44
MBV4410/9410 Fall 2017 Bioinformatics for Molecular Biology Variant Calling
| Date post: | 17-Jun-2018 |
| Category: |
Documents |
| Upload: | truongkhanh |
| View: | 215 times |
| Download: | 0 times |

MBV4410/9410 Fall 2017
Bioinformatics for Molecular Biology
Variant Calling

Today’s program
• Background• Mapping• Exercise• Re-alignment

Variant calling: Major goals for the lectures
• Gain some theoretical understanding of key HTS concepts
• Hands on experience with NGS data through a variant calling application
• NOT in-depth knowledge on the variant-calling algorithms.
• Algorithms and formats– Sequencing: fastq file– Mapping: SAM/BAM files
• The importance of summary statistics and inspecting data

Variant calling
• Using High throughput sequencing (HTS) data• Variant calling is determining how a DNA sample differs
from the reference genome• Comparative genomics
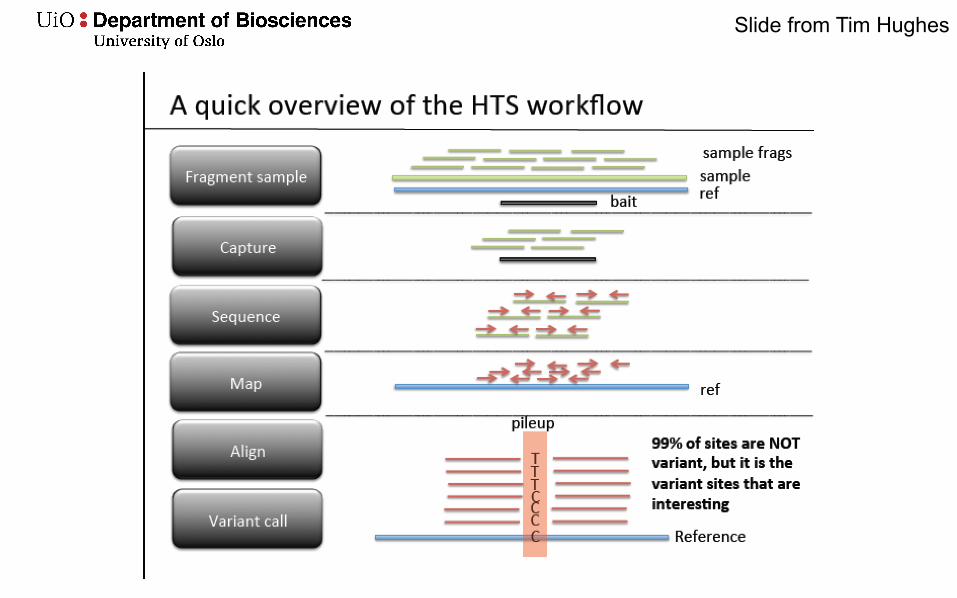
Slide from Tim Hughes

Remember that all our cells contain their own genome• But also different organs may be different• But also populations differ

• VC can help us understand demographic processes– Out-of-africa, bottlenecks, genetic variation between populations
• Medicine, genetic basis for diseases
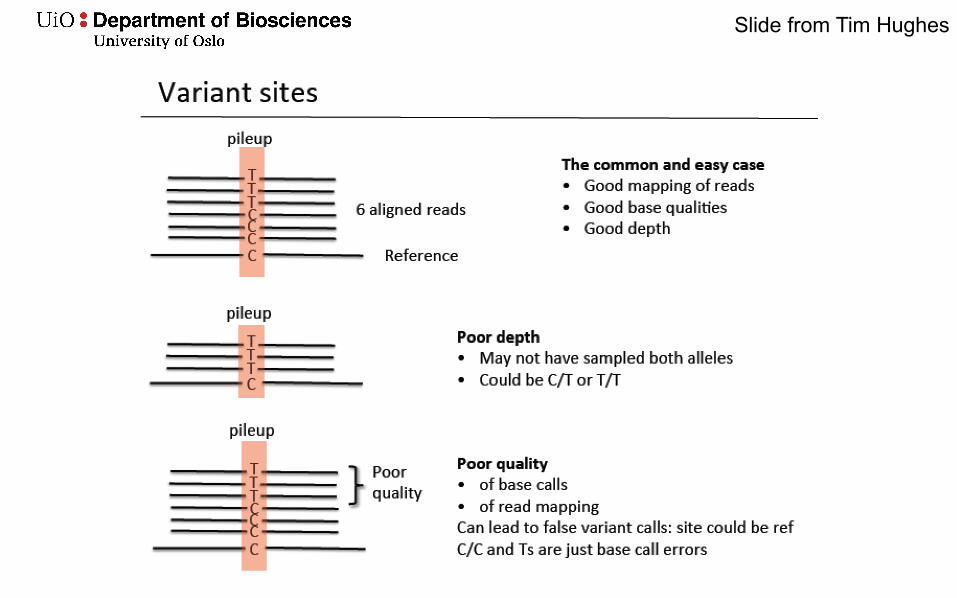
Slide from Tim Hughes

Slide from Tim Hughes

We need a reference genome
• We need to compare against a reference to detect variants
• We use the human genome, but can be your species of choice
• The reference genome may not represent the “true” human.
• GRCh37 – thirteen anonymous donors from New York (Wikipedia)
• Possible to use closely related species as reference

Variant calling not so easy to perform on non-model organisms
Need reference genome.Should have good idea of phenotypesFew bioinformatic tools developed for non-model.
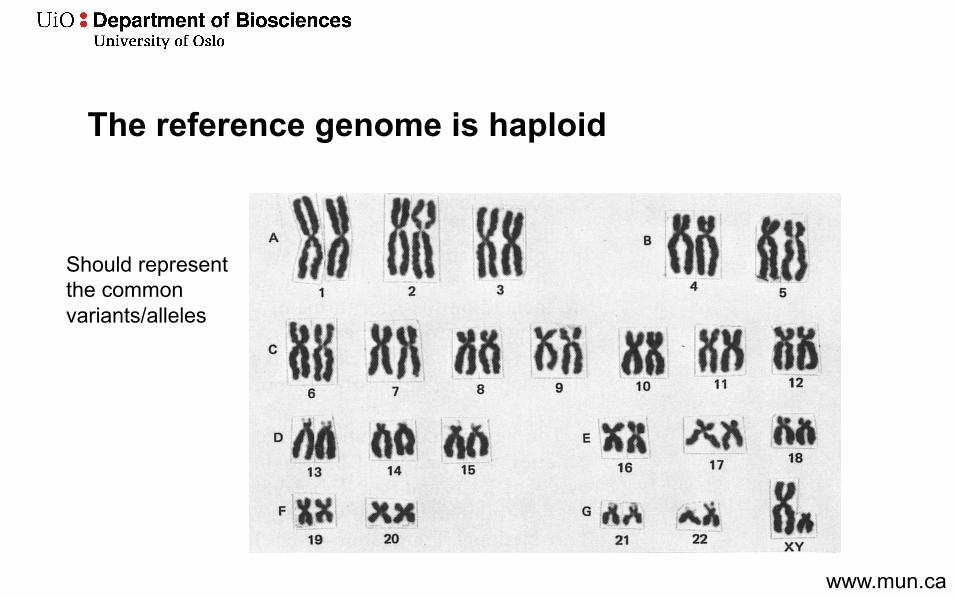
The reference genome is haploid
Should represent the common variants/alleles
www.mun.ca
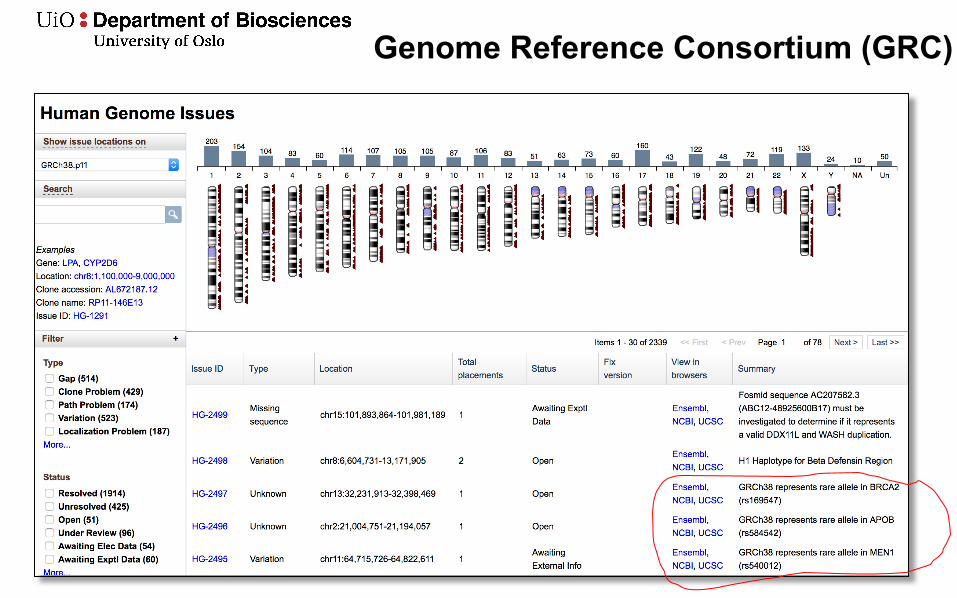
Genome Reference Consortium (GRC)

Different types of mutations
• Single nucleotide polymorphism (SNP)• A single base change (e.g. A to G)
• Indels (less than 100bp)• Insertion: bases added to the genome• Delete: bases removed from the genome
• Large indels are referred to as copy number variations• Other types of variation:
• Inversions• Translocations
• Whole chromosomes can be gained or lost (aneuploidy)• Most are lethal• Trisomy 21• Sex chromosomes (XXX, XYY…)

There is a lot of variation in the human genome!
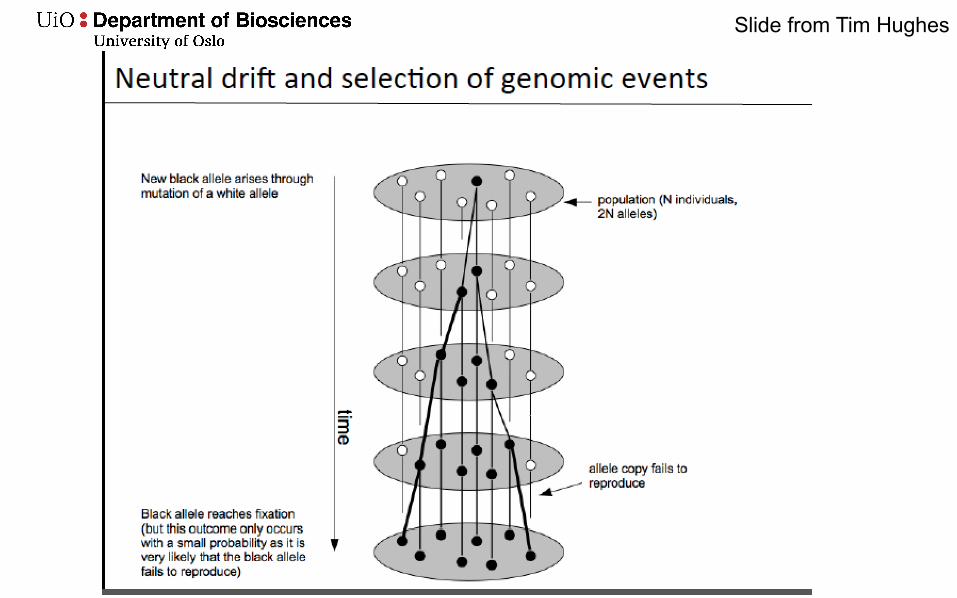
Slide from Tim Hughes
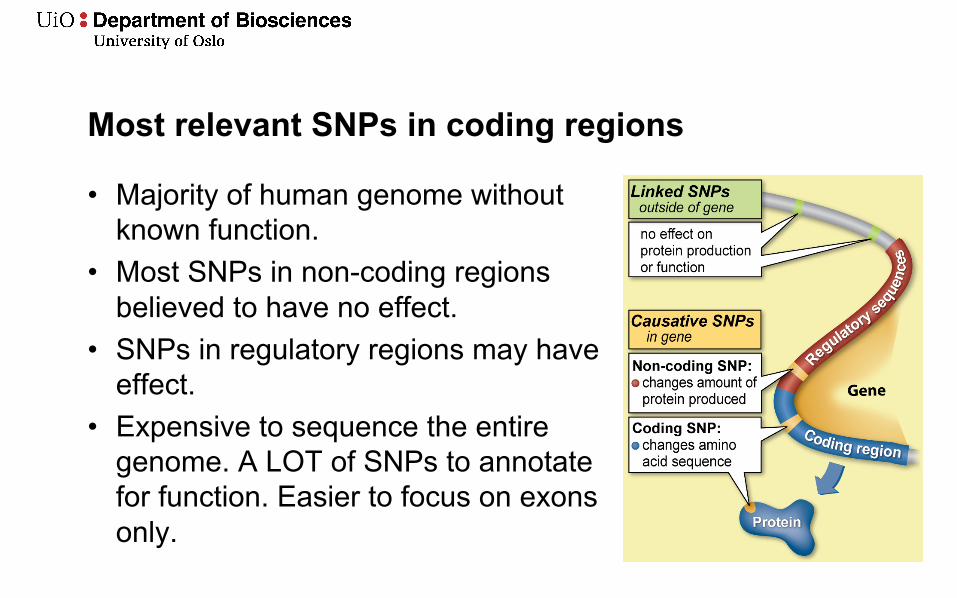
Most relevant SNPs in coding regions
• Majority of human genome without known function.
• Most SNPs in non-coding regions believed to have no effect.
• SNPs in regulatory regions may have effect.
• Expensive to sequence the entire genome. A LOT of SNPs to annotate for function. Easier to focus on exons only.
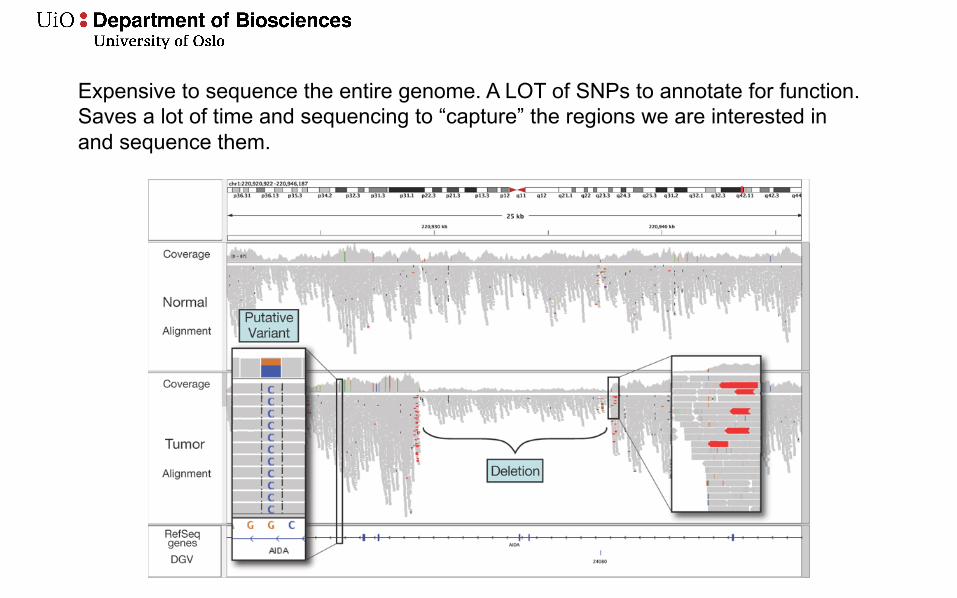
Expensive to sequence the entire genome. A LOT of SNPs to annotate for function. Saves a lot of time and sequencing to “capture” the regions we are interested in and sequence them.
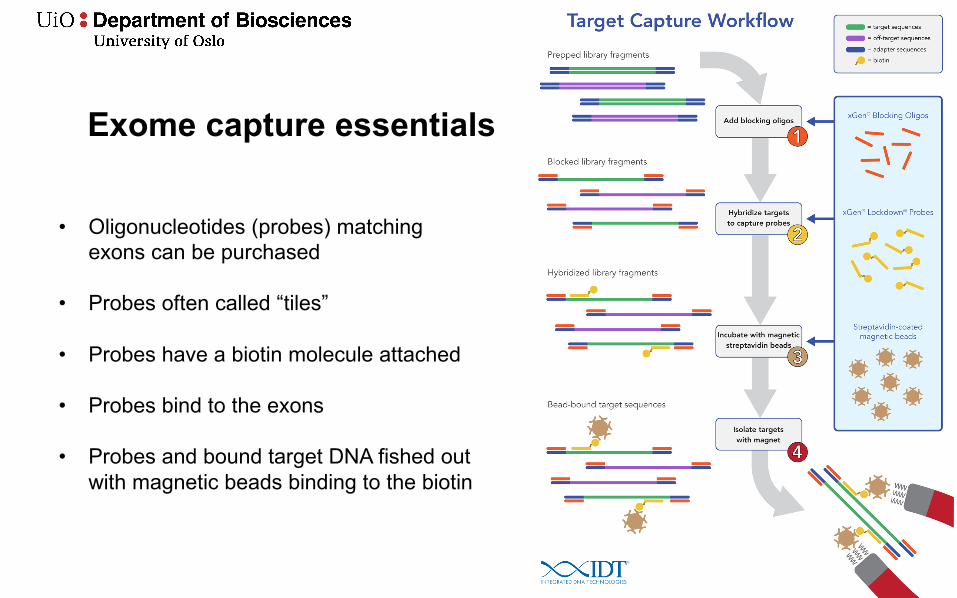
Exome capture essentials
• Oligonucleotides (probes) matching exons can be purchased
• Probes often called “tiles”
• Probes have a biotin molecule attached
• Probes bind to the exons
• Probes and bound target DNA fished out with magnetic beads binding to the biotin
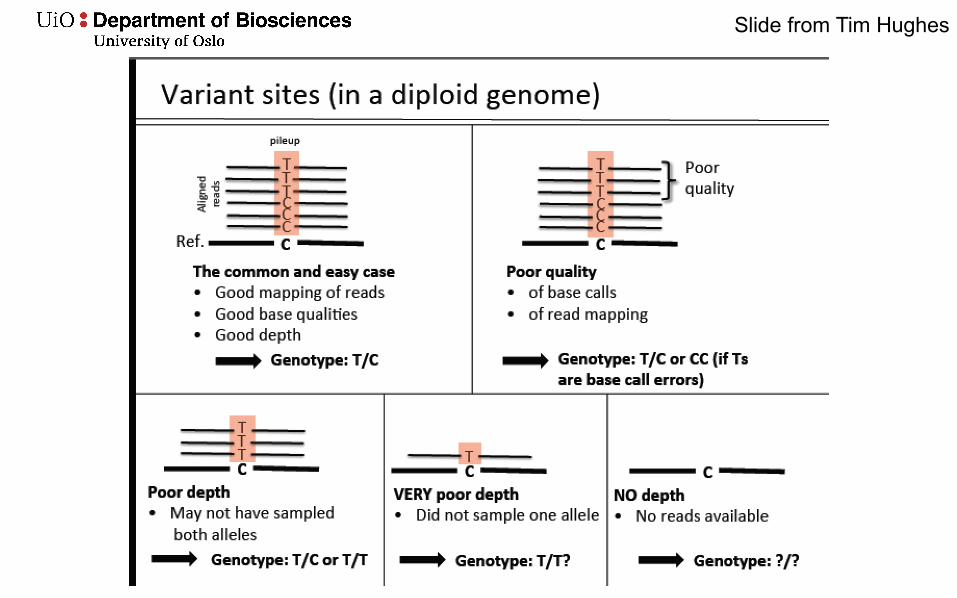
Slide from Tim Hughes
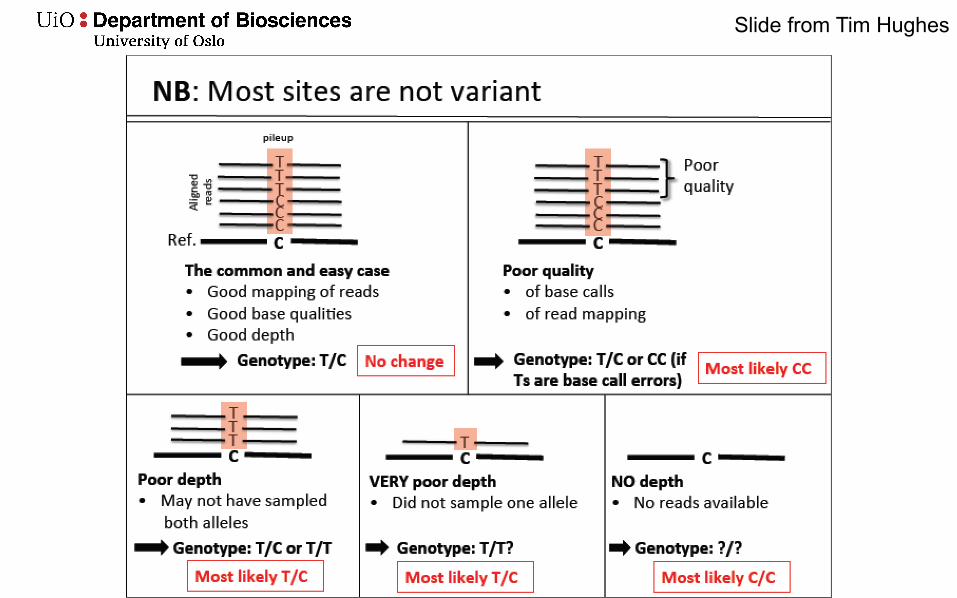
Slide from Tim Hughes

Ensembl – basis for variant effect prediction

Some human variant databases

Pause
• Deretter Mapping lecture + exerecises

Mapping reads to the genome
• Mapping is a very common task when working with NGS data
• Not specific to Variant calling. You will meet this also during transcriptomics session.

Why mapping?
• The biggest difference with Sanger• We did not design and use primers for sequence amplification• We randomly fragmented the DNA• We do not know where the reads “originate” from• We need to determine the likely origin for each read

What are desirable characteristics of a read mapper?• Accurately predict the source of a read
• But, not necessary to get the alignment exactly right as this can be done later using multiple sequence alignment (MSA)
Reference NNNNNCAAGNNNNSample NNNNNCAAAGNNN
• Provide an estimation of the quality, or accuracy, of the mapping
NNNNNCA_AGNNNNNNNNNCAAAGNNNN
Correct read alignment
NNNNNCAAGNNNNNNNNNCAAAGNNNN
Alternativealignment

Characteristics of read mapping
• We need to build an index of the genome to map (search) against• Reads are typically 100-150 bp• But the longer the string searched for the longer the search time. Thus
advantage to search with a shorter string.• There are 4 different nucleotides and ~3 bill bp (3E09)
– 4^16 = 4.0E09– 4^17 = 17.0E09
• 16, 17 bp should be enough for a unique match against the human genome• But
– Variation in sample relative to reference (can’t expect exact match)– Base call errors
• BWA searches with a “seed” of 32 and allows 2 differences to the reference

Many different mappers
• BWA• Bowtie• TopHat (uses Bowtie)• SOAP• Novoalign• …
Most based on BWT: Burrows-Wheeler Transform
A very neat computer algorithm for finding the location of substrings within a string.
Requires indexing of string/reference. Enables• Rapid search, necessary when mapping billions of
reads• Manageable memory (RAM) requirements. Usually
runs on an ordinary computer.
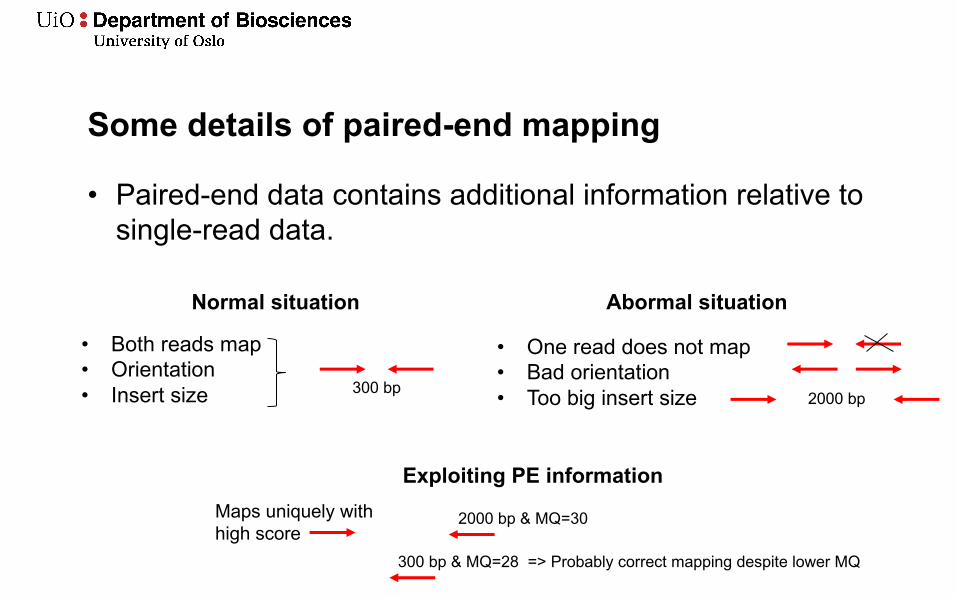
Some details of paired-end mapping
• Paired-end data contains additional information relative to single-read data.
Normal situation
• Both reads map• Orientation• Insert size 300 bp
Abormal situation
• One read does not map• Bad orientation• Too big insert size 2000 bp
Exploiting PE informationMaps uniquely with high score
2000 bp & MQ=30
300 bp & MQ=28 => Probably correct mapping despite lower MQ

BWA and Novoalign are most accurate.
Need short indels
Ruffalo et al. Bioinformatics. 2011

Ruffalo et al. Bioinformatics. 2011

Mapping vs. alignment
After mapping
Slide from Tim Hughes

Mapping vs. alignment
• Key component of alignment algorithm is the scoring• Negative contributions to score:
• Opening a gap• Extending a gap• Mismatches
• Positive contribution to score• Matches
• The total score determines which alignment is chosen• BWA uses the Smith-Waterman algorithm to extend the seed-alignments
Slide from Tim Hughes

• Longer reads makes it easier to correctly align
• Difficult when mismatch is at end of read
Slide from Tim Hughes

Some SNPs can be an indel
https://www.broadinstitute.org/files/shared/mpg/nextgen2010/nextgen_sivachenko.pdf

SAM/BAM format
• Most mappers output the results in a .sam file• These are often huge. Many Gb’s• Usually compressed to a binary .bam format• samtools is a nice toolkit for working on sam/bam files
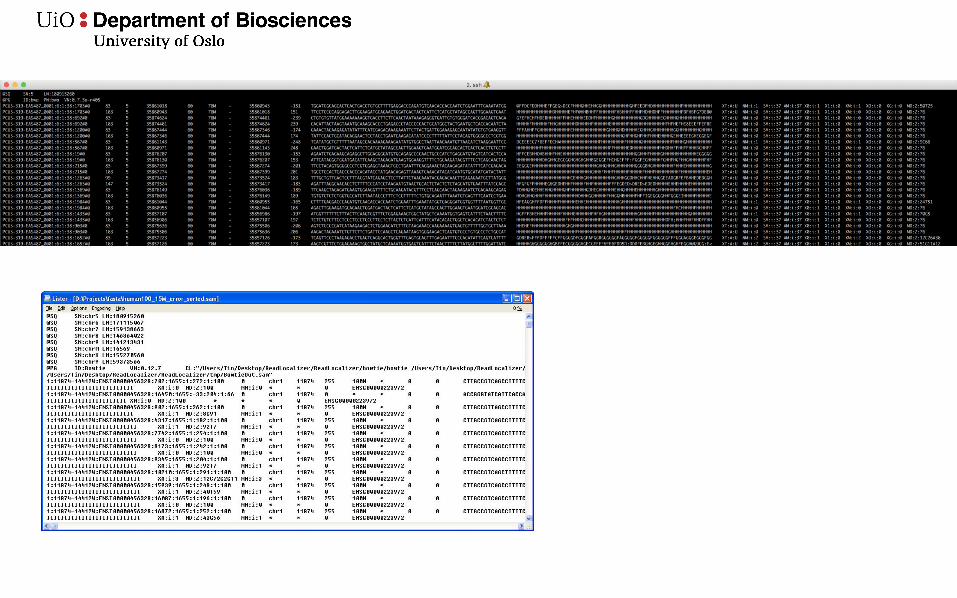

Header
Data lines(one line per read)

Header contains the reference sequence names with length information.
Here I mapped to chromosome 5 (only named 5) which is 180915260 bp long.
Also information on the program used and how it was run
More chromosomes

The headers can contain lots of additional information and be really, really long

WikipediaSam flags: https://broadinstitute.github.io/picard/explain-flags.html


Exercise
• Let’s map some reads and examine the results!
Go to:• https://github.com/jonbra/MBV-INFx410/blob/master/2017-
variant_calling_exercise.md
We do part 1 together

















