29
Trees, Stars, and Multiple Biological Sequence Alignment Jesse Wolfgang CSE 497 February 19, 2004
| Date post: | 24-Dec-2015 |
| Category: |
Documents |
| Upload: | ruth-oconnor |
| View: | 219 times |
| Download: | 2 times |

Trees, Stars, and Multiple Biological Sequence Alignment
Jesse Wolfgang
CSE 497
February 19, 2004

02/19/20042
Importance?
RNA folding (Trifonov, Bolshoi) Gene regulation (Galas et al.) Protein structure-function relationships(Wu, Kabat)
Molecular evolution (Dayhoff)

02/19/20043
Introduction
Original sequence unknown– Must consider all possible transformations– Including insertions, deletions, and replacements
Choose the most likely set of transformations– With a given model of protein evolution

02/19/20044
Sequences and Alignments
An alignment of the sequences
is written asnSS ,...,1
nSS ,...,1
K-sequence: sequence of k characters ),...,( 1 knnS =
Each is obtained from– Blanks are inserted in positions where some of the other
sequences have a nonblank character– At least one must be nonblank for each
is the length of the aligned sequences
iS iS
jS lj ,...,1=l

02/19/20045
Alignments
D Q L FD N V QQ G L
1S
2S
3S
D - - Q – L FD N V Q - - -- - - Q G L -
1S
2S
3S
Ex: sequences DQLF, DNVQ, QGL

02/19/20046
Lattices and Paths
– Cartesian product of strings of squaresn
A path between the sequences is a set of connected line segments (connected broken line)
),...,( 1 nSSγnSS ,...,1
A lattice of sequences with lengths),...,( 1 nSSL nSS ,...,1
nkk ,...,1
n
– Consists of -dimensional hypercubesn
– Forms an -dimensional parallelepipedn

02/19/20047
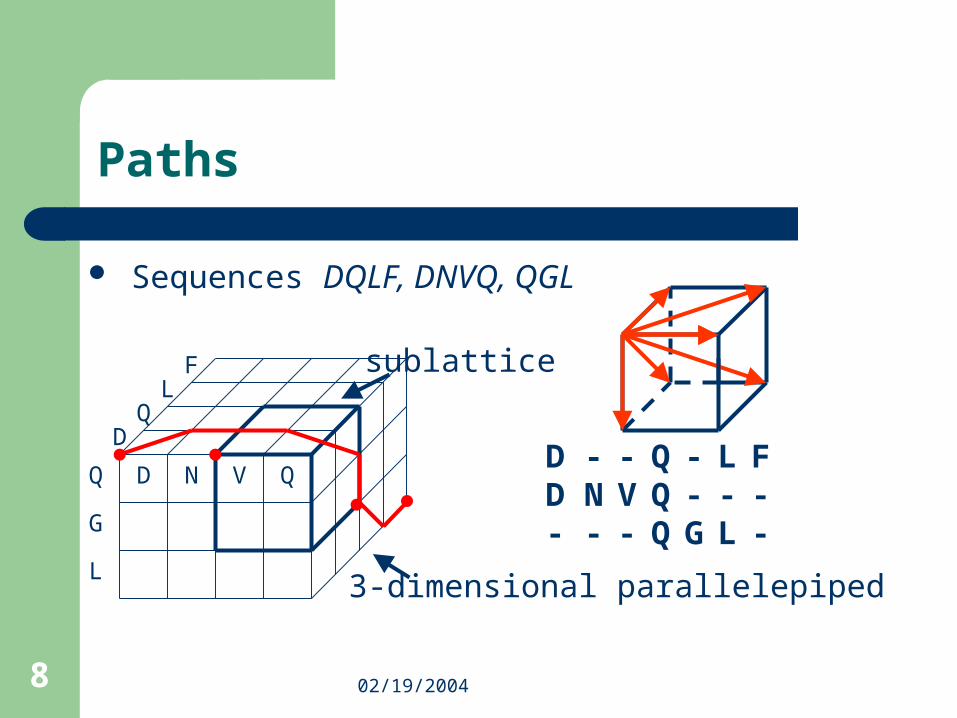
Paths
2 dimensions 3 dimensions
3 possible paths
7 possible paths
= 2n-1 = O(2n)
02/19/20048
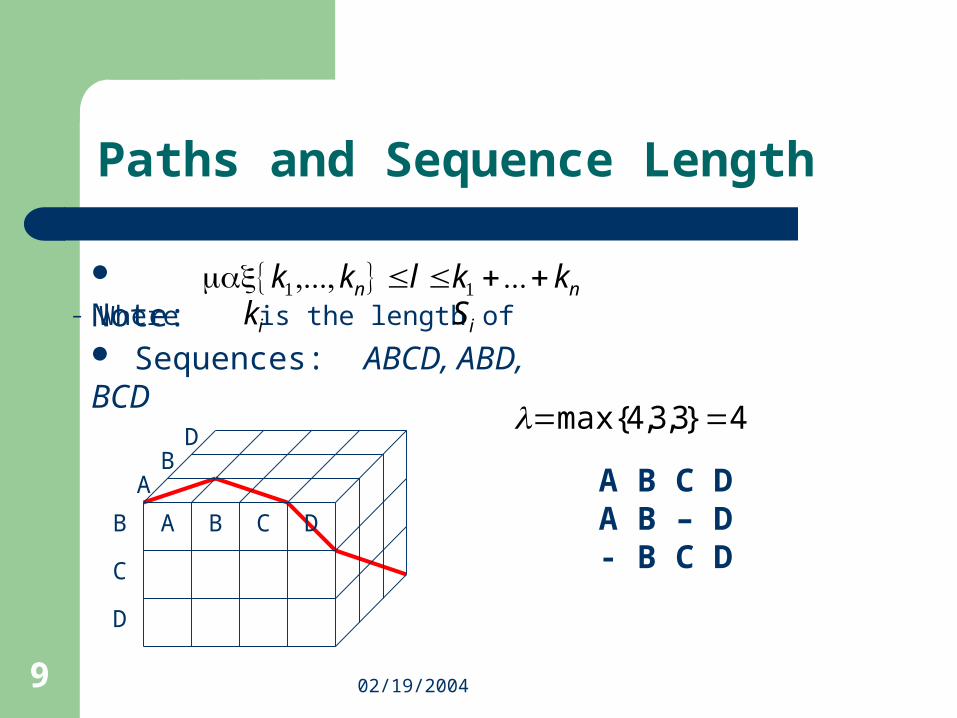
Paths
DQ
G
L
N V Q
DQ
LF
3-dimensional parallelepiped
sublattice
Sequences DQLF, DNVQ, QGL
DD-
-N-
QQQ
--G
L-L
F--
-V-
02/19/20049
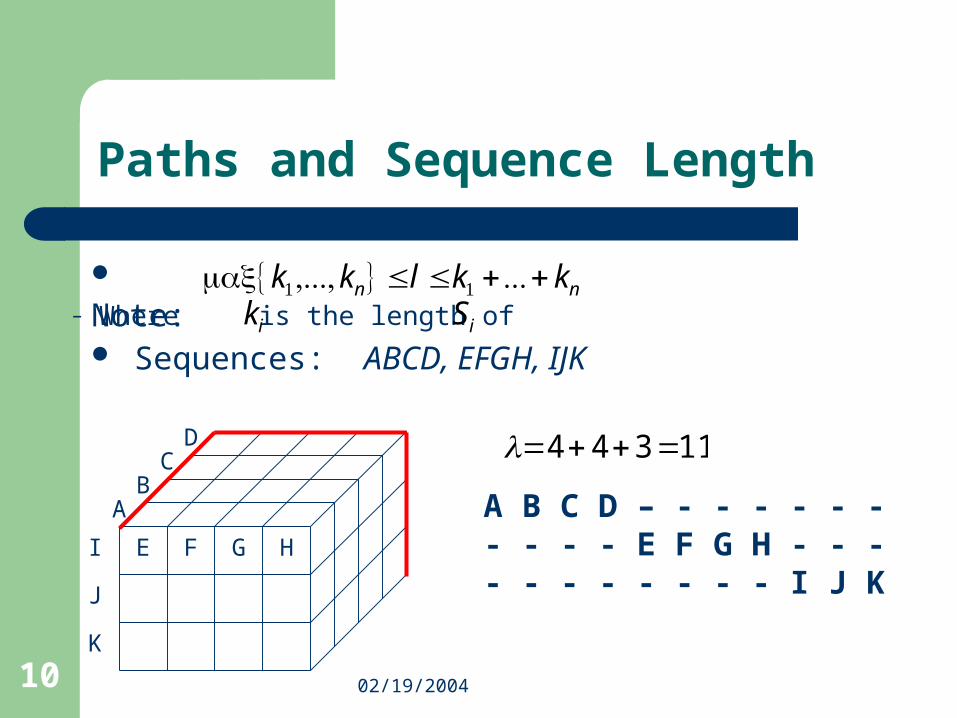
Sequences: ABCD, ABD, BCD
Paths and Sequence Length
Note:– Where is the length of
nn kklkk ++≤≤ ...},...,{max 11
ik iS
4}3,3,4max{ ==l
A B C DA B – D- B C D
A B C D
AB
D
B
C
D
02/19/200410
Sequences: ABCD, EFGH, IJK
Paths and Sequence Length
Note:– Where is the length of
nn kklkk ++≤≤ ...},...,{max 11
ik iS
EI
J
K
F G H
AB
CD 11344 =++=l
A B C D – - - - - - -- - - - E F G H - - -- - - - - - - - I J K
02/19/200411
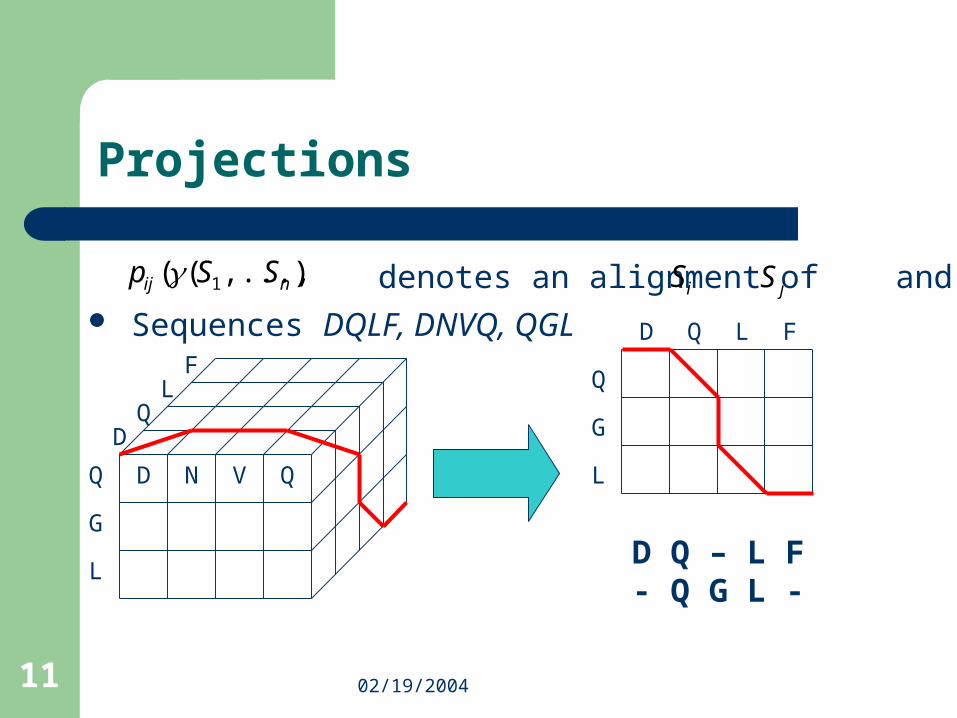
Sequences DQLF, DNVQ, QGL
Projections
DQ
G
L
N V Q
DQ
LF
denotes an alignment of and)),...,(( 1 nij SSp γiS jS
D Q – L F- Q G L -
D Q L F
Q
G
L

02/19/200412
Optimal Paths
is a measure assigned to)(γM γ– Measure of the similarity among based upon a particular metric
nSS ,...,1
For each measure there is at least one path with attaining a minimum value at , the optimal path
M),...,( 1*
nSSγ)(γM
*γ

02/19/200413
))(),...,(( 11 nn iSiSL
DQ
G
L
N V Q
DQ
LF
Each vertex in L is an end corner of the sublattice
Calculating Optimal Paths
First: compute score of each of the possible paths for the cube that has a vertex at the original corner Next: using this information, compute minimum score to reach the vertices of the adjacent cubes to the original corner

02/19/200414
Problems with This Algorithm
Calculates a weighted sum of its projected pairwise alignments– Called “Sum-of-the-Pairs” (SP)
Other methods fit biological intuition more closely

02/19/200415
Tree-Alignment
Treat sequences as leaves of an evolutionary tree
Reconstruct ancestral sequences which minimize the cost of the tree– Must assign sequences to internal nodes
Align the given and reconstructed sequences Star-alignment: only one internal node

02/19/200416
Tree-Alignment
Many different methods for calculating tree alignments
Discuss version used by ClustalX

02/19/200417
Tree-Alignment in ClustalX
Three main parts
1. Perform pairwise alignment on all sequences to calculate a distance matrix
2. Use distance matrix to calculate a guide tree
3. Sequences are progressively aligned using the branching order in the guide tree
http://bimas.dcrt.nih.gov/clustalw/clustalw.html

02/19/200418
Calculating Distance Matrix
Use standard dynamic programming to find the best alignment
– Gap penalties for opening a gap and continuing a gap (possibly different)
Divide number of matches by total number of residues compared (excluding gaps)
Convert to distances by dividing by 100 and subtracting from 1
Gives one entry in the n by n matrix

02/19/200419
Calculating Distance Matrix
Ex: sequences ATCG, ATCC, AGGC, AGCC
A T C GA T C C
= 3/4 = .75/100 = 1-.0075 = .9925
A T C GA G G C
= 1/4 = .25/100 = 1-.0025 = .9975

02/19/200420
Calculating Distance Matrix
ATCG ATCT AGGC GCAA
ATCG -- -- -- --
ATCT .9925 -- -- --
AGGC .9975 .9975 -- --
GCAA 1 1 1 --

02/19/200421
Calculating a Guide Tree
Using Nearest-Neighbor method to group sequences– Results in an unrooted tree– Branch lengths proportional to estimated
divergence “Mid-point” method used to determine root
– Means of the branch lengths to each side of the root are equal (or approximately equal)

02/19/200422
Calculating a Guide Tree
ATCG ATCT
ATCG AGGC
AGCC GCAA
AGAA
.9925.9925
.9975/2 .9975
1/3 1
ATCG = 1.8245ATCT = 1.8245
AGGC = 1.33081.6599
GCAA = 1

02/19/200423
Calculating a Guide Tree
ATCG = 1.4911ATCT = 1.4911
1.4911
AGGC = 1.4986GCAA = 1.4986
1.4986
ATCG ATCT
ATCG
AGGC
AGCC
GCAA
AGAA.9925.9925 1 1
.9975/2.9975/2

02/19/200424
Progressive Alignment
Perform a series of pairwise alignments– Slowly align larger and larger groups of
sequences
Follow the branching order of the tree– From leaves to root

02/19/200425
Progressive Alignment
ATCG ATCT
ATCG
AGCC
AGGC GCAA
AGAA
02/19/200426
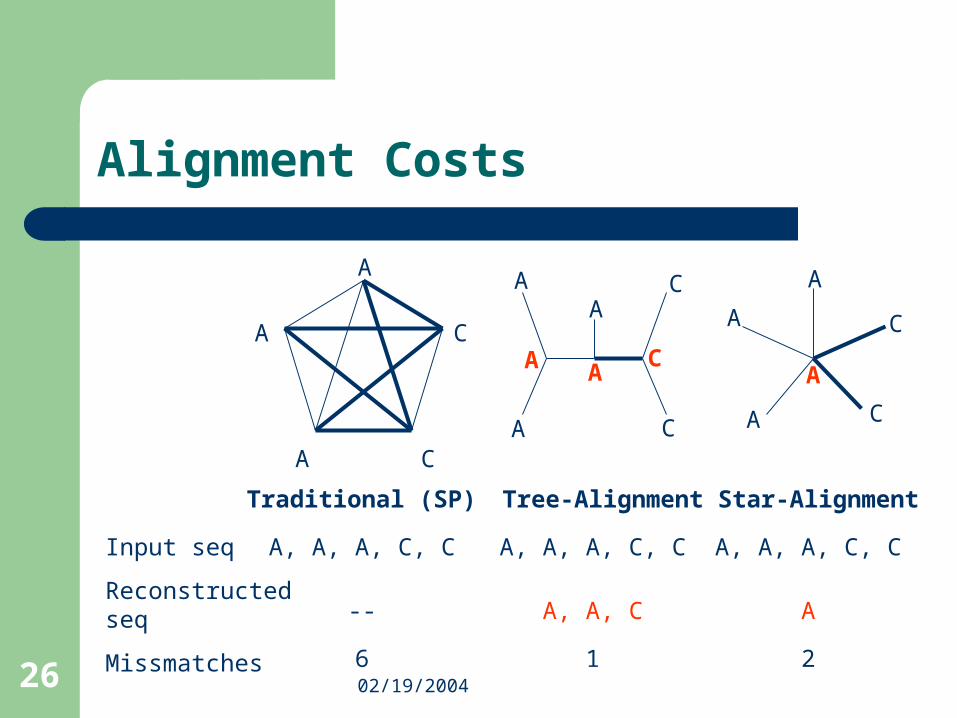
Alignment Costs
A C
A
A
C
A, A, A, C, C
--
6
A
A
A
A
AC
C
C
A, A, A, C, C
A, A, C
1
C
C
A
A
A
A
A, A, A, C, C
A
2
Traditional
Input seq
Reconstructedseq
Missmatches
Traditional (SP) Tree-Alignment Star-Alignment

02/19/200427
Alignment Inconsistencies
Different definitions of multiple alignments can yield different optimal alignments
Optimal tree-alignments minimize number of mutations from theorized common ancestors
SP-alignments maximize number of positions where aligned sequences agree– Sometimes makes more biological sense since
certain regions of proteins more likely to mutate

02/19/200428
Alignment Inconsistencies
Ex: cost of 1 for aligning two different letters, cost of 2 for aligning a letter with a null
Sequences: ACC, ACC, TCT, ATCT
Input sequences
Reconstructedsequences
- A C C- A C C- T C TA T C T
--
Traditional (SP)
A C C -A C C -T C T -A T C T
A C C -
Star-Alignment

02/19/200429
ClustalX Demo
Multiple sequence alignment program For more information on ClustalX
– http://www.at.embnet.org/embnet/progs/clustal/clustalx.htm

















