PLEASE SCROLL DOWN FOR ARTICLE This article was downloaded by: [Aghalayam, Preeti] On: 20 January 2010 Access details: Access Details: [subscription number 918703597] Publisher Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37- 41 Mortimer Street, London W1T 3JH, UK Catalysis Reviews Publication details, including instructions for authors and subscription information: http://www.informaworld.com/smpp/title~content=t713597232 Nitric Oxide Catalysis in Automotive Exhaust Systems Kathleen C. Taylor a a Physical Chemistry Department, General Motors Research and Environmental Staff, Warren, Michigan To cite this Article Taylor, Kathleen C.(1993) 'Nitric Oxide Catalysis in Automotive Exhaust Systems', Catalysis Reviews, 35: 4, 457 — 481 To link to this Article: DOI: 10.1080/01614949308013915 URL: http://dx.doi.org/10.1080/01614949308013915 Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf This article may be used for research, teaching and private study purposes. Any substantial or systematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply or distribution in any form to anyone is expressly forbidden. The publisher does not give any warranty express or implied or make any representation that the contents will be complete or accurate or up to date. The accuracy of any instructions, formulae and drug doses should be independently verified with primary sources. The publisher shall not be liable for any loss, actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directly or indirectly in connection with or arising out of the use of this material.
Transcript
PLEASE SCROLL DOWN FOR ARTICLE
This article was downloaded by: [Aghalayam, Preeti]On: 20 January 2010Access details: Access Details: [subscription number 918703597]Publisher Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
Catalysis ReviewsPublication details, including instructions for authors and subscription information:http://www.informaworld.com/smpp/title~content=t713597232
Nitric Oxide Catalysis in Automotive Exhaust SystemsKathleen C. Taylor a
a Physical Chemistry Department, General Motors Research and Environmental Staff, Warren,Michigan
To cite this Article Taylor, Kathleen C.(1993) 'Nitric Oxide Catalysis in Automotive Exhaust Systems', Catalysis Reviews,35: 4, 457 — 481To link to this Article: DOI: 10.1080/01614949308013915URL: http://dx.doi.org/10.1080/01614949308013915
Full terms and conditions of use: http://www.informaworld.com/terms-and-conditions-of-access.pdf
This article may be used for research, teaching and private study purposes. Any substantial orsystematic reproduction, re-distribution, re-selling, loan or sub-licensing, systematic supply ordistribution in any form to anyone is expressly forbidden.
The publisher does not give any warranty express or implied or make any representation that the contentswill be complete or accurate or up to date. The accuracy of any instructions, formulae and drug dosesshould be independently verified with primary sources. The publisher shall not be liable for any loss,actions, claims, proceedings, demand or costs or damages whatsoever or howsoever caused arising directlyor indirectly in connection with or arising out of the use of this material.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
458 TAYLOR
I. INTRODUCTION
This review covers the literature through 1991 on nitric oxide catalysis as applied to automobile exhaust systems. Attention is given to the three- way catalyst system which simultaneously promotes the reduction of nitro- gen oxides and the oxidation of carbon monoxide and hydrocarbons. These systems have been used on most passenger cars in the United States since 1982. Prior to 1980, emission control catalysts were oxidation catalysts, and reduction in exhaust nitric oxide was achieved using engine modifications (i.e., exhaust gas recirculation). This review focuses on catalytic control of NO, for gasoline-fueled vehicles (not diesels and alternate fuels) and pri- marily on developments reported since 1982. The term NO, refers to both N O and NOz. The reader is referred to an earlier publication by the author for a general review of automobile catalytic converters [l] and to a review by Egelhoff [ 2 ] on the nitric oxide literature through 1980. The recent literature on NO, reduction in lean exhaust is cited although such catalyst systems have not reached commercial application.
11. NO, AUTOMOBILE EMISSION REGULATIONS
In the United States exhaust emission standards (limits) have been set for passenger cars, light-duty trucks, and heavy-duty trucks (gasoline and diesel). The NO, regulations for passenger cars through 1993 are shown in Table 1. The regulations for the State of California for NO,, emissions have been more aggressive than the Federal regulations. Emissions are measured according to an EPA-prescribed driving cycle (Federal Test Procedure) and emissions sampling method. Exhaust emissions regulations have also been established for many parts of the world which have also led to the increased use of catalytic converters. These worldwide regulations are not summarized here.
The regulations and implementation schedule for 1993 through 2004 for passenger cars are given in Tables 2 and 3, respectively. NO, exhaust emission limits are reduced in several steps, phased in over several years. The Federal NO, standard goes from 1.0 g/mile (mi) to 0.4 g/mi starting with the 1994 model year vehicles for a minimum of 40% of the vehicles produced. The 0.4 g/mi regulation will apply to a minimum of 80% of the 1995 model year vehicles, and full production must be certified for the 0.4 g/mi NO, standard starting with the 1996 model year vehicles. The State of California’s regulations go to lower levels than the Federal limits and the implementation schedule is more aggressive. In fact, the California regulations call for a zero emission vehicle (ZEV) for a minimum of 2% of the 1998 model year vehicles. Zero emissions is not considered to be possible with an engine/emission control system, and electric vehicles are being pursued to meet this regulation.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 459
TABLE 1 US Passenger Car (light-duty vehicle) Emission
Standards for NO,, 1975 Through 1993"
Year Emissionb (g/mi)
1975 - 1976 3.1 (CA 2.0) 1977-1979 2.0 (CA 1.5) 1980 2.0 (CA 1.0) 1981-1982 1.0 (CA 0.7)' 1983-1988 1.0 (CA 0.4/0.7)d 1989- 1993 1.0 (CA 0.4)d *Measured according to the 1975 Federal test proce-
bCalifornia (CA) standards in parentheses. 'Federal standards optional for California for 1981, but
selection of 1981 Federal option required 1.0 C010.4 NO, for 1982.
d0.7 NO, standard optional in CA, but requires limited recall authority for 7 years/75,000 miles. Primary standard 0.4 gimi required on 50% of combined car and light-duty truck production in 1989, 90% in 1990-1993, and 100% thereafter.
dure.
TABLE 2 US Passenger Car Emission Standards and Categories for NO,, 1993 Through
2004 and Beyond
Certificationb NO, exhaust emission standards (g/mi) Emission category" 5 yr/50,000 mi 10 yr,100,000 mi 11 yr/120,000 mi
Fed. Tier 0 1 .0 Fed. Tier 1 0.4' 0.6d Fed. Tier 2 0.2' CA Tier 0 0.4 CA Tier 1 0.4 CA TLEV 0.4 0.6 CA LEV 0.2 0.3 CA ULEV 0.2 0.3 CA ZEV 0.0 0.0 0.0
bDifferent in-use exhaust standards may apply. =Diesel through 2003 MY - 1.0. dDiesel through 2003 MY - 1.25. 'Implementation based on need reported by EPA to Congress by June 1, 1997.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
460 TAYLOR
TABLE 3 US Passenger Car Exhaust Emission Standards Implementation Schedule for
NO,, 1993 Through 2004“
Emission standards compliance requirement by model year Emission categoryb 1993 1994 1995 1996-2003 2004
Fed. Tier 0 100% 60% max 20% max Fed. Tier 1 40% min 80% min 100% 100% Fed. Tier 2 100%‘ CA Tier 0 60% max CA Tier 1 40% min Undetermined CA TLEV Undetermined CA LEV Undetermined CA ULEV Undetermined
1998-2000 2001-2002 2003 2004
CA ZEV 2% min 5% min 10% min Undetermined
<‘Clean-fuel vehicle standards omitted from table. hAbbreviations: see note a to Table 2. ‘Tier 2 standards pending EPA study by 12/31/99
111. PROGRESS IN NO, CONTROL
The EPA “National Air Quality and Emissions Trends Report” reports that between 1980 and 1989, emissions of nitrogen oxides decreased overall by 5% [ 3 ] . All of this reduction was due to highway vehicles, which saw a 25% reduction in NO,, while the overall reduction in other sources was 0% [4]. For 1989 it is estimated that the highway vehicle contribution to the nationwide NO, inventory was 31%. Further decreases in the highway vehicle NO, inventory can be expected. Between 1975 and 1981 the nitric oxide emission standards for new light-duty vehicles decreased from 3.1 to 1.0 g/mi. The rate at which new vehicles are replacing older ones is low, and thus the rate at which vehicles meeting the 0.1 g/mi standard are ap- pearing is low. A consequence of this delay together with new stricter standards for NO, is that further reductions in the ambient concentration of NO, derived from vehicles can be expected beyond the end of the century. This projection allows for a 1.8% growth in vehicle miles traveled [ 5 ] . Except for the Los Angeles area, all areas in the United States are in compliance with the National Ambient Air Quality Standard. As of October 1991 only Pomona, California, was a nonattainment area for nitrogen diox- ide ambient levels, and the only exceedences of the standard were in this area. The National Ambient Air Quality Standard for NO2 is 0.053 ppm for an averaging time of 1 year.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 461
IV. EXHAUST COMPOSITION AND CONTROL STRATEGIES
Nitric oxide in engine exhaust is formed during combustion at high temperature and pressure by the reaction between nitrogen and oxygen in the intake air and by the oxidation of fuel-derived nitrogen-containing com- pounds. The most well-known chemical pathway for nitric oxide formation is the Zeldovich mechanism [6].
N, + O + N O + 0 N + 0,"O + 0 N + O H + N O + H
Assuming [O] is in equilibrium in the burnt gas regions of engines leads to a single equation for the rate of NO formation.
d"O1 ~ = 1.2 x 1014[0][N2]e~p[ -75.5 KcallRT]
dt
The Zeldovich mechanism dominates NO formation under most engine conditions. However, other pathways (involving superequilibrium 0 atoms, N,O intermediates [7], and fuel radicals such as CH [S]) can be significant, as suggested by detailed chemistry calculations in high pressure flames [9, 103. Lowering the combustion temperature by operating the engine fuel lean (excess air) or by recirculating exhaust gas through the engine is often used to lower the NO, emission from the engine, although these approaches are not as effective as the above equation implies.
The nitric oxide concentration in exhaust gas typically averages 1000 ppm, depending on driving conditions, or more correctly, on the engine combustion conditions such as temperature and aidfuel ratio. The lowest concentrations are observed immediately following start-up of the vehicle when the engine is cold. Highest concentrations occur under high-load conditions. Other important exhaust constituents are oxygen, carbon mon- oxide, hydrogen, and a mixture of hydrocarbons, as well as carbon dioxide, water vapor, and nitrogen. Typical exhaust concentrations of these gases are listed in Table 4.
The overall catalytic reactions promoted by the exhaust catalyst are listed in Table 5. The mechanism for catalytic removal of NO, from exhaust gas is based on the reduction of NO, to nitrogen. Optimum conditions for NO, reduction require that the overall stoichiometry of the exhaust be close to the stoichiometrically balanced composition since NO, reduction is re- tarded in the presence of excess oxygen. At the same time, oxidation of carbon monoxide and hydrocarbons is incomplete when the concentration of oxygen becomes limiting. The platinum-rhodium catalysts have the ca- pability to promote simultaneously the nearly complete reduction of NO, to nitrogen and the nearly complete oxidation of CO and hydrocarbons to CO, and H20 when the exhaust gas is close to its stoichiometrically balanced
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
462 TAYLOR
TABLE 4 Typical Concentrations of Exhaust Gas Constituents (average emissions over an
l T P test) ~ ~~ ~
HC 750 ppm" NO, 1050 ppm CO 0.68 vol% H, 0.23 vol%
CO, 13.5 vol% 0, 0.51 vol% H,O 12.5 vol%
OBased on C3.
TABLE 5 Overall Catalytic Reactions
~~
co + 1/2 0, + co, Hydrocarbons + 0, -+ H 2 0 + CO, NO, + CO and H, + N, + C 0 2 + H20
14.0 14.5 15.0 15.6
Simulated A/F Ratio
FIG. 1. Principle of 3-way exhaust emission control. Catalyst efficiencies measured in the laboratory with a steady feed stream composition at various sim- ulated aidfuel ratios. Catalyst: 0.042 wt% Pt/0.018 wt% Rh/alumina [ll].
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 463
loo
ae c
E 50 .- P C 0 0
0 14.0 14.5 15.0
Mean A/F Ratio 15.5
FIG. 2 . CO and NO conversions measured over a 0.042 wt% Pti0.018 wt% Rhialumina catalyst at various cycling amplitudes [ 111.
composition. The name “three-way catalyst” is based on the fact that the catalyst promotes the removal of all 3 pollutants. This property of a typical rhodium-containing catalyst is illustrated in Fig. 1 [11]. Best simultaneous conversion of CO, hydrocarbons, and NO, requires tight control of the air (A)/fuel (F) ratio (e.g., ? 0.3 A/F). Excursions to fuel-lean (net oxidizing) conditions result in a decrease in NO, reduction activity over the catalyst. Engine control systems which include an electronic control module and an exhaust oxygen sensor have been developed to control the aidfuel ratio and thereby keep the exhaust at the optimum composition for the catalysis (e.g., Refs. 12-14 and references therein). The oxygen sensor indicates whether excess oxygen is present in the exhaust. Because of inherent time delays in the A/F correction, the aidfuel ratio typically oscillates very rapidly about the stoichiometrically balanced composition, depending on vehicle speed. A typical oscillation frequency is 1 cycle per second. Minimizing the amplitude of these oscillations (e.g., A/F stays within t 0 . 3 A/F of the balanced composition) increases the effectiveness of the 3-way catalyst. Figure 2 illustrates how oscillations in the exhaust aidfuel ratio can result in a lowering of the peak conversion efficiency but at the same time broaden the control window [ll], so that the conversion remains high at A/F further away from the stoichiometrically balanced composition. Similar results have been reported for catalyst tests with engine exhaust [15].
A flow-through ceramic monolith type catalytic converter is shown in
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
464 TAYLOR
INSULATION COVER INSULATION
MAT \
\ CATALYST LOWER SHELL
FIG. 3 . Ceramic monolith catalytic converter
Fig. 3. The ceramic monolith is composed of corderite (2Mg0.2AI2O3 -5Si02) [16, 171. A typical honeycomb structure has 400 axial channel openings per in.* frontal area, and a monolith volume of 170 in.3 for a dual monolith converter and 85-110 in.3 for a single monolith converter. Metal foil mon- oliths are used in some applications. One example is small converters located close to the engine [18, 191. A thin washcoat of alumina (30-50 pm thick) is applied to the surface of the corderite or metal foil, which becomes the support for the noble metal catalyst. The operating temperature of the catalytic converter is determined significantly by the location of the catalytic converter on the vehicle. A temperature range of 350-650°C is within the desired range for the catalysis and below the temperature range where thermal deterioration of the catalyst or support becomes significant. Be- cause of the increasing need to control the CO and hydrocarbons emitted immediately following start-up of the vehicle, methods for quickly bringing the catalyst up to operating temperature are being explored. One method is the small converter located up close to the engine; another method being explored is to externally heat the converter [20, 211.
V. CATALYST COMPOSITION
The noble metal most closely associated with the catalytic reduction of NO, in exhaust is rhodium. Rhodium is the primary constituent in all 3-way catalysts for controlling NO, emissions. Rhodium has high activity for selectively reducing NO, to nitrogen with low ammonia formation. Rhodium also makes a significant contribution to CO oxidation. While platinum and palladium also catalyze simultaneous CO, hydrocarbon, and
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 465
NO, conversion under typical 3-way exhaust conditions, these metals are not as effective as rhodium. While replacing rhodium and/or platinum with less expensive metals such as palladium or copper might lower the cost of the catalysts, future stricter emissions regulations for NO, in the United States make wide adoption of rhodium replacements unlikely in the near future. The demand for rhodium for the autocatalyst (all vehicle exhaust control applications) reached 84% (300,000 oz t) (1 oz t = 31.103 g) of the total Western-world demand for rhodium in 1990 [22]. The increasing ap- plication of the rhodium containing 3-way catalytic converter worldwide and the use of rhodium in autocatalysts in excess of the naturally occurring noble metal ratio in South African mines has led to this high demand. Rhodium use in the catalytic converter is typically in the range of 0.005- 0.009 oz t; however, even higher amounts have been used on some vehicles.
Platinum is used in 3-way catalytic converters at loadings typically in the range 0.05-0.08 oz t. Studies of NO, control generally focus on rhodium catalysis. However, it is not possible to ascribe entirely independent func- tions to the two noble metals. Platinum is less expensive than rhodium and is more abundant. The demand for platinum for the autocatalyst in 1990 was 36% of the total Western-world demand for platinum [22]. Palladium use in autocatalysts has declined since the introduction of the 3-way catalytic converter. In 1990 the demand for palladium for autocatalysts was only 7% of the Western-world demand for palladium [22].
The noble metals are applied to the stabilized alumina coated monolith as a dilute solution of a salt of the noble metal. Precise control of noble metal uptake is necessary to ensure that strict converter performance re- quirements are met and that no metal is wasted.
In addition to the noble metals, autocatalysts contain several base metal additives which contribute significantly to catalyst performance and durability. The ceria content of current 3-way catalysts is 10-30 wt% (as cerium) of the washcoat. (The term “washcoat” refers to all the constituents placed on the corderite monolith.) The ceria has been shown to have mul- tiple functions [23]. One function is its ability to store oxygen (presumably by oxidation of the ceria) derived from NO, decomposition during fuel lean aidfuel ratios (net oxidizing) excursions and thereby enhance NO, conver- sion to N, [24]. Stored oxygen is then available for reaction with CO and hydrocarbons during subsequent fuel-rich aidfuel ratio excursions. Ceria has been shown to enhance the decomposition of NO by extending the time before the noble metal catalyst is deactivated by the accumulation of surface oxygen derived from NO decomposition [25,26]. That is, Rh/CeO, is deac- tivated more slowly than is Rh/AI2O3 during NO decomposition, probably due to oxygen spillover from the noble metal to the reduced ceria.
Other laboratory studies have demonstrated how ceria favorably alters the reaction kinetics of CO oxidation and NO, reduction over the ceria- containing rhodium catalyst [26, 271. This impact of ceria on the catalysis is probably one of the most significant functions of ceria. Ceria addition to
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
466 TAYLOR
an alumina-supported rhodium catalyst was shown to enhance NO reduction activity at low temperatures by decreasing the apparent activation energy for the reaction of CO with NO and by shifting to positive-order the de- pendence of the rate on NO partial pressure [27]. These results are shown in Fig. 4 and Table 6. Enhancement of catalyst performance at low tem- perature is needed in order to decrease the emissions immediately following start-up of the vehicle. Recently Nunan et al. [28] showed how, for a Pt/ ceria/alumina catalyst, a reduction in the particle size of ceria from 270 to 120 8, lowered the temperature for 25% NO reduction by 80°C in a
I .? 1.8 1.9 2.0 2.1
lOOW(K')
FIG. 4. Arrhenius plots for CO, production rates over 0.014 wt% Rh/A1,0, containing variable levels of Ce in a reactant mixture containing 1 vol% CO and 0.5 vol% NO [27].
TABLE 6 Apparent Activation Energy as a
Function of Ce Loading I271
Ce loading Activation energy (wt%) (kcaVmo1)
0 28, 38 0.5 29, 38 2 19 9 18
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
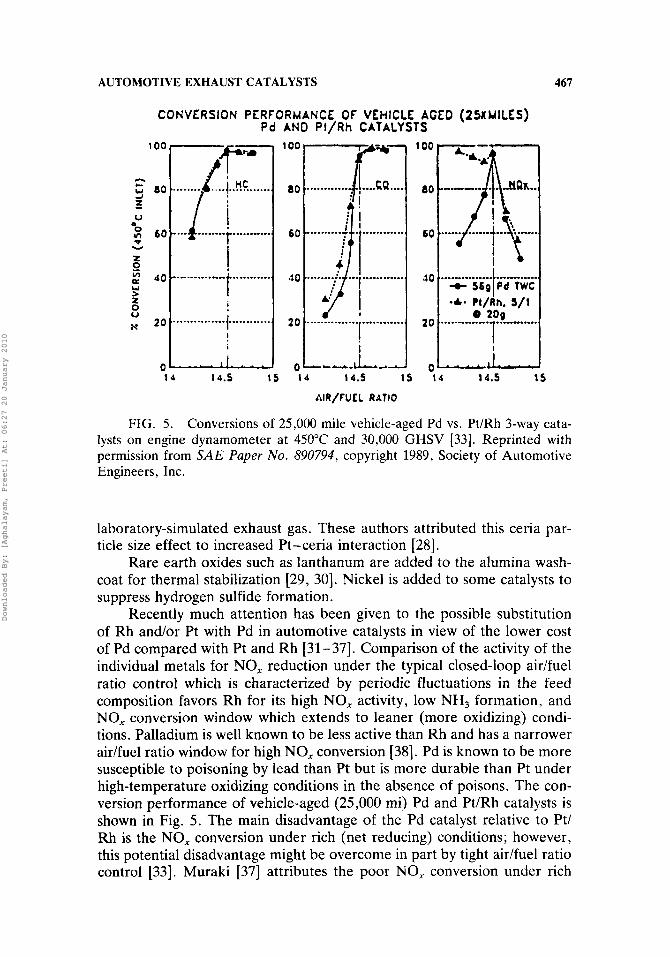
AUTOMOTIVE EXHAUST CATALYSTS 467
CONVERSION PERFORMANCE OF VEHICLE AGED (2% MILES) Pd AND Pt/Rh CATALYSTS
1
1 4 14.5 15 1 4 14.5 15
AlR/FUCL RATIO
14 14.5 1s
FIG. 5. Conversions of 25,000 mile vehicle-aged Pd vs. Pt/Rh 3-way cata- lysts on engine dynamometer at 450°C and 30,000 GHSV [33]. Reprinted with permission from SA E Paper No. 890794, copyright 1989, Society of Automotive Engineers, Inc.
laboratory-simulated exhaust gas. These authors attributed this ceria par- ticle size effect to increased Pt-ceria interaction [28].
Rare earth oxides such as lanthanum are added to the alumina wash- coat for thermal stabilization [29, 301. Nickel is added to some catalysts to suppress hydrogen sulfide formation.
Recently much attention has been given to the possible substitution of Rh and/or Pt with Pd in automotive catalysts in view of the lower cost of Pd compared with Pt and Rh [31-371. Comparison of the activity of the individual metals for NO, reduction under the typical closed-loop aidfuel ratio control which is characterized by periodic fluctuations in the feed composition favors Rh for its high NO, activity, low NH3 formation, and NO, conversion window which extends to leaner (more oxidizing) condi- tions. Palladium is well known to be less active than Rh and has a narrower aidfuel ratio window for high NO, conversion [38]. Pd is known to be more susceptible to poisoning by lead than Pt but is more durable than Pt under high-temperature oxidizing conditions in the absence of poisons. The con- version performance of vehicle-aged (25,000 mi) Pd and Pt/Rh catalysts is shown in Fig. 5. The main disadvantage of the Pd catalyst relative to Pt/ Rh is the NO, conversion under rich (net reducing) conditions; however, this potential disadvantage might be overcome in part by tight aidfuel ratio control [33]. Muraki [37] attributes the poor NO, conversion under rich
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
468 TAYLOR
conditions to poisoning by hydrocarbons but cites that NO, reduction ac- tivity is improved by the addition of La to the catalyst. Others have ex- amined palladium- tungsten [39] and platinum-molybdenum [40] with the objective decreasing Rh use.
VI. CATALYTIC REACTIONS
The catalytic reactions which result in the conversion of NO, to N, can in general be described as nitric oxide decomposition followed by re- moval of the oxygen from the catalyst surface via reaction with a reducing agent (e.g., CO).
Numerous studies have focused on the kinetics and mechanism of the NO-CO reaction and the adsorption and decomposition processes which participate in N2 formation. Application of various surface chemistry tech- niques to studies on polycrystalline and single-crystal surfaces has provided understanding of the elementary surface processes on an atomic scale. These techniques mostly require ultrahigh vacuum and are most suited for studying well-characterized low surface area metals, whereas the active metal on practical catalysts is dispersed on high surface area oxide supports.
The reaction mechanism of the NO-CO reaction over Rh can be rep- resented by the following elementary processes.
co -+ co, NO -+ NO,
NO, -+ N, + 0, Irreversible (6) NO, + N, --f N, + 0, 2N, -+ N, Irreversible
CO, + 0, -+ CO, NO, + N, + N 2 0
Irreversible
Irreversible
Irreversible
N 2 0 +- 0, + N, Irreversible (11) Evidence for stable molecular adsorption of N O on Rh is provided by infrared studies of NO adsorption on alumina and silica supported Rh [41- 431. Evidence for the dissociation of N O on Rh to form N, and 0, at elevated temperatures is provided by temperature-programmed desorption studies [44-471. Single reaction studies-for example, CO + N O (e.g., Refs. 43, 48-50) and N O reduction in competition with CO + 0,-have been reported (e.g., Ref. 51). Two different reaction pathways have been proposed for N, formation from the NO-CO reaction. These are N atom recombination [Eq. (S)] and the reaction of adsorbed N atoms with adsorbed NO (Eq. (97)]. The catalyst surface structure and reaction conditions which favor each pathway are discussed below.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 469
A . Structure Sensitivity of Rhodium
Studies have been reported which indicate that the kinetics of the NO- CO reaction over Rh are sensitive to changes in the surface morphology of the Rh. A comparative study by Oh et al. [48] of the kinetics of the NO- CO reaction over single-crystal Rh( 11 1) and Rh( loo), and over supported Rh/A1,03 catalysts revealed different kinetic behavior over the single-crystal and supported catalysts. Figure 6 compares the rates of the NO-CO reaction over Rh(ll1) and supported Rh/AI2O3 catalysts at Pco = PNo = 0.01 atm. The Rh single crystal exhibited substantially lower apparent activation ener- gies and higher specific rates than those over the supported Rh/A120, cat- alyst [48].
A mathematical model of the reaction, which accounts for the indi- vidual elementary reaction steps and which makes use of parameter values determined using bulk Rh metal, gives a good fit for the rate data for Rh(ll1) but not for the supported Rh catalyst [52]. A comparison of the kinetic parameters for the elementary reaction steps allows one to conclude that at 500 K NO dissociation and CO desorption are fast. The Rh(ll1) surface is predominately covered with nitrogen atoms, and nitrogen de-
Pco = PNo = 0.01 atm
R h ( 1 1 1 ) R h / A I 2 0 3 -.. 0 ----- Mode I . . . . . . . . .
.- vl
1 . 4 1.6 1 . 8 2 .o 1OOO/T ( K - l )
FIG. 6. Comparison of the specific rates of the NO-CO reaction measured over Rh(ll1) and Rh/A1203 at Pco = P,, = 0.01 atm. The predictions of the surface chemistry model follow closely the rate data for Rh(ll1) [48].
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
470 TAYLOR
sorption is the rate-determining step. Nitrogen atoms can be removed from the surface by the reaction of nitrogen atoms with adsorbed NO [Eq. (7)] and by nitrogen atom recombination [Eq. (S)]. At low temperature the reaction of adsorbed nitrogen atoms with adsorbed NO contributes to ni- trogen atom removal. At higher temperature nitrogen atom recombination determines the NO-CO reaction rate. These model calculations suggest that some of the elementary steps may proceed at different rates on supported catalysts; likely candidates include the NO dissociation and N2 desorption steps.
Further experiments aimed at clarifying the origins of this structure sensitivity revealed only a moderate effect of crystallographic orientation of the surface when kinetics of the NO-CO reaction were compared using Rh( 11 1) and Rh( 100) single crystals [53]. Comparisons carried out using a series of supported Rh catalysts revealed that the support material (Rh/ SOz , Rh/y-Al,O, or Rh/a-Al,O,) had only a minor effect on the kinetics of the NO-CO reaction [54]. The major factor influencing the kinetics of the NO-CO reaction with this series of Rh catalysts was metal particle size. For example, at 230°C a 45-fold increase in the specific rate of the NO-CO reaction was observed for an increase in the Rh particle size from <10 to 676 A [54]. See Table 7. Arrhenius plots for the rate data showed a dis- continuity with two distinct apparent activation energies, one for the lower- temperature regime and a second for the high-temperature regime. This increase in activation energy with temperature was attributed to a shift in the rate-controlling step. Kinetic analysis by Oh [54] suggests that the rate- controlling step may shift from the low-temperature N2/N20 formation path [Eqs. (7) and (lo)] to the high-temperature Nz formation path [Eq. (S)] with an increase in the catalyst temperature. For the large Rh particle sizes the Arrhenius plots exhibit a single apparent activation energy characteristic
TABLE 7 Turnover Number and Activation Energy for NO-CO Reaction as a Function of
Rh Particle Sizes [54]
Turnover number Activation
energy code size) 215°C 230°C (kcal/mol)
(s - '> Dispersion Sample (particle
C 100% (<10A) - 0.0166" 30, 39 D 26.4% (44 A) 0.0196 0.0535 32, 37 E 9.5% (121 A) 0.0320 0.0790 27, 36 F 4.0% (288 A) 0.123 0.386 32 G 1.7% (676 A) 0.253 0.741 33
"Calculated assuming 100% Rh dispersion.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 471
‘ A FILM
of those for the Rh(ll1) single crystal over the same temperature range used for the supported catalyst experiments [48, 531.
Oh et al. [54] offer the following explanation for these observations based on the NO temperature-programmed desorption results of Altman and Gorte [55]. Altman and Gorte [55] showed that for large Rh particles (Rh film), most N2 desorbs in a low-temperature peak near 190°C and only a small amount of N2 desorbs above 280°C (see Fig. 7 ) . They attribute the low-temperature N, peak to the NO, + N, reaction [Eq. (7)]. The low- temperature N2 desorption path (lower activation energy) would then dom- inate the kinetics of the NO-CO reaction and the reaction would exhibit a single activation energy. Altman and Gorte [55] further show that the rel- ative intensity of the low- and high-temperature N2 peaks change with Rh particle size such that the high-temperature N, desorption peak which is attributed to N, recombination [Eq. (7)] increases at the expense of the low-temperature N2 desorption peak as the Rh particle size is decreased. The structure sensitivity of the NO-CO reaction is then attributed to changes in the N2 desorption features with particle size. The slow removal of N2 from the small Rh particles at low temperatures results from the suppression of the NO, + N, reaction step [Eq. (8)], making the reaction rate lower for the small particles than for the large particles. One difference between Oh et al.’s work and Altman and Gorte’s work that could influence this
D <
n 2.6 nm
300 400 500 600 700 8 1 (K)
FIG, 7. Temperature-programmed desorption of N2 desorption as a func- tion of NO coverage for 2.6-nm Rh particles and a Rh film [%I. The top N2 TPD spectrum for the film and the 2.6 nm particles is for saturation coverage of NO.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
472 TAYLOR
comparison is that Altman and Gorte’s work was done in the absence of co.
B . N 2 0 Formation
N,O is a very minor exhaust constituent but is of interest because of its contribution to the “greenhouse effect.” Prigent and DeSoete [56] mon- itored N 2 0 in the exhaust gas from internal combustion engines and found the ratio of N 2 0 to N O concentrations to be less than 1%. The Pt-Rh catalyst forms some N 2 0 during NO reduction at low temperatures (just after light-off), whereas N2 formation dominates at higher temperatures [57-601. The catalyzed formation of N 2 0 and the reaction of N,O with CO to fo’rm N2 are important subreactions in the NO-CO reaction and have been included in the NO-CO reaction list [Eqs. (10)-(ll)].
In laboratory studies using an alumina-supported Rh catalyst, McCabe and Wong (601 determined the steady-state kinetics of the CO-N,O reaction between 500 and 700 K , and at CO and N 2 0 partial pressures between 0.6 and 7 torr. Comparison between the N20-CO kinetics and the kinetics of the NO-CO and CO-O2 reactions at 475 K showed that the N,O-CO reaction has a much lower rate. The ratio of the turnover frequencies was estimated to be 1 (N20-CO) vs. 7.1 x lo2 (NO-CO) and 4.4 X 10’ (CO-0,). They attributed this slow rate to a reduction mechanism involving CO reacting with N 2 0 dissociation products and to the slow dissociative adsorption of N 2 0 compared with the rates of dissociative O2 or N O adsorption. The apparent activation energy was reported to be 40 ~f: 2 kcal/gmol, and re- action orders were - 1 in CO pressure and 0.65 in N 2 0 pressure.
C. Interactions
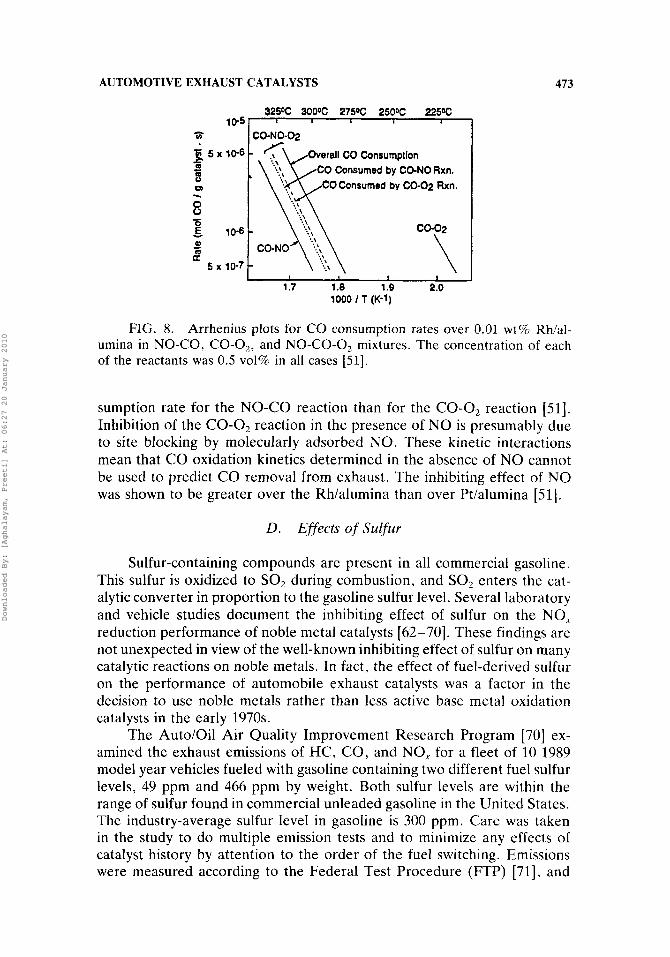
The complex composition of exhaust gas results in catalytic effects which are difficult to assess in detail and can be missed in laboratory studies using simplified feeds. One example is the effect of sulfur, which is discussed below. Another example is the influence of well-known catalyst poisons such as lead and phosphorus. The various reactive exhaust gas constituents can also influence one another. A strongly adsorbed species may retard the reaction of a more reactive species and delay the light-off temperature (the lowest temperature at which catalytic activity is observed; e.g., 25% con- version) until the desorption temperature of the strongly adsorbed species is reached. For example, the CO-O2 and NO-CO reactions, which are often studied separately over the exhaust catalyst, occur simultaneously in the presence of all the other exhaust constituents. Kinetic experiments using Pt/alumina [61] and Rh/alumina [51] catalysts have illustrated that NO has a strong inhibiting effect on the rate of the CO-O2 reaction until the extent of the NO-CO reaction becomes significant. Figure 8 shows that the CO consumption rate for the CO-NO-02 reaction is more like the CO con-
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 473
325OC 300OC 2750C 250DC 225% 10-5 I I I I I
CQNO-02 - Y)
CO Consumed by CO-NO Rxn. CO Consumed by CO-02 Rxn. V
cn z
- 8 '. \
,
'. \ 0
0,
g 10-6-
5 x 1 0 - 7 - I 4
1.7 1.8 1 .B 2.0 1000 I T (K-1)
FIG. 8. Arrhenius plots for CO consumption rates over 0.01 wt% Rhial- umina in NO-CO, CO-0,, and NO-CO-0, mixtures. The concentration of each of the reactants was 0.5 vol% in all cases [51].
sumption rate for the NO-CO reaction than for the CO-0, reaction [51]. Inhibition of the CO-0, reaction in the presence of NO is presumably due to site blocking by molecularly adsorbed NO. These kinetic interactions mean that CO oxidation kinetics determined in the absence of N O cannot be used to predict CO removal from exhaust. The inhibiting effect of N O was shown to be greater over the Rh/alumina than over Pt/alumina [51].
D. Effects of Sulfur
Sulfur-containing compounds are present in all commercial gasoline. This sulfur is oxidized to SO, during combustion, and SO, enters the cat- alytic converter in proportion to the gasoline sulfur level. Several laboratory and vehicle studies document the inhibiting effect of sulfur on the NO, reduction performance of noble metal catalysts [62-701. These findings are not unexpected in view of the well-known inhibiting effect of sulfur on many catalytic reactions on noble metals. In fact, the effect of fuel-derived sulfur on the performance of automobile exhaust catalysts was a factor in the decision to use noble metals rather than less active base metal oxidation catalysts in the early 1970s.
The Auto/Oil Air Quality Improvement Research Program [70] ex- amined the exhaust emissions of HC, CO, and NO, for a fleet of 10 1989 model year vehicles fueled with gasoline containing two different fuel sulfur levels, 49 ppm and 466 ppm by weight. Both sulfur levels are within the range of sulfur found in commercial unleaded gasoline in the United States. The industry-average sulfur level in gasoline is 300 ppm. Care was taken in the study to do multiple emission tests and to minimize any effects of catalyst history by attention to the order of the fuel switching. Emissions were measured according to the Federal Test Procedure (FTP) [71], and
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
474 TAYLOR
10 current model vehicles all equipped with 3-way catalysts were included in the study. The results of the study are listed in Table 8. Overall NO, emissions were 9% lower with the low-sulfur fuel. The lower NO, emissions were attributed to greater catalyst activity with the low-sulfur fuel. The mass emissions of NO, leaving the engine were reported to be unaffected by the fuel sulfur level in this study. The effect of fuel sulfur level on NO, conversion over the catalyst appeared to be reversible when the sulfur level was changed. This result is important since it suggests that a decrease in fuel sulfur levels could achieve reduction in NO, emissions from vehicles currently in use. Vehicle tests as reported for the Auto/Oil Program are very realistic results since all normal exhaust constituents and exhaust con- ditions are incorporated in the test. Any recommendation to lower fuel sulfur levels to obtain emission improvements is appropriately based on such vehicle tests.
Earlier laboratory studies examined the effects of sulfur on the con- version efficiency of 3-way catalysts [62-69]. Such studies offer more control over experimental parameters and possibly more understanding but are less definitive for use in the prediction of real-world impact. Monroe et al. [68] have reported that both the maximum conversion obtained with the 3-way catalysts and the low-temperature performance were deteriorated in the presence of SOz. See Fig. I). Differences were observed between pellet- and monolith-type catalysts in the reversibility of the effect which was at- tributed to the retention of sulfur by the pellets. The monolith-type catalyst rapidly recovered its lost activity when sulfur was removed from the inlet gas to the catalyst, whereas the pellet-type catalyst recovered only a portion of its lost activity. This difference was attributed to the ease of removal of sulfur from the monolith-type catalyst, which has less support volume. These observations pointed to the presence of ceria in the catalyst support as a factor in the ease of activity reversal since Ce-free pellet-type catalysts recovered lost activity quickly when sulfur was removed compared with ceria-containing pellet-type catalysts. The sulfur storage capacity of the catalyst support was enhanced by the presence of ceria and prolonged the
TABLE 8 Effects of Fuel Sulfur on Mass Exhaust Emissions: Current Vehicle Fleet [70]
High-S fuel Low-S fuel Constituent (gimi) ( g W Change (%)
HC 0.236 0.196 - 16.1 co 2.806 2.444 - 12.9 NO, 0.542 0.493 - 9.0
Nores: Mass emissions values are fleet averages. All changes in mass emissions are significant at the 9 5 8 confidence level. High-sulfur fuel: 446 ppm sulfur; low-sulfur fuel: 49 pprn sulfur.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS
Pelleted
0 180 360 ntx (MINUTES)
z 0 tn - F z N 8
Monolithic
loo l------
40 0 iao 3
TIME (MINUTES)
475
0
FIG. 9. The effect of SOz concentration on the conversion of NO over thermally aged (lOOO"C, cycled at 0.05 Hz between 5% O,/N, and 5% Hz/Nz for 5 h) pellet- and monolith-type 3-way Pt-Rhlalumina catalysts. Conversion was measured in cycled stoichiometric exhaust at 500°C. Sulfur dioxide was added to the feed after 60 min and remained in the feed for 180 min [68].
detrimental impact of sulfur on catalyst activity. Differences were also re- ported in the response of the noble metals to the reversibility of their sulfur- induced activity loss. Pt and Rh rapidly recovered all lost activity when sulfur was removed from the feed gas whereas Pd did not.
Laboratory studies have shown that the inhibiting effect of sulfur on the activity of 3-way catalysts is greater under net reducing conditions. Beck et al. [69] have recently reported how sulfur storage in the catalyst depends on the exhaust stoichiometry. Experiments were conducted in which cat- alysts were treated in a laboratory reactor under conditions closely simu- lating vehicle exhaust, and the amount and oxidation state of the sulfur on the catalyst was characterized using an ultrahigh vacuum instrument. Sulfur originating as SO2 is stored in the catalyst support as both sulfates and sulfites. Some stored sulfur was removed by exposure to a feed which did not contain any sulfur while the simulated exhaust gas was cycled between net reducing and net oxidizing conditions to simulate the aidfuel ratio fluc- tuations of an operating 3-way catalyst. The sulfur stored in the support was easily removed by treatment in hydrogen, particularly above 700°C. Treatment in a net oxidizing feed was not effective in removing sulfur from the catalyst support. The noble metal showed the opposite effect; sulfur was retained under reducing conditions and removed under net oxidizing conditions. Studies conducted with a Pt foil showed that SO2 under reducing conditions can dissociate on Pt at elevated temperatures to form adsorbed sulfur and adsorbed oxygen. This adsorbed elemental sulfur was easily removed by oxidation in O2 to form SOz and SO3. The interaction of sulfur
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
476 TAYLOR
with platinum and palladium is documented in the surface science literature [72-761.
E. Lean NO, Reduction
Recently, increased attention has been given to the catalytic reduction of nitric oxide under lean (net oxidizing) conditions. Such lean catalysts, if successful, could find application for NO, emission control for diesel-fueled engines and for lean-burn gasoline-fueled engines, which provide improved fuel economy but which present new challenges for emission control. New regulations for trucks equipped with diesel engines will require lower NO, emissions. Exhaust after treatment of NO, is being pursued for both of these applications.
The diesel and lean-burn engines operate at aidfuel ratios of greater than 17:l. While NO, emissions from the engine are considerably lower at these high aidfuel ratios, typical 3-way catalysts do not promote NO, re- duction, except in a narrow temperature window in the vicinity of catalyst light-off. Catalysts are being explored which have a wider temperature range for NO, reduction and exhibit higher lean NO, conversion than the Pt-Rh 3-way catalyst [77-871.
Held et al. [77] reported on the performance of a number of zeolites exchanged with various elements for reducing NO, in net oxidizing exhaust gas. In laboratory experiments using synthetic exhaust gas, these researchers reported 37% NO, conversion over copper-exchanged ZSMS using ethylene as the reducing gas at 350°C and in the presence of water vapor. Held et al. [77] further reported NO, conversion results for a lean-burn spark- ignition engine with the Cu-ZSMS catalyst, a catalyst temperature of 400°C, and 15,000 h- space velocity. The reduction of NO, was dependent on the hydrocarbon-to-NO, ratio. In steady-state tests, the highest conversion was found for the highest HCINO, ratios. For car tests conducted under transient driving conditions, Held et al. [77] reported 15% NO, conversion on the cold-start portion of the FTP (bag 1 of the Federal Test Procedure) and 30% NO, conversion on the hot start (bag 3). Unfavorable driving condi- tions for NO, reduction were cold start when the catalyst temperature was below the needed 350°C and during acceleration when the HC/NO, ratio was low. Other potential negatives cited were high space velocity and high exhaust gas temperatures ( >600"C) which accelerate catalyst deterioration. NO, reduction in diesel engine exhaust was hampered by the low concen- tration of reactive hydrocarbons relative to NO,. NO, conversion over the Cu-ZSM5 catalyst did not exceed 15% even when 4700 ppm ethylene was added to the diesel exhaust [77].
In the same time frame as Held's vehicle studies, several laboratory studies explored lean NO,r reduction in detail, namely, effects of catalyst support type and exchange level for various elements [78-851, effect of hydrocarbon type [78, 861, influence of SO2 [87], space velocity effects [81],
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 477
effect of temperature [78, 79, 811, effect of HC/NO, ratio [79], and effect of oxygen concentration [80, 861. Hamada et al. [78] observed that HC conversion was incomplete and that CO was formed via oxidation of the HC at the temperature of maximum NO, activity. No detailed mechanistic studies of NO, reduction by hydrocarbons over these zeolite-type catalysts have been reported.
At the time of this writing, it appears that the successful implemen- tation of a catalyst to reduce NO, in lean exhaust gas will require both temperature control and desirable HC-NO,-0, concentration ratios in order to have acceptable catalyst activity and durability. Catalysts beyond Cu- ZSMS need to be explored in view of the well-known instability of Cu zeolites under net reducing conditions and the low thermal stability of zeolites in comparison with the current stabilized alumina supports. While silica-alumina supports have been proposed [79, 831 , no vehicle emissions data with these alternative catalysts have been reported to date.
VII. CONCLUDING REMARKS
The catalyst technology described in this review is not a complete prescription for meeting future exhaust emission requirements. While cat- alysts will continue to be the focal point of exhaust emission control, greater attention will be given to the operating modes of the catalyst, especially the HC and CO emissions immediately following start-up of the vehicle. Adjusting the converter location or adding a second converter close to the engine makes greater use of the heat form the engine to warm the catalyst quickly. Also, converters are being modified so they can be heated exter- nally immediately following (or even before) start-up of the vehicle. Meeting the nitric oxide emissions regulations, however, cannot be achieved solely by these approaches aimed at reducing cold-start emissions since NO, emis- sions are highest with a fully warmed-up engine. Reducing engine-out NO, emissions should be pursued together with improved engine control systems and catalysts.
VIII. SUMMARY
Noble metal catalysts are used to reduce nitrogen oxides to nitrogen in automobile exhaust in order to meet demanding emission control re- quirements. Catalysts generally contain platinum and rhodium together with cerium oxide on alumina supports. The catalyst composition has been op- timized to meet the need for high activity and durability in the demanding exhaust environment. Engine control systems have been developed which keep the exhaust composition close to the stoichiometrically balanced com- position for simultaneous reduction of nitrogen oxide and oxidation of car-
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
478 TAYLOR
bon monoxide and hydrocarbons. This review covers the literature through 1991 on nitric oxide catalysis for automobile exhaust emission control.
ACKNOWLEDGMENTS
The author thanks Se Oh, David Monroe, and Marvin Jackson for their contributions to the preparation of this manuscript.
REFERENCES
1.
2.
3. 4. 5. 6. 7. 8.
9.
10. 11.
12.
13.
14. 15.
16. 17.
18. 19.
20.
21.
K. C . Taylor, Automobile catalytic converters, in Catalysis: Science and Tech- nology (J. R . Anderson, M. Boudart, eds.), Vol. 5, Springer-Verlag, Berlin, 1984. W. G . Egelhoff, Jr., in The Chemical Physics of Solid Surfaces and Hetero- geneous Catalysis (D. A. King and D . P. Woodruff, eds.), Vol. 4, p. 397, Elsevier, New York, 1982. EPA, “National Air Quality and Emissions Trends Report, 1989.” EPA, “National Air Pollution Emission Estimates 1940-1989.” K. C. Taylor, CHEMTECH, 20, 551 (1990). Y. B. Zeldovich, Acta Physiochem. URSS, 21, 577, 656 (1946). J. Wolfrum, Chemie Ingenieur Technik, 44 (1972). C. P. Fenimore, Thirteenth Symp. (lnt.) on Combustion, The Combustion Institute, 1971, p. 373. M. C . Drake, J , W. Ratcliffe, R. J. Blint, C . D. Carter, and N. M. Lauren- deau, Twenty- Third Symp. (Int.) on Combustion, The Combustion Institute, 1990, p. 387. M. C. Drake and R. J. Blint, Combustion Sci. Technol., 75, 261 (1991). J . C. Schlatter, R. M. Sinkevitch, and P. J . Mitchell, Ind. Eng. Chem. Prod. Res. Dev., 22, 51 (1983). R. P. Canale, S. R . Winegarden, C. R. Carlson, and D. L. Miles, Society of Automotive Engineers Publ. No. 780205. R. A. Grimm, R. J. Bremer, and S. P. Stonestreet, Society of Automotive Engineers Publ. No. 800053. L. L. Hegedus and J . J . Gumbleton, CHEMTECH, 10, 630 (1980). Y. Kaneko, H. Kobayashi, R . Komagome, 0. Hirako, and 0. Nakeyama, Society of Automotive Engineers Publ. No. 780607. S . T. Gulati, Society of Automotive Engineers Publ. No. 85013. S. T. Gulati and R . D . Sweet, Society of Automotive Engineers Publ. No. 900268. D. I . P. Oser, Society of Automotive Engineers Publ. No. 880319. S. Pelters, F. W. Kaiser, and M. Wolfgand, Society of Automotive Engineers Publ. No. 890488. W. A. Whittenberger and J. E. Kubsh, Society of Automotive Engineers Publ. No. Y00503. W. A. Whittenberger and J. E. Kubsh, Society of Automotive Engineers Publ. No. 910613.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 479
22. 23. 24.
25.
26. 27. 28.
29.
30. 31,
32. 33.
34.
35.
36. 37. 38. 39.
40.
41, 42. 43. 44. 45. 46. 47. 48.
49. 50. 51. 52. 53.
54. 55.
Platinum 1991, Johnson Matthey, London, 1991. B. Harrison, A. F. Diwell, and C. Hallett, Platinum Metals Rev., 32, 73 (1988). H. S. Gandhi, A. G. Piken, M. Shelef, and R. G. Delosh, Society of Auto- motive Engineers Publ. No. 760201. B. K. Cho and C. J. Stock, Transient Study of NO Decomposition over Supported Noble Metal Catalysts, General Motors Research Publ. GMR-5497, August, 1986, presented at the AIChE Meeting, Miami Beach, FL, November 1986. P. Loof, B. Kasemo, S. Anderson, and A. Frestad, J . Cutul., 130,181 (1991). S. H. Oh, J. Cutul., 224, 477 (1990). J. G. Nunan, H. J . Robota, M. J. Cohn, and S. H. Bradley, J. Cutul., 133, 309 (1992). H. Schaper, E. B. M. Doesburg, and L. L. Van Reijen, Applied Cutul., 7, 211 (1983). F. Oudet, P. Courtine, and A. Vejux, J . Cutul., 114, 112 (1988). J. C. Summers, W. B. Williamson, and M. G. Henk, Society ofAutomotive Engineers Publ. No. 880281. H. Muraki, K. Yokota, and Y. Fujitani, Appl. Cutul., 48, 93 (1989). J. C. Summers, J. J. White, and W. B. Williamson, Society of Automotive Engineers Publ. No. 890794. J. C. Summers, W. B. Williamson, and J. A. Scaparo, Society ofAutomotive Engineers Publ. No. 900495. R. J. Brisley, R. D. O’Sullivan, and A. J. J . Wilkins, Society ofAutomotive Engineers @bl. ,No. 920175. G. S. Sims, Society of Automotive Engineers Publ. No. 912369. H. Muraki, Society of Automotive Engineers Publ. No. 910842. K. C. Taylor and J. C. Schlatter, J. Cutul., 63, 53 (1980). K. M. Adams and H. S. Gandhi, Ind. Eng. Chem. Prod. Res. Dev.,22, 207 (1983). H. S. Gandhi, H. C. Yao, and H. K. Stepien, ACS Symposium Series No. 278, 1982, p. 143. H. Arai and H. Tominga, J . Cutul., 43, 131 (1976). F. Solymosi and J. Sarkany, Appl. Sur. Sci., 3, 68 (1979). W. C. Hecker and A. T. Bell, J . Catalysis, 84, 200 (1983). C. T. Campbell and J. M. White, Appl. Surf. Sci., 1 , 347 (1978). A. A. Chin and A. T. Bell, J. Phys. Chem., 87, 3700 (1983). R. J. Baird, R. C. Ku, and P. Wynblatt, Surf. Sci., 97, 346 (1980). T. W. Root, L. D. Schmidt, and G. B. Fisher, Surf. Sci., 134, 30 (1983). S. H. Oh, G. B. Fisher, J. E. Carpenter, and D. W. Goodman, J . Cutal., 200, 360 (1986). T. W. Root, L. D. Schmidt, and G. B. Fisher, Surf. Sci., 150, 173 (1985). D. Lorimer and A. T. Bell, J. Cutul., 59, 223 (1983). S. H. Oh and J. E. Carpenter, J. Cutul., 102, 114 (1986). T. W. Root, G. B. Fisher, and L. D. Schmidt, J. Chem. Phys., 85,4679 (1986). C. H. F. Peden, D. W. Goodman, D. S. Blair, P. J. Berlowitz, G. B. Fisher, and S. H. Oh, J . Phys. Chem., 92, 1563 (1988). S. H. Oh and C. C. Eickel, J. Cutul., 128, 526 (1991). E. I . Altman and R. J . Gorte, J . Cutul., 113, 185 (1988).
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
480 TAYLOR
56. 57.
58. 59. 60. 61.
62.
63. 64.
65.
66.
67.
68.
69.
70.
71.
72. 73. 74. 75.
76.
77.
78. 79.
80. 81.
82. 83.
M. Prigent and G . DeSoete, Society of Automotive Engineers Publ. No. 890492. G. L. Bauerle, G . R. Service, and K. Nobe, Ind. Eng. Chem. Prod. Res. Dev., 11, 54 (1972). N. K. Pande and A. T. Bell, Appl. Cutal., 20, 109 (1986). B. K. Cho, B. H. Shanks and J . E. Bailey, J . Card., 115, 486 (1989). R. W. McCabe and C. Wong, J . Cutul., 121, 422 (1990). S. E. Voltz, C. R. Morgan, D . Liederman, and S. M. Jacob, Znd. Eng. Chem. Prod. Res, Dev., 12, 294 (1973). H. S. Gandhi, H. C. Yao, H . K. Stepien, and M. Shelef, Society ofAutomotive Engineers Publ. No. 780606. J. C . Summers and K. Baron, J . Catal., 57, 380 (1979). W. B. Williamson, H. S. Gandhi, M. E. Heyde, and G . A. Zawacki, Society of Automotive Engineers Publ. No. 790942. G. C. Joy, G . R. Lester, and F. S. Molinaro, Society of Automotive Engineers Publ. No. 790943. W. B. Williamson, H. K. Stepien, and H. S. Gandhi, Environ. Sci. Technol., 14, 319 (1980). R. L. Furey and D. R. Monroe, Society of Automotive Engineers Publ. No. 81 1228. D. R. Monroe, M. H. Krueger, D. D. Beck, and M. J. D’Aniello, Jr., in Catalysis and Automotive Pollution Control I1 (A. Crucq, ed.), Vol. 71, p. 593, Elsevier, Amsterdam, 1991. D. D. Beck, M. H . Krueger, and D. R. Monroe, Society of Automotive Engineers Publ. No. 910844. J. D. Benson, V. Burns, R . A. Gorse, A. M. Hochhauser, W. J. Koehl, L. J. Painter, and R. M. Reuter, Society of Automotive Engineers Publ. No. 91 2323. Federal Register, New Motor Vehicles and New Motor Vehicle Engines- Control of Air Pollution, 37, Pt. 11, 221 (Nov. 15, 1972). U. Kohler and H. W. Wassmouth, Surf. Sci., 117, 668 (1982). U. Kohler and H. W. Wassermouth, Surf. Sci., 126, 448 (1983). R. C. Ku and P. Wynblatt, Appl. Surf. Sci., 8 , 250 (1981). C. R. Apesteguia, C. E. Brema, T. F. Garetto, A. Borgna, and J. M. Parera, J . Cutul., 89, 52 (1984). J . M. MacLaren, J . P. Pendry, and R. W. Joyner, Surf. Sci. Lett., 165, L80 (1986). W. Held, A. Konig, T. Richter, and L. Puppe, Society of Automotive En- gineers Publ. No. 900496. H. Hamada, Y. Kintaichi, M. Sasaki, and T. Ito, Appl. Cutul., 64, L1 (1990). Y. Kintaichi, H. Hamada, M. Tabata, M. Sasaki, and T. Ito, Catal. Lett., 6 , 239 (1990). H. Hamada, Y. Kinaichi, M. Sasaki, and T. Ito, Appl. Catal., 70, L15 (1991). S. Sato, Y. Yu-u, H. Yahiro, N. Mizuno, and M. Iwamoto, Appl. Catal., 70, L1 (1991). Y. Torikai, H. Yahiro, N. Mizuno, and M. Iwamoto, Cutal. Lett., 9,91(1991). H. Hosose, H . Yahiro, N. Mizuno, and M. Iwamoto, Chem. Lett., 1991, 1859.
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010
AUTOMOTIVE EXHAUST CATALYSTS 481
84. E. Kikuchi, K. Yogo, and M. Abe., Chem. Lett., 1991, 1063. 85. M. Misono and K. Kondo, Chem. Lett., 1991, 1001. 86. M. Iwamoto and H. Hamada, Catul. Today, 10, 57 (1991). 87. M. Iwamoto, H. Yahiro, S. Shundo, Y. Yu-u, and N. Mizuno, Appl. Catal.,
69, L15 (1991).
Downloaded By: [Aghalayam, Preeti] At: 06:27 20 January 2010