

The acrolein and acrylonitrile synthesis over a bismuthmolybdate catalyst : kinetics and mechanismCitation for published version (APA):Lankhuijzen, S. P. (1979). The acrolein and acrylonitrile synthesis over a bismuth molybdate catalyst : kineticsand mechanism. Eindhoven: Technische Hogeschool Eindhoven. https://doi.org/10.6100/IR1200
DOI:10.6100/IR1200
Document status and date:Published: 01/01/1979
Document Version:Publisher’s PDF, also known as Version of Record (includes final page, issue and volume numbers)
Please check the document version of this publication:
• A submitted manuscript is the version of the article upon submission and before peer-review. There can beimportant differences between the submitted version and the official published version of record. Peopleinterested in the research are advised to contact the author for the final version of the publication, or visit theDOI to the publisher's website.• The final author version and the galley proof are versions of the publication after peer review.• The final published version features the final layout of the paper including the volume, issue and pagenumbers.Link to publication
General rightsCopyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright ownersand it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights.
• Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal.
If the publication is distributed under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license above, pleasefollow below link for the End User Agreement:
www.tue.nl/taverne
Take down policyIf you believe that this document breaches copyright please contact us at:
providing details and we will investigate your claim.
Download date: 18. Feb. 2020


THE ACROLEIN AND ACRYLONITRILE SYNTHESIS OVER A BISMUTH MOLYBDATE CATALYST

' THE ACROLEIN AND ACRYLONITRILE SYNTHESIS
OVER A BISMUTH MOLYBDATE CATALYST
Kinetics and mechanism
PROEFSCHRIFT
TER VERKRIJGING VAN DE GRAAD VAN DOCTOR IN DE TECHNISCHE WETENSCHAPPEN AAN DE TECHNISCHE HOGESCHOOL EINDHOVEN, OP GEZAG VAN DE RECTOR MAGNIFICUS, .PROF. DR. P. VAN DER LEEDEN, VOOR EEN COMMISSIE AANGEWEZEN DOOR HET COLLEGE VAN DEKANEN IN HET OPENBAAR TE· VERDEDIGEN OP
VRIJDAG 22 JUNI 1979 TE 16.00 UUR
DOOR
SIMON PIETER LANKHUIJZEN
GEBOREN TE BREDA
ORUK: WIBRO HELMOND

Dit proefschrift is goedgekeurd door de promotoren:
Prof. drs. H. s. van der Baan, le promotor
Prof. dr. G. C. A. Schuit, 2e promotor

Aan Anny
Aan Jannelies
Gerdiene
Joanne
Machteld
Han

CONTENTS
1. Introduction
1.1. General
1.2. Acrylonitrile manufacture
1
2
1.3. The mechanism of the oxidation and arnmoxidation of
propene 3
1.4. Aim and outline of the present investigation 4
2. Literature
2.1. Introduction
2.2. Kinetics
2.3. Adsorption of reactants and products
2.4. Hydrocarbon surface intermediates
2.5. Nitrogen containing surface intermediates origi-
nating from ammonia
2.6. ~ole of oxygen
2.7. Catalyst
2.8. Models
3. Apparatus and analysis
3.1. Introduction
3.2. The flow reactor system
3.2.1. Analysis
3.3. The thermobalance
3.4. The pulse reactor system
3.4.1. Analysis
3.5. Safety
3. 5 .1. Toxicity
3.5.2. Flammability and explosive ranges
4. The catalyst
4.1. Introduction
7
8
9
12
13
14
15
16
21
23
27
32
33
34
35
35
37
39
4.2. The structure of the bismuth molybdate catalyst 40
4.3. Catalyst preparation 43
5. Experimental methods
5.1. Introduction 47

5.2. The rate of reaction 48
5.3. Factors governing the reactor behavioUr 49
5.3.1. Plug flow 49
5.3.2. Temperature gradients 51
5.3.3. Catalyst dilution 53
5.3.4. Mass and heat transfer 54
5.3.5. Pressure drop 57
5.4. Data handling and analysis of errors 57
6. Results and discussion
6.1. Introduction 61
6.2. Flow experiments 62
6.2.1. Introduction 62 6.2.2. Preliminary experiments 62
6.2.3. The oxidation of propene to acrolein 64
6.2.3.1. Experiments at 673 K 64
6.2.3.2. Experiments at other temperatures 68
6.2.4. The oxidation of ammonia to nitrogen 69
6.2.5. The ammoxidation of acrolein to acrylonitrile 70
6.2.5.1. Preliminary experiments 71
6.2.5.2. Experiments at 673 K 72
6.2.6. The ammoxidation of propene to acrylonitrile 74
6.2.6.1. Introduction and preliminary ex-
periments 74
6.2.6.2. Experiments at 673 K 76
6.2.6.3. Experiments at other temperatures 79
6.2.6.4. Experiments at non-initial con-
ditions 80 6.3. Thermobalance experiments 84
6.3.1. Introduction 84
6.3.2. Preliminary experiments 85 6.3.3. Reduction of the catalyst with propene 85
6.3.4. Reduction of the catalyst with hydrogen 90
6.3.5. Reoxidation of a reduced catalyst 93
6.3.6. The reduction of the catalyst with mixtures
of propene, nitrogen and small quantities
of oxygen 94

6.4. Pulse experiments 6.4.1. Introduction 100
6.4.2. Preliminary experiments 101
6.4.3. Experiments with propene-helium mixtures 103
6.4.4. Experiments with mixtures of propene and
oxygen 105
6.4.5. Experiments with mixtures of ammonia and
helium 107
6.4.6. Experiments with mixtures of propene, ammo-
nia and helium 107
7. Final discussion
7.1. Introduction
7.2. The mechanism of the catalytic reaction 113
113
7.3. Adsorption and adsorption sites 115
7.3.1. The adsorption of ammonia 115
7.3.2. The adsorption of propene 117
7.3.3. The adsorption of acrolein 120
7.3.4. The role of the catalyst in the activation
of molecular oxygen
7.4. The formation of the main products
7.4.1. The formation of acrolein
7.4.2. The formation of nitrogen
7.4.3. The formation of acrylonitrile
7.4.4. The formation of water
7.5. The kinetic model
7.6. Selectivities in the acrylonitrile synthesis reac-
tion
List of symbols
summary
Samenvatting Dankwoord
121
125
125
128
129
130
130
134
139
143
147
150

CHAPTER 1
INTRODUCTION
1.1. General
Since acrolein was discovered in 1843 by Redtenbacher
(1) and acrylonitrile was synthesized fifty years later
by Moureu (2) these compounds remained laboratory chemi
cals until the development of some large scale polymeri
zation processes. Preparative methods were replaced by
commercial catalytic processes and expensive chemicals by
cheaper raw materials to obtain these important unsatura
ted intermediates in chemical industry.
As a result of the development of certain heterogeneous
catalytic processes acrylonitrile is obtained nowadays
from propene, ammonia and air instead of acetylene and hydrogen cyanide. Contrary to the latter the former process
is based on catalytic oxidation, which is now a major tool
for the incorporation of carbonyl and nitrile groups in
hydrocarbons. The necessity of a catalytic oxidation re-'
action follows from thermodynamic calculations. The com-
plete oxidation of propene and ammonia to carbon dioxide
nitrogen and water at temperatures of interest is prefer
red to the formation of acrylonitrile or acrolein.
An intensive research effort has been directed towards
the development of selective catalysts for the partial
oxidation of hydrocarbons to a number of products. This
has led to the development of a number of selective oxi
dation catalysts made from transition metal oxides.
A major event in the history of oxidation catalysis was
the discovery of bismuth molybdate (3,4,5,6) as a selec
tive catalyst for the partial oxidation of propene and
also in an one-step operation for the ammoxidation of pro
pene. As other metal oxide combinations this catalyst is
1

very versatile, being effective in a number of processes,
e.g. the oxidative dehydrogenation of n•butenes and me
thyl-butene to butadiene and isoprene and the formation of
aromatic carbonyls and nitriles.
Although in the 1960's the catalytic ammoxidation of propene over bismuth molybdate became a commercial process,
the basic understanding of the role of the catalyst lagged
far behind the technological knowledge of the process.
Many research studies have been carried out to characte
rize the excellent catalytic properties of bismuth molybdate based catalysts. These studies have increased the un
derstanding of oxidation and.ammoxidation kinetics and
mechanisms, but the controlling parameters are not yet
completely understood.
Two basic principles however have become clear. Firstly
the oxygen atoms for the selective reaction come from the
catalyst. Secondly the metal oxide combinations must be
able to transfer oxygen by a redox reaction. The latter
requirement explains the suitability of transition metal
oxides in selective oxidation. The catalyst surface is
alternately in an oxidized and reduced state. Anion va
cancies play an important role.
1.2. Acrylonitrile manufacture
The impressive growth of the acrylonitrile production
(average annual growth between 16 and 20% from 1960-1975)
has resulted from the manufacturing technology that could
use cheaper raw materials at high selectivity. Older com
mercial processes were based on the reaction of acetylene with hydrogen cyanide or the reaction of ethylene oxide
with hydrogen cyanide followed by dehydration. Both pro
cesses required more expensive starting materials and more * extensive safety measures than the modern (SOHIO) process
which is based on the direct ammoxidation of propene, according to
(r 1.1)
2

The other advantages of this process are the high selecti
vity and the long lifetime and high activity of the cata
lyst.
After the use of a bismuth molybdate catalyst promoted
by phosphorus (composition 50% Bi9 P Mo12 o52/50% Si02 )
and an uranium-antimony oxide catalyst (composition usb3 o10 > nowadays the socalled multicomponent molybdates {MCM)
catalyst (SOHIO 41) is the most important catalyst (7,8).
It is composed of a variety of elements like nickel, cobalt, iron, manganese, potassium, phosphorus but always
contains bismuth and molybdenum. Today SOHIO's process
accounts for over 95% of the world's installed capacity
of 2.4 106 metric tons/year.
Some of the older ammoxidation processes used multitu
bular fixed bed reactors but all major modern processes
use fluidized bed reactors. The advantages of the fluid
bed are a better temperature control and a removal of the
limitations on propene and ammonia concentrations due to
the explosivity of the reactor feed (9), The process ope
rates at 1-3 bar and 673-773 K. The main feature of the
process is the high conversion obtained on a once-through
basis in the fluid bed, thanks to the high selectivity of the catalyst.
Over the past two decades the rapidly expanding market
for acrylonitrile has shifted more and more from the acry
lonitrile elastomers (NBR) to the acrylic fibers and re
sins (ABS and SAN). High impact resistance, low porosity
acrylic copolymers with over 75% acrylonitrile are a recent development in the manufacture of bottles and con
tainers. In the near future the demand for polyacrylamide
may capture a good deal of the acrylonitrile market (10).
1.3. The mechanism of the oxidation and ammoxidation of
propene
During the past ten years the commercial production of
acrylonitrile has been attended with an increasing number
of investigations dealing with the elucidation of the
3

mechanism of the ammoxidation reaction. Several reviews
have appeared about reaction mech~nisms of olefin oxida
tion { 11-18) •
In literature relatively little attention has been paid
to the mechanism of the ammoxidation reaction itself. On
the other hand research has been focussed mainly on the
behaviour of the catalyst under various conditions that
differ considerably from commercial ones.
Generally a relatively simple test reaction is used for
that purpose. It has been stated that in commercial multi
component catalysts bismuth molybdate performs the cataly
tic role. We decided to investigate the ammoxidation reac
tion itself in order to obtain further knowledge of the re
action kinetics and insight in the mechanism of the acry
lonitrile synthesis.
1.4. Aim and outline of the present investigation
It is the aim of this investigation to derive a reac
tion model based on kinetic results for the catalytic
ammoxidation of propene. Unsupported y-bismuth molybdate
is used as a catalyst.
In chapter 2 a survey is given of the literature with
respect to the subject of this investigation.
The apparatus and the methods of analysis for studying
the kinetics are described in chapter 3.
Chapter 4 deals with the preparation and the properties
of the catalyst.
Chapter 5 is devoted to the experimental methods
applied in the kinetic studies of heterogeneous catalysis.
Chapter 6 deals with the kinetic experiments carried
out in the different reactors. The kinetics of the propene
oxidation and ammoxidation reactions are studied to de
termine the conditions for the selective reaction proce
dures. The significance of acrolein in the reaction model
is investigated by means of its ammoxidation to acryloni
trile. Special attention is paid to the oxidation of ammo
nia to nitrogen.
4

Thermobalance and pulse reactor experiments are per
formed to investigate the reaction of propene and ammonia
with the catalyst in the absence of oxygen in the gas phase.
In this way the role of gasphase and catalyst oxygen is
studied during the catalytic reaction.
Finally in chapter 7 the reaction model for the diffe
rent oxidation and ~oxidation reactions based on the
experimental results is given. In a final discussion a mechanistic model is proposed which may contribute to the
understanding of the catalytic activity of bismuth molyb
date.
For the explanation of symbols, abbreviations and sub
scripts see List of Symbols.
References
1. Redtenbacher, J., Anw. Chern. Liebigs 47, 114 (1843)
2. Moureu, c., Bull. Soc. Chim. Fr. ~ (3) 424 (1893)
3. Idol, J.D. (Standard Oil Co.), u.s. Pat. 2.904.580
(Sept. 15, 1959)
4. Callahan, J.L., Foreman, R.W., Veatch, F. (Standard
Oil Co.) u.s. Pat. 3.044.966 (July 17, 1962)
5. Veatch, F., Callahan, J.L., Idol, J.D., Milberger,
E.C., Chern. Engng. Progr. 56 (10) 65 (1960)
6. Callahan, J.L., Grasselli, R.K., Milberger, E.C., ' Strecker, H.A., Ind. Engng. Chern. Prod. Res. Dev. ~.
134 (1970)
7. Krabetz, R., Chern. Irig. Techn. 46, 1029 (1974)
8. Wolfs, M.W.J., Thesis Eindhoven (1974)
9. Anon, Hydroc. Proc. 56 (11) 124 (1977)
10. Pujado, P.R., Vora, B.V., Krueding, A.P., Hydroc.
Proc. 56 (5) 169 (1977)
11. Sachtler, W.M.H., Catal. Rev. !• 27 (1970)
12. Margolis, L.Ya., Catal. Rev. !• 241 (1973)
5

13. Hucknall, D.J., Selective oxidation of hydrocarbons. Acad. Press, London (1974)
14. Skarchenko, V.K., Russ. Chem. Rev.~~ 731 (1977)
15. Schuit, G.C.A., J. Less. Com. Met: 36, 329 · (1974)
16. Gates, B.C., Katzer, J.R., Schuit, G.C.A., Chemistry
of Catalytic Processes, Ch. IV, McGraw Hill N.Y. (1979)
17. van der Wiele, K., van den Berg, P.J., in Bamford
C.H. and Tipper C.H.F. (Eds.) Comprehensive Chemical
Kinetic~ Vol. 20 Complex Catalytic Processes, Chapter
2 1 12~ Elsevier Publ. Cy Amsterdam 1978
18. Keulks, G.W., Adv. Catal. 27, 183 (1978)
6

CHAPTER 2
LITERATURE
2.1. Introduction
During the past two decades more than 400 papers and
reviews have been published about the selective oxidation of olefins in general and the ammoxidation of propene
over bismuth molybdates or bismuth molybdate containing
catalysts in particular.
In this chapter we will give a brief literature survey
to situate the subject of our investigation. It is not our
aim however to add a new comprehensive review of the li
terature to the excellent ones that have appeared already (1,2,3).
Catalytic oxidation reactions can be explained accor
ding to two different mechanisms, viz.
a) the reduction-oxidation mechanism, proposed by Mars
and van Krevelen (4) operating in the higher tempe
rature range;
b) the associative mechanism set up by Roiter (5) at
lower temperatures.
In the redox mechanism two separate steps are distinguish
ed: in the first step the hydrocarbon is oxidized with
lattice oxygen whereas in the second step the reduced
oxide is reoxidized by oxygen of the gasphase. In the
associative mechanism a reaction between adsorbed oxygen
species and the hydrocarbon occurs. Evidence for the two
mechanisms is obtained from i~otopic exchange experiments,
as has been pointed out by Boreskov (6) and Winter (7,8)
and from catalyst reduction experiments carried out by
Batist et al (9) and Sachtler et al (10).
Bismuth molybdate catalysts show a high activity in com
bination with a good selectivity both in the oxidation and
7

in the ammoxidation of propene. During the oxidation of
propene besides acrolein only small quantities of carbon dioxide, carbon monoxide, acetaldehyde and formaldehyde are
formed. Acrylonitrile is the main product of the propene
ammoxidation. Other products are acetonitrile, hydrogen cyanide, carbon dioxide and carbon monoxide whereas acrolein
is only a trace product. The stoichiometric equations are
(r 2.1)
(r 2.2)
2.2. Kinetics
Broadly there is a great similarity in the overall features of the oxidation and ammoxidation of propene over bismuth molybdate catalysts: the rates of oxidatiop and
ammoxidation are both first order with respect to propene
and zero order with respect to oxygen. The rate of ammoxi
dation is zero order in ammonia (11,12).
Activation enthalpies for the formation of acrolein and
acrylonitrile show a considerable spread mainly caused by
the different catalysts and temperature ranges as can be seen in table 2.1.
Eact (kJ/mol) Bi/Mo T(K) Ref. C3H40 C3H3N mol mol-l
84 38 2/1 670 (13)
54 - 1/1 650-825 (14)
121 - 1/1 670-730 (15)
159 71 1/1 650-750 (16)
71 - .74/1 700-775. (17)
104 104 .74/1 670 (13)
84 75 BigPMo12o52/Si02 670-700 (12)
Table 2.1. Activation enthalpies (kJ mol-l) for the forma~ tion of acrolein and acrylonitrile over different bismuth molybdate catalysts.
8

The rate of ammoxidation of acrolein, according to the
stoichiometric equation
(r 2.3)
is first order with respect to acrolein and zero order
both in ammonia and oxygen (12,13). The activation enthalpy is 29 kJ mol-l (12).
The rate of oxidation of ammonia over bismuth molybdate
according to the stoichiometric equation
+ (r 2 .4)
is first order with respect to ammonia and zero order with
respect to oxygen. The activation enthalpy is 155 kJ mol-l
( 18) •
If we compare the rate constant data presented by
Callahan et al (12) with those of Cathala et al (19)
carried out with slightly different catalysts it becomes
clear that the rate of propene ammoxidation at 700 K is
higher than the rate of propene oxidation. Callahan (12)
found the rate of acrolein ammoxidation at least twice as
high as the rate of propene ammoxidation. Contrary to
Shelstad et al (20), Callahan et al (12) conclude that
acrylonitrile is formed largely by a mechanism not in
volving acrolein as a vapour phase intermediate.
2. 3. AdsorE,t'i,on of reactants an:d ·products
The adsorption of the reactants and products of the
ammoxidation of propene on the catalyst has been studied
by Matsuura et al (18,21,22), who investigated not only the adsorption behaviour of a fully oxidized but also that
of a partly reduced catalyst and of Bi2o 3 and Moo3 • Mat
suura linked the adsorption data obtained at low pressures
and at temperatures between 325 and 475 K to the performance of some oxidation catalysts at atmospheric pressure
and temperatures above 673 K in order to develop a
9

reaction mechanism. He distinguishes between two types of
adsorption viz. the socalled A-type and the B-type adsorption.
The A-type is an activated, strong and slow adsorption, observed for butadiene, acrolein and ammonia on oxidized
Bi2Mo06 and for acrolein on Bi2o 3 • All adsorptions are of
the dual site type except the butadiene adsorption which
is a single site type. Enthalpies of adsorption are between 88 -1 .
and 100 kJ mol • Prereduction of the catalyst linearly de-creases the number of A-sites, so an A-site contains an
oxygen ion (OA). To allow for the two types of adsorption and for a similar adsorption of acrolein on Bi2o 3 it is
assumed that the A-site contains two anion vacancies (VBi)
located at two Bi-ions next to the oxygen ion OA. So the
A-site is VBiOAVBi' The B-type adsorption is a weak and fast adsorption
observed for butadiene, acrolein, olefins and ammonia.
This type of adsorption occurs on Bi 2Mo06 as well as on
Moo3 , but not on Bi2o 3 ~ On Bi2Moo6 all B-type adsorptions are of the dual site kind, except the ammonia adsorption. Enthalpies of adsorption are in the range of 25-50 kJ mol-l.
Previous reduction does not remove B .. sites, provided the
reduction temperature does not exceed 673 K. At tempera~
ture above 673 K Batist et al (23) found a rapid reduction
of the catalyst by butene-1 at degrees of reduction less than 8.3% and without loss of activity after reoxidation.
According to Matsuura (21) the reoxidation above 673 K is
first order in oxygen with an activation enthalpy of 72 kJ mol-1 •
The removal of B-sites, mentioned by Matsuura is probably connected with some rearrangement in the solid viz.
the formation of metallic Bi in a separate phase. This
phenomenon has been mentioned by Batist et al '(24,25).
B-sites are claimed to be combinations of an anion vacancy
(VM0
) and two oxygen ions (OB). So the B-site is OBVMooB. The A-type adsorption of ammonia and acrolein is strong
and the enthalpies of adsorption are so high that desorption
can only occur at reaction tP;mperatures. Adsorption of.
10

oxygen on non reduced catalysts does not occur. However, the catalyst shows some reversible dissociation when . the gas phase oxygen partial pressure is lower than the
equilibrium oxygen pressure (i.e. p02
eq (673 K) = 1.3 10-8
bar). According to Matsuura (21) the adsorption of oxygen
on partially reduced catalysts at room temperature is
small, rapid and independent of the degree of reduction
and does not lead to complete reoxidation.
Between 373 and 673 K the rate of reoxidation is zero order in oxygen. The enthalpy of activation is 113 kJ mol-1 ,
a value found also by Batist et al (9) for the reoxidation
of reduced Bi 2Mo 2o9 • Above 673 K the rate of reoxidation
becomes first order with respect to oxygen and has an
enthalpy of activation of- about 72 kJ mol-l, depending on the degree of reduction. According to Matsuura the acro
lein adsorption occurs on both A and B-sites. The strong and slow adsorption on site A, also observed on Bi2o3 is
a dual site adsorption. This acrolein adsorption fits the
adsorption model proposed by Sachtler et al (26}. This
dual site adsorption must influence the dual site adsorp
tion of propene on site B, which needs also OB' This could
be verified experimentally as the weak propene adsorptiqn
decreased after a pretreatment of the catalyst with acro
lein.
The adsorption behaviour of ammonia on an oxidized catalyst is very complicated. Matsuura (18) concludes that the
strong dual site adsorption is connected with the A-site,
with the donation of a proton to OB of the B-site. The ammonia adsorption on partially reduced catalysts is con
nected with a reduced A-site.
The weak and dual site adsorption of propene on site B
decreases after a pretreatment of the catalyst with ammo
nia. Experimental data of the strong adso~ption of ammonia
on Moo3 and Bi2o 3 are lacking because of the nitrogen formatio.n already occuring at low temperatures. Kfivanek et
al (27) calculated the enthalpy of adsorption of propene under
reaction conditions at 440 K on bismuth molybdate to be 130 kJ mol-1 .
11

2.4. Hydrocarbon surface intermediates
By the use of isotopic labels it is established by
Sachtler et al (10), McCain et al (28) and Adams et al
(29,30) that the oxidation of propene over bismuth molyb
date proceeds via the formation of the allylic interme
diate which is negatively charged. According to Schuit (2)
the proton is donated to an 0 2- ion at the surface, expe
rimentally confirmed by Beres et al (31), and the carbanion
is bonded to a metal ion at an anion vacancy. This mecha
nism resembles that taking place during the chemisorption
of benzaldehyde on a Sno2-v2o 5 catalyst, studied by
Sachtler (32).
Recent molecular orbital calculations by Haber et al
(33) carried out for different transition-metal cations
support the postulate that the IT-bonding electrons are
transferred from the allylic intermediate to the Mo 6+ ion.
The Mo 6+ is reduced to Mo 5+ or Mo 4+ and the positive char
ge on the c 3H5+-ion is concentrated on the terminal C
atoms in a symmetrical distribution. After the transfer of 2-electrons the allylic intermediate is cr-bonded to an 0
as was confirmed by Kondo et al (34). Dozono et al (35)
studied the ammoxidation of 3-13c propene at 450°C in the
presence of bismuth molybdate. Half of 3-13c in the acry
lonitrile was found to be in the CN-group. This points to
a symmetrical intermediate also in the acrylonitrile syn
thesis. The appearance of 13c in both the methyl- and the
cyanogroup of acetonitrile, although not completely dis
tributed (60/40 respectively) can only result from bond
rupture in the allylic intermediate rather than from the
breakage of a C = C bond in propene, acrylonitrile or
acrolein.
Further dehydrogenation must lead to a c 3H4-interme
diate and proton donation to another o 2- ion. Adams et al
(29,30) suggested that the allylic intermediate undergoes
this hydrogen abstraction before the incorporation of
oxygen which has been experimentally confirmed by means of
kinetic isotope effect measurements. Cathala et al (19)
12

connected this step with a parallel bond rupture which
gives rise to degradation products. Daniel and Keulks (36)
reported at 725 K an enhanced conversion of propene in a
reactor having a large post-catalytic volume. It appeared
that a surface-initiated homogeneous gas phase reaction
caused the formation of side products. Without the post
catalytic volume this formation disappeared, Recently
Kobayashi et al (37} have studied the mechanism of the
oxidation of propene by applying a transient response me
thod. It was found that a stable surface intermediate exists which can be formed either from propene or from
acrolein. Further dehydrogenation of the c 3H4 intermediate is highly
unlikely. In the case of ammoxidation Cathala et al (19)
supposed that dehydrogenation occurs after the formation
of allylidene-imine (C 3H4NH). This was also suggested by
Grassel+i et al (38) for the ammoxidation catalysed by Usb3o 10 • 1
2.5. Nitrogen containin<J surface intermediates ori<Jinatin<J from ammonia
The NH2-intermediate follows from the adsorption expe
riments of Matsuura {18). Ammonia is dissociatively ad
sorbed, according to Matsuura donating a proton to an
oxygen ion of the B-site. Ammonia adsorption on a reduced
catalyst is supposed to occur preferentially on the anion
vacancy left after reduction. Matsuura (18) and Cathala
et al (19) drew for mechanistic reasons a parallel between the dehydrogenation of the allylic intermediate and the
amide group and supposed the formation of allylidene
imine, synthesized by Bogdanovic et al (39), which proba
bly has adsorption properties comparable with acrolein and
butadiene. Germain et al (40) classified the oxides that
catalyse the oxidation of ammonia and postulated that the
imine-intermediate is a substitute for the double bonded
oxygen ion. He classified Moo 3 and not Bi 2o 3 among the
oxides that show moderate oxidation activity for ammonia.
13

As mentioned already for the c3H4 intermediate
further dehydrogenation of the imine is supposed to be
very unlikely.
2.6. Role of o~gen
It is generally assumed that the o2- ion on the surface
of the oxide catalyst is responsible for the oxidation of
the hydrocarbon.
Reoxidation by gas phase oxygen leads to the formation of
o2- but needs four electrons for every oxygen molecule,
as follows from the equation
o2 + 4 e + 2 c {r 2.5)
Gates et al (2) suggest a more stepwise donation of
electrons, viz. the formation of some intermediate oxygen - 2- ~ species e.g. o2 1 o2 and o at lower temperatures.
In that region the Mars van Krevelen mechanism does not
apply as was indicated by Boreskov et al (41) and Sancier
et al (42). The evidence of these intermediates is esta
blished by ESR spectroscopy (41). Van Hooff (44) suggested
that these intermediates lead to chain reactions. Haber
(45) assumed the oxygen intermediates to be electrophilic
reagents and the oxidizing species in the total oxidation
of hydrocarbons, whereas lattice oxygen ions are nucleo
philic reagents with non oxidizing properties. Van Dillen
(46) investigated the existence of these species extensi
vely. I 18 16 By means of 0 - 0 exchange, however, it is esta-
blished by Keulks (47) and Wragg et al (48) that bismuth
molybdate catalysts do not exchange with o2 at. temperatu
.res below 773 K in the absence of an oxidation reaction. . 18
Keulks (47) suggested from experiments with o2 gas phase oxygen and Bi 2Mo16o6 that during the oxidation of propene
at 698 K the oxygen of about 500 layers participated in
the reaction and that these layers were oxidized by a
rapid diffusion of oxygen from the bulk of the catalyst
14

rather than by gas phase oxygen. However the gas phase oxy
gen was gradually incorporated in the product. An immediate incorporation would be expected if the reaction with
catalyst oxygen was confined to the surface layer only on
which gas phase oxygen would be chemisorbed. Wragg et al
(48) with experiments at 748-773 K came to the same con
clusion. As also 180 is gradually incorporated in the
carbon dioxide Keulks assumed that the selective and com
plete oxidation of propene occurs at the same site.
Pendleton et al (49) studied the reaction between pro
pene and 18o2 over bismuth molybdate between 623 and 673 K. They showed the incorporation of lattice oxygen into the
acrolein, whereas oxygen for the carbon dioxide formation
in that temperature region comes from both the gas phase
and the lattice. Keulks et al (50} however in a later in
vestigation at 703 K concluded that there is no distinc
tion be~ween the lattice oxygen incorporated into carbon
dioxide and into acrolein.
Sancier et al (42) determined the relative contribution
of sorbed and lattice oxygen during propene oxidation over
silica supported bismuth molybdate between 590 and 670 K in a pulse reactor and concluded that above 623 K lattice
oxygen becomes more important whereas below 623 K the mo
bility of lattice oxygen is low and adsorbed oxygen takes
over the role. Recently van Oeffelen (51) found a rapid
increase of the electrical conductivity during the re
duction of Bi2Mo1 • 02o6 • 06 with propene at 673 K. He
ascribed this phenomenom to the formation of bismuth metal
particles on the surface. Similar evidence was also obtained by Peacock et al (52). E.s.r.-signals due to Mo5+ were detected when the catalyst was exposed to propene
but these signals were absent when oxygen was added (53).
Sancier et al (54) and Burlamacchi et al (55) obtained
the same results.
2.7. Catalyst
Significant contributions to our understanding of the
15

excellent catalytic properties of bismuth molybdate and the
nature of the active phase have been made by Schuit, Ba
tist and coworkers (2,9,56,58).
It would carry us too far to give a literature survey
about the structure of the active catalyst. We refer to
the recent review of Gates et al (2) and to chapter 4.
2.8. Models
Some authors have proposed models for the reaction me
chanism of the oxidation or ammoxidation of propene. These
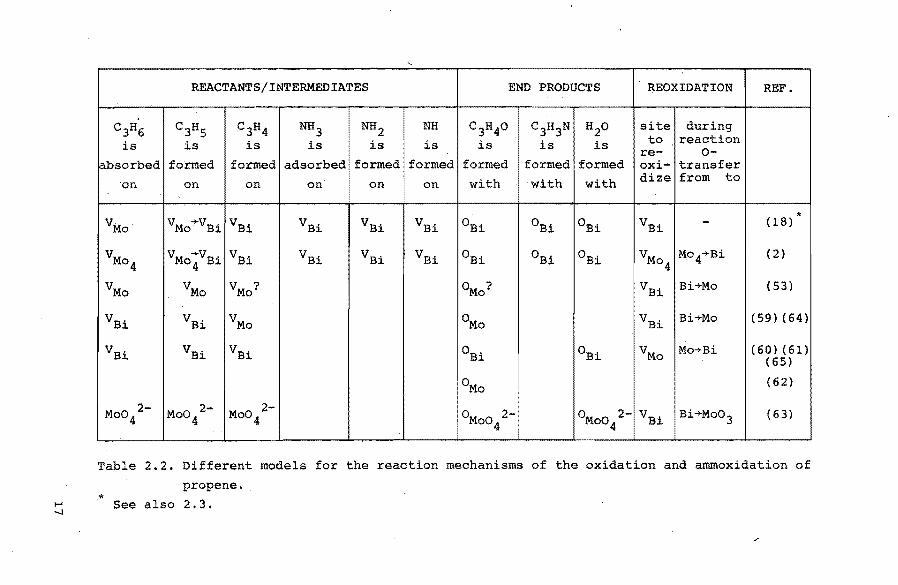
models are summarized in table 2.2 without detailed infor
mation. In chapter 7 these models will be discussed.
16
REACTANTS/INTERMEDIATES
C3H6 C3H5 is is
C3H4 is
NH3 is
NH2 is
NH
is
c3a4o is
END PRODUCTS
C~H3NI H~O ~s ~s
REOXIDATION
site during to . reaction
re- 0-!absorbed formed formedladsorbedlformedlformedlformed formed! formed
with with
oxi- transfer dize from to
on I on on
VM.o
VMo4
VMo
VBi
VBi
2-Mo04
VMo..,.VBiiVBi
VMo~VBiiVBi
VMo
VBi
VBi
2-Mo04
VMo?
VMo
VBi
2-Mo04
on
VBi
VBi
on on
VBi VBi
VBi VBi
with
OBi
OBi
0Mo?
0Mo
OBi
0Mo
2-0Mo04
OBi
OBi
OBi
OBi
OBi
VBi
VM04
Mo4->-Bi
VBi Bi+Mo
VBi IBi+Mo
VMo IMo+Bi
0Moo4
2-IVBi Bi->-Mo0 3
REF.
* (18)
(2)
(53)
(59) (64)
(60) (61) ( 6 5)
( 62)
( 63)
Table 2.2. Different models for the reaction mechanisms of the oxidation and ammoxidation of
propene. * ~ See also 2.3.
~

1. Hucknall, D.J., Selective oxidation of hydrocarbons,
Road Press London (1974)
2. Gates, B.C., Katzer, ~.R., Schuit, G.C.A., Chemistry
of Catalytic Processes, McGraw Hill, Ch. 4 (1979)
3. Vander Wiele, K., van den Berg, P.J., in Bamford C.H.
and Tipper C.F.H. (Eds.), Comprehensive Chemical Ki
netics, Vol. 20 Complex Catalytic Processes, Chapter
2, 123, Elsevier Publ. Cy. Amsterdam (1978)
4. Mars, J., van Krevelen, o.w., Chem. Eng. Sci. Suppl.
l, 41 (1954) 5. Roiter, V.A., Kin. i. Kat • .!_, 63 (1960)
6. Boreskov, G.K., Adv. Cat, 15, 285 (1964)
7. Winter, 8, Winter,
9. Batist,
G,C.A. I
E.R.S., Adv. Cat. 10, 196 (1958) E.R.S., J, Chem. Soc. A, 479 (1968)
Ph,A., Kapteijns, C.J., Lippens, B.C., Schuit,
J. Catal. z, 33 (1967) 10. Sachtler, W,M,H., Rec. Trav. Chim. 82, 243 (1963)
Sachtler, W.M,H., de Boer, N.H., Proc. 3rd Int. Congr.
Catal. Amsterdam 1964, Vol. I, 252, NH Publ. Co. Am
sterdam ( 1965)
11. Adams, C.R., Voge, H,H., Morgan, C.Z., Armstrong, W.E.,
J. Catal. l• 379 (1964) 12. Callahan, J.L., Grasselli, R.K., Milberger, E.C.,
Strecker, H.A., Ind. Engng. Chem. Prod. Res. Dev. 1• 134 (1970)
13. Wragg, R.D., Ashmore, P.G., Hockey, J.A., J. Catal.
ll· 293 (1973) 14. Gorshkov, A.P., Kolchin, I.K., Gribov, J.M., Margolis,
L.Ya,, Kin. i. Kat. 2• 1086 (1968)
15. Keulks, G.W., Rosynek, M.P., Daniel, c., Ind. Engng.
Chem. Prod. Res. Dev. 1Q, 138 (1971)
16. Cathala, M., Germain, J.E., Bull, Soc. Chim. Fr. 2167,
2174 (1971)
17. Peacock, J.M., Parker, A,J,, Ashmore, P.G., Hockey,
J .A., J. Catal, g, 398 (1969)
18. Matsuura, I,, J. Catal. 1.11 420 (1974)
18

19. Cathala, M., Germain, J.E., Bull. Soc. Chim. Fr. 4114
(1970)
20. Shelstad, K.A., Chong, T.C., Can. J. Chern. Engng. 47,
597 (1969)
21. Matsuura, I., Schuit, G.C.A,, J. Catal. ~, 19 (1971)
22. Matsuura, I., Schuit, G.C,A., J. Catal. ~, 314 (1972)
23. Batist, Ph.A., Prette, H.J., Schuit, G.C.A,, J. Catal.
ll· 267 ( 1969) 24. Batist; Ph.A., Bouwens, J.F.H., Schuit, G.C.A., J.
Catal. 25, 1 ( 1972)
25. Batist, Ph.A., Lankhuijzen, S.P., J. Catal. 28, 496
(1973)
26. Sachtler, W.M.H., Dorgelo, G.J.H., Fahrenfort, J.,
Voorhoeve, R.J.H., Proc. 4th Int. Congr. Catal. (1968)
(1), 454 (1971)
27. Krivanek, M., Jiru, P., z. phys. Chemie, Leipzig 256,
(1) 153 (1975)
28. McCain, c.c., Gough, G, 1 Godin, G.W., Nature, Lond.
198, 989 (1963)
29. Adams, C.R., Jennings, T.J., J. Catal. ~, 63 (1963)
30. Adams, C.R., Jennings, T.J., J. Catal. l• 549 (1964)
31. Beres, J., Bruckman, K., Haber, J,, Janas, J., Bull.
Acad. Pol. Sci. Ser. Sc. Chim. 20, (8) 813 (1972)
32. Sachtler, W.M.H., Catal. Rev. ! (1) 27 (1970}
33. Haber, J., Sochacka, M., Grzybowska, B., Golzbiewski,
A., J. Mol. Catal. l• 35 (1975)
34. Kondo, T., Saito, s., Tamaru, K., J, Am. Chern. Soc.
2§_, 6857 (1974)
35. Dozono, T., Thomas, D.W., Wise, H., J. Chern. Soc. Far.
Transa~t. I 69, 620 (1973)
36. Daniel, C., Keulks, G.W., J. Catal. ~, 529 {1972)
37. Kobayashi, M. , Futaya, R. , J. Catal. 56, 73 ( 1979)
38. Grasselli, R.K., Suresh, D.D., J. Catal. 25, 273 (1972)
39. Bogdanovic, B., Velie, M., Angew. Chern. 79, 818 (1967)
40. Germain, J.E., Perez, R., Bull. Soc. Chim. Fr. 2042
(1972)
41. Boreskov, G.K., 2nd Jap. Sov. Catal. Sem. Tokyo (1973)
42. Sancier, K,M., Wentreck, P.R., Wise, H., J. Catal. 39,
141 (1975)
19

43. Lunsford, J.H., Catal. Rev. l• 135 (1973) 44. van Hooff, J.H.C., Thesis, Eindhoven (1968)
45. Haber, J., 4th Roermond Conf. on Catal. (1978)
46. van Dillen, A.J., Thesis, Utrecht (1977)
47. Keulks, G,W,, J. Catal. ~' 232 (1970) 48. Wragg, R.D., Ashmore, P.G., Hockey, J.A., J. Catal.
22, 19 (1971)
49. Pendleton, P., Taylor, D01 J. Chem. Soc. Far. Trans.--I
72, 1114 (1976)
50. Keulks, G.W., Krenzke, L.D., Proc. 6th Int. Congr.
Catal. ~' 806 (1977) 51. van Oeffelen, D.A.G., Thesis, Eindhoven (1978)
52. Peacock, J.M., Parker, A.J., Ashmore, P.G., Hockey,
J.A., J. Catal. ~' 387 (1969) 53. Peacock, J.M., Sharp, M.J., Parker, A.J., Ashmore,
P.G., Hockey, J.A., J. Catal. ~· 379 (1969)
54. Sancier, K.M., Dozono, T., Wise, H., J .• Catal. ll' 270 ( 1971)
55. Burlamacchi, L., Martini, G., Ferroni, E., J. Chem.
Soc. Far. Trans. I ~' 1586 (1972) 56. Bleijenberg, A.C.A.M., Lippens, B.C., Schuit, G.C.A.,
J. Catal. !1 481 (1965)
57. Batist, Ph.A., Lippens, B.C., Schuit; G.C.A., J. Catal.
1· 55 (1966) 58. Batist, Ph.A., der Kinderen, A., Leeuwenburgh, Y.,
Metz, F., J. Catal. 12, 45 (1968)
59. Haber, J., Grzybowska, B., J. Catal. 28, 489 (1973)
60. Otsubo, T., Miura, H,, Morikawa, Y., Shirasaki, T., J.
Catal. ~· 240 (1975) 61. Miura, H., Otsubo, T., Shirasaki, T., Morikawa, Y,,
J. Catal. 56, 84 (1979)
62. Trifiro, F., Kubelkova, L., Pasquon, I., J. Catal. ~'
121 (1970)
63. Sleight, A.W., Adv. Mat, Catal. (eds. J.J. Burton,
R.L. Garten) Acad. Press N.Y. (1976)
64. Grzybowska, B., Haber, J., Janas, J., J. Catal. ~'
150 (1977)
65. Dadyburjor, D.B., Ruckenstein, E., J. Phys. Chem. 82, 1563 ( 1978)
20

CHAPTER 3
APPARATUS AND ANALYSIS
3-. 1. Introduction
It is generally accepted that the catalytic activity
of bismuth molybdate is closely related to its oxidizing
properties. In the absence of molecular oxygen for short
periods the catalytic activity and selectivity in the
oxidation and ammoxidation of propene are not affected
i.e. a reduction-oxidation mechanism is operative.
To study the behaviour of bismuth molybdate under stationary and non-stationary conditions three different
techniques have been used.
A. Reaction kinetics in a stationary state as carried
out in different plug flow fixed bed reactors,
operating under differential as well as under inte
gral conditions~
B. The behaviour of bismuth molybdate as an oxidant and the reoxidation of partially reduced bismuth molyb
date are studied in a thermobalance, acting as a
semi-batch reactor.
c. Additional information about the behaviour of the
catalyst under non-stationary conditions at a low
degree of reduction is gained with a pulse reactor
system.
In order to obtain reliable data the experiments have to meet certain requirements, such as:
- the experimental variables (temperature, flow and
reactant inlet concentrations) have to be measured
and controlled accurately~
- the concentration and temperature differences be
tween the bulk gas phase and the catalyst surface
should be as small as possible1
21

- the chemical analysis has to provide for a mass ba
lance over the whole range of experimental concentra
tions;
- isothermicity has to be pursued as much as possible;
- as the residence time distribution of the reaction
mixture generally has an effect on the conversion
level and on the selectivity of the reaction and more
over strongly depends upon the applied technique and
on the experimental variables this distribution
should be minimized and properly determined.
All reactors are connected to an on-line gaschromato
graphic analysis system for the determination of the
reaction components. However, since. such a GLC-analysis
takes at least 15 minutes and has only a moderate sensiti
vity it is less suitable for the examination of non-sta
tionary processes in which rapid change of the reaction
rate occurs.
To gather information about the rate of oxygen deple
tion of the oxidant, the thermobalance in combination with
a GLC apparatus with a flame ionization detector is suit
able because it gives additional information about the
weight of the oxidant. Moreover this apparatus is useful
for the study of the catalyst reduction and for the oxi~
dation of previously reduced samples. However the thermo~
balance has the drawback that the flow around the catalyst
is poorly defined and one has to keep in mind that the
concentrations at the catalyst surface can differ consi
derably from those in the bulk gasphase.
As our thermobalance is not resistant to ammonia vapour
the ammoxidation reactions could not be studied in this
apparatus. Additional information about these reactions
and about the behaviour of the catalyst has been obtained
with a pulse reactor. The pulse reactor is a good instru
ment to detect small changes in the catalyst properties
but, unless the concentrations of the pulse in the reactor
and its residence time are carefully studied the kinetic
information leaves much to desire.
22
The conversion level at which one performs the kinetic
experiments with the various techniques is a compromise
between the low level necessary for the study of the ki
netics at differential conditions and the higher level re
quired for reliable analytical data.
As we deal with moderately or strongly exothermic re
actions, the kinetic data can be affected by non-isother
mic conditions in the fixed bed reactors. We have re
pressed the axial and radial temperature gradients by
means of the dilution of the catalyst with silicon carbide
that has good heat conducting properties (A 673 K = 105 J s- 1 m- 1 K-1 ) (1). Although the commercial operation for
the acrylonitrile production takes place in a fluid bed
we have not used such reactors because of the unclear flow
pattern and the attrition of our unsupported catalyst.
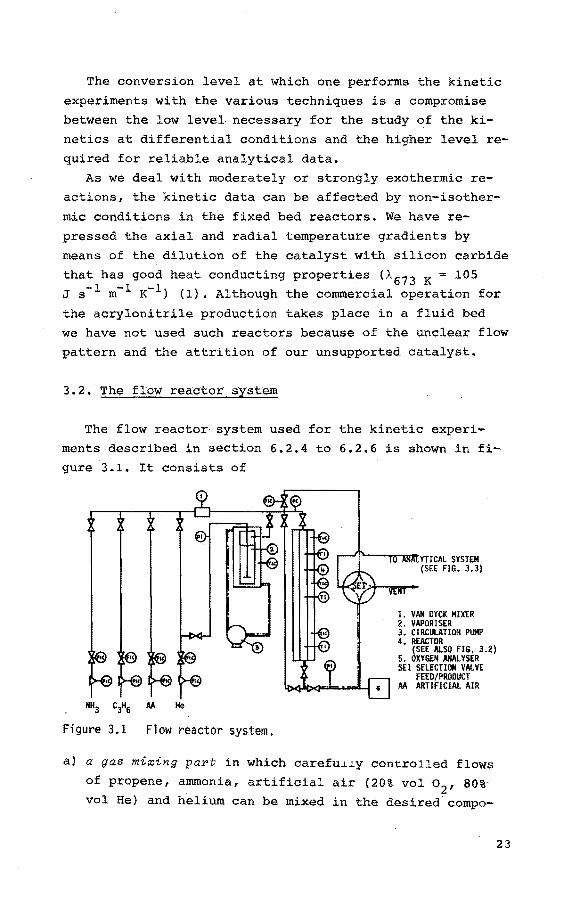
3. 2. The flow reactor system
The flow reactor system used for the kinetic experi
ments described in section 6.2.4 to 6.2.6 is shown in fi
gure 3.1. It consists of
NH3
C3H6 M He
Figure 3.1 Flow reactor system.
r--Jf'---r;orm111'yncAL SYSTEM (SEE FIG. 3.3)
1. VAN OYCK MIXER 2. VAPORISER 3. CIRCULATION PUMP 4. REACTOR
(SEE ALSO FIG. 3.2) 5. OXYGEN ANALYSER SEl SELECTION VALVE
FEED/PRODUCT M ARTIFICIAL AIR
a) a gas mixing part in which carefu~~y controlled flows
of propene, ammonia, artificial air (20% vol o2
, 80%
vol He) and helium can be mixed in the desired compo-
23

sitions. For the experiments involving a liquid reac
tant (acrolein) and for the determination of the sub
stance specific correction factors of the liquids in
the analysis of the feed and the product composition
helium can be passed through a double-walled thermostated vaporizer filled with the pure component in
question. The desired partial pressure of the reactant
can be established by adjusting and controlling the
temperature of the vaporizer. It has been ascertained
that the rising heli~ bubbles were completely satura
ted with vapour. We used the Fourier-number as a mea
sure for the saturation of the dispersed phase
Dt Fo = r2 (3.J.)
with D is the molecular diffusion coef.ficient (m2 s-1),
tis the residence time of the bubble in the liquid (s),
r is the radius of the bubble (m). We found Fo > 4,
whereas already at Fo = .5 for Biot numbers >> 10 (no concentration gradient in the continuous phase), the
concentration distribution over the bubble is practi
cally constant (2). Moreover we analysed the vapour
gaschromatographically at varying liquid levels in the vaporizer and we found a constant vapour concentration.
b) a tubular fixed bed reaator, which is made of AISI 321
stainless steel. Three reactors have been used for the
various reactions as can be seen in table 3.1.
24
Reactor B is shown in figure 3.2. An aluminium jacket
has been cast around the reactor tube to improve the
temperature profile in the reactor. This aluminium
jacket is divided in three sections that are indepen
dently heated. The temperature is measured at eight places, three in the catalyst bed and five in the
siliconcarbide bed under and above the catalyst section.
The temperature is controlled at the three sections
within 1 K with Eurotherm thyristor controllers.
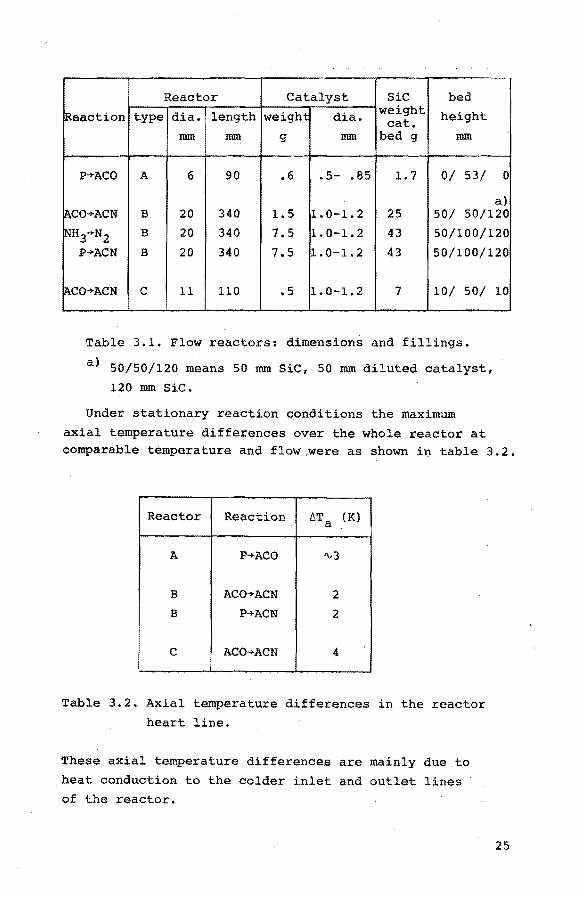
Reactor Catalyst SiC bed
Reaction type dia. length weight dia. weight height cat. lliiii lliiii g lliiii bed g lliiii
P-+ACO A 6 90 .6 • 5- • 85 1.7 0/ 53/ 0
a) jACO-+ACN B 20 340 1.5 1.0-1.2 25 50/ 50/120
INH3-+N2 B 20 340 7.5 1. 0-1.2 43 50/100/120 P-+ACN B 20 340 7.5 1. 0-1.2 43 50/100/120
IAco-+ACN c 11 110 .5 1.0-1.2 7 10/ 50/ 10
Table 3.1. Flow reactors: dimensions and fillings.
a) 50/50/120 means 50 mm SiC, 50 mm diluted catalyst,
120 mm SiC.
Under stationary reaction conditions the maximum
axial temperature differences over the whole reactor at comparable temperature and flow,were as shown in table 3.2.
Reactor Reaction t.Ta (K)
A P-+ACO 'V3
B ACO-+ACN 2 B P-+ACN 2
c ACO-+ACN 4
Table 3.2. Axial temperature differences in the reactor heart line.
These axial temperature differences are mainly due to
heat conduction to the colder inlet and outlet lines of the reactor,
25

II I ~ i
f;.
~ r
¥ ~
~ ~ ~
"' i~
fl-1-=~~~~~~:~~=--l ·~
H~.11
rg~~·i "'== • '
--'-.--J.I ~~"-'-'--· J
hfJ~(V/tf(l( .IVSI ZZt
{lllf¥,y·~s¥MMJrl«.
Figure 3.2 Flowreactor B.
26
Radial temperature profiles were measured in the cata
lyst section of reactor B and C during the ammoxidation of acrolein when the greatest differences could occur

and a temperature difference of not more than 1 K was
found in the radial direction.
c) an analysis system.
The feed or the product stream is introduced by
means of sampling valves in the analysis system, which
will be dealt with in the next section. The feed and
product lines are heated electrically and the tempera
ture of these lines is controlled at about 425 K to
prevent the condensation of water and hydrocarbons and
the polymerization of acrolein and acrylonitrile.
3.2.1. Anal:y:sis
All flow reactors are equipped with an on-line gas chroma
tograph. With this apparatus we can determine quantitatively the components
oxygen
nitrogen
carbon monoxide
carbon dioxide
ammonia
water
formaldehyde
acetaldehyde
acetonitrile
acrolein
propene acrylonitrile
During the catalytic oxidation of propene we used at
fLrst only one GLC-apparatus with katharometer detection
(3). For the separation of the components the column tem
perature had to be programmed in that case from 338 to
433 K with 12 K min-1 . With the introduction of ammonia
for the ammoxidation experiments however the reproducibility of the temperature programmed analysis decreased.
Crozat and Germain (4) analysed ammonia and water on two
columns, i.e. on Porapak Q at 360 K one peak for NHj+H2o was obtained, whereas on a PEG column at the same tempera
ture an inaccurate H2o determination was carried out.
With the introduction of two GLC's at constant tempera
ture (5) i.e. one for the analysis of the low boiling com
ponents and the other with a flame ionization detector for
the analysis of the combustible components we took advan
tage of the better separation of the low boiling compo-
27
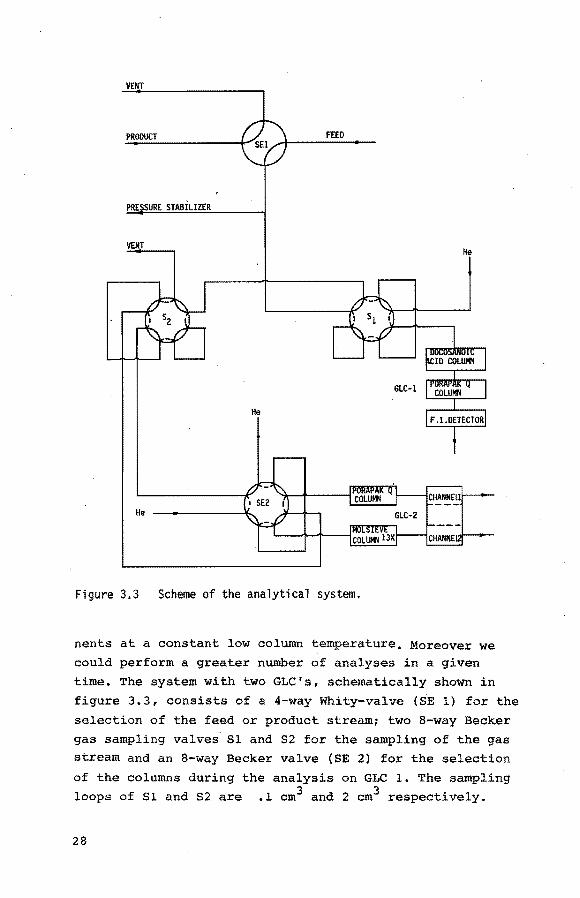
VENT
PRODUCT FEED
PRE SURE STABILIZER
VENT He
He
Figure 3.3 Scheme of the analytical system.
nents at a constant low column temperature, Moreover we could perform a greater number of analyses in a given
time. The system with two GLC's, schematically shown in
figure 3. 3, consists of a 4-way Whity-valve (S.E 1) for the
selection of the feed or product stream1 two 8-way Be.cker gas sampling valves S1 and S2 for the sampling of the gas stream and an 8-way Becker valve (SE 2) for the selection
of the columns during the analysis on GLC 1. The sampling
loops of S1 and S2 are .1 cm3 and 2 cro3 respectively.
28
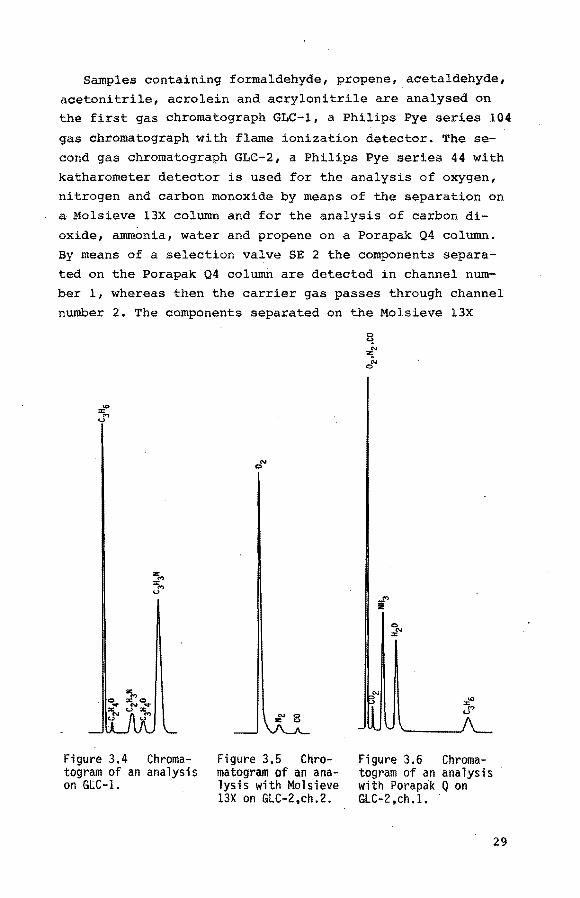
Samples containing formaldehyde, propene, acetaldehyde,
acetonitrile, acrolein and acrylonitrile are analysed on the first gas chromatograph GLC-1, a Philips Pye series 104
gas chromatograph with flame ionization detector. The se
cond gas chromatograph GLC-2, a Philips Pye series 44 with
katharometer detector is used for the analysis of oxygen, nitrogen and carbon monoxide by means of the separation on
a Molsieve l3X column and for the analysis of carbon di
oxide, ammonia, water and propene on a Porapak Q4 column.
By means of a selection valve SE 2 the components separa
ted on the Porapak Q4 column are detected in channel num
ber 1, whereas then the carrier gas passes through channel
number 2. The components separated on the Molsieve 13X
Figure 3.4 Chromatogram of an analysis on GLC-1.
a"'
Figure 3.5 Chromatogram of an analysis with Molsieve 13X on GLC-2,ch.2.
0
:rf''
Figure 3.6 Chromatogram of an analysis with Porapak Q on GLC-2 ,ch .1.
29

column are detected in channel number 2, whereas the
carrier gas passes through channel number 1. As the pro
pene peak is found in the chromatograms obtained with
GLC-1 as well as with GLC-2 a quantitative analysis of all
the components is feasible. For the prevention of a reaction between ammonia and acrolein in the Porapak Q4 column
of GLC-1 this column is preceded by a small column, filled
with docosanoic acid (c 21H43cooH, melting point 353 K)
which adsorbes ammonia completely. The only drawback is
the periodic regeneratio.n that is required for the Pora
pak Q4 column of GLC-2.
The analysing conditions are summarized in table 3.3,
whereas the chromatograms are shown in figure 3.4, 3.5
and 3.6.
GLC-1: Philips Pye 104, temperature 523 K with flame
ionization detector. Hydrogen 30 cm3 min-1 •
Air 50 cm3 min-1 •
CoZumn: Porapak Q4, 50-80 mesh
length: 3,5 m, i.d. 2 mm
temperature: 423 K carrier gas flow: 25 cm3 min- 1 He
analysing time: 3 minutes
column material: glass
GLC-2: Philips Pye 44, temperatu~e 523 K with katharometer detector. Bridge current 150 m A. CoZumna: a) Molsieve 13X, particle size .5-.7 mm
length: 2 m, i.d. 4 mm
temperature: 298 K carrier gas flow: 25 cm3 min-1 He
b) Porapak Q4, 50-80 mesh
length: 2.75 m, i.d. 2 mm
temperature: 333 K carrier gas flow: 25 cm3 min-1 He/NH3 total analysing time: 15 minutes
Table 3.3. Analysing conditions for the flow reactor system.
30

At low mole fractions the peak area of a component in .
a chromatogram is proportional to its mole fraction and
the quantitative analysis of the diluted product mixture
can be carried out using the relation
(3.2)
with XA mole fraction of component A
XC H : mole fraction of propene A 3 6. peak area of component A A •
AC H : peak area of propene f 3 6. substance specific correction factor of A •
component A
This equation is based on the assumption that fc H = 1. In order to determine the f-values of the variou~ 6
components, propene-helium gasmixtures of different com
positions are obtained with two plunger pumps (type Wosthoff) and analysed on the two gas chromatographs. The f
values of the gaseous components are obtained in the same way by mixing with propene/helium gasmixtures in the range
of the experimental mole fractions. The f-values of the
liquid components can be determined with the thermostated
vaporizer already mentioned in section 3.2. The f-value
of ammonia is obtained by means of titration. Peak areas
are determined with Infotronics model CRS 208 electronic
integrators. The reliability of the f-values was checked
periodically, because of the continuous ageing of the
columns. Although temperature programming to 423 K had a
favourable effect on the lifetime of the Porapak Q4 co-
lumn the isothermal method is preferred, as was already
stated above.
The slight increase in the number of moles as a result
of the oxidation and ammoxidation of propene can be ne
glected, especially because the reacting gas mixtures con
tain at least 80% vol He.
31
For the stability and activity of the catalyst the
gas mixtures must contain oxygen and therefore we analysed the oxygen content of the product continuously by means
of a Servomex oxygen analyser.
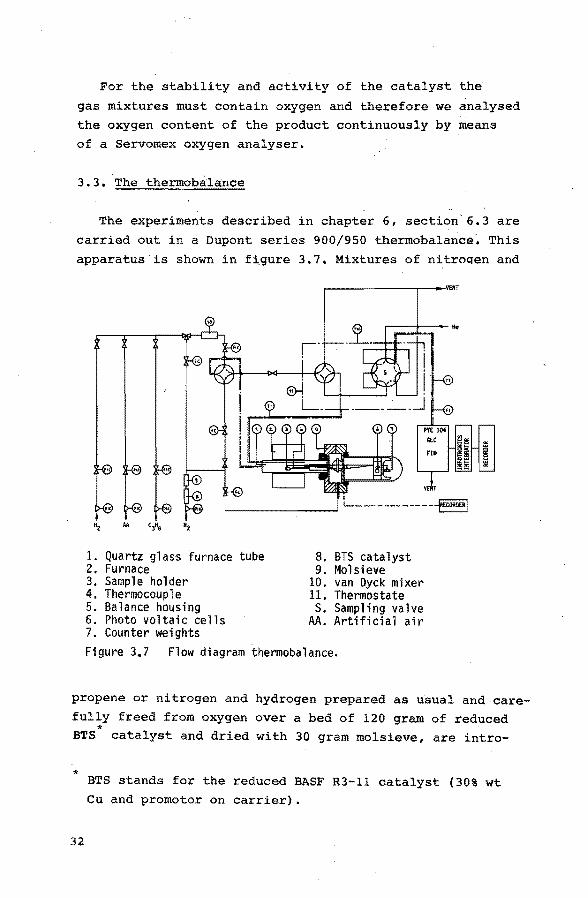
3.3. The thermoba:la:nce
The experiments described in chapter 6, section'6.3 are
carried out in a Dupont series 900/950 thermobalance. This
apparatus is shown in f i,gure 3. 7. Mixtures of ni troqen and
~----------~--·vm
"z AA Cff; "z
1. Quartz glass furnace tube 8. BTS catalyst 2. Furnace 9. Mol sieve 3. Sample holder 10. van Oyck mixer 4. Thermocouple 11. Thermos tate 5. Balance housing s. Sampling valve 6. Photo voltaic cells AA. Artificial air 7. Counter weights Figure 3.7 Flow diagram thermobalance.
propene or nitrogen and hydrogen prepared as usual and care
fully freed from oxygen over a bed of 120 gram of reduced * BTS catalyst and dried with 30 gram molsieve, are intra-
*
32
BTS stands for the reduced BASF R3-ll catalyst (30% wt Cu and promotor on carrier).

duced into the sample chamber of the thermobalance. This
sample chamber consists of a quartz tube with i.d. 2.1 em
heated by an electric furnace. The chamber is at atmos
pheric pressure. The experiments are carried out under
isothermal conditions. The temperature is measured with a
chromel-alumel thermocouple placed just above the 12.5 x
8.4 x 1.20 mm quartz glass sample bucket usually con
taining 75 mg of the oxidant sample. To avoid the presence
of reactants in the part of the balance where the weight
changes are recorded with a photoelectric cell, this side
of the system is continuously purged with nitrogen.
The accuracy of the temperature measurement is ± 1 K.
The sensitivity of the thermobalance is .01 mg, which
corresponds to an error in the degree of reduction of bis
muth molybdate of .OS%. Before the reduction experiment is
carried out the thermobalance is carefully freed from
oxygen by means of flushing the system for 15 minutes at
room temperature with pure and dry nitrogen. Subsequently the balance is flushed with nitrogen at reaction tempera
ture for one hour. We did not observe a weight
loss larger than .01 mg during this conditioning.period.
The effluent of the reduction experiment with propene
containing gas mixtures is analysed by means of gas chromatography as described in section 3.2.1 for the combustible
components.
3.4. The pulse reactor system
As shown in figure 3.8 a constant flow of helium passes
through the pulse reactor into an Hewlett Packard 5700 A
gas chromatograph with katharometer detector and further into a flame ionization detector. A pulse of a gasmixture
containing the reactants for the oxidation and the ammoxi
dation reactions is injected closely before the pulse reactor inlet. After the reaction in the catalyst bed the pulse
is subsequently analysed. The carrier gas is carefully
freed from oxygen and dried as described in section 3.3.
33

r-----IVEMT
1. Pulse reactor 2. Furnace 3. BTS - catalyst 4. Molsieve 5. van Oyck mixer 6. Thermostate S.· Samling valve V. Switching valve
AA. Artificial air
Figure 3.8 Pulse reactor system.
Th~ pulse reactor D is a micro reactor, inner diameter
5 mm, length 14.6 mm made of AISI 316 stainless steel.
100 mg Bismuth molybdate, particle size .3-.5 mm is placed· between two plugs of quartz wool. The pulse reactor is
heated by means of an electric furnace. The temperature is
continuously recorded with a chromel-alumel thermocouple
in the midale of the fixed bed. The temperature of the
furnace is controlled with an Eurotherm thyristor con
troller. The pulse volume is .155 cm3 NTP. The pressure
in the reactor is 2.5 bar and the carrier gas flow is
18 cm3 min-1 NTP. By means of an 8-way
Becker sampling valve s, which is switched pneumatically,
a pulse is introduced in the line to the reactor.
3.4.1. Analysis
The analysis of the pulse after reaction differs from that
of the flow reactor effluent because of the small sample
quantity, the maximum admissible pressure and the analysis
time. The separation of the components is obtained with a
temperature programmed Porapak Q4 column and the detection
occurs with a Hewlett Packard 5700 A katharometer. As the
quantities of the combustible p~oducts are very small the separated components subsequently pass through a flame
34

ionization detector of a Philips Pye 104 GLC for the determina
tion of the combustible components. The analysing condi-
tions are summarized in table 3.4.
Column: Porapak Q4, 80-100 mesh
length: 2.8 m i.d. 2 mm
temperature programs and analysing times:
a) p~opene/helium
333-393 K with 2 K min-1
analysing time: 50 minutes
b) p~opene/oreygen/helium
16 minutes on 333 K, 333-393 K with 16 K min-1
8 minutes on 393 K
analysing time: 40 minutes
c) ammonia/helium
333 K constant temperature
analysing time: 10 minutes d) p~opene/ammonia/helium
16 minutes on 333 K, 333-423 K with 16 K min-1
16 minutes on 423 K
analysing time: 38 minutes
Katha~omete~-deteato~: HP 5700 A, temperature 523 K
bridge current 150 m A
Flame ionization-deteato~: Philips Pye 104, temperature
523 K Hydrogen: 30 cm3 min- 1
Air: 50 cm3 min-1
Table 3.4. Analysing conditions for the pulse reactor
system.
3.5. Safety
3. 5. 1. Toxicity
As acrolein and acrylonitrile are highly toxic sub
stances (6), all experiments are carried out in a hood
35

with adequate exhaust ventilation. Due to its extreme
lachrymatory effect (the smelling limit is • 2 to • 4 ppm
(7)) acrolein serves as its own warning agent. It affects particularly the membranes of the eyes and respiratory
tract.
Acrylonitrile closely resembles hydrogen cyanide in its
toKic action. By inhibiting the respiratory enzymes of
tissue it renders the tissue cells incapable of oxygen
usage. In table 3.5 the Treshold Limit Values (time * ** weighted average) (TLC-TWA) and the LCLo values are
given.
TLV - TWA (8) LCLo (9)
ppm mg m- 3 ppm
c3H40 • 1 .25 150/10 min (inhalation human) 4. Qa) c3H3N 9.0 600/4 hrs (inhalation cat)
a) DuPont de Nemours has reduced the TLV-value to 2.0
ppm (I 0)
Table 3.5. Treshold Limit Values for acrolein and acrylo
nitrile.
_We calculated the mean concentration of acrolein in the
hood when condensation and subsequent destruction would
have been omitted as .19 mg m- 3• For acrylonitrile a value
of .18 mg m- 3 would have applied. These values are ·smaller
than the adopted TWA-values.
*
**
36
TLV-TWA = "the time weighted average concentration for
a normal 8 hour workday to which all workers
may be repeatedly exposed, day after day,
without adverse effect" (8).
LCLo = "the lowest lethal concentration of a sub-
stance in air, which has been reported to
have caused death for a given period of ex
posure" (11).
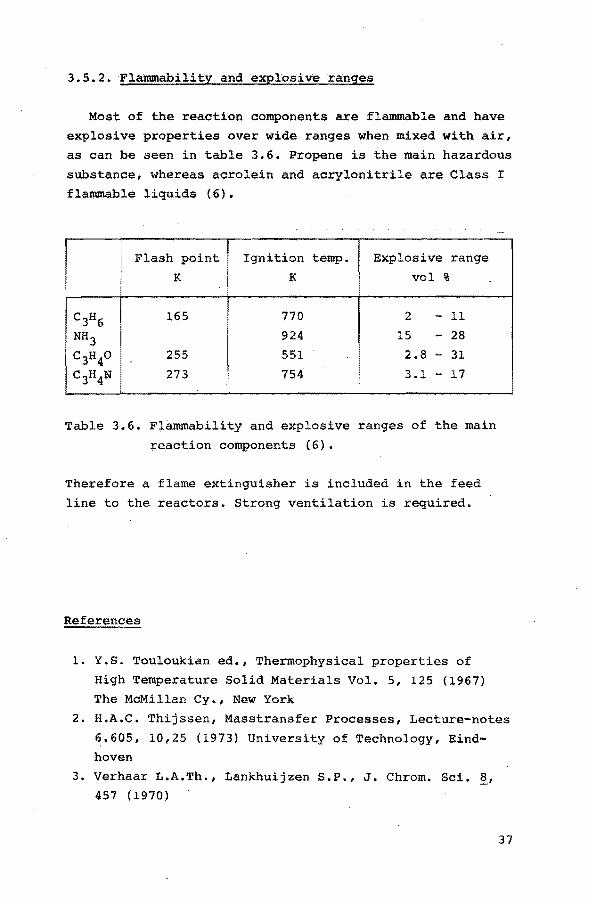
3.5.2. Flammability and explosive ranges
Most of the reaction components are flammable and have
explosive properties over wide ranges when mixed with air,
as can be seen in table 3.6. Propene is the main hazardous
substance, whereas acrolein and acrylonitrile are Class I
flammable liquids (6).
Flash point Ignition temp. Explosive range
K K vol %
C3H6 165 770 2 -11
NH3 924 15 - 28
c3H40 255 551 2.8 - 31
c3H4N 273 754 3.1 - 17
Table 3.6. Flammability and explosive ranges of the main
reaction components (6).
Therefore a flame extinguisher is included in the feed
line to the reactors. Strong ventilation is required.
References
1. Y.S. Touloukian ed., Thermophysical properties of
High Temperature Solid Materials Vol, 5, 125 (1967) The McMillan Cy., New York
2. H.A.C. Thijssen, Masstransfer Processes, Lecture-notes
6.605, 10,25 (1973) University of Technology, Eindhoven
3. Verhaar L.A.Th., Lankhuijzen S.P., J, Chrom, Sci.~, 457 (1970)
37

4. Crozat, M., Germain, J.E., Bull. Soc. Chim. Fr. 3526 (1972)
5. A.P.B. Sommen, Int. Report TC (1975) University of Technology, Eindhoven
6. Sax, N.I., Dangerous Properties of Industrial Mate
rials, 4th Ed., van Nostrand Reinhold Cy., N.Y. (1975)
7. Hommel, G., Handbuch der gefahrlichen Guter, 2 Aufl.,
Springer Verlag, Berlin (1973)
8. Association of American Governmental Industrial Hy
gienists, Index TLV, Am. Ind. Hyg. Ass. Journ. 37, 721 (1976)
9. The International Technical Information Institute;
Toxic and Hazardous Industrial Safety Manual, Tokyo (1977)
10. Anon, Chern. Weekblad 1.1 (22) 1 (1977)
11. Registry of Toxic Effects of Chemical Substances, u.s.
38
Dept. of Health, Education and Welfare, NIOSH (1977) Cincinnatti, Ohio

CHAPTER 4
THE CATALYST
4.1. Introduction
It seems to be the fate of every catalyst to be repla
ced by another more active and selective one. So the mul
tiphase Cu-cu2o-cuo catalyst introduced in 1948 by Hearne and Adams (1), showing only a yield of about 50% in the
oxidation of propene to acrolein has.been superseded in the sixties by the superior bismuth molybdate catalyst.
Callahan et al (2) claimed this catalyst to be useful not
only for the oxidation of propene but also for the dehy
drogenation of 1-butene to butadiene and even for the
ammoxidation of propene to acrylonitrile. Shortly after
the commercial realization of the acrylonitrile process
SOHIO developed its second process based on USb3o10 (31.
but nowadays these two component catalyst system have been
replaced by the so-called multi-component~molybdate (MCM)
catalyst which contains.besides bismuth and molybdenum a
variety of elements such as nickel, cobalt, iron, manganese, phosphorus and potassium.
Whichever oxide combination may be an active and selec~
tive catalyst for the incorporation of a hetero atom (0 or
N) or for the dehydrogenation of olefins, ~t became evi
dent that the superior catalysts are all oxidic combina
tions or compounds containing at least two different ele~
ments. One of these is always a transition metal and the
other belongs to the later row Sa elements. The group of
ammoxidation catalysts is characterized by a high selec
tivity for partial oxidation and its ability to supply
oxygen as a reactant for a selective fissure of the C-H
and N-H bonds.
39

4.2. The structure of the bismuthmolybdate, catalyst
Of the two components of the catalyst of our investi
gation, Bi2o 3 and Moo3 , the former shows at temperatures below 773 K a low ac~ivity, while Moo3 displays at these temperatures an even lower activity but a fair se
lectivity at those temperatures. In combination by means
of a proper preparation method, however, a conspicuously
active and selective catalyst emerges.
Many attempts have b~en made to determine the structure
of the active phase. Series of catalysts have been prepared with varying Bi/Mo atomic ratios in the range of 2/3
to 2/1. All the catalyst samples were found to be selec
tive but differently active (4). In this range three sta
ble compounds were found, viz.
- the a.-phase (Bi2Mo3o12 ) with a monoclinic structure (a = 7.89, b = 11.70, c = 12.24 10""10 m, ~ =.116° l2'l .• This
structure is related to the structure of Scheelite, mentioned by Mekhtiev (5). The x .. ray data of Aykan (6) were
in good agreement with those of E!leij~nberg et al {7)
although the pattern contained additional reflections,
which means that more than one phase was present. Single
crystal studies by van den Elzen and Rieck (8) have confirmed the monoclinic structure. The a.~phase is stable
and has a melting point of 949 K.
- the (3-phase (Bi2Mo2o 9) or the so .... called Erman phase (9)
has been studied in detail. However, there still remains a great deal of uncertainty with regard to its stability
in catalytic oxidation which depends on the applied pre~
parative technique. The solid state technique as used by
Erman (9) leads to different metastable phases such as
the high-temperature y'-bismuth molybdate in the temperature region of catalytic activity in oxidation reactions.
40
The precipitation technique as used by Grzybowska et
al (10), Trifiro et al (ll) and Batist et al (4) (12) is
influenced by factors as pH, concentration of reagents and the treatment of the precipitate. Moreover the calcination temperature is an important factor, because

Batist (12) stated that already at temperatures up to
773 K the ~-phase slowly disproportionates into the a
and the y-phase. It has been observed by Batist et al
(12)(13) that the pH during the precipitation has a
strong influence on the catalytic activity of the aphase. This phenomenon is connected with the equilibrium
between the Mo-O-octahedra and - tetrahedra and it is
assumed that in these preparations the y-phase is always
present. The discussion about the structure of the ~phase has not yet come to an end. As the Bi/Mo ratio 1/1
is frequently used in commercial catalysts the elucida
tion of the nature of the active phase is an interesting
issue.
the y-phase (Bi2Mo06) has an x-ray pattern similar to
that of the rare mineral bismuthrnolybdate named Koechli~
nite, as reported by Zemann (14). It has a layer struc~
ture made up of (Bi 2o2 )~+ and (Moo2 )~+ sheets connected
by o 2- in the arrangement:
The structure of the mineral Koechlinite is orthorhombic . ~0 .
(a = 5.50, b = 16,24, c = 5.49 10 m) which was con-
firmed by van de Elzen and Rieck (15) who found the
following parameters (a= 5.487, b = 16.226, c = 5.506
10-10 m). The structure was described as alternating
layers of (Bio);n and (Moo 42->n perpendicular to the y
direction. In figure 4.1 and figure 4.2 the corner sha
ring Me-octahedra and the Bi2o2-layer are shown respec
tively. These figures are based on van den Elzen's data (15).
The (MoO~-)n layer consists of Mo6+ ions in octahe
dral surrounding, the octahedra sharing corners in the
sheets and their apices point toward the (Bio);n layers.
The (Bio);n layers resemble the structure of BiOCl. In
Koechlinite the bond distances in the molybdenuro~xygen sheets are as follows: two at 1.76 10-10 m and two at
2.24 10-10 m. The molybdenum-oxygen bonds to the apex
41
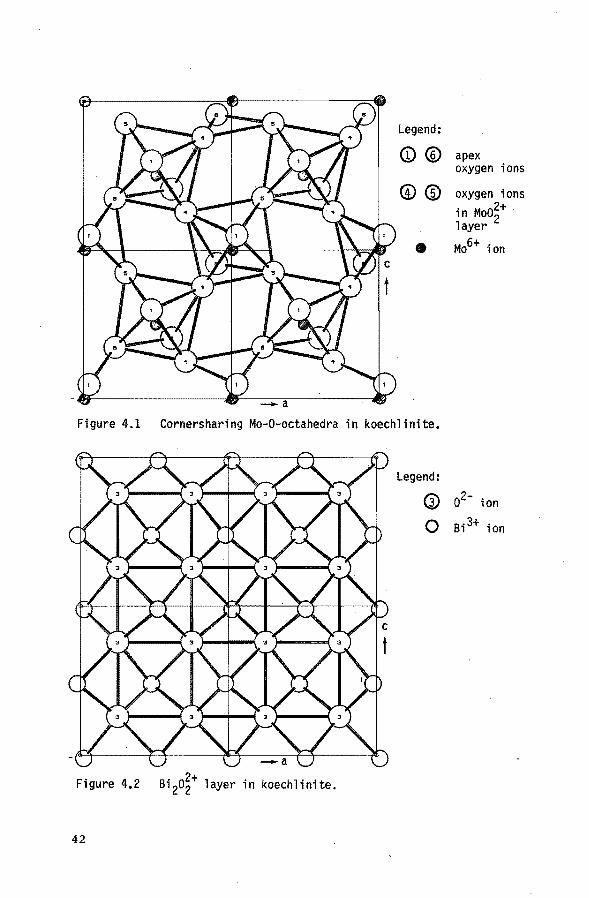
Legend:
<D® apex oxygen ions
®® oxygen ions in Moo~+ layer
• Mo6+ ion
Figure 4.1 Cornersharing Mo-O-octahedra in koechlinite.
Figure 4,2 Bi 2o~+ layer in koechlinite.
42
Legend:
G) o2- ion
0 Bi 3+ ion

o 2- ions have intermediate lengths: 1.86 10""10 and 1.93
10""10 m. The bismuth ion is bonded to six oxygen ions:
the four Bi-0 distances in the BiO-layer range from 2.15
to 2.50 10-10 m. The distances to the apical oxygens of the Mo octahedra are 2.33 and 2.67 10-10 m. As pointed
out by Schuit (16) the structure of the y-bismuth molyb..,
date can therefore be regarded as an intermediate be
tween one having a two dimensional Reo3 .... type of corner
sharing Moo6 octahedra and one having slightly distorted Moo 4 tetrahedra.
The y-phase is metastable and at temperature.s above
930 K it can be transformed to the y'~hase .with a te~ tragonal structure, reported by Blasse (17). The melting point is 1211 K (7), the density is 8.26 ~0 3 kg m~ 3 (6).
The catalytic activity of bismuth molybdate has been
connected by Schuit et al (4) with the presence of cornersharing oxo-molybdenum octahedra and it has been postulated that the active site for adsorption of pro..,.
pene is an oxygen anion vacancy on a mo~ybdenum ion still being present in a tetragonally pyramidal configu~
ration.
4.3. Catalyst preparation
y-Bismuth molybdate has been prepared according to the
method described by Batist et al (12), Batist (13) and
Konings et al (18) either starting with molybdic acid
(method A) or with ammonium heptamolybdate (method B). For
all preparations the basic chemicals were analysed care~ fully in order to be sure of the stoichiometry of the
catalyst. BismuthyJ nitrate (Merck, p.a.), containing 79.9
wt % Bi2o 3 , molybdic acid (BDH) with 87 .• 4% wt Moo3 and amrooniumheptaroolybdate (Merck, p.a.) containing 8~.8% wt
MoO 3 were used.
Method A
According to the method given by Konings et al (18) and based on the method described by Batist et al (12) 94.7 g
43
BiON03 are mixed with 27.3 g H2Moo4 and 2 liter distilled
water is added. Under vigorous stirring the slurry is
boiled continuously for 40 hours. The pH changes from 7 to 2 and within two hours the color of the suspension changes
from white into light yellow. After 10 hours the yellow
color becomes more intense. From time to time distilled
water is added to keep the volume of the slurry constant.
After filtration and drying at 393 K the material is cal
cined for two hours at 773 K. Great care is taken to keep
the calcination tempera~ure constant. In this w~y an
highly active and selective catalyst is prepared with a
stoichiometric excess of 2 mole % Mo. The specific surface area, varying from 3.0 to 3.4 m2 g~~ was determined with
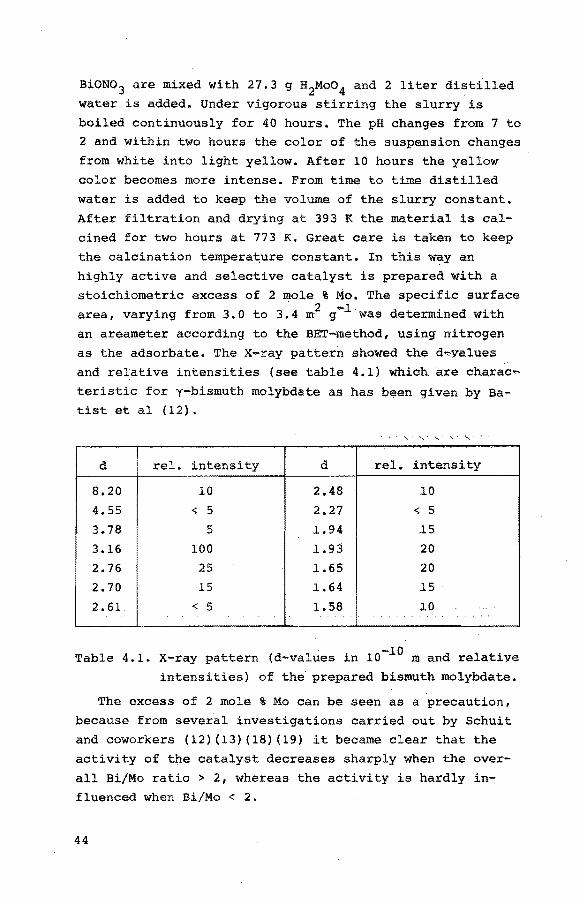
an areameter according to the BET-method, using nitro9en as the adsorbate. The x-ray pattern showed the d..,.yalues
and relative intensities (see table 4.1} which are charac..,_
teristic for y-bismuth molybdate as has been given by Batist et al (12).
d rel. intensity d rel. intensity
8.20 10 2.48 ~0
4.55 < 5 2.27 < 5
3.78 5 1.94 ~5
3.16 100 1.93 20
2.76 25 1.65 20
2.70 15 1.64 ~5
2. 61. < 5 1.58 ~0
Table 4.1. x-ray pattern (d-values in 10-~0 m and relative
intensities) of the prepared bismuth molybdate.
The excess of 2 mole % Mo can be seen as a precaution, because from several investigations carried out by Schuit
and coworkers (12)(13)(18)(19) it became clear that the
activity of the catalyst decreases sharply when the over
all Bi/Mo ratio > 2, whereas the activity is hardly in
fluenced when Bi/Mo < 2.
44

Method B
According to Batist's method (13), 29.3 g (NH4
) Mo7e24 •
4 H2o are dissolved in 2 1 distilled water and the pH of
the solution is lowered to 2.5 by careful addition of
nitric acid, keeping the molybdenum in the octahedral co
ordination. 94.7 g Bi0N03 are .added and under vigorous stirring the slurry is boiled continuously for 24 hours.
The same procedure is followed as at Method A. This cata
lyst showed a similar activity and selectivity as the catalyst prepared according to method A and has the same
specific surface area. The main stoichiometric equation of
the catalyst preparation is
(r 4 ~.11
Batist (13) assumed that the mass swelling during the
slurry reaction (method B) is a result of the decomposi~
tion of the heptamolybdate ion into molybdic acid, accor~
ding to the equation
+
followed by a penetration of solved molybdenum oxide oc
taeders into the layers of the solid BiON03 •
Both catalysts have the following stoichiometric bulk
composition: Bi2Mo1 • 02o6 • 06 as was checked by means of a.
weight analysis.
References
1. Hearne G.W., Adams M.L., u.s. Patent 2,451,485 (1948)
2. Callahan, J.L., Foreman, R.W., Veatch, F., u.s. Patent
3,044,966 (1962)
3. Callahan, J.L., Gertisser, B., u.s. Patent 3,198,750
(1965)
45

4. Batist, Ph.A'~ der Kinderen, A.H.W.M., Leeuwenburgh,
Y., Metz, F.A.M.G,, Schuit, G.C.A., J. Catal. g, 45
(1968)
5. Mekhtiev, K.M., Gamidov, R.S., Mamedov, Kh,S., Belov,
N.V., Dokl. Akad. Nauk. SSSR 162, 397 (1965)
6. Aykan, K., J. Catal. 12, 281 (1968)
7. Bleijenberg, A.C.A.M., Lippens, B.C., Schuit, G.C.A.,
J. Catal. i 1 581 (1965)
8. van den Elzen, A.F., Rieck, G.D., Acta Crystallogr.
Sect, B 29, 2433 (19.73)
9. Erman, L.Ya., Galgerin, E.L., Kolchin, I.K., Dobrzhan
skii, G.F., Chermyshev, K.S., Russ. J. Inorg. Chem, ir 1174 (1964)
10. Grzybowska, B., Haber, J., Komorek, J., J. Catal. ~,
25 (1972)
11. Trifiro, F., Hoser, H., Searle, R.D., J. Catal. 25, ~2 (1972)
12. Batist, Ph.A., Bouwens, J.F.H., Schuit, G.C.A., J. Catal. 25, 1 (1972)
13. Batist, Pn.A., to be published
14. Zemann, J., Heidelberger Beitr. Mineral Petrogr. ~~
139 (1956)
15. van den Elzen, A.F., Rieck, G.D., Acta Crystallogr.
Sect. B 29, 2436 (1973)
16. Gates, B.C., Katzer, J.R., Schuit, G.C.A., Chemistry of Catalytic Processes,·. Chapter 4, McGraw Hill,
New York (1979)
17. Blasse, G., J. Inorg. Nucl. Chem. 28, 1124 (1966)
18. Konings, A.J.A., Creemers, H.J.M., Batist, Ph.A., J.
Catal. 41, 333 (1976)
19. van Oeffelen, D.A.G., Thesis, University of Technology,
Eindhoven (1978)
46

CHAPTER 5
EXPERIMENTAL METHODS
s·.l. Introduction
Because a catalyst increases the rate of a reaction ki
netic experiments are among the key experimental methods
to investigate the behaviour of a catalyst. Kinetic mea
surements however have their limitations as they do not
provide enough information for a complete description of
the catalytic reaction sequence, since the kinetics prima
rily reflect the slowest step of the sequence of elemen
tary reactions.
The requirements for our kinetic experiments are
- an accurate and fast analysis of all reactants and pro-
ducts;
- the absence of heat and mass transfer limitations;
- an isothermal operation of the catalyst bed;
- a well defined flow pattern in the reactor;
- a constant catalyst activity.
We have chosen the tubular fixed bed reactor because
the flow pattern is well defined, we have no catalyst
attrition and the isothermicity can be obtained by apply
ing catalyst dilution. Reaction rate measurements in this
flow system were performed differentially and integrally.
A small fixed bed reactor was used ·for pulse experi
ments. Valuable information can be obtained with this
reactor type, although reliable quantitative rate data can
only be extracted from these experiments for simple first
order kinetics.
A thermobalance is a powerful instrument in heteroge
neous catalytic research. Because of the gasflow pattern
required the concentrations at the catalyst surface are
not well defined.
47

In this chapter the attention is directed to the main factors that determine the proper operation of the reactor
types that we have used during this investigation.
5.2. The rate of reaction
Before dealing with factors that determine the rate of formation of a product of a heterogeneous catalytic reac
tion, this rate of formation will be defined in connection
with the definition of the rate of reaction.
When the specific mass of the reaction mixture does not change (p is constant) the specific rate of formation of
component Ai of a Chemical reaction
(5.1)
is defined as
(5.2)
in which W is the catalyst weight (kg) and F is the molar flow rate {mol s-1 ). This. definition is related to there
commended definition for the rate of reaction by the IUPAC
(1) i.e. the rate of increase of the extent of the reaction~ of the reaction given by equation (5.1).
And
(5.3)
The specific rate of reaction of a heterogeneous catalytic reaction is defined as
(5.4)
with t the reaction time (s). However the stoichiometric
48

coefficients of an overall reaction are not defined unam
biguously like the coefficients -of an .elementary reaction
and therefore ~ and the rate of a non-elementary reaction
are not exactly defined either. For that reason we prefer
the definition of the rate of formation as given in equation (5.2) which can be calculated from measured data i.e.
the concentration, the amount of catalyst and the molar flow rate.
Note: The dimension of the rate of formation, [mol2 kg-1
m- 3 s-1], seems very complicated. If we would sub
stitute for W (kg catalyst) the number of active
sites, expressed in moles Ncr, then our reaction time
would have the dimension (S) again and the specific
rate of formation would revert to its normal dimen
sion [r] = [mol m- 3 s-1]. As the determination of Ncr
is not yet unambiguous we prefer the somewhat cum
bersome but straightforward quantity as defined in
the text. We will often use the terms "rate of for
mation" and "rate of consumption" instead of "spe
cific rate of formation" and "specific rate of con
sumption".
5. 3. Factors ·govern~n·g the re·a:ctor beha:Yiour
The interpretation of the kinetic results is very much
facilitated if the fluid flow in the reactor can be consi
dered to be an ideal plug flow and if the kinetic data are
obtained in a chemically controlled regime under isother
mal conditions •
.5. 3 .1. Plug flow
Danckwerts (2) pointed out that if the P~clet-number
for mass transport in axial direction based on the reactor
length
PemaL = iiL > 60 e:Da (5.5)
49

than the flow pattern will approach ideal plug flow. In
our fixed bed reactors the particle Reynolds number Redp is small and varies from .1 to 1.2. Edwards and Richardson
(3) have shown that at low Redp-values the axial disper
sion is only caused by molecular diffusion, thus Da = Dmol" The Peclet-number based on the particle diameter is
(5.6)
From the correlation given by equation (5.6) we calculate
the value of Pemadp in the order of .5. This means that at low Reynolds-numbers where equation (5.6) holds, it can be
deduced from equation (5.5) that axial mixing can be ne
glected if ~ > 120. This value is in line with the value p
of 100 calculated by Carberry and Wendel (4) for both an adiabatic and a fixed bed reactor based on an one dimen
sional model for a first order reac'tion and with the value
of 150 suggested by Finlayson (5) which is a more conser
vative value because the significance of axial heat trans
fer is taken into account. Mears (6) derived for non-first
order kinetics a plug flow criterion
:k...> d p
20 n Co ln Pemadp
(5.7)
with n is the order of the reaction and C0
and Ce are the
inlet and outlet concentrations respectively. This crite
rion shows that axial dispersion becomes more severe with increasing conversion and increasing order of the reaction.
Radial temperature gradients as a result of an exothermic
reaction are attended by radial dispersion, which further
improves the flow pattern in the direction of plug flow.
This improved flow pattern is known as the Taylor dispersion (7). During our experiments in the flow reactors the condition L/dp > 120 generally was satisfied. Measurements with respect to the residence time distribu
tion in the pulse reactor showed that even in that small
reactor the axial dispersion was almost absent.
50

Cpanneling and maldistribution in small reactors be
cause of wall effects can be dominant. Schwartz and Smith
(8) and Schertz and Bischoff (9) reported for reactor dia
meter to particle diameter D/dp ratios < 10 that point
velocities one particle diameter from the wall were 2 to
10 times as high as at the center line. A radial tempera
ture gradient as a result of an exothermic reaction gives
the highest figures bec~use of the corresponding radial
viscosity gradient. Carberry (10) and Bamford and Tipper
(11) recommend D/dp values > 10 for laboratory reactors.
5 •. 3. 2. Temperature 51radients
The reactions of our investigations are moderately or
strongly exothermic as can be seen in table 5.1.
stoichiometric equation (-l:IH~)6Zf K kJ mol
c3H6 + o2 + c3H4o + H2o 353 (r 5.1)
c3H6 + NH 3 + 1% 0 2 + c3H3N + 3 H2o 513 (r 5.2)
c3H4o + NH3 + ~ 02 + C3H3N + 2 H20 160 (r 5.3)
NH3 + ~ 0 2 + ~ N2 + 1~ H20 328 (r 5.4)
c3H6 + 4~ 0 2 + 3 C02 + 3 H20 1924 (r 5. 5)
C3H40 + 3% 0 2 + 3 C02 + 2 H20 1571 (r 5.6)
c3H3N + 3~ 0 2 + 3 C02 + % N2 + 1~ H20 1739 (r 5.7)
Table 5.1. Reaction enthalpies of the main reactions at
673 K in kJ mol-l of the first reactant in the
stoichiometric equation.
Therefore large reactor diameters must be avoided to pre
vent large radial temperature differences in a cooled
reactor. Consequently D/d ratios > 30 as suggested by ' p
Bamford and Tipper (11) are detrimental for the isothermal
51

behaviour of the fixed bed reactor. Because the enthalpies
of the various reactions are different the heat produced
at comparable conditions depends strongly on the integral selectivity of the reaction concerned and on the overall
reaction rate. From the experimental data we calculated at
a reactant inlet concentration of 1 mol m- 3 for the ammoxi
dation of propene at 673 K an initial heat production of
1.1 kJ kg- 1 s- 1 and for the amrnoxidation of acrolein 5,5 -1 -1 kJ kg s • This heat production at stationary conditions
causes the occurrence of temperature gradients, which can be divided in three types i.e.
- an intraparticle gradient within the catalyst particle;
- an interphase temperature difference between the exter-
nal surface of the particle and the adjacent gas;
- an interparticle temperature difference between the ca-
talyst particles.
According to Mears (12) the interparticle gradient has the
greatest heat transport resistance, followed by the inter
phase and the intraparticle gradients. During our experi
ments the intraparticle gradient is absent, as was calcu
lated for the ammoxidation of propene by means of Ander~
son's criterion (13). This criterion requires
< 3 RT Ea
(5.8)
for a deviation from the isothermal rate less than 5%. (~Hr) is the absolute value of the reaction enthalpy, r is
the observed rate of reaction, AP is the thermal conducti
vity of the particle and Ea is the activation enthalpy of the reaction. This criterion is amply satisfied, even for
the total oxidation reactions. The criterion for the onset
of an interphase heat transport limitation, given by Mears . ( 12) is
(5.9)
52

with h is the gas-solid heat transfer coefficient. During
our experiment this criterion is easily met and thus in
terphase heat transfer is not influencing the experimen
tal results.
The conditions in our fixed bed reactors make that in
terparticle heat transport limitations can be present. As
the axial heat transport in laboratory reactors is small
compared with the radial transport, both the effective
radial thermal conductivity {Aer) and the heat transfer at the reactor wall (aw) are important parameters. According to de Wasch and Froment (14) who derived a model for ra•
dial heat traasfer with these two parameters, ~ and A w er depend on the Reynolds number and on the D/dp ratio. It will be clear that low Re-values and large bed diameters
cause radial temperature differences in integral fixed bed
reactors. Isothermicity is favoured by catalyst dilution
with inert solids that have a good thermal conductivity
and by small reactor diameters.
5.3.3. Catalyst dilution
By means of dilution of the catalyst with inert parti
cles isothermal conditions can be obtained in integral
reactors. Caldwell and Calderbank (15) described strate
gies for optimizing reactor performance by varying the
dilution ratio with axial distance. They used a dilution
ratio which decreased linearly with conversion. Mears (6)
argues that dilution is advantageous in minimizing radial
temperature gradients only if the reactor operates at
Reynolds numbers sufficiently low, for the effective ther
mal conductivity to be relatively insensitive to the mass velocity.
The influence of catalyst dilution may be of a chemical
or a physical nature. The influence of a diluting material
on the kinetics of a catalytic reaction have been esta
blished by Mosely and Good (16) and by Nix and Weisz (17).
Dilution can further have an effect on the residence
time distribution (with respect to the catalyst) and on
53

the temperature distribution. Bypassing at a high dilu
tion ratio gives a lower conversion. If the dilution and
the catalyst particles have different particle size dis
tributions the bed void fraction is influenced. Van den Bleek et al (18) developed a stochastic model that descri
bes the influence of the bypassing effects on conversion.
Their criterion to determine the allowable degree of dilu
tion
L b -d > 250 7 p v
(5.10)
where b is the volumetric dilution ratio and o is the relative experimental error in the conversion, ensures that
the bypassing effect will be an order-of-magnitude smaller
than o. This model has the drawback not to distinguish between reactions that are influenced by diffusion limita
tions and reactions that are not.
During our experiments with the ammoxidation of acro
lein we found some dilution effect i.e. a decrease of the
conversion at higher dilution ratios, whereas with the
oxidation and ammoxidation of propene this phenomenom was not observed. This difference could be explained if we
considered the diffusion limitation of the reaction rate of the acrolein ammoxidation. In this case van den Bleek's
dilution criterion appeared to be insensitive.
5.3.4. Mass and heat transfer
An estimate of the mass and heat transfer coefficients
is required to decide whether external transport limitations influence the chemical reaction rate. The mass and
heat transfer coefficients kg and h are based on the film··
theory with the assumption that mass and heat transfer are caused by diffusion and conduction through a stagnant gas
film around the catalyst particle. As the film thickness depends on the Reynolds number, kg and h are similarly
54

related to Re. The Chilton and Colburn j 0 and jH relations
(19) offer the possibility to determine kg and h.
The following relations are often applied:
Yoshida et al (20) found for .01 < Re < 50
j 0 = .84 Re -.51 (5.11)
whereas Gamson et al (21) correlated jH and j0
(5.12)
For the mass transfer from the bulk to the catalyst sur
face the overall rate of reactions rA is related to the
concentration difference ([A]b-[A]s> of the reactant between the bulk gas phase and the catalyst surface,
according to
r = A (5.13)
in which kg is the mass transfer coefficient (m s-1), Sis the specific surface area of the catalyst (m2 kg-1), tfJ. is
the sphericity factor.
The Chilton and Colburn relation is
(5.14)
in which CfA is the concentration factor (mol m- 3), GM is the superficial molar flow rate (mol m- 2 s-1), pis the
-1 -1 viscosity of the fluid (kg m s ), Pf is the fluid den-sity (kg m~3) and Dmol is the molecular diffusivity of the reactant in the fluid (m2 s-1). As we know the j 0-value
from equation (5.11) kg can be calculated from equation
(5.14) and after substitution in equation (5.13)
C[A]b-[A]8
) is found. For the ammoxidation of propene we calculated at the most favourable conditions for mass
transfer limitation a concentration difference for propene between the bulk gas phase and the catalyst of 2.38 10-2
55

-3 mol m , which is 1.8% of the applied propene concentra-
tion. We conclude that the mass transfer limitation for
all propene ammoxidation experiments can be neglected. As the ammoxidation of acrolein is a very fast reaction in
comparison with the ammoxidation of propene, mass transfer
limitation of the overall rate can occur. We calculated
again at favourable conditions for mass transfer limitation an acrolein concentration difference of 1.16 10-1 mol
m- 3 which was 15% of the applied concentration of acrolein.
In a similar way the temperature differenc.e between the
catalyst surface and the bulk gas phase can be calculated,
using the following equations:
(5.15)
in which h is the heat transfer coefficient (J m- 2 s-1 K-1)
and (Ts-Tb) is the temperature difference (K) and .. the Chilton and ·Colburn relation for heat transfer
(5.16)
in which c is the heat capacity at constant pressure (J kg-1 K-~), Af is the thermal conductivity (J m- 1 s-1
K- 1 ) and us is the superficial velocity (m s-1) of the
fluid. jH is calculated from the equations (5.11) and
(5.12) and substituted in equation (5.16) whereas (Ts-Tb) is found from equation (5.15) after substitution of h.
For the flow reactor experiments at the most unfavourable conditions for heat transfer we calculated a temperature
difference between the catalyst surface and the gas.phase
of .15 K. Because the temperature measurements in a pulse reactor are less accurate than in a differential flow
reactor we have calcul~ted the temperature difference between the catalyst surface and the gas phase during the
pulse and found (Ts-Tb) to below .06 K. For the thermobalance we calculated in a similar way a temperature difference at the steady state below .04 K. We conclude that
56

the temperature measurements of the gas phase represent
very well the temperature at the catalyst surface,
A quantitative approach to the intraparticle heat and
mass transfer has been defined according to Thiele (22) in
terms of an effectiveness factor n, the ratio of the observed rate to the rate that would exist if all the cata
lyst surface were equally accessible~ The calculation of n
according to Weisz and Hicks (23) has shown that in all cases this. parameter has a value very close to one. This
has been verified experimentally with catalyst particles
of different sizes.
5. 3. 5. Pr·essu:re drop
According to Ergun (24) the pressure drop over a fixed
bed depends on the particle diameter d , the void fraction . p e, the viscosity of the fluid p and the mass velocity G. As we carried out the experiment at atmospheric pressure
the measurements at differential conditions (high mass
velocities) could give appreciable pressure differences.
However at the maximum attainable mass velocity in flow reactor B a pressure drop over the reactor of 8.10- 3 bar
was found, whereas in the other flow reactors this diffe
rence was smaller. We conclude that the influence of the
pressure variation on the rate of reaction can be neglected.
5. 4. Data: handling' a:nd a:na'lysis of errors
The analytical data obtained by means of GLC can easily
be converted to concentrations. From these values quanti
ties as conversion, space time and selectivities are cal
culated. As a complete analysis was carried out we used the additional information of the atombalances to improve the experimental data, according to the method given by
van der Grinten (25) and swenker (26). The results of the
raw concentration calculations are used in the adjustment
computing program. If the experimental errors are normally
57
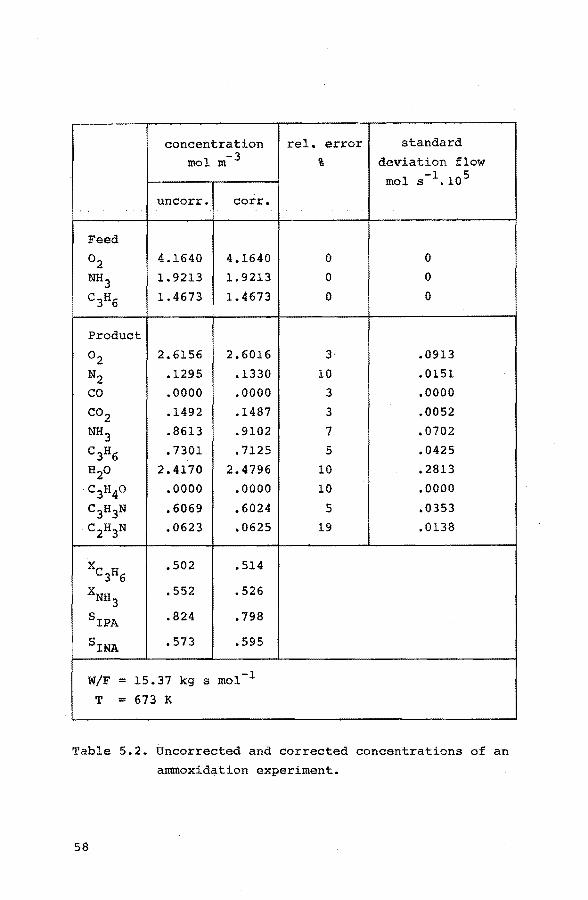
concentration rel. error standard
mol m- 3 % deviation flow -1 5 mol s ,10
uncorr. corr,
Feed
02 4.1640 4.1640 0 0
NH3 1.9213 1.9213 0 0
C3H6 1.4673 1.4673 0 0
Product
02 2.6156 2.6016 3 .0913
N2 .1295 .1330 10 .0151
co .0000 .0000 3 .0000
C02 .1492 .1487 3 .0052
NH 3 .8613 .9102 7 .0702
C3H6 .7301 • 7125 5 .0425
H2o 2.4170 2. 4796 10 .2813
·C3H4o .0000 .0000 10 .0000
C3H3N .6069 .6024 5 .0353
c2H3N ,0623 ,0625 19 .0138
X C3H6
.502 .514
XNH .552 .526 3
5 IPA .824 .798
5 INA .573 .595
W/F = 15.37 kg s mol-l
T = 673 K
Table 5.2. Uncorrected and corrected concentrations of an
ammoxidation experiment.
58

distributed the most probable value of the experimental
error can be calculated by means of a minimalisation of
the sum of squares of deviations. The problem of the non
constant error vari.ance was solved by the use of appro
priate weight factors, calculated from independent esti
mates of the experimental error variances as a function of
experimental data, In this way the corrected concentrations
of all components could be calculated and plotted as a
function of space time. An example of raw and corrected
data is given in table 5.2. The constants of the kinetic equations were calculated by linearization of the equation
or by means of the Newton Raphson iteration method. The
difference between the experimental and model values were
minimalized with the procedure Minifun (27).
References
1. Manual of Symbols and Terminology for physico chemical
Quantities and Units, Appendix II, Part II, Hetero
geneous Catalysis, Adv. Catal. 26, 351 (1977)
2. Danckwerts, P.V., Chern. Eng. Sci.~~ 1 (1953)
3. Edwards, M,F,, Richardson, J,F., Chern. Eng. Sci. 23,
U9 (1968)
4. Carberry, J.J., Wendel, M.M., A.I.Ch.E. Journ. 2• 129
(1963)
5. Finlayson, B.A., Catal. Rev. 10, 69 (1974)
6. Mears, D.E., Ind. Eng. Chern. Proc. Des. Dev. !Q, 541
(1971)
7. Taylor, G., Proc. Roy. Soc. (London) A219, 186 (1953)
8. Schwartz, C.E., Smith, J.M., Ind. Eng. Chern. 45, 1209
(1953)
9. Schertz, w.w., Bischoff, K.B., A.I.Ch.E. Journ. 15,
597 (1969) 10. Carberry, J.J., Ind. Eng. Chern. 56 (11), 39 (1964)
59

11. Bamford, C.H., Tipper, C.F.H., Comprehensive Chemical
Kinetics Vol. I. The practice of kinetics, Elsevier,
New York (1969)
12. Mears, D.E., J. Catal. £Q_, 127 (1971)
13, Anderson, J.B., Chern. Eng. Sci, 18, 147 (1963)
14. De Wasch, A.P., Froment, G.F., Chern. Eng. Sci. 27, 567
(1972)
15. Caldwell, A.D., Calderbank, P.H., Brit. Chern, Eng. 14,
1199 (1969)
16. Mosely, R.B., Good, G.M., J, Catal. !• 85 (1965)
17. Nix, P.S., Weisz, P.B., J, Catal. _!, 179 (1964}
18. Van den Bleek, C.M., van der Wiele, K., van den Berg,
P.J., Chern. Eng. Sci. 24, 681 (1969)
19. Chilton, T.H., Colburn, A.P., Ind. Eng. Chern. 1183
(1934)
20. Yoshida, F., Ramaswami, D., Hougen, O,A., A.I.Ch.E.
Journ. ~' 5 (1962}
21. Gamson, B.W., Thodos, G., Hougen, O.A., Trans. Am. Inst. Chern. Engrs. 39, 1 (1943)
22. Thiele, E.W., Ind. Eng, Chern. 21, 916 (1936)
23. Weisz, P.B., Hicks, J.S., Chern. Eng. Sci. 12, 265 (1962)
25. Vander Grinten, P.M.E.M., De Ingenieur ~ (21), 070
(1971)
26. Swenker, A.G., De Ingenieur ~ (21), 065 (1971)
27. Lootsma, F,A., Thesis, Eindhoven (1970)
60

CHAPTER 6
RESULTS AND DISCUSSION
6.1. Introduction
The aim of the experimental work presented in this
chapter is to develop a kinetic model for the catalytic
ammoxidation of propene, in order to contribute to the
elucidation of the reaction mechanism.
It is of technological significance to determine the
reaction kinetics of the acrylonitrile synthesis under con
ditions that are relevant for industrial circumstances. Therefore continuously operated fixed bed reactors are
used under differential as well as under integral steady
state conditions.
In the literature it is commonly accepted that the acry
lonitrile formation.mainly occurs by a direct ammoxidation
of propene and that the oxidation of. propene, followed by
the ammoxidation of acrolein is of minor importance. Less
attention has been paid to the kinetics of the oxidation
of ammonia. Therefore the kinetics of this combination of
parallel and consecutive reactions were studied in detail.
The experimental programme carried out with the fixed
bed reactors involved the following catalytic reactions:
a) the oxidation of propene to acrolein;
b) the oxidation of ammonia to nitrogen;
c) the ammoxidation of acrolein to acrylonitrile;
d) the ammoxidation of propene to acrylonitrile.
The kinetics of many oxidation reactions are described
by models based on the Mars-van Krevelen mechanism. Cor
respondingly the elucidation of the reaction mechanism of
the propene ammoxidation can be advanced by the study of
the role of bismuth molybdate as a reactant (i.e. an oxidant).
61

Two types of experiments at non-stationary conditions
have been applied to obtain information about the beha
viour of bismuth molybdate, i.e.
1. thermogravimetric experiments in a thermobalance in
combination with GLC-analysis1
2. pulse reactor experiments.
In the thermobalance reduction kinetics of bismuth mo
lybdate were studied using propene, propene-air mixtures
and hydrogen as reducing agents. Subsequently we investi
gated the kinetics of reoxidation of reduced molybdate
with different oxygen containing gas mixtures. Special
attention has been given to the adsorption of acrolein on
the reduced catalyst.
The pulse reactor is used in order to obtain more in
sight in the role of oxygen of the solid and the oxygen in
the gas phase in the different reactions and in the ini
tial activity of the catalyst under specific conditions.
6.2. Flow experiments
6.2.1. Introduction
In chapter 5 we have shown that the chosen types of
fixed bed reactors can be used to investigate the reaction
kinetics of the acrylonitrile synthesis reactions, provided one satisfies certain conditions. For a reliable
study of the reaction kinetics, one has to work under iso
thermal conditions. With our strongly exothermic reactions
this condition can only be attained by sufficient dilution of the catalyst. We have always used silicon carbide as
the diluting agent. The influence of this dilution has
been discussed in chapter 5 and will be dealt with in this chapter.
6.2.2. Preliminary experiments
- Catalyst stability and aativity The activity of the catalyst could be maintained at a
62

constant level during the entire experimental period
provided that the gas phase. is not depleted in oxygen.
Loss of activity is connected with a chemical desinte
gration of the bismuth molybdate due to reduction and formation of metallic bismuth droplets. Since a reoxida
tion carried out after a period produces the two oxides
Bi2o3 and Mo03 , the activity and selectivity of the catalyst is impaired. Experiments carried out with a low
oxygen content in a quartz glass reactor showed that the
catalyst changed in color from light-yellow in the upper
part of the reactor where oxygen is present to greenish
grey at the exit side of the reactor where all oxygen
had been consumed. A change in the oxygen content of the gas inlet stream was followed by a reversible change in
the color of the catalyst, provided the reduction time
was not too long. During the thermobalance experiments
we observed the same phenomenon as a .function of time. Reoxidation of a catalyst that has been reduced too far
results in.a loss of activity and selectivity. Especially
the integral selectivity for ammonia to acrylonitrile
remains low after reoxidation of a catalyst that has been
reduced too far. As the catalyst has a measurable oxy
gen pressure at reaction temperatures a small stream of
air is carried over the catalyst bed at night between
the experiments, whereas the temperature was kept at the
experimental level.
- External and internal mass and heat transfer resistances
With respect to film diffusion limitation preliminary
experiments under steady state conditions have shown
that varying the gas flow at constant mass space velo
city does not change the degree of conversion. Also a
calculation of the mass transfer coefficient for the
ammoxidation of propene at high temperatures showed only a very small difference between the surface and bulk gas phase concentration of propene.
From a calculation of the temperature difference be
tween the catalyst particle and the gas phase we draw
63

the conclusion that this temperature difference is very
small and that the temperature measured in the gas phase
represents the temperature of the catalyst surface.
The absence of pore diffusion limitation was confir
med by varying the average catalyst particle diameter,
i.e. no change of the degree of conversion was observed.
Also a calculation of the effectiveness factors n for
the different catalyst particles at isothermic condition,
according to Satterfield and Sherwood( 1) gives values
close to unity.
Axial and radial temperature profiles under steady
state conditions have been discussed in chapters 3 and 5.
- Catalyst dilution
As has been shown in chapters 3 and 5 great care has
been given to the selection of dilution materials for
controlling the temperature of the catalyst bed, thus
ascertaining a flat temperature profile over the reactor.
Application of silicon carbide was found to give a
strong decrease in carbon dioxide formation~ We ascribe
this to a suppression of the postcatalytic oxidation of
acrolein which can be noticed in empty reactors and to
a much smaller extent in a reactor filled with silicon
carbide.
6.2.3. The oxidation of propene to acrolein
6.2.3.1. Experiments at 673 K
The experimental data for the determination of the ki
netic parameters were obtained with flow reactor A (see
chapter 3, Table 3.1). This small reactor was chosen in
order to reduce the postcatalytic volume, and consequently
the non catalytic after reactions. The reactor was filled
with a mixture of 600 mg Bi2Mo1 •02o6 •06 prepared according
to method A and 1.7 g silicon carbide. The particle dia
meter range of the catalyst and of the silicon carbide was .50-.85 mm. The bed beight was 53 mm.
64
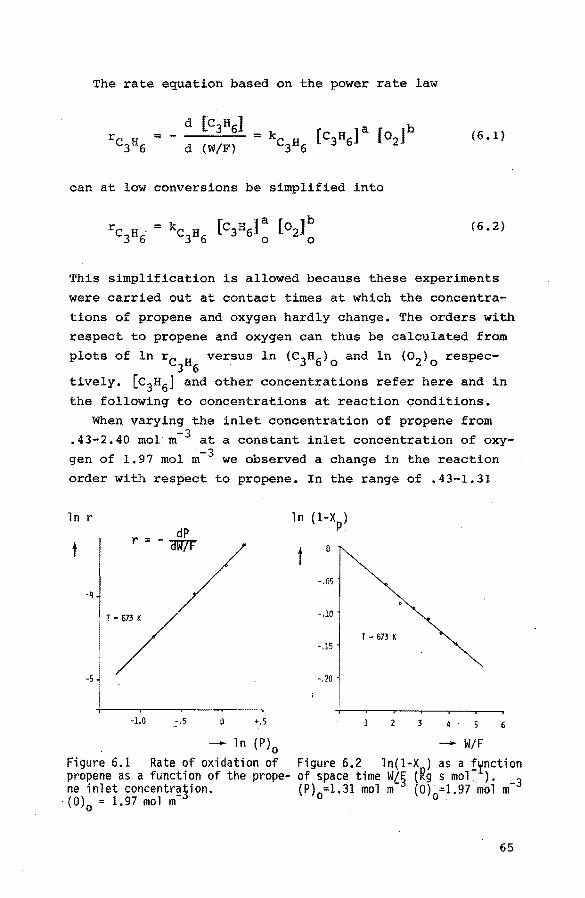
The rate equation based on the power rate law
(6 .1)
can at low conversions be simplified into
(6.2)
This simplification is allowed because these experiments
were carried out at contact times at which the concentra
tions of propene and oxygen hardly change. The orders with
respect to propene and oxygen can thus be calculated from
plots of ln rc H versus ln (C3H6)0
and ln (02)0
respec-3 6
tively. [c3H6] and other concentrations refer here and in
the following to concentrations at reaction conditions.
When varying the inlet concentration of propene from .43-2.40 mol m- 3 at a constant inlet concentration of oxy
gen of 1.97 mol m- 3 we observed a change in the reaction
order with respect to propene. In the range of .43-1.31
ln r
t
-4
-5
r = dP - awr
-1.0 :·5 +.5
- ln (P) 0 Figure 6.1 Rate of oxidation of propene as a function of the propene inlet concentra3ion.
· (0)0
= 1.97 mol m- ·
ln {1-Xp)
t 0
-.05
-.10
T = 673K -.15
-.20
2 6
__,.,. W/F
Figure 6.2 ln(l-X0
) as a f~nction of space time W[~ (Kg s mol- ). _3 (P) 0=1.31 mol m {0)
0=1.97 mol m
65
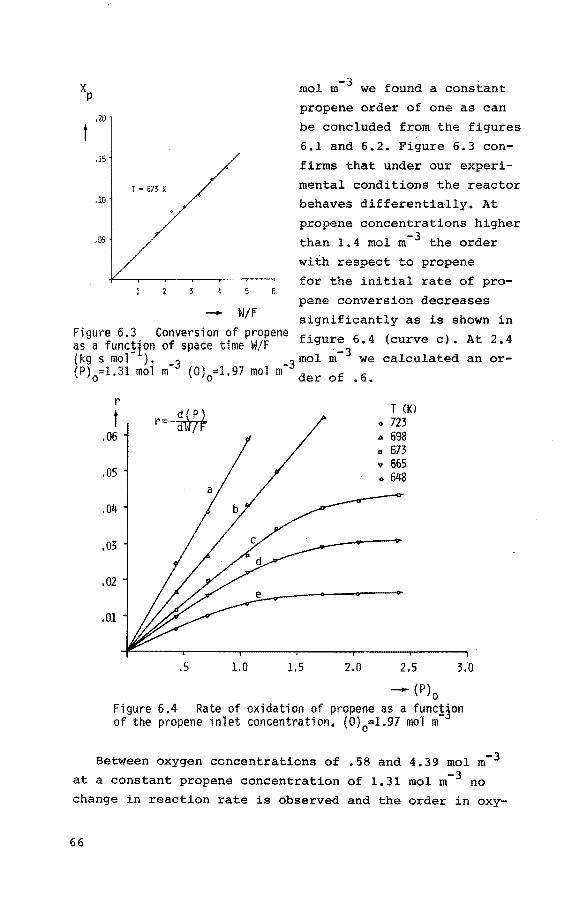
.20
t .15
.10
.05
mol m-3 we found a constant
propene order of one as can
be concluded from the figures
6.1 and 6.2. Figure 6.3 con
firms that under our experi
mental conditions the reactor
behaves differentia:lly. At
propene concentrations higher
than 1.4 mol m- 3 the order
with respect to propene
for the initial rate of pro-
pene conversion decreases
significantly as is shown in Figure 6. 3 Conversion of propene figure 6 • 4 (curve c) • At 2. 4 as a func!fon of space time W/F 3
- W/F
(kg s mol ) • mol m- we calculated an or-(P)0=1.31 mol m-3
(0) 0 =1.97 mol m~3 der of • 6 •
r
t r=-~ .06
.05
.04
.03
.02
.01
.5 1.0 1.5 2.0
T (K) 0 723 ... 698 a 673 v 665 0 648
2.5
- (P)
3.0
0 Figure 6.4 Rate of oxidation of propene as a func!~on of the propene inlet concentration. (0)
0=1.97 mol m
Between oxygen concentrations of .58 and 4.39 mol m-3
at a constant propene concentration of 1.31 mol m- 3 no
change in reaction rate is observed and the order in oxy-
66

gen is therefore zero for that concentration range. The
initial rate of acrolein formation at low propene inlet
concentrations is also first order with respect to pro
pene.'
As the decrease of the order with respect to propene
points either to a strong adsorption of propene or to an
adsorption of products (e.g. acrolein or water) we inves
tigated the influence of acrolein and water on the rate of
reaction. The partial pressures of acrolein and water in
the inlet gas stream are of the same order as obtained in
situ from the oxidation of propene. We observed a decrease
of the conversion of propene when acrolein was introduced
but water had no effect on the conversion, as is shown in
table 6.1.
Temperature (K) 673 648
Feed (mol m- 3 )
C3H6 1.310 1.310 1.310 1.341 1.341
02 1.970 1.970 1.970 1.985 1,985
c3H40 - .091 - - .042
H20 - - .061 - -Product (mol m- 3 }
C3H6 1.235 1.259 1.234 1.289 1.316
c3H4o .073 .136 .073 .050 .065
co2 .007 .012 .007 .003 .004
co .002 .004 .002 ,001 .002
c 2H40 trace .002 trace - -H20 .078 .064 .142 .048 .029
Conversion
C3H6 (%) 5.7 3,9 5.8 3.9 1.9
Space time W/F
(kg s mol-l) 1.68 1.68 1.68 2.70 2.70
Integral select!-
vity of propene to
· acrolein SIPACO .97 .93 .96 ,96 .92
Table 6.1. Influence of acrolein and water on the conver
sion of propene.
67

We conclude that acrolein hinders its own formation at
temperatures of 673 and below. As is illustrated by figure
6.5 the major part of the carbon dioxide and the carbon
monoxide formation occurs by means of a consecutive oxi
dation of acrolein.
(C)
t 1.35 C H .
1.30 c3H60 3 4
0 C02 1.25 . co 1.20 0 C2H40
(c) 1.15
1.10 .10 t .20 .08
.15 .06
.10 .04
.05 .02
---+- W/F Figure 6.5 Product concentration as _1 a function of space time W/F (kg s mol ).
6.2.3.2. Experiments at other temperatures
The rate of oxidation of propene at 698 K and 723 K is
first order with respect to propene and zero order in oxy
gen over the range of investigated concentrations and does
not show a substantial inhibition by acrolein as is shown
in figure 6.4 and 6.6. The rates of oxidation at 665 and
648 K are first order in propene only for low propene con
centrations. We confirmed that the decrease of the order
at the higher concentrations is also caused by acrolein
inhibition in the same way as described in the previous
section. The overall activation enthalpy for the oxidation
of propene to acrolein increases from 62 kJ mol-l at tem
peratures above 673 K to 102 kJ mol-l at temperatures be
low 673 K as depicted in figure 6.7.
68
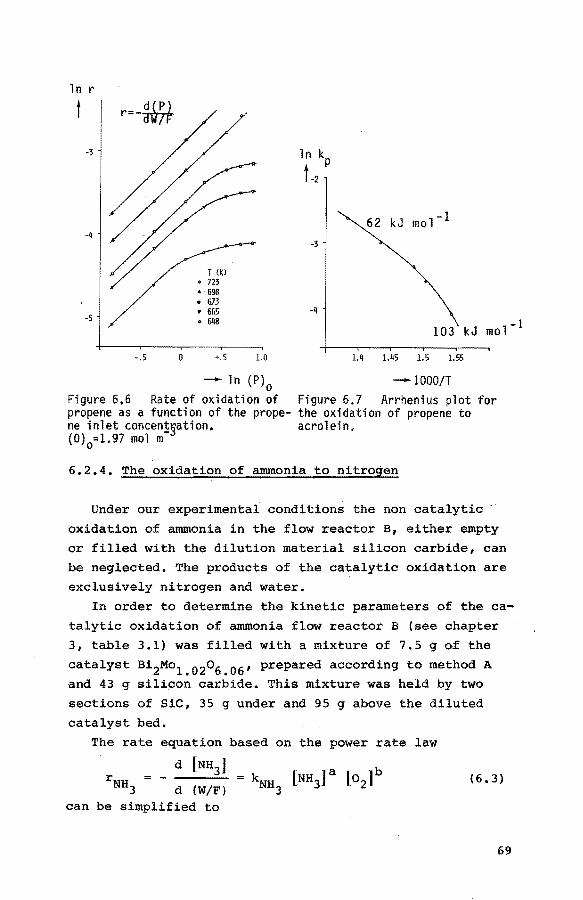
ln r
t r=-~
-3
-4
T (Kl • 723 A 698 • 673 • 665
62 kJ mol-l
-3
-4 -5 • 648
103 kJ mol-l
-.5 +,5 1.0
- ln (P)0
Figure 6.6 Rate of oxidation of propene as a function of the propene inlet concen!3ation. (0)
0=1.97 mol m
1.4 1.45 1.5 1.55
-1000/T
Figure 6.7 Arrhenius plot for the oxidation of propene to acrolein.
6.2.4. The oxidation of ammonia to nitrogen
Under our experimental conditions the non catalytic ·
oxidation of ammonia in the flow reactor B1 either empty
or filled with the dilution material silicon carbide, can
be neglected. The products of the catalytic oxidation are
exclusively nitrogen and water.
In order to determine the kinetic parameters of the ca
talytic oxidation of ammonia flow reactor B (see chapter
3, table 3.1) was filled with a mixture of 7.5 g of the
catalyst Bi2Mo1 •02o6 •06 , prepared according to method A and 43 g silipon carbide. This mixture was held by two
sections of SiC, 35 g under and 95 g above the diluted
catalyst bed.
The rate equation based on the power rate law
d [NH3] = kNH [NH3]a [o2]b
d (W/F) 3 (6.3)
can be simplified to
69

( 6. 4)
for differential conditions. As is shown in figure 6.8 and
6.9 the reaction rate is first order in ammonia and zero
order in oxygen.
1 n r
t -2 r=-Wf
T = 673 K
-3
- ln (N) 0 Figure 6.8 Rate of oxidation of ammonia as a function of the ammonia inlet conce~3ration. {0)
0=4.20 mol m
ln r
L2 r=-Wf
T = 673K
-3
4 4-----.----.-----r----~ 1.0 1.5 2.0 .5
-ln (0}0
Figur: 6.9 Rate of oxidation of ammon1a as a function of the oxygen inlet conce~3ration. (N)
0=1.55 mol m
Experiments carried out at 648, 698 and 723 K showed
the same kinetic relations. For the activation enthalpy a value of 159 kJ mol-l was found.
6,2,5. The ammoxidation of acrolein to acrylonitrile
6.2.5.1. Preliminary experiments
In literature the ideas of Shelstad et al (2), viz.
that acrolein is a necessary intermediate for the forma
tion.of acrylonitrile, made way for the view of Callahan
et al (3) and Cathala et al (4) that mainly oxygen free
intermediates are precursors for acrylonitrile and that
the slow formation of acrolein by a parallel oxidation
reaction is followed by a fast ammoxidation reaction.
70

--main
slow~ / fast
Experimentally we found the ammoxidation of acrolein to be
very fast.at 673 K, as can be seen in table 6.2.
k (mol kg -1 -1 s )
C3H6 + c3H40 .025
NH3
+ N2 .018
C3H6 + c3H3N .035
C3H40 + c3H3N .42
Table 6.2. Overall initial rate constants with respect to
the product at 673 K for the oxidation and
ammoxidation of propene, the oxidation of
ammonia and the ammoxidation of acrolein.
The main products besides acrylonitrile are water, carbon
dioxide and nitrogen, with only traces of other products.
Experiments at temperatures well above 673 K showed an
almost total conversion of acrolein even at the lowest
contact times attainable in the flow reactors. Experiments
at temperatures below 673 K gave severe blockage of the
product lines and frequent upsets of the analytical system
because of the presence of acrolein. So the ammoxidation
of acrolein has been studied at 673 K only.
As the reaction rate at 673 K is high, special attention
was paid to the possibility of diffusion limitation. The
experimental data that are used in this section are not
influenced by diffusion nor by dilution of the catalyst
bed by inertmaterial.
71
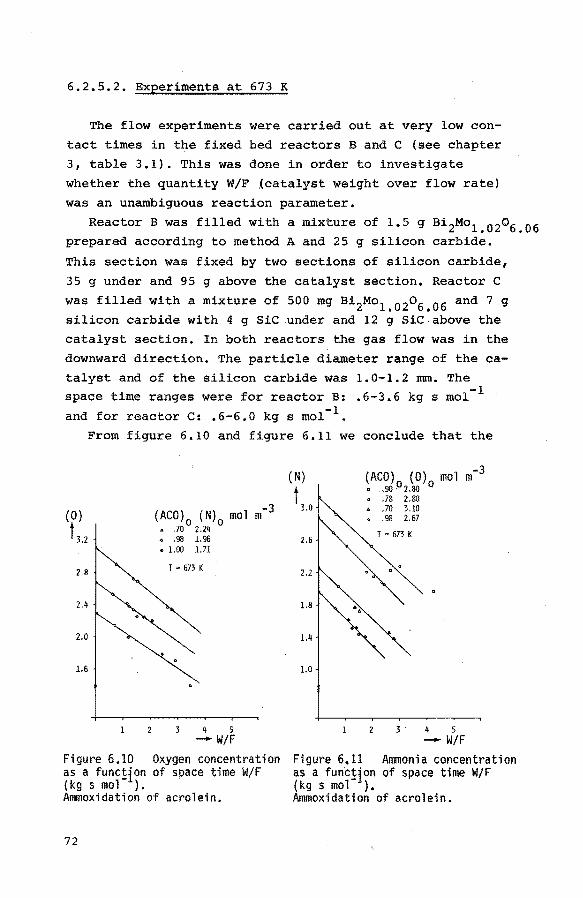
6.2.5.2. Experiments at 673 K
The flow experiments were carried out at very low con
tact times in the fixed bed reactors B and C (see chapter
3, table 3.1). This was done in order to investigate
whether the quantity W/F .(catalyst weight over flow rate)
was an unambiguous reaction parameter.
Reactor B was filled with a mixture of 1.5 g Bi2Mo1 •02o6 •06 prepared according to method A and 25 g silicon carbide.
This section was fixed by two sections of silicon carbide,
35 g under and 95 g above the catalyst section. Reactor C
was filled with a mixture of 500 mg Bi2Mo1 •02o6 •06 and 7 g
silicon carbide with 4 g SiC under and 12 g SiC above the
catalyst section. In both reactors the gas flow was in the
downward direction. The particle diameter range of the ca
talyst and of the silicon carbide was 1.0-1.2 mm. The space time ranges were for reactor B: .6-3.6 kg s mol-l
-1 and for reactor C: .6-6.0 kg s mol •
From figure 6.10 and figure 6.11 we conclude that the
(0)
t 3.2
2.8
2.4
2.0
1.6
2
-3 (AC0)0
(N)0
mol m • .70 2.2~ • ,98 1.96 • 1.00 1.71
T = 673 K
4 5 -W/F
Figure 6.10 Oxygen concentration as a func!fon of space time W/F (kg s mol ). Ammoxidation of acrolein.
72
( N)
t 3.0
2.6
2.2
1.8
1.4
1.0
-3 (AC0)0
(0)0
mol m • ,,90' 2.80 • .78 2.80 • .70 3.10 ~ .98 2.67
T=673K
4 5 -W/F
Figure 6.11 Ammonia concentration as a funcHon of space time W/F (kg s mol ). Ammoxidation of acrolein.
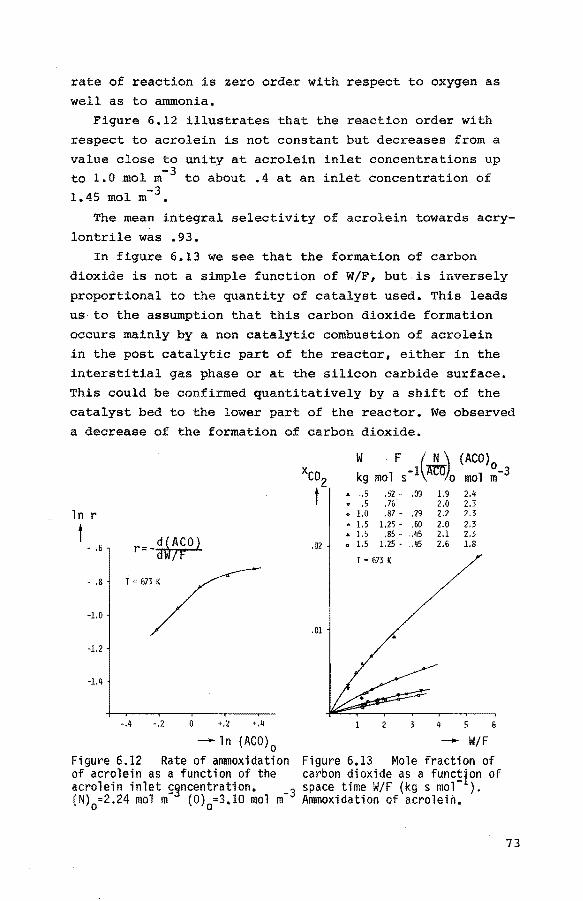
rate of reaction is zero order with respect to oxygen as
well as to ammonia.
Figure 6.12 illustrates that the reaction order with
respect to acrolein is not constant but decreases from a
value close to unity at acrolein inlet concentrations up to 1.0 mol m- 3 to about .4 at an inlet concentration of
-3 1,45 mol m •
The mean integral selectivity of acrolein towards acry
lontrile was .93.
In figure 6,13 we see that the formation of carbon
dioxide is not a simple function of W/F, but is inversely
proportional to the quantity of catalyst used. This leads
us to the assumption that this carbon dioxide formation
occurs mainly by a non catalytic combustion of acrolein
in the post catalytic part of the reactor, either in the
interstitial gas phase or at the silicon carbide surface.
This could be confirmed quantitatively by a shift of the
catalyst bed to the lower part of the reactor. We observed
a decrease of the formation of carbon dioxide.
xco 2
W . F ( N ~ (AC0)0 kg mol s-1 XCU'o mol m-3
t A .. 5 .52 .09 1.9 2.4 9 .5 .76 2.0 2.3
ln r • 1.0 .87 .29 2.2 2.3
- .6
• 1.5 1.25 .60 2.0 2.3 • 1.5 .85 .45 2.1 2.3
.02 • 1.5 1.25 - .. 45 2.6 1.8 t T = 673K
- .8 T 673K
-1.0
.01
-1.2
-1.4
-.4 -.2 +,2 +,4 2 3 4 6
___,..ln (AC0)0
- W/F Figure 6.12 Rate of ammoxidation Figure 6,13 Mole fraction of of acrolein as a function of the carbon dioxide as a function of acrolein inlet s~ncentration, _3 space time W/F (kg s mol- ). (N)
0=2.24 mol m (0)
0=3.10 mol m Ammoxidation of acrolein.
73

As compared to the ammoxidation of propene the formation
of nitrogen is now notably suppressed. The integral selec
tivity of ammonia towards nitrogen is .09-.32 at inlet
molar ratios of ammonia to acrolein of 1.3-3.7 mol mol-1 •
For the propene ammoxidation the integral ammonia selecti
vity towards nitrogen is at comparable conditions .60-.75
(see figure 6.24).
6.2.6. The ammoxidation of propene to acrylonitrile
6.2.6.1. Introduction and preliminary experiments
All reactions investigated in the preceding sections
appear to some extent during the ammoxidation of propene.
However for a selective catalyst it is expected that it
will not accelerate
- the parallel oxidation reactions of propene and ammonia
to acrolein, carbon dioxide, nitrogen and other oxida
tion products7 - the consecutive oxidation of acrylonitrile and acrolein
to carbon dioxide and other oxidation products.
It will be clear from the foregoing that besides the
main products acrylonitrile and water we will have a num
ber of byproducts. Of these nitrogen, carbon dioxide,
acetonitrile, carbon monoxide, acrolein and acetaldehyde
are of significance and generally found in modest quanti
ties in the reaction mixtures. At high conversions also
small quantities of ethene and of hydrogen cyanide have
been detected.
During the oxidation of propene the catalyst remains
active and selective for long periods of time, provided
gas phase oxygen is present. We calculated that more than
10 4 mol propene per kg catalyst were converted without
loss of activity and selectivity.
Preliminary experiments with water vapour in the feed
did not show a significant improvement of the integral
selectivity of propene towards acrylonitrile. As regards ammonia we observed some improvement of its integral se-
74
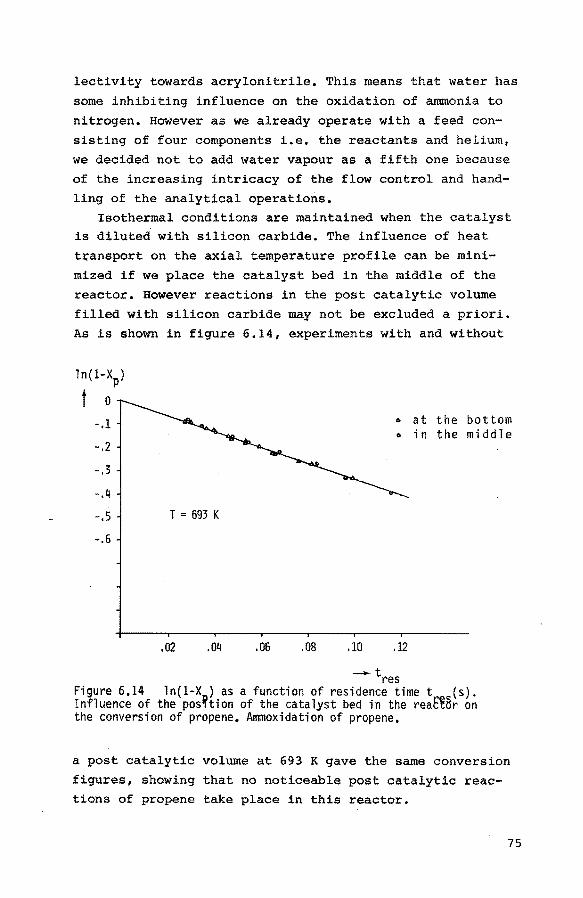
lectivity towards acrylonitrile. This means that water has
some inhibiting influence on the oxidation of ammonia to
nitrogen. However as we already operate with a feed con
sisting of four components i.e. the reactants and helium,
we decided not to add water vapour as a fifth one because
of the increasing intricacy of the flow control and hand
ling of the analytical operations. Isothermal conditions are maintained when the catalyst
is diluted with silicon carbide. The influence of heat
transport on the axial temperature profile can be mini
mized if we place the catalyst bed in the middle of the
reactor. However reactions in the post catalytic volume
filled with silicon carbide may not be excluded a priori.
As is shown in figure 6.14, experiments with and without
ln(l-Xp)
t 0
-.1 .,. at the bottom o in the middle
-.2
-.3
-.4
-.5 T = 693 K
-.6
.02 .OLJ .06 .08 .10 .12
-tres Figure 6.14 ln(l-X ) as a function of residence timet (s). Influence of the pos~tion of the catalyst bed in the reattBr on the conversion of propene. Ammoxidation of propene.
a post catalytic volume at 693 K gave the same conversion
figures, showing that no noticeable post catalytic reac
tions of propene take place in this reactor.
75

6.2.6.2. Experiments at 673 K
The experimental data were obtained with flow reactor
B (see chapter 3, table 3.1). The reaction conditions are
summarized in table 6.3.
[c3H6] .43 - 2.47 mol m -3
0 -3 (NH31 .78- 2.70 mol m
0 -3 [02] 2.58 - 4.51 mol m
0 -1 [c3H6/NH3] .21 - 2.05 mol mol 0
Pressure 1.0 bar
W/F 2.4 - 32.4 kg s mol -1
Table 6.3. Reaction conditions for the ammoxidation of
propene in flow reactor B at 673 K.
The catalysts prepared according to method A and B (see
chapter 4, section 4.3) did not show a difference in ac
tivity and selectivity. The product concentrations as a
function of the contact time W/F, which are shown in fi
gure 6.15, are representative for the kinetic experiments
of the propene ammoxidation at 673 K.
The initial rate of reaction is zero order with respect to oxygen, provided the oxygen is present in stoichiometric
excess (see figure 6.16). This also holds for the oxida
tion of propene and ammonia and for the ammoxidation of
acrolein, as is illustrated in section 6.2.3 to 6.2.5.
With respect to ammonia we observed a small decrease of
the rate of reaction at increasing ammonia concentrations.
For the rate of propene conversion we calculated an ammo
nia order of -.08 to -.14 at inlet propene concentrations
of .78-2.33 mol m- 3 • This is shown in figure 6.17. The
same influence of the ammonia concentration is found for the rate of acrylonitrile formation.
76

(C)
p.o
10 20
• C3H6 a NH3
A C-jl~ v 0)2 o N2 o c2H3N
T = 673 K
30 40 -W/F
{C02) (C)
.·4ot .2ot
.36 .18
.32 .16
.28 .14
.24 .12
.20 .10
.16 .08
.12 .06
.08 .04
.04 .02
Figure 6.15 Concentration~ 1of reactants_and products as a function of space time WL~ (kg s mol ). Ammoxidat1on of propene. (0) =4.51 mol m -3 o ln r
0 (P}
0mol m
r
f t • .78 ( ) : i:~ r o =- %£f o
T • 673K -2.0
.08
-2.5 .06
-3.0
-3.5 .02 T • 673 K
-4.0 +---~--.----r---r--~
1 .2 .4 .6 .8 1.0
- (0) - ln (N) Figure 6.16 Initial rate 8f am- Figure 6.17 Initial rates of Rmmoxidation of propene as a function moxidation of propene as a function of the oxygen c22centration. _3 of the ammonia i~let concentration. (P)
0=1.49 mol m (N)
0=1.55 mol m {0}
0=4.26 mol m
77
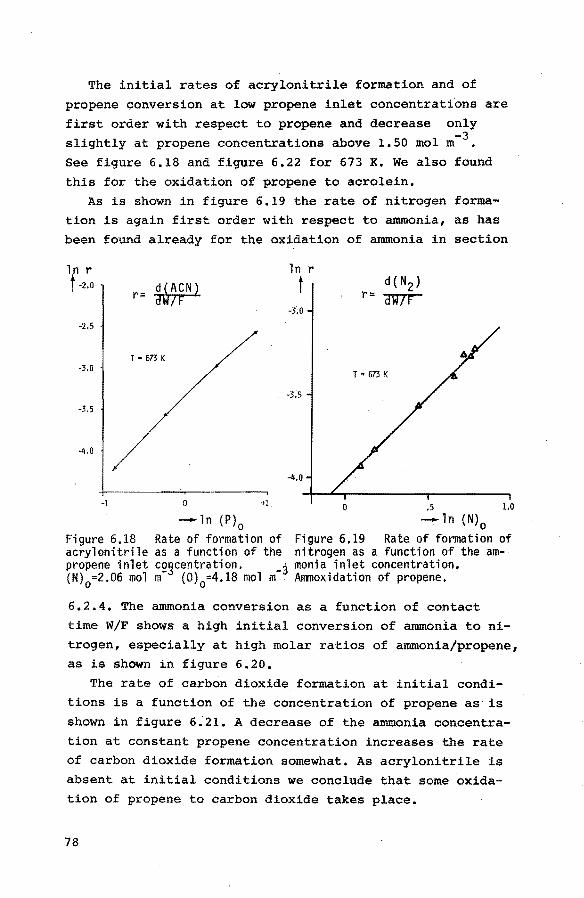
The initial rates of acrylonitrile formation and of
propene conversion at low propene inlet concentrations are
first order with respect to propene and decrease only -3 slightly at propene concentrations above 1.50 mol m
See figure 6.18 and figure 6.22 for 673 K. We also found
this for the oxidation of propene to acrolein.
As is shown in figure 6.19 the rate ofnitrogen forma
tion is again first order with respect to ammonia, as has
been found already for the oxidation of ammonia in section
ln r t ·2.0
-2.5
-3.0
-3.5
-4.0
-1
ln r
r=~ t d{N2) r= ifi17r
-3,0
T=673K
-3.5
-4.0
0 +1. 0 .5 1.0
-ln (P)0
-ln (N)0
Figure 6.18 Rate of formation of Figure 6,19 Rate of formation of acrylonitrile as a function of the nitrogen as a function of the am-propene inlet co~centration. -3 monia inlet concentration. {N)
0=2.06 mol m- (0)
0=4.18 mol m · Ammoxidation of propene.
6,2.4, The ammonia conversion as a function of contact
time W/F shows a high initial conversion of ammonia to ni
trogen, especially at high molar ratios of ammonia/propene, as is shown in figure 6.20.
The rate of carbon dioxide formation at initial condi
tions is a function of the concentration of propene as is
shown in figure 6.21. A decrease of the ammonia concentra
tion at constant propene concentration increases the rate
of carbon dioxide formation somewhat. As acrylonitrile is
absent at initial conditions we conclude that some oxida
tion of propene to carbon dioxide takes place.
78
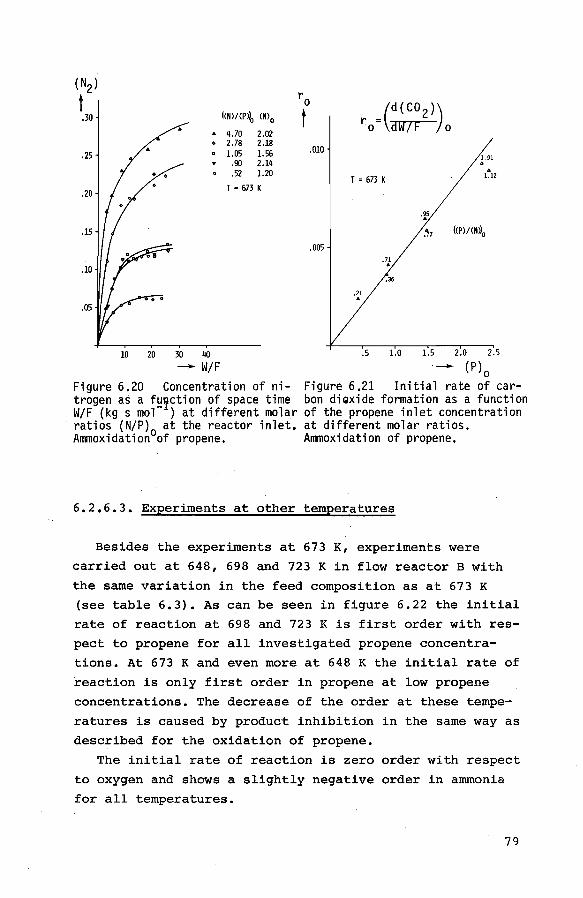
(N2) ro t r -C(co2)) .30 (<Nl/(Plb <Nl 0 t . 4.70 2.02
o- dW/F o . 2.78 2.18 .25 a 1.05 1.56 .010
.90 2.14 !.91
.52 1.20 . T = 673K 1.12
.20 T = 673 K
.95 . .15 .''n (<Pl/(N~0
.005 .71
. 10 . .36
.~1
.05
10 20 30 40 .5 1.0 1.5 2.0 2.5
-- W/F -- (P) 0 Figure 6.20 Concentration of ni- Figure 6.21 Initial rate of cartrogen as a fu~ction of space time bon diGxide formation as a function W/F (kg s mol- ) at different molar of the propene inlet concentration ratios {N/P) at the reactor inlet. at different molar ratios. Ammoxidation°of propene. Ammoxidation of propene.
6.2.6.3. Experiments at other temperatures
Besides the experiments at 673 K, experiments were
carried out at 648, 698 and 723 K in flow reactor B with
the same variation in the feed composition as at 673 K (see table 6.3). As can be seen in figure 6.22 the initial
rate of reaction at 698 and 723 K is first order with res
pect to propene for all investigated propene concentra
tions. At 673 K and even more at 648 K the initial rate of
reaction is only first order in propene at low propene
concentrations. The decrease of the order at these tempe
ratures is caused by product inhibition in the same way as
described for the oxidation of propene.
The initial rate of reaction is zero order with respect
to oxygen and shows a slightly negative order in ammonia
for all temperatures.
79
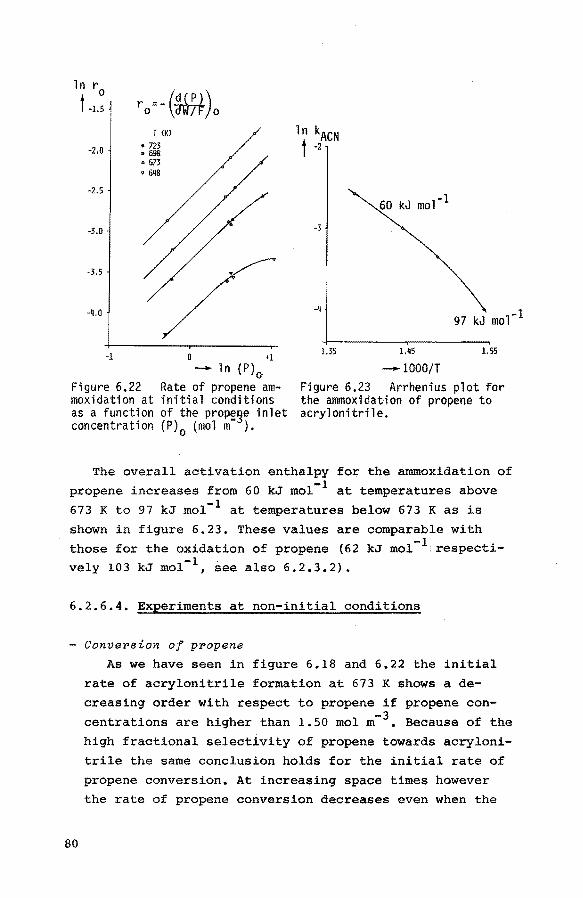
ln r0
t ·1.5
·2.0
·2.5
·3.0
-3.5
-1
T <K)
• 723 • 698 4 673 • 648
0 +1
- ln (P) 0
ln k.ACN t ·2
-3
-4
1.35
97 kJ mol-l
1.45 1.55
-1000/T Figure 6.22 moxidation at as a function concentration
Rate of propene aminitial conditions of the prop~~e inlet (P)
0 (mol m ) •
Figure 6.23 Arrhenius plot for the ammoxidation of propene to acrylonitrile.
The overall activation enthalpy for the arnmoxidation of -1 propene increases from 60 kJ mol at temperatures above
673 K to 97 kJ mol-l at temperatures below 673 K as is
shown in figure 6.23. These values are
those for the oxidation of propene (62 -1 . vely 103 kJ mol , see also 6.2.3.2).
comparable with -1 kJ mol respecti-
6.2.6.4. Experiments at non-initial conditions
- ConvePsion of ppopene
80
As we have seen in figure 6.18 and 6.22 the initial
rate of acrylonitrile formation at 673 K shows a de
creasing order with respect to propene if propene con
centrations are higher than 1.50 mol m- 3 • Because of the
high fractional selectivity of propene towards acryloni
trile the same conclusion holds for the initial rate of
propene conversion. At increasing space times however
the rate of propene conversion decreases even when the
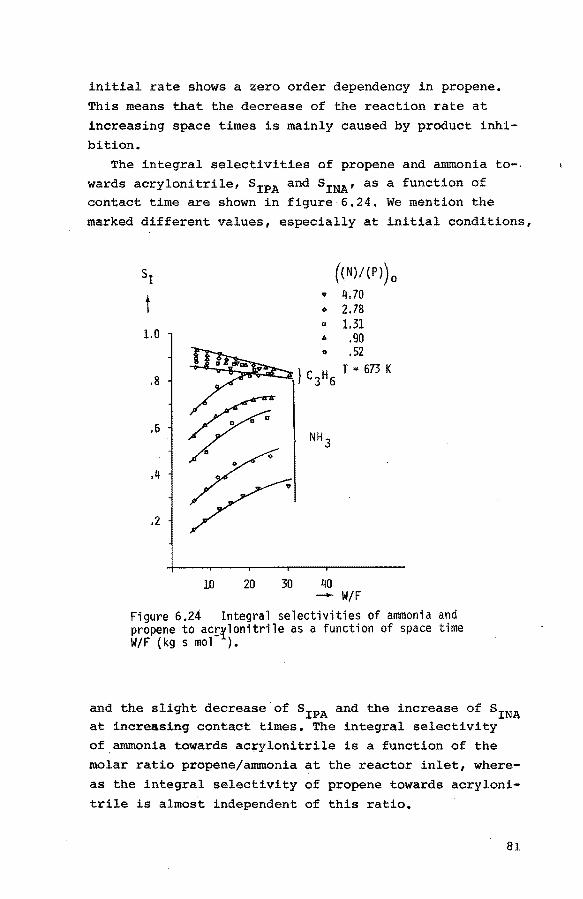
initial rate shows a zero order dependency in propene.
This means that the decrease of the reaction rate at
increasing space times is mainly caused by product inhi
bition.
The integral selectivities of propene and ammonia to-.
wards acrylonitrile, SIPA and SINA' as a function of contact time are shown in figure 6.24. We mention the
marked different values, e.specially at initial conditions,
t 1.0
.8
.6
.4
.2
((N)/ (P)) 0
v 4.70 0 2.78 II 1.31 I> .90 0 .52
.,._~~~~ } C H T "' 673 K 3 6
10 20 30 40 - W/F
Figure 6.24 Integral selectivities of ammonia and propene to acrrlonitrile as a function of space time W/F (kg s mol ).
and the slight decrease of SIPA and the increase of SINA at increasing contact times. The integral selectivity
of.ammonia towards acrylonitrile is a function of the
molar ratio propene/ammonia at the reactor inlet, where
as the integral selectivity of propene towards acryloni
trile is almost independent of this ratio.
81
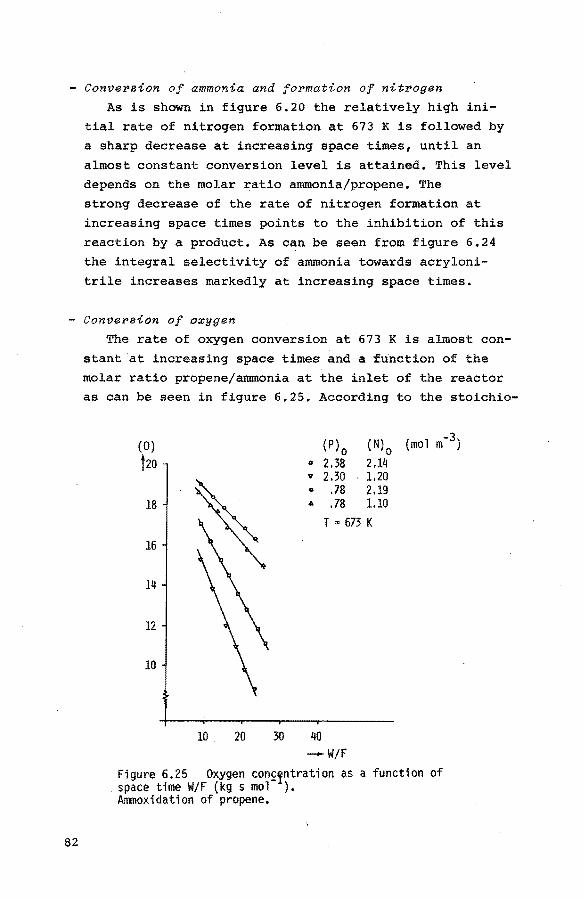
ConvePaion of ammonia and foPmation of nitPogen
As is shown in figure 6.20 the relatively high ini
tial rate of nitrogen formation at 673 K is followed by
a sharp decrease at increasing space times, until an
almost constant conversion level is attained. This level
depends on the molar ratio ammonia/propene. The
strong decrease of the rate of nitrogen formation at
increasing space times points to the inhibition of this
reaction by a product. As can be seen from figure 6.24
the integral selectivity of ammonia towards acryloni
trile increases markedly at increasing space times.
- ConvePaion of oxygen
82
The rate of oxygen conversion at 673 K is almost con
stant at increasing space times and a 'function of the
molar ratio propene/ammonia at the inlet of the reactor
as can be seen in figure 6.25. According to the stoichio-
(0) (P)o ( N} o (mol m-3}
ho D 2,38 2.1q
~ "' 2.30 1.20 0 .78 2.19
18 ... .78 1.10 T = 673 K
16
14
12
10
10. 20 30 40 -w;F
Figure 6.25 Oxygen con~!ntration as a function of space time W/F (kg s mol ). Ammoxidation of propene.
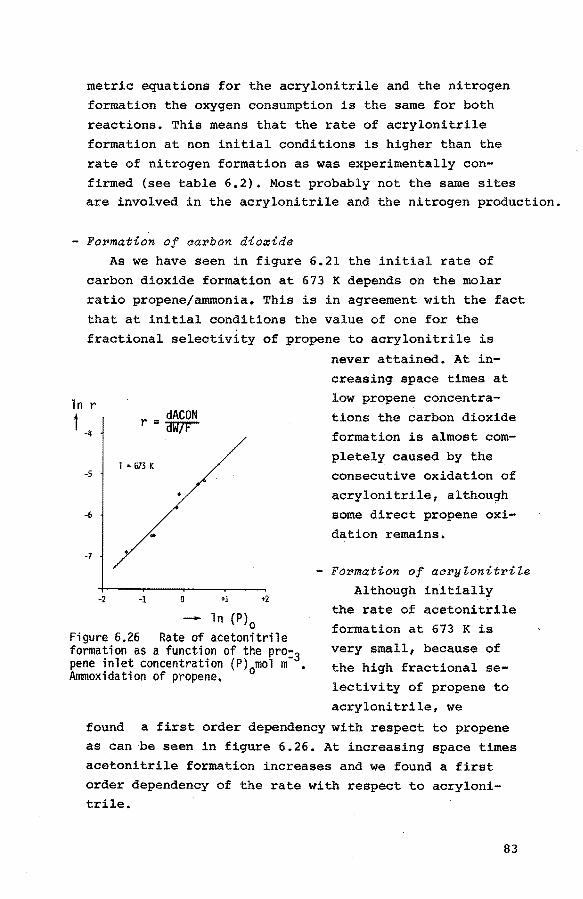
metric equations for the acrylonitrile and the nitrogen
formation the oxygen consumption is the same for both
reactions. This means that the rate of acrylonitrile
formation at non initial conditions is higher than the
rate of nitrogen formation as was experimentally con
firmed (see table 6.2). Most probably not the same sites are involved in the acrylonitrile and the nitrogen production.
- Formation of carbon dioreide As we have seen in figure 6.21 the initial rate of
carbon dioxide formation at 673 K depends on the molar
ratio propene/ammonia. This is in agreement with the fact
that at initial conditions the value of one for the
fractional selectivity of propene to acrylonitrile is
ln r
t -4 r _ dACON
- OA7t
T • 673K -5
-6
-7
-2 -1 +1 +2
- ln (P) 0 Figure 6.26 Rate of acetonitrile formation as a function of the pro" pene inlet concentration (P)
0mol m-3•
Ammoxidation of propene.
never attained. At in
creasing space times at
low propene concentra
tions the carbon dioxide
formation is almost com
plete!~ caused by the
consecutive oxidation of
acrylonitrile, althou~h
some direct propene oxi
dation remains.
- Formation of acryLonitrile Although initially
the rate of acetonitrile
formation at 673 K is
very small, because of
the high fractional se
lectivity of propene to
acrylonitrile, we
found a first order dependency with respect to propene
as can be seen in figure 6.26. At increasing space times
acetonitrile formation increases and we found a first
order dependency of the rate with respect to acrylonitrile.
83

- Other products
Under the conditions of our investigation into the
kinetics of the propene ammoxidation at 673 K products like carbon monoxide, ethene, acetaldehyde and hydrogen
cyanide are produced in such low quantities that con
clusions about their rate of formation would be specula
tive.
6.3. Thermobalance experiments
Catalyst reduction and propene oxidation.
6.3.1. Introduction
In a thermobalance the weight changes of a given amount
of catalyst under various reaction conditions can be stu
died as a function of reaction time. The weight changes
may be caused by reduction (oxygen depletion of the cata
lyst) and by adsorption of reactants and products.
As discussed in section 6.1 and 6.2 the kinetics of the
different reactions present a rather complicated picture.
The results as regards the influence of the concentrations are summarized in table 6.4.
Order with respect to Reaction
C3H6 NH3 c3H40 02
* C3H6 -+- c3H40 1-0 - - 0 * c3H40 -+- c3H3N - 0 1-0 0
* C3H6 -+- c3H3N 1-0 0 - 0 NH3 -+- N2 - 1 - 0
* depends on temperature and concentration
of propene or acrolein respectively
Table 6,4. Orders of the initial rates of various reactions.
84

Under similar conditions the rates of the oxidation of
propene, of the oxidation of ammonia, of the ammoxidation
of propene and of the ammoxidation of acrolein increase in this order, as was shown in table 6.2.
Below 673 K acrolein and acrylonitrile hinder the oxi
dation and ammoxidation of propene. Oxygen diffusion in the catalyst lattice to the reactive sites is also expected to influence the overall rate of reaction.
6.3.2. Preliminary experiments
For all thermobalance experiments a catalyst with a specific surface of 7.3 m2 g-1 was used, which was obtained
according to method B, but with a calcination temperature
of 753 K. Neither changes in gas flow rate (from 1-70 to 250 cm3
min-1 NTP) nor changes in catalyst.quantity (from 45 to 125 mg) nor changes in particle size (from .s to 1.4 mm)
had any influence on the rate of reduction of the cata
lyst. Calculations carried out according to the procedure of
Yoshida et al (5) showed that the tE;!mperature difference
across the boundary layer between the particle and the
gas phase can be neglected, so the temperature measured
above the sample is equal to the temperature of the particle.
6.3.3. Reduction of the catalyst with propene
In table 6.5 the reaction conditions are given for the reduction of bismuth molybdate with mixtures of propene and nitrogen.
Figure 6.27 shows for a number of temperatures the weight of a sample as a function of time for the first 400
seconds of exposure to diluted propene (mole fraction xp = .085). More prolonged exposure causes the weight to
increase again, due to acrolein adsorption (see this chapter, section 6.3.6). In section 6.4.3 we will.offer
85
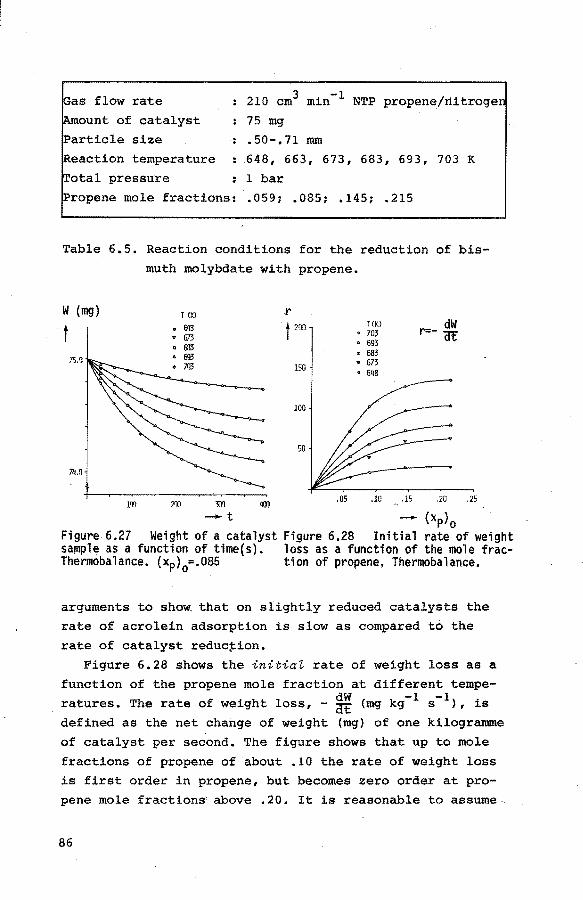
Gas flow rate
Amount of catalyst Particle size
Reaction temperature
Total pressure
210 cm3 min-1 NTP propene/nitrogen
75 mg
.50-.71 mm
648, 663, 673, 683, 693, 703 K
1 bar
Propene mole fractions: .059; .085; .145; .215
Table 6.5. Reaction conditions for the reduction of bis
muth molybdate with propene.
w {mg) T (Kl .r
t • 643 t zoo HKl dW 6 703 r=- at • 693 • 683
75.0 v 673 150 • 648
100
50
74.0
1?1 300 400 .05 .10 .15 .zo .25
- t - {xp) 0 Figure 6.27 Weight of a catalyst Figure 6.28 Initial rate of weight sa~ple as a function of time(s). loss as a function of the mole frac-Thermobalance. (xp)
0=.085 tion of propene, Thermobalance.
arguments to show that on slightly reduced catalysts the
rate of acrolein adsorption is slow as compared to the
rate of catalyst reduction.
Figure 6.28 shows the initiat rate of weight loss as a
function of the propene mole fraction at different tempe-dW -1 -1 ratures. The rate of weight loss,- dt (mg kg s ), is
defined as the net change of weight (mg) of one kilogramme
of catalyst per second. The figure shows that up to mole
fractions of propene of about .10 the rate of weight loss
is first order in propene, but becomes zero order at propene mole fractions above .20. It is reasonable to assume
86
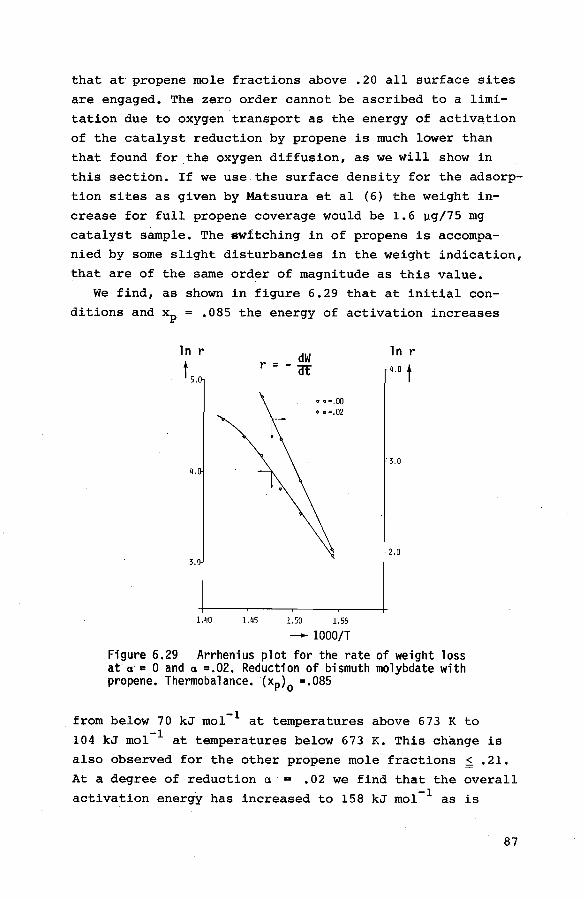
that at propene mole fractions above .20 all surface sites
are engaged. The zero order cannot be ascribed to a limi
tation due to oxygen transport as the energy of activation
of the catalyst reduction by propene is much lower than
that found for the oxygen diffusion, as we will show in
this section. If we use the surface density for the adsorp
tion sites as given by Matsuura et al (6} the weight in
crease for full propene coverage would be 1.6 ~g/75 mg
catalyst sample. The switching in of propene is accompa
nied by some slight disturbancies in the weight indication,
that are of the same order of magnitude as this value.
We find, as shown in figure 6.29 that at initial con
ditions and xp = .085 the energy of activation increases
ln r
t 5.0
3.
dW r = -at
1.50
• •=.00
1. 55
- 1000/T
3.0
2.0
Figure 6.29 Arrhenius plot for the rate of weight loss at·a·= 0 and a =.02. Reduction of bismuth molybdate with propene. Thermobalance. (xp)
0 =.085
from below 70 kJ mol-l at temperatures above 673 K to -1 104 kJ mol at temperatures below 673 K. This change is
also observed for the other propene mole fractions ~ .21.
At a degree of reduction ex = • 02 we find that the overall activation energy has increased to 158 kJ mol-l as is
87

shown by the curve for a = .02 in figure 6.29. The degree
of reduction a is defined as the fraction of the total
oxygen {six atoms per molecule bismuth molybdate) -that has
been removed i.e.
{6. 5)
This means that now another reaction step determines the
rate of reduction. We assume this to be the diffusion of
oxygen ions from the bulk to the surface of the catalyst
to reoxidize the reduced surface sites. If the diffusion
would be very fast the surface oxygen concentration would
be proportional to the bulk oxygen concentration and the
rate of reduction would be
dO - dt k 0 [Pj {6.6)
where 0 is the bulk concentration of oxygen. For constant
[P] we obtain from equation {6.6)
- ln
with k' = tion {6. 7)
0 k't 0 0
k [P] and t is
does not fit
the
the
{6.7)
reaction time. However equa
observed rate of weight loss.
We conclude therefore that the oxygen diffusion influences the observed rate of reaction.
A simplified model for such a combined surface oxidation
reaction and oxygen diffusion has been given by Batist et
al {7). They arrive after certain simplifications to the relation
a = -A + Bt~
that is valid for intermediate values of a and t. In equation {6.8) A and B are
88
{ 6. 8)

B 2 S p D~ -~
'!1'; s
(6.9)
(6.10)
with S is the specific surface area, p the density of the
catalyst, D the diffusion coefficient of oxygen ions in
the lattice, k the rate constant of the chemical reaction, X a socalled jump distance of the order of the lattice
constant and t the reaction time. Although the more gene
ral model of Steenhof de Jong (8) is based on the assump
tion that the chemical reaction step and the diffusion
step are not necessarily equal in rate and that the solid
is not a semi-infinite flat plate, the resulting equation
is rather difficult to handle and we decided to apply
Batist's relation. As is shown in figure 6.30 relation
t a
.OS
.04
.03
.02
.01
.00
-.01
T • 673 K Xp
•. 146 •• 081 •• 059
-.02 +--~--....---,--..-----., 10 15 20 25
- vt Figure 6.30 Degree of reduction as a function of~ Thermobalance.
(6.8) holds at 673 K and at different mole fractions of
propene for .01 < a. < .045 and 6 <It < 15 s~. Nearly the
same ranges are valid at the other temperatures. The de-
89

viation at the upper limit is caused by the adsorption of
acrolein but the values of A and B are still clearly de
fined. As we already know the value of the rate constant of
the chemical reaction on the catalyst surface and its
energy of activation we can use both the expression 6.9
and 6.10 to calculate the diffusion coefficient, its ener
gy of activation and the jump distance. For the diffusion
coefficient at 673 K and xp = .085 we found 3.3 l0-17 m2
s-1 . This value is comparable with that given by Batist
et al (7) for a less active catalyst at 723 K viz. 2.4 10-17 m2 s-1.
We calculate an activation energy from B of 168 kJ mol-l
and from A of 164 kJ mol-1 , assuming a jump distance A that is not dependent on the temperature. For the mean jump distance A we found a value of 3.8 l0-10 m for the
temperatures between 673 and 703 K. From the crystal struc
ture data determined by van den Elzen et al (9) (see fi
gure 4.1) we calculated a distance between two apex oxygen
ions of 3.64 10-10 m, which is in good agreement with the
value of the jump distance calculated from the experiments.
The energy of activation for the diffusion found in this
way fits very well with the results from the rates of re
duction at a= .02 viz. a value of 158 kJ mol-1 • We there
fore conclude that even at a low degree of reduction the
diffusion of oxygen in the bulk has a major influence on
the overall rate of reduction of the catalyst by propene
only. The above confirms that here the model of Batist et
al describes the process accurately.
6.3.4. Reduction of the catalyst with hxdrogen
From experiments carried out with propene, artificial
air and water in the fixed bed reactor we found that water
does not inhibit the formation of acrolein (see table
6.1). This means that also during the reduction of the
catalyst in the thermobalance inhibition by water is probably absent. The reduction of the catalyst with hydrogen
90
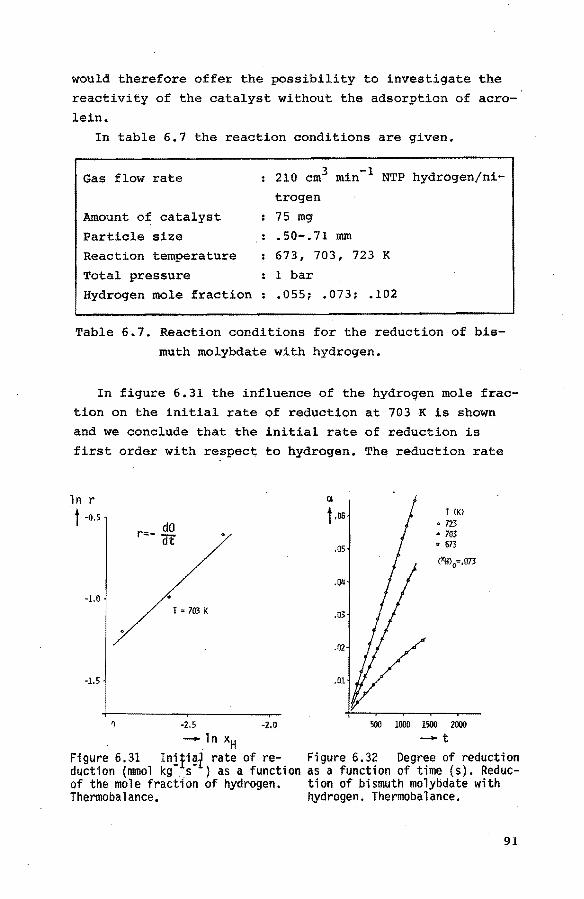
would therefore offer the possibility to investigate the
reactivity of the catalyst without the adsorption of acro
lein.
In table 6.7 the reaction conditions are given.
Gas flow rate 210 cm3 min-1 NTP hydrogen/ni
trogen
Amount of catalyst
Particle size
Reaction temperature
Total pressure
Hydrogen mole fraction
75 mg
.50-.71 mm
673, 703, 723 K
1 bar
.055~ .073~ .102
Table 6.7. Reaction conditions for the reduction of bis
muth molybdate with hydrogen.
In figure 6.31 the influence of the hydrogen mole frac
tion on the initial rate of reduction at 703 K is shown
and we conclude that the initial rate of reduction is
first order with respect to hydrogen. The reduction rate
ln r . t -0.5
-1.0
-1.5
_ dO r-- dt'
T = 703K
.OS
.03
.~II/ .Olr
T <Kl 0 723 6 703 • 673
(XHJ0=,073
q -2.5 -2.0 500 1000 1500 2000
-ln xH -t Figure 6.31 Initial rate of re- Figure 6.32 Degree of reduction duction (mmol kg-_s- } as a function as a function of time (s). Reduc;.. of the mole fraction of hydrogen. tion of bismuth molybdate with Thermobalance. · hydrogen. Thermobalance.
91

with hydrogen is much lower than with propene, as follows
from table 6.8.
dO -I -1 673 K 703 K -at {mmol kg s )
H2 .19 .37
C3H6 2.4 5.4
Table 6.8. Initial rates of reduction of bismuth molyb
date with hydrogen and propene.
XH = Xp = .073.
At 703 and 723 K the degree of reduction is a linear
function of time for the reduction range studied. At 673 K
this linear relationship is somewhat affected as is illu
strated in figure 6.32.
For the rate constant in the initial rate equation
(-~~) 0
(6.11)
we find an energy of activation of 85 kJ mol-l, as follows
from figure 6.33 (straight line). This value agrees with . -I
the value of 8I kJ mol given by Beres .et al {10). The
little curvature of the line in figure 6.32 for the re
duction at lower temperatures is ascribed to influences of the oxygen diffusion on the rate of reduction at a > 0.
As the mean overall energy of activation for the diffu--1 sion (E0 = 163 kJ mol ) is very much larger than the
energy of activation for the reduction with hydrogen the
influence of the diffusion will be felt only at tempera
tures below 673 K. This is illustrated in figure 6.33 by the dotted curve.
92
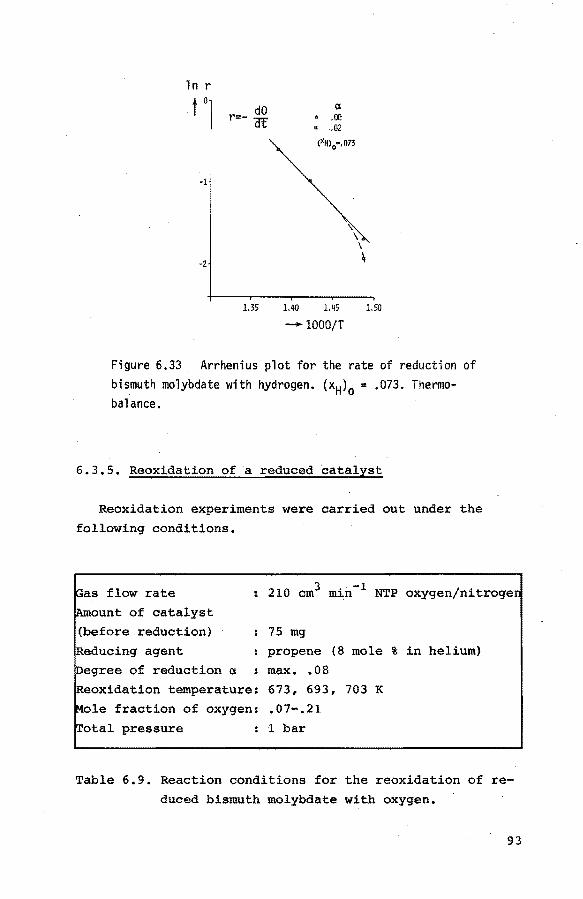
-1
-2
dO r=- df
1.35
(.l
• .00 • .02
(XHJ0=,073
'\ \ \
~
1.40 1.45 1.50
-1000/T
Figure 6.33 Arrhenius plot for the rate of reduction of bismuth molybdate with hydrogen. (xH)o = .073. Thermobalance.
6.3.5. Reoxidation of a reduced catalyst
Reoxidation experiments were carried out under the
following conditions.
Ga_s flow rate
Amount of catalyst (before reduction)
210 cm3 min-I NTP oxygen/nitroger
75 mg
Reducing agent propene (8 mole % in helium)
Degree of reduction a max •• 08
Reoxidation temperature: 673, 693, 703 K
Mole fraction of oxygen: .07-.21
Total pressure 1 bar
Table 6.9. Reaction conditions for the reoxidation of re
duced bismuth molybdate with oxygen.
93

The reoxidation of reduced bismuth molybdate is an extremely fast reaction in comparison with the reduction
reaction. At 673 K and a = .073 the initial rate of reoxi-1 -1 dation with air (x0 = .21) is 165 mmol 0 kg s , where-
2 as the maximum initial rate of reduction is only about 4 mmol 0 kg-1 s-1 •
The initial rate of reoxidation is first order with res
pect to oxygen as can be seen in figure 6.34. The rate de-
_ dO ln r
T (K) r-- (ff
t -1 " 703 • 673
·2 01 .081
·3
.q
-5
·5 .q ·3 -2 -1
-ln x 0
Figure 6.34 Initial rate of reoxidation (mmol kg-1 s-1} of reduced bismuth molybdate (a. = .081) as a function of the mole fraction of gas phase oxygen. Thermobalance.
pends on the degree of reduction. We conclude that at
these temperatures oxygen diffusion limitation is absent.
The calculated energy of activation is 64 kJ mol-1 • This value agrees with the value of 70 kJ mol-l given by Mat
suura and Schuit (1).
6.3.6. The reduction of the catalyst with mixtures of pro
pene, nitrogen and small quantities of oxx~en
The experiments were carried out at 673 K and 703 K
with gas mixtures having a constant mole fraction of pro
pene (xp = .08) and mole fractions of oxygen between .006
and .14.
94
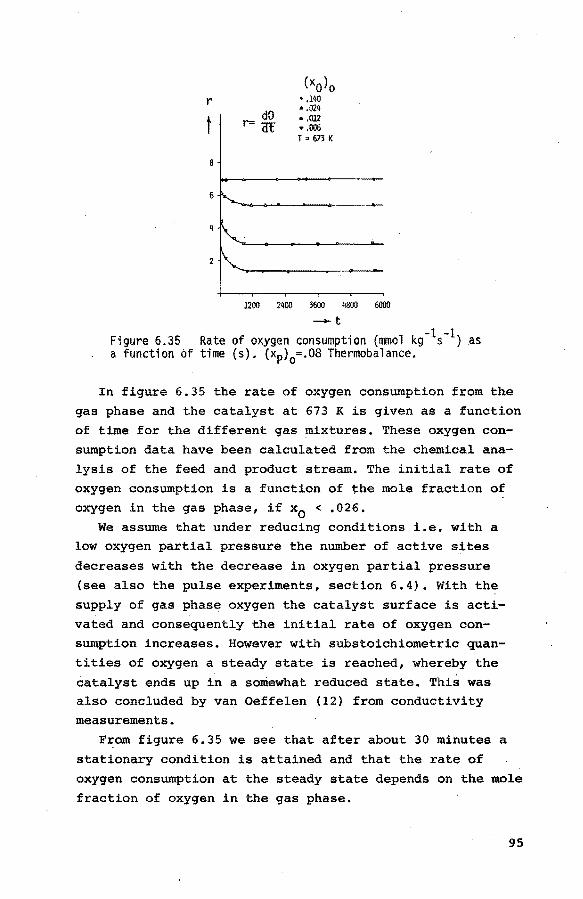
r
t
6
4
2
(xo)o ~ ,140
dO •• 024 •• 012
r=Of v,006 T~673K
1200 2400 3600 lj8IJO 6000
-t Figure 6.35 Rate of oxygen consumption (mmol kg -ls -l) .as a function of time (s). (xp}
0=.08 Thermobalance.
In figure 6.35 the rate of oxygen consumption from the gas phase and the catalyst at 673 K is given as a function
of time for the different gas mixtures. These oxygen consumption data have been calculated from the chemical ana
lysis of the feed and product stream. The initial rate of oxygen consumption is a function of the mole fraction of
oxygen in the gas phase, if x0 < .026.
We assume that under reducing conditions i.e. with a low oxygen partial pressure the number of active sites
decreases with the decrease in oxygen partial pressure (see also the pulse experiments, section 6.4}. With the
supply of gas phase oxygen the catalyst surface is activated and consequently the initial rate of oxygen consumption increases. However with substoichiometric quan
tities of oxygen a steady state is reached, whereby the
catalyst ends up in a somewhat reduced state. This was also concluded by van Oeffelen (12) from conductivity measurements.
From figure 6.35 we see that after about 30 minutes a
stationary condition is attained and that the rate of
oxygen consumption at the steady state depends on the mole fraction of oxygen in the gas phase.
95
After a reaction period of about 90 minutes the thermo
balance was flushed with nitrogen for 18 hours at 673 K.
Mass spectrometric analysis showed that acrolein is the
only desorbing product. This means that during the flush
ing period no further reduction of the catalyst takes
place. Subsequently the catalyst was reoxidized to the origi
nal weight with artificial air, containing 5 mole % o2 and
95 mole % He. As we did not detect carbon dioxide during
the reoxidation we conclude that after the flushing ex
periment no carbon deposit or carbon containing products
were present at the surface.
The experimental data are summarized in table 6.10.
weight increase weight decrease weight increase mole {mg/g} after 1~ (mg/g) after 18 {mg/g) after frac- hour reaction hour flushing reoxidation tion
02 673 K 703 K 673 K 703 K 673 K 703 K
* * * .006 2.47 2.93 3.24 5.50 .77 2.56 .012$ 1. 74 1. 31$ 2.29 3.27 • 55 1.96
.024 .77 .33 1.02 .80 .25 .47
.140 0 0 0 0 0 0
* A maximum weight decrease of .25 mg/g at 673 K and of 2.56 mg/g at 703 K was noticed after 5 minutes.
$ A maximum weight decrease of .28 mg/g at 703 K was noticed after 5 minutes.
Table 6.10. Changes of catalyst weight (mg/g) during pro
pene oxidation, flushing and reoxidation at
673 K and 703 K and at xc H = .oa. 3 6
From the legend of table 6.10 it will be clear that after
an initial weight decrease during the first 5 minutes of
the experiment the weight increases and after a time a
96
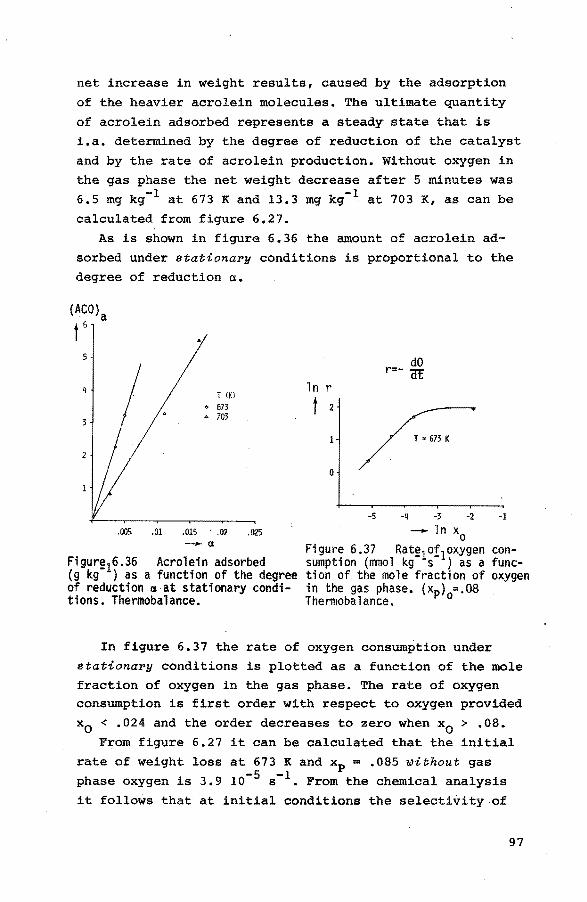
net increase in weight results, caused by the adsorption
of the heavier acrolein molecules. The ultimate quantity
of acrolein adsorbed represents a steady state that is
i.a. determined by the degree of reduction of the catalyst
and by the rate of acrolein production. Without oxygen in
the gas phase the net weight decrease after 5 minutes was 6.5 mg kg-l at 673 K and 13.3 mg kg-1 at 703 K, as can be
calculated from figure 6.27.
As is shown in figure 6.36 the amount of acrolein ad
sorbed under stationary conditions is proportional to the
degree of reduction a.
(ACO} .· a t 6
3
2
T (Kl . 673 . 703
.005 .01 .015 .• 02 .025 -a
Figure 6.36 Acrolein adsorbed (g kg-1) as a function of the degree of reduction a at stationary conditions. Thermobalance.
dO r=- at
ln r
t 2
T = 673K
0
-5 -4 -3 -2 -1
- ln x0 Figure 6.37 Rat!1of1oxygen consumption (mmol kg s ) as a function of the mole fraction of oxygen in the gas phase. (xp)
0=.08
Thermobalance.
In figure 6.37 the rate of oxygen consumption under
stationary conditions is plotted as a function of the mole fraction of oxygen in the gas phase. The rate of oxygen consumption is first order with respect to oxygen provided
x0 < .024 and the order decreases to zero when x0 > .08.
From figure 6.27 it can be calculated that the initial
rate of weight loss at 673 K and xp = .085 without gas phase oxygen is 3.9 10-5 s-1 • From the chemical analysis
it follows that at initial conditions the selectivity of
97

propene towards acrolein is .96 and that the other product
is carbon dioxide (see also the flow experiments, section
6.2.3.1, table 6.1 and the pulse experiments, section
6,4.3, figure 6,40).
The initial rate of acrolein formation was calculated
from these data, according to the stoichiometric equations.
(r 6. 1)
(r 6.2)
and a value of 6.3 10-5 s-1 was obtained. From the flow
experiments with gas phase oxygen and the same propene
mole fraction the initial rates of acrolein formations
were calculated (see figux-e 6.35 and table 6.11).
xo (\~co) 0
s -1 10 5
.140 16.9
.024 15.2
• 012 10.7
.006 7.0
Table 6.11. Initial rates of acrolein formation as a
function of x0 at 673 K and xp = .08.
One can conclude from these figures that the initial rate
of acrolein formation in the absence of gas phase oxygen
is smaller than in its presence. From a calculation of the number of acrolein molecules
adsorbed per m2 after the experiment at 703 K with x0 =
,006, i.e. 8.5 1018 m-2 and the area of one acrolein mole
cule (5 10-20 m2 ) it can be concluded that the reduced
catalyst surface is covered with 40 percent of a monolayer
of acrolein molecules.
98
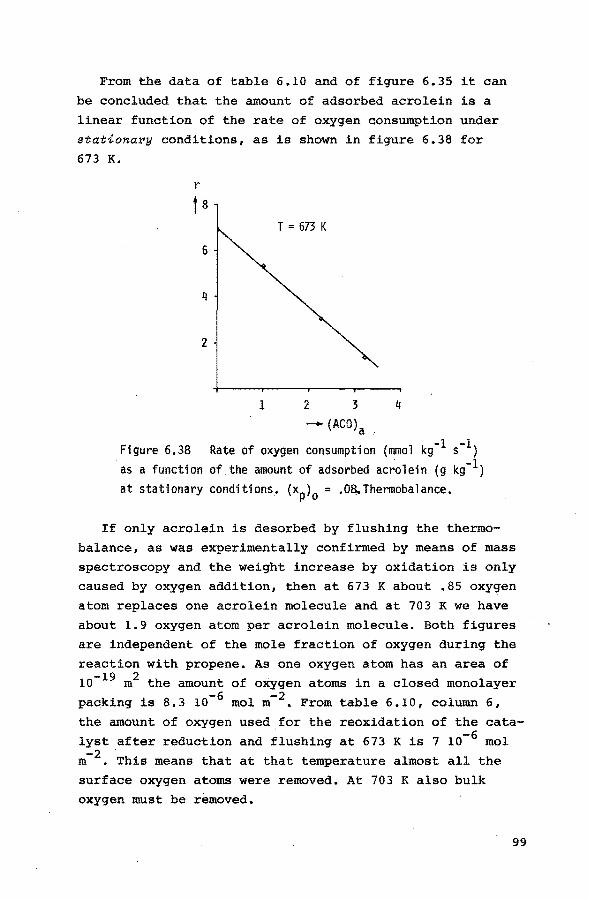
From the data of table 6.10 and of figure 6.35 it can
be concluded that the amount of adsorbed acrolein is a
linear function of the rate of oxygen consumption under stationary conditions, as is shown in figure 6.38 for
673 K.
r
t 8
T '" 673 K
6
4
2
1 2 4
-(AGO) a
Figure 6.38 Rate of oxygen consumption (mmol kg-1 s-1) as a function of the amount of adsorbed ac~olein (g kg-1) at stationary conditions.. (xp}o = .oa, Thermobalance.
If only acrolein is desorbed by flushing the thermobalance, as was experimentally confirmed by means of mass
spectroscopy and the weight increase by oxidation is only caused by oxygen addition, then at 673 K about .85 oxygen
atom replaces one acrolein molecule and at 703 K we have
about 1.9 oxygen atom per acrolein molecule. Both figures are independent of the mole fraction of oxygen during the
reaction with propene. As one oxygen atom has an area of
10-19 m2 the amount of oxygen atoms in a closed monolayer packing is 8.3 10-6 mol m- 2• From table 6.10, column 6,
the amount of oxygen used for the reoxidation of the catalyst after reduction and flushing at 673 K is 7 10-6 mol
m-2 • This means that at that temperature almost all the
surface oxygen atoms were removed. At 703 K also bulk oxygen must be removed.
99

From this experiment we conclude that the catalyst is
in a partly reduced state during the catalytic oxidation
of propene with substoichiometric quantities of oxygen,
even at a low propene conversion level.
6.4. Pulse experiments
6.4.1. Introduction
As we have seen the interpretation of the thermobalance
experiments was hampered by the fact that acrolein is ad
sorbed on the reduced catalyst. However if we accounted
for this adsorption the experimental observations showed
themseives to be useful for the development of a reaction
model for the oxidation of propene. We decided to check
the conclusions by an investigation in a pulse reactor.
With this type of reactor we expected to obtain additional
information about the oxidation and ammoxidation reaction
under initial conditions.
However a pulse reactor is not a very appropriate de
vice for a kinetic investigation for the following reasons:
1. For kinetic work the concentrations and the reaction
time must be known. But even with an ideal a-function
form of the input pulse the length of the pulse in the
reactor and the concentration distribution within the
pulse are rather difficult to assess;
2. Even when the information required sub 1 is available,
it is possible that the adsorption of the reactants is
not the rate determining step. If a fast adsorption
step is followed by a reaction step over a period which
can be longer than the residence time, the calculated
apparent reaction rate based on the contact time of the
pulse will be too high.
Therefore we checked the value of the residence time as
calculated from the inert gas flow by an experimental de
termination of the pulse shape.
100
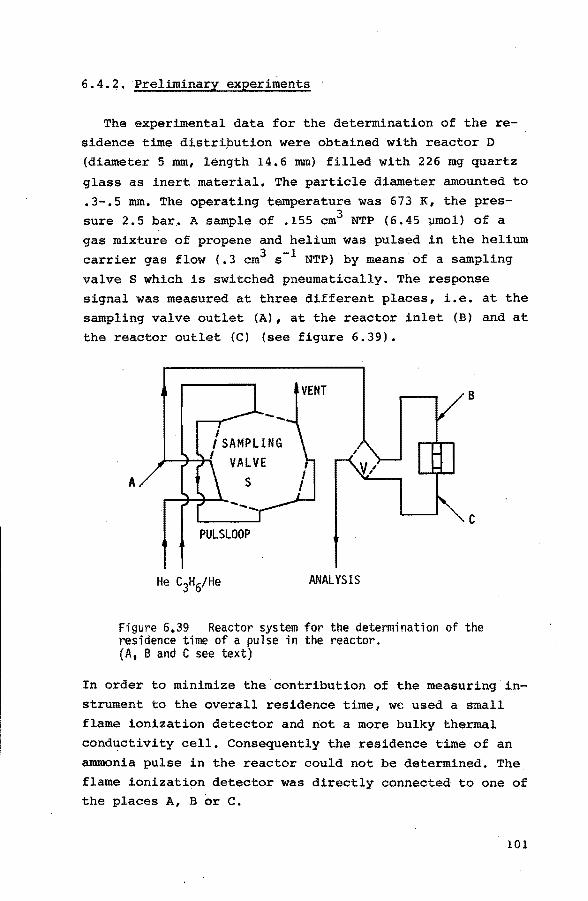
6.4.2. Preliminary experiments
The experimental data for the determination of the re
sidence time distripution were obtained with reactor D (diameter 5 mm, length 14.6 mm) filled with 226 mg quartz
glass as inert material. The particle diameter amounted to
.3-.5 mm. The operating temperature was 673 K, the pres
sure 2.5 bar. A sample of .155 cm3 NTP (6.45 ].!mol) of a gas mixture of propene and helium was pulsed in the helium
carrier gas flow (.3 cm3 s-1 NTP) by means of a sampling
valve S which is switched pneumatically. The response
signal was measured at three different places, i.e. at the sampling valve outlet (A), at the reactor inlet (B) and at
the reactor outlet {C) (see figure 6.39).
B
A
c PULSLOOP
ANALYSIS
Figure 6.39 Reactor system for the determination of the residence time of a pulse in the reactor. (A, B and C see text)
In order to minimize the contribution of the measuring instrument to the overall residence time, we used a small
flame ionization detector and n:ot a more bulky thermal
conductivity cell. Consequently the residence time of an
ammonia pulse in the reactor could not be determined. The
flame ionization detector was directly connected to one of the places A, B or C.
101

The increase of the pulse width is mainly caused hy the
axial diffusion in the line between the valve and the reac
tor and to a much smaller extent by the reactor itself. In particular valve V (see figure 6.39) and its connection to
the lines is responsible for a large increase of the re
sidence time.
As can be seen in table 6.12 the mean residence time
calculated at half height is about 6.6 s and independent
of the mole fraction of propene at the inlet of the flow
system, Consequently the gas mixtures will be more diluted
with helium at the reactor inlet.
p C3H6
inlet system (bar) .105 .298 .502
p C3H6
inlet reactor (bar) .025 .059 .086
crB 2 (s2) 5.04 7~63 8.63
ac 2 (s2) 7.04 8.49 8.01
tres (s) 6.2 6.7 6.8
Table 6,12. Data from the determination of the residence
time of a pulse in the reactor at 673 K.
Total pressure 2.5 bar abs.
The adsorption of reactants and products during a pulse
could be determined by means of the separate analysis of
the pulse and of the adsorbed products originating from
that pulse. To do that the reactor was closed by valve V
(see figure 6.39) after passage of the first pulse into
the gas chromatograph. The adsorbed products were given
time to desorb into the reactor gas phase and thereafter
flushed into the gas chromatograph. In this way we obtai
ned good carbon and nitrogen mass balances as can be seen from table 6.13.
From this table it follows that about 60% of the ammonia
remains adsorbed on the catalyst, As the product strip
102

c N
mmol kg -1 mmol kg -1
feed pulse 128.7 21.6
product pulse 15.6 5.4
unconverted pulse 110.0 3.5
pr<:>dU:ct strip 1.5 13.1 --- ---127.1 22.0
balance % 98.7 101.6
Table 6.13. Carbon and nitrogen mass balances for the re
action of a pulse containing 67 mole % c3a6 and 33 mole % NH3•
contains mainly N2 as a nitrogen containing product we
conclude that ammonia is easily oxidized by oxygen from
the catalyst. The quantity of ammonia adsorbed is equiva
lent to the number of surface Bi-sites if we use Matsu~ra's
data. This experiment further shows that of the first pul
se very little carbon containing material is adsorbed. We
decided not to reoxidiz~ the catalyst sample after each
series of pulses but to take a new quantity of catalyst prepared according to method A, for every new series.
6.4.3. Experiments with propene-helium mixtures
The experiments were carried out at 673 K in reactor D,
with 100 mg catalyst. As is shown in figure 6.40 for pure
propene in the sampling valve, the propene conversion de
creases from 8.3% at the first pulse to 5.0% at the tenth
puls~ with a sharp decrease from the first to the second
pulse. As from the second pulse onward the decay in conversion per pulse is small and constant, this decay is
ascribed to the oxygen loss of the catalyst.
103
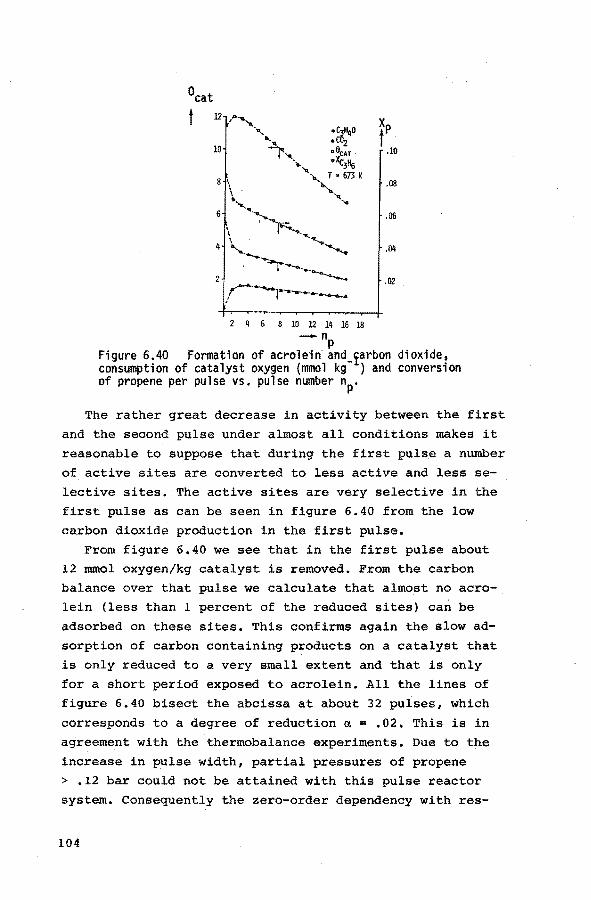
0cat
t 12 ,.....,_ '" oC~qO ...... ..c
10 r\.__ oOCAT. .. •XC3"o
8 \ "'·._ T • 673 K
~.. ... " .....
6
4
2
.. .... _ ..............
.. '~ .... ~-.. ~ .... ....__ r-··1 / ~
2 4 6 8 10 12 14 16 18 -n p
Xp t
.10
.08
.06
.04
.02
Figure 6.40 Formation of acrolein· and_yarbon dioxide! consumption of catalyst oxygen (mmol kg } and convers1on of propene per pulse vs. pulse number np.
The rather great decrease in activity between the first
and the second pulse under almost all conditions makes it
reasonable to suppose that during the first pulse a number
of active sites are converted to less active and less se
lective sites. The active sites are very selective in the
first pulse as can be seen in figure 6.40 from the low
carbon dioxide production in the first pulse.
From figure 6.40 we see that in the first pulse about
12 mmol oxygen/kg catalyst is removed. From the carbon
balance over that pulse we calculate that almost no acrolein (less than 1 percent of the reduced sites) can be
adsorbed on these sites. This confirms again the slow ad
sorption of carbon containing products on a catalyst that
is only reduced to a very small extent and that is only
for a short period exposed to acrolein. All the lines of
figure 6.40 bisect the abcissa at about 32 pulses, which
corresponds to a degree of reduction a = .02. This is in agreement with the thermobalance experiments. Due to the
increase in pulse width, partial pressures of propene
> .12 bar could not be attained with this pulse reactor
system. Consequently the zero-order dependency with res-
104
pect to propene of the rate of
in the thermobalance for Pc H
oxygen consumption as found
> .14 bar could only be 3 6
approached but not. obtained. The experimental data indi-
cate however that at a propene partial pressure of .115
almost full coverage of the active sites is attained, as
can be seen in table 6.14.
c3H6 partial pressures (bar) ocat-consumption 1st pulse
inlet system inlet reactor mmol 0 kg-1
.084 .021 4.0
.150 .035 6.9
.215 .047 7.3
.334 .066 9.5
.505 .087 11.1
1.000 .115 ll.5
Table 6.14 •. Catalyst reduction by propene/helium pulses as a function of the partial pressure of propene
at 673 K.
6.4.4. Experiments with mixtures of propene and. oxxgen
The experiments were carried out in reactor D at 673 K with a feed at the system inlet containing 50-99 mole %
propene and complementary percentages of oxygen. Due to
the increase of the pulse width the propene partial pressures at the reactor inlet were .086 to .114 bar, whereas
the oxygen partial pressures at the reactor inlet were .086 to .003 bar respectively. The total pressure was 2.5
bar. In figure 6.41 the amount of converted propene
(mmol kg-1 ) per pulse is shown as a function of the pulse
number. The lowest curve refers to the experiment without
gas phase oxygen. The conversion of propene per pulse in
creases with the partial pressure of oxygen at the pulse
reactor inlet up to p0 = .075 bar for all pulses. The in-
105

p
t
18
14
12
T = 673 K
(p0 )0
bar •• 000 •• 003 •• 024 • ,045 •• 062 •• 075 •• 086
..,..---.:&:l..-#..---'t.-.... ~ ........................
' 10 ...... _•-.-- ____ ...._ 0 ·--...... - .....
2
2 4 6 8 10 12 14 16 18 20
- n p Figure 6.41 Amount of Pr~pene converted per pulse (mmol kg ) vs. pulse number n
0• Oxidation of prope
ne with and wi~hout (6) gas phase oxygen.
ln ACO t3
-3
lstwthpulse
• • C#qO • • c~
T=673K
-2
- ln (p0)0 Figure 6.42 Rates of formation of acrolein and1carbon dioxide per pulse (mmol kg- ) as a function of the partial pressure of oxygen(bar) at the pulse reactor inlet.
crease of the conve~sion as explained for the thermoba
lance experiments (see section 6.3.6) can be ascribed to
a decrease of the number of reduced sites.
The rate of formation of acrolein is a function of th~ partial pressureof oxygen as is illustrated in figure 6.42
for the first and the tenth pulse at low partial pressures of oxygen the oxygen is completely consumed. Consequently
at low partial pressures of oxygen the rate of acrolein
formation decreases in the last part of the pulse and the
catalyst becomes reduced to a certain extent. This causes
the rate of acrolein formation to decrease from the first
to the tenth pulse. The rate of formation of carbon dioxide is also a function of the partial pressure of oxygen for
all pulses. The increase of the rate from the first to the
tenth pulse at the same partial pressure of oxygen is pre
sumably caused by an increase of the number of unselective
sites by reduction of the catalyst.
For all oxygen concentrations the selectivity of the first pulse was above .95.
106
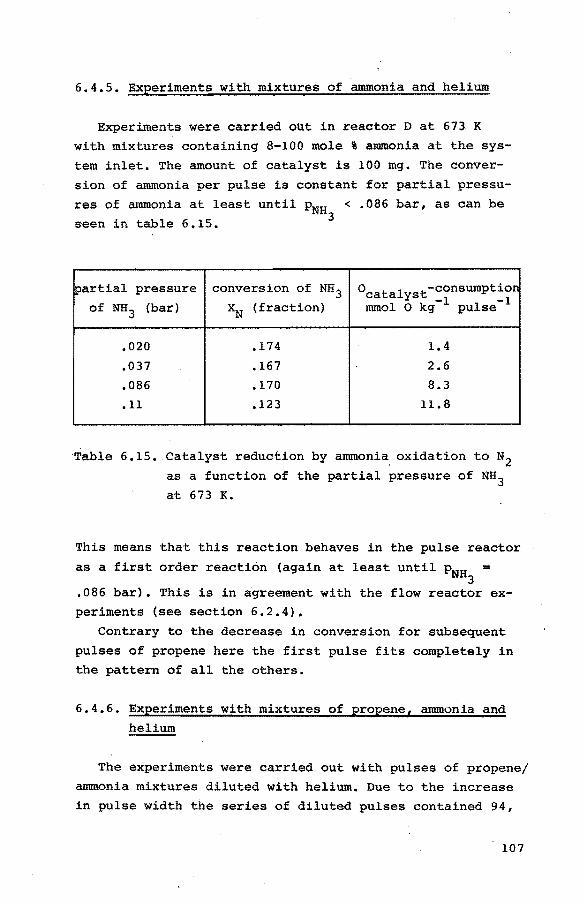
6.4.5. Experiments with mixtures of ammonia and helium
Experiments were carried out in reactor D at 673 K
with mixtures containing 8-100 mole % ammonia at the sys-
tern inlet. The amount of catalyst is 100 mg. The conver
sion of ammonia per pulse is constant for partial pressu
res of ammonia at least until pNH < .086 bar, as can be 3 seen in table 6.15.
partial pressure conversion of NH3 ocatalyst-consumpti~n of NH3 (bar) XN (fraction) mmol 0 kg-1 pulse-
.020 .174 1.4
.037 .167 2.6
.086 .170 8.3
.11 .123 11.8
Table 6.15. Catalyst reduction by ammonia oxidation to N2 as a function of the partial pressure of NH 3 at 673 K.
This means that this reaction behaves in the pulse reactor
as a first order reaction (again at least until pNH = 3
.086 bar). This is in agreement with the flow reactor ex-periments (see section 6.2.4).
Contrary to the decrease in conversion for subsequent
pulses of propene here the first pulse fits completely in
the pattern of all the others.
6.4.6~ Experiments with mixtures of gropene, ammonia and
helium
The experiments were carried out with pulses of propene/
ammonia mixtures diluted with helium. Due to the increase
in pulse width the series of diluted pulses contained 94,
107
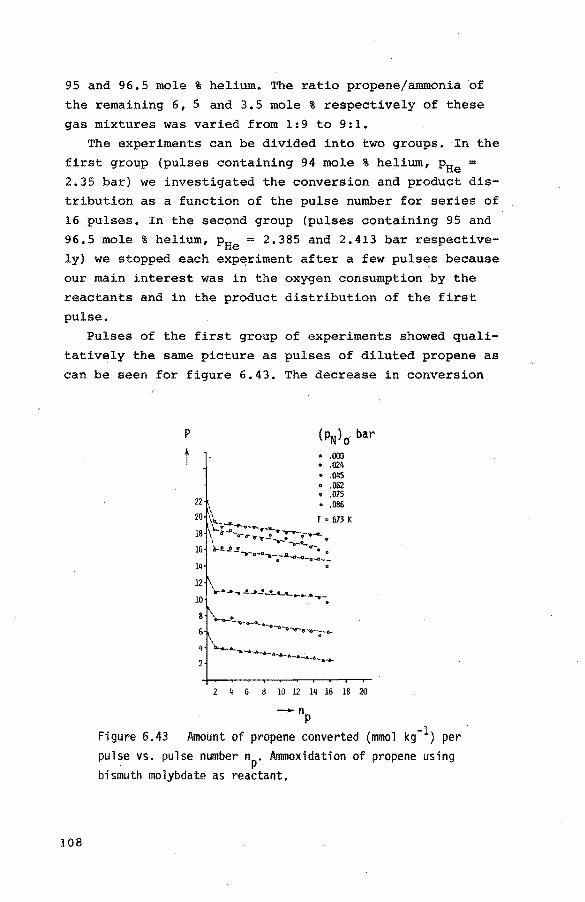
95 and 96.5 mole % helium. The ratio propene/ammonia of
the remaining 6, 5 and 3.5 mole % respectively of these
gas mixtures was varied from 1:9 to 9:1. The experiments can be divided into two groups. In the
first group (pulses containing 94 mole % helium, pHe = 2.35 bar) we investigated the conversion and product dis
tribution as a·function of the pulse number for series of
16 pulses. In the second group (pulses containing 95 and
96.5 mole % helium, pHe = 2.385 and 2.413 bar respectively) we stopped each experiment after a few pulses because
our main interest was in the oxygen consumption by the
reactants and in the product distribution of the first
pulse.
Pulses of the first group of experiments showed quali
tatively the same picture as pulses of diluted propene as
can be seen for figure 6.43. The decrease in conversion
108
p
t
1~
•• 000 •• 02~ • .!IllS •• 062 ... 075
.086
T = 673K
2 ~ 6 8 10 12 1~ 16 18 20
-n p
Figure 6.43 Amount of propene converted (mmol kg-1} per pulse vs. pulse number np. Ammoxidation of propene using bismuth molybdate as reactant.
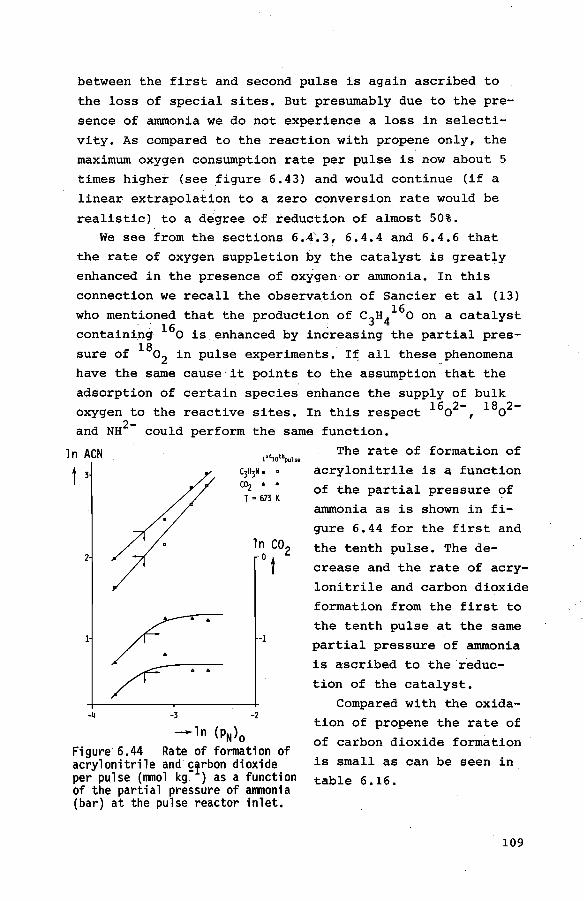
between the first and second pulse is again ascribed to
the loss of special sites. But presumably due to the pre
sence of ammonia we do not experience a loss in selecti
vity. As compared to the reaction with propene only, the
maximum oxygen consumption rate per pulse is now about 5
times higher (see figure 6.43) and would continue (if a
linear extrapolation to a zero conversion rate would be
realistic) to a degree of reduction of almost 50%. We see from the sections 6 .4'. 3, 6. 4. 4 and 6. 4. 6 that
the rate of oxygen suppletion by the catalyst is greatly
enhanced in the presence of oxygen or ammonia. In this
connection we recall the observation of Sancier et al (13)
who mentioned that the production of c3H416o on a catalyst
containing 16o is enhanced by increasing the partial pres
sure of 18o2 in pulse experiments. If all these phenomena
have the same cause it points to the assumption that the
adsorption of certain species enhance the supply of bulk 16 2- 18 2-oxygen to the reactive sites. In this respect 0 , 0
and NH2- could perform the same function.
ln ACN
t 3
2
15tlothpulse
C3H3N • a
c~ • • T = 673 K
-1
~ ~ ~
-ln (pN)o Figure·6.44 Rate of formation of acrylonitrile and·c,rbon dioxide per pulse (mmol kg- ) as a function of the partial pressure of ammonia (bar) at the pulse reactor inlet.
The rate of formation of
acrylonitrile is a function
of the partial pressure of
ammonia as is shown in fi
gure 6.44 for the first and
the tenth pulse. The de
crease and the rate of acry
lonitrile and carbon dioxide
formation from the first to
the tenth pulse at the same
partial pressure of ammonia
is ascribed to the r·educ
tion of the catalyst.
Compared with the oxida
tion of propene the rate of
of carbon dioxide formation
is small as can be seen in
table 6.16.
109

co2 mmol kg -1 pulse -1
Pulse composition
c 3H6-oxidation c 3H6-ammoxidation
pulse number pulse number
X xo ~H3 1 10 1 10 C3H6 2
1.0 - - .2 1.2 - -.9 .1 1.1 2.4 - -.6 .4 6.7 7.6 - -.9 - • 1 - - .07 .07
.6 - .4 - - ,19 .09
Table 6.16. Rate of carbon dioxide formation at 673 K for
the oxidation and ammoxidation of propene.
The product distributions obtained from the first pulse of
the various propene/ammonia/helium gas mixtures fit in
0cat t 16
14
.02 .04 .06 .08 .10 .02 ,04 .06 .08 .
- Pp - Pp Figure 6.45 A~~unt of catalyst Figure 6.46 ~~unt of catalyst oxygen (mmol kg ) used during the oxygen (mmol kg ) used during the first pulse for the formation of first pulse for the formation of the main products as a function of the main products as a function of !he partial pressure of propene(bar)~the partial pressure of propene(bar}. PHe=2.385 bar Pp+PN=.l15 bar PHe=2.413 bar Pp+PN=.087 bar
110

with those obtained from the first propene/helium and
ammonia/helium pulses as is shown in figure 6.45 and figure 6.46 where the rate of oxygen depletion of the cata
lyst for the formation of the main products acrylonitrile,
acrolein, acetonitrile, carbon dioxide and nitrogen is
plotted as a function of the partial pressures of propene and ammonia for two values of the partial pressures of
helium.
References
1. Satterfield, C.N., Sherwood, T.K., "The role of diffu
sion in catalysis", Addison-Wesley Publ. Cy. Inc., Reading (1963)
2. Shelstad, K.A., Chong, T.c., Can. J. Chem. Eng. !I• 598. (1969)
3. Callahan, J.L., Grasselli, R.K., Milberger, E.C.,
Strecker, H.A., Ind. Eng. Chem. Prod. Res. Dev. 2• 134
(1970)
4. Cathala, M., Germain, J.E., Bull. Chim. Soc. Fr. 2167
(1971)
5. Yoshida, F., Ramaswami, D., Hougen, O.A., A. I. Ch. E.
Journal ~. 5 (1962)
6. Matsuura, I., Schuit, G.C.A., J. Catal. 25, 314 (1972)
7. Batist, Ph.A., Kapteijns, c.J., Lippens, B.C., Schuit,
G.C.A., J. Catal. 2• 33 (1967)
8. Steenhof de Jong, J.G., Thesis, Eindhoven University of Technology (1972)
9. van den Elzen, A.F., Rieck, G.D., Acta Crystallogr. Sect. B. ~. 2433 (1973)
10. Beres, J., Bruckman, K., Haber, J., Janas, J., Bull. Acad. Pol. Sci. Ser. Sc. Chim. ~ (8) 813 (1972)
11. Matsuura, I., Schuit, G.C.A., J. Catal. 20, 19 (1971)
12. van Oeffelen, D.A.G., Thesis, Eindhoven University of Technology (1978)
13. Sancier, K.M., Wentreck, P.R., Wise, H., J. Catal. 39,
141 (1975)
111

112

CHAPTER 7
FINAL DISCUSSION
7.1. Introduction
It is the purpose of this chapter to put the experi
mental results described in chapter 6 in a more general
context. We will do that with the aid of two models, a
mechanistic model and a kinetic model. The mechanistic model will be a sketch of the network
of elementary steps that we presume to take place on the
catalyst and that lead from the reactants to the main
products and byproducts. This model will be based on li
terature data and arguments that can be developed from our
experimental work. It will remain sketchy and rather spe
culative in parts.
The kinetic model will have to conform with the mecha
nistic model but will contain the minimum number of the
reaction steps that are required to derive the kinetic
results described in chapter 6. The number of these re-.
action steps depends on the basic reaction concerned i.e.
- the oxidation of propene (P + ACO)
- the ammoxidation of pr.opene (P + ACN)
- the ammoxidation of acrolein (ACO + ACN)
- the oxidation of ammonia (N + N2}
Each sequence of reaction steps of a basic reaction in
cludes a rate determining reaction step, which will be
connected with the experimental kinetics of the proper re
action. The choice of the right rate determining steps
will be an important element in the derivation of the ki
netic models for the four basic reactions.
7.2. The mechanism of the catalrtic reaction
The mechanistic model comprises i.a. the following concepts:
113

a) Adsorption equilibria between the reactants (products)
in the gas phase and at the catalyst surface follow the
Langmuir adsorption theory;
b) Secondly we include in this mechanistic model a Marsvan Krevelen redox mechanism i.e. the catalyst oxidizes
the adsorbed species and the gas phase oxygen subse
quently oxidizes the reduced catalyst;
c) The active sites that are required for the various
elementary steps of the four basic reactions mentioned
in section 7.1 have to perform a number of functions
in order to catalyse the complete reaction sequence,
e.g. from propene, oxygen and ammonia to acrylonitrile
and water. In our mechanistic model we will endeavour to ascribe
elementary steps to the specific interaction of two sites.
To make this discussion more unambiguous we will use a
number of catalytic concepts that we have defined as
follows: - An aative site is a site, consisting of an coordinative
unsaturated atom, on which reactants or intermediates
can be adsorbed during the sequence of reaction steps;
- The expression active centre is used for a group of sur
face atoms that together bind one adsorbant entity;
- A reactive ensemble is a group of active sites involved
in a particular reaction step; - A reactive agg!'egate is the system of reactive ensembles,
which is required for the formation of one product mole
cule.
As the terms "active site" and "active centre" are often
used as synonyms, we prefer the term "reactive ensemble"
to "active centre", although according to the IUPAC ·(1)
both terms can be used to describe the place where a catalytic reaction step takes place.
For the Mars-van Krevelen mechanism the following terminology can be used:
- A redore component is a species or a vacancy required for
an elementary step in the redox mechanism;
- A redore ensemble is a set of redox components that can interact with each other;
114

- A ~edore system is the combination of redox ensembles required for the restoration of the oxidation activity of
the catalyst after one product molecule has been formed. The main questions to be answered for the development
of the reaction mechanisms are:
a) How are propene, ammonia and acrolein activated, which are the reactive ensembles and how are the products
formed? b) What is. the role of the catalyst in the activation of
molecular oxygen and what is the nature of the selective oxygen species?
Using the literature data presented in chapter 2 we focus our attention mainly on these questions.
From the extensive literature it became clear that on bismuth molybdate two different types of reactive ensem
bles are present 1} a reactive ensemble connected with molybdenum;
2) a reactive ensemble connected with bismuth. In the following we will derive arguments from our experi
ments to chose the active sites for propene and ammonia adsorption. The presence of ammonia as the third reactant
in the acrylonitrile synthesis is an important expedient
to achieve this object. In the following sections at first we will deal with the adsorption of ammonia and propene.
7.3. Adsorption and adsorEtion sites
7.3.1. Adsorption of ammonia
Although far more literature data are available for the adsorption of propene than for the adsorption of ammo
nia, we begin our discussion with the latter compound as
the results will be useful for the discussion of the ad
sorption of propene. Matsuura (2) assumed that NH2- species are formed on
an reactive ensemble A and adsorbed on Bi-ions. This .assumption resembles that of Grasselli et al (3) for the
activation of ammonia on the USb 3o10 catalyst during the
115

production of acrylonitrile. According to the latter 2-authors the proton is donated to the 0 ion of Matsuura's
a-ensemble, whereas Gates et al (4) postulated recently that the proton is ultimately donated to the o2- ion of
an A-ensemble. Weiss et al (5) suggested the formation of + an NH2 intermediate adsorbed on a molybdenum ion. In the
case of nitrogen production the formation of hydrazine as
an intermediate
step between an
Based on the
has been assumed according to a reaction + -NH2 and an NH2 intermediate.
kinetic .data of our investigation we
assume that the ammonia molecules are activated on t~o
different ensembtes: - first there is a nitrogen containing species (N
0) that
is required as a precursor for acrylonitrile. As the
rate of acrylonitrile formation is almost zero order in
ammonia we assume that the active centres that bind the
ammonia are fully occupied;
- secondly there is a nitrogen containing species (N1) that reacts with the species N
0 mentioned above to form
molecular nitrogen in a reaction that is first order in
ammonia. This can be understood if the degree of cove
rage of N1 is proportional to the ammonia pressure.
As we further have shown that in fact there is a
slight inhibiting effect of ammonia on the rate of acry
lonitrile formation we further assume that N1 and propene
compete for the same active centres. This then results in
the picture that ammonia is fully covering one type of
active centres, which we call "a" centres and is slightly
adsorbed on the other type of active centres, called the
"b" centres that also adsorb propene.
From the observation that reduction of the catalyst
leads to the formation of Bi0, a formation that can easily
be prevented by the presence of gas phase oxygen, we con
clude that oxygen is adsorbed fast and completely at bis
muth centres. In the pulse experiments we have seen that
the presence of ammonia acts in the same way as the pre
sence of oxygen. From this we would conclude that also
ammonia gives a fast and complete adsorption at bismuth
116

centres. If this conclusion is right our "a" centre will
be connected with bismuth, like Matsuura's A centre, but
then the "b" centre at which propene adsorbs will be com
parable to Matsuura's B centre.
7.3.2. Adsorption of propene
As_we have explained in chapter 2, it is now generally
assumed that the oxidation and the ammoxidation of pro
pene proceed via the formation of a negatively charged allylic intermediate. The first abstracted proton is do
nated to an oxygen ion at the surface, whereas the allylic
intermediate is bonded to a cation by means of a cr~-allyl
bond. The delocalized ~-bond system of an allylic inter
mediate, according to the simple Ruckel molecular orbital
theory, has three orbitals
(non-bonding)
with +i p orbitals on carbon atom i. It is generally
assumed that the plane of the carbon atoms is parallel to
the surface of the catalyst. The bonding orbital is in
the centre of the chain, whereas only in non-bonding or
bitals the electrons are localized at the terminal carbon atoms.
In the literature no agreement about· the adsorption
site for propene has yet been reached. Matsuura et al (6) (7) and Matsuura (2) postulated a model based on adsorp
tion experiments that start with the dissociative adsorp-6+ 2-tion of propene on Mo and 0 ·• Haber et al (8) (9) use
two sets of experiments to support the opinion that the
bismuth ion activates propene. One set of experiments
carried out by Beres et al (10) consists of the reduction
of a, B and y bismuth molybdate with hydrogen, yielding
117

different energies of activation, which reflects the
difference in the enthalpy of chemisorption of hydrogen
on the oxygen ions because of the different coordination
of molybdenum in the three molybdates. With propene no
correlation of the energies of activation with the diffe
rent coordination of molybdenum is obtained. In our
thermobalance experiments we also found a difference in
behaviour of hydrogen and propene in the reduction of our
catalyst. The rate of catalyst reduction with hydrogen
was low and represents a surface reaction,. whereas the
rate of reduction with propene was much higher and is
governed by the oxygen transport in the catalyst and does
not reflect the surface reaction.
Sakamoto et al (11), Swift et al {12), Haber et al (8)
and Boersma (13) have shown that propene can adsorb on
bismuth oxide and on bismuth containing oxides, yielding
i.a. 1,5-hexadiene. As however this reaction hardly takes
place below 773 K and during our experiments at tempera
tures between 648 and 723 K 1.5-hexadiene was never found
we conclude that the adsorption discussed by these authors
does not occur in our experiments. Grzybowska et al (14) did pulse experiments with allyl
iodide, using the oxides of·bismuth and of molybdenum and
bismuth molybdate as oxidizing agents, whereas Gamid-Zade
et al (15) oxidized allyl bromide over these compounds
with gas phase oxygen. The first hydrogen abstraction,
generally supposed to be the rate determining step was
bypassed as allyl radicals were formed rapidly. Molybde
num oxide, an inactive but selective catalyst for the
formation of acrolein from propene showed an increased
activity and especially at temperatures below 600 K a
high selectivity towards acrolein. On bismuth oxide non
oxygenated products were found below 673 K and 1,5-hexa
diene and propene were the main products. On bismuth
molybdate acrolein as well as non-oxygenated products
were obtained. The observation that only molybdenum oxide
produces acrolein shows that at least a part of the reaction takes place on reactive centres with molybdenum. The
118

fact that on bismuth molybdate acrolein and non-oxygenated
products are formed shows, when compared to the normal
propene oxidation that the bismuth related non-oxygenated
products are atypical for the reaction of propene on bis
muth molybdate. It is also possible that gas phase allyl
radicals play a role in the 1,5-hexadiene formation. Ex
perimental evidence for such a mechanism has been given
by Grasselli (16) who studied the reaction of azopropene
with bismuth molybdate at 600 K.
A model comparable with that of Matsuura has been pro-2-posed by Linn and Sleight (17). They used Moo4 groups
associated with a Bi-cation vacancy as active centres fo~
the propene adsorption. This leads to the formation of an
allylic intermediate on the Moo42- group and the donation
2-of a proton to the oxygen of a neighbouring Moo4 group.
Recently Matsuura (see (4) chapter 4) assumed that
Bi2Mo2o9 is the active phase and that the allyl formation
occurs on (Moo4 >4 clusters, according to the model pro
posed by Sleight (18) which is based on a scheelite struc
ture.
All models account for a dissociative adsorption, which
would lead to a half order dependency with respect to
propene if the two different active sites that form the
reactive ensemble are present in equal amounts and if the
adsorption is the rate determining step. However we have
found that the rates of acrolein and of acrylonitrile
formation are both first order with respect to propene for
the lower propene concentrations. This can only be achie
ved if either a) the number of o 2- ions for the adsorption
of the proton is much larger than the number of cations
for the adsorption of the allylic intermediate, or b) the
number of o 2- ions is constant as a result of a rapid
regeneration at the temperatures of this investigation.
A high ratio of o 2- ions to cations would in Matsuura's
low temperature adsorption studies result in single site
adsorption kinetics. Actually he finds that both propene
and ammonia adsorb according to a dissociative kinetic
model.
119

According to Peacock et al {19) the allyl intermediate
is bonded to the Mo6+ ion. The bismuth ion is assumed to
keep the molybdenum ion in the highest oxidation state,
either by accepting an electron directly from the allyl
intermediate or via the Mo5+ ion.
Haber et al {20) showed by means of semi empirical
quantum-chemical calculations that the TI allyl interme
diate is bonded to Mo6+ and that the TI allyl electrons
shift to the metal ion. These calculations are supported by the X-PS and U-PS measurements of Grzybowska et al {21)
that show the formation of Mo4+ and only in the later sta
ges the reduction of Bi3+.
It will be clear that from the discussion of the ammo
nia adsorption we also favour a propene adsorption con
nected with molybdenum cations.
7.3.3. Adsorption of acrolein
The activation of acrolein has been studied by means
of adsorption experiments carried out by Matsuura {2) and
has been discussed by Batist et al (22). The experiments
showed that acrolein is adsorbed on bismuth molybdate via two
or three different processes, viz. a fast, weak adsorption
similar to the butadiene adsorption and connected with the
B-ensembles and a slow and strong adsorption comparable to
the strong adsorption of butadiene and connected with the A-ensembles. Moreover there is a marked but not well under
stood adsorption on reduced bismuth molybdate. We assume
that the fast and weak adsorption of acrolein is the, for the propene conversion, slightly inhibiting adsorption
on molybdenum.
The slow and strong adsorption of acrolein, which is
non-inhibiting and reversible is assumed to occur on bismuth.
The third type of adsorption on a reduced catalyst may
be connected with the coverage of a large part of the catalyst or with the formation of acrolein polymers.
These adsorptions have been noticed in our experiments
120

in the flow reactors (inhibition, section 6.2.3.1) and in
the thermobalance (experiments with substoichiometric
amounts of oxygen, section 6.3.6).
7.3.4. The role of the catalyst in the activities of
molecular oxygen
Much attention has been paid in literature to the under
standing of the nature of the oxygen species that inter
acts with the reactants. According to the redox mechanism
proposed by Mars and van Krevelen {23) lattice oxygen of
active catalysts can be involved in the oxidation of
hydrocarbons. Aykan (24) found for the ammoxidation of
propene in the absence of oxygen on bismuth molybdate in
a fluid bed reactor at 723 K initially the same activity
and selectivity as in the presence of molecular oxygen.
Due to the increasing reduction of the bismuth molybdate
catalyst the activity of the oxidant decreases.
In our pulse experiments we also found a decreased ac
tivity with. increasing degree of reduction of the catalyst.
However we also found a higher initial activity in the
presence of gas phase oxygen than in the absence thereof.
The latter has been ascribed to the special conditions in
our pulse reactor.
As far as the structure of the catalyst surface is con
cerned we assume, in accordance with the work of Grzybow
ska et al (21) that during the reaction in the presence of
oxygen the surface Bi/0 and Mo/0 ratios are equal to
those of fresh samples and that only during reduction
changes in"the surface composition can take place.
From experiments carried out with labeled oxygen by
Keulks (25) and Wragg et al (26) in a recirculation reac
tor1 by Sancier et al (27) in a pulse reactor; by Pendleton
et al (28) in a batch reactor and by Keulks et al (29) in
an integral flow reactor it can be concluded that at tem
peratures above 648 K lattice oxygen is the only direct
source of oxygen for the formation of acrolein and acrylonitrile.
121

The hydrogenation steps and the ultimate selective oxi
dation use oxygen ions to accomodate protons. After that
the products are desorbed and the active surface is main
tained in a high degree of oxidation by diffusion of
oxygen ions through the lattice. This can only be realized
if the electrons produced can be transported simultaneous
ly in the opposite direction via positive ions.
In order to explain how the gas phase oxygen reaches
the ensembles active in the propene oxidation we will
assume that a reaction sequence ends after the desorption 4+ of the product with the presence of Mo ions on the sur-
face. These can be reoxidized to Mo6+, according to the
overall redox reaction:
3 Mo4+ + 2 Bi3+ (r 7.1)
as was earlier proposed by van Oeffelen (30)". This redox
reaction is also in agreement with the work of Grzybowska
et al (21) who have shown that the reduction of Bi 3+ only 4+ takes place after the formation of Mo •
Although no data for the different bismuth molybdate
phases are available, some indications may be found from
the thermodynamic data for Moo2 , Bi2o 3 , Moo3 and Bi (31,
32, 33). The necessary cp-values were calculated as a
function of temperature. The results are summarized in table 7 .1.
r.b02 Bi2o3 r.b03 Bi
-587.9
-578.0
-745.2
-
50.0
151.5
77.8 56.7
-533.1 -584.2
-496.0 -533.1
-668.1 -739.8
- 16.9 21.8
103.2
247.9
147.0
100.0
-465.6
-393.4
-573.4
- 45.5
Table 7.1. Thermodynamic values at 298.15 and 673 K for
the redox components of the bismuth molybdate catalyst.
122

We calculated the standard Gibbs free energy 6G
of the redox reaction
+ + 3 Mo03 + 2 Bi
r,673,Bi2Q3
(r 7.2)
to be -21.0 kJ mol-1 • The melting point of bismuth is 544.5 -1
K with 6HM = 10.9 kJ mol • ~he th~ee other redox compo-
nents are solids in the temperature range of 298 to 673 K.
This means that at 673 K in the absence of molecular oxygen
bismuth atoms will form and can agglomerate to liquid
droplets and thus leave the solid lattice. Actually we
have observed under reducing conditions at 673 K a con
densation of bismuth metal in those parts of the reactor
and outlet lines that were below 500 K (34). The equili
brium reaction (r 7.1) is shifted to the right hand side
and the catalyst will loose its activity. This can be
counteracted by the presence of sufficient gas phase oxy
gen, because then a second redox ensemble can become ac
tive:
+ + (r 7.3)
For this second redox ensemble surface vacancies Dr at the
reoxidation site of the catalyst are required. These
vacancies and the Bi0 atoms result from the diffusion of
o2- and electrons respectively through the lattice, the
o 2- ions going from the reoxidation sites to the reaction
sites and the electrons in the reverse direction.Miura et
al (35) have found from temperature programmed reoxidation
experiments indications that the reoxidation at bismuth
sites is preferred, i.e. it takes place at a temperature
that is 150 K below the temperature at which Moo2 is re
oxidized.
Much has been reported on the types of oxygen active in
propene oxidation and ammoxidation. Haber et al (9) rela
ted the increase in the activation energy from 81 to 136
kJ mol-l for the reduction of the series ,_,f bismuth molyb
dates with Bi/Mo ratios of 2, 1 and 2/3 respectively with
123

hydrogen, with the increase in tetrahedral surrounding of
molybdenum in these compounds. The fact that for propene
reduction the energies of activation were all the same
lead him to conclude that now oxygen from bismuth was ac
tive. However, the rate of reduction with propene was
about 50 times higher than with hydrogen and consequently
a completely different mechanism may be rate determining.
Moreover it must be mentioned that the energies of acti
vation for the reduction of Bi2o3 , Bi2Mo~~ and Mo03 were all found to be between 81 and 90 kJ mol · • Otsubo et al
(36) and Miura et al (37) prepared bismuth molybdates
(Bi/Mo = 2/1) by solid state reactions between bismuth
oxide and molybdenum oxide under vacuum at 823 K for 20
hour. The oxides were labeled by reduction and subsequent
reoxidation with 18o2 • The catalysts were reduced with
hydrogen and with propene. The 18o content of the water obtained from hydrogen reduction and of the· acrolein from
propene reduction was only initially somewhat higher for
the catalyst made with Bi218o3 • From these data the
authors conclude that the oxygen near bismuth is first
used in both reactions. This is not in agreement with the
work of Haber mentioned earlier, nor with the observation
of Grzybowska et al (21) who showed that the reduction of
bismuth is preceded by the reduction of molybdenum. In this connection it must be born in mind that the prolonged
vacuum treatment at high temperature may have lead to a
loss of slightly volatile molybdenum oxide and to a re
duction of the catalyst. The work of Bleijenberg et al
(38) and of Batist et al (39) has shown that the solid state reaction of the oxides leads to rather inactive
catalysts and that a complete reaction between the oxides
is difficult to achieve.
The information discussed above leads us to believe
that there is a st.rong interaction between oxygen ions, which possibly makes the assignment of certain reactive
oxygen ions as belonging exclusively to either bismuth or to molybdenum problematic.
124

7.4. The formation of the main products
7.4.1. The formation of acrolein
In section 7.3.2 we concluded that the dissociative
adsorption of Mo6+o 2-. The
propene occurs on the reactive ensemble
Mo6+ ions has no d electrons and if the ne-s gative allylcarbanion is placed at the empty site elec-
trans can be shared by cation and ligand, involving a
charge displacement to the cation. Preferentially the two
electrons in the non-bonding allyl orbital, concentrated
on the terminal carbon atoms are transferred, which re
sults in a partial positive charge on these atoms.
Additional information about the role of Mo6+ in the
oxidation of propene has been obtained by means of the
ESR technique applied by Peacock {19), Sancier et al {40)
and Burlamacchi et al {41). These experiments provide
evidence for the postulate that Mo6+ and o2- associated
with Mo6+ are essential for the propene oxidation and that
Mo5+ is an intermediate in the electron transfer process.
As the electron transfer process is not yet completely
understood we assume that two electrons are transferred to
one Mo6+ ion. This was experimentally confirmed by means
of X-PS measurements carried out by Grzybowska et al {21).
As has been suggested by Sachtler {42) the symmetric allyl intermediate can be of a transient nature only. By
means of the partial positive charges on the terminal car
bon atoms a carbon-oxygen bond can be formed, resulting
in an asymmetric a-allyl compound. Since Sachtler et al
{43) found the terminal carbon atoms undistinguishable, a
socalled dynamic a-allyl intermediate has been postulated
and experimentally proven.by Kondo et al {44). This was
achieved by means of H-D exchange in the olefin when o2o was added to the reaction mixture. The allyl is then a
fluxional ligand i.e. it can move from one oxygen to an
other presumably via a n-allyl intermediate on a cation.
According to Matsuura {7) the allyl intermediate moves
from this reactive ensemble B to the reactive ensemble A
125

("B-site" and "A-site" respectively in Matsuura's nomen
clature) and this step is supposed to be the rate deter
mining step. The abstraction of the second proton occurs
with the c3H4-intermediate staying on the reactive en-2-semble A whereas the proton is donated to an 0 of a
reactive ensemble B.
According to Haber et al {9) the second proton abstraction takes place on Mo6+ and the proton is again donated
2-to an neighbouring 0 ion. Matsuura's model is composed
of more elementary steps than Haber's model and moreover
the intermediates shift in the opposite direction, where
as the protons in Matsuura's case are ultimately donated
to his OA and in Haber's case to the oxygen near molyb
denum (Matsuura's OB). In a third model put forward by Otsubo et al (36) and
Miura et al (37) the second hydrogen abstraction occurs
also near bismuth, whereas the oxygen near molybdenum is
supplied to the anion vacancy near bismuth after the for
mation of the products. In this model the allyl interme
diate is not shifted.
Although Peacock et al (19, 45) do.not give a detailed
model one can conclude from their discussion that they
adhere to a view opposite to that of Otsubo and Miura i.e.
all elementary steps of the acrolein formation take place
on the reactive ensemble near molybdenum and the oxygen
near bismuth is supplied to the anion vacancy near molyb
denum. Linn and Sleight (17) donate also the second proton to
the oxygen of a neighbouring Moo42- group, whereas the
2- 2-C3H4 remains on the other Moo4 group. The formation of acrolein takes place by means of the donation of oxygen
2-from the Moo4 group. The electrons are donated to an
overlapping system of an empty Bi 6p conduction band and
to Mo 4d levels and are subsequently donated to an in
coming oxygen molecule near bismuth.
As we show in table 7.2 in two of the five models the
allyl intermediate is shifted to the reactive ensemble
containing the other cation, whereas in the other three
126
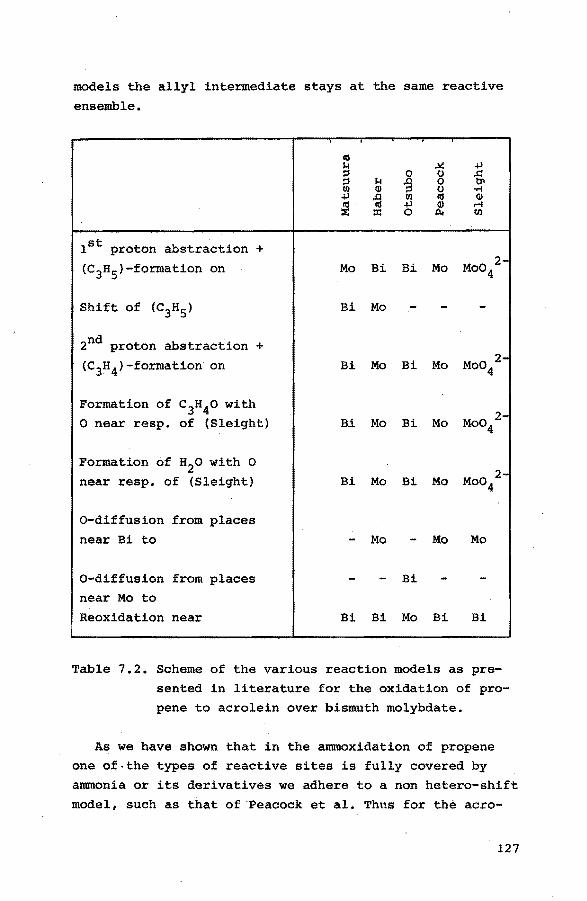
models the allyl intermediate stays at the same reactive
ensemble.
liS 1-1 .II: +l ~ 0 tl ..c: ~ 1-1 .Q 0 tF> !I) <II ~ tl ..... +l .Q !I) liS <ll
~ Ill +l Q) .-1 ::= 0 llo til
1st proton abstraction + Bi Bi 2-(C3H5)-formation on Mo Mo Moo4
Shift of (C3HS) Bi Mo - - -2nd proton abstraction + (C3H4)-formation Bi Mo Bi Mo 2-on Moo4
Formation of c3H40 with 2-0 near resp. of (Sleight) Bi Mo Bi Mo Moo4
Formation of H2o with 0 2-near resp. of (Sleight) Bi Mo Bi Mo Moo4
a-diffusion from places
near Bi to - Mo - Mo Mo
a-diffusion from places - - Bi - -near Mo to
Reoxidation near Bi Bi Mo Bi Bi
Table 7.2. Scheme of the various reaction models as presented in literature for the oxidation of propene to acrolein over bismuth molybdate.
As we have shown that in the ammoxidation of propene one of.the types of reactive sites is fully covered by
ammonia or its derivatives we adhere to a non he.tero-shift model, such as that of Peacock et al. Thus for the aero-
127

lein formation we assume the following sequence of stoi-
chiometric equations, all connected with moJybdenum
C3H6 + Mo6+ + OS 2-
+ 6+ -Mo (C3H5) + (OSH) (r 7.4)
6+ Mo (C3H5 ) + Mo4+cc3
H5
)+ (r 7.5)
Mo4+(C3H
5)+ 2- + Mo4+ + (OsC3H5) (r 7.6) + OS +-
4+ + Mo {C3H5 ) + 2-OS +
4+ Mo (C3H4} + (OsH) (r 7. 7)
The equations (r 7.4 tor 7.7) have been discussed above. 4+ In order to proceed from the.Mo (c3H4) species a number
of sequences is possible: e.g. either the Mo4+ of the
surface complex is oxidized by Bi3+ or the c 3H4 species
transfers to a Mo6+ site. In both cases electron transfer
can follow according
+ (r 7. 8)
This cc3H4) 2+ species then reacts with anos2- ion to form
acrolein that subsequently desorbs. The adsorbed acrolein
on molybdenum accounts for the inhibiting effect of acro
lein on the oxidation of propene.
There is also the possibility that we have a concerted
reaction,such as
+
(r 7. 9)
with subsequent desorption of acrolein and the same in
hibiting effect.
In all sequences we will end with a Mo4+ species that
will be reoxidized by means of the redox reaction (r 7.1).
7.4.2. The formation of nitro9en
We have discussed in section 7.3.1 that the nitrogen
128

containing species N0
and N1 are formed on the active
centres connected with bismuth and molybdenum respectively.
We suppose these species to be formed from ~mmonia by means of two separate dehydrogenation sequences.
The sequence on the active centre containing molybdenum
is comparable with the dehydrogenation of propene as given by the equations (r 7.4) to (r 7.8) for propene and ends
4+ 2+ with the species Mo (NH) • The sequence on the active centre containing bismuth
ends with the formation of the species Bi3+(NH) 2-. This
assumption is in agreement with that of Matsuura (2). The
abstraction of two protons is also mentioned by Gates et ,
al (4). Matsuura (2) assumed that the nitrogen formation occurs on an A-ensemble from two similar species Bi3+(NH) 2-.
We however postulate a reaction between the two intermediates formed on the different reactive ensembles viz. Bi3+(NH) 2- and Mo4+(NH) 2+. The formation of two different
species on different active sites is supported by our kinetic experiments. After the formation of the intermediate HN = NH proton abstraction and electron transfer leads to the formation of nitrogen. As was experimentally
found (see section 6.2.5.2) nitrogen formation is notably suppressed during the ammoxidation of acrolein. We a~cribe
4+ 2+ this phenomenon to the rapid formation of Mo (C3H4) and
Mo4+cc3H4NH) from acrolein, which results in a decrease
of the formation of the Mo4+(NH) 2+ intermediate.
7.4.3. The formation of acrylonitrile
Analogous to the.formation of acrolein Matsuura (2)
assumed that the greater part of the sequence that leads
to the formation of acrylonitrile from propene occurs o~ his A ensemble, whereas Gates et al (4) assume that the carbon-nitrogen bond is formed on this A ensemble before
the last two protons are abstracted. This also has been suggested by Cathala et al (46).
We also assume that acrylonitrile is formed via the formation of the intermediate c3H4NH, but from a
129

Mo4+cc3H4 ) 2+ and a Bi3+(NH) 2- species. This leads via a
rapid dehydrogenation to acrylonitrile, as was already
experimentally confirmed by Cathala et al (47), who found
the dehydrogenation of allyl amine over bismuth molybdate
to be a rapid and selective reaction.
The formation of acrylonitrile from acrolein at 673 K
is first order in acrolein for low acrolein concentrations
in the gas phase and zero order in ammonia. The rate of
acrolein formation is high in comparison with the rates of
oxidation and ammoxidation of propene, which points either
to a concerted reaction between N0
species and absorbed
acrolein or to a rapid formation of the intermediates 4+ 2+ 4+ .. Mo (C3H4) and Mo (C3H4NH). In th1.s respect we recall
the observation, mentioned in section 7.4.2 that the
nitrogen formation is notably suppressed, which means
that the intermediates are present on Mo4+. Further we mention the conclusion of Cathala et al (48) with respect
to the rapid formation of c3H4-species from acrolein. The
two possibilities are depicted in the scheme given in
section 7.5 by means of two dotted arrows.
7.4.4. The formation of water
The first order relation in propene for acrolein and
acrylonitrile formation requires that the proton abstrac
tion step is irreversible, which can be explained if the
conversion of surface hydroxyl groups to water is very
fast. This is confirmed by the observation that water va
pour does not inhibit the reaction. As discussed in sec
tion 7.3.4 we have no further evidence to offer as to what cations the hydroxyl groups are related.
7.5. The kinetic model
The experimental data given in chapter 6 show that
under comparable stationary conditions in the flow reac
tor the rate of propene oxidation is the smallest, direct
ly followed by the rate of propene ammoxidation, whereas
130

the rate of acrolein ammoxidation is much greater as can be seen in table 6.2, which is copied here for easy refe-
renee:
k (mol kg -1 -1 s )
p + ACO 0.025 p + ACN 0.035
ACO + ACN 0.42
Table 6.2. Reaction rate constants at 673 K.
The ratio between the rate constant of the ammoxidation
of propene and that of the oxidation of propene (R1), gi
ven in the literature and that calculated from this inves
tigation are given in table 7.3. The same is done for the
ratio between the rate constant of the ammoxidation of
acrolein and that of the ammoxidation of propene (R2).
R1 R2 T (K)
Cathala et al (48) .8 5.4 733
Kolchin et al (49) 1.8 - 750
Callahan et al (50) 1.8 2 698
Wragg et al (51) 1.9 26 673
this investigation 1.4 12 673
Table 7.3. Ratios of rate constants of the various reac
tions (for definition see text).
The bismuth molybdates used by these authors were dif
ferent in composition or prepared in different ways. Only
Wragg et al used a catalyst which was prepared according
to the same method A as our catalyst. The ratios R1 and R2
131
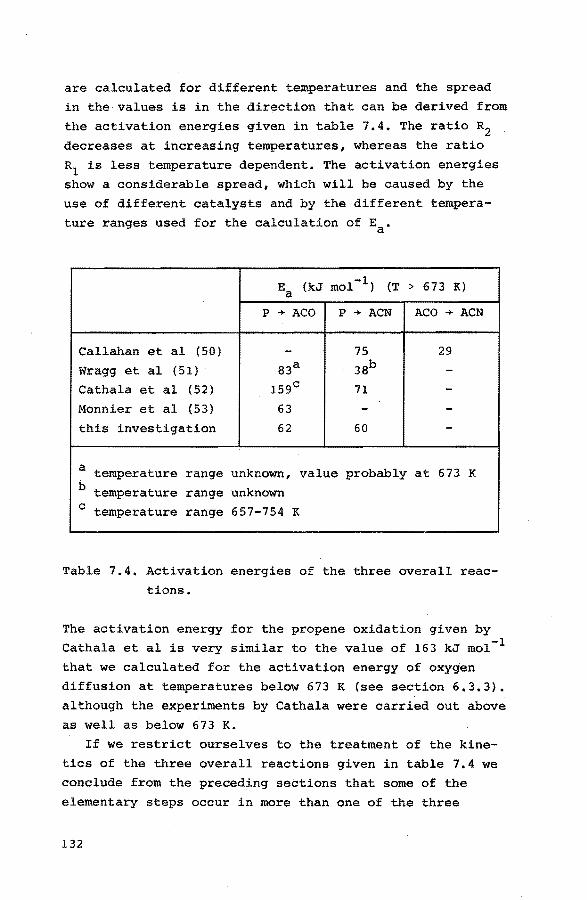
are calculated for different temperatures and the spread
in the values is in the direction that can be derived from
the activation energies given in table 7.4. The ratio R2 decreases at increasing temperatures, whereas the ratio
R1 is less temperature dependent. The activation energies
show a considerable spread, which will be caused by the
use of different catalysts and by the different tempera
ture ranges used for the calculation of Ea.
Ea (kJ mol-l) (T > 673 K)
p -~- ACO P -+ ACN ACO + ACN
Callahan et al (50) - 75 29
Wragg et al (51) 83a 38b -Cathala et al {52) 159c 71 -Monnier et al (53) 63 - -this investigation 62 60 -a temperature range unknown, value probably at 673 K b temperature range unknown c temperature range 657-754 K
Table 7.4. Activation energies of the three overall reac
tions.
The activation energy for the propene oxidation given by
Cathala et al is very similar to the value of 163 kJ mol-l
that we calculated for the activation energy of oxygen
diffusion at temperatures below 673 K (see section 6.3.3).
although the experiments by Cathala were carried out above
as well as below 673 K.
If we restrict ourselves to the treatment of the kine
tics of the three overall reactions given in table 7.4 we
conclude from the preceding sections that some of the
elementary steps occur in more than one of the three
132

overall reactions. This is shown in the following scheme,
which does not take account of all elementary steps nor
of the charges on the intermediates
1 2 3 4 5 C3H6 ~ (C3H6} a- (C3H5} a -_(C3H4} a:;:':: (C3H40} a~ C3H40
6 ~
7 ~
8 n
The first order in propene, both for oxidation and
ammoxidation requires that the surface coverage of the c3 species that enters the rate determining step is propor
tional to the propene gas phase concentration. This means
that this surface species must be formed via a reversible
reaction from the gas phase propene and the degree of
coverage for this species is relatively low. This indi
cates that reaction 2 of the scheme will be rate deter
mining. The fact that the overall rate constant for the
ammoxidation reaction is 1.4 times the rate constant for
the oxidation reaction is ascribed to a higher coverage
of the surface with acrolein than with acrylonitrile. This
is supported by the fact that the rate of the ammoxidation
reaction at 673 K remains first order in propene up to a -3 propene gas phase concentration of about 3 mol m , where-
as for the oxidation reaction deviation from the first
order dependency already begins at a propene concentration
of about 1.4 mol m- 3 (see figure 6.6 and 6.18}. Moreover
the enthalpies of activation for oxidation and ammoxida
tion have almost the same values i.e. 62-102 kJ mol-l and -1
60-97 kJ mol respectively for the same temperature
ranges (see figure 6.7 and 6.23}.
133

The observation that the rate of ammoxidation of acro
lein is more than one order of magnitude higher than that
of propene leads us to the conclusions that the rate of
adsorption of acrolein must be much greater than the ad
sorption of propene and further that the steps 7 and 8
also must be very fast and not rate determining in the
ammoxidation of propene. This is in agreement with the ob
servation that the ammoxidation of acrolein is only first
order in acrolein for an acrolein gas phase concentration
below 1 mol m- 3 (see figure 6.12), and that the inhibition
by acrylonitrile is smaller than the inhibition by acro
lein i.e. acrylonitrile desorbs much faster than acrolein.
As further little or no acrolein is found in the ammoxida
tion of propene, provided ammonia is present in excess,
we also conclude that the steps 7 and 8 are fast as well
in comparison with the steps 4 and 5, see also section 7.4.3,
7.6. Selectivities in the acrylonitrile synthesis reaction
The almost constant and high value of the integral pro
pene selectivity towards acrylonitrile (SIPA) and the
strong variation of the integral ammonia selectivity
(SINA)' as we have already mentioned in chapter 6, are
peculiar phenomena in this process. All other conditions
constant, the variation of SINA can be related to the molar ratio ammonia/propene Mat the reactor inlet (M ),
0 From experiments carried out at very short residence
times we calculated the initial rates of formation of the
nitrogen containing products at 673 K and from that the
fractional ammonia selectivity towards acrylonitrile
(SFNA) according to the definitions of the fractional
selectivity given by the IUPAC (1),
(7 .1)
could be calculated,
134
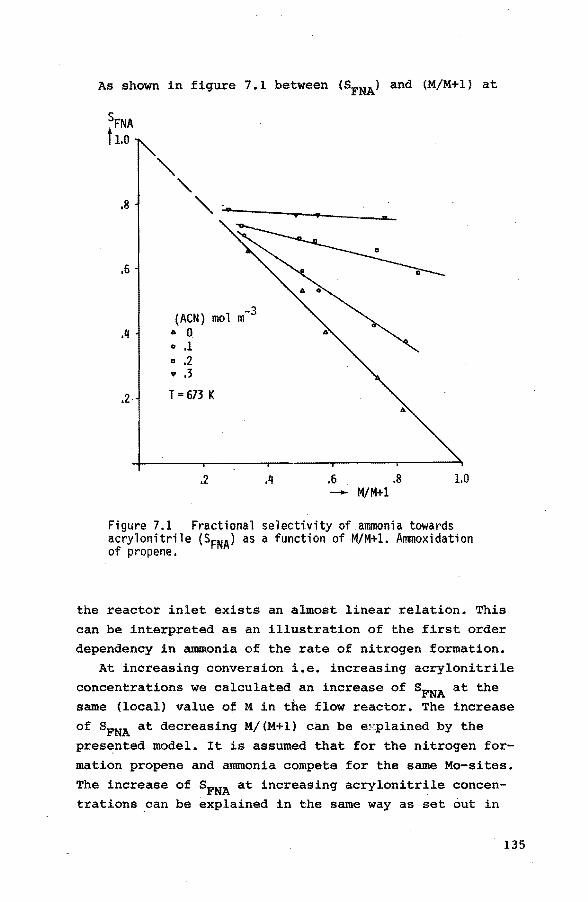
As shown in figure 7.1 between (SFNA) and (M/M+1) at
.6
(ACN} mol m-3
.4 A 0 0 .1 a ,2 9 .3
.2· T=673 K A
.2 .4 .6 .8 1.0 - M/M+l
Figure 7.1 Fractional selectivity of ammonia towards acrylonitrile (SFNA) as a function of M/M+l. Ammoxidation of propene.
the reactor inlet ~xists an almost linear relation. This can be interpreted as an illustration of the first order dependency in ammonia of the rate of nitrogen formation.
At increasing conversion i.e. increasing acrylonitrile
concentrations we calculated an increase of SFNA at the same (local) value of M in the flow reactor. The increase
of SFNA at decreasing M/(M+1) can be explained by the presented model. It is assumed that for the nitrogen for
mation propene and ammonia compete for the same Mo-sites.
The increase of SFNA at increasing acrylonitrile concentrations can be explained in the same way as set out in
135

section 7.4.2 for the influence of acrolein on the nitro
gen formation.
As the rate of nitrogen formation is first order in
ammonia it will be clear that the greatest increase of
SFNA by acrylonitrile will be obtained at high M/(M+1)values as was experimentally observed. However the frac
tional selectivity of ammonia towards acrylonitrile never
exceeds the value of .8 under the experimental conditions.
References
1. Manual of Symbols and Terminology for physico chemical Quantities and Units, Appendix II, Part II Hetero
geneous Catalysis, Adv. Catai. ~, 351 (1977)
2. Matsuura, I., J. Catal. 11• 420 (1974)
3. Grasselli, R.K., Suresh, D.D., J. Catal. ~. 273 (1972)
4. Gates, B.C., Katzer, J,R. and Schuit, G.C.A., "Chemi
stry of Catalytic Processes", chapter IV, McGraw Hill
N.Y. (1979)
5. Weiss, F., Marion, J.,. Metzger, J., Cognion, J .M.,
Kin. i. Kat. !!r 45 (1973)
6. Matsuura, I., Schuit, G.C.A., J. Catal. ~, 19 (1971)
7. Matsuura, I., Schuit, G.C.A., J. Catal. ~, 314 (1972)
8. Haber, J., z. Chern. ll• 241 (1973) 9. Haber, J., Grzybowska, B., J. Catal. .£!!, 489 (1973)
10. Beres, J., Bruckman, K., Haber, J., Janas, J., Bull.
Acad. Pol. Sci. Ser. Sci. Chim. ~, 813 (1972)
11. Sakamoto, T., Egashira, M., Seiyama, T., J. Catal. ~' 407 (1970)
12. Swift, H.E., Bozik, J.E., Ondrey, J.A., J. CataL £!_,
212 (1971)
13. Boersma, M.A.M., Thesis Eindhoven (1977)
14. Grzybowska, B., Haber, J., Janas, J., J. Catal. 49, 150 (1977)
15. Gamid-Zade, E.G., Kuliyev, A.R., Mamedov, E.A., Rizayev,
R.G., Solokovski, V.D., React. Kinet. Catal. Lett. l• i91 (1975)
136

16. Grasselli, R.K., 4th Roermond Conf. on Catal. (1978)
17. Linn, W.J., Sleight, A.W., Ann. N.Y. Acad. Sci.~' 22 (1976)
18. Sleight, A.w., in J.J. Burton and R.L. Garten (eds.)
"Advanced Materials in Catalysis" Acad. Press, N.Y •. in press
19. Peacock, J.M., Sharp, M.J., Parker, A.J., Ashmore, P.G.,
Hockey, J.A., J. Catal. ~, 379 (1969) 20. Haber, J. 1 Sochacka, M., Grzybowska, B., Golzbiewski,
A., J. Molec. Catal. ! 1 35 (1975)
21. Grzybowska, B., Haber, J., Marczew-ski, w., Ungier, L.,
J. Catal. ~' 327 (1976) 22. Batist, Ph.A., Bouwens, J.F.H., Matsuura, I., J. Catal.
,g, 362 (1974)
23. Mars, P., van Krevelen, D.W., Chem. Eng. Sci. Suppl.
lt 41 (1954) 24. Aykan, K., J. Catal. 12, 281 (1968)
25. Keulks, G.W., J. Catal. ~' 232 (1970) 26. Wragg, R.D., Ashmore, P.G., Hockey, J.A., J. Catal.
g, 49 (.1971)
27. Sancier, K.M., Wentreck, P.R., Wise, H., J. Catal.
l2_, 141 (1975)
28. Pendleton, P., Taylor, D., J. Chem. Soc. Far. Trans.
I. E, 1114 (1976) 29. Keulks, G.W., Krenzke, L.D., Proc. 6th Int. Congr.
Catal. London ~. 806 (1977) 30. van Oeffelen, D.A.G., Thesis Eindhoven (1978)
31. Landolt Bornstein' "Zahlenwerte und Funktionen" 4. Teil,
Kalorische zustandsgrossen, Springer Verlag Berlin 1961, p 226, 259
32. Gmelin's Handbuch der Anorganische Chemie 8. Aufl. 1964, Bismuth Syst. nr. 19, Erg. Band, 628-632, Verlag
Chemie GmbH Weinh,eim 33. JANAF Thermochemical Tables, 2nd ed., NSRDS-NBS 37,
US Government Printing Office, Washington DC 1971 34. Batist, Ph.A., Lankhuijzen, S.P., J. Catal. 28, 496
(.1973)
35. Miura, H., Morikawa,· Y. Shirasaki, T., J. Catal. 39, 22 (1975)
137

36. Otsubo, T., Miura, H., Morikawa, Y., Shirasaki, T.,
J. Catal. ~. 240 (1975) 37. Miura, H., Otsubo, T., Shirasaki, T., Morikawa, Y.,
J. Catal. 56, 84 (1979)
38. Bleijenberg, A.C.A.M., Lippens, B.C., Schuit, G.C.A.,
J. Catal. i• 581 (1965)
39. Batist, Ph.A., Lippens, B.C., Schuit, G.C.A., J. Catal. 2_, 55 (1966)
40. Sancier, K.M., Dozono, T., Wise, H., J. Catal. 23,
270 (1971)
41. Burlamacchi, L., Martini, G., Ferron!, E., J. Chern.
Soc. Far. Trans. I. ~, 1586 (1972)
42. Sachtler, W.M.H., Catal. Rev. i (I), 27 (1970)
43. Sachtler, W.M.H., de Boer, N.H., Proc. 3rd Int. Congr.
Catal. Amsterdam 1964, Vol. I, 252, NH Publ. Co.,
Amsterdam 1965
44. Kondo, T., Saito, s., Tamaru, K., J. Am. Chern. Soc.
96, 6857 (1974)
45. Peacock, J .M. I Parker, A.J •, Ashmore, p .G. I Hockey,
J .A., J. Catal. IS. 398 (1969)
46. Cathala, M, I Germain, J.E., Bull. Soc. Chim. Fr. 4114
(1970)
47. Cathala, M. I Germain, J.E., Bull. Soc. Chim. Fr. 2990
(1971)
48. Cathala, M. I Germain, J.E., Bull. Soc. Chim. Fr. 2167
(1971)
49. Kolchin, I .K. I Bobkov, s.s., Margolis, L. Ya.,
Neftekhimiya i• 301 (1964}
50. Callahan, J.L., Grasselli, R.K., Milberger, E.C.,
Strecker, H.A., Ind. Engng. Chern. Prod. Res. Dev. !• 134 (1970)
51. Wragg, R.D. 1 Ashmore, P.G., Hockey, J .A. I J. Catal.
l!,, 293 (1973)
52. Cathala, M. I Germain, J.E., Bull. Soc. Chim. Fr. 2174 (1971)
53. Monnier, J.R., Keulks, G.W., Paper Div. Petr. Chern.
138
Joint Meeting Am. Chern. Soc. and Chern. Soc. Japan
Honolulu, April 1-6 (1979)

List of symbols
Latin symbols
A
A
component of a chemical reaction
peak areas in chromatogram
A constant in a-It relation
[A],(A) concentration of A
a reaction order
Units
av B
surface area catalyst per bed volume m2 m-3
b
b
C, (C)
constant in a-It relation s-~ reaction order
volumetric dilution ratio
concentration
concentration factor
mol m-3
mol m-3 cf
cp D
heat capacity at constant pressure
diffusion coefficient
J kg-1 K-1
m2 s-1
Da
dp
Ea F
f
axial dispersion coefficient
particle diameter
activation enthalpy
molar gasflow rate
substance specific correction factor
2 -1 m s
m
kJ mol-l
mol s-1
G mass velocity kg m- 2 s-1
GM
t.Go
t.G o
superficial molar flow rate mol m-2 s-1
r
t.H o f
standard Gibbs free energy kJ mol-l
standard Gibbs free energy of reac-
tion kJ mol-l
standard enthalpy of formation kJ mol-l
standard enthalpy of reaction kJ mol-l t.H o r
h heat transfer coefficient J m- 2 s-1 K-l
j 0 Chilton and Colburn factor for
mass transfer
jH Chilton and Colburn factor for
heat transfer
K adsorption equilibrium constant
139

reaction rate constant
rate constant in eq. 6.9
mass transfer coefficient
length of catalyst bed
M molar ratio ammonia/propene
n number of moles
np pulse number
n reaction order
0 oxygen content of catalyst,
oxygen consumption per pulse
p pressure
R gas constant
r radius
r rate of reaction
rspec specific rate of reaction
S specific surface area
S selectivity
u
v w
standard entropy
temperature
residence time
reaction time
superficial gas velocity
anion vacancy
catalyst weight
X conversion
x mole fraction
Greek symbols
ct degree of reduction
ctw heat transfer coefficient
at the wall
t: porosity
n effectiveness factor
e fraction of sites
I. jump distance
140
2-n 3n-n -1 -1 mol m kg s -1 s
m s-1
m
mol
mmol kg-1
bar J mol-l K- 1
m
mol s-1
mol m- 3 s- 1
mol kg-l s-1
m2 kg-1
J mol-l K-1
K
s
s -1 m s
kg
m

A effective radial thermal conduct!-er -1 -1 K-1 vity J m s
-1 -1 K-1 Af thermal conductivity of fluid J m s
-1 s-1 K-1 thermal conductivity of particle Jm '-p -1 -1 ll dynamic viscosity kg m s
\) coefficient in stoichiometric
equation
~ extent of reaction
p density cr2 variance
41 sphericity factor
Abbpeviations
A acrylonitrile
ABS acrylonitrile-butadiene-styrene
ACO ACON
ACN BET
copolymer
acrolein acetonitrile
acrylonitrile
Brunauer-Emmet-Teller (determina-
tion method of catalyst surface area)
Fo Fourier number
H hydrogen IUPAC International Union of Pure and
Applied Chemistry LCLo lowest lethal concentration
N ammonia
NBR nitrile-butyl-rubber NTP normal temperature and pressure
o oxygen P propene
P~ P~clet number ppm parts per million
mol kg m-3
s2
ppm
Re = ~. Reynolds number (equation 5.11) avq>u
141

pud Redp = ~ Reynolds number with ref. to particle
SAN
Sc SOHIO
TLV
TWA
Subsar>ipts
a
b
cat e
eq
f
F
I
L
0
p
r
s
t
Symbol
c
142
diameter
styrene-acrylonitrile copolymer
Schmidt number Standard Oil Company of Ohio
Treshold Limit Value
time weighted average
activation, adsorbed, axial
bulk
catalyst
effective, reactor outlet equilibrium
fluid
fractional
integral
liquid state, bed length
initial value, reactor inlet
particle, pressure, pulse
reaction, reoxidation, radial
surface, superficial at time t
2-0 vacancy
ppm

Summary
This thesis deals with the synthesis of acrolein and
acrylonitrile over a bismuth molybdate catalyst. During
the last twenty years catalysts based on this compound
played an important role both in the commercial production
of acrylonitrile and in catalytic research with respect to
the elucidation of the mechanism of the selective oxida
tion of olefins over oxide catalysts.
Bismuth molybdate is not only a catalyst for the oxi
dation and ammoxidation of propene but catalyzes at the
same time also the oxidation of ammonia to nitrogen and
the ammoxidation of acrolein to acrylonitrile.
In order to contribute to the elucidation of the reac
tion mechanism we have investigated the kinetics of these
four reactions. The experimental work can be divided into
two parts:
1. The determination of the kinetics of these four reac
tions in continuously operating fixed bed flow reactors
under differential and integral conditlons at tempera
tures between 648 and 723 K and at atmospheric pressure;
2. An investigation into the behaviour.of bismuth molyb
date acting as oxidant in the absence of oxygen and in
the presence of substoichiometric amounts of oxygen ' and ammonia. These reactions have been studied in a
thermobalance and in a socalled pulse reactor.
At temperatures above 673 K and in the range of the
concentrations of this investigation the oxidation and
ammoxidation reactions are all first order in the organic
reactant and zero order in oxygen. At temperatures below
673 K the order in the organic reactant decreases with
increasing concentrations. At these temperatures the rate
of propene conversion is also inhibited by acrolein. The
ammoxidation of propene has a slightly negative order in
ammonia. The catalyst shows initially a high activity for
the oxidation of ammonia to nitrogen, which reaction is
first order in ammonia. The ammoxidation of acrolein is
zero order in ammonia. The activation enthalpies for the
143

oxidation and the ammoxidation of propene are very similar -1 . .
and increase from about 60 kJ mol at temperatures above 673 K to about 100 kJ mol-l at temperatures below 673 K.
Experiments in a thermobalance in the same temperature
range have shown that the initial rate of catalyst weight
loss is a function of the propene concentration. At tem
peratures below 673 K even at a low degree of reduction
the oxygen diffusion in the catalyst has a major influence
on the overall rate of reduction of the catalyst. The ad
sorption of acrolein on a partially reduced catalyst
appears to be a relatively slow process. The reoxidation
of a reduced catalyst with gas phase oxygen is an extreme
ly fast reaction and is first order in oxygen at tempera
tures above 673 K. The oxidation of propene with substoi
chiometric amounts of oxygen and at low propene conversion
can be carried out under stationary conditions with the catalyst operating in a partially reduced state, Under
these conditions the rate of reaction is first order in
oxygen for low oxygen concentrations and the amount of
acrolein adsorbed is a function of the degree of reduction and of the temperature.
Pulse experiments with propene-helium gas mixtures con
firmed the adsorption behaviour of partially reduced bis
muth molybdate for acrolein. When small quantities of
oxygen are added the rate of acrolein formation per pulse is again a function of the partial pressure of oxygen.
From experiments with ammonia-propene-helium gas mixtures
without oxygen it became clear that the rate of oxygen
suppletion by the catalyst is enhanced in the presence of
ammonia in the same way as in the presence of gas phase . 2- 2-
oxygen and it has been assumed that 0 and NH species perform the same function.
The presence of ammonia as the third reactant in the acrylonitrile synthesis is an important expedient to de
rive a mechanistic model from the experiments and from
literature data. We have concluded that ammonia molecules
are activated on two different active centres in two different ways. The activation for the ammoxidation of
144

propene and acrolein occurs on the active centre connected with bismuth, whereas that for the oxidation of ammonia
occurs on two different active centres connected with
molybdenum and with bismuth. From this we have concluded that propene is activated on molybdenum. The activation of oxygen takes place after the reduction of the catalyst
to some extent, according to the reduction-oxidation me
chanism proposed by Mars and van Krevelen and is related
to the diffusion of oxygen in the catalyst. From the kinetic data we have concluded that the rate
determining step of the oxida~ion and ammoxidation of
propene is the abstraction of the first proton of propene.
The acrolein ammoxidation may either proceed via a con
certed mechanism or via an adsorbed c3H4 intermediate.
145

146

Samenvatting
In dit proefschrift wordt de synthase van acroleine en
acrylonitril behandeld. Bismuth molybdaat wordt gebruikt
als katalysator. Gedurende de laatste twintig jaar hebben
katalysatoren op basis van deze verbinding niet alleen
een belangrijke rol gespeeld in deproduktie van acrylo
nitril maar ook heeft bismuth molybdaat in het fundamen
teel katalytisch onderzoek naar het mechanisme van de
selectieve oxidatie van olefinen met behulp van oxidische
katalysatoren een centraleplaats ingenomen.
Bismuth molybdaat is niet alleen een katalysator voor
de oxidatie en ammoxidatie van propeen, maar versnelt
tegelijkertijd ook de oxidatie van ammoniak tot stikstof
en de ammoxidatie van acroleine tot acrylonitril.
Om een bijdrage te leveren aan de opheldering van het
reactiemechanisme hebben we deze vier reacties aan een
kinetisch onderzoek onderworpen. Het experimentele werk
kan in twee stukken worden onderverdeeld:
1. De bepaling van de kinetiek van deze vier reacties in
continu werkende reactoren met een vast katalysatorbed
zowel onder differentiele als onder integrale kondities
bij temperaturen tussen 648 en 723 K en onder atmos
ferische druk;
2. Een onderzoek naar het gedrag van bismuth molybdaat
als oxidatiemiddel in afwezigheid van zuurstof en bij
gebruik van ondermaat zuurstof of ammoniak. Deze reac
ties zijn bestudeerd met gebruikmaking van een thermo
balans en een zogenaamde pulsreaktor.
Bij temperaturen boven 673 K zijn in het onderzochte
concentratie gebied de oxidatie en ammoxidatie reacties
eerste orde in de organische reaktant en nulde orde in
zuurstof. Beneden 673 K daalt de orde in de organische
reaktant bij toenemende concentraties. Bij deze tempera
turen wordt de omzettingvan propeen door acroleine geremd.
De ammoxidatie van propeen vertoont een kleine negatieve
orde in ammoniak. De katalysator is initieel zeer actief
in het oxideren van ammoniak tot stikstof, een reactie die
117

eerste orde in anunoniak is. De anunoxidatie van acroleine
is nulde orde in ammoniak. De activeringsenthalpieen voor
de oxidatie en de ammoxidatie van propeen zijn vrijwel
gelijk en nemen toe van ongeveer 60 kJ mol-l bij tempera•
turen hoven 673 K tot ongeveer 100 kJ mol-l bij tempera
turen beneden 673 K.
Experimenten met propeen-helium gasmengsels in het
zelfde temperatuurgebied in een thermobalans hebben aan
getoond dat de initiele snelheid waarmee het katalysator
gewicht afneemt een functie is van de propeen concentra
tie. Bij temperaturen beneden 673 K en zelfs bij lage
reduktiegraden heeft de zuurstofdiffusie in de katalysator
een grote invloed op de reduktiesnelheid van de katalysa
tor. De adsorptie van acroleine op een gedeeltelijk gere
duceerde katalysator blijkt vrij langzaam te verlopen. De
reoxidatie van een gereduceerde katalysator met zuurstof
is een zeer snelle reactie die bij temperaturen hoven 673
K een eerste orde gedrag in zuurstof vertoont. De oxida
tie van propeen met ondermaat zuurstof kan bij een lage
propeen conversie stationnair verlopen, waarbij de kata
lysator zich in een gedeeltelijk gereduceerde toestand
bevindt. Onder deze omstandigheden is de reactie bij lage.
zuurstofconcentraties eerste orde in zuurstof. De gead
sorbeerde hoeveelheid acroleine onder stationnaire om
standigheden is een funktie van de reduktiegraad en van
de temperatuur.
Puls experimenten met propeen-helium gasmengsels hebben
bevestigd dat gedeeltelijk gereduceerd bismuth molybdaat
acroleine adsorbeert. Als kleine hoeveelheden zuurstof
worden meegepulst is de vormingssnelheid van acroleine een
funktie van de partiele druk van zuurstof. Experimenten
met ammoniak-propeen-helium gasmengsels zonder zuurstof
hebben aangetoond dat de zuurstof afgiftesnelheid van de
katalysator in aanwezigheid van ammoniak op dezelfde wijze
wordt verhoogd als die in aanwezigheid van zuurstof zonder
ammoniak tijdens de oxidatie van propeen. Er wordt verondersteld dat o2- en NH2- species dezelfde funktie
vervullen bij de synthese van acroleine respectievelijk acrylonitril.
148

De aanwezigheid van ammoriiak als de derde reaktant in
de acrylonitril synthase is een belangrijk middel om een
mechanistisch model af te leiden uit de experimenten en de literatuurgegevens. We hebben geconcludeerd dat ammo
niak op twee verschillende manieren wordt geactiveerd. De
activering van ammoniak in de ammoxidatie van propeen en acroleine gebeurt op een "active centre" samenhangend met
bismuth, terwijl die voor de oxidatie van ammoniak tot
stikstof op twee verschillende "active centres" plaats
vindt, nl. op een bismuth- en op een molybdeen-centre.
Propeen wordt op het laatste "active centre" geactiveerd. De activering van zuurstof vindt plaats nadat de kataly
sator enigszins gereduceerd is volgens het Mars-van Kre
velen redox mechanisme en de inbouw van zuurstof in de
produkten geschiedt via een diffusie door het katalysator rooster.
Uit de kinetische gegevens hebben we eveneens gecon
cludeerd dat de snelheidsbepalende stap van de oxidatie
en ammoxidatie van propeen de eerste proton afsplitsing
van propeen is. De ammoxidatie van acroleine verloopt via een concerted mechanisme of via een geadsorbeerd c3H4 intermediair.
149

Dankwoord
Het onderzoek weergegeven in dit proefschrift is tot
stand gekomen da~ij de medewerking van velen. In het
bijzonder geldt dit voor de leden van de vakgroep Chemi
sche Technologie van de Afdeling der Scheikundige Techno
logie. Ik dank speciaal de heren D. Francois, J. van
Hettema, w. van Lith, G. van de Put, A. Sommen, L. Verhaar
en R. van der Wey, die steeds bereid waren de experimenten
te laten slagen.
Veel dank ben ik verschuldigd aan de afstudeerders, de
heren E. van Poelvoorde, P. Derks, J. van Laarhoven, P.
Linders, J. Voet, A. Tjang, R. van Collenburg, N. Willem
sen, P. Evertse, P. Florack, F. Hautus, A. de Laet, F.
Kroes, H. Houben, F. Douven, J. Spork, P. Oostveen en H.
van Liempt, die enthousiast aan het onderzoek hebben mee
gewerkt. Anderen die hebben bijgedragen dank ik in de
persoon van de heer R. Poulina, die tijdens twee stages
metingen heeft verricht.
Mijn dank gaat uit naar mijn oud-collega's van de werk
groep Gasfase reacties dr. J. Steenhof de Jong, dr. H.
Heynen en dr. M. Boersma met wie ik over dit onderzoek
interessante discussies h.Elb gevoerd. De heer Ph. Batist, die op kenmerkende wijze dit werk gestimuleerd heeft dank
ik zeer. Voorts dank ik dr. G. Visser voor de wijze waarop hij heeft bijgedragen tot de verdieping van het inzicht in
de katalysator structuur en mijn collega's van andere vak
groepen voor de nuttige wetenschappelijke informatie die
zij mij hebben verschaft.
Bij de vormgeving van het proefschrift heb ik de onmis
bare steun gekregen van de heren W. van Lith en R. van der
Wey. Mevr. E. Eichhorn-Meijers heeft op concientieuze
wijze het typewerk verricht. Hen allen dank ik zeer.
Gaarne maak ik van deze gelegenheid gebruik om ook
anderen, buiten mijn gezin en mijn werkverband, te danken
voor hun stimulerende bijdragen.
150

STELLINGEN
1 Otsubo et al en Miura et al houden - gelet op de kondi
ties waaronder zij de katalysatoren bereiden - bij de
verklaring van het mechanisme van de acroleine synthese
over bismuth molybdaat onvoldoende rekening met de mo
gelijke aanwezigheid van vrije oxiden en met de beweeg
lijkheid van de zuurstof in de katalysator.
T. Otsubo, H. Miura, Y. Morikawa, T. Shirasaki,
J. Catal. 240 {1975)
H. Miura, T. Otsubo, T. Shirasaki, Y. Morikawa,
J. Catal. 56, 84 (1979)
Dit proefschrift, hoofdstuk 7
2 Door voorbij te gaan aan o.a. het sterisch effekt van
de substituenten komen van Krevelen en Chermin met hun
berekeningsmethode voor de plafond temperatuur van een
polymerisatie reactie voor verschillende op dezelfde
plaats gesubstitueerde monomeren tot dezelfde uitkomst,
hetgeen in strijd is met de experimenteel gevonden
waarden.
D.W. van Krevelen, Properties of Polymers, 2nd ed.
Elsevier, Amsterdam 1976
R.W. Lenz, Organic Chemistry of Synthetic High
Polymers, Interscience, New York 1967
J. :&J:"andru:;>, E.H. Immergut, Polymer Handbook,
John Wiley, New York 1965
3 De onduidelijkheid met betrekking tot het begrip selec
tiviteit in de chemie is door de IUPAC slechts gedeel
telijk weggenomen. Het onderdeel integrale selectivi
teit voor een reactie uitgevoerd in een continu bedreven
reactor moet nog worden gedefinieerd.
Manual of Symbols and Terminology for physico che
mical Quantities and Units, Appendix II, Part II,
Heterogeneous Catalysis, Pure & Appl. Chern~ 46, 71 (1976)

4 Voor een juistE;! toepassing van primaire, droge batte
rijen is het noodzakelijk, zowel ten aanzien van energie
gebruik als ten aanzien van milieu aspekten dat de eti
kettering in plaats van de weinig zeggende informatie
"medium duty", "transistor batterij" of "long life" dui
delijke gegevens verstrekt over de scheikundige samen
stelling, kapaciteit, belastbaarheid en houdbaarheid van
de batterij.
Consumentengids 24, 259 (1976)
Consumentengids 25, 14 (1977)
5 De interpretatie van de veranderingen in het 13c NMR
spectrum van 1-buteen door chernisorptie op zeolieten en
de vertaling daarvan naar elektronen dichtheden c.q. verschuivingen leidt tot verkeerde conclusies.
D. Denney, W.M. Mastikhin, s. Narnba, J. Turkevich,
J. Phys, Chern. 82, 1752 (1978)
6 De bewering van Wauters et al dat gesilyleerde polyolen
in het algerneen geen (geprotoneerde) molekulaire ionen
geven in de chernische ionisatie rnassaspektrornetrie met isobutaan als reaktiegas, is onjuist.
E. Wauters, F. Vangaever, P. Sandra, M. Verzele,
J. Chrornatogr. 170, 133 {1979)
A.C. Schoots, F.E. Mikkers, C.A.M.G. Cramers,
s. Ringoir, J. Chromatogr. Biomedical Appl. (verschijnt binnenkort)
7 Minstens vier van de diffusiepaden in het systeem
Cu-Ni-Zn die Wirtz en Dayananda presenteren als resul
taat van hun onderzoek zijn op grond van de wet van be
houd van rnassa onmogelijk. Hun theoretische beschouwingen omtrent het verloop van de diffusie in dit systeern komen
hierdoor op losse schroeven te staan.
L.E. Wirtz, M.A. Dayananda, Metal. Trans. A 8A, 567 {1977)

8 Aangezien vele chemische en fysische wegen voor milieu
belastende stoffen naar de oceaan leiden, verdient het
aanbeveling de bestudering van bodem-, lucht- en water
verontreiniging uit te voeren in samenhang met de
oceanografie.
9 Hoewel het een goed gebruik is experimenten reeksen
zorgvuldig te plannen, moet de betekenis van het gelso
leerde, ad hoc experiment niet worden onderschat.
P.C. Sander, Intreerede, T.H. Eindhoven (1978)
10 Door de toepassing van zig-zag kanalen in een centri
fugaal-zifter wordt het principe van het zuivere meer
trapsproces, zoals dit geldt in een zig-zag zifter in
het zwaartekrachtsveld, nadelig beinvloed.
F. Kaiser, Chern. Ing. Techn. ~~ 273 (1963)
M.M.G. Senden, Proefschrift, T.H. Eindhoven (1979)
11 De bewering dat de kostprijs voor gas verkregen door
ondergrondse vergassing van steenkoollagen met 80 % kan
worden gereduceerd door verdubbeling van de afstand der
boorgaten,is bij gebrek aan uitkomsten van een onder
zoek naar de invloed van deze afstand op het rendement,
ongegrond.
P.N. Thompson, Endeavour~ (2), 93 (1978)
12 Kinderen met een lichte achterstand in de ontwikkeling
van de fijne motoriek kunnen baat vinden bij het be
spelen van een muziekinstrument, waarbij de gewenste
toonhoogte wordt verkregen door een juiste plaatsing
van de vingers. Vooral een zelfgemaakte bamboefluit
is hiervoor geschikt, omdat bij het intoneren de
vingers in volgorde bij het spel betrokken raken.

13 De periodieke betrouwbaarheidstest van de sirenes in
de gebouwen van de afdeling der Scheikundige Technolo
gie onder werktijd is zonder een evacuatie van het
personeel ongewenst, aangezien deze test nadelige bij
verschijnselen zoals gehoor irritatie, gehoor beschadiging of verlies van arbeidstijd veroorzaakt.
Eindhoven, 22 juni 1979 S.P. Lankhuijzen







