UNIVERSITY OF NOVA GORICA GRADUATE SCHOOL ROLE OF AUTOPHAGY AND ITS ENHANCING IN THE CLEARANCE OF TDP-43 AGGREGATES DISSERTATION Naj e t e Safini Mentor: Dr.Sergio G. Tisminetzky Nova Gorica, 2014
Transcript
UNIVERSITY OF NOVA GORICA GRADUATE SCHOOL
R O L E O F A U T OPH A G Y A ND I TS E N H A N C IN G IN T H E C L E A R A N C E O F T DP-43 A G G R E G A T ES
DISSERTATION
Najete Safini
Mentor: Dr.Sergio G. Tisminetzky
Nova Gorica, 2014
UNIVERZA V NOVI GORICI
FAKULTETA ZA PODIPLOMSKI !TUDIJ
VLOGA AVTOFAGIJE IN NJEN VPLIV NA IZBOLJ!ANJE
"I!"ENJA AGREGATOV TDP-43
DISERTACIJA
Najete Safini
Mentor: dr. Sergio G. Tisminetzky
Nova Gorica, 2014
2
A BST R A C T
Neurodegenerative diseases, such as Amyotrophic Lateral Sclerosis (ALS) and
Fronto-Temporal Lobar Degeneration (FTLD), are characterized by imbalance
between generation and degradation of misfolded TDP-43 protein, resulting in the
formation of cytoplasmic inclusions followed by a loss of nuclear TDP-43 in affected
neuronal cells. Previously established cell-based TDP-43 aggregation model, where
tandem repeats carrying the C-terminal Gln/Asn-rich region of TDP-43 (EGFP-
12XQ/N) were able to trigger the formation of phosphorylated and ubiquitinated
aggregates, have been used in this study to investigate the autophagy-endolysosomal
cell pathway response. Cotransfection with EGFP-12xQ/N and HcRed-hLC3 (LC3
Light chain 3is marker of autophagosome and autophagy induction), immunostaining
with anti-p62 (p62 is an autophagy adaptor) and anti-Lamp1 (Lamp1 is marker of
late endosome/lysosome marker) show that EGFP-12xQ/N is able to generate
cytoplasmic aggregates that have the ability to induce an autophagy-endolysosomal
pathway cell response. Observed autophagic response incidence in EGFP-12XQ/N
aggregates led us to try Dextran Sulfate 5000 Da (DS), previously associated with
the autophagy induction, as a therapeutic effector aimed at reducing the aggregation
phenomenon. We found that DS treatment enhances inclusions clearance and
significantly increase the proportion of the LC3-positive aggregates, suggesting an
improved autophagic response of the cell. In addition, it was shown that DS
decreases the amount of flag-TDP43 wild type in the insoluble fraction suggesting its
release from the aggregates sequestration. Observed DS-triggered clearance is in
correlation with the increased LC3-II/LC3-I ratio, as well as with the significant p62
decay in the treated cells. It has been demonstrated that DS-induced EGFP-12xQ/N
clearance was abolished in an autophagy-deficient cells confirming DS clearing
potential through the autophagy pathway activation. Furthermore, we have identified
the histone deacetylase-6 (HDAC6), as an important player of DS clearance action.
Indeed, DS treatment failed to clear EGFP-12xQ/N in the HDAC6 deficient cells,
while by re-introducing HDAC6 wild type protein DS-triggered clearance has been
restored. Moreover, it has been demonstrated that DS-induced EGFP-12xQ/N
product decay requires catalytically active HDAC6.
Our study suggests that DS may help the cell to clear TDP-43 aggregates, in this
model, through the autophagy resulting in the more efficient cargo degradation in
3
HDAC6-dependent manner, thus revealing DS therapeutic potential in TDP-43
proteinopathies.
K eywords: Dextran sulfate (DS), Autophagy, TDP-43 proteinopathies, Amyotrophic Lateral
Sclerosis (ALS) and Frontotemporal Lobar Degeneration (FTLD).
4
Izvleček
Nevrodegenerativna obolenja, kot so amiotrofična lateralna skleroza (ALS) in
frontotemporalna lobarna degeneracija (FTLD), so posledica neravnovesja med
tvorbo in degradacijo nepravilno zvitega TDP-43 proteina, kar vodi do kopičenja
TDP-43 v citoplazmi v obliki vključkov in izgube v jedrih prizadetih nevronskih
celic. V raziskavah sem za študij odgovora avtofagne-endolizosomalne celične poti
uporabila predhodno uveljavljen celični model tvorbe agregatov TDP-43, kjer
skleroza (ALS) in frontotemporalna lobarna degeneracija (FTLD).
6
A C K N O W L E D G M E N TS
I would like to express my gratitude to my Thesis mentor, Dr. Sergio Tisminetzky,
for allowing me to work on this project in his lab and for his great supervision and
support throughout my PhD experience.
A very special thank go out to Dr. Natasa Skoko, for her understanding, and
patience. I appreciate her vast knowledge and skill in many areas, and her close
assistance on the bench and also in writing reports (i.e., proposals, and this
thesis),which without her this work would have never happened.
Prof. Francesco Baralle, and Dr. Marco Baralle for allowing me the opportunity to
use their aggregation model (EGFP-12xQ/N construct), and for their very helpful
discussions and suggestions.
The members of the commity for reading carefully this thesis and offering their
precious time to make my dissertation better. I really appreciate the time you have
taken to evaluate this work.
I am deeply grateful to all the current biotechnology development group members for
their encouragement, support, and help.
I would like to thank Mojca Tajnik for the translation of my abstract to Slovinien,
Thank you so much (hvala).
Thanks to Dr. Maurizio Budini for the pEGFP-12xQ/N construct, for Dr. Francesca
Arnoldi for the siATG7, for Dr Francesca DeMarchi and Dr Isei Tanida for the
HcRed-LC3 plasmid.
I would like to extend my acknowledgments to all wonderful people and friends I
had the honor to meet them during my stay in ICGEB. Especially, mio Sylvia
DellaMea and her family, Fatemeh, Cristiana, Maureen, and Betul. Your support and
friendship have been priceless.
7
And finally, I would like to thank deeply my parent, and sisters for their
unconditional love and support, which made all of this possible. And also, for my ex-
boyfriend Mohammad, you were really a good person that marked my life and will
stay in my heart, thanks for your support, I will never forget that.
8
Abbreviations ALS Amyotrophic lateral sclerosis AD Alzheimer’s disease ALIS Aggresome-like inducible structures ALS Amyotrophic lateral sclerosis a-Syn alpha-Synuclein Atg Autophagy-related genes CMA Chaperone-mediated autophagy Cvt Cytoplasm to vacuole targeting ESCRT Endosomal sorting complex required for transport FIP200 Focal adhesion kinase family interacting protein of 200 kDa FUS/TLS fused in sarcoma/translocated in liposarcoma HD Huntington’s diseases HDAC6 Histone deacetylase 6 HDL high-density lipoprotein KO Knock out LAMP2A Lysosome-associated membrane protein type-2A LDL low-density lipoprotein LC3 Light chain 3 LIR LC3-interacting region mAtg Mammalian Autophagy geneMTOC Microtubule-organizing centre MVB Multivesicular body NBR1 Neighbour of BRCA1 gene 1 NES Nuclear export sequence ROS Oxygene species SCA Spinocerebellar ataxia SOD1 Cu/Zn superoxide dismutase 1 SQSTM1 Sequestome 1 TOR Target of rapamycin TORC1/2 TOR complex 1 or 2 TARDBP TDP-43 Tar DNA-binding protein UBA Ubiquitin-associated ULK1 Unc-51-like kinase 1 UPS Ubiquitin-proteasome system UVRAG Ultraviolet irradiation resistance-associated gene VCP Valosin-containing protein VLDL very low-density lipoprotein
9
C O N T E N TS
A BST R A C T ....................................................................................... 2
3.9. DS-induced E G FP-12xQ /N clearance requires H D A C6 ...... 99
3.10. Mouse motor neuronal-like cell line NSC-34 model of
E G FP-12xQ/N aggregation .......................................................... 103
3.12. Dextran sulfate 5000 Da is not toxic for the motor
neuronal-like cell line NSC-34 ..................................................... 104
4. D ISC USSI O N ............................................................................ 106
5.C O N C L USI O NS ........................................................................ 119
6. B IB L I O G R APH Y .................................................................... 120
12
L ist of figures
F igure 1. Intracellular protein degradation via two main proteolytic systems: the ubiquitin-proteasome system (UPS) and the lysosomal autophagy pathway………18 F igure 2. Schematic representation of macroautophagy……………………..........20 F igure 3. Molecular pathway of autophagy initiation……………………………..23 F igure 4. Autophagy elongation………………………...........................................25 F igure 5. Atg12 and Atg8/LC3 ubiquitin-like conjugation pathways required for autophagosome formation……………………………………………………….....25 F igure 6. Molecules involved in the maturation step of autophagy………………..28 F igure 7. Aggrephagy main players ……………………………………………......34 F igure 8. TDP-43 proteinstructure…………………………..…………………….43 F igure 9. Chemical structure of dextran sulfate……………………………………56 F igure 10. EGFP-12x Q/N aggregates are labeled with LC3, p62 and Lamp1……75 F igure 11. Dextran sulfate 5000 Da decreases the number of cells containing EGFP-12xQ/N aggregates………………………………………………………………….78 F igure 12. Positive trend in autophagic response to the aggregation upon DS treatment………………………………………………………………….................80 F igure 13. EGFP-12xQ/N aggregates formed in HEK293 Flp-In T-Rex stable cell line after tetracycline induction…………………………………………………… 82 F igure 14. Dextran sulfate 5000 Da (DS) enhances the clearance of EGFP-12xQ/N aggregates…………………………………………………………………...............86 F igure 15. DS enhances the clearance of EGFP-12xQ/N aggregates…………….. 85 F igure 16. DS enhances the clearance of EGFP-12xQ/N aggregates through the autophagy……………………………………………………………………………87 F igure 17. Autophagy inhibition with siRNA against Atg7 counteracts the clearance action of DS…………………………………………………………………………89 F igure 18. EGFP-12xQ/N aggregates colocalize with autophagy marker LC3 in stable cell line. …………………………………………………………………… 90
13
F igure 19. EGFP-12xQ/N aggregates may entrap endogenous TDP-43 and flag-TDP-43 wild type…………………………………………………………………95 F igure 20. EGFP-12xQ/N aggregation model is able to produce truncation products of TDP-43…………………………………………………………………………94 F igure 21. DS decreases the capture of flag-TDP43 wild type in the insoluble fraction……………………………………………………………………………..95 F igure 22. DS enhances EGFP-12xQ/N aggregates clearance and decreases TDP-43 wild type retention in the cytoplasm………………………………………………97 F igure 23. DS is decreasing acetylated tubulin level in HEK293 cells forming EGFP-12xQ/N aggregates…………………………………………………………99 F igure 24. DS action on EGFP-12xQ/N clearance is HDAC6 dependent……….101 F igure 25. EGFP-12xQ/N aggregates are co-localizing with autophagosome marker LC3………………………………………………………………………………..102 F igure 26. Dextran sulfate 5000 Da is not toxic for the neuronal-like cell line NSC-34…………………………………………………………………………………..104 Tables Table 1.Some genes and loci for familial ALS ……………………………………40 Table 2.Chemical structure and mechanism of action of compounds reported to treat ALS disease ………………………………………………………………………..53
14
1. IN T R O DU C T I O N
1.1. Protein degradation
Eukaryotic cells are continuously synthesizing proteins in order to endure, to
survive, and to maintain proper homeostasis. In physiological conditions, cells need
to eliminate misfolded/unfolded proteins. However, an imbalance between synthesis
and mis/unfolded proteins degradation is frequently observed. Cells accumulating
unfolded proteins have an impaired internal homeostasis that leads subsequently to
their death. Thus, it is essential to identify the mechanisms behind protein formation
and degradation, in order to propose a specific therapeutic strategy to target disease
which is characterized by an abnormal accumulation of mis/unfolded proteins.
Since early 1898, Hahn and colleagues have described cellular proteolytic activities,
using yeast(Hahn 1898). Later on, De Duve and Novikoff were able to obtain the
first electron micrographs of the partially purified hepatic lysosomes and have shown
the localization of acid phosphatase activity in these organelles(Essner E. &
Novikoff 1961).In the following years, researchers have studied different types of
cells using electron microscope and discovered a wide variety of vesicles involved in
the proteolytic pathway of the cell. As some of the vesicles contained engulfed
cytoplasmic material, it was suggested that these particular vesicles were pre-
lysosomes. These pre-lysosomes were found to be formed de novo in the cytoplasm
from a cup-shaped membrane called a phagophore. The extremities of the
phagophore expand while becoming spherical until they seal, enclosing the engulfed
pieces of cytoplasm with whatever might lie inside, and giving rise to a double-
membrane vesicle. Farquhar, for the first time, observed these closed vesicles, which
are later on called autophagosomes. Autophagosomes take up damaged molecules or
organelles and carry this cargo to the lysosomes. When De Duve observed
autophagosomes, he realized that cells could degrade their own components and
named the process "autophagy" (Smith & Farquhar 1966; De Duve Christian 1965).
Few years later, it was shown that the degradation of the proteins can happen not
only inside lysosomes, but also outside these structures in an energy-dependent
manner through the proteasomes(Coux et al. 1996; Hilt & Wolf 1996; Hochstrasser
1995; Peters 1994; Rubin & Finley 1995).
15
Autophagy seems to be active in the degradation of long-lived proteins and was in
the beginning identified as a bulk degradation mechanism for protein turnover during
periods of nutrient starvation (Klionsky & Emr 2000). In contrast to autophagy,
degradation depending on proteasome removes selectively ubiquitinated, and
aberrant short-lived proteins(Heinemeyer et al. 1991), thus it is denoted as the
ubiquitin-proteasome system (UPS). Since many of these short-lived proteins have
essential cellular regulatory roles; UPS plays a major role in different cellular
activities such as in the regulation of the cell cycle, and gene expression, in response
to variant cellular stresses, and in apoptosis (Attaix et al. 2001; Hershko et al. 2000).
Nevertheless, both UPS and autophagy are involved in the removal of misfolded
protein that fails to be refolded by chaperones, as well as different cytoplasmic
cargoes under various conditions (Figure 1).
1.1.1 UPS pathway:
The UPS machinery is composed of the proteasome and the enzymatic cascade,
through which the targeted ubiquitinated substrates are catalysed for the degradation.
First of all, the unfolded protein is tagged for proteasomal degradation with a chain
of 4 or more ubiquitin units by ubiquitin ligases. Ubiquitin (Ub) is a highly
conserved protein of 76 amino acids that is covalently linked to lysine residue(s) of
the targeted protein. Further on, these units form polyubiquitin chains that are bound
to each other through seven internal lysines (K6, K11, K27, K29, K33, K48, and
K63). It was reported that degradation by the UPS depends mainly on K48-linked
polyubiquitination of the targeted misfolded substrate(Komander 2009). Although
K48 represent the canonical proteasomal degradation tag, yet some substrates tagged
with K63-linked polyubiquitin can be also degraded through the
proteasome(Komander 2009). Next, these tagged substrates are delivered to the
proteasome where they will be degraded to short peptides and reusable ubiquitin.
The proteasome that is exclusively used in mammals is the cytosolic 26S
proteasome. It is a barrel-shaped structure with a molecular weight of about 2000
kDa, containing one 20S proteolytic core particle and two 19S regulatory particles.
26S proteasome structure has two openings at the ends where the target protein is
allowed to enter. 19S regulatory particle contains at least 18 subunits with the
16
multiple ATPase active sites and ubiquitin binding sites. This structure is responsible
for the recognition of the polyubiquitinated proteins, their unfolding and
translocation into the catalytic core. Catalytic core is composed of four rings that
form a central pore. The two inner rings are made of seven β subunitsthat contain
three to seven protease active sites. The target protein must enter the central pore
through these sites to be degraded. The two other outer rings have seven α
subunitswhose function is to maintain the pore entry through which proteins enter the
barrel (Komander 2009).
Autophagy (discussed fully in the following chapter) and UPS are both critical in the
maintenance of cellular homeostasis suggesting that their activity should be highly
orchestrated. Nevertheless, it was reported a complex and often an unexpected
interplay between these two cellular waste conveyors.
The narrow size of the proteasomal catalytic pore suggests that protein substrates
need to be partially-unfolded prior to their entry into the proteasome. Thus, protein
complexes and aggregates that have bigger size are reported to be poor proteasome
substrates (Nandi et al. 2006).In contrast to the UPS, macroautophagy is capable of
degrading a much wider spectrum of substrates, which, on average, tend to be
longer-lived and bulkier. These substrates include misfolded soluble proteins, protein
complexes, and aggregates. Ubiquitination appears to be a universal tag targeting
substrates for destruction via both catabolic systems, yet the exact type of
modification recognised by each pathway appears to be different. While K48-linked
polyubiquitin chains are employed by the UPS, substrates recognised by
autophagosome-lysosome pathway are thought to be modified either by K63-linked
chains (adopting a more open conformation than K48 chains), or might be just
monoubiquitylated (Welchman et al. 2005).
Several studies have focused on how changes in the activity of one of the degradative
pathways affect the flux through the other system (Ding et al. 2007).
Impairment of the UPS leads to an increase of the autophagic function.
Autophagy here is considered as a compensatory mechanism, since it is
allowing cells to reduce the accumulated UPS substrates. Indeed, in cells and
17
mice, where proteasome activity was inhibited using lactacystin, it was
shown that treating these cells and mice with rapamycin (inducer of
autophagy) tends to protect them against cell death by reducing the
accumulated UPS substrates (Pan et al. 2008). It was also shown that
upregulation of autophagy has a protective role when the UPS is impaired in
Drosophila (Pandey et al. 2007). Moreover, this protective effect is reported
to be HDAC6 (histone deacetylase 6) dependent (Pandey et al. 2007; Iwata et
al. 2005). However, the role of HDAC6 in this process is to ensure an
efficient delivery of autophagic machinery substrates for degradation.
HDAC6 was earlier found to regulate the formation of ubiquitinated inclusion
bodies, called aggresomes (see further in aggrephagy session) (Kawaguchi et
al. 2003). These aggresomes has been hypothesised to be formed in order to
be degraded more efficiently by autophagy (Iwata et al. 2005). These
molecular mechanisms may not be mutually exclusive and may be of
different importance in different cell types after the proteasome inhibition.
Thus more studies have to be done to identify different players connecting
two cellular proteolytic pathways.
On the other hand, when autophagy is inactivated in vivo and in vitro by the
knockout of essential autophagic genes (Atg5 or Atg7), significant
accumulation and aggregation of ubiquitinated proteins was reported (Hara et
al. 2006; Komatsu et al. 2006).Autophagy pathway clearance impairment
leads to the dysfunctional UPS. Indeed, several studies support this claim, as
it was shown that impaired autophagy also leads to the impaired degradation
of specific UPS substrates (Korolchuk, Mansilla, et al. 2009)
Decreased UPS flux in autophagy-compromised cells was not due to
impaired catalytic activity of proteasomes isolated from these cells, but due to
the accumulation of p62 protein that acts as an autophagy receptor and
connects ubiquitinated protein aggregates to the autophagic machinery.In
fact, when p62 was knocked down, the levels of UPS substrates in these
autophagy-deficient cells was rescued(Korolchuk, Menzies, et al. 2009).
Thus, lack of compensation for autophagy dysfunction by the UPS is in
agreement with the fact that p62, when accumulates, oligomerizes and is
therefore too bulky to be a good substrate for the proteasome with its narrow
18
catalytic pore. Yet, further studies are recommended to be done in order to
understand more, the molecular cross talk between the two main degradative
pathways.
F igure 1. Intracellular protein degradation via two main proteolytic systems: the ubiquitin-proteasome system (UPS) and the lysosomal-autophagy pathway. Delivery of cytoplasmic material to the lysosomes by autophagy can occur by three different pathways: (1) macroautophagy, which involves the sequestration of cytoplasmic components by a membrane forming an autophagosome, which fuses with the lysosome (2) microautophagy, which involves engulfment of small volumes of cytoplasm by a direct invagination of the lysosomal membrane (3) Chaperone-Mediated Autophagy (C M A), a process by which soluble substrates associated with a specific chaperone complex are translocated into the lysosome through the LAMP-2A lysosomal receptor. Proteins tagged with the polyubiquitin chain can be targeted by both the UPS and autophagy. Adapted from (Nedelsky et al. 2008).
19
1.1.2 Autophagy:
The term autophagy (originated from Greek word: auto phagin) means self-eating or
self-cannibalism. The term “autophagy” was first introduced by Christian De Duve
in mid-sixties to define the digestion of endogenous cellular material. Autophagy is
an evolutionarily conserved process through which the intracellular cytoplasmic
material (cargo) is delivered to lysosomes for degradation. Three major types of
autophagy in eukaryotes are known: chaperone-mediated autophagy (CMA),
microautophagy and macroautophagy (Figure 1).
Chaperone-mediated autophagy (C M A)Two main properties differentiate
chaperone-mediated autophagy (CMA) from the other types of autophagy in
mammalian cells: (1) the selectivity towards a particular pool of cytosolic proteins
and (2) the mechanism of delivery of the substrate proteins to the lysosomes. Mostly,
proteins bearing a targeting motif in their amino acid sequence, biochemically related
to the pentapeptide KFERQ, are selectively recognized by the Heat shock cognate
protein of 70 kDa (Hsc70), the chaperone that mediates their delivery to lysosomes
for degradation via CMA. It is estimated that about 30% of soluble cytosolic proteins
contain this CMA-targeting motif. After this targeting step, the substrate protein–
chaperone complex enclosures at the lysosomal membrane through interaction with
the cytosolic end the lysosome associated membrane protein type 2A (LAMP-2A),
which acts as a receptor for this autophagic pathway. Translocation of the substrate
across the lysosomal membrane also requires the presence of a luminal form of
Hsc70 (lysHsc70). After translocation, substrate proteins are rapidly degraded by the
abundant array of lysosomal hydrolases(Chiang et al. 1989; Cuervo 2010; Cuervo &
Dice 1996).
Microautophagy was first described in 1966 by De Duve and Wattiaux (De Duve
& Wattiaux 1966). Microautophagy is the non-selective lysosomal degradative
process. This process translocates cytoplasmic materials into the lysosome or
endosomes for degradation by direct invagination, protrusion, or septation of the
lysosomal or vacuolar membrane (Ahlberg & Glaumann 1985).With its constitutive
characteristics, microautophagy of soluble substrates can be induced by amino-acids
starvation or rapamycin via regulatory signaling complex pathways (Dubouloz et al.
2005). The maintenance of the organelles size, membrane homeostasis, and cell
survival under nutrients restriction are the main functions of microautophagy. In
20
addition, microautophagy is coordinated and complements macroautophagy and
chaperone-mediated autophagy (Dubouloz et al. 2005; Mijaljica et al. 2011). The
exact mechanism and molecular core machinery involved in the microautophagy in
mammalian cells is still unknown. Reviewed in Ref.(Li et al. 2012).
Macroautophagy, referred hereafter as autophagy, is the most studied form of
autophagy. Autophagy is a process of self-degradation of cellular components in
which double-membrane autophagosomes sequester organelles or portions of cytosol
and fuse with lysosomes or vacuoles for degradation. Autophagy is induced in
response to extra- or intracellular stress and signals such as starvation, growth factor
deprivation, ER stress, and pathogen infection. Nevertheless, autophagy can also be
observed at basal level in the cell, since cells have to maintain its integrity by
selectively removing unfolded proteins, damaged organelles, or invasive pathogens.
Cargo-specific names have been given to describe these various forms of selective
autophagy (e.g. mitophagy and pexophagy for the selective degradation of damaged
mitochondria and peroxisomes, respectively) (Klionsky et al. 2007; Mizushima et al.
2008).
F igure 2:Schematic representation of macroautophagy. Autophagosomes form from a pre-autophagic structure (phagophore) of unknown origin. The phagophore extends to envelop cytoplasmic constituents and damaged organelles. This structure elongates generating autophagosomes that are directed to the endocytic/lysosomal compartments that form autolysosome where engulfed constituents are degraded.
21
The most typical inducer of autophagy is nutrient starvation; in this sense, lack of
any type of essential nutrient can induce autophagy. In yeast, nitrogen starvation is
the most potent stimulus, but withdrawal of other essential factors such as carbon,
auxotrophic amino acids and nucleic acids, and even sulphate can induce autophagy.
In mammals, it is well known that serum starvation can induce autophagy in many
types of cultured cell. Amino acid and insulin/growth factor signals are thought to
turn on mTOR (mammalian target of rapamycin), which is a master regulator of
nutrient signalling (Yorimitsu & Klionsky 2005). Indeed, treatment with inhibitors of
mTOR such as rapamycin induces autophagy. In addition to insulin and amino acid
signaling, the involvement of many other factors in autophagy regulation has
recently been reported. These include Bcl-2, reactive oxygen species (ROS), AMP-
activated protein kinase (AMPK), and myo-inositol-1,4,5-triphosphate (IP3).
More than 30 different genes regulating autophagy (Atgs) have been identified in
yeast, and many of these have mammalian orthologs. These genes are involved at
key stages of the autophagy pathway: initiation, elongation, and maturation/ fusion
with the lysosomes (Figure 2)(Harding 1995; Thumm et al. 1994; Tsukada &
Ohsumi 1993).
A . Initiation of autophagy
Autophagy is initiated by the formation of an essential vesicle that is typical for
autophagy: the autophagosome. Membrane dynamics during autophagy are highly
conserved from yeast to plants and animals. Autophagy initiation starts by the
autophagosome formation, where cytoplasmic constituents, including organelles are
sequestered by a unique membrane called the phagophore or isolation membrane.
Where and how autophagosomes emerge has been a major question. It has been
hypothesized that autophagosomes can either be generated de novo from pre-existing
intracellular precursor molecules, or could arise from other intracellular
membranestructures like the endoplasmic reticulum (ER). The latter hypothesis has
recently been supported by more evidence suggesting that ER could contribute to
autophagosome formation, as an accumulation of vesicles associated with the ER
22
outer surface was seen in cells that had a block in maturation of pre-autophagosomal
structures. Thus, ER may be one of the membrane sources contributing to the
formation of pre-autophagosomal structures, so-called phagophores.The formation of
new autophagosomes requires the activity of (1) the class III phosphatidylinositol 3-
kinase complex (PI3K) (also known as Vps34/Beclin1 complex), and (2) FIP200-
ULK1/Atg1 complex (Figure 3).
Vps34 is the only identified PI3K in yeast so far, and its essential role in vacuolar
protein delivery was initially described through yeast genetics studies (Herman &
Emr 1990). The essential role of Vps34 in autophagy has been established largely
through the use of the pharmacological inhibitors wortmannin and 3-methyladenine
(3-MA), which have been used to suppress autophagy in many studies.Moreover, it
was shown that inhibition of Vps34 activity with wortmannin leads to
autophagosome formation inhibition. Furthermore, Vps34 is activated upon its
interaction with Beclin 1 that is the orthologus gene of Atg6 in yeast and is one of the
main pro-autophagic genes. In fact, if Vps34/Beclin1 interaction is disrupted, the
autophagosome formation is impaired. Upon Vps34 and Beclin1 interaction, Vps34
phosphorylates a phosphatidylinositol. Phosphatidylinositol-3-phosphate (PI-3-P) is
the major product of this activity. PI-3-P plays an essential role in the early stages of
the autophagy pathway. Recent studies have identified strong colocalization of early
autophagosome markers in PI-3-P-enriched structures that were formed upon
starvation(Kim et al. 2013).
A second macromolecular complex associated with the initiation step of
autophagosome formation is the FIP200 -ULK1/Atg1 complex. Upon starvation,
Atg13 binds to the mammalian Atg1 homolog ULK1 and mediates their interaction
with FIP200, thereby forming a ULK1-Atg13-FIP200 stable complex that triggers
the autophagic response downstream of mTOR. Under nutrient-rich conditions,
mTORC1 suppresses autophagy through direct interaction with this complex and
mediates phosphorylation-dependent inhibition of the kinase activities of Atg13 and
ULK1. Under starvation conditions or rapamycin treatment, mTOR dissociates from
the complex, resulting in the inhibition of mTOR-mediated phosphorylation of Atg13
and ULK1. This leads to dephosphorylation-dependent activation of ULK1 and
23
ULK1-mediated phosphorylations of Atg13, FIP200, and ULK1 itself, which triggers
autophagy. Therefore, the ULK1-Atg13-FIP200 complex acts as an integrator of the
autophagy signals downstream of mTORC1 (Hosokawa et al. 2009). However, it is
not clear yet how phosphorylation of these proteins regulates their activities (Figure
3).
F igure 3. Molecular pathway of autophagy initiation:the first step of the
autophagy pathway is its initiation (shown inside the frame) the initiation step starts
when Vps34 interacts with Beclin 1, in one hand and with FIP200-ULK1 complex,
on the other hand initiating the phagophore formation, once sensing the presence of
autophagic cargos in the cytoplasm.
B . E longation
The elongation of the membrane sac called: isolation membrane involved many Atg
proteins. Atg9 is one of the proteins that plays an essential role in phagophore
elongation. Atg9 is a transmembrane protein that cycles between the trans-Golgi
network and endosomes, probably carrying membrane for expansion of phagophore
24
(Figure 4). Furthermore, it has been reported that this process requires different other
Atgs, that are involved and grouped in two ubiquitin-like reactions (Figure 5).
(1) In the first cascade of the reactions, the ubiquitin-like protein Atg12 is covalently
tagged to Atg5. Atg12 is first activated by Atg7 (E1 ubiquitin activating enzyme-
like) and then transferred to Atg10 (E2 ubiquitin conjugating enzyme-like). Atg12 is
then linked by its C-terminal glycine to an internal lysine residue of Atg5. The
Atg12-Atg5 later on forms a conjugate with Atg16L1 (Atg12-Atg5-Atg16L1),
resulting in an 800-kDa complex tetramers. This complex is essential for the
elongation of the pre-autophagosomal membrane, but it dissociates once the fully
autophagosomes are formed (Mizushima et al. 1998).
(2) The second cascade ubiquitin-like reaction involves the protein microtubule-
associated protein 1 light chain 3 (MAP1-LC3/LC3/Atg8). LC3 is synthesized as a
precursor form and is cleaved at its C- terminus by the protease Atg4B, resulting in
the cytosolic isoform LC3-I. LC3-I is conjugated to PhosphatidylEthanolamine (PE)
in a reaction involving Atg7 (E1-like) and Atg3 (E2-like) to form LC3-II. LC3-II is
specifically targeted to the elongating autophagosome membrane and, unlike the
Atg12-Atg5-Atg16L1 complex, remains on completed autophagosomes until fusion
with the lysosomes. After fusion LC3-II on the cytoplasmic face of autolysosomes
can be delipidated by Atg4 and recycled. In addition, LC3 is also found on the
internal surface of autophagosomes that is degraded in the autolysosomes. The
association of LC3-II with autophagosomes makes it a specific marker for studying
autophagy. Upon vesicle completion and engulfing the sequestered substrates,
autophagosomes are delivered to lysosomes in the maturation step.
25
F igure 5. A tg12 and Atg8/L C3 ubiquitin-like conjugation pathways
required for autophagosome formation. Atg4 encodes a cysteine protease that
cleaves Atg8/LC3. Atg7 is similar to an E1-like protein, and Atg10 and Atg3
encode E2-like proteins. Atg5, Atg12 and Atg16 are physically associated with
the isolation membrane, whereas Atg8/LC3 is directly conjugated to the lipid
PhosphatidylEthanolamine (PE) that is inserted in the isolation membrane.
Adapted from (Geng & Klionsky 2008).
F igure 4.Autophagy elongation:Atg9 exists already on the surface of circulating
vesicles inside cytoplasm, once the pre-autophagosomal structure is formed, Atg9
vesicles fuse with these structures, and subsequently Atg9 becomes integrated into
the outer autophagosomal membrane. The autophagosomal membrane elongation
requires also Atg8 (LC3) and the complex formed by Atg16/Atg5/Atg12.
26
C . Maturation and fusion
Nascent autophagosomes undergo a stepwise maturation, resulting in the creation of
amphisomes and autolysosomes by fusion with multiple endocytic compartments,
such as early endosomes, multivesicular bodies (MVBs), late endosomes, and
lysosome (Tooze et al. 1990). Amphisomes, an intermediate hybrid vesicular
compartment, contain both autophagosomal and endosomal contents, while
autolysosomes are formed either from amphisomes or directly from autophagosomes
by fusion with lysosomes.These degrading structures are generally called
“autolysosomes” or “autophagolysosomes.”
While in yeast the autophagosome fuses directly with the vacuole, in higher
eukaryotes the process seems to be more complicated. Endocytosis is a process
involving a simple invagination of plasma membrane when the extracellular material
is internalized, whereas, the fusion step of autophagy involves proteins such as
endosomal sorting complex ESCRT, Rab7 and UVRAG/VPS34 protein (Figure 5).
ESCRTs are necessary for formation of multivesicular bodies (MVBs) and they are
indeed involved in sorting ubiquitylated membrane proteins into multivesicular
bodies (Simonsen & Tooze 2009). UVRAG, a Beclin 1 interacting protein, is also
involved in the maturation step by regulating positively the maturation of both
autophagosomes and endosomes. This function is independent of its interaction with
Beclin 1 (an ortholog of Atg6 and is a positive regulator of autophagy). UVRAG
recruits the VPS34 and via this interaction activates Rab7. Rab7 is found to be
important in the maturation of the autophagosome (Gutierrez et al. 2004; Stein et al.
2005). It is involved in vesicle transport from early endosomes to late
endosomes/lysosomes as well as in the fusion between autophagosomes and
lysosomes. Moreover, Rab7 is associated with mature autophagosomes and
autolysosomes, as well as with LC3, which preferentially labels immature
autophagosomes. Indeed, Rab7 is not essential for the initial step of autophagosome
maturation, but is involved in the final step of the maturation of late autophagic
vacuoles, possibly in the fusion with lysosome.
In mammalian cells, autophagosomes/lysosomes fusion is facilitated by microtubules
and appears to necessitate dynein. While, in yeast autophagosome/vacuole fusion
seems not to require these structures (Aplin et al. 1992; Fass et al. 2006; Fengsrud et
27
al. 1995; Kirisako et al. 1999; Köchl et al. 2006; Punnonen & Reunanen 1990;
Ravikumar et al. 2005; Webb et al. 2004). In mammalian cells, the destabilization of
microtubules by either vinblastin or nocodazole blocks the maturation of
autophagosomes, whereas their stabilization by taxol increases the fusion between
autophagic vacuoles and lysosomes. Autophagosomes move bidirectionally along
microtubules. Their centripetal movement is dependent on the dynein motor.
Dyneins are the motors that move intracellular cargos (including organelles and
vesicles) along microtubule tracks. It was suggested that dyneins could simply act by
moving autophagosomes to perinuclear locations where the lysosomes are
concentrated. Yet, the exact mechanism by which dyneins facilitate
autophagosomes/lysosomes fusion require further studies.
Another molecule recently added to the core machinery of maturation step is histone
deacetylase-6 (HDAC6), an ubiquitin-binding deacetylase, which selectively targets
the ubiquitinated proteins to autophagosomes. It has been shown that knockdown of
HDAC6 leads to the accumulation of autophagosomes, suggesting that HDAC6
controls the autophagosomematuration rather than theautophagosome formation.
Moreover, it has been demonstrated that HDAC6 role in this step is to control the
actin cytoskeleton (Lee et al. 2010). Despite of such recent progress, molecular
mechanisms underlying coordinated regulation of multiple maturation steps by these
factors are still incompletely understood. The maturation step is ended by lysosomal
degradation of cargos. The autophagosome content is degraded inside autolysosome
structures that contain different hydrolytic enzymes, such as proteases, lipases, and
nucleases that are capable of breaking down all types of biological polymers.
Here, it is also essential to mention that lysosomal positioning is dynamically
regulated by nutritional conditions, hence by autophagy. Starvation induces
preferential re-localization of lysosomes from cell peripheries to juxtanuclear regions
close to the microtubule-organizing center (MTOC), thus regulating the autophagic
flux in cells (Maday et al. 2012). It is important to mention that in neurons,
bidirectional movements of lysosomes within axons are observed, while
autophagosomes and endosomes are also bidirectionally moved mainly in distal
axons. They are then transported exclusively in retrograde direction after fusion with
lysosomes to the cell body of the neuron for complete degradation of their cargos.
28
Taken together, lysosomal degradation of engulfed cargos in neurons could be
strictly dependent on retrograde transport and late autophagosome/lysosomal
trafficking (Katsumata et al. 2010).
F igure 6. Molecules involved in the maturation step of autophagy:
UVRAG interacts independently with Vps 34 and ESCRT complex to promote
autophagosome maturation that is later on fused either with endosome or lysosome to
generate autolysosome, UVRAG-Vps34 is also involved in the endosome–lysosome
transition by activation of Rab7. Adapted from (Liang et al. 2008).
1.1.2.1 Physiological role of Autophagy:
To understand the various roles of autophagy, it may be useful to subclassify
macroautophagy into “induced autophagy” and “basal autophagy” (Mizushima
2005). The former is used to produce amino acids following starvation, while the
latter is important for constitutive turnover of cytosolic components. However, this
distinction is too simplified and cannot be applied to more complicated issues.
Autophagy has a greater variety of physiological roles than expected, such as
starvation adaptation, intracellular protein and organelle clearance, development,
anti-aging, elimination of microorganisms, cell death, and the antigen presentation.
The physiological roles of autophagy are based on the respective processes involved
29
in, such as usage of degradation products, elimination of macromolecules and
organelles, and sequestration/packing.
A-U tilization of degradation products:
Under normal conditions and during very short periods of starvation, maintenance of
the amino acid pool seems to rely primarily on the ubiquitin–proteasome system
rather than autophagy (Vabulas & Hartl 2005). However, during starvation that
persists for several hours, necessary amino acids are produced by autophagy, which
is up-regulated as an adaptive response. It is important to emphasize that excess
production of amino acids by autophagy is an acute response or emergency action.
Therefore, induction of autophagy can support cell survival only for a short time. For
example, during cell growth, autophagy is activated at initial stages, but returns to
basal levels after a normal conditions are stablished (Degenhardt et al. 2006). In
contrast, little is known about how useful autophagy is in overcoming chronic
starvation.
B-Elimination of macromolecules and organelles:
The second purpose of autophagy is the elimination of cytoplasmic contents.
Although this role has been thought to be the specialty of the ubiquitin–proteasome
system, many recent studies have shown that autophagy also participates in
intracellular clearance or protein/organelle quality control. The most direct evidence
is the accumulation of abnormal proteins and organelles in autophagy-deficient
hepatocytes, neurons, and cardiomyocytes even in the absence of any
disease(Komatsu et al. 2005; Hara et al. 2006; Nakai et al. 2007), ubiquitin-positive
inclusion bodies, and deformed organelles accumulate in these cells. Since induced
autophagy is not observed in the brain during starvation, low levels of basal
autophagy are likely sufficient for quality control.Some types of induced autophagy
are aimed at the elimination of excess or unneeded organelles. For example,
peroxisomes induced by metabolic demand are selectively degraded primarily by
microautophagy(Sakai et al. 1998).The elimination of cytoplasmic contents by
autophagy is so important that defects cause various cellular malfunctions. One of
the possible outcome of autophagy defects is neurodegeneration. Indeed, The
accumulation of autophagic vacuoles has been observed in many human
30
neurodegenerative diseases, including Alzheimer’s disease for instance (Okamoto et
al. 1991). It remains largely unknown whether these represent up-regulation of
autophagy or blockage of autophagic flux.
C-Sequestration/packing:
In some cases, sequestration in autophagic membranes, even without degradation,
seems to be important to exert special functions. Autophagy can be induced by
several stresses, including ER stress. ER stress-induced autophagy is basically
protective against cell death in both yeast and mammals(Bernales et al. 2006; Ogata
et al. 2006). How autophagy protects cells during ER stress is not exactly known, but
it was suggested that sequestration of ER into autophagosomes might be sufficient to
relieve ER stress(Bernales et al. 2006).Moreover, autophagy has been suggested in
the context of neurodegenerative disease. Autophagy likely has a beneficial role in
the clearance of misfolded or other harmful proteins. However, if autophagic
degradation is not rapid enough, sequestration of cytoplasm might rather have an
adverse effect. It was proposed that autophagosome maturation into autolysosomes is
impaired in Alzheimer’s disease brains for instance(Yu et al. 2005).
In fact, defective physiological autophagic role plays a significant effect in human
pathologies, neurodegeneration diseases, for instance.
1.1.2.2. Selective autophagy
Although autophagy is historically described as a non-selective bulk protein
degradation system, recent findings confirm that it can be also a highly selective
process. Selective autophagy is mediated by both autophagy receptors and adaptor
proteins that link the cargo with the core autophagic machinery. The term selective
autophagy refers to the selective degradation of organelles, bacteria, ribosomes,
specific proteins, and protein aggregates by autophagy. When autophagy is
selectively recognizing a specific cargo, its name is attributed according to the
targeted cargo such as aggrephagy (aberrant protein aggregates), mitophagy
(damaged mitochondria), reticulophagy (ER) and xenophagy (invasive pathogenes)
(Klionsky et al. 2007). Selective autophagy functions as a quality control system, but
the signals involved in recognition of selective cargo for autophagy degradation are
31
still poorly understood. Autophagy receptors, such as p62 and NBR1 are the main
proteins involved in the selective autophagy of protein aggregates, so-called
aggrephagy.
Furthermore, autophagy receptors could also bindto other specific adaptors, which
function as scaffolding proteins that selectively bring the cargo-receptor complex in
contact with the core autophagic machinery to allow sequestration of the substrate. In
addition to the autophagy receptors and specific adaptors, such as Atg19, Atg34, p62,
NBR1, NDP52 (nuclear dot protein 52 kDa for bacteria recognition), OPTN
(optineurin), Nix (NIP3-like protein X), and ATG32, selective autophagy in general
relies on the same molecular core machinery of non-specific autophagy.
The protein aggregates represent intermediates in autophagic degradation of
aggregation-prone proteins. The assembly of autophagy substrates into larger
aggregates or clustered structures is a common feature of selective autophagy. Their
assembly may facilitate uptake into autophagosomes, and aggregates may work as
nucleation sites for the phagophore. Damaged proteins are recognized and sorted by
chaperone and co-chaperone complexes containing chaperone-assisted ubiquitin E3
ligases to three different degradation pathways: UPS, CMA, and/or aggrephagy. In
the following section, current knowledge about aggrephagy will be discussed.
1.1.2.3. Aggrephagy
To accomplish their normal cellular function, proteins have to be correctly folded. In
fact, the accumulation of misfolded or unfolded proteins into protein aggregates is a
pathological trait of many clinical disorders. The term aggrephagy was introduced
for the first time by Per O. Seglen to describe the selective sequestration of protein
aggregates by autophagy(Øverbye et al. 2007). Generally, protein aggregation is
caused by an abnormal protein conformation, leading to the formation of oligomeric
intermediates, and further on to the small protein aggregates(Merlini et al. 2001).
These small protein aggregates can again form variety of structures (Dobson 2003),
termed as intracellular inclusions bodies (Grune et al. 2004; Kopito 2000). Larger
cytoplasmic inclusions can develop further and fuse to form an aggresome, a
32
pericentriolar, membrane-free cytoplasmic inclusion formed specifically at
themicrotubule organizing center (MTOC) containing misfolded, and ubiquitinated
proteins (Johnston et al. 1998; Kopito 2000).
Aggresome is thought to be a protective cellular structure, since it is made by
sequestered misfolded proteins that cannot be degraded by the proteasome and that
could be toxic circulating in the cytoplasm (Johnston et al. 1998; Kopito 2000). On
this way, the misfolded proteins inside the aggresome are straightly transported
through autophagy for degradation. Protein aggregates are able to be sequestrated
inside the cell because of a variety of cellular stressors, such as mis/unfolded protein
formation, impaired proteasomes, oxidative stress, or aging (Kopito 2000).
Misfolded proteins generally become poly-ubiquitinated. Such proteins are normally
degraded by the UPS, while aggregate-prone proteins are poor substrates for
proteasomal degradation as they are highly insoluble and too big to pass through the
narrow proteasome pore (Stefanis et al. 2001; Verhoef et al. 2002). It has been also
shown that the ubiqutin linked chain K48 is a classical signal for UPS dependent
degradation and it has been suggested that autophagic substrates present a modified
K63-linked ubiquitin chains (Tan et al. 2008). Moreover, it has been demonstrated
that the autophagy receptors p62 and NBR1 recognize K63-linked ubiquitin
chains(Kirkin et al. 2009; Long et al. 2008; Wooten et al. 2008) and inclusions
containing K63-linked ubiquitin chains and target them for autophagic degradation
(Tan et al. 2008).
p62/A170/ SQSTM1 (hereafter referred to as ‘p62’) and NBR1 (neighbor of BRCA1
gene 1) are the components of the ubiquitin-positive inclusion bodies found in some
neurodegenerative and liver diseases. These molecules have unique features, N-
terminal Phox and Bem1p domain (PB1), which has the ability in self-
oligomerization, C-terminal ubiquitin associated domain (UBA) capable of
interaction with ubiquitinated proteins and LC3-interacting region (LIR) responsible
for the interaction with ATG8 family proteins. Both p62 and NBR1 mediate
aggregate formation, also called p62 bodies, sequestosomes or aggresome-like
inducible structures (ALIS) (Bjørkøy et al. 2005; Szeto et al. 2006; Clausen et al.
2010)and thus connect ubiquitinated protein aggregates to the autophagic machinery
players(Bjørkøy et al. 2005; Lelouard et al. 2002; Szeto et al. 2006) (Figure 7).
33
It was shown through two main studies that p62 is critical for protein aggregates
formation and their clearance by autophagy. In the first study, in p62 deficient
models, it was observed that the aggresome-like inclusion bodies formation is
notably impaired (Pankiv et al. 2007; Clausen et al. 2010). In accordance with this,
using Atg7 knock-out (KO) mice or Atg8 mutant flies, it was confirmed that large
ubiquitin-positive protein aggregates accumulate in these mutated models, and no
longer persist when P62 is impaired(Komatsu et al. 2007; Nezis et al. 2008).
Accordingly, over-expression of p62 leads to accumulation of ubiquitinated protein
aggregates(Bjørkøy et al. 2005; Seibenhener et al. 2004). In the second study, using
electronic microscopy p62 was localized inside double membrane vesicles (Bjørkøy
et al. 2005). It is important to point out that p62 was also identified as one of the
specific substrates that are degraded through the autophagy–lysosomal pathway.
Recently, the ubiquitin-binding histone deacetylase 6 (HDAC6) is reported to be a
key player in recruiting ubiquitinated, misfolded proteins to the aggresome (Iwata et
al. 2005; Kawaguchi et al. 2003; Olzmann et al. 2007). While most members of the
histone deacetylase (HDAC) family are localized in the nucleus, HDAC6 is found
exclusively inside cytoplasm and it has been shown to contain an ubiquitin-binding
domain (BUZ finger). As it has been demonstrated that HDAC6 may bind to the
microtubule motor protein dynein as well, it was proposed that HDAC6 can facilitate
transport of aggregates to the MTOC in order to form the aggresome (Kawaguchi et
al. 2003). In HDAC6 deficient cells dispersed micro-aggregates were formed inside
the cytoplasm, while formation of aggresomes was impaired suggesting a failure in
aggregates transport to the MTOC region (Kawaguchi et al. 2003).
34
F igure 7. Aggrephagy main players. P62 and NBR1 mediate aggresome/inclusion
body formation and are substrates prone to autophagosome engulfement and
degradation inside the autolysosome. In addition, aggresome formation is
coordinated by HDAC6 and is directly carried to lysosome via microtubules (e.g:
dynein), adopted from(Kraft et al. 2010).
1.2. Autophagy and proteinopathies Proteinopathies belong to conformational diseases that are caused by the failure of a
protein to attain or maintain correct conformation which is usually due to genetic
mutations. With aging, an increase in intracellular oxidative molecules leads to the
accumulation of unfolded proteins in cells, too. Misfolded proteins are engaged in
inappropriate interactions with other cellular components and can accumulate in
potentially toxic protein inclusions(Lashuel & Lansbury 2006). A number of studies
have identified small oligomeric aggregates as species linked to toxicity(Kayed et al.
2003). For example, in mammalian cell culture and yeast, accumulation of small
oligomeric Huntingtin aggregates has shown to correlate with toxicity(Kitamura et
al. 2006; Finkbeiner et al. 2006).
On the other hand, some studies have shown that soluble oligomers coming out from
proteasomal degradation of misfolded proteins in neurons could be more toxic than
their sequestration into aggregates. So, formation of aggregates might prevent their
toxicity awaiting for their transport to lysosomes for a complete degradation via
autophagy(Arrasate et al. 2004; Szeto et al. 2006; Tanaka et al. 2004). In the same
context, it has been suggested that formation of insoluble amyloid inclusions can also
promote cell survival and may serve as a protective mechanism by sequestering
potentially harmful aggregates from the cytosol. Several studies suggest that the
formation of tightly packed Huntingtin deposits is beneficial for cell survival. When
Greenberg and colleagues have transfected with mutant Huntingtin primary striatal
neuron, they could induce the formation of inclusions. These inclusions resembled
the protein deposits found in the brains of Huntington patients, as they were
intranuclear and ubiquitinylated. But these inclusions were not sufficient to induce
apoptosis(Saudou et al. 1998). Incorporation of toxic oligomers into protective
amyloid-like protein inclusions has been observed to reduce toxicity of Huntingtin
35
expressed in mammalian cells and mouse models(Cohen et al. 2009; Cheng et al.
2007). These studies support the hypothesis that sequestration into an
aggresomeremoves toxic misfolded species from the cellular environment.
Under normal conditions the cell can efficiently degrade misfolded/unfolded proteins
through targeted proteolysis. The UPS and autophagy are the two major intracellular
protein degradation pathways. While UPS is mediating degradation of most normal
proteins after performing their normal functions as well as the removal of the
abnormal soluble proteins, autophagy is mainly responsible for the degradation of
defective organelles and the bulk of aggregated proteins. UPS proteolytic function
often becomes inadequate in proteinopathies, which leads to activation of autophagy
for the removal of abnormal proteins especially the aggregated forms. Enhancement
of autophagy via pharmacological intervention has shown great promises in relieving
proteinopathies in the cell. Even though few different strategies are employed to
increase proteasome function, it seems that UPS is not as efficient as autophagy in
alleviating proteinopathies (Díaz-Hernández et al. 2003).
Proteinopathies are exemplified mainly by human neurodegenerative diseases such
Table 2: Chemical structure and mechanism of action of compounds reported to treat A LS disease. Adopted and reviewed in details in (Limpert et al. 2013)
Several other drugs are targeting dysfunctional mitochondria such as: olesoxime and
dexpramipexole. Both are involved in the prevention of mitochondrial dysfunction
and maintenance of energy production in stressed mitochondria within motor
neurons. Eventhough, dexpramipexole gave some promising results in the preclinical
trials, yet it failed in phase III of the clinical trial (Limpert et al. 2013; Glicksman
2011; Dunkel et al. 2012).
1.5 A LS treatment through autophagy induction
As many neurodegenerative diseases are described as proteinopathies and face a
problem with the intracytoplasmic protein inclusions, a viable therapeutic strategy is
PBA sodium phenylbutyrate
HDAC inhibitor (Ryu et al. 2005)
Trichostatin A 7-[4-(dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxohepta-2,4-dienamide
HDAC inhibitor (Yoo & Ko 2011)
DCA dichloroacetate
inhibits the pyruvate dehydrogenase enzyme, and modulates mitochondrial activity
(Miquel et al. 2012)
54
needed to overcome the aggregation problem. Indeed, promoting the clearance of
aggregate-prone proteins via pharmacological induction of autophagy-endolysosomal
pathway has proved to be a useful mechanism protecting against neuronal loss.
Autophagy is shown to be critical for motor neuron survival in many
neurodegenerative disorders such as spinal and bulbar muscular atrophy (SBMA,
Kennedy’s disease). Clinical symptoms and the histopathology of SBMA show many
similarities with ALS.SBMA is a progressive neuromuscular disorder in which
degeneration of lower motor neurons results in proximal muscle weakness, and
muscle atrophy. SBMA is among polyglutamine (polyQ) diseases that are caused by
the expansion in specific genes of a trinucleotides repeat, cytosine–adenine–guanine
(CAG), which encodes glutamine. CAG repeat expansion in the androgen receptor
(polyQ AR) accumulates in ubiquitin-positive aggregatesinside affected neurons of
SBMA patients(La Spada et al. 1991). In 2009, it has been demonstrated that
autophagy was able to degrade the aberrant proteins (polyQ AR) retained inside the
cytoplasm in mouse model of SBMA, thus pointing out the importance of the
autophagy activation in prevention of the motor neuron death (Montie et al. 2009).
In different ALS and TDP-43 models, similar studies have shown that autophagy
seems to have a major protective role in spinal motor neurons against
neurodegeneration. Full-length TDP43 has an exceptionally long half-life of 12–34 h
in most cell types, whereas TDP43 C-terminal fragments (CTFs) have a half-life of
only ~4 h. It is believed that both UPS and autophagic degradation pathways are
involved in maintaining the normal levels of TDP-43 inside cell and also the removal
of CTFs. Disruption of the UPS and autophagy might contribute to increased levels
of ubiquitylated TDP43 in ALS and FTLD-TDP. Indeed, inhibition of the UPS and
autophagy leads to increased levels of phosphorylated TDP43 aggregates in cultured
cells(Winton et al. 2008). Investigating the proteolytic pathways especially
autophagy, could be a promising strategy to treat TDP-43 aggregated disorders.
In fact, in primary motor neuron cultures, obtained from SOD1-G93A transgenic
mice (model of ALS), several autophagy substrates such as SOD1, ubiquitin, and
alpha-synuclein were significantly cleared when autophagy was induced using
lithium or rapamycin (Fornai, Longone, Cafaro, et al. 2008). Moreover, Kabuta and
55
colleagues reported that using wild-type and mutant SOD1 in neuronal and non-
neuronal cells, autophagy decreases mutant SOD1-mediated toxicity and its mutant
SOD1 protein aggregates levels (Kabuta et al. 2006).In line with this, Caccamo and
co-workers have overexpressed 25-kDa C-terminal fragment of TDP-43, that tends to
accumulate together with endogenous full-length TDP-43 in the cell, and were able
to modulate this accumulation and TDP-43 localization with the autophagy
activation(Caccamo et al. 2010). Wang et al., overexpressed TDP-43 and its
pathogenic form TDP-25 in HEK 293 cells and have demonstrated that the protein
levels of TDP-43 and TDP-25 were increased in cells treated with an autophagy
inhibitor, 3-MA, whereas, they were decreased in cells treated with an enhancer of
autophagy, trehalose(Wang et al. 2012).
Another study also confirmed the effect of rapamycin, as well as other autophagy
inducers, such as: spermidine, carbamazepine, and tamoxifen, in rescuing the motor
dysfunction of TDP-43 affected mice through autophagy pathway.Using FTLD-U
mouse model with TDP-43 proteinopathy, Wang et al., have shown that rapamycin
treatment effectively rescues the learning/memory impairment of these mice at 3
months of age, and it significantly slows down the age-dependent loss of their motor
function. These behavioral improvements upon rapamycin treatment are
accompanied by decreased level of caspase-3 and a reduction of neuron loss in the
brain of these mice. Furthermore, the number of cells with cytosolic TDP-43 positive
inclusions and the amounts of full-length TDP-43 as well as its cleavage products (35
kDa and 25 kDa) are significantly decreased upon rapamycin treatment. All these
studies suggest that autophagy induction may be a valid therapeutic target for TDP-
43 proteinopathies (Wang et al. 2012).
1.6 Dextran sulfate as inducer of autophagy
Dextran sulfate (DS) is a synthetic branched polyanionic sulfated compound derived
from purified dextran, that is originally synthesized by bacteria Leuconostoc
mesenterioides and then synthetically sulphated. Dextran sulfate can reach the level
of sulfur between 17 and 20 % of the total molecule. Once after being sulfated, every
glucose residue will present between 2 and 3 sulfate groups per glucosyl residue,
normally localized on carbons 2 and 4. 95% of the linkages between monomeres are
α-D-(16), and the other 5% are bindings of the type α-D-(13), directly related to
56
the chain branching of DS (Figure 9). DS is available commercially, with different
molecular weights, depending on the total amount of type α-D-(13) bindings
present and they can range from 5000 Da to 500.000 Da.
DS is a well-known compound used routinely in the biological field for decades. It
has been shown that low concentrations of DS can be used to selectively precipitate
lipoproteins such as VLDL and LDL, and higher concentrations of DS can also be
used to precipitate HDL. DS is in use as an agent able to separate DNA from histone
complexes, inhibit the binding of DNA with ribosomes, inhibit ribonucleases and it
has also been proven to inhibit initial binding of several viruses with cells, such as
the herpes virus and the HIV virus. Sulfated glycosaminoglycans, such as DS, have
been widely used in biotechnology as cell culture media additives, as they have
shown to promote cell growth, disperse cell clumps, increase viable cell yield, and
overall recombinant protein productivity.
Furthermore, for DS it has been shown to be safe for human consumption and indeed
DS has been used as an alimentary supplement for more than 20 years(Gräßler et al.
2013). It was originally discovered that DS possesses an anticoagulant capacity. Oral
F igure 9.Chemical structure of dextran sulfate
57
intake of DS is responsible for the reduced levels of LDL-cholesterol, HDL-
cholesterol, and triglyceride, confirming its action in reducing lipid metabolites in
humans. In this manner, DS has been used to prevent the progression of
atherosclerosis through its action as a competitor for the binding of modified LDL
with scavenger receptors in macrophages (Tsubamoto et al. 1994). Moreover, it has
also been reported that DS suppresses the expression of adhesion factors involved in
metastases, conferring anti-metastatic capacity to this polyanion (Takagi et al. 2005).
Furthermore, DS was able to work as a cytoprotectant of myocardial ischaemic and
damaged endothelium (Banz et al. 2005).
It has been proven that DS 5.000 Da, due to the presence of the sulfated groups, has
the capacity of mimicking endogenous glycosaminoglycans, thus showing many
effects on cells.Recently, DS was shown to act as an anti-apoptotic agent on CHO
cells, under staurosporine-induced apoptosis (Jing et al. 2011).They have shown that
dextran sulfate action involves mitochondrial pathway, since they were able to
measure a significant decrease of pro-caspase-9 activation, cleaved caspase-3 levels,
cytochrome c release into the cytosol and mitochondrial transmembrane potential
collapse.
In this note, our group have shown that DS5000 Da (DS5000) is able to prolong the
life of cells interfering with the apoptotic pathway (Menvielle et al. 2013). At a
molecular level, we show that DS inhibits apoptosis by DNA fragmentation delay
and decrease of annexinV-labeled cells, causes a G0/G1 cell-cycle arrest, decreases
p53 expression and increases the pro-survival factor Hsc70 expression. Moreover,
we were able to demonstrate that DS treatment also resulted in an enhanced LC3-I to
LC3-II conversion and increased autophagosomes formation employing tagged-LC3.
The experimental evidence provided in this work indicates that DS 5000 Da
treatment in low doses is able to induce autophagosome formation most likely in a
cell type independent manner. Nonetheless, our findings strongly suggest that
sulfated glycosaminoglycans should be re-considered, in this new context, as
autophagy inducers.
Aim of the study:
58
Previously established cell-based TDP-43 aggregation model, where tandem repeats
carrying the C-terminal Q/N-rich region of TDP-43 were able to trigger the
formation of phosphorylated and ubiquitinated aggregates, will be used in this study
to investigate the autophagy-endolysosomal cell pathway response.
As there is continuous need for the new and more effective agents in the treatment of
proteinopathies, it would be interesting to investigate if TDP-43 12xQ/N cell-based
model of aggregation could benefit from the autophagy induction and if DS
treatment is able to enhance inclusions clearance and modulate subcellular
distribution of TDP-43.
With such approach we hope to gain more knowledge about new therapeutic
strategies/effectors, which might have a broad therapeutic potential in human TDP-
43 proteinopathies.
59
2. M A T E RI A LS A ND M E T H O DS 2.1. Chemical reagents:
General chemicals were purchased from Sigma Chemical Co., Merck, Gibco
BRL, Boehringer Mannheim, Carlo Erba and Riedel-de Haen.
Dextran sulfate sodium salt 5000 Da (DS, Sigma) was dissolved in water and
sterilized through 0.22 µm filter.
Trichostatin A (TSA, from Sigma) was dissolved in DMSO, Four hours before
harvesting, cells were treated with 5 µM final concentration of TSA, and the
same concentration of TSA was kept in the cell lysis buffer, DMSO (Riedel-de
Haen) was used as vector control.
2.2. Standard solutions:
All solutions are identified in the text except for the following:
TE: 10 mM Tris-HCl (pH 7.4), 1 mM EDTA (pH 7.4)
PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4
fusion, and/or ineffective autophagosome clearance. In Alzheimer disease, it was
reported that autophagosome maturation was impaired (Yu et al. 2005). In
Huntington disorder, macroautophagy was shown to be impaired in early stage of the
disease (Pasquali et al. 2009). Autophagy progression seems to be impaired in ALS
patients, as sized autophagosomes and autolysosomes were found inside diseased
neurons of the ALS patients(Sasaki 2011), in agreement with the similar findings in
110
the animal model of ALS(Li, Liang; Zhang, Xiaojie; Le 2008). There is a solid
evidence that links directly a defective autophagy to ALS. This was for the first time
demonstrated by Fornai et al. (Fornai, Longone, Cafaro, et al. 2008; Fornai,
Longone, Ferrucci, et al. 2008), in SOD1 mice model aggregates and firmly
confirmed by other subsequent work (Laird et al. 2008). In fact, autophagy inhibitors
worsen the viability of motor neurons in a variety of ALS models while the
stimulation of autophagy alleviates motor neuron degeneration(Wang et al. 2012).
Autophagy inducers such as trehalose (Gomes et al. 2010), rapamycin (Berger et al.
2006),SMERs(Sarkar et al. 2007), lithium(Fornai, Longone, Cafaro, et al. 2008), and
resveratrol (Kim et al. 2007) have shown beneficial effects by decreasing protein
aggregation through the autophagy pathway activation and promoting neuronal
survival in different neurodegenerative diseases. Therefore, it is reasonable to expect
that tuning autophagy to enhance autophagy mediated clearance may have an actual
therapeutic impact to alleviate the central cause of degeneration: accumulation of
protein aggregates.
Yet, one has to take into consideration that induction of autophagy might also show
to be non beneficial. Indeed, Zhang et al., have shown that in the SOD1G93A mice,
treatment with autophagy enhancer rapamycin accelerates the MNs degeneration,
shortens the life span of the ALS mice, and did not have any positive effects on the
reduction of SOD1 aggregates (Zhang et al. 2011). Rapamycin treatment augments
motor neuron degeneration in SOD1G93A mouse model of amyotrophic lateral
sclerosis.). Since this negative effect was reported up to now just on rapamycin,
another work could explain why this effect was negatively affecting SOD1
aggregates clearance. In fact, rapamycin could suppress protective immune responses
while enhancing protective autophagy reactions during the ALS disease process.
Their results indicate that maximal therapeutic benefit may be achieved through the
use of compounds that enhance autophagy without causing immune
suppression(Staats et al. 2013).
Interestingly, it was shown that autophagy itself is regulated by TDP-43. Indeed,
Bose et al, have shown that TDP-43 functions as maintenance factor of the
autophagy system. Moreover, they have shown that the depletion of TDP-43 with the
consequent loss of the Atg7 mRNA/ATG7 protein causes impairment of the
111
autophagy and facilitates the accumulation of TDP-43 inclusions (Bose et al.
2011)(Tdp- et al. 2011).
Taking all this facts together, we have decided to investigate the autophagy role in
TDP-43 aggregation model previously established in the lab (Budini et al. 2012),
where 12 repetitions of TDP-43 amino-acid sequence 331-369 are able to induce
cytoplasmic aggregates formation. It has been previously shown by Budini et al that
the EGFP-12xQ/N aggregates are able to sequester full-length TDP-43 wild type and
are showing ubiquitinated and phosphorylated features. Thus the first thing to
investigate was if these TDP-43 inclusions may colocalize with some markers of
autophagosome-lysosome pathway. We have shown that EGFP-12xQ/N inclusions
were able to colocalize with some players of the autophagy-lysosome pathway in
transfected non-neuronal cells U2OS and mouse motor neuron-like cells NSC-34.
This observation of colocalization of EGFP-12xQ/N inclusions with autophagic
markers LC3 and p62 (sequestosome-1 (SQSTM1) (Figure 10 Panel A, B, and C)
and with lysosomal marker Lamp1 (Figure 10 Panel D) is in aggreement with the
data published by Sasaki et al, where the round bodies and skein-like inclusions from
degenerated motor neurons of ALS patients were immunostained for LC3 and for
p62(Sasaki 2011). P62 is one of the first proteins recognized as an adaptor for
delivering cargo marked by ubiquitination to the autophagic organelles(Bjørkøy et al.
2005). P62 binds ubiquitin and polyubiquitinated proteins via its C-terminal UBA
domain and it bridges the cargo and autophagic machinery by binding directly to the
autophagic effector protein LC3 Several other studies have shown an evidence that
neuronal inclusions were p62-positive and that p62 binds directly to TDP-43 in
brains of FTLD patients with TDP-43 inclusions, and furtheremore the aggregation
of TDP-43 C-terminal fragments was reported to be regulated by p62
phosphorylation through autophagy and proteasome-mediated degradation pathways
(King et al. 2011; Tanji et al. 2012; Al-Sarraj et al. 2011; Brady et al. 2011). Hence,
these results suggest that autophagy-endolysosomal pathway markers LC3, Lamp 1
and the adaptor protein p62 label EGFP-12xQ/N aggregates, probably targeting them
for degradation through the autophagy-lysosome pathway.
Inducing autophagy-lysosome pathway has proved to be an useful mechanism
against protein aggregation in different neurodegenerative diseases such as
huntington disorder (Qin et al. 2003). One of the well-known inducer of autophagy,
112
rapamycin has been used to induce TDP-43 clearance and the correction of the TDP-
43 mislocalization through the autophagy in ALS and FTD models(Wang et al. 2012;
Caccamo et al. 2009). Therefore, there is still a great interest and need to investigate
possible drug candidates able to reduce TDP-43 aggregates. We have previously
shown in our lab that the sulfated polyanion dextran sulfate 5000 Da (DS) in low
concentration favors autophagy (Menvielle et al. 2013).Therefore, we have tested if
DS could help the cell to clear the aggregates in our TDP-43 aggregation model. We
have shown that upon DS treatment, less cells were possessing dot-like structure
inside the cytoplasm and more diffused GFP signal in comparison with the control
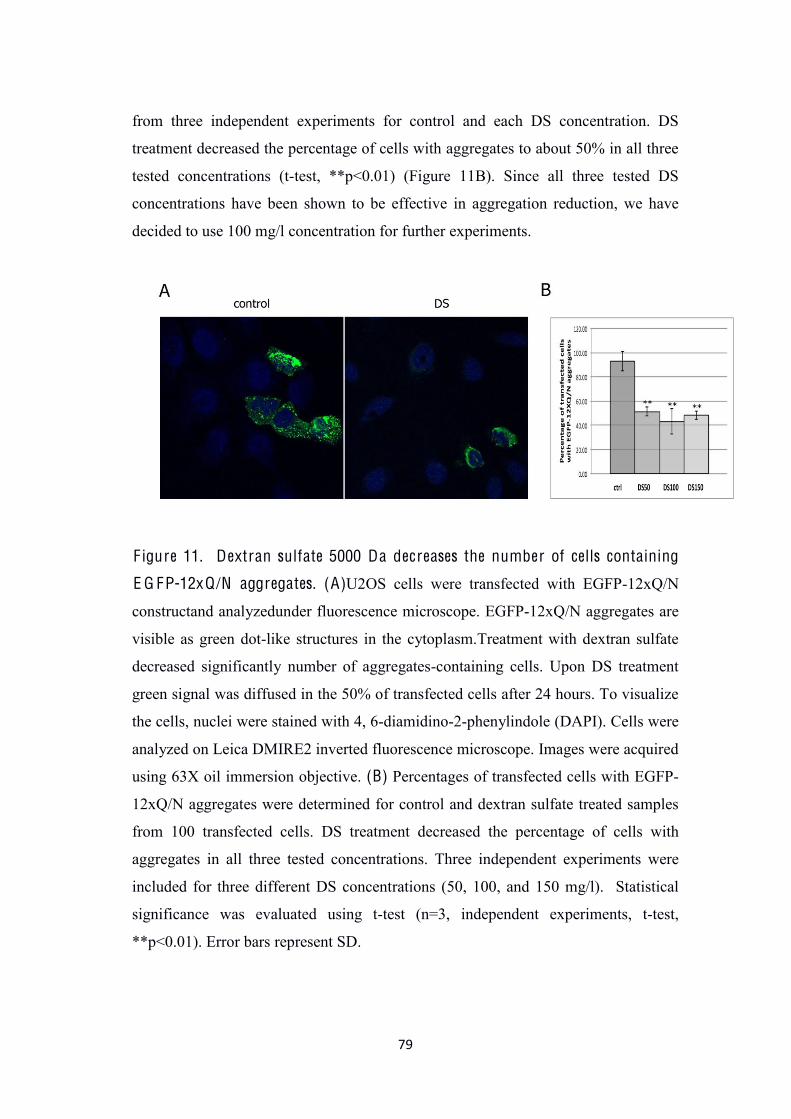
(Figure 11A and B). This result encouraged us to investigate DS-related aggregates
LC3-labelling. When U2OS cells were co-transfected with both EGFP-12xQ/N and
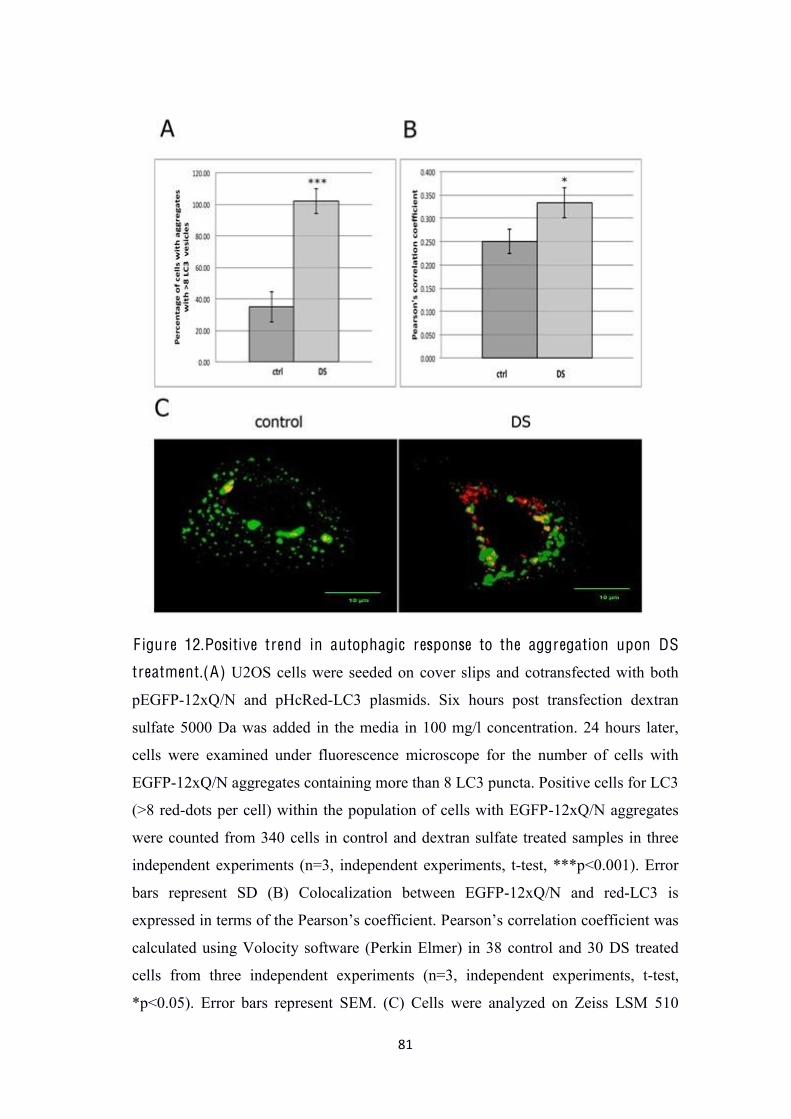
HcRed-hLC3 plasmids we noted that most of DS-treated cells with EGFP-12xQ/N
aggregates were LC3 positive (cells containing more than 8 LC3 red-dots) (Figure
12A). Moreover, DS treatment increased colocalization of EGFP-12xQ/N and red-
LC3 compared with control (Figure12B and 12C). This result is suggesting that there
was more LC3 recruitment (autophagosomes) to the EGFP-12xQ/N aggregates in DS
treated cells when compared to the control.
In order to obtain more consistent and regulated expression system we then
developed a stable HEK293 cell line expressing EGFP-12xQ/N under tetracycline
inducible promoter. We have demonstrated that our inducible stable cell system is
able to reproduce the dot-like EGFP-12xQ/N inclusions in the cytoplasm (Figure 13).
Next, we confirmed that DS increased the EGFP-12xQ/N product clearance as the
product amount decays when its synthesis is stopped in comparison to the control.In
addition as mentioned in the Results section we have observed significantvariability
of the aggregate clearance efficiency using different DS batches andcurrent
experiments are trying to pin down the cause.(Figure 14A, 14B, and 15A). We
further measured the effect of DS on EGFP-12xQ/N insoluble fraction by filter-trap
assay, where insoluble aggregates are not able to pass through the membrane unlike
the soluble GFP used as a negative control (Figure15B). DS-treated sample showed
aggregate content reduction of 0.6-fold in comparison to the control (Figure 15B,
GFP line) giving the confirmation of the DS induces aggregates clearance.
As the EGFP-12xQ/N aggregates were shown to be labelled with autophagosome-
lysosome markers, we aimed at investigation if autophagy is involved in enhanced
113
aggregates clearance upon DS addition. We checked the autophagy marker LC3-I to
LC3-II conversion, as an indicator of the autophagic activity in DS treated cells in
comparison to the untreated ones. In fact, we observed an increase of the autophagic
response upon DS treatment (Figure 16, LC3-I/II). As mentioned before, another
important player in autophagy is p62/SQSTM1, an aggregate bound protein that
recruits the autophagy machinery to the aggregates(Myeku & Figueiredo-Pereira
2011). Moreover, p62 is degraded by autophagy whereas, it is found accumulated
when the autophagic flux is inhibited. P62 level was significantly decreased in DS
samples compared to the control (Figure 16, p62). Taken together our data supports
the idea that DS plays an active role in the clearance of TDP-43 aggregates through
the adaptor protein p62 within the autophagy.
Suppression of the autophagy promoting genes such as Atg7 or Atg5, could be a tool
to check the direct involvement of autophagy in the DS enhenced EGFP-12xQ/N
clearance. For this purpose, we silenced one of the essential proteins for mammalian
autophagy, autophagy related gene 7 (Atg7), and we were able to abolish the DS
clearance action, compared to the respective control (siAtg7 ctrl in Figure 17) giving
the final evidence of the autophagy related DS clearance action.
As mentioned before, “loss of function” theory is putting the TDP-43 nuclear
depletion (as the consequence of TDP-43 retention, aggregation in the cytoplasm) in
the first line for the cause of the neuronal death in TDP-43 proteinopathies. Budini et
al. have shown before that some EGFP-12xQ/N aggregates are able to colocalize
with endogenous TDP-43 wild type suggesting the sequestration of the wild type
protein within the aggregates. In agreement with this, we were able to show
overlapping of the EGFP-12xQ/N signal with the endogenous TDP-43 (Figure 19A)
as well as with the overexpressed flag-TDP-43 wild type (Figure 19B) in some cells.
We therefore confirmed the ability of EGFP-12xQ/N to sequester TDP-43 wild type
in our stable inducible system. We further examined if DS treatment was able to
reduce this sequestration and to release TDP-43 wild type from the insoluble
fraction. We expressed simultaneously EGFP-12xQ/N and the exogenous flag-TDP-
43 wt in the clearance assay and separated the soluble (S) and the insoluble (P)
fractions and immunobloted with antibodies against GFP, flag, actin and tubulin. We
have shown that DS decreased the amount of flag-TDP-43 wt in the insoluble
fraction (P) compared to the control (0.4-fold change), consistenly with the EGFP-
114
12xQ/N aggregates behavior (0.5-fold change). This observation led us to propose
the model where DS treatment enhances the clearance of the aggregated EGFP-
12xQ/N through the autophagy leading to the decreased entrapment of TDP-43 wt
inside the insoluble fraction in the cell.
We further hypothesized that the enhanced clearance of TDP-43 aggregates by DS
could lower the capture of the cytoplasmic TDP-43 wild type, thus compensating its
loss from the nucleus, as TDP-43 is normaly nuclear protein. It was previously
shown the direct involvement of autophagy in rescuing the TDP-43 mislocalization
using rapamycin, suggesting that the cytoplasmic TDP-43 is normally degraded by
autophagy(Caccamo et al. 2009). In the same study it was given an in vivo evidence
that supports the hypothesis that accelerating autophagy may alleviate TDP-43-
mediated toxicity(Caccamo et al. 2009). Thus, we have decided to perform
nuclear/cytoplasmic fractionation to evaluate the level of TDP-43 wild type trapped
in the cytoplasm inuntreated and DS treated cells. We observed an enhanced TDP-43
wild type level in the nuclear fraction upon DS treatment when compared to the
control (Figure 22). Our findings support the scenario where low doses of DS 5000
Damay increase the efficacy of the main degradation pathway for TDP-43 aggregates
and decrease TDP-43 wild type sequestration in the cytoplasm. Projecting this model
to the ALS/FTD patient’s affected neurons where endogenous TDP-43 is captured by
cytoplasmic aggregates, leading to its nuclear loss of function, we suggest that DS
promoted autophagy would decrease TDP-43 sequestration in the cytoplasm and
restore normal nuclear level. This observation leads DS toward its possible
therapeutic use.
Interestingly, when we overexpressed TDP-43 wild type in our aggregation TDP-43
model, we noticed the appearance of small fragments of TDP-43 that correspond to
C-terminal fragments: 35 kDa and 25 kDa. Recent findings have shown that TDP-43
C-terminal fragments form cytoplasmic aggregates and cause cytotoxicity(Zhang et
al. 2009; Nonaka et al. 2009; Igaz et al. 2009; Johnson et al. 2008). Thus, TDP-43
truncation may play an important role in the pathogenesis of ALS, FTLD-U and
other TDP-43 proteinopathies. The degradation of TDP-43 by caspases was reported
to be responsible for generation of the C-terminal fragments due to the intrinsic
caspase cleavage sites in its primary sequence(Zhang et al. 2007; Rohn 2008; Rohn
& Kokoulina 2009). We aimed to confirm the caspase involved TDP degradation in
115
our model by the help of the pan caspase inhibitor Z-VAD (OMe)-FMK. The
generation of proteolytic fragments was inhibited by this caspase inhibitor as shown
in the Figure 20. After confirming that the fragments seen correspond to TDP-43 C-
terminal fragments, we checked if DS could decrease TDP-43 C-fragments amount
compared to the control. We have seen a slight decrease of the TDP-43 C-terminal
fragments in the cytoplasmic fraction in the DS sample compared to the control
(Figure22, anti TDP-43deltaQ/N). Yet, it is important to stand out that DS also could
have such action by decreasing caspase-3 action on TDP-43 degradation. In fact, it
was shown by Zhang et al., that the treatment with staurosporine (1µM), a potent
inducer of apoptosis and caspase-3 activator, leads to the high degradation of TDP-
43 and hence the presence of C-terminal fragments in the cytoplasm(Zhang et al.
2007). It was also reported that DS inhibits staurosporine-induced apoptosis via
decreasing the pro-caspase-9 activation, cleaved caspase-3 levels and the
cytochrome c release into the cytosol(Jing et al. 2011). Nevertheless, more
experiments are required to reveal the mechanism by which DS interferes with the
caspase action on TDP-43 or whether DS while enhancing EGFP-12xQ/N aggregates
clearance clear the C-terminal fragments too.
We further aimed to investigate the molecular bases by which DS would be acting as
an enhancer of EGFP-12xQ/N clearance through the autophagy. HDAC6 has
emerged as an important player in the cellular management of protein
aggregates(Rodriguez-Gonzalez et al. 2008). It is now well known that histone
deacetylase-6 (HDAC6) recruits and binds the polyubiquitinated misfolded protein
cargo to dynein motors for transportation to the aggresome(Kawaguchi et al. 2003), a
process required for autophagic degradation of aberrant aggregated proteins like
Huntingtin(Iwata et al. 2005). In addition, several studies showed that HDAC6
mutations reduce the transportation of autophagosomes to perinuclear regions where
lysosomes are more abundant, resulting in the impairment of autophagosome-
lysosome fusion and reduced clearance of aggregate-prone proteins(Lee et al. 2010).
HDAC6 also colocalizes with α-syn and ubiquitin in the Lewy bodies of PD and
Dementia with Lewy Bodies (Kawaguchi et al. 2003; Boyault et al. 2006). Therefore,
HDAC6 may be a key regulator in the cellular clearance of abnormal proteins.
Here,we sought to investigate whether HDAC6 regulates the aggresome-autophagic
clearance of EGFP-12xQ/N and if DS may interfere with HDAC6. Despite the fact
116
that no HDAC6 protein expression level difference upon DS treatment in comparison
with the control was noticed (Figure 24, lane 1 and lane 3, HDAC6), we decided to
check if HDAC6 activity could be altered by DS. It is known that HDAC6 works as a
tubulin deacetylase protein(Zhang et al. 2003), so we looked if the level of acetylated
tubulin was altered in the DS treated cells in comparison to the non treated control.
In fact, upon DS treatment, we could see reduction of the acetylated tubulin (figure
24, ac-tubulin) that could indicate the effect of DS on HDAC6 activity, rather than
protein expression level. Inhibiting HDAC6 with Trichostatin A (TSA) abolished the
effect of DS on tubulin acetylation (Figure 24, lane 4 and 3, anti ac-tubulin). We
found that both pharmacological inhibition of HDAC6 as well as its depletion with
specific siRNA enhance microtubule acetylation, and eliminate DS clearance effect
on EGFP-12xQ/N aggregates (shown in Figure 23). In order to confirm that DS
action on EGFP-12xQ/N clearance is HDAC6 activity dependent, we reintroduced
wild type (HDAC6-wt) or the catalytically inactivate HDAC6 (HDAC6-DC) into the
HDAC-6 silenced cells. We actually used expression plasmids with silent mutations
in the sequences targeted by the HDAC6 siRNA in order to protect them from
degradation(Kawaguchi et al. 2003). We have shown that adding back HDAC6 wt in
the silenced cells restored the DS clearance effect in the treated cells compared to its
respective control, whereas, catalytically inactive HDAC6 mutant had no effect
(Figure 24, line 7 and line 8). Our findings demonstrated that the observed DS
induced EGFP-12xQ/N product decay is mediated by HDAC6. Moreover, this DS-
mediated autophagy action requires a catalytically deacetylase activity of HDAC6. In
fact, it was suggested that HDAC6 also plays a role in the eventual clearance of the
aggresomes in an autophagy-dependent manner, implying a functional connection
between HDAC6 and autophagy(Iwata et al. 2005; Pandey et al. 2007). In addition,
for HDAC6 has been shown to promote fusion of autophagosomes and lysosomes
and facilitates clearance of protein aggregates(Lee et al. 2010). Even more
specifically HDAC6 catalytic activity has been shown to be essential for turnover of
abberant proteins by autophagy(Pandey et al. 2007).As HDAC6 seems to play a
regulatory role in the clearance of misfolded proteins, activation of HDAC6
deacetylase function could be a promising strategy in processing aberrant protein
aggregation in ALS and other proteinopathies. It seems that in general
pharmacological activation of deacetylation reactions in the cytoplasm is positively
117
correlated with the autophagy induction. Indeed, for resveratrol and a panel of
phenolic compounds was demonstrated to stimulate the deacetylation of cytoplasmic
proteins and protects against neurotoxicity in AD and ALS mouse models through
the autophagic flux promotion(Kim et al. 2007; Pietrocola et al. 2012).
Interestingly, it has been suggested that the defect in autophagosome-lysosome
fusion may be an important factor in the development of neurodegenerative disease,
giving abnormal autophagic structures found in Alzheimer’s disease patients
neurons(Nixon et al. 2005). Taking together DS clearance action through the
HDAC6 and HDAC6 involvement in the autophagosome-lysosome fusion one could
investigate a possible link of DS and the maturation step of autophagy. The construct
ptfLC3 that contains mRFP-EGFP tandem fluorescent-tagged LC3 is showing a GFP
and mRFP signal within autophagosomes, but only mRFP signal after
autophagosomes fusion with lysosomes due to the GFP lost in the acidic lysosomal
environment (Kimura et al. 2007). The number of yellow puncta (colocalization of
GFP and mRFP signals) is reflecting the number of autophagosomes per cell,
whereas the red puncta (mRFP signal) is reflecting the number of lysosomes per cell.
On this way mRFP-EGFP-LC3 reporter could be very useful to discriminate between
acidic (autolysosomes) and neutral (autophagosomes) LC3-positive vesicles. The use
of EGFP-12xQ/N construct in this assay is not possible due to the green signal
overlap with GFP fluorescence from LC3. Our preliminary data with HeLa cells
transfected with mRFP-EGFP-LC3 reporter and treated with DS are showing the
lower number of autophagosomes in favor of autolysosomes (red puncta) in the DS
treated samples in comparison to the control conditions (data not shown). This result
may suggest that autophagic turnover in the cell could be faster upon DS treatment.
This observation is leading to the interpretation where DS may help the cell to clear
TDP-43 aggregates through the more effective autophagosomes fusion to lysosomes
resulting in the more efficient cargo degradation. Further experiments are necessary
to confirm and analyze in details DS tendency toward autophagy flux promotion and
link with HDAC6, but the autophagosome-lysosome fusion stimulation, rather than
autophagy activation is doubtless an attractive therapeutic approach for
neurodegenerative disease.
On the base of our data, we are suggesting the DS as a potential therapeutic agent for
TDP-43 proteinopathies. In this respect, we have decided to investigate DS toxicity
118
on the motor neuronal-like cells. NSC-34 cell line was proposed to be a good model
for neurotoxicity testing (Durham et al. 1993). In order to assess DS toxicity, many
assays could be used, such as cell viability, cell morphology, cell proliferation and
cellular membrane damage. We decided to use the most standardized test that assess
cell viability, MTT reduction assay as an indicator of cell redox activity. NSC-34
cells viability was assessed 48 hours after DS 5000 Da (100 mg/l) addition into the
medium, in the presence or absence of EGFP-12xQ/N aggregates. In three
independent experiments, DS did not show any toxicity in NSC-34 cells (Figure 26A
and 26B). This is in the agreement with our previously published data obtained with
HeLa and CHO cells (Menvielle et al. 2013), where we showed that DS is able to
enhance the cells survival through autophagy induction and apoptosis inhibition. DS-
mediated apoptosis inhibition was also shown in staurosporine-induced apoptosis
model in the cells(Jing et al. 2011).
Dextran sulfate is a semisynthetic analog of the glycosaminoglycan family, which
includes heparin, heparan sulfate, dermatan sulfate, and chondroitin sulfate. For
some low molecular weight heparins was shown to be able to pass through the blood-
brain barrier (Leveugle et al. 1998; Ma et al. 2002) and act as neuroprotective agents
(Pérez et al. 1998; Stutzmann et al. 2002; Ma et al. 2007). Dextran sulfate 5000 Da is
not toxic for the motor neuronal-like cell line NSC-34, that together with the fact that
DS already passed the test of time for human use, experiencing over 40 years of its
consumption in Japan to treat arteriosclerosis and high cholesterol, may be
considered as a promising therapeutic agent to treat TDP-43 proteinopathies in
humans.
119
5. C O N C L USI O NS
Protein clearing systems play a key role in TDP-43 proteinopathies. In particular,
autophagy represents the main pathway which through the clearance of the misfolded
proteins removes the potentially toxic material countering to cell death. There is now
a solid evidence that links directly a defective autophagy to ALS. Many studies have
shown that increasing autophagic flux with pharmacological inducers of autophagy
could counteract the pathophysiology of ALS.
EGFP-12xQ/N model of TDP-43 aggregation is a valuable tool to investigate cellular
protein degradation responses. We have shown that some EGFP-12xQ/N aggregates
were labeled with autophagy-lysosomal pathway markers: LC3, p62, and Lamp1
targeting them for degradation through the autophagy. Next, we have demonstrated
that dextran sulfate 5000 Da was able to promote the clearance of the EGFP-12xQ/N
aggregates via autophagy stimulation. Projecting this model to the ALS patient's
affected neurons where endogenous TDP-43 is captured by cytoplasmic aggregates
leading to its nuclear loss, we have demonstrated DS-induced lower TDP-43 wild
type sequestration inside the insoluble fraction. Additionally, we observed a
reduction of acetylated tubulin in DS-treated cells through the higher HDAC6
activity, indicating the role of HDAC6 in DS-mediated TDP-43 EGFP-12xQ/N
aggregates clearance.
120
In conclusion, we propose a model where DS leads to autophagy activation through
the stimulation of HDAC6 activity. As HDAC6 seems to play a regulatory role in the
clearance of misfolded proteins, its pharmacological activation could be a promising
strategy in processing aberrant protein aggregation in ALS, as it was shown before
for Huntington's disease model(Iwata et al. 2005; Pandey et al. 2007).
Data obtained in this study leads DS toward its possible therapeutic use. We have
shown here that DS 5000 Da is not toxic per se for the motor neuronal-like cell line
NSC-34 and may be considered as a promising therapeutic agent to treat TDP-43
proteinopathies in humans.
6. BIB L I O G R APH Y Ahlberg, J. & Glaumann, H., 1985. Uptake--microautophagy--and degradation of
exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Experimental and molecular pathology, 42(1), pp.78–88.
Al-Sarraj, S. et al., 2011. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta neuropathologica, 122(6), pp.691–702.
Andersen, P.M. et al., 2003. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World F ederation of Neurology, Research Group on Motor Neuron Diseases, 4(2), pp.62–73.
Andersson, M.K. et al., 2008. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC cell biology, 9(1), p.37.
Aplin, A. et al., 1992. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. Journal of cellular physiology, 152(3), pp.458–66.
Arai, T. et al., 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochemical and biophysical research communications, 351(3), pp.602–11.
121
Arrasate, M. et al., 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431(7010), pp.805–10.
Attaix, D. et al., 2001. Regulation of proteolysis. Current opinion in clinical nutrition and metabolic care, 4(1), pp.45–9.
Ayala, Y.M. et al., 2005. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. Journal of molecular biology, 348(3), pp.575–88.
Ayala, Y.M. et al., 2008. Structural determinants of the cellular localization and shuttling of TDP-43. Journal of cell science, 121(Pt 22), pp.3778–85.
Balls, M. & Horner, S.A., 1985. The FRAME interlaboratory programme on in vitro cytotoxicology. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association, 23(2), pp.209–13.
Banks, G.T. et al., 2008. TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mammalian genome : official journal of the International Mammalian Genome Society, 19(5), pp.299–305.
Banz, Y. et al., 2005. Locally targeted cytoprotection with dextran sulfate attenuates experimental porcine myocardial ischaemia/reperfusion injury. European heart journal, 26(21), pp.2334–43.
Barmada, S.J. & Finkbeiner, S., 2010. Pathogenic TARDBP mutations in amyotrophic lateral sclerosis and frontotemporal dementia: disease-associated pathways. Reviews in the neurosciences, 21(4), pp.251–72.
Berger, Z. et al., 2006. Rapamycin alleviates toxicity of different aggregate-prone proteins. Human molecular genetics, 15(3), pp.433–42.
Bernales, S., McDonald, K.L. & Walter, P., 2006. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS biology, 4(12), p.e423.
Bjørkøy, G. et al., 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of cell biology, 171(4), pp.603–14.
Del Bo, R. et al., 2009. TARDBP (TDP-43) sequence analysis in patients with familial and sporadic ALS: identification of two novel mutations. European journal of neurology : the official journal of the European Federation of Neurological Societies, 16(6), pp.727–32.
Boland, B. et al., 2008. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience, 28(27), pp.6926–37.
122
Borroni, B. et al., 2009. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Human mutation, 30(11), pp.E974–83.
Boyault, C. et al., 2007. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene, 26(37), pp.5468–76.
Boyault, C. et al., 2006. HDAC6-p97/VCP controlled polyubiquitin chain turnover. The EMBO journal, 25(14), pp.3357–66.
Brady, O. a et al., 2011. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. Journal of neurochemistry, 116(2), pp.248–59.
Brooks, B.R. et al., 2000. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases, 1(5), pp.293–9.
Brooks, B.R., 1994. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and th. Journal of the neurological sciences, 124 Suppl, pp.96–107.
Bruijn, L.I. et al., 1998. Aggregation and Motor Neuron Toxicity of an ALS-Linked SOD1 Mutant Independent from Wild-Type SOD1. Science, 281(5384), pp.1851–4.
Bruijn, L.I., Miller, T.M. & Cleveland, D.W., 2004. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annual review of neuroscience, 27, pp.723–49.
Budini, M. et al., 2012. Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. The Journal of biological chemistry, 287(10), pp.7512–25.
Buratti, E. et al., 2005. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. The Journal of biological chemistry, 280(45), pp.37572–84.
Buratti, E. & Baralle, F.E., 2001. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. The Journal of biological chemistry, 276(39), pp.36337–43.
Buratti, E. & Baralle, F.E., 2008. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Frontiers in bioscience : a journal and virtual library, 13, pp.867–78.
123
Caccamo, A. et al., 2010. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. The Journal of biological chemistry, 285(17), pp.13107–20.
Caccamo, A. et al., 2009. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. The Journal of biological chemistry, 284(40), pp.27416–24.
Chen, Y. et al., 2011. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein & cell, 2(6), pp.477–86.
Cheng, I.H. et al., 2007. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. The Journal of biological chemistry, 282(33), pp.23818–28.
Chen-Plotkin, A.S., Lee, V.M.-Y. & Trojanowski, J.Q., 2010. TAR DNA-binding protein 43 in neurodegenerative disease. Nature reviews. Neurology, 6(4), pp.211–20.
Chiang, H.L. et al., 1989. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science (New York, N.Y.), 246(4928), pp.382–5.
Chung, C.T., Niemela, S.L. & Miller, R.H., 1989. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proceedings of the National Academy of Sciences, 86(7), pp.2172–2175.
Clausen, T.H. et al., 2010. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy, 6(3), pp.330–44.
Cohen, E. et al., 2009. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell, 139(6), pp.1157–69.
Corrado, L. et al., 2009. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Human mutation, 30(4), pp.688–94.
Coux, O., Tanaka, K. & Goldberg, A.L., 1996. Structure and functions of the 20S and 26S proteasomes. Annual review of biochemistry, 65, pp.801–47.
Cozzolino, M., Ferri, A. & Carrì, M.T., 2008. Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxidants & redox signaling, 10(3), pp.405–43.
Da Cruz, S. & Cleveland, D.W., 2011. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Current opinion in neurobiology, 21(6), pp.904–19.
124
Cuervo, A.M. et al., 2005. Autophagy and Aging: The Importance of Maintaining “Clean” Cells. Autophagy, 1(3), pp.131–141.
Cuervo, A.M., 2010. Chaperone-mediated autophagy: selectivity pays off. Trends in endocrinology and metabolism: TEM, 21(3), pp.142–50.
Cuervo, A.M. & Dice, J.F., 1996. A receptor for the selective uptake and degradation of proteins by lysosomes. Science (New York, N.Y.), 273(5274), pp.501–3.
D’Ambrogio, A. et al., 2009. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic acids research, 37(12), pp.4116–26.
Daoud, H. et al., 2009. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. Journal of medical genetics, 46(2), pp.112–4.
Van Deerlin, V.M. et al., 2008. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet neurology, 7(5), pp.409–16.
Degenhardt, K. et al., 2006. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer cell, 10(1), pp.51–64.
Dewey, C.M. et al., 2012. TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain research, 1462(null), pp.16–25.
Díaz-Hernández, M. et al., 2003. Neuronal induction of the immunoproteasome in Huntington’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience, 23(37), pp.11653–61.
Ding, W.-X. et al., 2007. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. The American journal of pathology, 171(2), pp.513–24.
Dobson, C.M., 2003. Protein folding and misfolding. Nature, 426(6968), pp.884–90.
Dreyfuss, G. et al., 1993. hnRNP proteins and the biogenesis of mRNA. Annual review of biochemistry, 62, pp.289–321.
Dubouloz, F. et al., 2005. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Molecular cell, 19(1), pp.15–26.
Dunkel, P. et al., 2012. Clinical utility of neuroprotective agents in neurodegenerative diseases: current status of drug development for Alzheimer’s, Parkinson's and Huntington's diseases, and amyotrophic lateral sclerosis. Expert opinion on investigational drugs, 21(9), pp.1267–308.
Durham, H.D., Dahrouge, S. & Cashman, N.R., 1993. Evaluation of the spinal cord neuron X neuroblastoma hybrid cell line NSC-34 as a model for neurotoxicity testing. Neurotoxicology, 14(4), pp.387–95.
125
De Duve, C. & Wattiaux, R., 1966. Functions of lysosomes. Annual review of physiology, 28, pp.435–92.
De Duve Christian, 1965. the lysosome. Scientific American, (208), pp.64–78.
Essner E. & Novikoff, A.B., 1961. Localization of acid phosphatase activity in hepatic lysosomes by means of electron microscopy. The Journal of biophysical and biochemical cytology, 9, pp.773–84.
Estes, P.S. et al., 2011. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Human molecular genetics, 20(12), pp.2308–21.
Fass, E. et al., 2006. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. The Journal of biological chemistry, 281(47), pp.36303–16.
Feiguin, F. et al., 2009. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. F EBS letters, 583(10), pp.1586–92.
Fengsrud, M. et al., 1995. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: effects of vinblastine and asparagine on vacuole distributions. Experimental cell research, 221(2), pp.504–19.
Fiesel, F.C. et al., 2010. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. The EMBO journal, 29(1), pp.209–21.
Finkbeiner, S. et al., 2006. -Disease modifying pathways in neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience, 26(41), pp.10349–57.
Fornai, F., Longone, P., Ferrucci, M., et al., 2008. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy, 4(4), pp.527–30.
Fornai, F., Longone, P., Cafaro, L., et al., 2008. Lithium delays progression of amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America, 105(6), pp.2052–7.
Fuentealba, R.A. et al., 2010. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. The Journal of biological chemistry, 285(34), pp.26304–14.
Geng, J. & Klionsky, D.J., 2008. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. “Protein modifications: beyond the usual suspects” review series. EMBO reports, 9(9), pp.859–64.
Geser, F. et al., 2009. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. Journal of neurology, 256(8), pp.1205–14.
126
Gidalevitz, T. et al., 2009. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. H. Orr, ed. PLoS genetics, 5(3), p.e1000399.
Giordana, M.T. et al., 2010. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain pathology (Zurich, Switzerland), 20(2), pp.351–60.
Gitcho, M.A. et al., 2009. TARDBP 3’-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta neuropathologica, 118(5), pp.633–45.
Glicksman, M.A., 2011. The preclinical discovery of amyotrophic lateral sclerosis drugs. Expert opinion on drug discovery, 6(11), pp.1127–38.
Gomes, C., Escrevente, C. & Costa, J., 2010. Mutant superoxide dismutase 1 overexpression in NSC-34 cells: effect of trehalose on aggregation, TDP-43 localization and levels of co-expressed glycoproteins. Neuroscience letters, 475(3), pp.145–9.
Gong, Y.H. et al., 2000. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience, 20(2), pp.660–5.
Gräßler, J. et al., 2013. Differential effects of lipoprotein apheresis by lipidfiltration or dextran sulfate adsorption on lipidomic profile. Atherosclerosis. Supplements, 14(1), pp.151–5.
Grune, T. et al., 2004. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and “aggresomes” during oxidative stress, aging, and disease. The international journal of biochemistry & cell biology, 36(12), pp.2519–30.
Gutierrez, M.G. et al., 2004. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. Journal of cell science, 117(Pt 13), pp.2687–97.
Habib, A.A. & Mitsumoto, H., 2011. Emerging drugs for amyotrophic lateral sclerosis. Expert opinion on emerging drugs, 16(3), pp.537–58.
Hahn, 1898. Das proteolytische Enzym des Hefepresssaftes. Berichte der Deutschen Chemischen Gesellschaft, 31, pp.200–201.
Hanson, K.A. et al., 2010. Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). The Journal of biological chemistry, 285(15), pp.11068–72.
127
Hara, T. et al., 2008. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. The Journal of cell biology, 181(3), pp.497–510.
Hara, T. et al., 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature, 441(7095), pp.885–9.
Harding, T.M., 1995. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. The Journal of Cell Biology, 131(3), pp.591–602.
Hars, E.S. et al., 2007. Autophagy regulates ageing in C. elegans. Autophagy, 3(2), pp.93–5.
Haverkamp, L.J., Appel, V. & Appel, S.H., 1995. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain : a journal of neurology, 118 ( Pt 3, pp.707–19.
Hayes-Punzo, A. et al., 2012. Gonadectomy and dehydroepiandrosterone (DHEA) do not modulate disease progression in the G93A mutant SOD1 rat model of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis : official publication of the World F ederation of Neurology Research Group on Motor Neuron Diseases, 13(3), pp.311–4.
Heinemeyer, W. et al., 1991. Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. The EMBO journal, 10(3), pp.555–62.
Herman, P.K. & Emr, S.D., 1990. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Molecular and cellular biology, 10(12), pp.6742–54.
Hershko, A., Ciechanover, A. & Varshavsky, A., 2000. Basic Medical Research Award. The ubiquitin system. Nature medicine, 6(10), pp.1073–81.
Hilt, W. & Wolf, D.H., 1996. Proteasomes: destruction as a programme. Trends in Biochemical Sciences, 21(3), pp.96–102.
Hochstrasser, M., 1995. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Current opinion in cell biology, 7(2), pp.215–23.
Hosokawa, N. et al., 2009. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular biology of the cell, 20(7), pp.1981–91.
Huang, C.-C. et al., 2014. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. Journal of cell science, p.jcs.136150–.
128
Hubbard, V.M. et al., 2012. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology, 13(1), pp.21–35.
Ichimura, Y. et al., 2008. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy, 4(8), pp.1063–6.
Igaz, L.M. et al., 2009. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. The Journal of biological chemistry, 284(13), pp.8516–24.
Iwata, A. et al., 2005. HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated Huntingtin * . , 280(48), pp.40282–40292.
Jaeger, P.A. & Wyss-Coray, T., 2009. All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Molecular neurodegeneration, 4(1), p.16.
Jing, Y. et al., 2011. Dextran sulfate inhibits staurosporine-induced apoptosis in Chinese hamster ovary (CHO) cells: Involvement of the mitochondrial pathway. Process Biochemistry, 46(1), pp.427–432.
Johansen, T. & Lamark, T., 2011. Selective autophagy mediated by autophagic adapter proteins. Autophagy, 7(3), pp.279–96.
Johnson, B.S. et al., 2008. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America, 105(17), pp.6439–44.
Johnson, B.S. et al., 2009. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. The Journal of biological chemistry, 284(30), pp.20329–39.
Johnston, J.A. et al., 2000. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America, 97(23), pp.12571–6.
Johnston, J.A., Ward, C.L. & Kopito, R.R., 1998. Aggresomes: a cellular response to misfolded proteins. The Journal of cell biology, 143(7), pp.1883–98.
Kabashi, E. et al., 2010. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Human molecular genetics, 19(4), pp.671–83.
Kabashi, E. et al., 2008. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nature genetics, 40(5), pp.572–4.
129
Kabeya, Y. et al., 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO journal, 19(21), pp.5720–8.
Kabuta, T., Suzuki, Y. & Wada, K., 2006. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. The Journal of biological chemistry, 281(41), pp.30524–33.
Kalmar, B. et al., 2008. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. Journal of neurochemistry, 107(2), pp.339–50.
Katsumata, K. et al., 2010. Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy, 6(3), pp.378–85.
Kawaguchi, Y. et al., 2003. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell, 115(6), pp.727–38.
Kayed, R. et al., 2003. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science (New York, N.Y.), 300(5618), pp.486–9.
Kim, D. et al., 2007. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. The EMBO journal, 26(13), pp.3169–79.
Kim, J. et al., 2013. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell, 152(1-2), pp.290–303.
Kimura, S., Noda, T. & Yoshimori, T., 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy, 3(5), pp.452–60.
King, A. et al., 2011. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathology : official journal of the Japanese Society of Neuropathology, 31(3), pp.239–49.
Kirisako, T. et al., 1999. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. The Journal of cell biology, 147(2), pp.435–46.
Kirkin, V. et al., 2009. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Molecular cell, 33(4), pp.505–16.
Kitamura, A. et al., 2006. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nature cell biology, 8(10), pp.1163–70.
130
Klionsky, D.J. et al., 2007. How shall I eat thee? Autophagy, 3(5), pp.413–6.
Klionsky, D.J. & Emr, S.D., 2000. Autophagy as a Regulated Pathway of Cellular Degradation. Science, 290(5497), pp.1717–21.
Köchl, R. et al., 2006. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic (Copenhagen, Denmark), 7(2), pp.129–45.
Komander, D., 2009. The emerging complexity of protein ubiquitination. Biochemical Society transactions, 37(Pt 5), pp.937–53.
Komatsu, M. et al., 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell, 131(6), pp.1149–63.
Komatsu, M. et al., 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. The Journal of cell biology, 169(3), pp.425–34.
Komatsu, M. et al., 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature, 441(7095), pp.880–4.
Kopito, R.R., 2000. Aggresomes, inclusion bodies and protein aggregation. Trends in cell biology, 10(12), pp.524–30.
Korolchuk, V.I., Mansilla, A., et al., 2009. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Molecular cell, 33(4), pp.517–27.
Korolchuk, V.I., Menzies, F.M. & Rubinsztein, D.C., 2009. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy, 5(6), pp.862–3.
Korolchuk, V.I., Menzies, F.M. & Rubinsztein, D.C., 2010. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. F EBS letters, 584(7), pp.1393–8.
Kraft, C., Peter, M. & Hofmann, K., 2010. Selective autophagy: ubiquitin-mediated recognition and beyond. Nature cell biology, 12(9), pp.836–41.
Kroemer, G., Mariño, G. & Levine, B., 2010. Autophagy and the integrated stress response. Molecular cell, 40(2), pp.280–93.
Kühnlein, P. et al., 2008. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Archives of neurology, 65(9), pp.1185–9.
Kuo, P.-H. et al., 2009. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic acids research, 37(6), pp.1799–808.
131
Kwiatkowski, T.J. et al., 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, N.Y.), 323(5918), pp.1205–8.
Kwong, L.K. et al., 2007. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta neuropathologica, 114(1), pp.63–70.
Lagier-Tourenne, C., Polymenidou, M. & Cleveland, D.W., 2010. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Human molecular genetics, 19(R1), pp.R46–64.
Laird, A.S. et al., 2010. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PloS one, 5(10), p.e13368.
Laird, F.M. et al., 2008. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. The Journal of neuroscience : the official journal of the Society for Neuroscience, 28(9), pp.1997–2005.
Lamark, T. & Johansen, T., 2012. Aggrephagy: selective disposal of protein aggregates by macroautophagy. International journal of cell biology, 2012, p.736905.
Lashuel, H.A. & Lansbury, P.T., 2006. Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Quarterly reviews of biophysics, 39(2), pp.167–201.
Lee, J.-Y. et al., 2010. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. The EMBO journal, 29(5), pp.969–80.
Lelouard, H. et al., 2002. Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature, 417(6885), pp.177–82.
Leveugle, B. et al., 1998. Heparin oligosaccharides that pass the blood-brain barrier inhibit beta-amyloid precursor protein secretion and heparin binding to beta-amyloid peptide. Journal of neurochemistry, 70(2), pp.736–44.
Li, W., Li, J. & Bao, J., 2012. Microautophagy: lesser-known self-eating. Cellular and molecular life sciences : CMLS, 69(7), pp.1125–36.
Li, Y. et al., 2010. A Drosophila model for TDP-43 proteinopathy. Proceedings of the National Academy of Sciences of the United States of America, 107(7), pp.3169–74.
132
Li, Liang; Zhang, Xiaojie; Le, W., 2008. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy, 4(3), pp.290–3.
Liang, C. et al., 2008. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature cell biology, 10(7), pp.776–87.
Limpert, A.S., Mattmann, M.E. & Cosford, N.D.P., 2013. Recent progress in the discovery of small molecules for the treatment of amyotrophic lateral sclerosis (ALS). Beilstein journal of organic chemistry, 9, pp.717–32.
Logroscino, G. et al., 2010. Incidence of amyotrophic lateral sclerosis in Europe. Journal of neurology, neurosurgery, and psychiatry, 81(4), pp.385–90.
Long, J. et al., 2008. Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. The Journal of biological chemistry, 283(9), pp.5427–40.
Lu, Y., Ferris, J. & Gao, F.-B., 2009. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Molecular brain, 2, p.30.
Ma, Q. et al., 2007. Heparin oligosaccharides as potential therapeutic agents in senile dementia. Current pharmaceutical design, 13(15), pp.1607–16.
Ma, Q. et al., 2002. The blood-brain barrier accessibility of a heparin-derived oligosaccharides C3. Thrombosis research, 105(5), pp.447–53.
Mackenzie, I.R.A., 2007. The neuropathology of FTD associated With ALS. Alzheimer disease and associated disorders, 21(4), pp.S44–9.
Maday, S., Wallace, K.E. & Holzbaur, E.L.F., 2012. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. The Journal of cell biology, 196(4), pp.407–17.
Martinez-Vicente, M. et al., 2008. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. The Journal of clinical investigation, 118(2), pp.777–88.
McCord, J.M. & Fridovich, I., 1969. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). The Journal of biological chemistry, 244(22), pp.6049–55.
McDonnell, M.E. et al., 2012. Riluzole prodrugs for melanoma and ALS: design, synthesis, and in vitro metabolic profiling. Bioorganic & medicinal chemistry, 20(18), pp.5642–8.
McEwan, D.G. & Dikic, I., 2011. The Three Musketeers of Autophagy: phosphorylation, ubiquitylation and acetylation. Trends in cell biology, 21(4), pp.195–201.
133
Menvielle, J.P. et al., 2013. Dual role of dextran sulfate 5000 Da as anti-apoptotic and pro-autophagy agent. Molecular biotechnology, 54(2), pp.711–20.
Merlini, G. et al., 2001. Protein aggregation. Clinical chemistry and laboratory medicine : CCLM / FESCC, 39(11), pp.1065–75.
Mijaljica, D., Prescott, M. & Devenish, R.J., 2011. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy, 7(7), pp.673–82.
Miller, R.G. et al., 2007. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane database of systematic reviews (Online), (1), p.CD001447.
Miquel, E. et al., 2012. Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. S. T. Ferreira, ed. PloS one, 7(4), p.e34776.
Mitsumoto, H., 1997. Can we treat amyotrophic lateral sclerosis? Rinshō shinkeigaku = Clinical neurology, 37(12), p.1150.
Mitsumoto, H., 1997. Diagnosis and progression of ALS. Neurology, 48(Issue 4, Supplement 4), p.2S–8S.
Mizushima, N. et al., 1998. A protein conjugation system essential for autophagy. Nature, 395(6700), pp.395–8.
Mizushima, N. et al., 2008. Autophagy fights disease through cellular self-digestion. Nature, 451(7182), pp.1069–75.
Mizushima, N., 2004. Methods for monitoring autophagy. The international journal of biochemistry & cell biology, 36(12), pp.2491–502.
Mizushima, N., 2005. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell death and differentiation, 12 Suppl 2, pp.1535–1541.
Montie, H.L. et al., 2009. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Human molecular genetics, 18(11), pp.1937–50.
Myeku, N. & Figueiredo-Pereira, M.E., 2011. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. The Journal of biological chemistry, 286(25), pp.22426–40.
Nakai, A. et al., 2007. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nature medicine, 13(5), pp.619–24.
Nandi, D. et al., 2006. The ubiquitin-proteasome system. Journal of biosciences, 31(1), pp.137–55.
134
Nedelsky, N.B., Todd, P.K. & Taylor, J.P., 2008. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochimica et biophysica acta, 1782(12), pp.691–9.
Neumann, M. et al., 2009. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta neuropathologica, 117(2), pp.137–49.
Neumann, M. et al., 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, N.Y.), 314(5796), pp.130–3.
Nezis, I.P. et al., 2008. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. The Journal of cell biology, 180(6), pp.1065–71.
Nixon, R.A., 2006. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends in neurosciences, 29(9), pp.528–35.
Nixon, R.A. et al., 2005. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. Journal of neuropathology and experimental neurology, 64(2), pp.113–22.
Nonaka, T. et al., 2009. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Human molecular genetics, 18(18), pp.3353–64.
Oeda, T. et al., 2001. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Human molecular genetics, 10(19), pp.2013–23.
Ogata, M. et al., 2006. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and cellular biology, 26(24), pp.9220–31.
Okamoto, K. et al., 1991. Reexamination of granulovacuolar degeneration. Acta Neuropathologica, 82(5), pp.340–345.
Olzmann, J.A. et al., 2007. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. The Journal of cell biology, 178(6), pp.1025–38.
Otomo, A., Pan, L. & Hadano, S., 2012. Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurology research international, 2012, p.498428.
Ou, S.H. et al., 1995. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. Journal of virology, 69(6), pp.3584–96.
135
Øverbye, A., Brinchmann, M.F. & Seglen, P.O., 2007. Proteomic Analysis of Membrane-Associated Proteins from Rat Liver Autophagosomes. Autophagy, 3(4), pp.300–322.
Pan, T. et al., 2008. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiology of disease, 32(1), pp.16–25.
Pandey, U.B. et al., 2007. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature, 447(7146), pp.859–63.
Pankiv, S. et al., 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. The Journal of biological chemistry, 282(33), pp.24131–45.
Pasquali, L. et al., 2009. Autophagy, lithium, and amyotrophic lateral sclerosis. Muscle & nerve, 40(2), pp.173–94.
Pérez, M. et al., 1998. Sulphated glycosaminoglycans prevent the neurotoxicity of a human prion protein fragment. The Biochemical journal, 335 ( Pt 2, pp.369–74.
Pesiridis, G.S., Lee, V.M.-Y. & Trojanowski, J.Q., 2009. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Human molecular genetics, 18(R2), pp.R156–62.
Peters, J.-M., 1994. Proteasomes: protein degradation machines of the cell. Trends in Biochemical Sciences, 19(9), pp.377–382.
Phillips, J.P., 1989. Null Mutation of Copper/Zinc Superoxide Dismutase in Drosophila Confers Hypersensitivity to Paraquat and Reduced Longevity. Proceedings of the National Academy of Sciences, 86(8), pp.2761–2765.
Pickford, F. et al., 2008. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. The Journal of clinical investigation, 118(6), pp.2190–9.
Pietrocola, F. et al., 2012. Pro-autophagic polyphenols reduce the acetylation of cytoplasmic proteins. Cell cycle (Georgetown, Tex.), 11(20), pp.3851–60.
Polymenidou, M. et al., 2011. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience, 14(4), pp.459–68.
Punnonen, E.L. & Reunanen, H., 1990. Effects of vinblastine, leucine, and histidine, and 3-methyladenine on autophagy in Ehrlich ascites cells. Experimental and molecular pathology, 52(1), pp.87–97.
Qin, Z.-H. et al., 2003. Autophagy regulates the processing of amino terminal huntingtin fragments. Human molecular genetics, 12(24), pp.3231–44.
136
Ravikumar, B., 2002. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Human Molecular Genetics, 11(9), pp.1107–1117.
Ravikumar, B. et al., 2005. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nature genetics, 37(7), pp.771–6.
Ravikumar, B. et al., 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nature genetics, 36(6), pp.585–95.
Ritson, G.P. et al., 2010. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. The Journal of neuroscience : the official journal of the Society for Neuroscience, 30(22), pp.7729–39.
Rodriguez-Gonzalez, A. et al., 2008. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer research, 68(8), pp.2557–60.
Rohn, T.T., 2008. Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer’s disease. Brain research, 1228, pp.189–98.
Rohn, T.T. & Kokoulina, P., 2009. Caspase-cleaved TAR DNA-binding protein-43 in Pick’s disease. International journal of physiology, pathophysiology and pharmacology, 1(1), pp.25–32.
Rosen, D.R. et al., 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 362(6415), pp.59–62.
Rothstein, J.D., Martin, L.J. & Kuncl, R.W., 1992. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. The New England journal of medicine, 326(22), pp.1464–8.
Rowland, L.P., 1998. Diagnosis of amyotrophic lateral sclerosis. Journal of the neurological sciences, 160 Suppl , pp.S6–24.
Rowland, L.P., 2001. How amyotrophic lateral sclerosis got its name: the clinical-pathologic genius of Jean-Martin Charcot. Archives of neurology, 58(3), pp.512–5.
Rubin, D.M. & Finley, D., 1995. Proteolysis: The proteasome: a protein-degrading organelle? Current Biology, 5(8), pp.854–858.
Rubinsztein, D.C., 2006. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature, 443(7113), pp.780–6.
Rutherford, N.J. et al., 2008. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. G. A. Cox, ed. PLoS genetics, 4(9), p.e1000193.
137
Ryu, H. et al., 2005. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. Journal of neurochemistry, 93(5), pp.1087–98.
Sakai, Y. et al., 1998. Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. The Journal of cell biology, 141(3), pp.625–36.
Sarkar, S. et al., 2005. Lithium induces autophagy by inhibiting inositol monophosphatase. The Journal of cell biology, 170(7), pp.1101–11.
Sarkar, S. et al., 2007. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nature chemical biology, 3(6), pp.331–8.
Sasaki, S., 2011. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. Journal of neuropathology and experimental neurology, 70(5), pp.349–59.
Saudou, F. et al., 1998. Huntingtin Acts in the Nucleus to Induce Apoptosis but Death Does Not Correlate with the Formation of Intranuclear Inclusions. Cell, 95(1), pp.55–66.
Schmid, B. et al., 2013. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proceedings of the National Academy of Sciences of the United States of America, 110(13), pp.4986–91.
Scotter, E.L. et al., 2014. Differential roles of the ubiquitin proteasome system (UPS) and autophagy in the clearance of soluble and aggregated TDP-43 species. Journal of cell science, p.jcs.140087–.
Seibenhener, M.L. et al., 2004. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Molecular and cellular biology, 24(18), pp.8055–68.
Sephton, C.F. et al., 2010. TDP-43 is a developmentally regulated protein essential for early embryonic development. The Journal of biological chemistry, 285(9), pp.6826–34.
Shaw, C.E., Al-Chalabi, A. & Leigh, N., 2001. Progress in the pathogenesis of amyotrophic lateral sclerosis. Current Neurology and Neuroscience Reports, 1(1), pp.69–76.
Shaw, P.J., 2005. Molecular and cellular pathways of neurodegeneration in motor neurone disease. Journal of neurology, neurosurgery, and psychiatry, 76(8), pp.1046–57.
Shiina, Y. et al., 2010. TDP-43 dimerizes in human cells in culture. Cellular and molecular neurobiology, 30(4), pp.641–52.
138
Simonsen, A. et al., 2008. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy, 4(2), pp.176–84.
Simonsen, A. & Tooze, S.A., 2009. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. The Journal of cell biology, 186(6), pp.773–82.
Smith, R.E. & Farquhar, M.G., 1966. Lysosome function in the regulation of the secretory process in cells of the anterior pituitary gland. The Journal of cel l biology, 31(2), pp.319–47.
La Spada, A.R. et al., 1991. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature, 352(6330), pp.77–9.
Spilman, P. et al., 2010. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. P. F. Ferrari, ed. PloS one, 5(4), p.e9979.
Staats, K.A. et al., 2013. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Molecular neurodegeneration, 8(1), p.31.
Stefanis, L. et al., 2001. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience, 21(24), pp.9549–60.
Stein, M.-P. et al., 2005. Interaction and functional analyses of human VPS34/p150 phosphatidylinositol 3-kinase complex with Rab7. Methods in enzymology, 403, pp.628–49.
Strong, M.J. et al., 2007. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Molecular and cellular neurosciences, 35(2), pp.320–7.
Stutzmann, J.-M. et al., 2002. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS drug reviews, 8(1), pp.1–30.
Szeto, J. et al., 2006. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy, 2(3), pp.189–99.
Takagi, T. et al., 2005. Dextran sulfate suppresses cell adhesion and cell cycle progression of melanoma cells. Anticancer research, 25(2A), pp.895–902.
Tan, J.M.M. et al., 2008. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Human molecular genetics, 17(3), pp.431–9.
139
Tanaka, M. et al., 2004. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. The Journal of biological chemistry, 279(6), pp.4625–31.
Tanji, K. et al., 2012. p62/sequestosome 1 binds to TDP-43 in brains with frontotemporal lobar degeneration with TDP-43 inclusions. Journal of neuroscience research, 90(10), pp.2034–42.
Tdp-, P. et al., 2011. Regulation of autophagy by neuropathological protein TDP-43. The Journal of biological chemistry, 286(52), pp.44441–8.
Thumm, M. et al., 1994. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. F EBS letters, 349(2), pp.275–80.
Tooze, J. et al., 1990. In exocrine pancreas, the basolateral endocytic pathway converges with the autophagic pathway immediately after the early endosome. The Journal of cell biology, 111(2), pp.329–45.
Tsubamoto, Y. et al., 1994. Dextran sulfate, a competitive inhibitor for scavenger receptor, prevents the progression of atherosclerosis in Watanabe heritable hyperlipidemic rabbits. Atherosclerosis, 106(1), pp.43–50.
Tsukada, M. & Ohsumi, Y., 1993. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. F EBS Letters, 333(1-2), pp.169–174.
Udan, M. & Baloh, R.H., 2011. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion, 5(1), pp.1–5.
Urushitani, M. et al., 2010. Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. Journal of neuroscience research, 88(4), pp.784–97.
Vabulas, R.M. & Hartl, F.U., 2005. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science (New York, N.Y.), 310(5756), pp.1960–3.
Vance, C. et al., 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (New York, N.Y.), 323(5918), pp.1208–11.
Verdel, A. & Khochbin, S., 1999. Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. The Journal of biological chemistry, 274(4), pp.2440–5.
Verhoef, L.G.G.C. et al., 2002. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Human molecular genetics, 11(22), pp.2689–700.
140
Voigt, A. et al., 2010. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. M. B. Feany, ed. PloS one, 5(8), p.e12247.
Wang, H.-Y. et al., 2004. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics, 83(1), pp.130–9.
Wang, I.-F. et al., 2012. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proceedings of the National Academy of Sciences of the United States of America, 109(37), pp.15024–9.
Wang, I.-F., Wu, L.-S. & Shen, C.-K.J., 2008. TDP-43: an emerging new player in neurodegenerative diseases. Trends in molecular medicine, 14(11), pp.479–85.
Wang, X. et al., 2010. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neuroscience letters, 469(1), pp.112–6.
Ward, W.F., 2002. Protein degradation in the aging organism. Progress in molecular and subcellular biology, 29, pp.35–42.
Warraich, S.T. et al., 2010. TDP-43: a DNA and RNA binding protein with roles in neurodegenerative diseases. The international journal of biochemistry & cel l biology, 42(10), pp.1606–9.
Watanabe, M. et al., 2001. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiology of disease, 8(6), pp.933–41.
Watson, M.R. et al., 2008. A Drosophila Model for Amyotrophic Lateral Sclerosis Reveals Motor Neuron Damage by Human SOD1. Journal of Biological Chemistry, 283(36), pp.24972–24981.
Webb, J.L. et al., 2003. Alpha-Synuclein is degraded by both autophagy and the proteasome. The Journal of biological chemistry, 278(27), pp.25009–13.
Webb, J.L., Ravikumar, B. & Rubinsztein, D.C., 2004. Microtubule disruption inhibits autophagosome-lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. The international journal of biochemistry & cell biology, 36(12), pp.2541–50.
Welchman, R.L., Gordon, C. & Mayer, R.J., 2005. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nature reviews. Molecular cell biology, 6(8), pp.599–609.
Winton, M.J. et al., 2008. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. The Journal of biological chemistry, 283(19), pp.13302–9.
Wong, E. & Cuervo, A.M., 2010. Autophagy gone awry in neurodegenerative diseases. Nature neuroscience, 13(7), pp.805–11.
141
Wooten, M.W. et al., 2008. Essential Role of Sequestosome 1/p62 in Regulating Accumulation of Lys63-ubiquitinated Proteins. Journal of Biological Chemistry, 283(11), pp.6783–6789.
Worms, P.M., 2001. The epidemiology of motor neuron diseases: a review of recent studies. Journal of the neurological sciences, 191(1-2), pp.3–9.
Xilouri, M. & Stefanis, L., 2011. Autophagic pathways in Parkinson disease and related disorders. Expert reviews in molecular medicine, 13, p.e8.
Yim, M.B. et al., 1996. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proceedings of the National Academy of Sciences of the United States of America, 93(12), pp.5709–14.
Yoo, Y.-E. & Ko, C.-P., 2012. Dihydrotestosterone ameliorates degeneration in muscle, axons and motoneurons and improves motor function in amyotrophic lateral sclerosis model mice. L. Mei, ed. PloS one, 7(5), p.e37258.
Yoo, Y.-E. & Ko, C.-P., 2011. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Experimental neurology, 231(1), pp.147–59.
Yorimitsu, T. & Klionsky, D.J., 2005. Autophagy: molecular machinery for self-eating. Cell death and differentiation, 12 Suppl 2(S2), pp.1542–52.
Yu, W.H. et al., 2005. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. The Journal of cell biology, 171(1), pp.87–98.
Zhang, T. et al., 2012. Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. The Journal of biological chemistry, 287(11), pp.8371–82.
Zhang, X. et al., 2011. Rapamycin treatment augments motor neuron degeneration in SOD1 G93A mouse model of amyotrophic lateral sclerosis. Autophagy, 7(4), pp.412–425.
Zhang, Y. et al., 2003. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. The EMBO journal, 22(5), pp.1168–79.
Zhang, Y. et al., 2007. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. The Journal of neuroscience : the official journal of the Society for Neuroscience, 27(39), pp.10530–4.
Zhang, Y.-J. et al., 2009. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America, 106(18), pp.7607–12.
142
Zhang, Y.-J. et al., 2010. Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. Molecular neurodegeneration, 5, p.33.