edge serves as the from-node for possible second edges ie F2 5 2)and so forth
The algorithm starts from each of n nodes and searches to a xed depth of p for a pattern of p edgesexamining only the edges from a speci ed node at each step A bound d on the maximum degree of eachnode permits a bound on the complexity of the search we perform n searches each of size Odp
Claim 1 (Computational Complexity of Fragment Pattern Identi cation) Given an interaction graphwith n nodes and maximum degree d instances of a fragment pattern involving p edges can be identi edin time Ondp
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 545
FIG 5 JIGSAW algorithm overview (a) identify graph fragments (b) merge them sequentially and (c) collect theminto complete secondary structure graphs Only correct fragments are shown here Graphs from experimental data alsogenerate a large number of incorrect fragments but mutual inconsistencies prevent them from forming either longsequences or large secondary structure graphs
In practice (as demonstrated in Table 5 below) the interaction graph constraints greatly restrict thesearch pruning most paths before they reach a depth of p
We assume that the fragment patterns generate a complete set of fragments That is any secondarystructure graph Gcurren for a given interaction graph G can be formed from a union of the fragments identi edin G Due to the large number of incorrect edges there can also be many incorrect fragments It remainsfor the subsequent processing stages (below) to eliminate them
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
546 BAILEY-KELLOGG ET AL
FIG 6 Interaction graph fragment patterns in (a) reg-helices and (b) macr-sheets
322 Merge sequentially-consistent fragments Given a set of fragment ldquojigsaw piecesrdquo F JIGSAWstarts solving the puzzle of secondary structure by nding sequences of fragments whose union de neseither an reg-helix or two neighboring strands of a macr-sheet and is consistent with the interaction graphconstraints To reduce the computational cost it is possible to identify a set of root fragments F 0 sup3 F thatsatisfy stronger constraints and to root the sequences at these fragments
Table 1 Pseudocode for JIGSAW Graph Fragment Identi cation1
Function fragmentsG p1 p F T
Set G not For each v 2 V G
Let G1 5 ffv toeg feg j frome 5 vgFor i 5 2p1
Let Gi 5 fV G [ ftoeg EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFig
For i 5 p1 1 1p
Let Gi 5 fV G EG [ feg j G 2 Gi iexcl 1 ^ frome 5 V GFi ^ toe 5 V GTi
gSet G not G [ Gp
return G
1Add edges to the growing fragments such that each additional edge is from the speci ed already-found vertex(up to p1) or between the speci ed already-found vertices (after p1) Other constraints (eg type and match score) lter the sets but are not shown here for simplicity
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 547
Table 2 Pseudocode for JIGSAW Fragment Sequence Growth1
Function sequencesF F0Set S not F0
While S continues to growSet S not fV S [ V F ES [ EF j S 2 S ^ F 2 F ^ S [ F connected ^ S [ F consistentg
return S
1Add fragments to the growing sequences such that each additional fragment is connected to the left or right end of asequence (ie to a node with no forward or no backward adjacency) and the new sequence satis es the interaction graphconstraints
De nition 7 (Rooted Fragment Sequence) Given a set of fragments F for an interaction graph G anda set of chosen root fragments F 0 sup3 F a rooted fragment sequence F is a subgraph of G consistent withthe interaction graph constraints for either a single reg-helix or a pair of adjacent macr-strands and formedfrom the union of a set of n fragments F 5 ff1 f2 fng raquo F where f1 2 F 0
Fragment sequences are computed by a straightforward exhaustive search from the root fragments(Table 2 provides pseudocode) In the worst case there are an exponential number of sequencesmdashif anyfragment can connect to any other then there are jFj possible such sequences However as with fragmentpattern identi cation the interaction graph constraints limit the possible sequences and as Table 5 willillustrate the number of sequences generated from an initial fragment is much less than this upper bound
The completeness of fragment sequences follows immediately from the assumed completeness of frag-ments if there is at least one root fragment per helix or strand pair We state the claim here for completenessof exposition
Claim 2 (Completeness of Fragment Sequences) Any secondary structure graph Gcurren for a giveninteraction graph G is a union of the fragment sequences for the fragments F in G
323 Collect consistent sequences To obtain an entire consistent secondary structure graph for theprotein JIGSAW forms unions of consistent fragment sequences (see Table 3 for the speci cation) Im-posing directionalitymdash rst identifying sequences and then joining themmdashgreatly reduces the size andredundancy of the search space While the merging step is worst-case exponential in the number of frag-ment sequences again in practice the interaction graph constraints keep the search subexponential (aswill be shown in Table 5) and allow the algorithm to run in only minutes
As with fragment sequences the completeness result follows immediately from the de nition and isstated here for purposes of formalization
Claim 3 (Completeness of Secondary Structure Graphs) JIGSAW nds all consistent secondary struc-ture graphs Gcurren for a given interaction graph G
324 Identify best secondary structure graphs The nal step in the JIGSAW graph search is to identifythe best secondary structure graphs from the set of collected possibilities Intuitively the algorithm shouldproduce a large graph reaching all the vertices expected to belong to the given secondary structure typeSmaller graphs probably were not expanded due to inconsistencies Furthermore as many of the expectededges as possible should belong to the graph (vertices should have high degree) and should have goodmatch scores
Table 3 Pseudocode for JIGSAW Sequence Collection1
Function secondary_structuresS
return fG0 5 [
G2S
V G[
G2S
EG j S sup3 S ^ G0 consistentg
1Find subsets of the fragment sequences such that the resulting graphsatis es the interaction graph constraints
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
548 BAILEY-KELLOGG ET AL
This intuition is formalized with a probabilistic measure of a graphrsquos correctness For simplicity weassume a Gaussian a priori probability that an edge e indicates the correct interaction represented bya spectral peak based on comparison of 1H chemical shifts (recall that the match score me encodesthe differencemdashsee De nition 1) it remains interesting future work to incorporate actual spectral ldquolineshapesrdquo (Koradi et al 1998) into this analysis Normalization over all edges generated for the peak yieldsthe probability that that edge is a good explanation for its peak This yields a higher probability when apeak closely matches and when it does not have many good competitors
P interactione 5 Gfrac34 me (1)
P goode 5P interactione
X
e02Ce
P interactione0(2)
where Gfrac34 denotes a Gaussian of width frac34 and C denotes the set of edges generated for the peak ofa given edge
The correctness probability for a secondary structure graph Gcurren depends the goodness of its edges
P correctGcurren 5 1 iexclY
e2Gcurren
1 iexcl P goode (3)
The correctness probability can be applied during fragment sequence enumeration (Section 322) andsecondary structure graph construction (Section 323) in order to prune graphs with too little support(correctness probability too low for the graph size)
33 Experimental results
JIGSAW was tested on experimental data for Human Glutaredoxin (huGrx) (Sun et al 1997) CoreBinding Factor-Beta (CBF-macr) (Huang et al 1998) and Vaccinia Glutaredoxin-1 (vacGrx) (Kelley andBushweller 1998)6 The 15N-edited HSQC HNHA 80 ms TOCSY and NOESY spectra were collectedon a 500MHz Varian spectrometer at Dartmouth and processed with the program PROSA (Guumlntert et al1992) Peaks were picked manually and in a semi-automated fashion with the program XEASY (Bartelset al 1995) JIGSAW was invoked with the appropriate primary sequences and ASCII peak lists referencedacross spectra7 In order to distinguish the dependence on HNHA from the dependence on NOESYJIGSAW was run with two spectral suites the rst with simulated J-coupling constants set at the nominalvalues for the correct secondary structure type and the second with J-coupling constants computed fromthe experimental HNHA data all other spectra were the same in the two suites JIGSAW used the patternsof Figure 4 with a set of generic constraints on match score and atom distance Computation took aboutone to ten minutes depending on the protein
Figures 7 8 and 9 depict the reg-helices discovered by JIGSAW in CBF-macr huGrx and vacGrx respec-tively with both suites of spectra The results are similar for both suites except that reg-helices in suite 2sometimes extend past or fail to reach the end of an reg-helix or macr-strand due to misleading J constantsIn vacGrx under suite 2 an additional potential rigid piece of secondary structure is uncovered extendingfrom residue 48 to residue 51
Figures 10 and 11 show the macr-sheets uncovered by JIGSAW in CBF-macr and huGrx respectively usingsuite 2 The results for CBF-macr with suite 1 are the same as in Figure 10 but with the correct edges toresidue 100 rather than the incorrect edges to 101 and 71 The results for huGrx with suite 1 are identicalin both cases connectivity in the lower two strands of huGrx is too sparse for JIGSAW Figure 12 showsthat the NOESY connectivities for macr-sheets in vacGrx are too sparse for general-purpose JIGSAW patternsto detect These test cases demonstrate that JIGSAW correctly uncovers a signi cant portion of the macr
structure particularly in well-connected portions of the graph Note that macr-sheets are tertiary structureindicating more than just the sequentiality of their strands
The purpose of the graph search is to identify the small fraction of edges in the NOESY interaction graphthat are actually involved in secondary structure Ultimately this means that the algorithm is identifying
6While huGrx and vacGrx have similar structures their experimental spectra have signi cant differences7For CBF-macr JIGSAW uses manually computed J-constants following the NMR protocol of Huang et al (1998)
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 549
FIG 7 reg-helices of CBF-macr computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative X 5 false positive Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
for each NOE peak which putative residues are interacting to cause that peak Thus appropriate metrics forJIGSAWrsquos graph search performance are the numbers of correct and incorrect edgespeaks identi ed basedon the actual assignments known from the literature Table 4 summarizes the results for all three proteins Italso includes the number of ldquoextrardquo edges that are not considered part of the secondary structure elementsbut are still sequentially correct With spectral suite 2 JIGSAW is less accurate about the extent of a helixor strand however the actual extent is ambiguous and extending to additional sequentially-connectedresidues can be bene cial by providing additional assignments The macr-sheet peaks for both huGrx andvacGrx are so sparse that JIGSAW identi es little to no macr structure In general it is much harder touncover macr-sheets than reg-helices since macr-strand sequentiality is speci ed by the noisier Hreg region of thespectrum We expect proteins with signi cant macr-sheet content such as CBF-macr to have enough connectivityto support the mutually con rming JIGSAW graph patterns
Table 5 demonstrates that due to the interaction graph constraints the actual combinatorics of JIGSAWare much better than the worst-case exponential possibility Notice that JIGSAW ef ciently explores oneto two thousand edges (Table 5 line 1) to nd less than one hundred correct ones (Table 4 lines 1ndash2)
4 FINGERPRINT-BASED SEQUENCE ALIGNMENT
Fingerprint-based sequence alignment nds sets of sequential residues in the protein sequence corre-sponding to the vertex sequences identi ed by the JIGSAW graph search algorithm This process utilizesthe TOCSY ngerprints introduced in Section 2
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
550 BAILEY-KELLOGG ET AL
FIG 8 reg-helices of huGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The BioMagResBank (BMRB) has collected statistics from a large database of observed chemical shifts(Seavey et al 1991) Figure 13 shows the mean chemical shifts for the protons of the 20 different aminoacid types The chemical shifts are affected by local chemical environment which includes amino acidtype and secondary structure The chemical shift index (CSI) has successfully used this information topredict secondary structure type given chemical shift and amino acid type (Wishart et al 1992) JIGSAWtakes a different approach it ldquoinvertsrdquo the BMRB to predict amino acid type given chemical shift andsecondary structure type
The rst step in alignment is to match each vertexrsquos ngerprint with the canonical BMRB ngerprintsDue to extra and missing peaks only a partial match might be possible
De nition 8 (Partial Fingerprint Match) A partial ngerprint match between vertex ngerprint Sv andBMRB amino acid ngerprint Sa (a 2 A 5 fAla Arg g) is a bijection m Sv
0 Sa0 between subsets
Sv0 sup3 Sv and Sa
0 sup3 Sa
Partial ngerprint matches are scored based on how well corresponding points match together withpenalties for extra and missing points Assuming Gaussian noise around the expected chemical shift withstandard deviation frac34a for amino acid type a the match score is de ned as follows
partialSv0 Sa
0 5 c0jSv iexcl Sv0j 1 c1jSa iexcl Sa
0j 1 c2
Y
p2Sv0
Gfrac34ap iexcl mp (4)
where c0 c1 c2 are weighting factors
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 551
FIG 9 reg-helices of vacGrx computed by JIGSAW using spectral suites 1 and 2 Edges solid 5 correct dotted 5false negative Vertices solid 5 correct empty 5 sequentially correct but not in reg-helix
The match score for a vertex and amino acid type is de ned as the best partial ngerprint match scorenormalization yields the probability that a vertex is of a given amino acid type
matchSv Sa 5 maxSv
0raquoSv Sa0raquoSa
partialSv0 Sa
0 (5)
P typev a 5matchSv Sa
X
b2A
matchSv Sb(6)
Then the probability that a sequence of vertices V 5 v1 v2 vn aligns at position r in the primarysequence L (where r micro jLj iexcl jV j) is the joint type probability over corresponding vertices and amino acidtypes The best alignment for a sequence of vertices V relative to a primary sequence s is the position r
maximizing the probability
P alignV s r 5nY
i 5 1
P typevi sr 1 i iexcl 1 (7)
alignmentV s 5 argmaxrmicrojLjiexcl jV j
P alignV s r (8)
This alignment process aligns each secondary structure element separately As it is this approach providesa basic algorithm for spectral interpretation explaining peaks in a TOCSY spectrum by identifying which
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
552 BAILEY-KELLOGG ET AL
FIG 10 macr-sheets of CBF-macr computed by JIGSAW Edges solid 5 correct dotted 5 false negative X 5 falsepositive
side-chain protons of a particular amino acid type could have caused them However in order to achievethe additional goal of nding a complete secondary structure assignment it is necessary to ensure that noalignments con ict This problem is similar to that of protein threading (Lathrop and Smith 1996) wherea novel primary sequence must be matched up against secondary structure elements from a known globalfold However in our case the ordering of the secondary structure elements is unknown (and of course thescoring function is different) It remains future work to extend threading algorithms to handle this hardertask
41 Experimental results
The purpose of the alignment process is to interpret a TOCSY spectrum by identifying a substringof amino acid types in the primary sequence such that the peaks expected for the side chain protonscan explain the observed peaks The performance of this spectral interpretation process was tested byseparately aligning each secondary structure element Tables 6 7 and 8 detail the results of ngerprint-based alignment for the TOCSY shifts of known reg-helices and macr-strands in CBF-macr huGrx and vacGrxrespectively Table 9 summarizes the number of correct alignments for all three proteins The simulatedTOCSY is produced from the average chemical shifts of the side-chain protons entered in the BMRBfor the given protein Since the simulated ngerprints are from data correlated among many spectra theyare much more complete and indicative of the amino acid types than are the single experimental TOCSYspectra While experimental TOCSY yields good alignment results the simulated results demonstrate thatas pulse sequences improve (see eg Zhu et al (1999a) and Zhu et al (1999b)) the experimental resultsshould get even better In general long sequences align better than short ones although unusually noisydata can disrupt the alignment
FIG 11 macr-sheets of huGrx computed by JIGSAW using spectral suite 2 Edges solid 5 correct dotted 5 falsenegative
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 553
FIG 12 Known macr-sheet connectivities in vacGrx The connectivitiesare too sparse for the generic JIGSAW algorithmto uncover much structure
Table 4 Summary of Results for JIGSAW SecondaryStructure Discovery ((a) reg-helices and (b) macr-sheets)
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
554 BAILEY-KELLOGG ET AL
FIG 13 BMRB mean 1H chemical shifts over different amino acid types These shifts de ne ldquo ngerprintsrdquo for theexpected TOCSY peaks of different amino acid types the ngerprint for H is isolated as an example
As discussed in the preceding section a generalized threading approach will be necessary in order todetermine a consistent alignment for all secondary structure elements of a protein We tried a simple testin order to evaluate the potential for success of such an algorithm This test found the top ve alignmentsfor each secondary structure element of huGrx took the cross product to identify sets of alignments onefor each element eliminated the members that included overlapping alignments and scored the remainingalignment sets with the product of probabilities for the individual member alignments The correct alignmentset received the best score by a factor of 100 This motivates the hope that while individual alignmentsmight not always score best determining a complete alignment set will correct individual mistakes byeliminating sets with inconsistent members
Table 6 Fingerprint-Based Alignment Results forreg-helices and macr-strands of CBF-macr with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg110ndash16 1 9 104 1 3 102
reg218ndash23 1 2 104 17 4 10 iexcl 6
reg334ndash36 1 4 101 3 7 10 iexcl 2
reg443ndash52 1 1 1013 1 2 104
reg5131ndash140 1 7 1014 1 1 1019
macr1127ndash31 1 4 103 5 3 10 iexcl 2
macr1255ndash60 1 2 106 1 2 104
macr1365ndash68 1 2 101 1 1 103
macr2196ndash104 1 2 101 1 7 102
macr22108ndash117 1 4 1010 11 3 10 iexcl 5
macr23122ndash130 1 3 104 5 1 10 iexcl 1
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 555
Table 7 Fingerprint-Based Alignment Results forreg-helices and macr-strands of huGrx with bothSimulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg14ndash9 1 7 107 1 1 105
reg225ndash34 1 5 1017 1 8 106
reg354ndash65 1 1 1016 1 9 1013
reg483ndash91 1 4 105 1 2 104
reg594ndash100 1 2 107 2 2 10iexcl 1
macr1143ndash47 1 1 103 3 7 10iexcl 3
macr1215ndash19 1 2 103 1 3 103
macr1372ndash75 1 1 103 4 2 10iexcl 2
macr1478ndash80 2 2 10 iexcl 1 4 4 10iexcl 2
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 8 Fingerprint-Based Alignment Results forreg-helices and macr-strands of vacGrx with both
Simulated and Experimental TOCSY Data1
Simulated Experimental
Sequence Rank frac12 Rank frac12
reg13ndash8 1 2 1010 5 3 10iexcl 2
reg225ndash34 1 1 1011 2 3 10iexcl 1
reg354ndash63 1 1 1032 1 2 103
reg483ndash91 1 7 1013 4 5 10iexcl 3
reg594ndash101 1 1 105 3 2 10iexcl 2
macr1142ndash47 1 4 101 1 2 101
macr1214ndash20 1 3 103 15 3 10iexcl 8
macr1372ndash74 1 4 102 10 5 10iexcl 4
macr1478ndash80 12 2 10 iexcl 3 1 1 103
1frac12 indicates the relative score of the alignmentmdashrelative to either thebest alignment if the correct one is not best or else to the second-bestalignment
Table 9 Fingerprint-Based Alignment Results Summary forboth Simulated and Experimental TOCSY Data
This paper has described the JIGSAW algorithm for automated high-throughput protein structure de-termination JIGSAW uses a novel graph formalization and new probabilistic methods to nd and alignsecondary structure fragments in protein data from a few key fast and cheap NMR spectra A set of rst-principles graph consistency rules allows JIGSAW to manage the search space and prevent combinatorial
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
556 BAILEY-KELLOGG ET AL
explosion JIGSAW has proven successful in structure discovery and alignment with experimental data forthree different proteins
JIGSAW offers a novel approach to the automated assignment of NMR data and the determination ofprotein secondary structure Since JIGSAW uses only four spectra and 15N-labeled protein it is applicablein a much higher throughput fashion than traditional techniques and could be useful for applications suchas quick structural assays and SAR by NMR It demonstrates the large amount of information available in afew key spectra Finally JIGSAW formalizes NMR spectral interpretation in terms of graph algorithms andprobabilistic reasoning techniques laying the groundwork for theoretical analysis of spectral information
We are developing a random graph analysis of the complexity correctness and completeness of JIGSAWThis analysis uses a statistical model of the noise (extra and missing edges) in an interaction graph tocompute the probability of false positives and false negatives for fragments fragment sequences andsecondary structure graphs This is an important direction for future work
JIGSAW has only been run on the three proteins reported above We plan to apply JIGSAW to experi-mental data for additional proteins and to extend the techniques to analysis of DNA NMR data We invitestructural biologists desiring a fast structural assay to contact us if they wish to run JIGSAW We anticipatethat since larger proteins have more NOEs JIGSAW will have to handle more incorrect edges and thuswill require increased computational cost An accurate noise model will provide a better indication of thedependence of computational complexity on spectrum size We believe that as long as the noise edgesare randomly distributed and do not achieve an overwhelming density only correct graphs will be able toconnect a large number of nodes with a dense set of consistent edges There are also interesting possibleconnections between JIGSAW and approaches to computing structures of large proteins via deuterationand sparse NOEs the introduction discusses the synergy in more detail
An iterative deepening approach (Russell and Norvig 1995 70ndash71) could be incorporated into JIGSAWby noticing incompleteness of a secondary structure graph and restarting with looser constraints for frag-ment generation For example circular dichroism data provides an accurate estimate of the total amountsof reg-helical and macr-sheet structure in a protein (Galat 1996) By converting the secondary structure per-centage to a count of the number of vertices involved JIGSAW could recognize that a secondary structuregraph was incomplete Similarly statistical secondary structure predictors (eg Dealeage et al (1987) andCuff et al (1998)) predict the number of residues participating in separate secondary structure elementsJIGSAW could recognize and analyze the difference between its results and such a prediction
The JIGSAW technique could be extended to assign HNndash1H NOESY peaks on the side chains andto compute the global fold of a protein Spectral referencing between TOCSY and NOESY gives anindication of which NOESY peaks belong to a given residue additional interresidue interactions couldthen be identi ed in the NOESY and used to constrain the global geometry of reg-helices and macr-sheets Whilesuch interactions will be sparse in purely 15N-labeled protein they might be suf cient to aid threadingtechniques that utilize secondary structure and sparse NOEs (eg Ayers et al (1999) and Xu et al (2000))or structure determination algorithms from sparse NOE sets (eg Standley et al (1999)) Finally JIGSAWcould be re-targeted to include data from 13C-labeled proteins in order to attack larger proteins while stillrequiring smaller sets of data than traditional approaches
ACKNOWLEDGMENTS
We are very grateful to Xuemei Huang and Chaohong Sun for contributing their NMR data on huGrxand CBF-macr to this project and for many helpful discussions and suggestions and to Xuemei for runningan invaluable new 15N-TOCSY experiment for us We would also like to thank Cliff Stein Tomaacutes Lozano-Peacuterez Chris Langmead Ryan Lilien and all members of Donald Lab for their comments and suggestionsFinally we would like to thank the anonymous reviewers for a number of very helpful comments
This research is supported by the following grants to BRD from the National Science Foundation NSFII-9906790 NSF EIA-9901407 NSF 9802068 NSF CDA-9726389 NSF EIA-9818299 NSF CISECDA-9805548 NSF IRI-9896020 NSF IRI-9530785 and by an equipment grant from Microsoft Research
REFERENCES
Ayers DJ Gooley PR Widmer-Cooper A and Torda AE 1999 Enhanced protein fold recognitionusing secondarystructure information from NMR Protein Science 8 1127ndash1133
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
THE NOESY JIGSAW 557
Bartels C Guumlntert P Bileter M and Wuumlthrich K 1997 GARANTmdasha general algorithm for resonance assignmentof multidimensional nuclear magnetic resonance spectra Journal of Computational Chemistry 18 139ndash149
Bartels C Xia T-H Billeter M Guumlntert P and Wuumlthrich K 1995 The program XEASY for computer-supportedNMR spectral analysis of biological macromolecules Journal of Biomolecular NMR 5 1ndash10
Cavanagh J FairbrotherW Palmer III A and Skelton N 1996 Protein NMR SpectroscopyPrinciples and PracticeAcademic Press Inc
Chen T Filkov V and Skiena S 1999 Identifying gene regulatory networks from experimental data in ProcRECOMB 94ndash103
Chen Y Reizer J Saier Jr MH Fairbrother WJ and Wright PE 1993 Mapping of the binding interfaces ofthe proteins of the bacterial phosphotransferase system HPr and IIAglc Biochemistry 32 1 32ndash37
Croft D Kemmink J Neidig K-P and Oschkinat H 1997 Tools for the automated assignment high-resolutionthree-dimensional protein NMR spectra based on pattern recognition techniques Journal of Biomolecular NMR 10207ndash219
Cuff J Clamp M Siddiqui A Finlay M and Barton G 1998 JPRED A consensus secondary structure predictionserver Bioinformatics 14 892ndash893
Dealeage G Tinland B and Roux B 1987 A computerized version of the Chou and Fasman method for predictingthe secondary structure of proteins Analytical Biochemistry 163(2) 292ndash297
Englander S and Wand A 1987 Main-chain directed strategy for the assignment of 1H NMR spectra of proteinsBiochemistry 26 5953ndash5958
Galat A 1996 A note on circular-dichroic-constrained prediction of protein secondary structure European Journalof Biochemistry 236 428ndash435
Gardener K Rosen MK and Kay LE 1997 Global folds of highly deuterated methyl-protonated proteins bymultidimensional NMR Biochemistry 36 1389ndash1401
Gronenborn A Bax A Wing eld P and Clore G 1989 A powerful method of sequential proton resonanceassignment in proteins using relayed 15N-1H multiple quantum coherence spectroscopy FEBS Letters 243 93ndash98
Grzesiek S Wing eld P Stahl S Kaufman JD and Bax A 1995 Four-dimensional 15N-separated NOESY ofslowly tumbling predeuterated 15N-enriched proteins application to HIV-1 Nef Journal of the American ChemicalSociety 117 37 9594ndash9595
Guumlntert P Doumltsch V Wider G and Wuumlthrich K 1992 Processing of multi-dimensional NMR data with the newsoftware PROSA Journal of Biomolecular NMR 2 619ndash629
Hajduk P Meadows R and Fesik S 1997 Drug design Discovering high-af nity ligands for proteins Science 278497ndash499
Hare B and Wagner G 1999 Application of automated NOE assignment to three-dimensional structure re nementof a 28 kD single-chain T cell receptor Journal of Biomolecular NMR 15 103ndash113
Hartuv E Schmitt A Lange J Meier-Ewert S Lehrach H and Shamir R 1999 An algorithm for clusteringcDNAs for gene expression analysis In Proc RECOMB 188ndash197
Huang X Speck N and Bushweller J 1998 Complete heteronuclear NMR resonance assignments and secondarystructure of core binding factor macr (1-141) Journal of Biomolecular NMR 12 459ndash460
Karp R Stoughton R and Yeung K 1999 Algorithms for choosing differential gene expression experiments InProc RECOMB 208ndash217
Kay LE 1998 Protein dynamics from NMR Nature Structural Biology 5 Suppl 513ndash517Kelley III J and Bushweller J 1998 1H 13C and 15N NMR resonance assignments of vaccinia glutarexdoxin-1 in
the fully reduced form Journal of Biomolecular NMR 12 353ndash355Koradi R Billeter M Engeli M Guumlntert P and Wuumlthrich K 1998 Automated peak picking and peak integration
in macromolecular NMR spectra using AUTOPSY Journal of Magnetic Resonance 135 288ndash297Lathrop RH and Smith TF 1996 Global optimum protein threading with gapped alignment and empirical pair
score functions J Mol Biol 255 651ndash665Leutner M Gschwind R Liermann J Schwarz C Gemmecker G and Kessler H 1998 Automated backbone
assignment of labeled proteins using the threshold accepting algorithm Journal of Biomolecular NMR 11 31ndash43Lukin J Gove A Talukdar S and Ho C 1997 Automated probabilistic method for assigning backbone resonances
of (13C 15N)-labeled proteins Journal of Biomolecular NMR 9 151ndash166Mumenthaler C and Braun W 1995 Automated assignment of simulated and experimental NOESY spectra of
proteins by feedback ltering and self-correcting distance geometry J Mol Biol 254 465ndash480Mumenthaler C Guumlntert P Braun W and Wuumlthrich K 1997 Automated combined assignment of NOESY spectra
and three-dimensional protein structure determination Journal of Biomolecular NMR 10 351ndash362Nelson S Schneider D and Wand A 1991 Implementation of the main chain directed assignment strategy Bio-
physical Journal 59 1113ndash1122Palmer III AG 1997 Probing molecular motion by NMR Current Opinion in Structural Biology 7 732ndash737Palmer III AG Williams J and McDermott A 1996 Nuclear magnetic resonance studies of biopolymer dynamics
Journal of Physical Chemistry 100 13293ndash13310Pearlman D 1999 Automated detection of problem restraints in NMR data sets using the FINGAR genetic algorithm
method Journal of Biomolecular NMR 13 325ndash335
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department
Hanover NH 03755
E-mail brdcsdartmouthedu
558 BAILEY-KELLOGG ET AL
Russell S and Norvig P 1995 Arti cial Intelligence A Modern Approach Prentice-Hall Englewood Cliffs NJSeavey B Farr E Westler W and Markley J 1991 A relational database for sequence-speci c protein NMR data
Journal of Biomolecular NMR 217ndash236Shuker S Hajduk P Meadows R and Fesik S 1996 Discovering high-af nity ligands for proteins SAR by NMR
Science 274 1531ndash1534Standley DM Eyrich VA Felts AK Friesner RA and McDermott AE 1999 A branch and bound algorithm
for protein structure re nement from sparse NMR data sets J Mol Biol 285 1691ndash1710Stefano DD and Wand A 1987 Two-dimensional 1H NMR study of human ubiquitin a main-chain directed
assignment and structure analysis Biochemistry 26 7272ndash7281Sun C Holmgren A and Bushweller J 1997 Complete 1H 13C and 15N NMR resonance assignments and
secondary structure of human glutaredoxin in the fully reduced form Protein Science 6 383ndash390Third meeting on the critical assessment of techniques for protein structure prediction 1999 Proteins Structure
Function and Genetics S3Venters RA Metzler WJ Spicer LD Mueller L and Farmer BT 1997 Use of H1
N-H1N NOEs to determine
protein global folds in predeuterated proteins Journal of the American Chemical Society 117 37 9592ndash9593Wishart D Sykes B and Richards F 1992 The chemical shift index A fast and simple method for the assignment
of protein secondary structure through NMR spectroscopy Biochemistry 31 6 1647ndash1651Wuumlthrich K 1986 NMR of Proteins and Nucleic Acids John Wiley and Sons New YorkXu Y Xu D Crawford OH Einstein JR and Serpersu E 2000 Protein structure determination using protein
threading and sparse NMR data In Proc RECOMB 299ndash307Zhu G Kong X and Sze K 1999a Gradient and sensitivity enhancement of 2D TROSY-based experiments Journal
of Biomolecular NMR 13 3ndash10Zhu G Xia Y Sze K and Yan X 1999b 2D and 3D TROSY-enhanced NOESY of 15N-labeled proteins Journal
of Biomolecular NMR 14 377ndash381Zimmerman D Kulikowsi C Huang Y Feng W Tashiro M Shimotakahara S Chien C Powers R and
Montelione G 1997 Automated analysis of protein NMR assignments using methods from arti cial intelligenceJ Mol Biol 269 592ndash610
Address correspondence toBruce Randall Donald
6211 Sudikoff LaboratoryDartmouth Computer Science Department