101


UNIVERSITÉ DE SHERBROOKE
Pathogenic Role of IL-15 in Non-Alcoholic Fatty Liver Disease
Written by: Yuneivy Cepero Donates
Department Pediatrics, Division of Immunology
Master’s thesis presented to the Faculty of Medicine and Health Sciences in view of obtaining a Master of Science (MSc.) in Immunology
September, 2014

Résumé
UNIVERSITÉ DE SHERBROOKE
RÔLE PATHOGÉNIQUE DE l’IL-15 DANS LA STÉATOSE HÉPATIQUE
Yuneivy Cepero Donates Département de pédiatrie, Service d’immunologie
Mémoire présenté en Juillet 2014 à la Faculté de Médecine en vue de l’obtention du
grade de Maître en Sciences (MSc.) en Immunologie
RÉSUMÉ
Les cytokines pro-inflammatoires jouent un rôle important dans la pathogenèse de l’obésité et la stéatose hépatique. L'IL-15 est une cytokine pro-inflammatoire qui est trans-présentée par l'IL-15Rα aux chaines IL-2/IL-15Rβ et γc. La fonction de l'IL-15 a été largement décrite dans les cellules immunitaires, mais ses fonctions dans d'autres tissus sont moins connues. Le but de ce mémoire est d'élucider le rôle de l'IL-15 dans la stéatose hépatique. Les souris C57BL/6 de type sauvage (WT) et Il15-/- ont été soumises à un régime hyperlipidique (HFD) ou à un régime normal. Après 16 semaines, le poids corporel, la masse hépatique, l'accumulation de lipides dans le foie, les taux de lipides sériques et l'expression des différents gènes reliés à l’inflammation et au métabolisme dans le foie ont été évalués. Les lymphocytes intra-hépatiques (IHL) ont été également étudiés. Des hépatocytes primaires ont été stimulés avec IL-15, et l'expression génique de chimiokines a été déterminée. Les populations de IHLs ont été également caractérisées chez les souris WT, Il15-/- et Il15ra-/-, ainsi que chez des souris dont la déficience dans l’expression d’IL-15Rα est ciblée aux macrophages ou aux hépatocytes. Nos résultats montrent que la déficience en IL-15 empêche l'accumulation de lipides dans le foie. Les taux de cholestérol et d’acides gras non estérifiés dans le sang étaient élevés chez les souris WT, mais pas chez les souris Il15-/- . L'expression hépatique des chimiokines Ccl2, Ccl5, Cxcl10 et des marqueurs de macrophages était augmentée chez les souris WT sous HFD, mais pas chez les souris Il15-/-. La stimulation des hépatocytes primaires avec l'IL-15 induit l'expression des gènes des chimiokines chez les hépatocytes WT, mais pas chez les Il15ra-/-. En outre, nous avons trouvé une infiltration réduite des cellules NK et NKT dans le foie des souris déficientes en Il15ra-/- dans les hépatocytes, ce qui suggère que l'expression d’IL15Rα chez les hépatocytes est nécessaire au recrutement des cellules NK, NKT et / ou à leur maintien. En conclusion, nous proposons que l’IL-15 favorise l'accumulation de lipides dans le foie, et que ceci est associée à une réponse inflammatoire accrue. La disponibilité accrue de l'IL-15 dans l'obésité pourrait stimuler les hépatocytes à secréter des chimiokines ce qui favorise l'inflammation hépatique et conduirait à la stéatose hépatique. L’expression de l'IL-15Rα dans les hépatocytes semble jouer un rôle principal dans l’infiltration des cellules NK, NKT et iNKT dans le foie. Mots clés : Stéatose hépatique non-alcoolique (NAFLD), lymphocytes intra-hépatiques (IHL), régime hyperlipidique (HFD), IL-15, chimiokines, inflammation, IL-15Rα.

Abstract
UNIVERSITÉ DE SHERBROOKE
PATHOGENIC ROLE OF IL-15 IN NON-ALCOHOLIC FATTY LIVER DISEASE
Yuneivy Cepero Donates
Department Pediatrics, Division of Immunology
Master’s thesis presented to the Faculty of Medicine and Health Sciences in view of obtaining a Master of Science (MSc.) in Immunology
ABSTRACT
Pro-inflammatory cytokines play a key role in pathogenesis of obesity and non-alcoholic fatty liver disease (NAFLD). IL-15 is a pro-inflammatory cytokine, which signals through a receptor complex composed of the IL-15 receptor (IL-15R) alpha chain, the IL-2/IL-15R beta chain and the common gamma chain. The functions of IL-15 have been extensively described in immune cells but less is known about its functions in others tissues such as the liver. The aim of this thesis is to investigate the role of IL-15 in fatty liver disease. C57BL/6 wildtype (WT) and IL-15 knockout (Il15-/-) mice were maintained on high fat diet (HFD) or normal control diet (NCD). After 16 weeks, body weight, liver mass, fat accumulation in the liver, serum lipid levels and gene expression in the liver were evaluated. Intrahepatic lymphocytes (IHL) were also analysed. Primary hepatocytes were stimulated with IL-15 and chemokines gene expression was studied. IHLs were examined in WT, Il15-/- and Il15ra-/-, as well as in macrophage- and hepatocyte-specific Il15ra-/- mice. We found that IL-15 deficiency prevents weight gain and accumulation of lipids in the liver. Circulating levels of cholesterol and non-esterified fatty acids were elevated in WT mice but not in Il15-/- mice. Hepatic expression of chemokines such as Ccl2, Ccl5 and Cxcl10 was increased in WT mice under HFD, but not in Il15-/- mice. The livers of Il15-/- and Il15ra-/- mice also showed decreased expression of Tnfa and iNOS, and macrophage markers Cd68 and F4/80. Accordingly, stimulation of primary hepatocytes with IL-15 induced chemokine gene expression in WT but not in Il15ra-/- hepatocytes. Furthermore, hepatocyte-specific ablation of IL-15Rα reduced infiltration of NK and NKT cells in the liver, suggesting that IL15Rα expression in the hepatocytes is needed for the recruitment and/or maintenance of the NK cell population in the liver. In conclusion, IL-15 promotes fat accumulation in the liver, and this is associated with increased inflammatory response in the liver. Increased availability of IL-15 in obesity may stimulate hepatocytes to secrete chemokines that promote hepatic inflammation resulting in fatty liver disease. IL-15Rα expression in hepatocytes appears to play a role in the maintenance of NK, NKT and iNKT cells.
Keywords: non-alcoholic fatty liver disease (NAFLD), intra-hepatic lymphocytes (IHL), high fat diet (HFD), IL-15, chemokines, inflammation, IL-15Rα.

Table of contents
iv
RÉSUMÉ ................................................................................................................................ ii
ABSTRACT ........................................................................................................................... iii
LIST OF FIGURES AND TABLES ......................................................................................... vi
LIST OF ABBREVIATIONS................................................................................................... viii
1. INTRODUCTION ................................................................................................................ 1
1.1. Structure and functions of the liver ......................................................................... 1
1.1.1. Structure of the liver ......................................................................................... 1
1.1.2. Metabolic functions of the liver ......................................................................... 2
1.1.3. Liver as part of the immune system.................................................................. 4
1.2. Obesity ................................................................................................................... 5
1.2.1. Obesity-associated inflammation ..................................................................... 6
1.3. Fatty liver diseases ................................................................................................. 7
1.3.4. Mouse models of NAFLD ................................................................................12
1.4. Cytokines that signal via the common γ-chain (γc; CD132) receptor ......................12
1.5. Interleukin-15 (IL-15) .............................................................................................15
1.5.1. Structure .........................................................................................................15
1.5.2. IL-15 receptor .................................................................................................16
1.5.3. IL-15 signalling ................................................................................................17
1.5.4. Mechanisms mediating IL-15 responses .........................................................18
1.5.5. Function of IL-15 in T cells ..............................................................................21
1.5.6. IL-15 in NK cell biology ...................................................................................21
1.5.7. The role of IL-15 in iNKT cells .........................................................................23
1.5.8. IL-15 functions in non-immune cells ................................................................24
1.5.9. Functional dichotomy between IL-2 and IL-15 .................................................26
1.6. The thesis premises ..............................................................................................27
1.7. Hypothesis ............................................................................................................28
1.8 Objectives ..............................................................................................................28
2. MATERIALS and METHODS ............................................................................................ 29
2.1. Mice ......................................................................................................................29
2.1.1 Induction of NAFLD in mice .............................................................................29
2.2. Isolation of intrahepatic lymophocytes (IHL) ..........................................................30
2.3. Isolation of splenocytes .........................................................................................30

Table of contents
v
2.4. FACS analyses......................................................................................................31
2.5. Isolation of primary hepatocytes ............................................................................31
2.6. Stimulation of hepatocytes ....................................................................................32
2.7. Assessment of mitochondrial respiration in hepatocytes ........................................32
2.7.1 Fatty acids oxidation test ..................................................................................33
2.8. Tissues processing for histology ............................................................................34
2.9. Histology and lipids detection ................................................................................35
2.10. Immunofluorescence microscopy ........................................................................35
2.11. RNA isolation ......................................................................................................36
2.12. Quantitative PCR .................................................................................................36
2.13. Graphs and statistical analysis ............................................................................37
3. RESULTS ......................................................................................................................... 38
3.1. IL-15 promotes weight gain and fat accumulation in the liver .................................38
3.2. IL-15 deficiency reduces Pparg and increases Ppara induction after HFD .............40
3.3. IL-15 regulates β-oxidation ....................................................................................41
3.4. IL-15 suppresses mitochondrial respiration in mice primary hepatocytes ..............43
3.5 HFD induces Il15 expression in the liver .................................................................46
3.6. HFD promotes the expression of pro-inflammatory mediators in the liver. .............46
3.7. HFD-induced macrophage infiltration in the liver is reduced in the absence of IL-15
.....................................................................................................................................47
3.8. HFD increases immune cells infiltration in mice liver .............................................48
3.9. HFD induced chemokine gene expression in the liver is mediated by IL-15. ..........51
3.10. IL-15 induces chemokines expression in mice primary hepatocytes ....................53
3.11. IL-15 and IL-5Rα are required for the maintenance of NK populations in the liver
.....................................................................................................................................54
3.12. IL-15Rα expression in both macrophages and hepatocytes contribute to NK cell
maintenance in the liver ...............................................................................................56
3.13 Summary of results ..............................................................................................59
4. DISCUSSION ................................................................................................................... 60
5. CONCLUSIONS ............................................................................................................... 75
6. ACKNOWLEDGEMENTS ................................................................................................. 77
7. REFERENCES ................................................................................................................. 78

List of figures and tables
vi
LIST OF FIGURES AND TABLES
FIGURES Page
Figure 1.1 Figure 2.1:
Principals mechanism for IL-15 delivery
XF Cell Mito Stress Test Profile
20
33
Figure 3.1: A) IL-15 deficiency prevents weight gain in the liver and hepatic fat
accumulation.
39
B) HFD results in increased circulating levels of cholesterol and NEFA
in wild type mice, but not in Il15 deficient mice
39
C) IL-15 deficiency prevents hepatic fat accumulation 40
Figure 3.2: IL-15 deficiency reduces Pparg and increases Ppara induction after
HFD
41
Figure 3.3: A) HFD induces the expression of Cd36 in the liver 42
B) Cpt1a levels are increased in Il15-/- mice in NCD 42
C) HFD induces Acadm expression in the liver 43
Figure 3.4: A) IL-15 deficiency does not affect cytochrome c oxidase levels 44
B) IL-15 deficiency increases mitochondrial respiration 45
C) IL-15 deficiency enhances fatty acid oxidation in hepatocytes 45
Figure 3.5: Il15 mRNA levels are increased in WT mice in HFD 46
Figure 3.6: HFD induces Tnfα and iNOS expression in the liver 47
Figure 3.7: A) HFD increases macrophages infiltration in the liver 48
B) Expression of macrophages markers is increased in WT mice
maintained on HFD
48
Figure 3.8: A) HFD induce lymphocytes infiltration in the liver 50
B) HFD induces CD8+ T cell activation and NK cell infiltration in the
liver
51
Figure 3.9: HFD induced chemokine mRNA expression in the liver requires IL-15 52
Figure 3.10: IL-15 induces Ccl5 and Cxcl10 expression in mouse primary
hepatocytes
53

List of figures and tables
vii
Figure 3.11:
A) Splenic but not liver CD8+ T cells are dependent on IL-15 signaling
54
B) NK and NKT cells are reduced in the absence of IL-15 or IL-15Rα 55
C) IL-15 and IL-15Rα are needed for iNKT cell maintenance in the liver 55
Figure 3.12: A) IL-15Rα expression in macrophages is required for NKT cells
maintenance in the liver
57
B) IL-15Rα expression in the hepatocytes is required for the
maintenance of NK, NKT and iNKT cells in the liver
58
Figure 5.1: IL-15 regulates immune cells recruitment and homeostasis in the liver 76
TABLES
Table 2.1: List of murine oligonucleotide sequences 37

List of abbreviations
viii
LIST OF ABBREVIATIONS
Acadm Acyl-Coenzyme A dehydrogenase, C-4 to C-12 straight chain
acetyl-CoA Acetylcoenzyme A
ACK Ammonium-chloride-potassium, lysing buffer
AICD Activation-induced cell death
APPs Acute-phase proteins
Bcl2 B-cell lymphoma 2
BM Bone marrow
BMI Body mass index
BSA Bovine serum albumin
Cepba CCAAT/enhancer binding protein
Cox4i1 Cytochrome c oxidase
Cpt1a Carnytyl palmitoyl transferase
DCs Dendritic cells
DIO Diet-induced obesity
DMEM-F12 Dulbecco’s modified eagle medium: nutrient mixture f-12
ER Endoplasmic reticulum
FA Fatty acids
FACS Fluorescence-activated cell sorting
FCCP Carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone
FCS Fetal calf serum
GFP Green fluorescent protein
HBSS Hank’s buffered salt solution
HCC Hepatocellular carcinoma
HDL High-density lipoprotein cholesterol
HFD High fat diet
HGF Hepatocyte growth factor
HSCs Hepatic stellate cells

List of abbreviations
ix
IELs Intraepithelial lymphocytes
IFN-γ Interferon gamma
IGF-1 Insulin growth factor-1
IHL Intrahepatic lymphocytes
IL- Interleukin
IL-15Rα IL-15 receptor alpha
iNKT Invariant Natural killers T cells
iNOS Inducible nitric oxide synthase
IP-10 Interferon gamma-induced protein 10
IRS1 Insulin receptor substrate 1
JAK Janus kinase
JNK1 C-Jun N-terminal kinase 1
KCs Kupffer cells
KHB Krebs-Henseleit buffer
KO Knockout
KRB Krebs-ringer-buffer
Lck Lymphocyte-specific protein tyrosine kinase
LPL Lipoprotein lipase
LSEC Liver sinusoidal endothelial cells
LSP Long signal peptide
M1 Pro-inflammatory macrophages
M2 Anti-inflammatory macrophages
MCP-1 Monocyte chemotactic protein-1
MRC Maximal respiratory capacity
mRNA Messenger ribonucleic acid
mTECs Medullary thymic epithelial cells
NAFLD Non-alcoholic fatty liver disease
NASH Non-alcoholic steatohepatitis
NCD Normal control diet

List of abbreviations
x
NEFA Non-esterified fatty acids
NK Natural killer cells
NKp NK precursors
NKT Natural killer T cells
ns Not significant
PBMCs Peripheral blood mononuclear cells
PBS Phosphate buffered saline
PCD Programmed cell death
PFA Paraformaldehyde
Pgc1a PPARγ coactivator 1-alpha
PPAR Peroxisome proliferator-activated receptor
RA Rheumatoid arthritis
RNA Ribonucleic acid
ROS Reactive oxygen species
SRC Spare respiratory capacity
SREBP1c Sterol regulatory element-binding protein 1c
Srebpf1 Sterol-regulatory-element-binding protein
SSP Short signal peptide
STAT Signal Transducer and Activator of Transcription
Syk Spleen Tyrosine kinase
TCR T-cell receptor
TECs Tubular epithelial cells
TG Triacylglycerol
TGF Transforming growth factor
Th1 T helper 1
Th2 T helper 2
TLRs Toll-like receptors
TM Transmembrane region
Tnfa Tumor necrosis factor -α

List of abbreviations
xi
Treg Regulatory T cells
TSLPR Thymic stromal lymphopoietin receptor
Tw Tween®20
VLDL Very-low-density lipoprotein
VSV Vesicular stomatitis virus
WHO World health organization
WT Wild type
XSCID X-linked severe combined immunodeficiency

Introduction
1
1. INTRODUCTION
1.1. Structure and functions of the liver
1.1.1. Structure of the liver
Liver is one of the most important metabolic organs in the body and plays a central role in
the synthesis of many plasma proteins and carbohydrates, storage of minerals and
vitamins and detoxifying toxins that enter portal vein circulation (Fausto, N. et al., 2006).
Liver cells are classified into two main groups: epithelial cells and mesenchymal cells. In
the epithelial cells group, the major subtype is hepatocytes, which represents more than
90% of the liver parenchyma (Papoulas, M. and Theocharis, S., 2009). Cholangiocytes
also belong to this group, and are located in the intra-hepatic and extra-hepatic biliary duct
system. Moreover, cholangiocytes release bicarbonate and water, which modifies the bile
produced by hepatocytes (O'Hara, S.P. et al., 2013). Hepatic non-parenchymal cells
include endothelial cells, Kupffer cells (KCs), lymphocytes, hepatic stellate cells (HSCs)
and biliary ductal cells (Lemoinne, S. et al., 2013). KCs reside in liver sinusoids and are
derived from circulating monocytes. They constitute approximately 20% of hepatic non-
parenchymal cells (Ruck, P. and Xiao, J.C., 2002)
Hepatic stellate cells produce hepatocyte growth factor (HGF), store retinoids and serve
as precursors of liver myofibroblasts (Friedman, S.L., 2008). Myofibroblasts are the main
fibrogenic effector cells and are absent in the healthy liver. However, following liver
damage, HSCs differentiate and start producing extracellular matrix and collagen
(Lemoinne, S. et al., 2013). Liver sinusoidal endothelial cells (LSEC) line the capillaries
and sinusoids and differ from the endothelium of big vessels in lacking basement
membrane and containing the fenestrae structures. Moreover, a subset of LSECs is
derived from the bone marrow and, like HSCs, is one of the main sources of HGF
(DeLeve, L.D., 2013). Liver lymphocytes are primarily located around the portal tracts.

Introduction
2
This distribution of lymphocytes in the liver aids in the rapid removal of gastrointestinal
antigens from the circulation (Zhan, Y.T. and An, W., 2010).
1.1.2. Metabolic functions of the liver
Liver is a major metabolic organ in the body and plays important roles in protein,
carbohydrate and lipid metabolic pathways.
In proteins metabolism, liver participates in amino acids degradation to supply both,
carbohydrates and fatty acids (FAs) metabolic pathways, and protein synthesis. In the
human serum, the most abundant protein is albumin, which is synthetized by hepatocytes
and represents the 15 percent of the total hepatic protein synthesis. Albumin’s half-life of
is around 20 days, so that a decrease in serum albumin is unlikely to occur within a short
time of acute liver injury. In patients with ascites and chronic liver disease the serum level
of albumin is often low (Rothschild, M.A. et al., 1969). Various coagulation factors such as
II, VII, IX, X, V, XI, XII, and XIII and fibrinogen are synthesized in the liver. As coagulation
factors have short life-span, defects in coagulation quickly become apparent in acute liver
damage. Some inhibitors of coagulation and fibrinolysis are also synthesized in the liver.
Moreover, in liver disease, the prolongation of prothrombin time results from a deficiency
in factors II, V, VII, and X; and can be used as a predictive factor of acute liver failure
(Aranha, G.V. and Greenlee, H.B., 1986). Transamination and oxidative deamination of
amino acids takes place in the hepatocytes and leads to nitrogenous excretory products,
which enter in the Krebs-Henseleit cycle producing urea (Zieve, L., 1979).
In carbohydrates metabolism, liver regulates glucose levels in the blood by controlling
glucose uptake after meals and releasing glucose during fasting. Carbohydrate
metabolism in the liver is proportional to the degree of hypoglycemia or hyperglycemia. In
hypoglycemic condition, glucose synthesis from endogenous sources is increased, for

Introduction
3
example, endogenous glycogen is catabolized into glucose that enters the blood (Stanley,
J.C., 1981). This process is controlled by several hormones, such as catecholamines that
stimulate the rate of gluconeogenesis via cyclic adenosine monophosphate (Fausto, N. et
al.)-dependent (beta-mimetic) and cAMP-independent (alpha-mimetic) mechanisms
(Hems, D.A. and Whitton, P.D., 1980). On the other hand, insulin antagonizes the action
of both glucagon and catecholamines on hepatic gluconeogenesis.
Fat containing chylomicrons reach the liver via lymph and the blood. Degradation of fat to
acetyl coenzyme A (acetyl-CoA), a main step in the metabolic integration of different
pathways, occurs in the liver. Acetyl-CoA can enter into the tricarboxylic acid cycle or
participate in the synthesis of triglyceride phospholipid, cholesterol and lipoproteins. Fatty
acids in the liver can be metabolized by esterification or oxidation. Glucagon markedly
stimulates the rate of FA oxidation, whereas insulin inhibits it. The beta oxidation of FAs
results in the production of acetyl-CoA in the mitochondria, where the acetyl-CoA is further
oxidized to carbon dioxide and water or is converted to ketone bodies (Voet, J.G. and
Voet, D., 2000).
FA synthesis from excess glucose also occurs in the liver. FAs are esterified with glycerol
in the liver to form triglycerides, which are then incorporated into lipoproteins, principally
very-low-density lipoprotein (VLDL), that are secreted by the liver. The principal factor
affecting VLDL production is the amount of free FAs reaching the liver. Secretion of VLDL
is also stimulated by insulin (Kamagate, A. and Dong, H.H., 2008). Hepatic lipid synthesis
is stimulated by insulin after food intake. Interestingly, in metabolic disease (insulin
resistant state), liver and plasma triglycerides are increased. Different studies propose a
model where distinct insulin signalling pathways independently modulate glucose and lipid
metabolism (Brown, M.S. and Goldstein, J.L., 2008). In insulin-resistant subjects, whereas
insulin fails to adequately augment hepatic glucose uptake or suppress hepatic glucose
production, hepatic lipogenesis and triglycerides accumulation remain elevated and

Introduction
4
contribute to hypertriglyceridemia. The major metabolic pathways disrupted in insulin
resistance condition are: hepatic glucose uptake and glycogen deposition (impaired);
gluconeogenesis and de novo lipogenesis (active) and FA delivery and triglyceride
esterification and secretion (accelerated). The collective effects of these dysregulated
pathways in multiple tissues precipitates the metabolic syndrome phenotype (Otero, Y.F.
et al., 2014).
1.1.3. Liver as part of the immune system
Recent evidence suggests that liver can also be considered as part of the immune system
as it plays a major role in innate immunity (Dong, Z. et al., 2007; Gao, B. et al., 2008). The
innate immune system is the first barrier in the host defense during an infection. Innate
immunity consists of innate lymphocytes, phagocytic cells, physical and chemical barriers
and humoral factors. Between 80%-90% of innate protein biosynthesis takes place in the
liver, for example acute-phase proteins (APPs), complement factors and secreted pattern
recognition receptors. Complement system components are synthesized in the liver and
represent about 5% of the globulin fraction of blood plasma. The complement system is
involved in the development of many liver disorders, including liver fibrosis and alcoholic
liver disease (Zhan, Y.T. and An, W., 2010).
The proportion of innate immune cells is much higher in the liver compared to other
organs such as spleen. Due to its anatomical location, liver immune cells are exposed to
large amounts as well as a wide variety of antigens and toxins. KCs are the main
phagocytic cells in the liver and constitute almost the 80%-90% of the total tissues
macrophages in the body (Doherty, D.G. and O'Farrelly, C., 2000). As liver macrophages,
KCs possess scavenger receptors that eliminate blood-borne pathogens and bacteria
from blood stream (Lemoinne, S. et al., 2013). They also secrete pro-inflammatory

Introduction
5
cytokines and reactive oxygen species (ROS) (Nagy, L.E., 2003). The immune response
induced by these mediators promotes hepatocytes injury. Natural killers (NK) and natural
killers T cells (NKT) cells are the major subsets of intra-hepatic lymphocytes and they
account for 10%-30% in mouse and between 30%-50% in rat and human livers (Exley,
M.A. and Koziel, M.J., 2004).
The adaptive immune cells of the liver have been also studied, mostly in the context of
viral infections. CD8+ T cells are the main effector cells that control viral infections via
cytotoxic activity and cytokine secretion, as observed in acute hepatitis C virus infection
(Sung, P.S. et al., 2014). It has also been reported that liver-resident memory CD8+ T
cells are required for protection against liver-stage Plasmodium infection (Van Braeckel-
Budimir, N. and Harty, J.T., 2014).
1.2. Obesity
Sedentary lifestyle and increased energy intake are the major causes of obesity and the
associated metabolic syndrome (Otero, Y.F. et al., 2014). Other contributing factors of
obesity include genetic susceptibility, endocrine disorders, medications and psychiatric
illnesses. The excessive accumulation of body fat also lead to decreased mobility and
other obesity-associated pathologies that reduce life expectancy (Haslam, D.W. and
James, W.P., 2005). Obesity is expressed as body mass index (BMI), a measurement
obtained by dividing a person's weight by the square of the person's height. Values
greater than 30 kg/m2 are considered overweight (WHO 2000, p.9).
Obesity is characterized by altered glucose homeostasis, hyperinsulinemia and
hypertriglyceridemia. Hyperinsulinemia is thought to be driven by the accompanying
insulin resistance in multiple tissues including liver, muscle, adipose tissue, vasculature
and the brain (Carey, M. et al., 2013). Furthermore, obesity leads to many co-morbidities,

Introduction
6
particularly heart disease, hypertension, type 2 diabetes, obstructive sleep apnea, certain
types of cancer, and osteoarthritis (Haslam, D.W. and James, W.P., 2005). For example,
hypertension incidence is increased five-fold in obese patients compared to those with
normal weight (Haslam, D.W. and James, W.P., 2005). Adipocytes from obese individuals
secrete an angiotensin precursor, which increases blood pressure. Profibrinogen and
plasminogen activator inhibitor 1 are also secreted by adipocytes and are responsible for
the change in blood viscosity (Haslam, D.W. and James, W.P., 2005).
The most common consequence of obesity is type 2 diabetes. Almost 90% of individuals
who develop type 2 diabetes have an elevated BMI. There are others factors that increase
the susceptibility of obese patients to develop diabetes, such as family history of diabetes,
mothers who had gestational diabetes, age, etc. (Haslam, D.W. and James, W.P., 2005).
Fat accumulation in different tissues leads to the development of tissue specific obesity-
associated pathologies.
1.2.1. Obesity-associated inflammation
Fat deposition in obesity is also characterized by increased infiltration of pro-inflammatory
immune cells into adipose tissues causing chronic, low-grade inflammation (Sell, H. et al.,
2012). Adipose tissue dysfunction during obesity is a consequence of inflammatory
response in the visceral compartment (Despres, J.P. and Lemieux, I., 2006). The link
between obesity and inflammation remains unclear, and different mechanisms have been
proposed. One of first hypotheses proposed is that macrophage infiltration is initiated
following death of adipocytes as a consequence of increased production of chemokines
and adipokines. During obesity, adipocyte hypertrophy results in necrosis-like death
leading to increased macrophage recruitment. Cinti et al. report that these infiltrating
macrophages form crown-like structures around necrotic adipocytes (Cinti, S. et al.,
2005). On the other hand, apoptosis, a physiological process during normal adipocyte

Introduction
7
turnover leads to accumulation of M2-type macrophages, which are anti-inflammatory.
(Sell, H. et al., 2012). Thus, the inflammation in the adipose tissues is determined by the
fate of adipocytes.
It has been suggested that the phenotype switching of the adipose tissue-associated
macrophages from anti-inflammatory M2 type to pro-inflammatory M1 type plays a central
role in obesity-associated inflammation (Sell, H. et al., 2012). The M2 macrophage
population, predominantly anti-inflammatory, decreases during obesity with a concomitant
increase in the pro-inflammatory M1 macrophages. The presence of M1 macrophages
correlates with insulin resistance and states of over-nutrition (Lumeng, C.N. et al., 2007).
A complex crosstalk between adipocytes, macrophages and other immune cells from both
innate and adaptive immune systems results in adipose tissue inflammation (Sun, K. et
al., 2011).
1.3. Fatty liver diseases
In developed countries, the increase in obesity is also related to the increased prevalence
of non-alcoholic fatty liver disease (NAFLD). Fatty liver diseases progress from benign
fatty changes to cirrhosis, portal hypertension, and hepatocellular carcinoma (Haslam,
D.W. and James, W.P., 2005). Among fatty liver patients, 50% develop fibrosis, 30%
cirrhosis, and 3% will develop liver failure and with a requirement for transplantation. Fatty
liver is one of the most common causes of end-stage liver failure. The main predisposing
factors to NAFLD are obesity, diabetes, hyperlipidemia, and hypertension. The disease is
mainly asymptomatic. The first clinical indication is the increase in the concentration of γ-
glutamyl transpeptidase and alanine aminotransferase, and to a lesser extent, aspartate
aminotransferase and alkaline phosphatase (Haslam, D.W. and James, W.P., 2005). The

Introduction
8
main treatment for NAFLD is weight reduction by regulated diet and physical activity,
insulin sensitizers and medications that reduce oxidative stress (Tapia, N.C. et al., 2006).
In 1998, Day et al., describe the pathogenesis of NAFLD as two-step pathology. Fat
accumulation is considered the first one and makes the liver susceptible to the injurious
effects of others factors, while the second one promotes the development of inflammation
(NASH: non-alcoholic steatohepatitis) and fibrosis (Day, C.P. and James, O.F., 1998).
These factors include cytokine overproduction, lipid peroxidation, hepatocyte organelle
(particularly mitochondria) malfunction, ROS and peroxisome proliferator-activated
receptor (PPAR) dysfunction in the nucleus (Zhan, Y.T. and An, W., 2010).
In the context of insulin resistance, the development of NAFLD depends on the functional
crosstalk between the liver and peripheral tissues, including the skeletal muscle and
adipose tissues, but the underlying molecular mechanisms have not been fully elucidated.
In insulin-resistant subjects, increased lipolysis in the adipose tissues increases the
circulating free-FAs that are incorporated into hepatic triglyceride (Adams, L.A. et al.,
2005). De novo hepatic lipogenesis is also upregulated by the activation of several
lipogenic transcription factors, including sterol regulatory element-binding protein 1c
(SREBP1c) and carbohydrate response element binding protein (also known as MLXIPL).
Insulin-mediated activation of SREBP1c promotes malonyl-CoA increase, which also
inhibits FA oxidation (Browning, J.D. and Horton, J.D., 2004).
Free-fatty acid toxicity has been partially explained by the endoplasmic reticulum (ER)
stress and apoptosis, induced by metabolites, including ceramides and diacyglycerols. ER
stress as well as serum-free FAs, cytokines, etc., can activate c-Jun N-terminal kinase 1
(JNK1) (also known as MAPK8), which phosphorylates insulin receptor substrate 1 (IRS1)
resulting in its inhibition and induces pro-inflammatory cytokines in target cells such as
macrophages leading to insulin resistance (Smith, B.W. and Adams, L.A., 2011).

Introduction
9
NASH and insulin resistance are extensively related to a cytokine imbalance towards pro-
inflammatory microenvironment in the liver. Even if NASH is not classically considered to
be a T helper 1(Th1)-polarized disease, recent studies show that the increase in pro-
inflammatory Th1 cytokines and a decrease in anti-inflammatory cytokines can affect fatty
liver disease progression (Li, Z. and Diehl, A.M., 2003; Maher, J.J. et al., 2008).
FAs from the diet, adipose tissue lipolysis and intestinal bacteria activate toll-like receptors
(TLRs) expressed on immune cells in the liver, resulting in the activation of innate immune
system (Wolowczuk, I. et al., 2008). Moreover, the adipose tissue-derived cytokines can
also activate immune cells in the liver. In NAFLD, KCs become activated by
proinflammatory cytokines such as tumor necrosis factor-α (TNFα) (Fan, J. et al., 2001;
Su, G.L., 2002), and their inactivation could prevent the development of alcoholic fatty
liver and NAFLD (Zhan, Y.T. and An, W., 2010). Moreover, different cell types within the
liver can promote each other in perpetuating the inflammatory response. For instance,
KCs stimulate NK activity by direct interaction or indirectly through interleukin (IL)-12, IL-
18 and TNF-α (Hou, X. et al., 2009).
Similarly to macrophages, KCs can switch from anti-inflammatory state (M2) to pro-
inflammatory activated (M1) phenotype. Activated KCs play an important role in the
development of NAFLD by producing TNF-α, IL-12, IL-6, and ROS. Particularly, TNF-α
exerts a principal role in the development of NAFLD. Thus anti-TNF-α treatment improves
high fat diet-induced NAFLD (Li, Z. et al., 2003). Besides TNF-α, several other pro-
inflammatory cytokines have been studies in the context of NAFLD and insulin resistance.
IL-6 is also implicated in promoting hepatic inflammation and fibrosis. Moreover, in the
liver, IL-6 mediates insulin resistance while in the muscle it promotes the insulin-regulated
glucose metabolism (Zhan, Y.T. and An, W., 2010). Another study showed that IL-6
treatment is beneficial in NAFLD in mice, even though the mechanism is not clear (Hong,
F. et al., 2004). In addition to the proinflammatory cytokines, activated KCs also generate

Introduction
10
ROS that promote insulin resistance and are believed to play an important role in the
conversion of simple hepatic steatosis to NASH. However, the molecular mechanisms
involved in the production of ROS by KCs is not clear (Maher, J.J. et al., 2008).
Transforming growth factor (TGF)-β1 secreted by KCs, hepatic stellate cells and
sinusoidal endothelial cells, is one of the main fibrogenic factors in NASH (Zhan, Y.T. and
An, W., 2010).
Natural killer (NK) and NK cells expressing TCR (NKT) are implicated in the pathogenesis
of fatty liver diseases. NK cells originate from the bone marrow and undergo a complex
maturation process, resulting in the acquisition of their effector functions. They redistribute
from the bone marrow and lymph nodes to blood, spleen, liver and lung (Gregoire, C. et
al., 2007). NK cells are abundant in the liver sinusoids. NK cells have been shown to
contribute to the pathogenesis of liver injury, fibrosis and regeneration (Sun, R. and Gao,
B., 2004; Melhem, A. et al., 2006; Chen, Y. et al., 2007). NK cells are also implicated and
in the development of NAFLD (Lamas, O. et al., 2004). In contrast, the cytotoxic activity of
NK cells was observed to be significantly decreased in rats maintained on high fat diet
when compared to control rats (Lamas, O. et al., 2004). Also, obese patients had
significantly lower circulating NK (CD56+CD3-) cells (7.6% of all lymphocytes) when
compared with lean healthy controls (16.6% of all lymphocytes) (O'Shea, D. et al., 2010).
Another study reported an increase in hepatic NK cells in patients with NASH, but not in
patients with NAFLD, while they are barely detectable in healthy controls (Kahraman, A. et
al., 2010).
Two different roles have been proposed for hepatic NK cells in the context of liver injury.
First, NK cells exert an anti-fibrotic function by inducing cell cycle arrest and apoptosis in
HSCs. Second, interferon-γ (IFN-γ) secreted by NK cells can induce the apoptosis of
hepatocytes, which is one of the prominent feature of liver injury during the pathogenesis
of NASH (Zhan, Y.T. and An, W., 2010). NK cell-regulating cytokines are involved in

Introduction
11
NASH; for example, IL-18 as well as IL-12 are the most important KC-derived cytokines,
which promote NK cell activation (Diehl, A.M., 2002). Another important cytokine in NK
cell homeostasis and proliferation is IL-15, which will be discussing in later sections.
NKT cells, co-express T-cell receptor (TCR) and NK cells markers (Balato, A. et al.,
2009). In the fatty liver of leptin-deficient ob/ob mice, NKT cells were selectively reduced
(Guebre-Xabier, M. et al., 2000). However, recent studies show that NKT cells accumulate
in progressive fatty liver disease. For example hepatic CD3+CD56+ NKT cells increased
during progression of NAFLD (Tajiri, K. et al., 2009). In patients with chronic liver disease,
NKT cell accumulation coincides with the development of cirrhosis (Syn, W.K. et al.,
2010). However, the role of NKT cells in NAFLD is not clear. Recently Lynch, L. et al.,
showed that invariant NKT (iNKT) cells are decreased in mice maintained on HFD. The
same group also reported a reduction in circulating iNKT population in obese patients
(Lynch, L. et al., 2012). The iNKT cells (also known as classical, conventional or class-I
NKT cells) express a restricted TCR repertoire composed of Va14–Ja18 and
Vb8.2/Vb7/Vb2 chains in mice and homologous Va24–Ja18 and Vb11 chains in humans,
and recognize glycolipid antigens in the context of the MHC-I-related protein CD1d
(Bendelac, A. et al., 2007).
The reported differences in the NKT cells infiltration in the fatty liver in several studies may
be explained by the different fatty liver models used and the variety of markers that have
been used to identify NKT cells. The role of the different subsets of NKT cells in fatty liver
disease is not clear at present. Activation of NKT results in the upregulation of FAS ligand
on their cell surface. FAS ligand can interact with FAS that is expressed on hepatocytes
inducing their apoptosis (Swain, M.G., 2008). Based on this observation, it has been
proposed that, therapies aimed at reducing the accumulation of NKT cells in the liver
could be a therapeutic strategy to minimize liver damage in patients with NASH.

Introduction
12
1.3.4. Mouse models of NAFLD
The published models for fatty liver disease can be divided into two main groups: those
caused by genetic mutations and those with an acquired phenotype produced by dietary
modifications or pharmacological treatment. These models reproduce some of the
features of human fatty liver disease, but not the complete phenotype (Anstee, Q.M. and
Goldin, R.D., 2006). In the first group, leptin deficient mice (ob/ob) are is the most widely
used mouse model. These mice became hyperphagic, inactive, obese and severely
diabetic with marked hyperglycemia. Young ob/ob mice develop steatosis and become
obese even on normal chow. Histological analysis of the liver tissues revealed fat
deposition. Similar phenotype is also observed in mice and rats carrying a genetic
invalidating mutations in the leptin receptor (db/db mouse and fa/fa rat) (Anstee, Q.M. and
Goldin, R.D., 2006).
In diet-induced models, the mice are maintained on diet that is high on carbohydrate (65%
sucrose) or fat (high fat diet, HFD). By 8 weeks into these diets, mice develop hepatic
steatosis with obesity, insulin resistance and macrovesicular steatosis (Anstee, Q.M. and
Goldin, R.D., 2006). Mice on HFD gradually develop visceral adiposity, hyperglycemia,
insulin and leptin resistance, as well as hepatic steatosis. On the other hand, mice fed
with low fat diet do not gain weight, are euglycemic, have normal insulin and leptin levels,
and do not develop hepatic steatosis (Collins, S. et al., 2004). As obesity is the main
cause of fatty liver in the western countries, diet induced obesity (DIO) is a good model to
study NAFLD.
1.4. Cytokines that signal via the common γ-chain (γc; CD132) receptor
Cytokines are a diverse group of soluble proteins, peptides, and glycoproteins that act as
hormonal regulators of the immune system. They regulate proliferation, differentiation and

Introduction
13
survival of immune cells. Type I cytokines include many interleukins, as well as some
growth and hematopoietic factors. They have a common structure that contains four α-
helical bundles (Rochman, Y. et al., 2009). The cytokines that signal via the common γ-
chain (γc; also known as IL-2Rγ and CD132) constitute an important family cytokines.
These include interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15 and IL-21 (Leonard, W.J., 2001).
The γ-chain was first described as a part of IL-2 receptor, which is the prototypic member
of this family (Takeshita, T. et al., 1992). In human patients the deficiency in the gene
encoding this receptor subunit leads to X-linked severe combined immunodeficiency
(XSCID). Phenotypic characterization of immune cells from those patients revealed a lack
of T cells and NK cells (Noguchi, M. et al., 1993). Moreover, the immune defect in those
patients is much more severe than in humans or mice lacking IL-2 only, suggesting that
this receptor can be shared by others cytokines, as described later (Leonard, W.J., 2001).
IL-2, also known as T cell growth factor, has an important role in the activation of NK and
B cells (Kim, H.P. et al., 2006). It also promotes peripheral T cell tolerance by controlling
the development of regulatory T cells (Treg), as well as regulating the proliferation and
apoptosis of activated T cells (Lenardo, M.J., 1991; D'Souza, W.N. and Lefrancois, L.,
2003). The high-affinity receptor for IL-2 is composed of three chains (IL-2Rα, IL-2Rβ and
γc) (Takeshita, T. et al., 1992).
IL-4 is a classical T helper 2 (Th2) cytokine that is required for the development and
function of TH2 cells. In allergy and asthma, as well as in immunoglobulin class switching,
IL-4 has been reported to play a critical role (Holgate, S.T. and Polosa, R., 2008). IL-7,
another member of the IL-2 family of cytokines, is required during the early stages of T
cell development in the thymus (Mazzucchelli, R. and Durum, S.K., 2007). It is also
required for the survival of mature T and B cells. However, NK cell development and
acquisition of effector functions is perfectly normal in the absence of IL-7 (Surh, C.D. and
Sprent, J., 2008). It should be noted that IL-7Rα is used in combination with the Cytokine

Introduction
14
Receptor-like factor 2 to form the thymic stromal lymphopoietin receptor (TSLPR). IL-7 is
also required for the development of B cells in mice but it is not necessary for B cell
development in humans (Rochman, Y. et al., 2009).
IL-9 production was first associated with the Th2 phenotype, and many of the preliminary
functions of IL-9 were tested in models of Th2-associated immunity (Gessner, A. et al.,
1993). IL-9 is produced by a subset of activated CD4+ T cells and it induces the activation
of epithelial cells, B cells, eosinophils and mast cells (Hauber, H.P. et al., 2004). Other Th
subsets also appear to have the potential to produce IL-9. Th17 cells, which are
characterized by the secretion of IL-17A and IL-17F, may also secrete IL-9 in vitro and ex
vivo (Elyaman, W. et al., 2009). IL-9R has two subunits: the α-chain (IL-9Rα) and the
common γ-chain receptor (Renauld, J.C. et al., 1992), and is expressed on T cell lines
and effector T cells but not naive T cells (Cosmi, L. et al., 2004).
Interleukin-21 (IL-21), the most recently discovered IL-2 family member, is believed to be
a key factor in the transition between innate and acquired immunity. IL-21 is secreted by
activated NKT cells, CD4+, but not CD8+ T cells, CD19+ B cells, CD14+ monocytes and
dendritic cells (Brandt, K. et al., 2003). IL-21 is preferentially expressed in Th2 cells in
murine models, whereas in humans IL-21 mRNA was detected in Th1 cells and in
follicular helper T cells (Pelletier, M. and Girard, D., 2007). A more detailed study showed
that human IL-21 is mainly expressed by activated CD4+ central and effector memory T
cells, some activated Th1-polarized cells, but not Th2-polarized cells (Onoda, T. et al.,
2007). IL-15 is essential for the development of NK cells the homeostasis of CD8+ T cells
(Rochman, Y. et al., 2009). Given that this cytokine is the main subject of this thesis, it is
discussed in detail in the following section.

Introduction
15
1.5. Interleukin-15 (IL-15)
1.5.1. Structure
IL-15 is a 14–15kDa cytokine containing 114 amino acids and is located in human
chromosome 4q31, and the central region of mouse chromosome 8 (Waldmann, T.A.,
2006). The genomic structure of human IL-15 contains 9 exons (7 coding exons), with a
similar intron/exon structure (Anderson, D.M. et al., 1995a). The overall intron/exon
structure of IL-15 is similar to that of the IL-2 gene and other 4 α-helix bundle cytokines.
However, at the nucleotide or protein level, there is minimal homology between IL-2 and
IL-15 (Fehniger, T.A. and Caligiuri, M.A., 2001).
IL-15 precursor protein contains a long 48–amino acid leader peptide and a 114-AA
mature protein (Nagarajan, S. et al.). Identification of human, simian, and murine IL-15
indicated that this cytokine was conserved between species (97% identity between human
and simian; 73% identity between human and murine) (Grabstein, K.H. et al., 1994). An
alternative IL-15 precursor protein expresses a 21-AA short signal peptide (SSP)
compared to the originally discovered IL-15 precursor with 48-AA long signal peptide
(LSP). However, both IL-15 isoforms encode an identical mature IL-15 protein in human
and mouse (Fehniger, T.A. and Caligiuri, M.A., 2001). Both LSP–IL-15 and SSP–IL-15
appear to have 2 to 3 log-fold less secretion than IL-2. Studies with IL-15–green
fluorescent protein (GFP) fusion protein, show that LSP–IL-15 was targeted to the
secretory pathway (ER/ Golgi apparatus), whereas SSP–IL-15 appeared to be restricted
to the cytoplasm and nucleus (Gaggero, A. et al., 1999). SSP–IL-15 mRNA is expressed
in the heart, thymus, appendix, and testis, whereas LSP–IL-15 is in skeletal muscle,
placenta, heart, lung, liver, thymus, and kidney. The biologic significance of these different
IL-15 isoforms is not clear (Gaggero, A. et al., 1999).

Introduction
16
1.5.2. IL-15 receptor
The structure of the IL-15/IL-15R quaternary complex bears similarity to that of the two
other γc-containing cytokine-receptor complexes reported so far, IL-2 and IL-4 (Wang, X.
et al., 2005; LaPorte, S.L. et al., 2008). The IL-15 quaternary complex, containing its own
α-receptor subunit and the shared signalling receptors IL-2Rβ (CD122) and γc, assembles
in a way nearly identical to that of the IL-2 quaternary complex (Protein Data Bank
accession code, 2B5I), with IL-2Rβ binding to site I on the cytokine and γc binding to site II
(Ring, A.M. et al., 2012). The IL-2R/15Rβ receptor contain a 214 amino acid extracellular
segment, a 25 amino acids transmembrane region (Balato, A. et al.), and a 286-amino
acid cytoplasmic domain (Hatakeyama, M. et al., 1989). The human γc consists of a 233-
amino acid extracellular domain, a 28-amino acid TM domain, and an 86-amino acid
cytoplasmic region (Takeshita, T. et al., 1992). IL-15Rα is structurally similar to IL-2Rα
with a conserved extracellular protein-binding Sushi domain. IL-15Rα has a 173-amino
acid extracellular domain, a single 21-amino acid TM region, and a 37 amino acid
cytoplasmic domain (Takeshita, T. et al., 1992).
The gene coding for IL-15Rα (Il15ra) is located on human chromosome 10 and contains
seven exons. Alternative splicing leads to eight different isoforms of IL-15Rα (Dubois, S.
et al., 1999). In humans there are three types of splicing events: (i) alternative usage of
exon 7 or 7’; (ii) deletion of exon 3 encoding the linker region, and (iii) deletion of exon 2,
which encodes the Sushi domain and thus cannot bind IL-15 (Dubois, S. et al., 1999). IL-
15Rα alone is sufficient for high-affinity (Kd greater than or equal to 10-11 M) binding of IL-
15, but, like IL-2Rα, it plays no role in signal transduction. Thus, the signal transduction
can happen only in the presence of the IL-2/15Rβ and γc, even if IL-15Rα binds IL-15 with
high affinity. IL-15, like IL-2, may also bind and signal through the heterodimeric IL-
2/15Rβγc with intermediate affinity (Kd approximately 10-9 M) in the absence of IL-15Rα
(Armitage, R.J. et al., 1995).

Introduction
17
Transcript levels of full-length IL-15Rα are found in numerous tissues and cell lines (Steel,
J.C. et al., 2010). In certain tissues such as brain, intestine, liver, peripheral blood
mononuclear cells [PBMCs], the expression of all 8 IL-15Rα isoforms was observed;
however, the relative expression of each isoform varied (Anguille, S. et al., 2009). IL-15Rα
mediates proliferative and homing functions that are essential for the homeostasis of
mature lymphocytes. Lodolce et al. (Lodolce, J.P. et al., 1998), show that Il15-/-, Il15ra-/-,
Il2rb-/- and Il2rg-/- mice are deficient in NK, NKT, and TCRγδ intraepithelial lymphocytes
(IELs). Thus, they proposed that all the 3 subunits of IL15R are required for IL-15-
mediated signals during the development of these cell types. The dependence of NKT
lymphocytes and TCRγδ IELs on IL-15Rα appears to be intermediate between NK cells
and CD4+ T lymphocytes, suggesting that other cytokines (e.g., IL-7) may partly
compensate for IL-15 in the differentiation of NKT lymphocytes and TCRγδ IELs (Lodolce,
J.P. et al., 1998). CD8 single positive (SP) thymocytes were reduced in IL-15RαKO mice
indicating that intrathymic T cell differentiation of CD8 SP, but not CD4 SP thymocytes is
dependent on IL15Rα (Lodolce, J.P. et al., 1998).
1.5.3. IL-15 signalling
IL-15R signalling leads to activation of the Janus kinase (Sabatti, C. et al.) and Signal
Transducer and Activator of Transcription (STAT) pathway. CD122 recruits JAK1, which
leads to the phosphorylation of STAT3. The γc recruits and phosphorylates JAK3, which
leads to the phosphorylation of STAT5. Once phosphorylated, STAT3 and STAT5 form
homo- or hetero-dimers and translocate to the nucleus where they are responsible for the
activation of certain genes (Johnston, J.A. et al., 1995). The IL-15 signalling pathway can
also induce phosphorylation of Lck and spleen Tyr kinase (Syk) kinase (Ratthe, C. and
Girard, D., 2004; Uhlin, M. et al., 2005).

Introduction
18
1.5.4. Mechanisms mediating IL-15 responses
As IL-15 was initially identified through its ability to mimic IL-2–induced T-cell proliferation,
the biochemical and functional relationship between these two cytokines were examined
in detail. IL-2 and IL-15 signalling starts when the cytokines bind IL-2Rα or IL-15Rα,
respectively, and are presented to IL-2Rβ and γc. IL-15 can also be presented in trans, by
cells expressing IL-15Rα, to IL-15-responsive cells expressing IL-2Rβ and γc (Ring, A.M.
et al., 2012). In 2001, it was reported that IL-15 response in T cells did not required IL-
15Rα expression in the lymphocytes, but was absolutely dependent on the IL-15Rα
expression by the non-hematopoietic cells (Lodolce, J.P. et al., 2001). Given that IL-15
was previously shown to have direct effect on T cells, other investigations were performed
in order to elucidate the role of IL-15Rα expression in others cells. In 2002, Dubois et al.
(Dubois, S. et al., 2002) proposed the theory of trans-presentation based on the effect of
IL-15R component on responding T cells and in monocytic cell lines. In this study, they
showed that the effect of IL-15 on T cells was longer than that of IL-2 since IL-15R
allowed for the continued presence of IL-15 on the cell surface of monocytes. Moreover,
IL-15 and its α-receptor associate in the endoplasmic reticulum (ER) (Dubois, S. et al.,
2002). The finding that IL-15R shuttles IL-15 to the cell surface suggest that IL-15 is not
secreted, providing an explanation for the inability to detect IL-15 in biological fluids.
Overall, trans-presentation was proposed as a mechanism to explain how the expression
of IL-15Rα by neighbouring cells was crucial for IL-15 to signal through the βγc (Dubois, S.
et al., 2002).
The theory of IL-15 trans-presentation was postulated based on in vitro experiments, but
subsequent in vivo results provided additional evidence. The generation of antigen-
specific memory CD8+ T cells and their homeostatic proliferation in vivo was independent
of IL-15Rα expression in T cells, but the response required IL-15Rα expression by the
host cells (Schluns, K.S. et al., 2004a). Moreover, similar results were found in NK cells

Introduction
19
development and homeostasis. Using specific combinations of cell transfers and bone
marrow (BM) chimeras, Ma, A. et al., proved that NK cells require IL-15R expression by
the cells in their environment but did not need self-expression (Koka, R. et al., 2004).
IL-15 trans-presentation is not the result of surface-capture effect mediated by IL-15
binding to IL-15Rα in the same cell, as is the case for IL-2. Nevertheless, trans-
presentation has proven to be a major mechanism of IL-15 action in vivo, which suggests
that IL-15Rα may have other IL-15-sensitizing functions in addition to surface capture. For
example, Aaron et al., suggested that IL-15Rα might stabilize a conformation of IL-15 that
is more able to bind IL-2Rβ, akin to the effect of IL-2Rα for IL-2 (Ring, A.M. et al., 2012).
IL-15Rα binding increased the affinity of IL-15 for IL-2Rβ approximately 150-fold (Ring,
A.M. et al., 2012). IL-15Rα is widely expressed in the tissues, IL-15 is believed to exist in
the body mainly in a complex with IL-15Rα and is therefore primed for trans-presentation
to cells that express IL-2Rβ and γc (Stonier, S.W. and Schluns, K.S., 2010). The
cytoplasmic domain of IL-15Rα, like that of IL-2Rα, appears to be dispensable for
signalling. Indeed, it has been reported that high concentration of IL-15 can bind and
transduce a signal in cells expressing only the IL-2Rβ and γ chains, as can IL-2
(Anderson, D.M. et al., 1995b).
Other groups have proposed alternative mechanisms for IL-15 responses and delivery.
Following Vesicular stomatitis virus (VSV) infection, it was found that CD8+ T cell
expansion was partially defective in the absence of IL-15 but was not defective in the
absence of IL-15Rα (Schluns, K.S. et al., 2002). This suggested that in an inflammatory
context, an abundance of sIL-15 (define soluble or surface, as sIL-15 can mean both) may
be available to act through the βγc complex and bypass IL-15Rα trans-presentation. In this
scenario, IL-15Rα is completely dispensable. As described above, numerous studies
postulate that the expression of IL-15Rα is not required on IL-15-dependent cells (i.e.

Introduction
20
CD8+ T cells, NK cells, and IELs). However, lymphocytes express some of the highest
levels of IL-15Rα, the significance of which is not known (Schluns, K.S. et al., 2004b).
Crystal structure of IL-15Rα revealed a threonine/proline-rich region between the trans-
membrane and the IL-15 binding domain, that confers flexibility. This suggests that IL-
15Rα can also present IL-15 to βγc in the same cell, in a process called as cis-
presentation (Olsen, S.K. et al., 2007). The physiological relevance of this process has not
been yet elucidated. Transfection experiments with IL-15Rα in naïve CD8+ T cells showed
that the response to IL-15 is enhanced in transfected cells versus non-transfected
counterparts (Rowley, J. et al., 2009). Another group reported that IL-15Rα knock-in in T
cells had slightly increased levels of homeostatic proliferation (Wu, Z. et al., 2008). Taken
together, these observations reveal that cis-presentation can also mediate IL-15-induced
responses. In the Fig 1.1 we summarise the main mechanisms for IL-15 delivery to the
responsive cells.
Figure 1.1: Principals mechanism for IL-15 delivery. A) Trans-presentation: IL-15Rα and IL-15 are synthetized in the same cell and transported to the cell surface where the cell surface complex can stimulate neighboring cells through the IL-15R βγc. B) Cis-presentation: IL-15 is presented by IL-15Rα on the same cell; these mechanisms may utilize IL-15 derived from autocrine or paracrine sources (Stonier, S.W. and Schluns, K.S., 2010).

Introduction
21
1.5.5. Function of IL-15 in T cells
IL-15 induces proliferation of naïve CD8+ and memory CD4+ and CD8+ T cells (Kanegane,
H. and Tosato, G., 1996; Stoklasek, T.A. et al., 2006). In the presence of IL-15, CD4+ and
CD8+ T cells resist the regulatory effects of Tregs (Ben Ahmed, M. et al., 2009). IL-15 also
has demonstrated chemotactic activity towards CD4+ and CD8+ T cells (Wilkinson, P.C.
and Liew, F.Y., 1995).
The anti-apoptotic effects of IL-15 in lymphocytes are mediated by the upregulation of
anti-apoptotic proteins of the Bcl-2 family. Stimulation with IL-15 increases the levels of
anti-apoptotic proteins BCL-2, BCL-xL, MCL-1 (Lodolce, J.P. et al., 1998; Becker, T.C. et
al., 2002) and downregulates pro-apoptotic proteins BAX, BID, BIM, NOXA and PUMA
(Van Belle, T. and Grooten, J., 2005; Huntington, N.D. et al., 2007). IL-15 can also
prevent apoptosis by activating NF-kB (Hoontrakoon, R. et al., 2002), and inhibiting
caspase-3 and -8 (Bouchard, A. et al., 2004). Long-term survival, proliferation and
renewal of memory CD8+ T cells by IL-15 are mediated by the upregulation of Bcl-2
(Lodolce, J.P. et al., 1998; Becker, T.C. et al., 2002). As a result, Il15-/- and Il15ra-/- mice
lack memory CD8+ T cells (Lodolce, J.P. et al., 1998; Kennedy, M.K. et al., 2000).
1.5.6. IL-15 in NK cell biology
Natural killer cells (also known as NK cells, K cells, and killer cells) are a type of
lymphocytes that play a major role in elimination of both tumours and viral infected cells.
Their functions do not require priming. While the earliest stages of NK cells development
is confined to the BM, the later stages of development is a continuous process that occurs
in both the BM and peripheral tissues, such as the spleen and liver. In the BM, the
common lymphoid progenitor differentiates into NK precursors (NKp), which express IL-
15Rβ (Schluns, K.S. et al., 2004a). In mice, transition from NKp to immature NK cells is

Introduction
22
marked by the expression of NKG2D, NK1.1 and the CD94/NKG2 heterodimeric complex.
This is followed by the increased expression of both activating and inhibitory Ly49
receptors (Ly49R) (Kim, S. et al., 2002). Maturation of NK cells is characterized by
increased killing capacity and pro-inflammatory cytokine secretion, as well as the
expression of CD49b (DX5) (Yajima, T. et al., 2001). These functional attributes become
enhanced with the upregulation of CD11b and CD43, which identifies the second stage of
maturation (Kim, S. et al., 2002). There are many transcription factors involved in NK cell
differentiation and functional maturation, such as Id2, Id3, Ikaros, Runx3, E4bp4, Gata-3,
T-bet and Eomesodermin (Boggs, S.S. et al., 1998; Gordon, S.M. et al., 2012).
IL-15 is indispensable for NK cell development. The absence of IL15 or its receptor
components results in a decrease of this cell population (DiSanto, J.P. et al., 1995;
Lodolce, J.P. et al., 1998; Kennedy, M.K. et al., 2000). IL-15-mediated upregulation of the
expression of anti-apoptotic molecules Bcl-2 and Mcl-1 and the downregulation of pro-
apoptotic molecules Bim and Noxa also mediate the survival of NK cells (Huntington, N.D.
et al., 2007; Nakazato, K. et al., 2007). Activation of NK cells is also dependent on IL-15.
Accumulating evidence suggests that trans-presentation of IL-15 is essential for the
activation of NK cells. In lymphopenic recipients, adoptively transferred IL-15Rα-deficient
NK cells can survive, but this response requires IL-15Rα expression by the recipients
(Koka, R. et al., 2003). Interestingly, the lack of IL-15Rα in non-hematopoietic cells does
not affect NK cell numbers. However, trans-presentation of IL-15 by hematopoietic cells is
more efficient than that by non-hematopoietic cells. Limiting IL-15Rα expression to
hematopoietic cells alone is sufficient to generate normal NK cell numbers in the BM,
while their numbers are only slightly reduced in the periphery (Schluns, K.S. et al., 2004b).

Introduction
23
1.5.7. The role of IL-15 in iNKT cells
iNKT cells are an NKT cell subset that expresses an invariant TCR that recognizes
galactoceramide and do not require antigen priming for activation. Development of iNKT
cells in the thymus and their homeostasis in the periphery are dependent on IL-15
(Kennedy, M.K. et al., 2000; Matsuda, J.L. et al., 2002). Il15ra-/- as well as Il15-/- mice
present similar defects in iNKT cells (Matsuda, J.L. et al., 2002; Matsuda, J.L. et al.,
2006). In the thymus, IL-15 enhances the survival of developing iNKT cells (Castillo, E.F.
et al., 2009; Chang, C.L. et al., 2011; Gordy, L.E. et al., 2011). Moreover, it has been
shown that IL-15 upregulates anti-apoptotic factors, such as Bcl-2, BclxL and Mcl-1 and
downregulates proapoptotic factors like Bim (Huntington, N.D. et al., 2007). Conversely, in
the periphery, IL-15 regulates both proliferation and survival of iNKT cells. IFN-γ
production by iNKT cells following a-galactosylceramide stimulation is deficient in Il15ra-/-
mice suggesting that IL-15 signalling also regulates their activation (Chang, C.L. et al.,
2011; Gordy, L.E. et al., 2011). Unlike NK cells, which require IL-15Rα expression on
hematopoietic cells, IL-15Ra expressed on non-hematopoietic cells is sufficient for thymic
iNKT development (Castillo, E.F. et al., 2010; Chang, C.L. et al., 2011).
In the thymus, selective expression of IL-15Rα in dendritic cells (DCs) had no impact on
iNKT development whereas IL-15 trans-presentation by medullary thymic epithelial cells
(mTECs) seems to play a major role in the development and functional maturation of iNKT
cells (Castillo, E.F. et al., 2010). In the periphery, studies using BM chimeras and
transgenic models suggest that IL-15Rα expression is equally important in both
hematopoietic and non-hematopoietic cells (Castillo, E.F. et al., 2010). IL-15Rα
expression only by hematopoietic cells or DCs lead to a defective thymic iNKT
development while differentiation and expansion of hepatic iNKT cells is relative normal,
demonstrating the existence of IL-15-mediated extrathymic iNKT cell development
(Castillo, E.F. et al., 2010).

Introduction
24
In the liver, IL-15 is produced by liver resident DCs, macrophages (KCs) and hepatic
stellate cells (non-hematopoietic origin). DC-mediated IL-15 trans-presentation generates
functionally mature iNKT cells (Castillo, E.F. et al., 2010). As the role of KCs and HSCs
cells in IL-15 trans-presentation is less clear, future studies using different IL-15Rα-
conditional KO mice are needed to elucidate their role. Overall these studies suggest that
IL-15 trans-presentation by the tissue microenvironment is absolutely required for the
maintenance of iNKT cells in peripheral tissues.
1.5.8. IL-15 functions in non-immune cells
Mesenchymal stem cells, osteoblasts, adipocytes, endothelial cells and myoblasts
express high amounts of IL-15 mRNA (Nilsen, E.M. et al., 1998; Satoh, J. et al., 1998;
Silva, W.A., Jr. et al., 2003), suggesting that IL-15 could have some effects on these non-
immune cells. For example, fibroblasts are the main source of increased levels of IL-15 in
the synovium of rheumatoid arthritis (RA) patients (Miranda-Carus, M.E. et al., 2004).
Moreover, apoptosis of synovial fibroblasts as well as TNFα-induced apoptosis in mouse
L929 fibroblasts is inhibited by IL-15 (Yang, L. et al., 2002; Budagian, V. et al., 2005). IL-
15 stimulates the formation of osteoclast-like cells in rat bone-marrow cultures in vitro.
Also, stimulation with IL-15 induces the expression of calcitonin receptor mRNA in
preosteoclasts (Ogata, Y. et al., 1999). In RA patients IL-15 aggravates bone destruction
by stimulating excessive bone resorption by osteoclasts (Ogata, Y. et al., 1999; Miranda-
Carus, M.E. et al., 2006).
Epithelial cells lines such as human keratinocytes and immortalized HaCaT keratinocytes
express both IL-15 and IL-15Rα (Ruckert, R. et al., 2000). IL-15 inhibits apoptosis in
keratinocytes (Ruckert, R. et al., 2000; Yano, S. et al., 2003). Additionally, IL-15 induces
epithelial cells proliferation through the activation of ERK1/2, PI3K and Akt pathways

Introduction
25
(Yano, S. et al., 2003). Tubular epithelial cells (TECs) are a rich source of IL-15 that
stimulates intratubular CD8+ T cells (Robertson, H. and Kirby, J.A., 2003). Also IL-15 is an
autocrine survival factor for TECs, protecting them from apoptosis and inhibiting
manifestation of nephrotoxic serum-induced glomerulonephritis (Shinozaki, M. et al.,
2002).
Extravasation of activated T cells is stimulated by IL-15 since this cytokine induces
hyaluronan expression by endothelial cells. Interaction between cell-surface glycoprotein,
CD44, and hyaluronan is important for T cells adhesion and recruitment to the blood
vessel wall (Estess, P. et al., 1999). Endothelial cell-derived IL-15 induces
transendothelial migration of T cells by activating the binding capacity of LFA-1 integrin,
and increases T-cell motility (Oppenheimer-Marks, N. et al., 1998).
Muscles express high levels of IL-15 mRNA (Grabstein, K.H. et al., 1994). IL-15 has
anabolic effects in the muscle in vitro, as described for insulin growth factor-1 (IGF-1)
(Quinn, L.S. et al., 1995). Overexpression of IL-15 induces skeletal muscle hypertrophy in
vitro (Quinn, L.S. et al., 2002). The general reduction of proteolysis induced by IL-15 could
be the main mechanism involved in its anabolic effects (Busquets, S. et al., 2005). IL-15
has been also described as a myokine (cytokines which are secreted by muscle cells) with
effects on adipose tissue. Muscle-to-fat endocrine axis is responsible for fat body
composition and insulin sensitivity (Carbo, N. et al., 2001; Quinn, L.S. et al., 2005).
Interesting, IL-15 sensitivity differ in different adipocyte subpopulations and there are also
species- and developmental stage- dependent differences. For example in 3T3-L1
preadipocytes, IL-15 inhibits lipid deposition while it has no effect on lipid deposition in
fully differentiated 3T3-L1 cells (Quinn, L.S. et al., 2005).
The wide range of expression of IL-15 and its receptor also occurs in the central nervous
system. IL-15 mRNA is detected in microglia, astrocytes and neuronal cell lines. Low

Introduction
26
doses of IL-15 support microglial cell growth, attenuats their nitric oxide production and
induce JAK1 phosphorylation (Hanisch, U.K. et al., 1997; Kurowska, M. et al., 2002).
1.5.9. Functional dichotomy between IL-2 and IL-15
IL-15 and IL-2 have similar biologic properties in vitro, consistent with their shared
receptor signalling components (IL-2/15Rβγc). Like IL-2, IL-15 stimulates lymphocyte
activation and proliferation in vitro (Bamford, R.N. et al., 1994; Armitage, R.J. et al., 1995).
This stimulation occurs via both promotion of cell proliferation and protection against
apoptosis. Despite the apparent similarities, in vivo experiments with IL-2 and IL-2RαKO
mice indicate that IL-2R and IL-15R are not redundant proteins (Lodolce, J.P. et al.,
1998). IL-2 and IL-2Rα mice suffer severe lymphadenopathy and autoimmunity
suggesting the critical role of IL-2 in the regulation of immune response (Sadlack, B. et al.,
1993). IL-2R functions cannot be compensated by IL-15R or by other cytokines of the Ɣc
receptors family. Thus, IL-15R likely performs distinct immune functions from IL-2R in
vivo. Accordingly, IL15Rα-deficient mice show lymphopenia and innate immune deficiency
(Lodolce, J.P. et al., 1998). Moreover, the cellular distribution of the distinct IL-15Rα and
IL-2Rα chains might also contribute to the temporal and spatial distinction between the IL-
2 and IL-15 induced activation of the via βγc-dependent signalling pathways (Fehniger,
T.A. and Caligiuri, M.A., 2001).

Introduction
27
1.6. The thesis premises
Obesity is considered as a major problem in this decade. This condition is frequently
associated with the increased prevalence of fatty liver disease. Liver diseases can evolve
from benign non-alcoholic fatty changes to NASH, and in the long-run to hepatocellular
carcinoma (Haslam, D.W. and James, W.P., 2005). Chronic low-grade inflammation is a
common feature of obesity and NAFLD. Under obese conditions, inflammatory cells
infiltrate the adipose tissues and liver. In addition to activation of resident immune cells,
inflammatory cells are also recruited from the circulation. In the normal liver, immune cells
are mainly represented by NK, NKT and CD8+ T cells, whereas in fatty liver disease there
is an increase in the infiltration and activation of these cells, in addition to the activation of
liver-specific macrophages (KCs) and Th1 cells (Li, Z. and Diehl, A.M., 2003; Zhan, Y.T.
and An, W., 2010). NASH and NAFLD are extensively related to an imbalance in the
cytokines in the liver towards pro-inflammatory microenvironment. KCs, stellate cells and
other cells can produce the inflammatory cytokines TNF-α, IL-12 and IL-6 upon activation
(Li, Z. et al., 2003). IL-15 is another pro-inflammatory cytokine, which is indispensable for
the development and homeostasis of CD8+ T cells, NK, NKT and iNKT cells (Kennedy,
M.K. et al., 2000). As these cells are implicated in perpetuation of inflammation in obesity,
and the influence of IL-15 on these cells in the liver under conditions of obesity is unclear,
I have addressed these issues using IL-15 knockout mice under conditions of diet-induced
obesity.

Introduction
28
1.7. Hypothesis
Based on the above premises, we hypothesize that IL-15 signalling in liver cells
promotes the development of NAFLD by inducing hepatic inflammation.
1.8 Objectives
The specific aims of my research project are:
1. To evaluate the role of IL-15 in the development of NAFLD
a. Characterize the induction of NAFLD by HFD in WT and Il15-/- mice using
macroscopic and microscopic analysis of the liver and serum lipids levels.
b. Evaluate the expression of genes involved in metabolism in the livers of
WT and Il15-/- after HFD.
c. Elucidate the role of IL-15 in the metabolic behavior of primary murine
hepatocytes
2. To characterize HFD-induced inflammation in the liver
a. Evaluate intrahepatic lymphocytes (NK, NKT, iNKT, T cells) and
macrophages infiltration after HFD.
b. Evaluate inflammation related genes expression in liver samples and
primary hepatocytes.
3. To define the role of IL-15 and IL-15Rα in the maintenance of NK and NKT
subsets in the liver
a. Characterize IHLs in WT, Il15-/- and Il15ra-/-
b. Characterize IHLs in macrophages and hepatocyte tissue-specific
Il15ra deficient mice

Materials and Methods
29
2. MATERIALS and METHODS
2.1. Mice
All the mice used were in C57BL/6 background. Mice were maintained in filter-topped
cages in a specific pathogen-free facility and fed with standard chow diet and water unless
specified otherwise. All experiments were carried out with the approval of the institutional
ethics committee.
Wild type (WT) C57BL/6 mice were from the Jackson laboratory. Il15-/- mice have been
already described (Ramanathan, S. et al., 2006). Il15ra-/- mice were purchased from
Charles River and bred into C57BL/6 background for more than ten generations.
Conditional Il15ra-floxed mice, with loxP sites were inserted in the first and third intron of
Il15ra locus (Il15rafl/fl), (Mortier, E. et al., 2009) were a generous gift from Dr. Averil Ma,
University of California at San Francisco, USA. Tissue specific deletion of Il15ra was
achieved by crossing these mice with Alb-Cre or LysM-Cre transgenic mice (purchased
from the Jackson Laboratory) to ablate the IL-15Rα gene specifically in hepatocytes
(Il15rafl/fl Alb-Cre+) or macrophages (Il15rafl/fl LysM-Cre+) respectively.
2.1.1 Induction of NAFLD in mice
To induce hepatic steatosis, 8-weeks old WT, Il15-/- and Il15ra-/- mice, were maintained on
high fat diet (HFD) (Product Data-D12492: 20%kcal protein, 20% carbohydrate and 60%
fat). Mice fed with normal control diet (NCD) were used as controls. These experiments
were performed in male mice. Female mice were not used as they show hormone-
mediated resistance to obesity induction (Hong, J. et al., 2009). Mice were maintained on
NCD or HFD for 16 weeks before sacrifice. The body weight and the weight of liver and
adipose tissues were measured at sacrifice. Aliquots of tissues were stored in formalin for

Materials and Methods
30
histology, in OCT (optimal cutting temperature gel-like medium consisting of polyethylene
glycol and polyvinyl alcohol) for immunohistochemistry and snap frozen in liquid nitrogen
for RNA and protein extractions.
2.2. Isolation of intrahepatic lymophocytes (IHL)
Mice were sacrificed and the livers were collected and rinsed with Krebs-Ringer-Buffer
(KRB, 154 mM NaCl, 5.6 mM KCl, 5.5 mM Glucose, 20.1 mM HEPES, 25 mM NaHCO₃,
pH 7.4). The liver tissues were digested in pre-warmed KRB supplemented with 2mM
CaCl₂, 2mM MgCl₂, 300 CDU (Collagenase digestion unit)/ mL Collagenase IV
(Worthington) and 150 U/mL DNase I (Sigma) using gentleMACS Dissociator (Miltenyi
Biotec) according to the instructions of the manufacturer. Next, the homogenized liver
samples were gently agitated on a rocking shaker for 30 minutes at room temperature.
The tubes were left on a stand for 1 minute to precipitate undigested liver tissue and the
supernatants were passed through 40µm cell strainers. Cells were resuspended in 25 ml
of cold PEB buffer (0.5% Bovine serum albumin and 2mM EDTA in Phosphate buffered
saline (PBS)) and centrifuged at 50×g for 5 minutes at 4 °C to eliminate contaminating
hepatocytes. The supernatant was centrifuged at 300×g for 10 minutes at 4 °C to collect
the lymphocytes. Cells were resuspended in a buffered solution of ammonium chloride-
potassium (ACK) (155 mM NH4Cl, 1mM KHCO3, 0.5 mM EDTA, pH 7.2) to lyse the red
blood cells. The remaining cells were washed twice with PEB buffer and used to prepare
the single cell suspension for FACS analysis.
2.3. Isolation of splenocytes
Single cell suspensions were prepared by teasing the spleens in PBS containing 2%
foetal calf serum (FCS). The suspension was centrifuged at 300×g and the supernatant
was discarded. Red blood cells were lysed by resuspending in ACK solution for one

Materials and Methods
31
minute. After washing with PBS-FCS 2%, the cells were counted and resuspended in
PBS-FCS 2%, for FACS staining and analyses.
2.4. FACS analyses
IHL and splenocytes were stained with the corresponding antibodies (Armitage, R.J. et al.)
at RT, 15 minutes and washed with PBS before analyses. Abs against mouse CD3,
CD8α, CD4, CD44, CD62L, NK1.1 conjugated to PE-Cy5.5, PE, APC-Cy7, FITC and APC
were purchased from BD Biosciences (San Jose, CA), Biolegend (San Diego, CA) or
eBioscience (San Diego, CA). Mouse CD1d tetramer pre-loaded with α-GalCeramide
(CD1d*) conjugated to PE was a kind gift of Professor Yi-Guang Chen, Department of
Pediatrics, Max McGee National Research Center for Juvenile Diabetes, Medical College
of Wisconsin, Milwaukee, USA. Data was acquired on FACS Canto flow cytometer (BD
Biosciences San Diego, CA) and was analyzed using FlowJo software from TreeStar Inc
(Ashland, OR).
2.5. Isolation of primary hepatocytes
Primary hepatocytes isolation was carried out in 6-8 weeks-old mice as described
previously (Gui, Y. et al., 2011). Intra-peritoneal injection of a mixture of Ketaset® (100
mg/kg, Wyeth, Guelph, Canada) and Xylazine (10 mg/kg) was used as anesthetic. Liver
perfusion was performed through the portal vein, the thoracic segment of the inferior vena
cava was clamped and the abdominal segment was cut, allowing a closed circulation in
the liver. The liver was first perfused with 25 ml Ca2+/Mg2+-free Hank’s buffered salt
solution (HBSS) containing 0.5 mM EGTA, kept warm in 37˚ water bath, at the rate of 7
ml/min. After, the collagen digestion was made with 25 ml of collagenase type IV
(100U/ml, Worthington, Lakewood, NJ, USA) in HBSS supplemented with CaCl2 (1.8 mM)
via the same route. After slicing the liver, hepatocytes were sedimented twice by
centrifugation at 50 g at 4˚C to eliminate the non-parenchymal cells. Viability was

Materials and Methods
32
assessed by trypan blue exclusion and 0.5x106 hepatocytes in DMEM-F12 (Dulbecco’s
Modified Eagle Medium: Nutrient Mixture F-12) (GibcoBRL, Burlington, ON, Canada) with
10% FCS were seeded in collagen I (Sigma)-coated 35 mm culture dishes.
2.6. Stimulation of hepatocytes
Lyophilized recombinant human IL-15 (Peprotech) was reconstituted according to the
manufacturer’s instructions and stored at -80˚C until used. Primary hepatocytes were
starved overnight (ON) in DMEM-F12 with 0.1% FCS and stimulated with hIL-15 (20ng/ml)
for 2 hours. Subsequently, the cells were washed twice and RNA extraction was
performed.
2.7. Assessment of mitochondrial respiration in hepatocytes
Primary hepatocytes from WT and Il15-/- mice were isolated as described above and
seeded in a 96 wells plate. Sensor cartridges were pre-incubated ON in XF96 calibrating
solution in a non-CO2 incubator at 37ºC and XF96 Analyzer temperature was kept at 37ºC
during the assay. Hepatocytes were kept in culture for 4 days until they acquired the
appropriate morphology and reached approximately 90% confluence. On the day of the
assay, cells were washed 3 times with XF Assay media (non-buffered media to accurately
measure proton production rate and extra cellular acidification rate) and incubated in 150
μl of the same medium in a non-CO2 incubator at 37ºC for 20 minutes before
measurement the extracellular flux analyzer (XF96 Analyzer). The sensor cartridge was
loaded with 15μl of pre-warmed respiratory chain inhibitors (diluted in XF Assay media) at
pH 7.4 into appropriate ports. Then, the XF96 Analyzer software was run using the
optimized protocol. While the protocol is running, the inhibitors are injected in sequence
automatically, in order to evaluate the basal respiration, the maximum respiratory capacity
and non-mitochondrial respiration in the cells. For example, oligomycin inhibits ATP
synthase and consequently the oxygen consumption rate decreases to minimal level. The
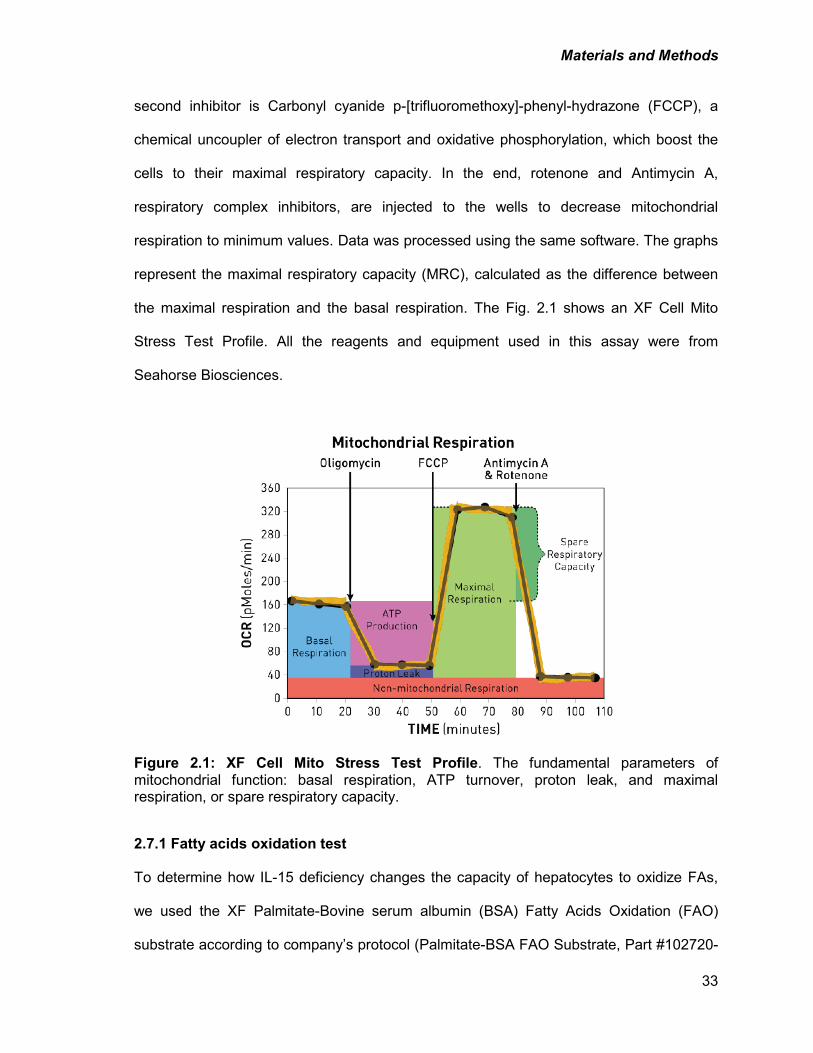
Materials and Methods
33
second inhibitor is Carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone (FCCP), a
chemical uncoupler of electron transport and oxidative phosphorylation, which boost the
cells to their maximal respiratory capacity. In the end, rotenone and Antimycin A,
respiratory complex inhibitors, are injected to the wells to decrease mitochondrial
respiration to minimum values. Data was processed using the same software. The graphs
represent the maximal respiratory capacity (MRC), calculated as the difference between
the maximal respiration and the basal respiration. The Fig. 2.1 shows an XF Cell Mito
Stress Test Profile. All the reagents and equipment used in this assay were from
Seahorse Biosciences.
Figure 2.1: XF Cell Mito Stress Test Profile. The fundamental parameters of mitochondrial function: basal respiration, ATP turnover, proton leak, and maximal respiration, or spare respiratory capacity.
2.7.1 Fatty acids oxidation test
To determine how IL-15 deficiency changes the capacity of hepatocytes to oxidize FAs,
we used the XF Palmitate-Bovine serum albumin (BSA) Fatty Acids Oxidation (FAO)
substrate according to company’s protocol (Palmitate-BSA FAO Substrate, Part #102720-

Materials and Methods
34
100). Palmitate-BSA FAO integrates the XF Cell Mito Stress Test with the BSA Control
and XF Palmitate reagent. Primary hepatocytes were isolated as described in Section 3.5
and seeded in a 96 well plate. The day previous to the assay, the growth medium was
exchanged for substrate-limited medium (DMEM, 0.5 mM Glucose, 1.0 mM GlutaMAXTM
(life technologies), 0.5 mM carnitine and 1% FCS) and incubated ON. Next, cells were
kept in FAO medium (Krebs-Henseleit Buffer (KHB) with 2.5 mM glucose, 0.5 mM
carnitine and 5 mM HEPES) for 45 min and BSA control or Palmitate-BSA substrate was
added. XF Cell Mito Stress Test analysis was performed following the protocol described
in the previous section.
2.8. Tissues processing for histology
Mice were sacrificed and the tissues were sectioned and fixed in a 10% formalin solution.
Fixative volume was approximately 20 times that of tissue on a weight per volume. The
tissues were fixed for a minimum of 48 hours at room temperature and then kept at 4˚C.
For paraffin infiltration the tissues were dehydrated through a series of graded ethanol
baths to displace the water, and then infiltrated with paraffin. The infiltrated tissues were
then embedded into paraffin blocks. Tissues were sectioned into 5 µm thick sections.
Some tissue sections were kept frozen for immunohistochemistry or lipids staining. To
preserve tissue morphology and retain the antigenicity of the target molecules, the tissues
were fixed in paraformaldehyde (PFA)/PBS 4%, 5 min and kept ON at 4°C in 4%
sucrose/PBS solution. The sucrose solution was changed daily to increase sucrose
percentage up to 30%. Tissues were mounted in OCT embedding compound and frozen
at -80°C. Tissue sections (7 µm) were cut using a cryostat and conserved at -20°C until
use.

Materials and Methods
35
2.9. Histology and lipids detection
Liver sections were de-paraffinized and rehydrated in serial dilutions of ethanol and water
before staining. Slides were stained with Harris’s hematoxylin solution for 5 min, then
washed in tap water and fixed in acid-water solution (1% acetic acid) followed by rinsing in
saturated aqueous lithium carbonate. The slides were washed and stained with Eosin for
1 minute and then washed in 50% ethanol before being dehydrated in increasing
percentages of ethanol solutions and xylene. Subsequently the samples were
coverslipped using Permount™ Mounting Medium (SP15-500, Fisher Scientific).
Lipid staining was carried out using Sudan Black, a basic dye that combine with acidic
groups in lipids compound, including phospholipids. Frozen sections were fixed with 10%
formalin and immersed in 100% propylene glycol, two changes, 5 minutes each. Fat
staining was made by 7 min incubation in Sudan Black (7% m: v in Propylene glycol)
followed by two washes in Propylene glycol. The slides were washed in water and
mounted with aqueous mounting media (VectaMount™ from Vector Labs). Images were
taken using automatic tissue slide scanning (NanoZoomer Digital Pathology (NDP)
system from Hamamatsu Photonics).
2.10. Immunofluorescence microscopy
Fresh liver tissue samples from WT mice in NCD or HFD were examined for macrophages
infiltration. Slices were fixed in cold acetone 10 min and washed three times in PBS and
blocked with 2% BSA in PBS for 45 minutes at 37˚C. Endogenous avidin and biotin were
blocked using Avidin/Biotin Blocking kit (Invitrogen). All the steps were intervened with
three times PBS-0.5% TWEEN®20(PBS/Tw) wash and were carried out at room
temperature. Tissues were incubated ON at 4˚C with primary anti-CD68 biotinylated
antibody (MCA 1957B, Serotec), washed in PBS/Tw and then incubated with streptavidin
coupled to Alexa-488 Fluor and the nuclear stain DAPI for 1 hour at 4˚C. Indirect

Materials and Methods
36
immunofluorescence was examined without counterstain using an Axioskop 2 phase-
contrast/epifluorescence microscope (Carl Zeiss, Inc., Thornwood, NY, USA) equipped
with a band pass filters for fluorescence of DAPI (excitation D360/40; emission D460/50)
and FITC (excitation D480/30; emission. D535/40) (Chroma Technology Corp.).
Photomicrographs of 1392 x 1040 pixels were captured using 40x objective and Retiga
SRV cooled color digital camera (Qimaging, Burnaby, BC, Canada).
2.11. RNA isolation
Liver samples were snap frozen and kept at -80˚C until use. Liver samples were
homogenized in TRIzol® (Life Technologies) using mixer mill MM 400 (Retsch, Hann,
Germany). The cultured hepatocytes were directly lysed in TRIzol®. Chloroform was
added for 10 min to homogenized samples which were then centrifuged to separate the
phases. Subsequently, the aqueous phase was collected and isopropanol was added to
precipitate the RNA. This step was followed by washing the RNA pellet in 75% ethanol
and re-suspension in RNAse-free water. RNA purity was evaluated by measuring the
260/280 nm and 260/230 nm absorption ratios and RNA quality was confirmed by running
1 µg RNA on the denaturing formaldehyde-agarose gel.
2.12. Quantitative PCR
The first strand was synthesized from 1 µg total RNA using Quantitect® (Qiagen,
Mississauga). The selected primers were examined for the efficiency and melting curve.
Primer sequences are shown in table 2.1. Gene expression was evaluated with MyQi5®
cycler (Bio-Rad) using SYBR Green Supermix (Bio-Rad). Fold induction was calculated
based on the ribosomal gene 36B4 expression, as the internal control and WT mice on
NCD as the control group for all in vivo experiments. For in vitro experiments, we used the
same housekeeping gene and the controls were non-stimulated primary cells from WT
mice.
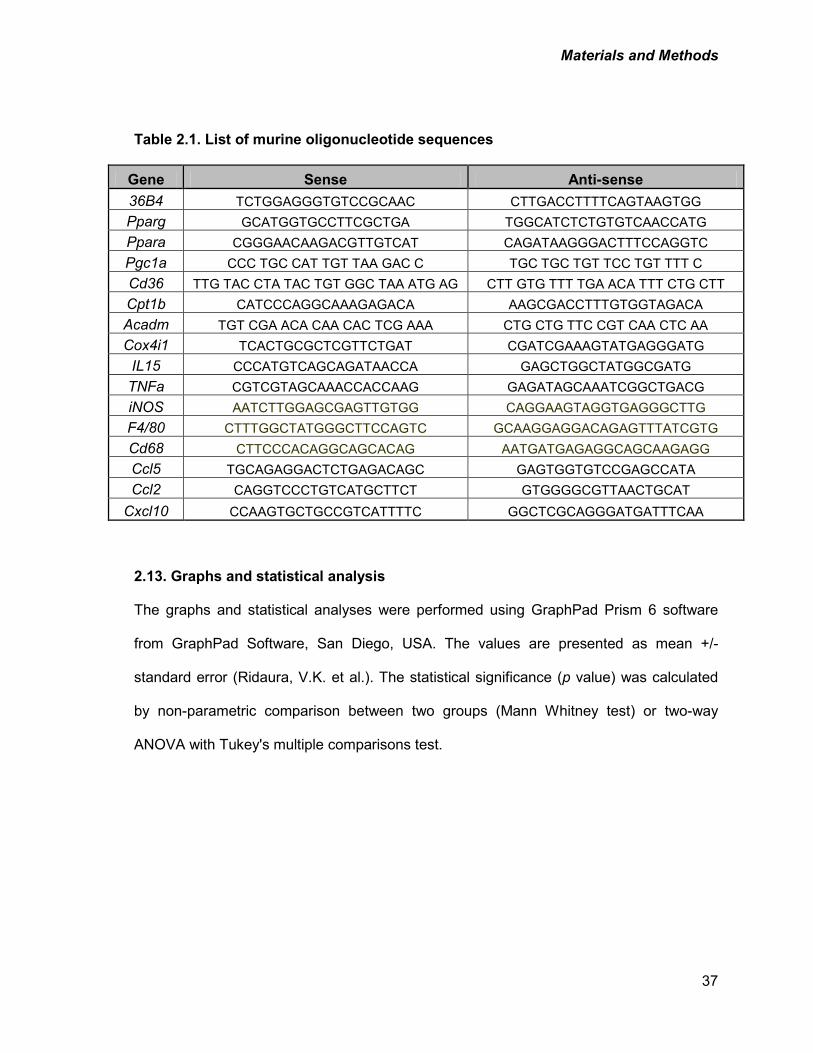
Materials and Methods
37
Table 2.1. List of murine oligonucleotide sequences
Gene Sense Anti-sense 36B4 TCTGGAGGGTGTCCGCAAC CTTGACCTTTTCAGTAAGTGG Pparg GCATGGTGCCTTCGCTGA TGGCATCTCTGTGTCAACCATG Ppara CGGGAACAAGACGTTGTCAT CAGATAAGGGACTTTCCAGGTC Pgc1a CCC TGC CAT TGT TAA GAC C TGC TGC TGT TCC TGT TTT C Cd36 TTG TAC CTA TAC TGT GGC TAA ATG AG CTT GTG TTT TGA ACA TTT CTG CTT Cpt1b CATCCCAGGCAAAGAGACA AAGCGACCTTTGTGGTAGACA Acadm TGT CGA ACA CAA CAC TCG AAA CTG CTG TTC CGT CAA CTC AA Cox4i1 TCACTGCGCTCGTTCTGAT CGATCGAAAGTATGAGGGATG
IL15 CCCATGTCAGCAGATAACCA GAGCTGGCTATGGCGATG TNFa CGTCGTAGCAAACCACCAAG GAGATAGCAAATCGGCTGACG iNOS AATCTTGGAGCGAGTTGTGG CAGGAAGTAGGTGAGGGCTTG F4/80 CTTTGGCTATGGGCTTCCAGTC GCAAGGAGGACAGAGTTTATCGTG Cd68 CTTCCCACAGGCAGCACAG AATGATGAGAGGCAGCAAGAGG Ccl5 TGCAGAGGACTCTGAGACAGC GAGTGGTGTCCGAGCCATA Ccl2 CAGGTCCCTGTCATGCTTCT GTGGGGCGTTAACTGCAT
Cxcl10 CCAAGTGCTGCCGTCATTTTC GGCTCGCAGGGATGATTTCAA
2.13. Graphs and statistical analysis
The graphs and statistical analyses were performed using GraphPad Prism 6 software
from GraphPad Software, San Diego, USA. The values are presented as mean +/-
standard error (Ridaura, V.K. et al.). The statistical significance (p value) was calculated
by non-parametric comparison between two groups (Mann Whitney test) or two-way
ANOVA with Tukey's multiple comparisons test.

Results
38
3. RESULTS
3.1. IL-15 promotes weight gain and fat accumulation in the liver
Given the proposed role of inflammation in NAFLD development, we examined the role of
IL-15 in the development of hepatic steatosis. To this end, aged-matched wild type and
Il15-/- mice were fed either NCD or HFD for 16 weeks. HFD is widely used to promote
hepatic steatosis and NASH in rodents (Nakamura, A. and Terauchi, Y., 2013). We first
evaluated whether HFD induces weight gain and fat accumulation in the liver. While the
weight gain in the WT mice was significantly higher when maintained on HFD, Il15-/- mice
did not show significant differences in body weight or liver mass when maintained on
HFD. These observations suggest that IL-15 contributes to the diet-induced increase in
body weight and liver mass in WT mice (Fig.3.1.A).
Serum levels of cholesterol and non-esterified fatty acids (NEFA) are used as clinical
indices of NAFLD severity (Okada, Y. et al., 2013). The levels of circulating cholesterol
and NEFA were increased in WT-HFD mice, but not in Il15 null mice (Fig.3.1.B).
Moreover, livers from mice fed with NCD exhibited normal hepatic architecture whereas
the livers from HFD fed mice, revealed extensive microvesicular and macrovesicular
steatosis. In contrast, lipid deposition in the liver parenchyma was absent in Il15-/- mice
after HFD (Fig.3.1.C).
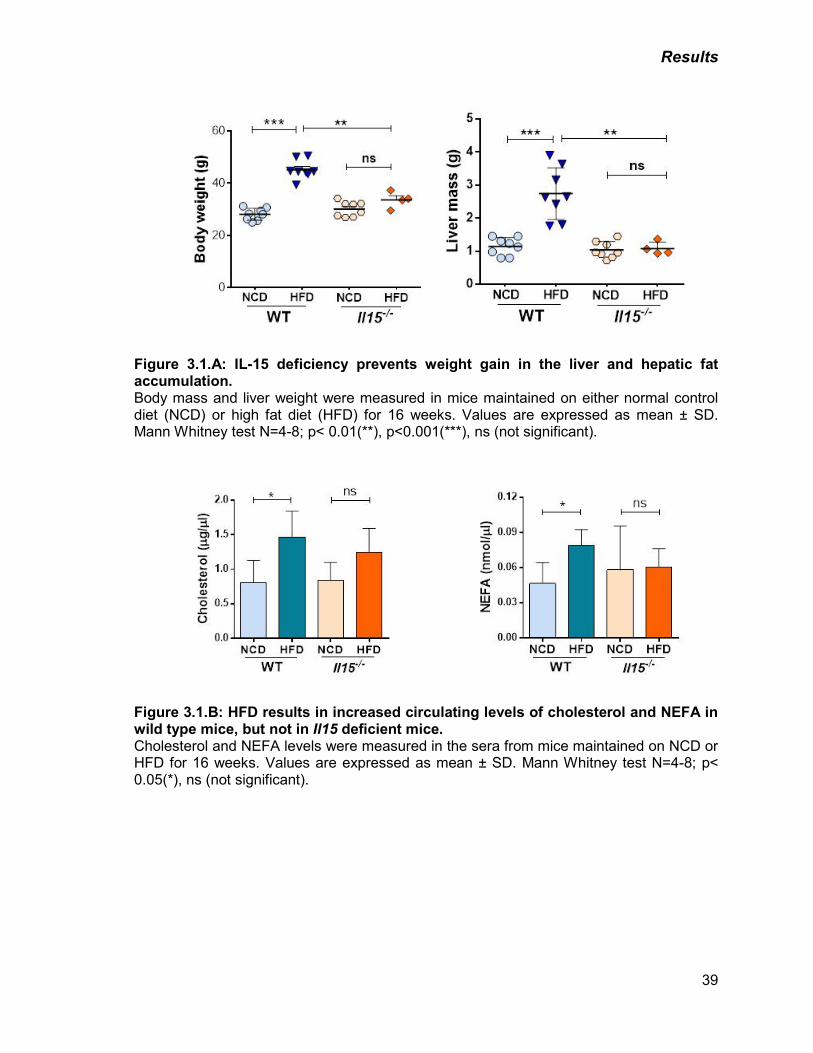
Results
39
Figure 3.1.A: IL-15 deficiency prevents weight gain in the liver and hepatic fat accumulation. Body mass and liver weight were measured in mice maintained on either normal control diet (NCD) or high fat diet (HFD) for 16 weeks. Values are expressed as mean ± SD. Mann Whitney test N=4-8; p< 0.01(**), p<0.001(***), ns (not significant).
Figure 3.1.B: HFD results in increased circulating levels of cholesterol and NEFA in wild type mice, but not in Il15 deficient mice. Cholesterol and NEFA levels were measured in the sera from mice maintained on NCD or HFD for 16 weeks. Values are expressed as mean ± SD. Mann Whitney test N=4-8; p< 0.05(*), ns (not significant).
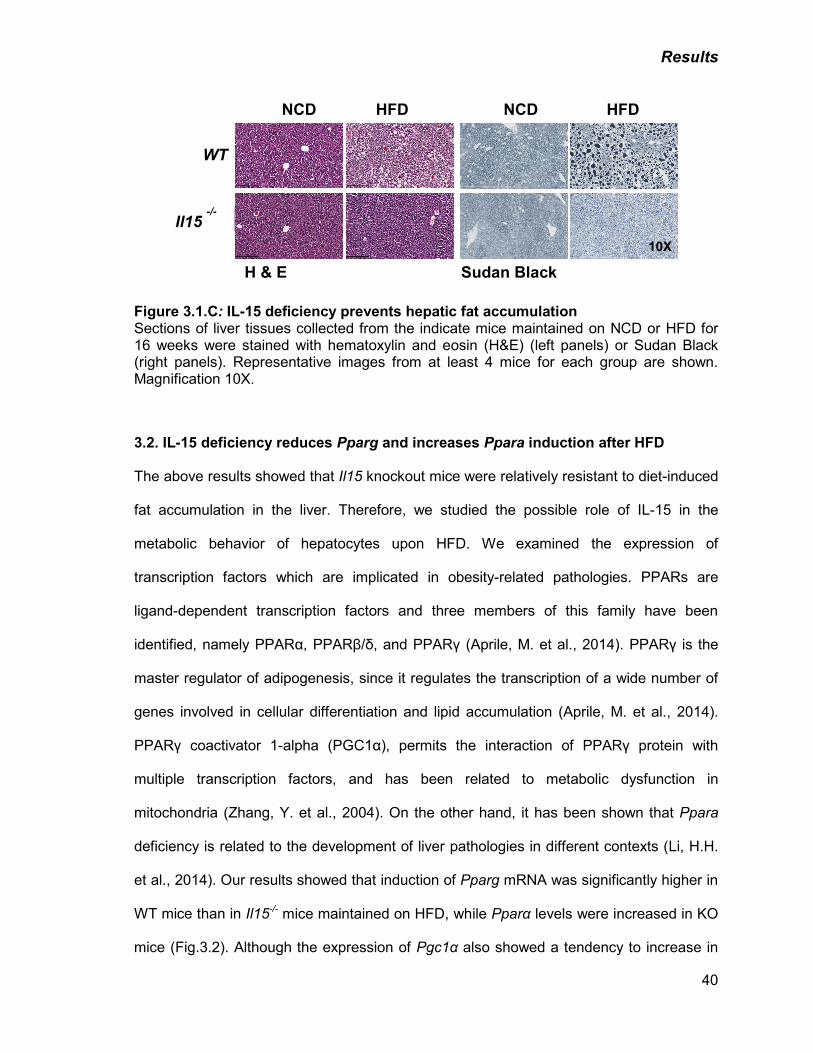
Results
40
Figure 3.1.C: IL-15 deficiency prevents hepatic fat accumulation Sections of liver tissues collected from the indicate mice maintained on NCD or HFD for 16 weeks were stained with hematoxylin and eosin (H&E) (left panels) or Sudan Black (right panels). Representative images from at least 4 mice for each group are shown. Magnification 10X.
3.2. IL-15 deficiency reduces Pparg and increases Ppara induction after HFD
The above results showed that Il15 knockout mice were relatively resistant to diet-induced
fat accumulation in the liver. Therefore, we studied the possible role of IL-15 in the
metabolic behavior of hepatocytes upon HFD. We examined the expression of
transcription factors which are implicated in obesity-related pathologies. PPARs are
ligand-dependent transcription factors and three members of this family have been
identified, namely PPARα, PPARβ/δ, and PPARγ (Aprile, M. et al., 2014). PPARγ is the
master regulator of adipogenesis, since it regulates the transcription of a wide number of
genes involved in cellular differentiation and lipid accumulation (Aprile, M. et al., 2014).
PPARγ coactivator 1-alpha (PGC1α), permits the interaction of PPARγ protein with
multiple transcription factors, and has been related to metabolic dysfunction in
mitochondria (Zhang, Y. et al., 2004). On the other hand, it has been shown that Ppara
deficiency is related to the development of liver pathologies in different contexts (Li, H.H.
et al., 2014). Our results showed that induction of Pparg mRNA was significantly higher in
WT mice than in Il15-/- mice maintained on HFD, while Pparα levels were increased in KO
mice (Fig.3.2). Although the expression of Pgc1α also showed a tendency to increase in
Il15 -/-
NCD HFD
WT
NCD HFD
10X
H & E Sudan Black
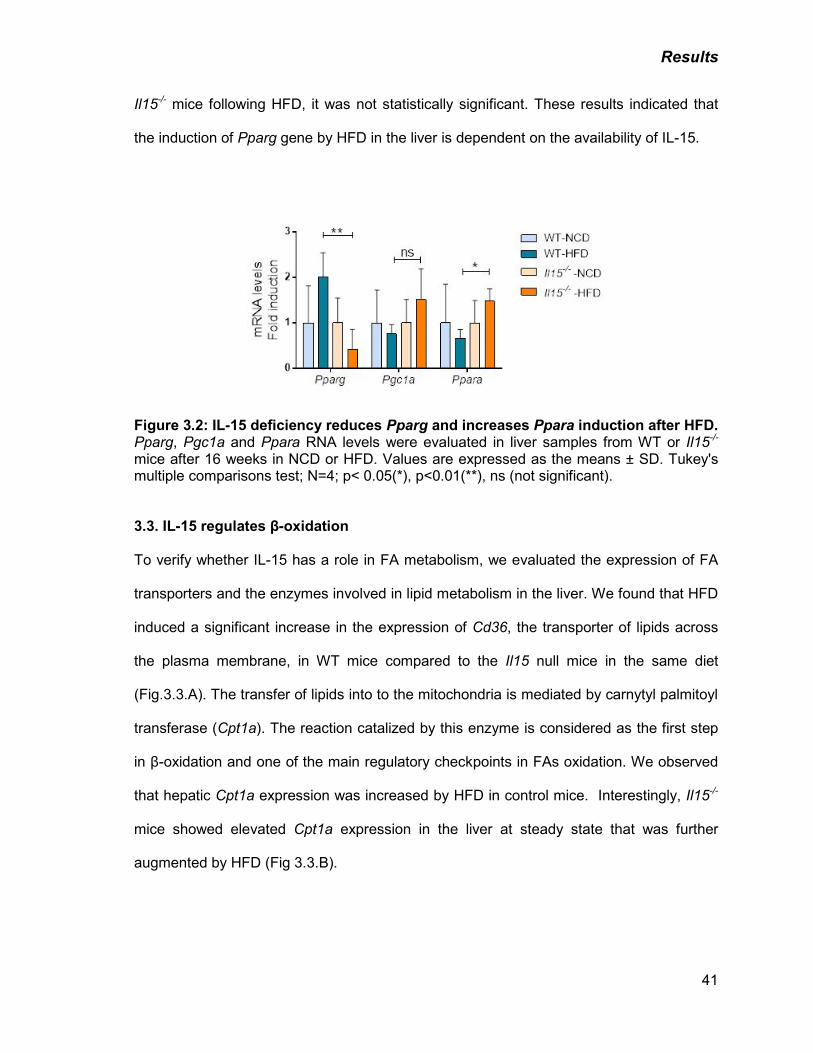
Results
41
Il15-/- mice following HFD, it was not statistically significant. These results indicated that
the induction of Pparg gene by HFD in the liver is dependent on the availability of IL-15.
Figure 3.2: IL-15 deficiency reduces Pparg and increases Ppara induction after HFD. Pparg, Pgc1a and Ppara RNA levels were evaluated in liver samples from WT or Il15-/-
mice after 16 weeks in NCD or HFD. Values are expressed as the means ± SD. Tukey's multiple comparisons test; N=4; p< 0.05(*), p<0.01(**), ns (not significant).
3.3. IL-15 regulates β-oxidation
To verify whether IL-15 has a role in FA metabolism, we evaluated the expression of FA
transporters and the enzymes involved in lipid metabolism in the liver. We found that HFD
induced a significant increase in the expression of Cd36, the transporter of lipids across
the plasma membrane, in WT mice compared to the Il15 null mice in the same diet
(Fig.3.3.A). The transfer of lipids into to the mitochondria is mediated by carnytyl palmitoyl
transferase (Cpt1a). The reaction catalized by this enzyme is considered as the first step
in β-oxidation and one of the main regulatory checkpoints in FAs oxidation. We observed
that hepatic Cpt1a expression was increased by HFD in control mice. Interestingly, Il15-/-
mice showed elevated Cpt1a expression in the liver at steady state that was further
augmented by HFD (Fig 3.3.B).
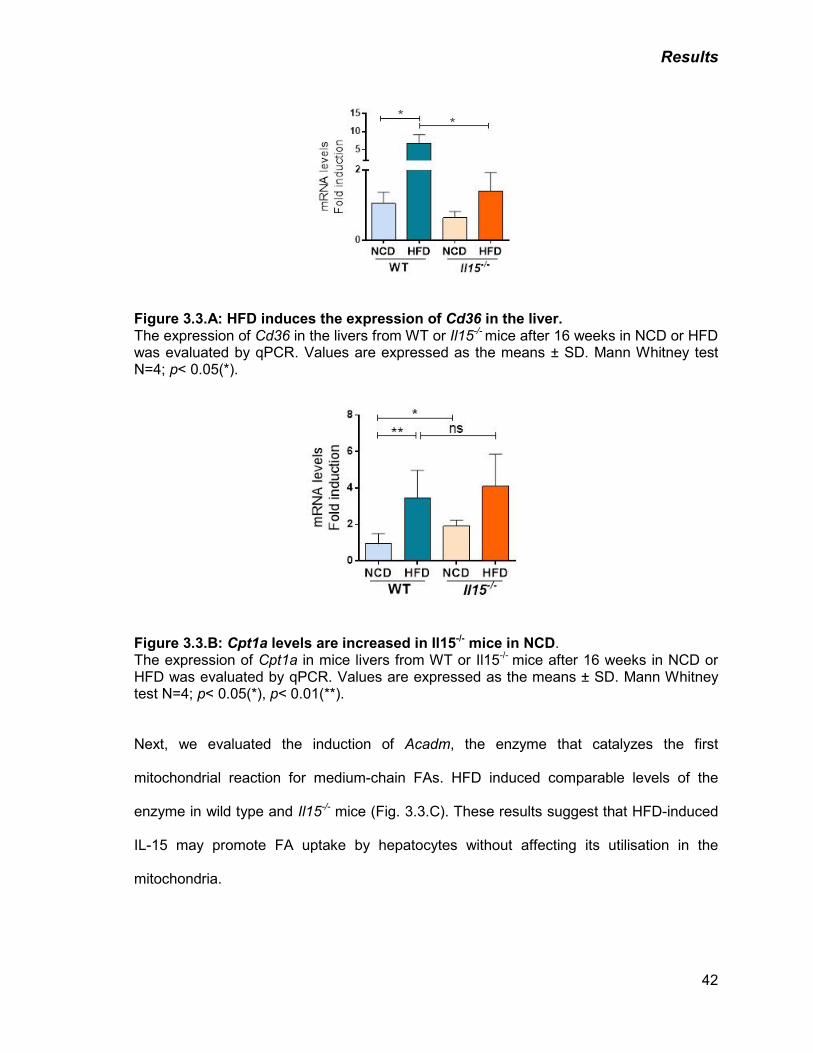
Results
42
Figure 3.3.A: HFD induces the expression of Cd36 in the liver. The expression of Cd36 in the livers from WT or Il15-/- mice after 16 weeks in NCD or HFD was evaluated by qPCR. Values are expressed as the means ± SD. Mann Whitney test N=4; p< 0.05(*).
Figure 3.3.B: Cpt1a levels are increased in Il15-/- mice in NCD. The expression of Cpt1a in mice livers from WT or Il15-/- mice after 16 weeks in NCD or HFD was evaluated by qPCR. Values are expressed as the means ± SD. Mann Whitney test N=4; p< 0.05(*), p< 0.01(**).
Next, we evaluated the induction of Acadm, the enzyme that catalyzes the first
mitochondrial reaction for medium-chain FAs. HFD induced comparable levels of the
enzyme in wild type and Il15-/- mice (Fig. 3.3.C). These results suggest that HFD-induced
IL-15 may promote FA uptake by hepatocytes without affecting its utilisation in the
mitochondria.
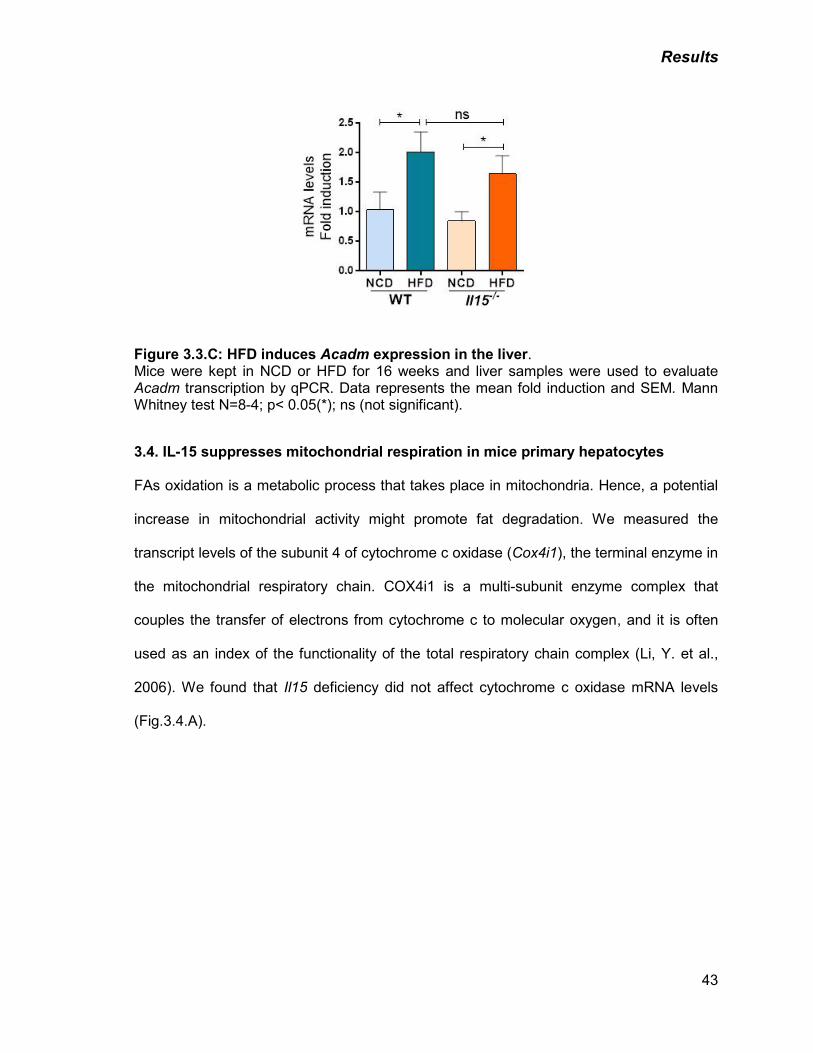
Results
43
Figure 3.3.C: HFD induces Acadm expression in the liver. Mice were kept in NCD or HFD for 16 weeks and liver samples were used to evaluate Acadm transcription by qPCR. Data represents the mean fold induction and SEM. Mann Whitney test N=8-4; p< 0.05(*); ns (not significant).
3.4. IL-15 suppresses mitochondrial respiration in mice primary hepatocytes
FAs oxidation is a metabolic process that takes place in mitochondria. Hence, a potential
increase in mitochondrial activity might promote fat degradation. We measured the
transcript levels of the subunit 4 of cytochrome c oxidase (Cox4i1), the terminal enzyme in
the mitochondrial respiratory chain. COX4i1 is a multi-subunit enzyme complex that
couples the transfer of electrons from cytochrome c to molecular oxygen, and it is often
used as an index of the functionality of the total respiratory chain complex (Li, Y. et al.,
2006). We found that Il15 deficiency did not affect cytochrome c oxidase mRNA levels
(Fig.3.4.A).
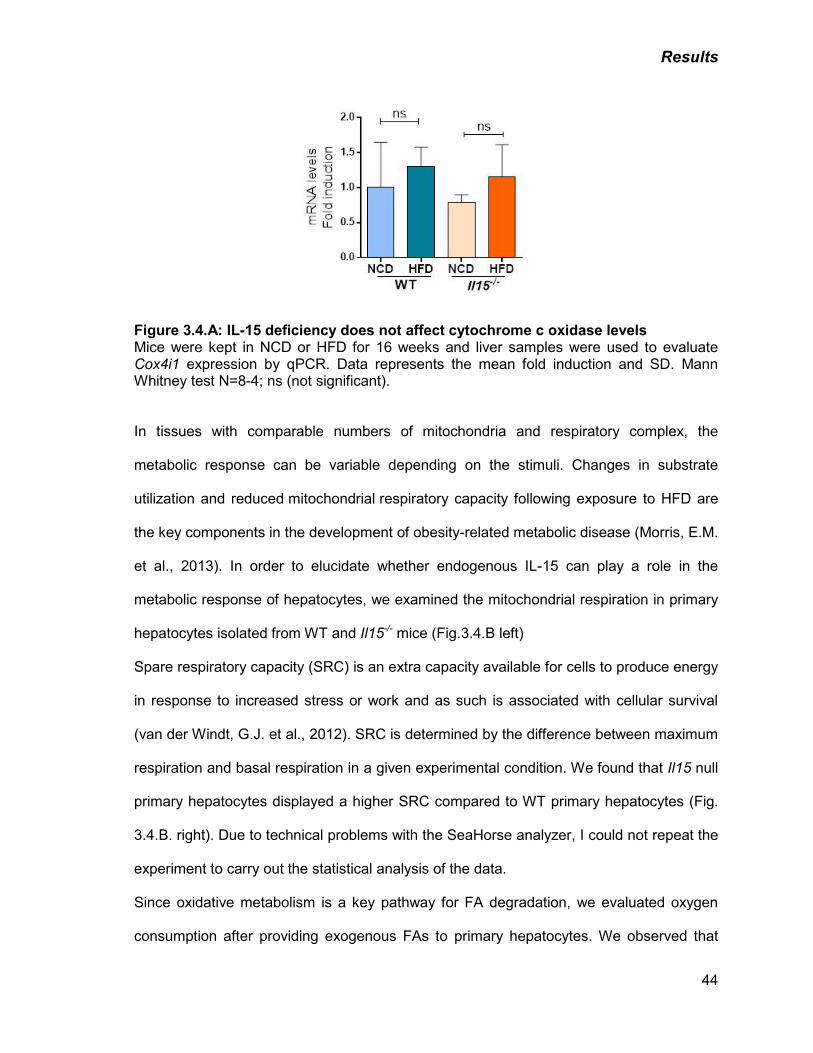
Results
44
Figure 3.4.A: IL-15 deficiency does not affect cytochrome c oxidase levels Mice were kept in NCD or HFD for 16 weeks and liver samples were used to evaluate Cox4i1 expression by qPCR. Data represents the mean fold induction and SD. Mann Whitney test N=8-4; ns (not significant). In tissues with comparable numbers of mitochondria and respiratory complex, the
metabolic response can be variable depending on the stimuli. Changes in substrate
utilization and reduced mitochondrial respiratory capacity following exposure to HFD are
the key components in the development of obesity-related metabolic disease (Morris, E.M.
et al., 2013). In order to elucidate whether endogenous IL-15 can play a role in the
metabolic response of hepatocytes, we examined the mitochondrial respiration in primary
hepatocytes isolated from WT and Il15-/- mice (Fig.3.4.B left)
Spare respiratory capacity (SRC) is an extra capacity available for cells to produce energy
in response to increased stress or work and as such is associated with cellular survival
(van der Windt, G.J. et al., 2012). SRC is determined by the difference between maximum
respiration and basal respiration in a given experimental condition. We found that Il15 null
primary hepatocytes displayed a higher SRC compared to WT primary hepatocytes (Fig.
3.4.B. right). Due to technical problems with the SeaHorse analyzer, I could not repeat the
experiment to carry out the statistical analysis of the data.
Since oxidative metabolism is a key pathway for FA degradation, we evaluated oxygen
consumption after providing exogenous FAs to primary hepatocytes. We observed that
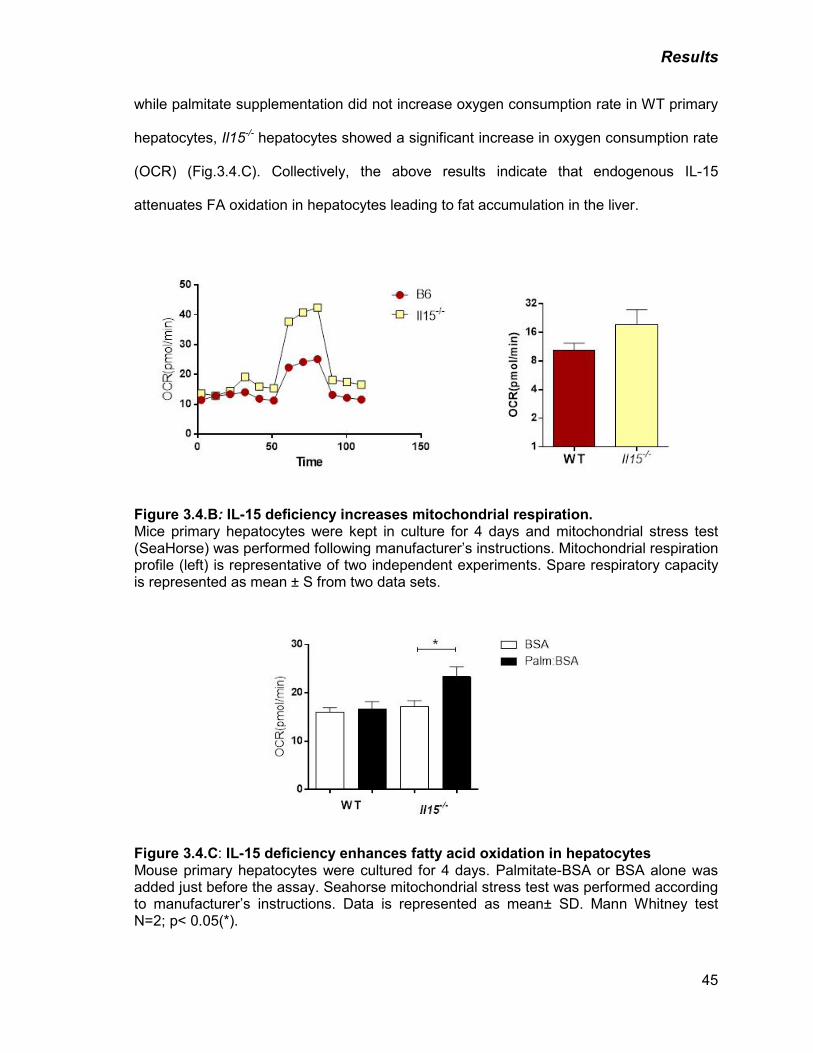
Results
45
while palmitate supplementation did not increase oxygen consumption rate in WT primary
hepatocytes, Il15-/- hepatocytes showed a significant increase in oxygen consumption rate
(OCR) (Fig.3.4.C). Collectively, the above results indicate that endogenous IL-15
attenuates FA oxidation in hepatocytes leading to fat accumulation in the liver.
Figure 3.4.B: IL-15 deficiency increases mitochondrial respiration. Mice primary hepatocytes were kept in culture for 4 days and mitochondrial stress test (SeaHorse) was performed following manufacturer’s instructions. Mitochondrial respiration profile (left) is representative of two independent experiments. Spare respiratory capacity is represented as mean ± S from two data sets.
Figure 3.4.C: IL-15 deficiency enhances fatty acid oxidation in hepatocytes Mouse primary hepatocytes were cultured for 4 days. Palmitate-BSA or BSA alone was added just before the assay. Seahorse mitochondrial stress test was performed according to manufacturer’s instructions. Data is represented as mean± SD. Mann Whitney test N=2; p< 0.05(*).
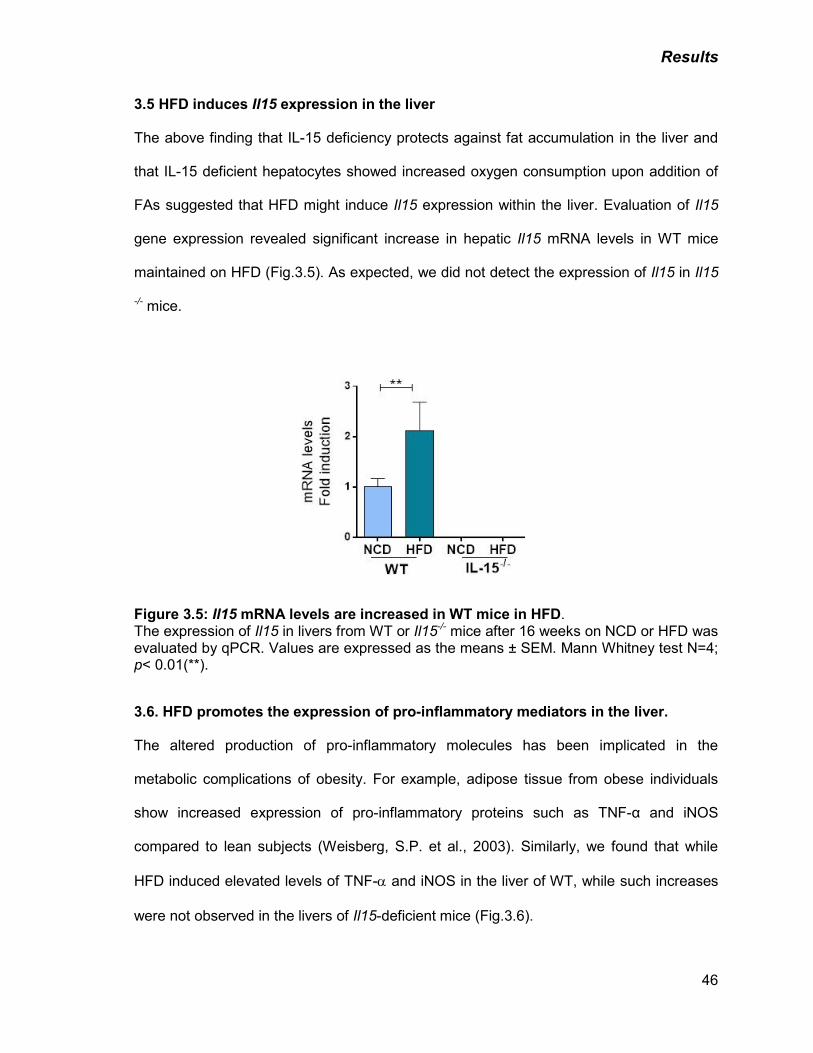
Results
46
3.5 HFD induces Il15 expression in the liver
The above finding that IL-15 deficiency protects against fat accumulation in the liver and
that IL-15 deficient hepatocytes showed increased oxygen consumption upon addition of
FAs suggested that HFD might induce Il15 expression within the liver. Evaluation of Il15
gene expression revealed significant increase in hepatic Il15 mRNA levels in WT mice
maintained on HFD (Fig.3.5). As expected, we did not detect the expression of Il15 in Il15
-/- mice.
Figure 3.5: Il15 mRNA levels are increased in WT mice in HFD. The expression of Il15 in livers from WT or Il15-/- mice after 16 weeks on NCD or HFD was evaluated by qPCR. Values are expressed as the means ± SEM. Mann Whitney test N=4; p< 0.01(**).
3.6. HFD promotes the expression of pro-inflammatory mediators in the liver.
The altered production of pro-inflammatory molecules has been implicated in the
metabolic complications of obesity. For example, adipose tissue from obese individuals
show increased expression of pro-inflammatory proteins such as TNF-α and iNOS
compared to lean subjects (Weisberg, S.P. et al., 2003). Similarly, we found that while
HFD induced elevated levels of TNF-α and iNOS in the liver of WT, while such increases
were not observed in the livers of Il15-deficient mice (Fig.3.6).
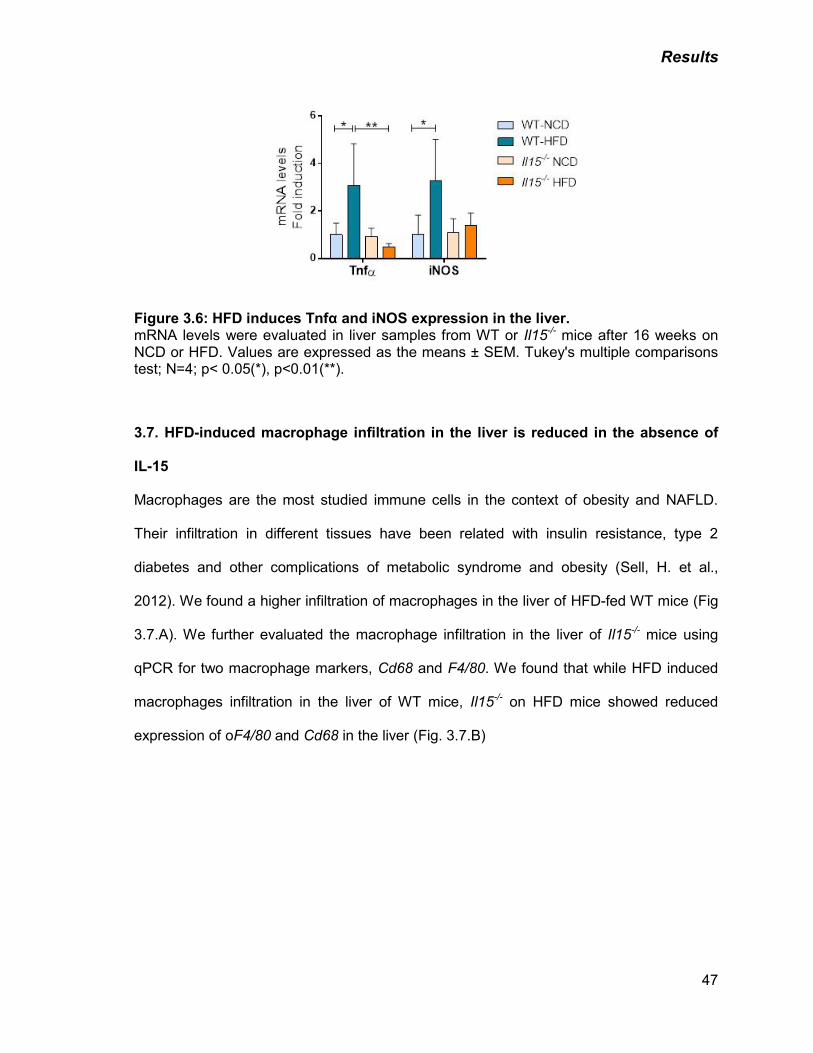
Results
47
Figure 3.6: HFD induces Tnfα and iNOS expression in the liver. mRNA levels were evaluated in liver samples from WT or Il15-/- mice after 16 weeks on NCD or HFD. Values are expressed as the means ± SEM. Tukey's multiple comparisons test; N=4; p< 0.05(*), p<0.01(**).
3.7. HFD-induced macrophage infiltration in the liver is reduced in the absence of
IL-15
Macrophages are the most studied immune cells in the context of obesity and NAFLD.
Their infiltration in different tissues have been related with insulin resistance, type 2
diabetes and other complications of metabolic syndrome and obesity (Sell, H. et al.,
2012). We found a higher infiltration of macrophages in the liver of HFD-fed WT mice (Fig
3.7.A). We further evaluated the macrophage infiltration in the liver of Il15-/- mice using
qPCR for two macrophage markers, Cd68 and F4/80. We found that while HFD induced
macrophages infiltration in the liver of WT mice, Il15-/- on HFD mice showed reduced
expression of oF4/80 and Cd68 in the liver (Fig. 3.7.B)
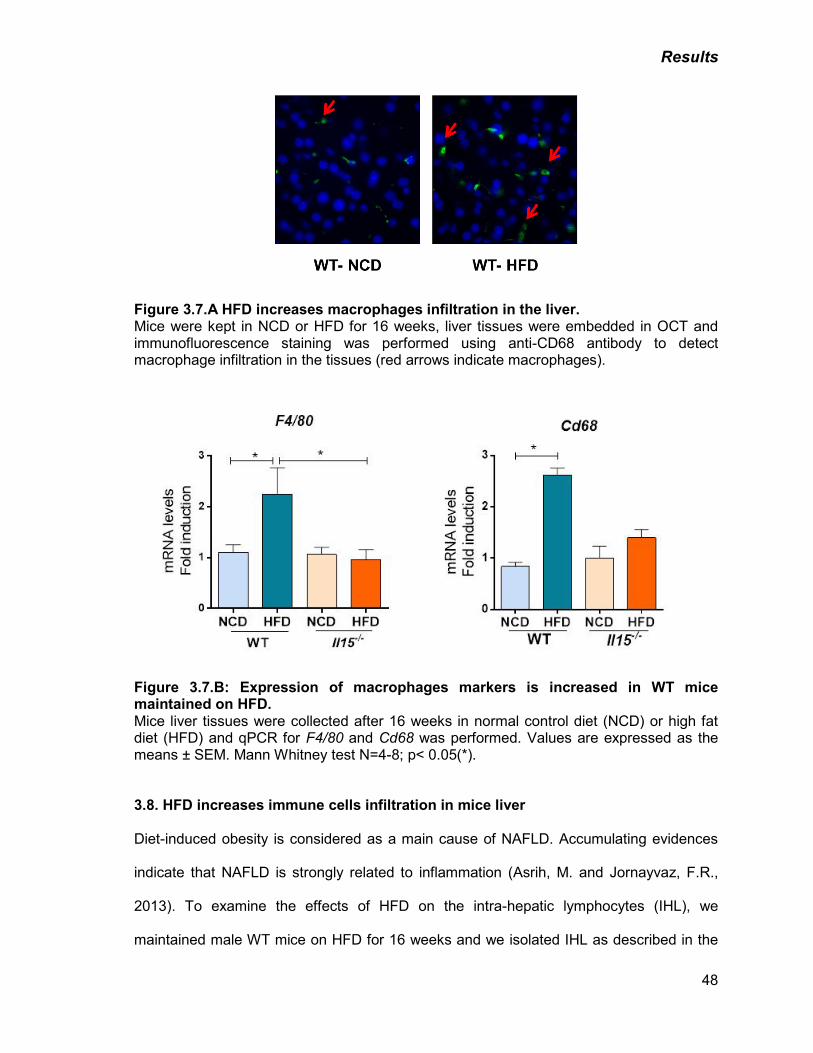
Results
48
Figure 3.7.A HFD increases macrophages infiltration in the liver. Mice were kept in NCD or HFD for 16 weeks, liver tissues were embedded in OCT and immunofluorescence staining was performed using anti-CD68 antibody to detect macrophage infiltration in the tissues (red arrows indicate macrophages).
Figure 3.7.B: Expression of macrophages markers is increased in WT mice maintained on HFD. Mice liver tissues were collected after 16 weeks in normal control diet (NCD) or high fat diet (HFD) and qPCR for F4/80 and Cd68 was performed. Values are expressed as the means ± SEM. Mann Whitney test N=4-8; p< 0.05(*). 3.8. HFD increases immune cells infiltration in mice liver
Diet-induced obesity is considered as a main cause of NAFLD. Accumulating evidences
indicate that NAFLD is strongly related to inflammation (Asrih, M. and Jornayvaz, F.R.,
2013). To examine the effects of HFD on the intra-hepatic lymphocytes (IHL), we
maintained male WT mice on HFD for 16 weeks and we isolated IHL as described in the

Results
49
Methods sections (Chapter 2). Due to problems with the availability of sufficient numbers
of male mice, we did not carry out the analysis of livers of Il15-/- mice on HFD. First, we
determined the effect of HFD on the total number of IHL in the liver. WT mice fed with
HFD showed a significant increase in total IHL compared with those on NCD (Fig. 3.8.A).
To identify the lymphocytes subsets that were increased in mice maintained on HFD, we
carried out a phenotypic analysis of the IHLs. Splenocytes from the same mice served as
controls. In this experimental model, we observed that there was no significant difference
in the total numbers or the frequency of CD4+, CD8+ T cells, NK cells and NKT cells
between the spleens of mice maintained on NCD or HFD (Fig. 3.8.B left panel). Although
the total number of IHLs was increased in the mice maintained on HFD, we did not
observe any significant differences in the relative percentage of intra-hepatic CD4+ and
CD8+ T cell subsets. However, within the CD8+ T cells, the frequency of naïve CD8+ T cell
subset (CD62Lhigh, CD44low) was decreased with a concomitant increase in the effector
CD8+ population (CD62Llow, CD44high) in the livers of mice maintained on HFD (Fig.3.8.B ,
right panel).
NK cells constitute a major proportion of mononuclear cells in the liver (Li, Z. and Diehl,
A.M., 2003). We assessed whether differences in the NK and NKT cell subsets
contributed to the observed increase in the total number of IHLs in HFD fed mice. The
total number of innate NK cells (NK1.1+ CD3-) was increased in the liver of WT mice
maintained on HFD. While the frequency of NKT cells (NK1.1+ CD3+) was not different,
their total numbers were increased (Fig.3.8.A and Fig.3.8.B right panel). The frequency of
iNKT (CD3+ CD1d: galcer tetramer+) cell subset, that has been shown to be decreased in
the peripheral circulation of obese patients (Lynch, L. et al., 2012), was decreased in the
livers of WT mice maintained on HFD. (Fig.3.8.B, right panel). Despite the differences in
the frequency of the various subsets mentioned above, the total number of cells from each
subset, except iNKT, showed a significant increase in the liver of HFD-fed mice
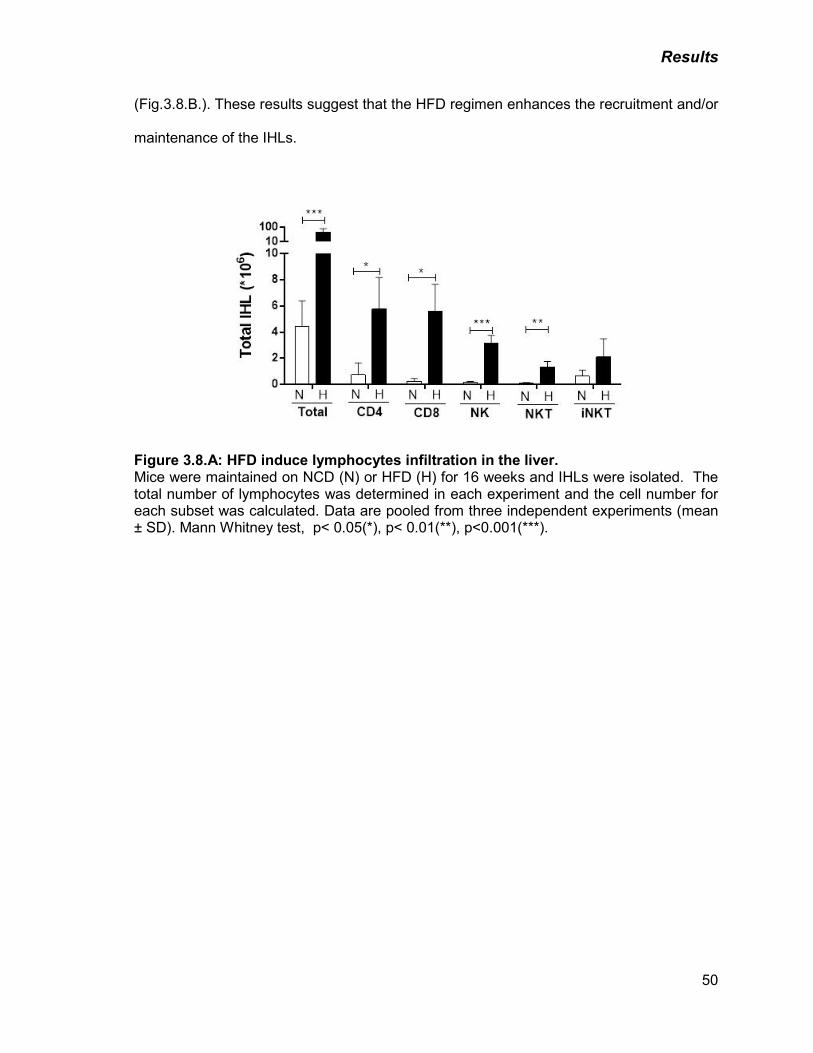
Results
50
(Fig.3.8.B.). These results suggest that the HFD regimen enhances the recruitment and/or
maintenance of the IHLs.
Figure 3.8.A: HFD induce lymphocytes infiltration in the liver. Mice were maintained on NCD (N) or HFD (H) for 16 weeks and IHLs were isolated. The total number of lymphocytes was determined in each experiment and the cell number for each subset was calculated. Data are pooled from three independent experiments (mean ± SD). Mann Whitney test, p< 0.05(*), p< 0.01(**), p<0.001(***).
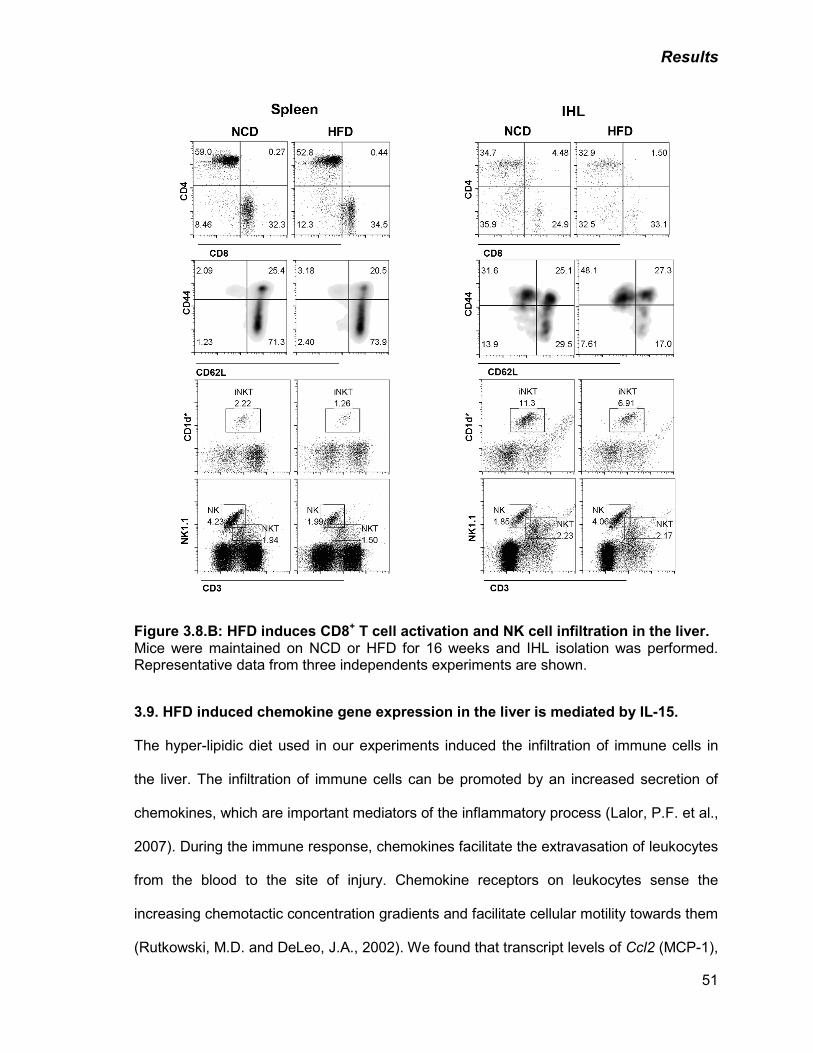
Results
51
Figure 3.8.B: HFD induces CD8+ T cell activation and NK cell infiltration in the liver. Mice were maintained on NCD or HFD for 16 weeks and IHL isolation was performed. Representative data from three independents experiments are shown.
3.9. HFD induced chemokine gene expression in the liver is mediated by IL-15.
The hyper-lipidic diet used in our experiments induced the infiltration of immune cells in
the liver. The infiltration of immune cells can be promoted by an increased secretion of
chemokines, which are important mediators of the inflammatory process (Lalor, P.F. et al.,
2007). During the immune response, chemokines facilitate the extravasation of leukocytes
from the blood to the site of injury. Chemokine receptors on leukocytes sense the
increasing chemotactic concentration gradients and facilitate cellular motility towards them
(Rutkowski, M.D. and DeLeo, J.A., 2002). We found that transcript levels of Ccl2 (MCP-1),
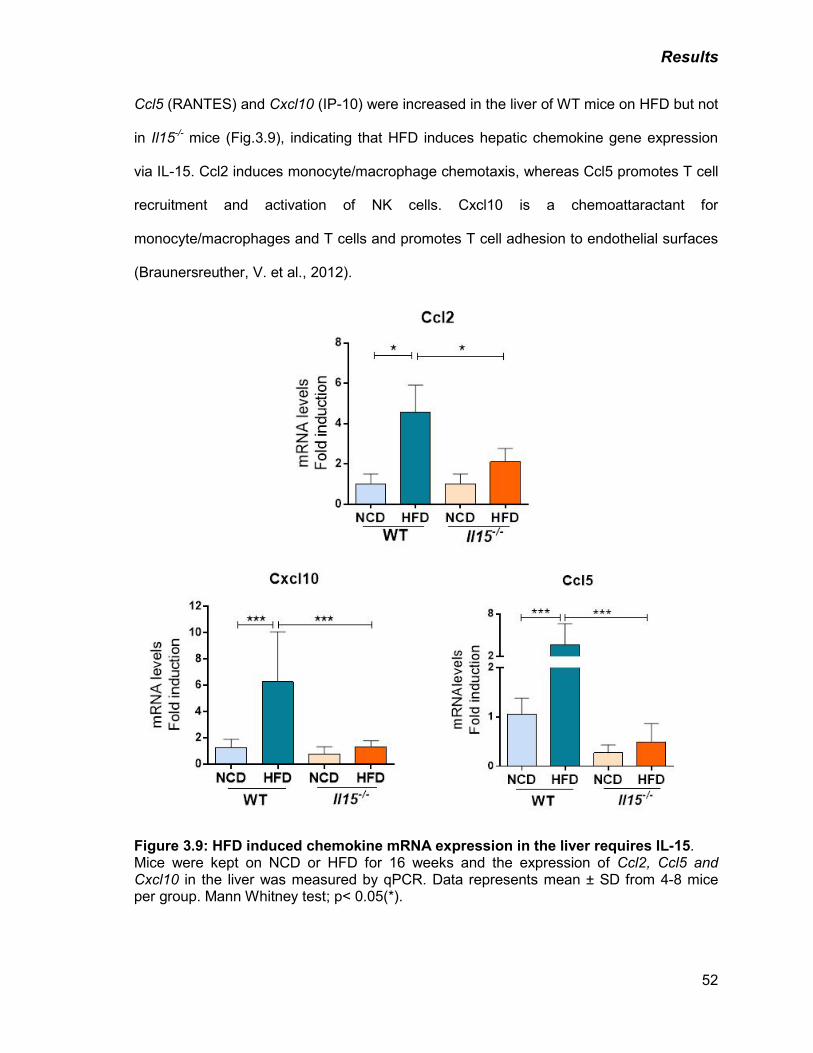
Results
52
Ccl5 (RANTES) and Cxcl10 (IP-10) were increased in the liver of WT mice on HFD but not
in Il15-/- mice (Fig.3.9), indicating that HFD induces hepatic chemokine gene expression
via IL-15. Ccl2 induces monocyte/macrophage chemotaxis, whereas Ccl5 promotes T cell
recruitment and activation of NK cells. Cxcl10 is a chemoattaractant for
monocyte/macrophages and T cells and promotes T cell adhesion to endothelial surfaces
(Braunersreuther, V. et al., 2012).
Figure 3.9: HFD induced chemokine mRNA expression in the liver requires IL-15. Mice were kept on NCD or HFD for 16 weeks and the expression of Ccl2, Ccl5 and Cxcl10 in the liver was measured by qPCR. Data represents mean ± SD from 4-8 mice per group. Mann Whitney test; p< 0.05(*).
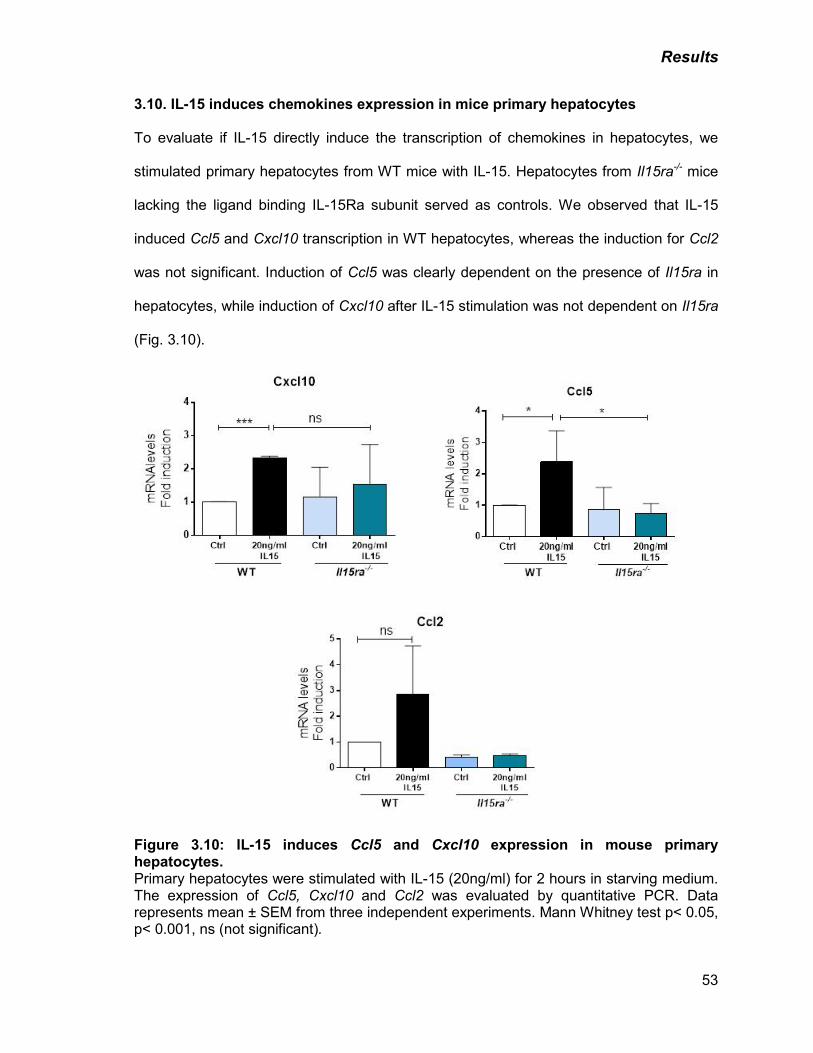
Results
53
3.10. IL-15 induces chemokines expression in mice primary hepatocytes
To evaluate if IL-15 directly induce the transcription of chemokines in hepatocytes, we
stimulated primary hepatocytes from WT mice with IL-15. Hepatocytes from Il15ra-/- mice
lacking the ligand binding IL-15Ra subunit served as controls. We observed that IL-15
induced Ccl5 and Cxcl10 transcription in WT hepatocytes, whereas the induction for Ccl2
was not significant. Induction of Ccl5 was clearly dependent on the presence of Il15ra in
hepatocytes, while induction of Cxcl10 after IL-15 stimulation was not dependent on Il15ra
(Fig. 3.10).
Figure 3.10: IL-15 induces Ccl5 and Cxcl10 expression in mouse primary hepatocytes. Primary hepatocytes were stimulated with IL-15 (20ng/ml) for 2 hours in starving medium. The expression of Ccl5, Cxcl10 and Ccl2 was evaluated by quantitative PCR. Data represents mean ± SEM from three independent experiments. Mann Whitney test p< 0.05, p< 0.001, ns (not significant).

Results
54
3.11. IL-15 and IL-5Rα are required for the maintenance of NK populations in the
liver
It is well established that the maintenance of the CD8+ T and NK cell subsets in the liver is
mediated by IL-15. Therefore, we examined whether the absence of IL-15 signalling would
affect lymphocyte homeostasis in the liver. To this end, we characterized the IHL
populations in age-matched WT, Il15-/- and Il15ra-/- mice. In agreement with previous
reports, splenic CD8+ T cells were reduced in the absence of IL-15 or its receptor alpha
subunit. Interestingly, there were no differences in the percentage of intra-hepatic CD8+ T
cells (Fig.3.11.A). However, there was a 6-fold reduction in the NK cells in the livers of
Il15-/- and Il15ra-/- mice. A similar reduction was also observed in the intra-hepatic NKT
and iNKT cell subsets (Fig.3.11.B and C)
Figure 3.11.A: Splenic but not liver CD8+ T cells are dependent on IL-15 signaling. Spleen and IHL isolation was performed in age matched WT, Il15-/- and Il15ra-/- mice. T cells were characterized by FACS analyses. Data represent four independents experiments.
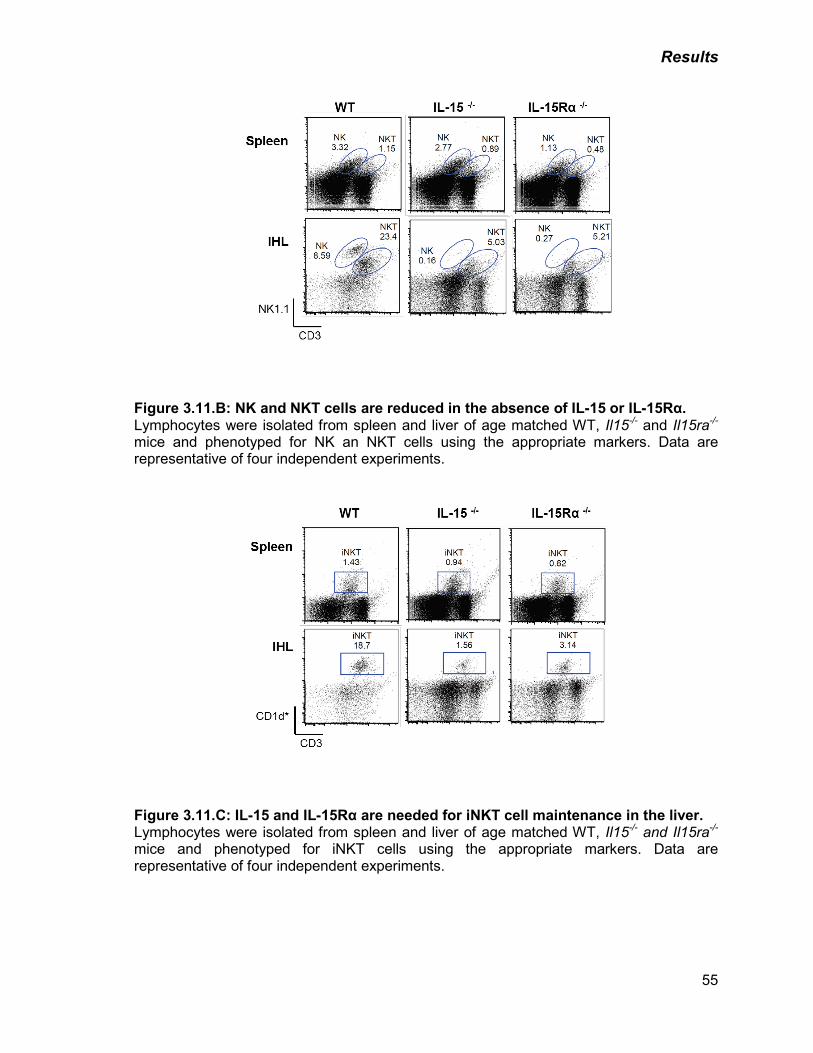
Results
55
Figure 3.11.B: NK and NKT cells are reduced in the absence of IL-15 or IL-15Rα. Lymphocytes were isolated from spleen and liver of age matched WT, Il15-/- and Il15ra-/- mice and phenotyped for NK an NKT cells using the appropriate markers. Data are representative of four independent experiments.
Figure 3.11.C: IL-15 and IL-15Rα are needed for iNKT cell maintenance in the liver. Lymphocytes were isolated from spleen and liver of age matched WT, Il15-/- and Il15ra-/- mice and phenotyped for iNKT cells using the appropriate markers. Data are representative of four independent experiments.

Results
56
3.12. IL-15Rα expression in both macrophages and hepatocytes contribute to NK
cell maintenance in the liver
IL-15Rα, the high affinity receptor for IL-15, increases the biological activity of IL-15. The
presence of IL-15Rα in different cells types can influence the homeostasis of immune
populations mainly TCD8 and NK (Mortier, E. et al., 2009). In order to elucidate the
specific cell types that express IL-15Rα and contribute to NK cell maintenance in the liver,
we evaluated NK cell infiltration in the absence of IL-15Rα in the macrophages. NKT cells
population in the liver was significantly lower in the absence of Il15ra in the macrophages,
but the reduction in NK or iNKT was not significant (Fig. 3.12.A). Spleen was used as a
control, and a subtle reduction in NK and NKT cells was observed in the absence of IL-
15Ra in macrophages.
Hepatocytes represent around the 80% of total cells in the liver (Okada, Y. et al., 2013).
Thus, we evaluated the impact of IL-15Rα expression in hepatocytes on the homeostasis
of NK, NKT and iNKT cells. Remarkably, we observed that IL-15Rα in the hepatocytes
was indispensable for NK, NKT and iNKT maintenance in the liver (Fig. 3.12.B)
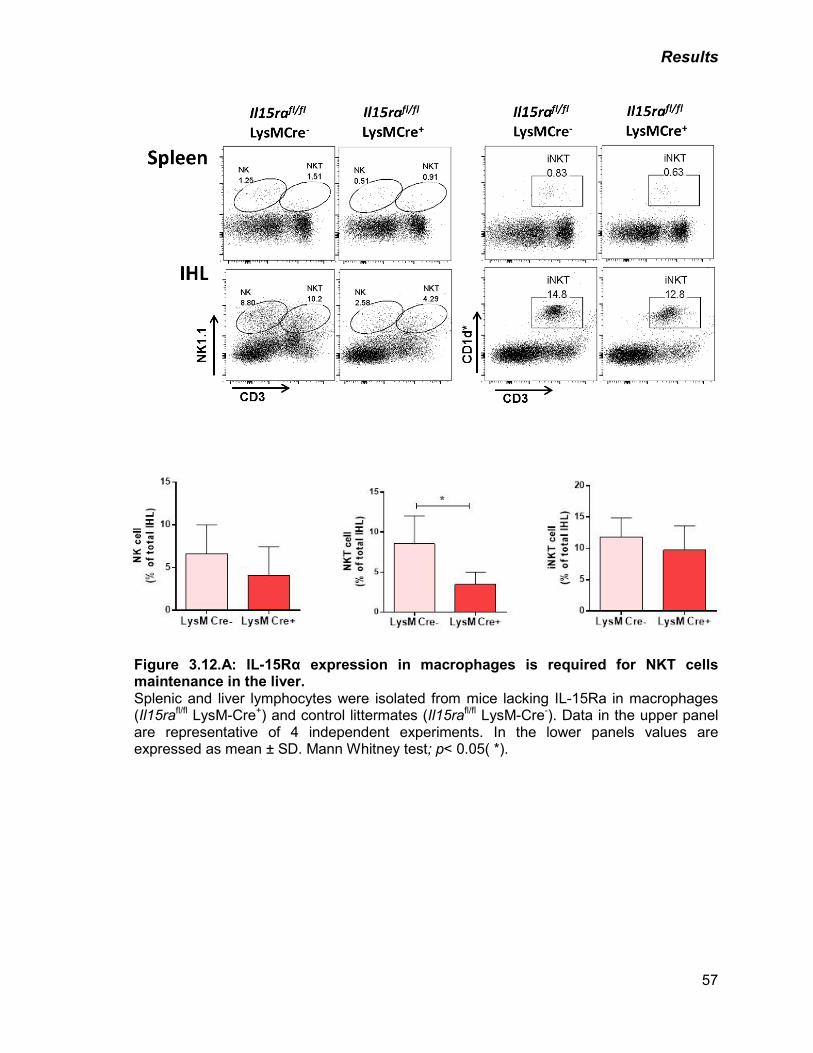
Results
57
Figure 3.12.A: IL-15Rα expression in macrophages is required for NKT cells maintenance in the liver. Splenic and liver lymphocytes were isolated from mice lacking IL-15Ra in macrophages (Il15rafl/fl LysM-Cre+) and control littermates (Il15rafl/fl LysM-Cre-). Data in the upper panel are representative of 4 independent experiments. In the lower panels values are expressed as mean ± SD. Mann Whitney test; p< 0.05( *).
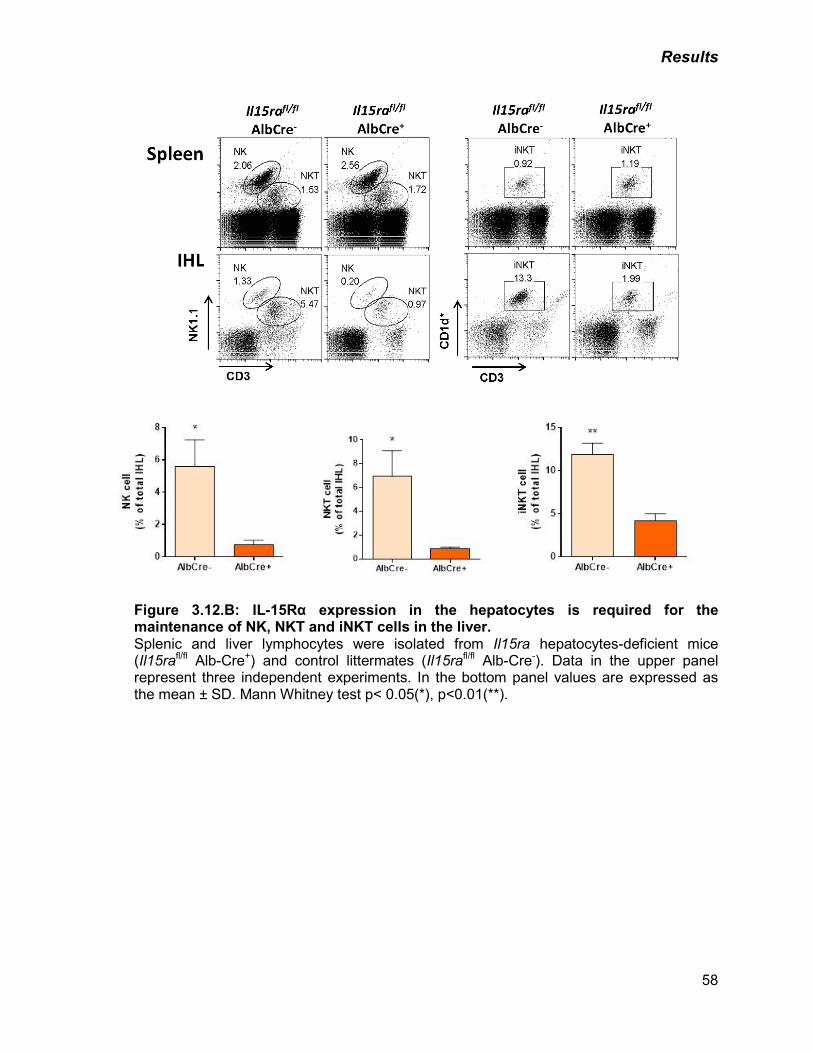
Results
58
Figure 3.12.B: IL-15Rα expression in the hepatocytes is required for the maintenance of NK, NKT and iNKT cells in the liver. Splenic and liver lymphocytes were isolated from Il15ra hepatocytes-deficient mice (Il15rafl/fl Alb-Cre+) and control littermates (Il15rafl/fl Alb-Cre-). Data in the upper panel represent three independent experiments. In the bottom panel values are expressed as the mean ± SD. Mann Whitney test p< 0.05(*), p<0.01(**).

Results
59
3.13 Summary of results
In this thesis we described a new role for IL-15 in fatty liver disease. We found that IL-15
promotes weight gain and fat accumulation in the liver by modulating liver metabolism.
Moreover, we showed that IL-15 decreased mitochondrial FAs oxidation in hepatocytes.
Furthermore, in the context of NAFLD, we showed the role of IL-15 in induction of
chemokines in the liver. Additionally, we found that IL-15Ra expression in hepatocytes is
indispensable for NK cells homeostasis in the liver. Overall, our results contribute to a
better understanding of obesity-associated inflammation.

Discussion
60
4. DISCUSSION
Obesity-associated metabolic syndrome represents a cluster of metabolic diseases
including type 2 diabetes mellitus, cardiovascular disease and NAFLD. Sedentary lifestyle
and increased food intake are the major predisposing factors in obesity. Obesity
associated inflammation is characterized by immune cell infiltration in the affected tissues,
which induces local and systemic increase in pro-inflammatory cytokine levels, eventually
leading to the development of insulin resistance (Sell, H. et al., 2012).
NAFLD is considered as the most common cause of chronic liver disease in western
countries and is associated with systemic and hepatic insulin resistance. NAFLD has a
wide histological spectrum ranging from ‘simple’ steatosis to nonalcoholic NASH, which
may progress to cirrhosis (Zhan, Y.T. and An, W., 2010). In this thesis we evaluated the
role of IL-15, an immuno-inflammatory cytokine in the progression of the liver
manifestation of obesity, namely NAFLD.
Following the HFD regimen, body and liver weight are increased in WT but not in Il15-/-
mice, indicating that IL-15 plays a crucial role in diet-induced obesity and fatty liver
disease. The HFD regimen also increased the circulating levels of cholesterol and NEFA
in WT mice but not in IL-15 deficient mice. Elevated level of cholesterol is one of the
manifestations of disorders in lipoprotein metabolism know as dyslipidemia, which
promotes insulin resistance. Other parameters used to characterize dyslipidemia are the
elevated low-density lipoprotein cholesterol (LDL) and triglyceride levels, or decreased
high-density lipoprotein cholesterol (HDL) (Plana, N. et al., 2014). An increase in
circulating NEFA may contribute to NAFLD development in obese subjects. Triacylglycerol
(TG) stored in adipose tissue is hydrolyzed to NEFA to be transported to target tissues for
utilization. In the fasting state, plasma NEFAs are derived mainly from hydrolysis of TG in
adipocytes. After a meal, lipoprotein lipase (LPL) in the capillaries of adipose tissue

Discussion
61
hydrolyses circulating TG, mainly the dietary fat carried in the chylomicrons, and this is an
additional route of generation of plasma NEFA (Karpe, F. et al., 2011). NEFAs from
adipose tissue can be used as an energy source by many tissues, including liver and
skeletal muscle. In hepatocytes, their fate differs depending on energy needs, hormone
balance and substrate availability, i.e. they can be re-packaged into TGs and exported as
very low density lipoproteins (VLDL), stored within the liver, or converted to ketones
(Nguyen, P. et al., 2008). The excess of TG produced in the liver are stored as fat within
hepatocytes, leading to lipid deposition observed in fatty liver diseases (Browning, J.D.
and Horton, J.D., 2004). The increased microvesicular and macrovesicular fat deposition
in the liver parenchyma of WT mice after HFD, but not in Il15-/- mice indicates that IL-15
promotes lipid deposition in the liver under dietary conditions that promote obesity.
Day and James have proposed that while lipid deposition in the liver is the first of the 2
‘hits’ needed for NASH development, the second hit represents all the factors that
contributes to liver inflammation (Day, C.P. and James, O.F., 1998). Our results show that
IL-15 promotes diet-induced NAFLD by modulating lipid metabolism in the liver, as well as
by perpetuating the inflammatory responses in the liver.
Lipid metabolism is controlled by different transcription factors. These include Srebpf1
(sterol-regulatory-element-binding protein), Cepba (CCAAT/enhancer binding protein) and
factors that regulate with lipid storage and utilization, namely Pparg, which promotes lipid
storage, and Ppara, which directs FA oxidation (Wheeler, M.C. and Gekakis, N., 2014).
FAs, absorbed from diet or generated in adipose tissues lipolysis, as well as
prostaglandins are natural ligands of PPAR family of transcription factors (Vega, R.B. et
al., 2000). We found a reduction in Pparg expression in Il15-/- mice after HFD compared to
WT mice. Previous reports showed that hepatocyte- or macrophage- specific deletion of
Pparg protects mice against diet-induced hepatic steatosis, suggesting a pro-steatotic role
of PPARγ in parenchymal and non-parenchymal cells (Moran-Salvador, E. et al., 2011).

Discussion
62
Therefore, the reduced Pparg expression in the liver of IL-15 deficient mice on HFD could
contribute, at least partly, to reduced hepatic lipid storage in these mice.
In contrast to Pparg, Ppara gene expression is increased in the livers of IL-15 deficient
mice under HFD regimen, suggesting a possible increase in FAs oxidation in the liver of
KO mice. Metabolically active tissues such as liver, muscle, intestine, and brown adipose
tissue expresses high levels of PPARα. Specifically in hepatocytes, PPARα expression
levels are very high, possibly to increase β-oxidation that reduces lipid storage in the liver
(Lefebvre, P. et al., 2006). Methionine choline-deficient diet results in liver injury similar to
human NASH. Ppara deficient mice fed with this diet develop severe hepatic steatosis and
steatohepatitis. Consequently, PPARα agonist treatment reverses steatohepatitis in mice
with established NASH (Lefebvre, P. et al., 2006). Hence, increased Ppara expression in
the liver of IL-15 KO mice on HFD, could act in synergy with reduced Pparg expression to
prevent hepatic fat deposition.
PPARα has also been described as a transcriptional regulator of inflammatory response in
various tissues, including liver by inhibiting the inflammatory genes induced by NF-κB
(Tailleux, A. et al., 2012). The increase in Ppara levels that we observe the KO mice can
also control the inflammatory response by IL-15 in NAFLD.
We also evaluated the expression of Pgc1a, the PPARγ co-activator, which allows the
interaction of PPARγ with multiple transcription factors (Zhang, Y. et al., 2004). This co-
activator was initially described as a positive regulator of PPARγ target genes expression
(Puigserver, P. et al., 1998). However, we did not find any significant increase in Pgc1a
expression in the livers of WT mice under HFD regimen, despite a significant increase in
Pparg expression. Pgc1a is also capable of co-activating Ppara in the transcriptional
control of genes regulating enzymes involved in FA oxidation (Vega, R.B. et al., 2000).
The livers of IL-15 deficient mice on HFD also showed a tendency to upregulate Pgc1a,
although this was not statistically significant. Thus, the decreased hepatic lipid

Discussion
63
accumulation in Il15-/- mice maintained on HFD could results from the combined effects of
reduced lipid uptake due to downregulation of Pparg and increased FAs degradation
stimulated by Ppara expression, and a possible co-operation between Ppara and Pgc1a.
Hepatic steatosis can be a consequence of increased lipid synthesis and/or lipid uptake,
via overexpression of the FAs transporters. CD36 is a plasma membrane lipid transporter
is under the transcriptional control of PPARγ, and has been related to fat accumulation in
the liver (Wheeler, M.C. and Gekakis, N., 2014).
WT mice in HFD showed high expression of Cd36, consistent with lipid deposition within
the hepatocytes, also known as microvesicular fat deposition. However, Cd36 increase
after the HFD regimen was significantly less in Il15-/- mice than in WT mice, suggesting
that IL-15 promotes FAs intake by liver cells.
Within cells, FAs are catabolized by β-oxidation mainly in the mitochondria and also in
peroxisomes to generate energy. β-oxidation of the bulk of short-, medium-, and long-
chain FAs derived from diet take place in mitochondria, and the main enzymes
responsible for this process are localized in the cristae and in the matrix (Reddy, J.K. and
Hashimoto, T., 2001). The genes encoding enzymes involved in the beta-oxidation
pathway in liver are transcriptionally regulated by PPARα. The initial step in mitochondrial
FAs oxidation is the translocation of FA from cytosol to the matrix. This step is catalyzed
by sequential enzymatic reactions mediated by CPT1, CPTII and carnitine-acylcarnitine
translocase. Cpt1a is a key enzyme in the carnitine-dependent FA transport across the
mitochondrial inner membrane and its deficiency results in a decreased rate of FA β-
oxidation (Assimacopoulos-Jeannet, F. et al., 1997). Whereas HFD induced the
expression of the liver-specific isoform of Cpt1a in WT mice, the IL-15 KO mice showed
an elevated basal expression even under normal diet. This observation suggests that IL-
15 is available even under normal steady state, and that it attenuates FA oxidation in the
liver at the level of FA import into the mitochondria.

Discussion
64
β-oxidation of FA within mitochondria is initiated by acyl-CoA dehydrogenases. Deficiency
of Acadm, a medium-chain acyl-CoA dehydrogenase induces different metabolic
abnormalities, such as hepatic dysfunction and fasting hypoglycemia. (Matern, D. and
Rinaldo, P., 1993). Lipid-rich diet induced Adcam mRNA in both WT and IL-15 KO mice to
a comparable level. Collectively, IL-15 appears to attenuate FA utilization at the level of
lipid transport through plasma membrane (Cd36) and the FAs transfer into the
mitochondria (Cpt1a).
Lipid oxidation within mitochondria is dependent on mitochondrial respiratory capacity,
which is controlled by several factors, including the synthesis, assembly and functioning of
the respiratory chain complex. For example, cytochrome c oxidase exerts a tight control
on mitochondrial function. The COX IV protein is essential for the assembly of cytochrome
c oxidase complex (Li, Y. et al., 2006). Both WT and IL-15 KO mice expressed
comparable levels of Cox4i1 mRNA, suggesting that IL-15 does not affect the assembly of
cytochrome c oxidase complex. However, direct measurement of the mitochondrial
respiration profile of primary hepatocytes (SeaHorse), showed a higher maximum
respiratory capacity in IL-15 deficient hepatocytes compared to WT hepatocytes (Fig.
3.4.B). This observation indicates that the loss of IL-15 enhances the cellular capacity to
increase oxidative metabolism under certain conditions, by boosting the oxygen
consumption rate. One of those conditions could be the excess lipids availability during
the HFD regimen. Energy starvation enables primary hepatocytes to use exogenous FAs
such as palmitate as energy source, via beta oxidation followed by Acetyl-CoA
metabolism in the tricarboxylic acid cycle and oxidative phosphorylation. Under these
conditions, the maximum oxygen consumption was increased in Il15-/- primary hepatocytes
(Fig. 3.4.C)
Collectively our results show that IL-15 deficiency attenuates the HFD-induced expression
of genes involved in lipid uptake (Pparg, Cd36), but enhances the expression of genes

Discussion
65
implicated in lipid utilization (Ppara) and FA transport into mitochondria (Cpt1a) as well as
the basal and FA-induced OCR. These findings indicate that Il-15 induced during HFD
regimen not only enhances hepatic lipid uptake but also attenuates its degradation via
beta-oxidation and oxidative phosphorylation.
Obesity is a low-grade inflammatory disease (Sell, H. et al., 2012). Several pro-
inflammatory cytokines have been associated with the metabolic complications involved in
this pathology. IL-15 has been extensively studied as an inflammatory cytokine, critical for
the development, homeostasis and functioning of the cells of the innate and adaptive
immune system (Kanegane, H. and Tosato, G., 1996; Stoklasek, T.A. et al., 2006).
Besides phagocytic cells, skeletal muscles express high levels of IL-15 mRNA. It
suggested that IL-15 may function as a muscle-derived endocrine factor, or “myokine”,
which can modulate body composition (Quinn, L.S. and Anderson, B.G., 2011). Other
reports have also suggested that IL-15 is produced by the liver, in hepatocytes cell lines
and by hepatoma cell lines (Golden-Mason, L. et al., 2004; Correia, M.P. et al., 2009). In
this work, we show that IL-15 is constitutively expressed in the liver and is upregulated
after 16 weeks in HFD. In agreement with these results, Quinn et.al., have shown that in
mice susceptible to oxidative stress, obesigenic diet with high calcium significantly
increased IL-15 mRNA expression in the visceral fat and skeletal muscle tissues (Quinn,
L.S. and Anderson, B.G., 2011), but the cell types involved were not identified. Similarly,
very little information is available on the expression of IL-15 at the mRNA and protein
levels in different tissues under physiological conditions. Additionally, in agreement with a
pathogenic role of IL-15 in obesity, circulating levels of IL-15 are increased in obese
insulin resistant subjects and decreased after weigh loss (Christiansen, T. et al., 2010).
In contrast to the above studies and our own findings, another study has reported a
beneficial role of IL-15 in obesity reported before. Barra et al., found that Il15-/- mice

Discussion
66
exhibit higher amounts of body fat than control mice (Barra, N.G. et al., 2010). Although
the reasons for these completely contradictory conclusions are unclear, it is noteworthy
that Barra et la., have observed anti-obesity effect of IL-15 on female IL-15 KO mice fed
with NCD, whereas we observed the pro-obesity role of IL-15 in male IL-15 KO mice fed
with HFD. As both mice strains are in C57BL/6 background, a possible explanation for
these different findings could be the gender of the mice, the food regimen used, and
influence by the gut microbiota. Diet is an important modulator of microbial diversity in the
gut. Changes in gut microbiota are not only the consequence of obesity, but can also be a
contributing factor because the obese phenotype can be transposed by gut microbiota
transplantation, (Ridaura, V.K. et al., 2013). Nonetheless, as described earlier our in vitro
data on isolated primary hepatocytes strongly support a pro-obesity role of IL-15, at least
under conditions of excess dietary fat.
Most of the studies on the influence of pro-inflammatory cytokines in obesity-associated
pathologies have focussed on the adipose tissue. For example, TNF-α, IL-6, iNOS, TGF-
β1, C-reactive protein, soluble ICAM, and monocyte chemotactic protein-1 (MCP-1) are
elevated in adipose tissues from obese subjects when compared to lean ones. Skeletal
muscle tissue in obesity also produces elevated amounts of TNF-α and iNOS (Weisberg,
S.P. et al., 2003). Deficiency in Tnfa or iNOS was reported to have a beneficial role on the
insulin sensitivity in obese mice (Weisberg, S.P. et al., 2003). In the liver, it was reported
that treatment with insulin-like growth factor (IGF) prevented liver failure by inhibiting TNF-
α production and reducing the induction of iNOS (Hijikawa, T. et al., 2008). We observed
an upregulation of Tnfa and iNOS expression in the liver of WT obese mice that occurred
concomitantly with Il15 gene induction. This upregulation of Tnfa and iNOS genes did not
occur in the liver of IL-15 KO mice maintained on HFD (Fig. 3.6), suggesting that IL-15
acts upstream of TNFa and iNOS in obesity-associated changes in gene expression in the
liver.

Discussion
67
Tissue macrophages play an important role in the development of obesity-associated
inflammation. Several studies reveal a pathogenic role for adipose tissue-associated
macrophages in metabolic abnormalities related to over nutrition (Sell, H. et al., 2012).
Even though the role of liver macrophages (KCs) in fatty liver disease has not been
extensively studied, high-fat or high-sucrose diet-induced steatosis and hepatic insulin
resistance were reported to be prevented following depletion of KCs in the liver (Huang,
W. et al., 2010). Moreover, this study also showed that TNF-α is partially responsible for
KC-mediated alterations in hepatocyte FA oxidation, triglyceride accumulation, and insulin
responsiveness (Huang, W. et al., 2010).
We observed an increase in the macrophage infiltration in the liver parenchyma in WT
mice fed with lipid-rich diet, and analysis of mRNA expression revealed low levels of
macrophage markers (Cd68 and F4/80) in IL-15 KO mice on HFD (Fig. 3.7.B), suggesting
that IL-15 may play a role in the recruitment of macrophages to the liver during the
inflammatory conditions such as NAFLD. Accordingly, we observed upregulation of two
macrophage chemotactic factors Ccl2 and Cxcl10 in WT mice fed with HFD but not in IL-
15 deficient mice. IL-15 is also known to activate macrophages; however the underlying
mechanisms are unclear. Earlier reports showed that the effects of IL-15 on macrophages
are dependent on the cytokine concentration (Alleva, D.G. et al., 1997). Whereas high IL-
15 concentrations enhanced pro-inflammatory (i.e., TNF-α, IL-1, and IL-6) and anti-
inflammatory (i.e., IL-10) cytokine production by macrophages, low concentrations
suppressed pro-inflammatory, but not anti-inflammatory, cytokine release. Suppression
induced by low IL- 15 concentrations is mediated by the high affinity IL-I5Rα, and high
doses effects were mediated by the IL-2/IL-15Rβ (Alleva, D.G. et al., 1997). Clearly,
further studies are needed to delineate the role of IL-15 in macrophage functions.

Discussion
68
Healthy liver contains large number of lymphocytes, which include not only NK and NKT
cells but also CD4 and CD8 T cells (Exley, M.A. and Koziel, M.J., 2004). In pathological
conditions, there is an increase in lymphocytes in the liver parenchyma. Lymphocytes are
mainly found at the sites of active damage and fibrogenesis in progressive disease (Lalor,
P.F. et al., 2007). We found a massive increase in the total IHL induced by HFD in WT
mice (Fig.3.8.B). While the frequencies of CD4+ and CD8+ T cell populations are not
modified in the liver of WT mice after HFD, their total numbers are significantly increased
as a consequence of lipid-rich diet (Fig.3.8). T cells are critical to the progression of liver
disease (Lalor, P.F. et al., 2007). Even though CD8 and CD4 T cells are found in areas of
parenchymal inflammation and fibrosis in steatohepatitis, their antigen specificity is not
known. Some reports suggest that T cells recognize liver antigens generated by oxidative
stress–induced modification and others postulate that T cells are derived from the
circulation and have their own specificity (Lalor, P.F. et al., 2007). We found that effector
CD8+ T cells (CD44high CD62Llow) are increased in the livers of WT mice fed with HFD,
with the concomitant reduction in naïve population (CD44low CD62Lhigh) which was not
reflected in the spleen of the same mice (Fig.3.8). These results suggest that under the
experimental conditions of HFD-induced liver pathologies, liver-derived neo-antigens
might activate CD8+ IHL. Alternatively, it is possible that these CD8+ T cells with activated
phenotype are terminally differentiated effector cells that die in the liver by apoptosis
(Mehal, W.Z. et al., 1999). Nevertheless, HFD promotes the infiltration of activated CD8+
T cells in the liver and these cells, in turn, can contribute to the inflammatory process.
Given that HFD diet induces Il15 gene expression in the liver and that IL-15 is implicated
in the homeostatic expansion of CD8+ T cells with memory phenotype, it is also possible
that IL-15 may also be involved in their activation. The effect of IL-15 deficiency on the
activation phenotype of intrahepatic CD8 T cells in HFD-fed mice remains to be studied.

Discussion
69
In human subjects, hepatic NK cells were reported to be increased in NASH, but in
established NAFLD their numbers were reduced (Kahraman, A. et al., 2010). The
frequency and the total numbers of NK cells are increased in the liver of HFD-fed mice
(Fig.3.8), suggesting these mice developed NASH. In the pathogenesis of NAFLD, hepatic
NK cells may have two different roles. First, an anti-fibrotic effect mediated by the
elimination of HSCs and second, activated NK cell-mediated killing of hepatocytes and
cholangiocytes via TRAIL (tumor necrosis factor-related apoptosis-inducing ligand).
Hepatocytes injury is one of the principal features of NASH, and is considered as one of
the primary events mediating liver damage in this pathology (Zhan, Y.T. and An, W.,
2010).
NKT cells are often divided into two groups: type I NKT cells, which express an invariant
TCR (Vα14/ Jα18) and are readily detectable by α-galactosylceramide (α-GalCer)-loaded
CD1d tetramers, and type II NKT cells which express a more diverse T-cell receptor
repertoire and cannot be directly identified (Martin-Murphy, B.V. et al., 2014). In our
experiments, we identified type I NKT cells as iNKT (CD3+ α-GalCer/CD1d tetramer +)
and type II as NKT (CD3+ NK1.1+). Our results showed no changes in the frequency of
NKT cells within IHL, but their total numbers are increased. Additionally, iNKT population
is decreased within the CD3+ cells but the total numbers did not change after HFD (Fig.
3.8). Published data about NKT infiltration in fatty liver is contradictory. In obese, leptin-
deficient ob/ob mice, NKT cells were reduced in the liver (Guebre-Xabier, M. et al., 2000).
Other reports suggest that hepatic NKT cells can skew the local cytokine production
towards Th2 cytokines rather than Th1, and as a consequence, depletion of NKT cells
leads to Th1 polarization of hepatic cytokine production in a mouse model of obesity
(Kremer, M. and Hines, I.N., 2008). Conversely, in patients with advanced NAFLD an
increase in liver NKT cells has been observed (Tajiri, K. et al., 2009). Therefore, both

Discussion
70
suppressive and inflammatory functions have been ascribed to NKT cells in an obese
state.
Recently, Martin-Murphy et al. reported that CD1d-/- mice displayed increased adiposity
and greater induction of inflammatory genes in the liver (Martin-Murphy, B.V. et al., 2014).
This study did not distinguish among NKT subtypes. However, as CD1d is required for
positive selection of both Type I and Type II NKT cells in the thymus (Martin-Murphy, B.V.
et al., 2014), CD1d−/− mice will lack both NKT subsets. The role of iNKT in obesity is also
controversial. In adipose tissue of HFD-fed mice, iNKT cell activation causes impairment
of metabolic functions through the release of pro-inflammatory cytokines and the
recruitment of other pathogenic immune cells (Wu, L. et al., 2012). In contrast to this,
iNKT cells were decreased after HFD in a mouse model, and in the circulation in obese
patients (Lynch, L. et al., 2012). In line with this report the reduction in iNKT cells in the
liver of HFD fed-mice, may suggest a protective role for this immune population in the
basal state.
Until now, we have shown that HFD increases the infiltration of many types of immune
cells in the livers of WT mice, but not in IL-15-deficient mice. As memory (CD62Llow, CD44
high) CD8+T cells, NK and NKT cells are dependent on IL-15 and are not detected in the
secondary lymphoid organs of IL-15 deficient mice (Ring, A.M. et al., 2012), the reduction
of these cell types in the liver is a direct consequence of IL-15 deficiency. In line with this
assumption, the number of these cells is reduced in IL-15-deficient mice maintained on
normal diet. Nonetheless, as IL-15 is induced by HFD, it is reasonable to propose that
HFD-induced IL-15 within the liver and consequent modulation of IHL population would
impact on the NAFLD. The phenotype of the infiltrating cells in the liver of IL-15 deficient
mice on HFD needs to be evaluated in order to elucidate their contribution to NAFLD.
Our finding that macrophages are also reduced in the livers of IL-15 KO mice needs to be
explored further, as the role of IL-15 in the homeostasis of macrophages has not yet been

Discussion
71
reported. Infiltration of immune cells to the site of inflammation (here the liver) is initiated
by the expression of a variety of chemokines (Rutkowski, M.D. and DeLeo, J.A., 2002).
For example, in alcoholic hepatitis, the expression of several CC and CXC chemokines
including CCL2, CCL3, CCL4, CCL5, CXCL1, CXCL5 and CXCL8, has been reported
(Maltby, J. et al., 1996). On the other hand, chemokine and chemokines receptors
expression in NAFLD and NASH is not well characterized, even though the inflammatory
conditions are similar and likely to induce a similar spectrum of chemokines.
We observed that increased levels of Ccl2, Ccl5 and Cxcl10 mRNA in the livers of mice
fed with HFD (Fig.3.9). Indeed, serum CCL2/MCP1 levels are reported to be increased in
steatosis, and Ccl2-deficient mice are protected from liver injury and fibrosis (Zamara, E.
et al., 2007) in agreement with our results in WT mice maintained on HFD (Fig.3.9).
Similarly, CCL5/RANTES plays an important role in the progression of hepatic
inflammation, and elevated hepatic Ccl5 expression was reported in a dietary model
of NAFLD. Moreover, in patients with ultrasound-diagnosed NAFLD, CCL5 serum levels
were elevated (Kirovski, G. et al., 2010). Expression of Cxcl10/IP10 is associated with
accelerated progression of inflammatory liver disease following HCV infection (Lalor, P.F.
et al., 2007). During NASH development, TNFα-producing KCs also produce Cxcl10 and
Ccl2 promoting blood monocyte infiltration (Tosello-Trampont, A.C. et al., 2012). Our
finding that the HFD-induced expression of hepatic Ccl2, Ccl5 and Cxcl10 mRNA did not
occur in Il15-/- mice (Fig. 3.9) indicates that immune cell infiltration of the liver during HFD
regimen is mediated by IL-15.
In vitro study in primary human hepatocytes stimulated with palmitic acid showed a dose-
dependent lipid accumulation, and corresponding dose-dependent induction of Ccl5
(Kirovski, G. et al., 2010). Others showed that incubation of irradiated hepatocytes in vitro
with TNF-alpha or IL-1beta, led to the up-regulation of Ccl2 or Cxcl10 and Ccl2,
respectively (Moriconi, F. et al., 2008). We also observed an increase in the expression of

Discussion
72
Ccl5 and Cxcl10 in isolated primary hepatocytes following stimulation with IL-15,
indicating that at least part of their expression in HFD-fed WT mice could be derived from
hepatocytes. Whereas induction of Cxcl10 occurred in both WT and IL-15Ra KO
hepatocytes, Ccl5 induction occurred only in WT primary hepatocytes (Fig.3.10). Hence, it
appears that differential IL-15 signalling can occur in the presence or absence of IL-15Ra,
leading to differential chemokine gene expression and possibly other functions. In contrast
to Ccl5 and Cxcl10, IL-15 stimulation did not induce any significant expression Ccl2,
suggesting that the expression of this chemokine in the total liver is mediated by other
liver resident cells or is stimulated by another factors induced by IL-15 rather than directly
by IL-15.
Liver immune populations are enriched in NK and NKT cells, and to a lesser extent in
CD8α+ T cells (Exley, M.A. and Koziel, M.J., 2004). As IL-15 is implicated in the
development and homeostasis of these lymphoid cells, we hypothesize that IL-15
availability and its signalling within the liver would profoundly influence the immune cell
composition in the liver and that this would in turn modulate the inflammatory responses in
the liver. Intriguingly, we observed that IL-15 or IL-15Rα deficiency reduced CD8+ T
lymphocytes in the spleen as described previously (Kanegane, H. and Tosato, G., 1996;
Lodolce, J.P. et al., 1998; Stoklasek, T.A. et al., 2006), but did not affect hepatic CD8+
cells (Fig. 3.11.A). Studies on hepatic CD8+ T lymphocytes in Il15-/- or Il15ra-/- mice are
very limited. Moreover, evaluation of CD8+ T cells in the liver is confounded by the fact
that they migrate to the liver following systemic activation, where they are trapped and
eventually eliminated (John, B. and Crispe, I.N., 2004). The importance of IL-15 signaling
in the maintenance of memory cells in the liver has been documented and memory CD8+
T cell cluster formation in the liver is dependent on IL-15 and IL-15Ra, like in bone marrow
(Su, Y.C. et al., 2010). It remains to be studied whether Il-15 deficiency selectively affects
the memory CD8 T cell subsets in the liver.

Discussion
73
We showed that NK, NKT and iNKT cells are reduced in the livers of Il15-/- or Il15ra-/- mice
when compared to WT mice (Fig 3.11.B & 3.11.C). These results are concurrent with the
requirement of IL-15 for NK cell homeostasis and activation in lymphoid organs (DiSanto,
J.P. et al., 1995; Lodolce, J.P. et al., 1998; Kennedy, M.K. et al., 2000; Gordy, L.E. et al.,
2011). Recently, Pek et al., showed that administration of recombinant human IL-15 (rhIL-
15) or Ad-vector expressing hIL-15 to humanized mouse significantly enhanced NK cell
development and maturation, particularly in bone marrow and liver (Pek, E.A. et al., 2011).
Previous studies using macrophage specific Il15ra-/- mice reported that macrophage-
derived IL-15Rα maintains NK cells (NK1.1+ CD3-) homeostasis by supporting proliferation
and maturation (Mortier, E. et al., 2009). We also evaluated NK cells in the liver of the
same conditional KO mice, but additionally we characterized NKT and iNKT cells. We
observed that while the NK and NKT populations in the liver were diminished in the
absence of IL-15Ra expression in macrophages, iNKT cells did not (Fig. 3.12.A.). On the
other hand, we observed that IL-15Ra deficiency in hepatocytes caused profound
reduction in NK, NKT and iNKT cells in the liver (Fig. 3.12.B.). An in vitro study had shown
that hepatocytes as well as IL-15 can induce antigen independent survival of peripheral
blood cells (Correia, M.P. et al., 2009). Previous studies in monocytes showed that trypsin
treatment caused a decrease in membrane bound IL-15 and the loss of IL-15R complex
ability to bind IL-15-IgG2b fusion protein (Musso, T. et al., 1999). We suggest that IL-15 in
hepatocytes is possibly bound to IL-15Rα, which trans-present IL-15 to effector cells.
Thus, in this study we have uncovered an unexpected function for hepatocytes in
maintaining the homeostasis of all NK subsets in the liver. Further studies are needed to
determine how hepatocyte-derived IL-15 promotes the maintenance of NK cell
populations in the liver.

Discussion
74
As discussed earlier, obesity-associated pathologies decrease life expectancy and quality.
Even though lifestyle changes, such as lower calorie intake and physical activity are quite
helpful, medical and surgical interventions still remain as options for some people with
morbid obesity. Moreover, NAFLD is treated by weight loss and therapies against insulin
resistance or dyslipidemia. This means to treat either directly the hepatic steatosis or the
pathological consequences. Of importance, although NAFLD is the most common liver
disease in the US, no pharmacological therapies have been approved by the FDA so
far(Tolman, K.G. and Dalpiaz, A.S., 2007).
Our results propose a pathogenic role for IL-15 in NAFLD. Therefore, IL-15 could be
potentially a novel target for developing new therapies against NAFLD. However, IL-15 is
a non-dispensable player of antimicrobial immunity and any treatment affecting IL-15
biology should take precautions into account (Kanegane, H. and Tosato, G., 1996;
Stoklasek, T.A. et al., 2006). Lymphocytic infiltration in liver parenchyma and fibrogenesis
characterizes most of liver disorders (Lalor, P.F. et al., 2007). Notably, we found that IL-15
could contribute significantly to lymphocyte infiltration in fatty liver by modulating various
chemokines. Given that chemokines are induced by IL-15 in hepatocytes in an IL-15Rα-
dependent mechanism, we propose that IL-15Rα in hepatocytes could be considered as a
potential therapeutic target for NAFLD.

Conclusions
75
5. CONCLUSIONS
Inflammation is frequently considered as a hallmark of obesity and non-alcoholic liver
diseases. Several pro-inflammatory cytokines have been related to the development of
obesity-associated pathologies. The current study was carried out to elucidate the role of
IL-15 in the development of NAFLD. IL-15 is a pro-inflammatory cytokine, which is
indispensable for the homeostasis of NK, NKT and memory CD8+ T cells. We showed that
the gain in body weight and liver mass after 16 weeks of HFD are significantly reduced in
Il15 null mice, which also displayed significantly reduced hepatic steatosis. Moreover,
HFD increased the hepatic expression of IL-15 as well as others pro-inflammatory
mediators, such as Tnfa and iNOS. In addition to reduced inflammatory response, Il-15
deficient livers showed FA oxidation contributing to the observed decrease in fat
deposition.
Infiltration of immune cells plays an important role in liver inflammation and the
development of NAFLD. HFD-induced macrophage infiltration of the liver was significantly
decreased in the livers of IL-15 KO mice, which could be attributed to reduced hepatic
expression of genes coding for the macrophage chemokines Ccl2 and Cxcl10. We also
found that IL-15 stimulation induced the expression of Ccl5 and Cxcl10 genes in primary
hepatocytes. These results suggest that the increased levels of IL-15 found in the liver of
HFD-fed mice might stimulate hepatocytes to secrete chemokines, leading to immune cell
recruitment and hepatic inflammation.
We observed that HFD caused a significant increase in the total numbers of IHLs, and the
NK cell populations and macrophage-derived IL-15Rα is needed for NKT cells
homeostasis. Interestingly, we found that hepatocyte-specific expression of IL-15 Rα is
indispensable for NK, NKT and iNKT cells maintenance in the liver. These findings
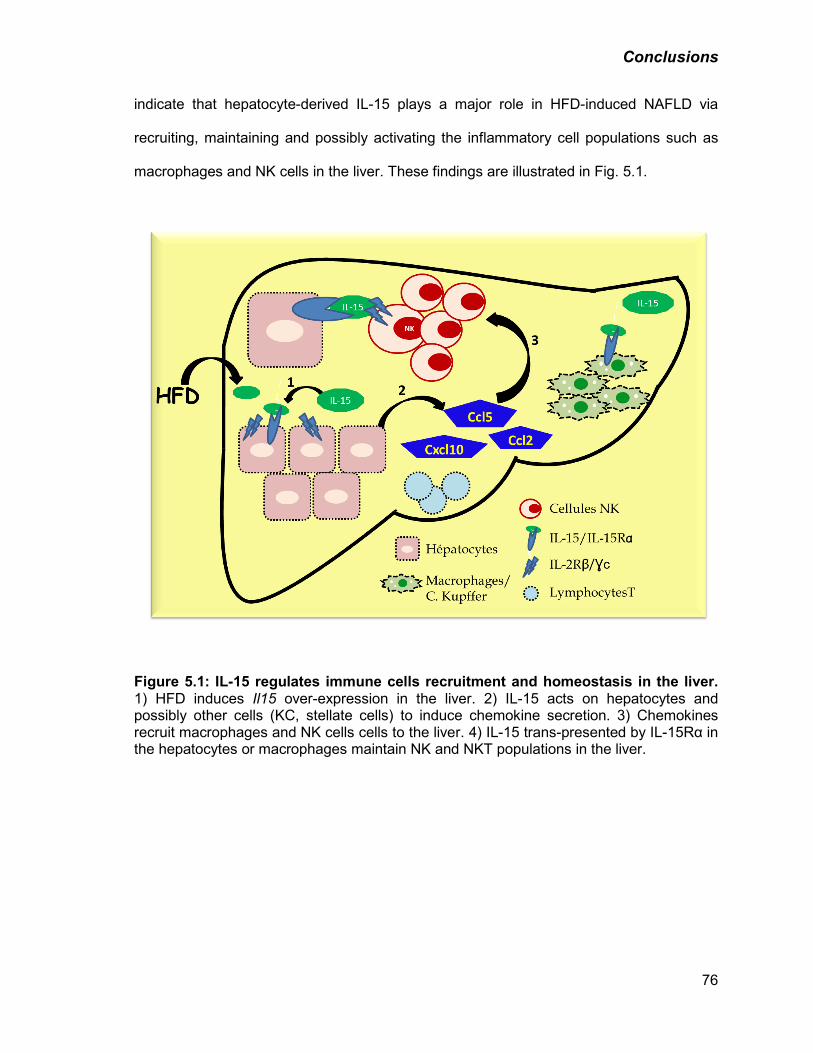
Conclusions
76
indicate that hepatocyte-derived IL-15 plays a major role in HFD-induced NAFLD via
recruiting, maintaining and possibly activating the inflammatory cell populations such as
macrophages and NK cells in the liver. These findings are illustrated in Fig. 5.1.
Figure 5.1: IL-15 regulates immune cells recruitment and homeostasis in the liver. 1) HFD induces Il15 over-expression in the liver. 2) IL-15 acts on hepatocytes and possibly other cells (KC, stellate cells) to induce chemokine secretion. 3) Chemokines recruit macrophages and NK cells cells to the liver. 4) IL-15 trans-presented by IL-15Rα in the hepatocytes or macrophages maintain NK and NKT populations in the liver.

Acknowledgements
77
6. ACKNOWLEDGEMENTS
I would first like to thank my supervisors Dr. Ilangumaran and Dr. Ramanathan for the
opportunity to complete a MSc. in your lab. You have always encouraged me to do better
experiments each time and to critical think about the results. I also appreciate the support
you both gave me when I arrived here.
I would also like to thank Marian Mayhue for being my support in the lab every day; it is
really a pleasure to work with. I would like to give thanks to Mehdi Yeganeh as well, for
show me almost all the technics I know now in the liver work, also for the helpful
discussion about my project.
I would also like to thank the others members of my lab and the lab of Dr. Ramanathan:
Diwakar, Yirui, Xi Lin, Galaxia, Daniel, Rajani and Alberto, I have had a great time
spending these two years in the lab with you.
I would also like to thank the short-term students we had in our lab and worked with me,
Veronique and Maude, as well as Leonid Volkov for his instructions on how to use the
microscope.
Lastly I would like to thank my evaluators Dr. Caroline Saucier and Dr. Claire Dubois for
taking the time to evaluate my thesis.

References
78
7. REFERENCES
• Adams, L.A., P. Angulo and K.D. Lindor (2005). "Nonalcoholic fatty liver disease." CMAJ 172(7): 899-905.
• Alleva, D.G., S.B. Kaser, M.A. Monroy, M.J. Fenton and D.I. Beller (1997). "IL-15 functions as a potent autocrine regulator of macrophage proinflammatory cytokine production: evidence for differential receptor subunit utilization associated with stimulation or inhibition." J Immunol 159(6): 2941-2951.
• Anderson, D.M., L. Johnson, M.B. Glaccum, N.G. Copeland, D.J. Gilbert, et al. (1995a). "Chromosomal assignment and genomic structure of Il15." Genomics 25(3): 701-706.
• Anderson, D.M., S. Kumaki, M. Ahdieh, J. Bertles, M. Tometsko, et al. (1995b). "Functional characterization of the human interleukin-15 receptor alpha chain and close linkage of IL15RA and IL2RA genes." J Biol Chem 270(50): 29862-29869.
• Anguille, S., E.L. Smits, N. Cools, H. Goossens, Z.N. Berneman, et al. (2009). "Short-term cultured, interleukin-15 differentiated dendritic cells have potent immunostimulatory properties." J Transl Med 7: 109.
• Anstee, Q.M. and R.D. Goldin (2006). "Mouse models in non-alcoholic fatty liver disease and steatohepatitis research." Int J Exp Pathol 87(1): 1-16.
• Aprile, M., M.R. Ambrosio, V. D'Esposito, F. Beguinot, P. Formisano, et al. (2014). "PPARG in Human Adipogenesis: Differential Contribution of Canonical Transcripts and Dominant Negative Isoforms." PPAR Res 2014: 537865.
• Aranha, G.V. and H.B. Greenlee (1986). "Intra-abdominal surgery in patients with advanced cirrhosis." Arch Surg 121(3): 275-277.
• Armitage, R.J., B.M. Macduff, J. Eisenman, R. Paxton and K.H. Grabstein (1995). "IL-15 has stimulatory activity for the induction of B cell proliferation and differentiation." J Immunol 154(2): 483-490.
• Asrih, M. and F.R. Jornayvaz (2013). "Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance." J Endocrinol 218(3): R25-36.
• Assimacopoulos-Jeannet, F., S. Thumelin, E. Roche, V. Esser, J.D. McGarry, et al. (1997). "Fatty acids rapidly induce the carnitine palmitoyltransferase I gene in the pancreatic beta-cell line INS-1." J Biol Chem 272(3): 1659-1664.
• Balato, A., D. Unutmaz and A.A. Gaspari (2009). "Natural killer T cells: an unconventional T-cell subset with diverse effector and regulatory functions." J Invest Dermatol 129(7): 1628-1642.
• Bamford, R.N., A.J. Grant, J.D. Burton, C. Peters, G. Kurys, et al. (1994). "The interleukin (IL) 2 receptor beta chain is shared by IL-2 and a cytokine, provisionally designated IL-T, that stimulates T-cell proliferation and the induction of lymphokine-activated killer cells." Proc Natl Acad Sci U S A 91(11): 4940-4944.
• Barra, N.G., S. Reid, R. MacKenzie, G. Werstuck, B.L. Trigatti, et al. (2010). "Interleukin-15 contributes to the regulation of murine adipose tissue and human adipocytes." Obesity (Silver Spring) 18(8): 1601-1607.
• Becker, T.C., E.J. Wherry, D. Boone, K. Murali-Krishna, R. Antia, et al. (2002). "Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells." J Exp Med 195(12): 1541-1548.

References
79
• Ben Ahmed, M., N. Belhadj Hmida, N. Moes, S. Buyse, M. Abdeladhim, et al. (2009). "IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway." J Immunol 182(11): 6763-6770.
• Bendelac, A., P.B. Savage and L. Teyton (2007). "The biology of NKT cells." Annu Rev Immunol 25: 297-336.
• Boggs, S.S., M. Trevisan, K. Patrene and K. Geogopoulos (1998). "Lack of natural killer cell precursors in fetal liver of Ikaros knockout mutant mice." Nat Immun 16(4): 137-145.
• Bouchard, A., C. Ratthe and D. Girard (2004). "Interleukin-15 delays human neutrophil apoptosis by intracellular events and not via extracellular factors: role of Mcl-1 and decreased activity of caspase-3 and caspase-8." J Leukoc Biol 75(5): 893-900.
• Brandt, K., S. Bulfone-Paus, D.C. Foster and R. Ruckert (2003). "Interleukin-21 inhibits dendritic cell activation and maturation." Blood 102(12): 4090-4098.
• Braunersreuther, V., G.L. Viviani, F. Mach and F. Montecucco (2012). "Role of cytokines and chemokines in non-alcoholic fatty liver disease." World J Gastroenterol 18(8): 727-735.
• Brown, M.S. and J.L. Goldstein (2008). "Selective versus total insulin resistance: a pathogenic paradox." Cell Metab 7(2): 95-96.
• Browning, J.D. and J.D. Horton (2004). "Molecular mediators of hepatic steatosis and liver injury." J Clin Invest 114(2): 147-152.
• Budagian, V., E. Bulanova, Z. Orinska, L. Thon, U. Mamat, et al. (2005). "A promiscuous liaison between IL-15 receptor and Axl receptor tyrosine kinase in cell death control." EMBO J 24(24): 4260-4270.
• Busquets, S., M.T. Figueras, S. Meijsing, N. Carbo, L.S. Quinn, et al. (2005). "Interleukin-15 decreases proteolysis in skeletal muscle: a direct effect." Int J Mol Med 16(3): 471-476.
• Carbo, N., J. Lopez-Soriano, P. Costelli, B. Alvarez, S. Busquets, et al. (2001). "Interleukin-15 mediates reciprocal regulation of adipose and muscle mass: a potential role in body weight control." Biochim Biophys Acta 1526(1): 17-24.
• Carey, M., S. Kehlenbrink and M. Hawkins (2013). "Evidence for central regulation of glucose metabolism." J Biol Chem 288(49): 34981-34988.
• Castillo, E.F., L.F. Acero, S.W. Stonier, D. Zhou and K.S. Schluns (2010). "Thymic and peripheral microenvironments differentially mediate development and maturation of iNKT cells by IL-15 transpresentation." Blood 116(14): 2494-2503.
• Castillo, E.F., S.W. Stonier, L. Frasca and K.S. Schluns (2009). "Dendritic cells support the in vivo development and maintenance of NK cells via IL-15 trans-presentation." J Immunol 183(8): 4948-4956.
• Chang, C.L., Y.G. Lai, M.S. Hou, P.L. Huang and N.S. Liao (2011). "IL-15Ralpha of radiation-resistant cells is necessary and sufficient for thymic invariant NKT cell survival and functional maturation." J Immunol 187(3): 1235-1242.
• Chen, Y., H. Wei, R. Sun, Z. Dong, J. Zhang, et al. (2007). "Increased susceptibility to liver injury in hepatitis B virus transgenic mice involves NKG2D-ligand interaction and natural killer cells." Hepatology 46(3): 706-715.
• Christiansen, T., S.K. Paulsen, J.M. Bruun, S.B. Pedersen and B. Richelsen (2010). "Exercise training versus diet-induced weight-loss on metabolic risk factors and inflammatory markers in obese subjects: a 12-week randomized intervention study." Am J Physiol Endocrinol Metab 298(4): E824-831.

References
80
• Cinti, S., G. Mitchell, G. Barbatelli, I. Murano, E. Ceresi, et al. (2005). "Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans." J Lipid Res 46(11): 2347-2355.
• Collins, S., T.L. Martin, R.S. Surwit and J. Robidoux (2004). "Genetic vulnerability to diet-induced obesity in the C57BL/6J mouse: physiological and molecular characteristics." Physiol Behav 81(2): 243-248.
• Correia, M.P., E.M. Cardoso, C.F. Pereira, R. Neves, M. Uhrberg, et al. (2009). "Hepatocytes and IL-15: a favorable microenvironment for T cell survival and CD8+ T cell differentiation." J Immunol 182(10): 6149-6159.
• Cosmi, L., F. Liotta, R. Angeli, B. Mazzinghi, V. Santarlasci, et al. (2004). "Th2 cells are less susceptible than Th1 cells to the suppressive activity of CD25+ regulatory thymocytes because of their responsiveness to different cytokines." Blood 103(8): 3117-3121.
• D'Souza, W.N. and L. Lefrancois (2003). "IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion." J Immunol 171(11): 5727-5735.
• Day, C.P. and O.F. James (1998). "Steatohepatitis: a tale of two "hits"?" Gastroenterology 114(4): 842-845.
• DeLeve, L.D. (2013). "Liver sinusoidal endothelial cells and liver regeneration." J Clin Invest 123(5): 1861-1866.
• Despres, J.P. and I. Lemieux (2006). "Abdominal obesity and metabolic syndrome." Nature 444(7121): 881-887.
• Diehl, A.M. (2002). "Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines." Am J Physiol Gastrointest Liver Physiol 282(1): G1-5.
• DiSanto, J.P., W. Muller, D. Guy-Grand, A. Fischer and K. Rajewsky (1995). "Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain." Proc Natl Acad Sci U S A 92(2): 377-381.
• Doherty, D.G. and C. O'Farrelly (2000). "Innate and adaptive lymphoid cells in the human liver." Immunol Rev 174: 5-20.
• Dong, Z., H. Wei, R. Sun and Z. Tian (2007). "The roles of innate immune cells in liver injury and regeneration." Cell Mol Immunol 4(4): 241-252.
• Dubois, S., F. Magrangeas, P. Lehours, S. Raher, J. Bernard, et al. (1999). "Natural splicing of exon 2 of human interleukin-15 receptor alpha-chain mRNA results in a shortened form with a distinct pattern of expression." J Biol Chem 274(38): 26978-26984.
• Dubois, S., J. Mariner, T.A. Waldmann and Y. Tagaya (2002). "IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells." Immunity 17(5): 537-547.
• Elyaman, W., E.M. Bradshaw, C. Uyttenhove, V. Dardalhon, A. Awasthi, et al. (2009). "IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells." Proc Natl Acad Sci U S A 106(31): 12885-12890.
• Estess, P., A. Nandi, M. Mohamadzadeh and M.H. Siegelman (1999). "Interleukin 15 induces endothelial hyaluronan expression in vitro and promotes activated T cell extravasation through a CD44-dependent pathway in vivo." J Exp Med 190(1): 9-19.
• Exley, M.A. and M.J. Koziel (2004). "To be or not to be NKT: natural killer T cells in the liver." Hepatology 40(5): 1033-1040.
• Fan, J., L. Zhong, G. Wang, X. Wu, M. Li, et al. (2001). "The role of Kupffer cells in non-alcoholic steatohepatitis of rats chronically fed with high-fat diet." Zhonghua Gan Zang Bing Za Zhi 9(1): 16-18.

References
81
• Fausto, N., J.S. Campbell and K.J. Riehle (2006). "Liver regeneration." Hepatology 43(2 Suppl 1): S45-53.
• Fehniger, T.A. and M.A. Caligiuri (2001). "Interleukin 15: biology and relevance to human disease." Blood 97(1): 14-32.
• Friedman, S.L. (2008). "Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver." Physiol Rev 88(1): 125-172.
• Gaggero, A., B. Azzarone, C. Andrei, Z. Mishal, R. Meazza, et al. (1999). "Differential intracellular trafficking, secretion and endosomal localization of two IL-15 isoforms." Eur J Immunol 29(4): 1265-1274.
• Gao, B., W.I. Jeong and Z. Tian (2008). "Liver: An organ with predominant innate immunity." Hepatology 47(2): 729-736.
• Gessner, A., H. Blum and M. Rollinghoff (1993). "Differential regulation of IL-9-expression after infection with Leishmania major in susceptible and resistant mice." Immunobiology 189(5): 419-435.
• Golden-Mason, L., A.M. Kelly, D.G. Doherty, O. Traynor, G. McEntee, et al. (2004). "Hepatic interleuklin 15 (IL-15) expression: implications for local NK/NKT cell homeostasis and development." Clin Exp Immunol 138(1): 94-101.
• Gordon, S.M., J. Chaix, L.J. Rupp, J. Wu, S. Madera, et al. (2012). "The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation." Immunity 36(1): 55-67.
• Gordy, L.E., J.S. Bezbradica, A.I. Flyak, C.T. Spencer, A. Dunkle, et al. (2011). "IL-15 regulates homeostasis and terminal maturation of NKT cells." J Immunol 187(12): 6335-6345.
• Grabstein, K.H., J. Eisenman, K. Shanebeck, C. Rauch, S. Srinivasan, et al. (1994). "Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor." Science 264(5161): 965-968.
• Gregoire, C., L. Chasson, C. Luci, E. Tomasello, F. Geissmann, et al. (2007). "The trafficking of natural killer cells." Immunol Rev 220: 169-182.
• Guebre-Xabier, M., S. Yang, H.Z. Lin, R. Schwenk, U. Krzych, et al. (2000). "Altered hepatic lymphocyte subpopulations in obesity-related murine fatty livers: potential mechanism for sensitization to liver damage." Hepatology 31(3): 633-640.
• Gui, Y., M. Yeganeh, S. Ramanathan, C. Leblanc, V. Pomerleau, et al. (2011). "SOCS1 controls liver regeneration by regulating HGF signaling in hepatocytes." J Hepatol 55(6): 1300-1308.
• Hanisch, U.K., S.A. Lyons, M. Prinz, C. Nolte, J.R. Weber, et al. (1997). "Mouse brain microglia express interleukin-15 and its multimeric receptor complex functionally coupled to Janus kinase activity." J Biol Chem 272(46): 28853-28860.
• Haslam, D.W. and W.P. James (2005). "Obesity." Lancet 366(9492): 1197-1209. • Hatakeyama, M., M. Tsudo, S. Minamoto, T. Kono, T. Doi, et al. (1989). "Interleukin-2
receptor beta chain gene: generation of three receptor forms by cloned human alpha and beta chain cDNA's." Science 244(4904): 551-556.
• Hauber, H.P., C. Bergeron and Q. Hamid (2004). "IL-9 in allergic inflammation." Int Arch Allergy Immunol 134(1): 79-87.
• Hems, D.A. and P.D. Whitton (1980). "Control of hepatic glycogenolysis." Physiol Rev 60(1): 1-50.

References
82
• Hijikawa, T., M. Kaibori, Y. Uchida, M. Yamada, K. Matsui, et al. (2008). "Insulin-like growth factor 1 prevents liver injury through the inhibition of TNF-alpha and iNOS induction in D-galactosamine and LPS-treated rats." Shock 29(6): 740-747.
• Holgate, S.T. and R. Polosa (2008). "Treatment strategies for allergy and asthma." Nat Rev Immunol 8(3): 218-230.
• Hong, F., S. Radaeva, H.N. Pan, Z. Tian, R. Veech, et al. (2004). "Interleukin 6 alleviates hepatic steatosis and ischemia/reperfusion injury in mice with fatty liver disease." Hepatology 40(4): 933-941.
• Hong, J., R.E. Stubbins, R.R. Smith, A.E. Harvey and N.P. Nunez (2009). "Differential susceptibility to obesity between male, female and ovariectomized female mice." Nutr J 8: 11.
• Hoontrakoon, R., H.W. Chu, S.J. Gardai, S.E. Wenzel, P. McDonald, et al. (2002). "Interleukin-15 inhibits spontaneous apoptosis in human eosinophils via autocrine production of granulocyte macrophage-colony stimulating factor and nuclear factor-kappaB activation." Am J Respir Cell Mol Biol 26(4): 404-412.
• Hou, X., R. Zhou, H. Wei, R. Sun and Z. Tian (2009). "NKG2D-retinoic acid early inducible-1 recognition between natural killer cells and Kupffer cells in a novel murine natural killer cell-dependent fulminant hepatitis." Hepatology 49(3): 940-949.
• Huang, W., A. Metlakunta, N. Dedousis, P. Zhang, I. Sipula, et al. (2010). "Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance." Diabetes 59(2): 347-357.
• Huntington, N.D., H. Puthalakath, P. Gunn, E. Naik, E.M. Michalak, et al. (2007). "Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1." Nat Immunol 8(8): 856-863.
• John, B. and I.N. Crispe (2004). "Passive and active mechanisms trap activated CD8+ T cells in the liver." J Immunol 172(9): 5222-5229.
• Johnston, J.A., C.M. Bacon, D.S. Finbloom, R.C. Rees, D. Kaplan, et al. (1995). "Tyrosine phosphorylation and activation of STAT5, STAT3, and Janus kinases by interleukins 2 and 15." Proc Natl Acad Sci U S A 92(19): 8705-8709.
• Kahraman, A., M. Schlattjan, P. Kocabayoglu, S. Yildiz-Meziletoglu, M. Schlensak, et al. (2010). "Major histocompatibility complex class I-related chains A and B (MIC A/B): a novel role in nonalcoholic steatohepatitis." Hepatology 51(1): 92-102.
• Kamagate, A. and H.H. Dong (2008). "FoxO1 integrates insulin signaling to VLDL production." Cell Cycle 7(20): 3162-3170.
• Kanegane, H. and G. Tosato (1996). "Activation of naive and memory T cells by interleukin-15." Blood 88(1): 230-235.
• Karpe, F., J.R. Dickmann and K.N. Frayn (2011). "Fatty acids, obesity, and insulin resistance: time for a reevaluation." Diabetes 60(10): 2441-2449.
• Kennedy, M.K., M. Glaccum, S.N. Brown, E.A. Butz, J.L. Viney, et al. (2000). "Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice." J Exp Med 191(5): 771-780.
• Kim, H.P., J. Imbert and W.J. Leonard (2006). "Both integrated and differential regulation of components of the IL-2/IL-2 receptor system." Cytokine Growth Factor Rev 17(5): 349-366.
• Kim, S., K. Iizuka, H.S. Kang, A. Dokun, A.R. French, et al. (2002). "In vivo developmental stages in murine natural killer cell maturation." Nat Immunol 3(6): 523-528.

References
83
• Kirovski, G., E. Gabele, C. Dorn, L. Moleda, C. Niessen, et al. (2010). "Hepatic steatosis causes induction of the chemokine RANTES in the absence of significant hepatic inflammation." Int J Clin Exp Pathol 3(7): 675-680.
• Koka, R., P. Burkett, M. Chien, S. Chai, D.L. Boone, et al. (2004). "Cutting edge: murine dendritic cells require IL-15R alpha to prime NK cells." J Immunol 173(6): 3594-3598.
• Koka, R., P.R. Burkett, M. Chien, S. Chai, F. Chan, et al. (2003). "Interleukin (IL)-15R[alpha]-deficient natural killer cells survive in normal but not IL-15R[alpha]-deficient mice." J Exp Med 197(8): 977-984.
• Kremer, M. and I.N. Hines (2008). "Natural killer T cells and non-alcoholic fatty liver disease: fat chews on the immune system." World J Gastroenterol 14(3): 487-488.
• Kurowska, M., W. Rudnicka, D. Maslinska and W. Maslinski (2002). "Expression of IL-15 and IL-15 receptor isoforms in select structures of human fetal brain." Ann N Y Acad Sci 966: 441-445.
• Lalor, P.F., J. Faint, Y. Aarbodem, S.G. Hubscher and D.H. Adams (2007). "The role of cytokines and chemokines in the development of steatohepatitis." Semin Liver Dis 27(2): 173-193.
• Lamas, O., J.A. Martinez and A. Marti (2004). "Energy restriction restores the impaired immune response in overweight (cafeteria) rats." J Nutr Biochem 15(7): 418-425.
• LaPorte, S.L., Z.S. Juo, J. Vaclavikova, L.A. Colf, X. Qi, et al. (2008). "Molecular and structural basis of cytokine receptor pleiotropy in the interleukin-4/13 system." Cell 132(2): 259-272.
• Lefebvre, P., G. Chinetti, J.C. Fruchart and B. Staels (2006). "Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis." J Clin Invest 116(3): 571-580.
• Lemoinne, S., A. Cadoret, H. El Mourabit, D. Thabut and C. Housset (2013). "Origins and functions of liver myofibroblasts." Biochim Biophys Acta 1832(7): 948-954.
• Lenardo, M.J. (1991). "Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis." Nature 353(6347): 858-861.
• Leonard, W.J. (2001). "Cytokines and immunodeficiency diseases." Nat Rev Immunol 1(3): 200-208.
• Li, H.H., J.B. Tyburski, Y.W. Wang, S. Strawn, B.H. Moon, et al. (2014). "Modulation of Fatty Acid and Bile Acid Metabolism By Peroxisome Proliferator-Activated Receptor alpha Protects Against Alcoholic Liver Disease." Alcohol Clin Exp Res.
• Li, Y., J.S. Park, J.H. Deng and Y. Bai (2006). "Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex." J Bioenerg Biomembr 38(5-6): 283-291.
• Li, Z. and A.M. Diehl (2003). "Innate immunity in the liver." Curr Opin Gastroenterol 19(6): 565-571.
• Li, Z., S. Yang, H. Lin, J. Huang, P.A. Watkins, et al. (2003). "Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease." Hepatology 37(2): 343-350.
• Lodolce, J.P., D.L. Boone, S. Chai, R.E. Swain, T. Dassopoulos, et al. (1998). "IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation." Immunity 9(5): 669-676.
• Lodolce, J.P., P.R. Burkett, D.L. Boone, M. Chien and A. Ma (2001). "T cell-independent interleukin 15Ralpha signals are required for bystander proliferation." J Exp Med 194(8): 1187-1194.

References
84
• Lumeng, C.N., J.L. Bodzin and A.R. Saltiel (2007). "Obesity induces a phenotypic switch in adipose tissue macrophage polarization." J Clin Invest 117(1): 175-184.
• Lynch, L., M. Nowak, B. Varghese, J. Clark, A.E. Hogan, et al. (2012). "Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production." Immunity 37(3): 574-587.
• Maher, J.J., P. Leon and J.C. Ryan (2008). "Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis." Hepatology 48(2): 670-678.
• Maltby, J., S. Wright, G. Bird and N. Sheron (1996). "Chemokine levels in human liver homogenates: associations between GRO alpha and histopathological evidence of alcoholic hepatitis." Hepatology 24(5): 1156-1160.
• Martin-Murphy, B.V., Q. You, H. Wang, B.A. De La Houssaye, T.P. Reilly, et al. (2014). "Mice lacking natural killer T cells are more susceptible to metabolic alterations following high fat diet feeding." PLoS One 9(1): e80949.
• Matern, D. and P. Rinaldo (1993). Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. GeneReviews(R). R. A. Pagon, M. P. Adam, H. H. Ardingeret al. Seattle (WA).
• Matsuda, J.L., L. Gapin, S. Sidobre, W.C. Kieper, J.T. Tan, et al. (2002). "Homeostasis of V alpha 14i NKT cells." Nat Immunol 3(10): 966-974.
• Matsuda, J.L., Q. Zhang, R. Ndonye, S.K. Richardson, A.R. Howell, et al. (2006). "T-bet concomitantly controls migration, survival, and effector functions during the development of Valpha14i NKT cells." Blood 107(7): 2797-2805.
• Mazzucchelli, R. and S.K. Durum (2007). "Interleukin-7 receptor expression: intelligent design." Nat Rev Immunol 7(2): 144-154.
• Mehal, W.Z., A.E. Juedes and I.N. Crispe (1999). "Selective retention of activated CD8+ T cells by the normal liver." J Immunol 163(6): 3202-3210.
• Melhem, A., N. Muhanna, A. Bishara, C.E. Alvarez, Y. Ilan, et al. (2006). "Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC." J Hepatol 45(1): 60-71.
• Miranda-Carus, M.E., A. Balsa, M. Benito-Miguel, C. Perez de Ayala and E. Martin-Mola (2004). "IL-15 and the initiation of cell contact-dependent synovial fibroblast-T lymphocyte cross-talk in rheumatoid arthritis: effect of methotrexate." J Immunol 173(2): 1463-1476.
• Miranda-Carus, M.E., M. Benito-Miguel, A. Balsa, T. Cobo-Ibanez, C. Perez de Ayala, et al. (2006). "Peripheral blood T lymphocytes from patients with early rheumatoid arthritis express RANKL and interleukin-15 on the cell surface and promote osteoclastogenesis in autologous monocytes." Arthritis Rheum 54(4): 1151-1164.
• Moran-Salvador, E., M. Lopez-Parra, V. Garcia-Alonso, E. Titos, M. Martinez-Clemente, et al. (2011). "Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts." FASEB J 25(8): 2538-2550.
• Moriconi, F., H. Christiansen, D. Raddatz, J. Dudas, R.M. Hermann, et al. (2008). "Effect of radiation on gene expression of rat liver chemokines: in vivo and in vitro studies." Radiat Res 169(2): 162-169.
• Morris, E.M., M.R. Jackman, G.M. Meers, G.C. Johnson, J.L. Lopez, et al. (2013). "Reduced hepatic mitochondrial respiration following acute high-fat diet is prevented by PGC-1alpha overexpression." Am J Physiol Gastrointest Liver Physiol 305(11): G868-880.
• Mortier, E., R. Advincula, L. Kim, S. Chmura, J. Barrera, et al. (2009). "Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets." Immunity 31(5): 811-822.

References
85
• Musso, T., L. Calosso, M. Zucca, M. Millesimo, D. Ravarino, et al. (1999). "Human monocytes constitutively express membrane-bound, biologically active, and interferon-gamma-upregulated interleukin-15." Blood 93(10): 3531-3539.
• Nagarajan, S., D. Amir, A. Grupi, D.P. Goldenberg, A.P. Minton, et al. (2011). "Modulation of functionally significant conformational equilibria in adenylate kinase by high concentrations of trimethylamine oxide attributed to volume exclusion." Biophys J 100(12): 2991-2999.
• Nagy, L.E. (2003). "Recent insights into the role of the innate immune system in the development of alcoholic liver disease." Exp Biol Med (Maywood) 228(8): 882-890.
• Nakamura, A. and Y. Terauchi (2013). "Lessons from mouse models of high-fat diet-induced NAFLD." Int J Mol Sci 14(11): 21240-21257.
• Nakazato, K., H. Yamada, T. Yajima, Y. Kagimoto, H. Kuwano, et al. (2007). "Enforced expression of Bcl-2 partially restores cell numbers but not functions of TCRgammadelta intestinal intraepithelial T lymphocytes in IL-15-deficient mice." J Immunol 178(2): 757-764.
• Nguyen, P., V. Leray, M. Diez, S. Serisier, J. Le Bloc'h, et al. (2008). "Liver lipid metabolism." J Anim Physiol Anim Nutr (Berl) 92(3): 272-283.
• Nilsen, E.M., F.E. Johansen, F.L. Jahnsen, K.E. Lundin, T. Scholz, et al. (1998). "Cytokine profiles of cultured microvascular endothelial cells from the human intestine." Gut 42(5): 635-642.
• Noguchi, M., H. Yi, H.M. Rosenblatt, A.H. Filipovich, S. Adelstein, et al. (1993). "Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans." Cell 73(1): 147-157.
• O'Hara, S.P., J.H. Tabibian, P.L. Splinter and N.F. LaRusso (2013). "The dynamic biliary epithelia: molecules, pathways, and disease." J Hepatol 58(3): 575-582.
• O'Shea, D., T.J. Cawood, C. O'Farrelly and L. Lynch (2010). "Natural killer cells in obesity: impaired function and increased susceptibility to the effects of cigarette smoke." PLoS One 5(1): e8660.
• Ogata, Y., A. Kukita, T. Kukita, M. Komine, A. Miyahara, et al. (1999). "A novel role of IL-15 in the development of osteoclasts: inability to replace its activity with IL-2." J Immunol 162(5): 2754-2760.
• Okada, Y., K. Yamaguchi, T. Nakajima, T. Nishikawa, M. Jo, et al. (2013). "Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats." Liver Int 33(2): 301-311.
• Olsen, S.K., N. Ota, S. Kishishita, M. Kukimoto-Niino, K. Murayama, et al. (2007). "Crystal Structure of the interleukin-15.interleukin-15 receptor alpha complex: insights into trans and cis presentation." J Biol Chem 282(51): 37191-37204.
• Onoda, T., M. Rahman, H. Nara, A. Araki, K. Makabe, et al. (2007). "Human CD4+ central and effector memory T cells produce IL-21: effect on cytokine-driven proliferation of CD4+ T cell subsets." Int Immunol 19(10): 1191-1199.
• Oppenheimer-Marks, N., R.I. Brezinschek, M. Mohamadzadeh, R. Vita and P.E. Lipsky (1998). "Interleukin 15 is produced by endothelial cells and increases the transendothelial migration of T cells In vitro and in the SCID mouse-human rheumatoid arthritis model In vivo." J Clin Invest 101(6): 1261-1272.
• Otero, Y.F., J.M. Stafford and O.P. McGuinness (2014). "Pathway-Selective Insulin Resistance and Metabolic Disease: The Importance of Nutrient Flux." J Biol Chem.

References
86
• Papoulas, M. and S. Theocharis (2009). "Primary liver tumors: origin and target therapy." Expert Opin Ther Targets 13(8): 957-965.
• Pek, E.A., T. Chan, S. Reid and A.A. Ashkar (2011). "Characterization and IL-15 dependence of NK cells in humanized mice." Immunobiology 216(1-2): 218-224.
• Pelletier, M. and D. Girard (2007). "Biological functions of interleukin-21 and its role in inflammation." ScientificWorldJournal 7: 1715-1735.
• Plana, N., D. Ibarretxe, A. Cabre, E. Ruiz and L. Masana (2014). "Prevalence of atherogenic dyslipidemia in primary care patients at moderate-very high risk of cardiovascular disease. Cardiovascular risk perception." Clin Investig Arterioscler.
• Puigserver, P., Z. Wu, C.W. Park, R. Graves, M. Wright, et al. (1998). "A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis." Cell 92(6): 829-839.
• Quinn, L.S. and B.G. Anderson (2011). "Interleukin-15, IL-15 Receptor-Alpha, and Obesity: Concordance of Laboratory Animal and Human Genetic Studies." J Obes 2011: 456347.
• Quinn, L.S., B.G. Anderson, R.H. Drivdahl, B. Alvarez and J.M. Argiles (2002). "Overexpression of interleukin-15 induces skeletal muscle hypertrophy in vitro: implications for treatment of muscle wasting disorders." Exp Cell Res 280(1): 55-63.
• Quinn, L.S., K.L. Haugk and K.H. Grabstein (1995). "Interleukin-15: a novel anabolic cytokine for skeletal muscle." Endocrinology 136(8): 3669-3672.
• Quinn, L.S., L. Strait-Bodey, B.G. Anderson, J.M. Argiles and P.J. Havel (2005). "Interleukin-15 stimulates adiponectin secretion by 3T3-L1 adipocytes: evidence for a skeletal muscle-to-fat signaling pathway." Cell Biol Int 29(6): 449-457.
• Ramanathan, S., J. Gagnon, C. Leblanc, R. Rottapel and S. Ilangumaran (2006). "Suppressor of cytokine signaling 1 stringently regulates distinct functions of IL-7 and IL-15 in vivo during T lymphocyte development and homeostasis." J Immunol 176(7): 4029-4041.
• Ratthe, C. and D. Girard (2004). "Interleukin-15 enhances human neutrophil phagocytosis by a Syk-dependent mechanism: importance of the IL-15Ralpha chain." J Leukoc Biol 76(1): 162-168.
• Reddy, J.K. and T. Hashimoto (2001). "Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system." Annu Rev Nutr 21: 193-230.
• Renauld, J.C., C. Druez, A. Kermouni, F. Houssiau, C. Uyttenhove, et al. (1992). "Expression cloning of the murine and human interleukin 9 receptor cDNAs." Proc Natl Acad Sci U S A 89(12): 5690-5694.
• Ridaura, V.K., J.J. Faith, F.E. Rey, J. Cheng, A.E. Duncan, et al. (2013). "Gut microbiota from twins discordant for obesity modulate metabolism in mice." Science 341(6150): 1241214.
• Ring, A.M., J.X. Lin, D. Feng, S. Mitra, M. Rickert, et al. (2012). "Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15." Nat Immunol 13(12): 1187-1195.
• Robertson, H. and J.A. Kirby (2003). "Post-transplant renal tubulitis: the recruitment, differentiation and persistence of intra-epithelial T cells." Am J Transplant 3(1): 3-10.
• Rochman, Y., R. Spolski and W.J. Leonard (2009). "New insights into the regulation of T cells by gamma(c) family cytokines." Nat Rev Immunol 9(7): 480-490.
• Rothschild, M.A., M. Oratz, D. Zimmon, S.S. Schreiber, I. Weiner, et al. (1969). "Albumin synthesis in cirrhotic subjects with ascites studied with carbonate-14C." J Clin Invest 48(2): 344-350.

References
87
• Rowley, J., A. Monie, C.F. Hung and T.C. Wu (2009). "Expression of IL-15RA or an IL-15/IL-15RA fusion on CD8+ T cells modifies adoptively transferred T-cell function in cis." Eur J Immunol 39(2): 491-506.
• Ruck, P. and J.C. Xiao (2002). "Stem-like cells in hepatoblastoma." Med Pediatr Oncol 39(5): 504-507.
• Ruckert, R., K. Asadullah, M. Seifert, V.M. Budagian, R. Arnold, et al. (2000). "Inhibition of keratinocyte apoptosis by IL-15: a new parameter in the pathogenesis of psoriasis?" J Immunol 165(4): 2240-2250.
• Rutkowski, M.D. and J.A. DeLeo (2002). "The Role of Cytokines in the Initiation and Maintenance of Chronic Pain." Drug News Perspect 15(10): 626-632.
• Sabatti, C., S.K. Service, A.L. Hartikainen, A. Pouta, S. Ripatti, et al. (2009). "Genome-wide association analysis of metabolic traits in a birth cohort from a founder population." Nat Genet 41(1): 35-46.
• Sadlack, B., H. Merz, H. Schorle, A. Schimpl, A.C. Feller, et al. (1993). "Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene." Cell 75(2): 253-261.
• Satoh, J., K. Kurohara, M. Yukitake and Y. Kuroda (1998). "Interleukin-15, a T-cell growth factor, is expressed in human neural cell lines and tissues." J Neurol Sci 155(2): 170-177.
• Schluns, K.S., K.D. Klonowski and L. Lefrancois (2004a). "Transregulation of memory CD8 T-cell proliferation by IL-15Ralpha+ bone marrow-derived cells." Blood 103(3): 988-994.
• Schluns, K.S., E.C. Nowak, A. Cabrera-Hernandez, L. Puddington, L. Lefrancois, et al. (2004b). "Distinct cell types control lymphoid subset development by means of IL-15 and IL-15 receptor alpha expression." Proc Natl Acad Sci U S A 101(15): 5616-5621.
• Schluns, K.S., K. Williams, A. Ma, X.X. Zheng and L. Lefrancois (2002). "Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells." J Immunol 168(10): 4827-4831.
• Sell, H., C. Habich and J. Eckel (2012). "Adaptive immunity in obesity and insulin resistance." Nat Rev Endocrinol 8(12): 709-716.
• Shinozaki, M., J. Hirahashi, T. Lebedeva, F.Y. Liew, D.J. Salant, et al. (2002). "IL-15, a survival factor for kidney epithelial cells, counteracts apoptosis and inflammation during nephritis." J Clin Invest 109(7): 951-960.
• Silva, W.A., Jr., D.T. Covas, R.A. Panepucci, R. Proto-Siqueira, J.L. Siufi, et al. (2003). "The profile of gene expression of human marrow mesenchymal stem cells." Stem Cells 21(6): 661-669.
• Smith, B.W. and L.A. Adams (2011). "Nonalcoholic fatty liver disease and diabetes mellitus: pathogenesis and treatment." Nat Rev Endocrinol 7(8): 456-465.
• Stanley, J.C. (1981). "The regulation of glucose production. The role of liver glycogen and gluconeogenesis in the liver and kidney cortex." Br J Anaesth 53(2): 137-146.
• Steel, J.C., C.A. Ramlogan, P. Yu, Y. Sakai, G. Forni, et al. (2010). "Interleukin-15 and its receptor augment dendritic cell vaccination against the neu oncogene through the induction of antibodies partially independent of CD4 help." Cancer Res 70(3): 1072-1081.
• Stoklasek, T.A., K.S. Schluns and L. Lefrancois (2006). "Combined IL-15/IL-15Ralpha immunotherapy maximizes IL-15 activity in vivo." J Immunol 177(9): 6072-6080.
• Stonier, S.W. and K.S. Schluns (2010). "Trans-presentation: a novel mechanism regulating IL-15 delivery and responses." Immunol Lett 127(2): 85-92.
• Su, G.L. (2002). "Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation." Am J Physiol Gastrointest Liver Physiol 283(2): G256-265.

References
88
• Su, Y.C., C.C. Lee and J.T. Kung (2010). "Effector function-deficient memory CD8+ T cells clonally expand in the liver and give rise to peripheral memory CD8+ T cells." J Immunol 185(12): 7498-7506.
• Sun, K., C.M. Kusminski and P.E. Scherer (2011). "Adipose tissue remodeling and obesity." J Clin Invest 121(6): 2094-2101.
• Sun, R. and B. Gao (2004). "Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-gamma)." Gastroenterology 127(5): 1525-1539.
• Sung, P.S., V. Racanelli and E.C. Shin (2014). "CD8 T-Cell Responses in Acute Hepatitis C Virus Infection." Front Immunol 5: 266.
• Surh, C.D. and J. Sprent (2008). "Homeostasis of naive and memory T cells." Immunity 29(6): 848-862.
• Swain, M.G. (2008). "Hepatic NKT cells: friend or foe?" Clin Sci (Lond) 114(7): 457-466. • Syn, W.K., Y.H. Oo, T.A. Pereira, G.F. Karaca, Y. Jung, et al. (2010). "Accumulation of
natural killer T cells in progressive nonalcoholic fatty liver disease." Hepatology 51(6): 1998-2007.
• Tailleux, A., K. Wouters and B. Staels (2012). "Roles of PPARs in NAFLD: potential therapeutic targets." Biochim Biophys Acta 1821(5): 809-818.
• Tajiri, K., Y. Shimizu, K. Tsuneyama and T. Sugiyama (2009). "Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease." Eur J Gastroenterol Hepatol 21(6): 673-680.
• Takeshita, T., H. Asao, K. Ohtani, N. Ishii, S. Kumaki, et al. (1992). "Cloning of the gamma chain of the human IL-2 receptor." Science 257(5068): 379-382.
• Tapia, N.C., F.I. Avila, J.G. Leiva, M.H. Ramos, J.F. Avila, et al. (2006). "[Therapeuric aspects of NAFLD. A literature review]." Rev Gastroenterol Mex 71(4): 487-495.
• Tolman, K.G. and A.S. Dalpiaz (2007). "Treatment of non-alcoholic fatty liver disease." Ther Clin Risk Manag 3(6): 1153-1163.
• Tosello-Trampont, A.C., S.G. Landes, V. Nguyen, T.I. Novobrantseva and Y.S. Hahn (2012). "Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production." J Biol Chem 287(48): 40161-40172.
• Uhlin, M., M.G. Masucci and V. Levitsky (2005). "Regulation of lck degradation and refractory state in CD8+ cytotoxic T lymphocytes." Proc Natl Acad Sci U S A 102(26): 9264-9269.
• Van Belle, T. and J. Grooten (2005). "IL-15 and IL-15Ralpha in CD4+T cell immunity." Arch Immunol Ther Exp (Warsz) 53(2): 115-126.
• Van Braeckel-Budimir, N. and J.T. Harty (2014). "CD8 T-cell-mediated protection against liver-stage malaria: lessons from a mouse model." Front Microbiol 5: 272.
• van der Windt, G.J., B. Everts, C.H. Chang, J.D. Curtis, T.C. Freitas, et al. (2012). "Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development." Immunity 36(1): 68-78.
• Vega, R.B., J.M. Huss and D.P. Kelly (2000). "The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes." Mol Cell Biol 20(5): 1868-1876.
• Voet, J.G. and D. Voet (2000). "Biochemistry and Molecular Biology Education (BAMBEd)." Biochem Educ 28(3): 124.

References
89
• Waldmann, T.A. (2006). "The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design." Nat Rev Immunol 6(8): 595-601.
• Wang, X., M. Rickert and K.C. Garcia (2005). "Structure of the quaternary complex of interleukin-2 with its alpha, beta, and gammac receptors." Science 310(5751): 1159-1163.
• Weisberg, S.P., D. McCann, M. Desai, M. Rosenbaum, R.L. Leibel, et al. (2003). "Obesity is associated with macrophage accumulation in adipose tissue." J Clin Invest 112(12): 1796-1808.
• Wheeler, M.C. and N. Gekakis (2014). "Hsp90 modulates PPARgamma activity in a mouse model of non-alcoholic fatty liver disease." J Lipid Res.
• Wilkinson, P.C. and F.Y. Liew (1995). "Chemoattraction of human blood T lymphocytes by interleukin-15." J Exp Med 181(3): 1255-1259.
• Wolowczuk, I., C. Verwaerde, O. Viltart, A. Delanoye, M. Delacre, et al. (2008). "Feeding our immune system: impact on metabolism." Clin Dev Immunol 2008: 639803.
• Wu, L., V.V. Parekh, C.L. Gabriel, D.P. Bracy, P.A. Marks-Shulman, et al. (2012). "Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice." Proc Natl Acad Sci U S A 109(19): E1143-1152.
• Wu, Z., H.H. Xue, J. Bernard, R. Zeng, D. Issakov, et al. (2008). "The IL-15 receptor {alpha} chain cytoplasmic domain is critical for normal IL-15Ralpha function but is not required for trans-presentation." Blood 112(12): 4411-4419.
• Yajima, T., H. Nishimura, R. Ishimitsu, K. Yamamura, T. Watase, et al. (2001). "Memory phenotype CD8(+) T cells in IL-15 transgenic mice are involved in early protection against a primary infection with Listeria monocytogenes." Eur J Immunol 31(3): 757-766.
• Yang, L., S. Thornton and A.A. Grom (2002). "Interleukin-15 inhibits sodium nitroprusside-induced apoptosis of synovial fibroblasts and vascular endothelial cells." Arthritis Rheum 46(11): 3010-3014.
• Yano, S., M. Komine, M. Fujimoto, H. Okochi and K. Tamaki (2003). "Interleukin 15 induces the signals of epidermal proliferation through ERK and PI 3-kinase in a human epidermal keratinocyte cell line, HaCaT." Biochem Biophys Res Commun 301(4): 841-847.
• Zamara, E., S. Galastri, S. Aleffi, I. Petrai, M. Aragno, et al. (2007). "Prevention of severe toxic liver injury and oxidative stress in MCP-1-deficient mice." J Hepatol 46(2): 230-238.
• Zhan, Y.T. and W. An (2010). "Roles of liver innate immune cells in nonalcoholic fatty liver disease." World J Gastroenterol 16(37): 4652-4660.
• Zhang, Y., L.W. Castellani, C.J. Sinal, F.J. Gonzalez and P.A. Edwards (2004). "Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism by activation of the nuclear receptor FXR." Genes Dev 18(2): 157-169.
• Zieve, L. (1979). "Amino acids in liver failure." Gastroenterology 76(1): 219-221.

















