Free-energy landscape of protein oligomerization from atomistic simulations Alessandro Barducci a,b,1 , Massimiliano Bonomi c , Meher K. Prakash d , and Michele Parrinello b,d,1 a Laboratoire de Biophysique Statistique, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland; b Department of Chemistry and Applied Biosciences, Eidgenössische Technische Hochschule Zürich, CH-8006 Zurich, Switzerland; c Department of Bioengineering and Therapeutic Sciences, California Institute for Quantitative Biosciences, University of California, San Francisco, CA 94158; and d Facoltà di Informatica, Istituto di Scienze Computazionali, Università della Svizzera Italiana, CH-6900 Lugano, Switzerland Contributed by Michele Parrinello, October 28, 2013 (sent for review August 19, 2013) In the realm of protein–protein interactions, the assembly process of homooligomers plays a fundamental role because the major- ity of proteins fall into this category. A comprehensive understand- ing of this multistep process requires the characterization of the driving molecular interactions and the transient intermediate spe- cies. The latter are often short-lived and thus remain elusive to most experimental investigations. Molecular simulations provide a unique tool to shed light onto these complex processes complementing experimental data. Here we combine advanced sampling techni- ques, such as metadynamics and parallel tempering, to characterize the oligomerization landscape of fibritin foldon domain. This system is an evolutionarily optimized trimerization motif that represents an ideal model for experimental and computational mechanistic studies. Our results are fully consistent with previous experimen- tal nuclear magnetic resonance and kinetic data, but they provide a unique insight into fibritin foldon assembly. In particular, our simulations unveil the role of nonspecific interactions and suggest that an interplay between thermodynamic bias toward native structure and residual conformational disorder may provide a kinetic advantage. molecular dynamics | enhanced sampling | fly-casting mechanism | conformational selection I n the cell, proteins most often exist and perform their functions as complexes. In particular, a large percentage of proteins self- associate into homooligomers to gain functional advantages, such as improved thermodynamic stability and regulation of ac- tivity (1, 2). Understanding how these oligomeric complexes are assembled is thus a central topic in biophysics and molecular biology. Several studies have investigated the kinetics and ther- modynamics of protein oligomerization, and different kinetic mechanisms have been identified (3, 4). However, current un- derstanding of the assembly mechanism is still limited. Indeed, a comprehensive description of these reactions would require the identification and structural characterization of transient inter- mediates that remain elusive to most experimental techniques. Molecular dynamics (MD) simulations are an extremely useful tool for unraveling interesting biophysical processes. However, the time scales accessible to MD simulations based on accurate atomistic models are limited by the computational resources currently available. Although great progress has been made with the introduction of distributed computing platforms (5) and specialized hardware (6), the characterization of oligomerization free-energy landscapes with straightforward MD still remains an extremely challenging task, even for dimers (7, 8). This limitation may be circumvented by using advanced sampling algorithms such as metadynamics (9, 10) (MetaD). MetaD is based on the introduction of a history-dependent bias potential that simulta- neously enables enhanced sampling and estimation of the free- energy surface (FES) as a function of a few selected collective variables (CVs). Furthermore, MetaD can be seamlessly in- tegrated with other advanced sampling algorithms, such as par- allel tempering (PT) (11), with great benefits when studying complex biomolecular processes and large systems. Here we focus on the oligomerization process of the fibritin foldon domain. Fibritin is a trimeric rod-like structural protein from bacteriophage T4 with a multidomain architecture (12). A small C-terminal globular domain is essential for fibritin folding and assembly both in vivo and in vitro (12, 13). This domain, dubbed “foldon domain,” is an evolutionarily optimized trime- rization motif with remarkable properties: it can act as a chap- erone during the assembly of trimeric proteins (14), and it can even induce the trimerization of other proteins (15–17). The foldon domain trimer is extremely stable, and its structure has been solved by means of nuclear magnetic resonance (NMR) spectroscopy (18) (Fig. 1A). Each 27-residue monomer is com- posed of an N-terminal extended structure, a β-hairpin, and a small 3 10 helical turn (Fig. 1B). The trimeric structure is sta- bilized by several intermolecular interactions including hydro- phobic packing, backbone–backbone hydrogen bonds (H bonds), and salt bridges. The small size of the foldon domain, its simple fold, and its high stability make it an ideal model for detailed mechanistic studies of an assembly process both experimentally and computationally. The kinetics of fibritin foldon domain assembly has been characterized by Kiefhaber and coworkers by monitoring Trp fluorescence in stopped-flow experiments (18). At physiological concentration, the oligomerization is a multistep process initi- ated by the submillisecond formation of an intermediate mo- nomeric species which then dimerizes and finally reaches the native trimeric state through the addition of a third monomer to the preformed dimer. Remarkably, the dimerization and trime- rization steps are extremely fast compared with other bio- Significance Oligomeric proteins, comprising two or more associating poly- peptide chains, represent a large fraction of cellular proteins. In particular, many proteins self-associate into homooligomers to gain functional advantages. Our understanding of the oligo- merization at the molecular level is currently limited because intermediates that occur in the process are short-lived in most occasions, precluding a direct experimental characterization. Using molecular dynamics simulations, enhanced by a sampling method developed in our group, we obtained an atomistic description of the assembly of an evolutionary-optimized tri- meric protein. Our results are in excellent agreement with available experimental data and extend the current view of oligomerization by showing the importance of the apparently contradictory requirements of monomer preorganization and conformational flexibility. Author contributions: A.B., M.B., M.K.P., and M.P. designed research, performed research, analyzed data, and wrote the paper. The authors declare no conflict of interest. 1 To whom correspondence may be addressed. E-mail: [email protected] or alessandro.barducci@epfl.ch. This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1320077110/-/DCSupplemental. www.pnas.org/cgi/doi/10.1073/pnas.1320077110 PNAS Early Edition | 1 of 6 BIOPHYSICS AND COMPUTATIONAL BIOLOGY PNAS PLUS
Transcript
Free-energy landscape of protein oligomerization fromatomistic simulationsAlessandro Barduccia,b,1, Massimiliano Bonomic, Meher K. Prakashd, and Michele Parrinellob,d,1
aLaboratoire de Biophysique Statistique, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland; bDepartment of Chemistry and AppliedBiosciences, Eidgenössische Technische Hochschule Zürich, CH-8006 Zurich, Switzerland; cDepartment of Bioengineering and Therapeutic Sciences, CaliforniaInstitute for Quantitative Biosciences, University of California, San Francisco, CA 94158; and dFacoltà di Informatica, Istituto di Scienze Computazionali,Università della Svizzera Italiana, CH-6900 Lugano, Switzerland
Contributed by Michele Parrinello, October 28, 2013 (sent for review August 19, 2013)
In the realm of protein–protein interactions, the assembly processof homooligomers plays a fundamental role because the major-ity of proteins fall into this category. A comprehensive understand-ing of this multistep process requires the characterization of thedriving molecular interactions and the transient intermediate spe-cies. The latter are often short-lived and thus remain elusive to mostexperimental investigations.Molecular simulations provide a uniquetool to shed light onto these complex processes complementingexperimental data. Here we combine advanced sampling techni-ques, such as metadynamics and parallel tempering, to characterizethe oligomerization landscape of fibritin foldon domain. This systemis an evolutionarily optimized trimerization motif that representsan ideal model for experimental and computational mechanisticstudies. Our results are fully consistent with previous experimen-tal nuclear magnetic resonance and kinetic data, but they providea unique insight into fibritin foldon assembly. In particular, oursimulations unveil the role of nonspecific interactions and suggestthat an interplay between thermodynamic bias toward nativestructure and residual conformational disorder may provide akinetic advantage.
In the cell, proteins most often exist and perform their functionsas complexes. In particular, a large percentage of proteins self-
associate into homooligomers to gain functional advantages,such as improved thermodynamic stability and regulation of ac-tivity (1, 2). Understanding how these oligomeric complexes areassembled is thus a central topic in biophysics and molecularbiology. Several studies have investigated the kinetics and ther-modynamics of protein oligomerization, and different kineticmechanisms have been identified (3, 4). However, current un-derstanding of the assembly mechanism is still limited. Indeed,a comprehensive description of these reactions would require theidentification and structural characterization of transient inter-mediates that remain elusive to most experimental techniques.Molecular dynamics (MD) simulations are an extremely useful
tool for unraveling interesting biophysical processes. However,the time scales accessible to MD simulations based on accurateatomistic models are limited by the computational resourcescurrently available. Although great progress has been made withthe introduction of distributed computing platforms (5) andspecialized hardware (6), the characterization of oligomerizationfree-energy landscapes with straightforward MD still remains anextremely challenging task, even for dimers (7, 8). This limitationmay be circumvented by using advanced sampling algorithmssuch as metadynamics (9, 10) (MetaD). MetaD is based on theintroduction of a history-dependent bias potential that simulta-neously enables enhanced sampling and estimation of the free-energy surface (FES) as a function of a few selected collectivevariables (CVs). Furthermore, MetaD can be seamlessly in-tegrated with other advanced sampling algorithms, such as par-allel tempering (PT) (11), with great benefits when studyingcomplex biomolecular processes and large systems.
Here we focus on the oligomerization process of the fibritinfoldon domain. Fibritin is a trimeric rod-like structural proteinfrom bacteriophage T4 with a multidomain architecture (12). Asmall C-terminal globular domain is essential for fibritin foldingand assembly both in vivo and in vitro (12, 13). This domain,dubbed “foldon domain,” is an evolutionarily optimized trime-rization motif with remarkable properties: it can act as a chap-erone during the assembly of trimeric proteins (14), and it caneven induce the trimerization of other proteins (15–17). Thefoldon domain trimer is extremely stable, and its structure hasbeen solved by means of nuclear magnetic resonance (NMR)spectroscopy (18) (Fig. 1A). Each 27-residue monomer is com-posed of an N-terminal extended structure, a β-hairpin, anda small 310 helical turn (Fig. 1B). The trimeric structure is sta-bilized by several intermolecular interactions including hydro-phobic packing, backbone–backbone hydrogen bonds (H bonds),and salt bridges. The small size of the foldon domain, its simplefold, and its high stability make it an ideal model for detailedmechanistic studies of an assembly process both experimentallyand computationally.The kinetics of fibritin foldon domain assembly has been
characterized by Kiefhaber and coworkers by monitoring Trpfluorescence in stopped-flow experiments (18). At physiologicalconcentration, the oligomerization is a multistep process initi-ated by the submillisecond formation of an intermediate mo-nomeric species which then dimerizes and finally reaches thenative trimeric state through the addition of a third monomer tothe preformed dimer. Remarkably, the dimerization and trime-rization steps are extremely fast compared with other bio-
Significance
Oligomeric proteins, comprising two or more associating poly-peptide chains, represent a large fraction of cellular proteins. Inparticular, many proteins self-associate into homooligomers togain functional advantages. Our understanding of the oligo-merization at the molecular level is currently limited becauseintermediates that occur in the process are short-lived in mostoccasions, precluding a direct experimental characterization.Using molecular dynamics simulations, enhanced by a samplingmethod developed in our group, we obtained an atomisticdescription of the assembly of an evolutionary-optimized tri-meric protein. Our results are in excellent agreement withavailable experimental data and extend the current view ofoligomerization by showing the importance of the apparentlycontradictory requirements of monomer preorganization andconformational flexibility.
Author contributions: A.B., M.B., M.K.P., and M.P. designed research, performed research,analyzed data, and wrote the paper.
molecular folding reactions. The great efficiency of this processwas attributed to the formation of an intermediate monomericspecies that is structurally similar to the monomer conformationin the native trimeric structure, thus providing a template forrapid assembly during the dimerization and trimerization steps.Similar mechanisms based on templated growth have been pro-posed, in the more general context of protein aggregation, torationalize fibril formation kinetics (19–21).In the case of fibritin foldon domain, this intuition was sup-
ported by the NMR structural characterization of the E5R fol-don mutant which has a monomeric fold similar to the monomerconfiguration in the wild-type trimeric structure (22).In this study, we used a combination of MetaD and PT
(PTMetaD) (23) to fully characterize the assembly of fibritinfoldon domain by means of atomistic MD simulations. To makethis challenging problem computationally tractable, we tookadvantage of the multistep nature of the process as revealed bythe experimental kinetics data. Thus, we adopt a hierarchicalapproach, and we divide the assembly of the foldon trimer fromunfolded monomers into three different simulations: (i) mono-mer folding (U → M), (ii) dimerization (M + M → D), and (iii)trimerization (D + M → T). The FES obtained in each of thesesimulations was used to understand the detailed molecularmechanism of each of these stages of oligomerization.
ResultsMonomer Folding. The simulation of the fibritin foldon monomerwas performed using the PTMetaD protocol with MetaD biasapplied to three CVs: the radius of gyration (Rgyr), the totalnumber of backbone–backbone H bonds (Hb), and the numberof native Cα–Cα contacts defined according to the experimentalstructure of the wild-type trimer ðNMÞ. The FES at 300 K asa function of Rgyr and NM is characterized by the presence ofthree main basins (Fig. 2A): the unfolded state (U), a partiallyfolded intermediate (P), and a folded state (N). N is the globalfree-energy minimum, whereas U and P are slightly less popu-lated (ΔFNP ’ 0:9 kcal=mol, ΔFNU ’ 0:6 kcal=mol). The un-folded basin ðNM < 5Þ contains disordered, yet rather compact,structures. The intermediate basin ð8<NM < 13Þ corresponds toconfigurations in which the C-terminal hairpin is partially formedbut does not interact with the yet unstructured, detached N-terminal tail. The global free-energy minimum encompassesconformations which are structurally similar to the monomer inthe trimeric wild-type experimental structure (rmsd < 0.2 nm).However, this basin is characterized by a significant degree offlexibility of the N-terminal tail which samples multiple con-formations that deviate from the ensemble of the NMR con-formers determined for the E5R mutant (Fig. 2B).
The picture of the monomer landscape described above isperfectly consistent with several experimental evidences includingthe paucity of NOE signals measured for the E5R mutant in theN-terminal region (22) and the temperature-dependent behaviorof chemical shifts and residual dipolar couplings of wild-typefibritin trimer (24). To quantify the agreement of our results withNMR experiments on the E5R monomer, we back-calculated thechemical shifts from the PTMetaD simulation using two differentalgorithms [SHIFTX (25) and CamShift (26)]. These observablesare particularly sensitive to variations in the local chemical en-vironment, and they play an important and ever-increasing rolein the structural determination of disordered and partially dis-ordered proteins (27–29). A reweighting algorithm (30) was usedto recover the correct expectation values of the experimental ob-servables from the biased PTMetaD simulations (31). The resultsfor the Hα nuclei showed an excellent agreement between thesimulated and the experimental chemical shifts (Table 1). Thequality of this agreement does not change if we consider onlythe structural ensemble corresponding to basin N. Remarkably,the simulations reproduce the experimental chemical shifts betterthan the NMR-derived structures of the E5Rmutant deposited inthe Protein Data Bank (PDB) database (PDB code: 2KBL).
Dimerization. The dimerization process was studied by com-plementing the PTMetaD scheme with the well-tempered ensem-ble (WTE) approach (32). This combined algorithm (PTMetaD-WTE) allowed us to further reduce the high computational cost ofsimulating protein–protein binding with an explicit-solvent model
Fig. 1. (A) Top view of the fibritin foldon domain native structure (PDBcode: 1RFO) which is composed of three separate chains. Each chain of thehomotrimer is colored differently. (B) Side view of the fibritin foldon domainnative structure. One monomer is colored according to its secondary struc-ture (β-structure in magenta and 310 helix in orange).
Rgy
r [n
m]
[kcal/mol]
NM
A
B
NI
Fig. 2. Monomer folding. (A) FES at 300 K as a function of the total numberof native contacts ðNMÞ and the Rgyr. Isoenergy lines are drawn every 0.6kcal/mol. Structures representative of the two free-energy basins I and N(gray) are superimposed on the monomer conformation in the trimeric ex-perimental structure (red). (B) The bundle of the NMR conformers obtainedfor E5R mutant (blue) is compared with the conformational ensemble cor-responding to the N basin (green). An additional cartoon representation ofa single NMR conformer is added as a guide for the eye. All of the structuresare aligned by superimposing the β-hairpin region (residues 13–23).
2 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1320077110 Barducci et al.
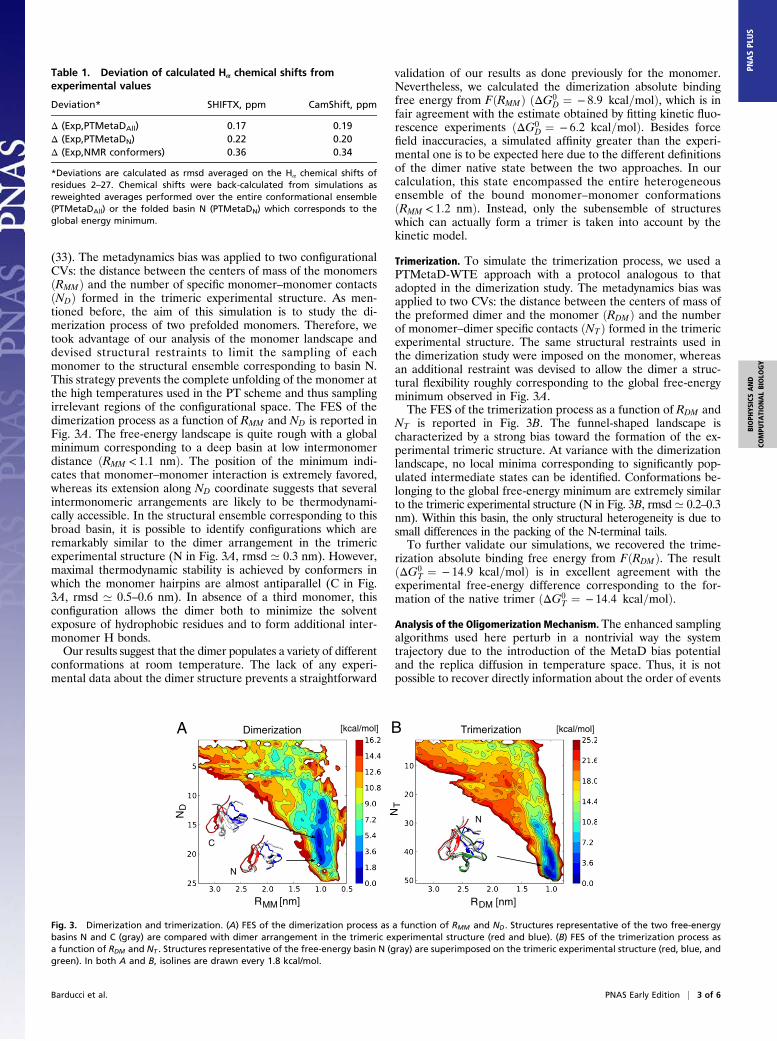
(33). The metadynamics bias was applied to two configurationalCVs: the distance between the centers of mass of the monomersðRMMÞ and the number of specific monomer–monomer contactsðNDÞ formed in the trimeric experimental structure. As men-tioned before, the aim of this simulation is to study the di-merization process of two prefolded monomers. Therefore, wetook advantage of our analysis of the monomer landscape anddevised structural restraints to limit the sampling of eachmonomer to the structural ensemble corresponding to basin N.This strategy prevents the complete unfolding of the monomer atthe high temperatures used in the PT scheme and thus samplingirrelevant regions of the configurational space. The FES of thedimerization process as a function of RMM and ND is reported inFig. 3A. The free-energy landscape is quite rough with a globalminimum corresponding to a deep basin at low intermonomerdistance ðRMM < 1:1 nmÞ. The position of the minimum indi-cates that monomer–monomer interaction is extremely favored,whereas its extension along ND coordinate suggests that severalintermonomeric arrangements are likely to be thermodynami-cally accessible. In the structural ensemble corresponding to thisbroad basin, it is possible to identify configurations which areremarkably similar to the dimer arrangement in the trimericexperimental structure (N in Fig. 3A, rmsd ’ 0.3 nm). However,maximal thermodynamic stability is achieved by conformers inwhich the monomer hairpins are almost antiparallel (C in Fig.3A, rmsd ’ 0.5–0.6 nm). In absence of a third monomer, thisconfiguration allows the dimer both to minimize the solventexposure of hydrophobic residues and to form additional inter-monomer H bonds.Our results suggest that the dimer populates a variety of different
conformations at room temperature. The lack of any experi-mental data about the dimer structure prevents a straightforward
validation of our results as done previously for the monomer.Nevertheless, we calculated the dimerization absolute bindingfree energy from FðRMMÞ ðΔG0
D ¼ − 8:9 kcal=molÞ, which is infair agreement with the estimate obtained by fitting kinetic fluo-rescence experiments ðΔG0
D ¼ − 6:2 kcal=molÞ. Besides forcefield inaccuracies, a simulated affinity greater than the experi-mental one is to be expected here due to the different definitionsof the dimer native state between the two approaches. In ourcalculation, this state encompassed the entire heterogeneousensemble of the bound monomer–monomer conformationsðRMM < 1:2 nmÞ. Instead, only the subensemble of structureswhich can actually form a trimer is taken into account by thekinetic model.
Trimerization. To simulate the trimerization process, we used aPTMetaD-WTE approach with a protocol analogous to thatadopted in the dimerization study. The metadynamics bias wasapplied to two CVs: the distance between the centers of mass ofthe preformed dimer and the monomer ðRDMÞ and the numberof monomer–dimer specific contacts ðNTÞ formed in the trimericexperimental structure. The same structural restraints used inthe dimerization study were imposed on the monomer, whereasan additional restraint was devised to allow the dimer a struc-tural flexibility roughly corresponding to the global free-energyminimum observed in Fig. 3A.The FES of the trimerization process as a function of RDM and
NT is reported in Fig. 3B. The funnel-shaped landscape ischaracterized by a strong bias toward the formation of the ex-perimental trimeric structure. At variance with the dimerizationlandscape, no local minima corresponding to significantly pop-ulated intermediate states can be identified. Conformations be-longing to the global free-energy minimum are extremely similarto the trimeric experimental structure (N in Fig. 3B, rmsd’ 0.2–0.3nm). Within this basin, the only structural heterogeneity is due tosmall differences in the packing of the N-terminal tails.To further validate our simulations, we recovered the trime-
rization absolute binding free energy from FðRDMÞ. The resultðΔG0
T ¼ − 14:9 kcal=molÞ is in excellent agreement with theexperimental free-energy difference corresponding to the for-mation of the native trimer ðΔG0
T ¼ − 14:4 kcal=molÞ.Analysis of the Oligomerization Mechanism. The enhanced samplingalgorithms used here perturb in a nontrivial way the systemtrajectory due to the introduction of the MetaD bias potentialand the replica diffusion in temperature space. Thus, it is notpossible to recover directly information about the order of events
Table 1. Deviation of calculated Hα chemical shifts fromexperimental values
*Deviations are calculated as rmsd averaged on the Hα chemical shifts ofresidues 2–27. Chemical shifts were back-calculated from simulations asreweighted averages performed over the entire conformational ensemble(PTMetaDAll) or the folded basin N (PTMetaDN) which corresponds to theglobal energy minimum.
[kcal/mol]Dimerization TrimerizationA B
R [nm]MM R [nm]DM
ND NT
[kcal/mol]
N
C
N
Fig. 3. Dimerization and trimerization. (A) FES of the dimerization process as a function of RMM and ND. Structures representative of the two free-energybasins N and C (gray) are compared with dimer arrangement in the trimeric experimental structure (red and blue). (B) FES of the trimerization process asa function of RDM and NT . Structures representative of the free-energy basin N (gray) are superimposed on the trimeric experimental structure (red, blue, andgreen). In both A and B, isolines are drawn every 1.8 kcal/mol.
Barducci et al. PNAS Early Edition | 3 of 6
BIOPH
YSICSAND
COMPU
TATIONALBIOLO
GY
PNASPL
US
in the different stages of the multimerization process which arelikely to occur through multiple microscopic pathways.Nevertheless, we could reconstruct the unbiased distribution
of any configurational variable by means of the reweighting al-gorithm mentioned above. This allowed us to shed light on thedimerization and trimerization mechanisms by monitoring theexpectation value of properly chosen quantities as a function ofRMM and RDM . A similar approach was used to characterize themechanism of urea-driven denaturation of the GB1 C-terminalβ-hairpin (34).We first investigated the interplay between the formation of
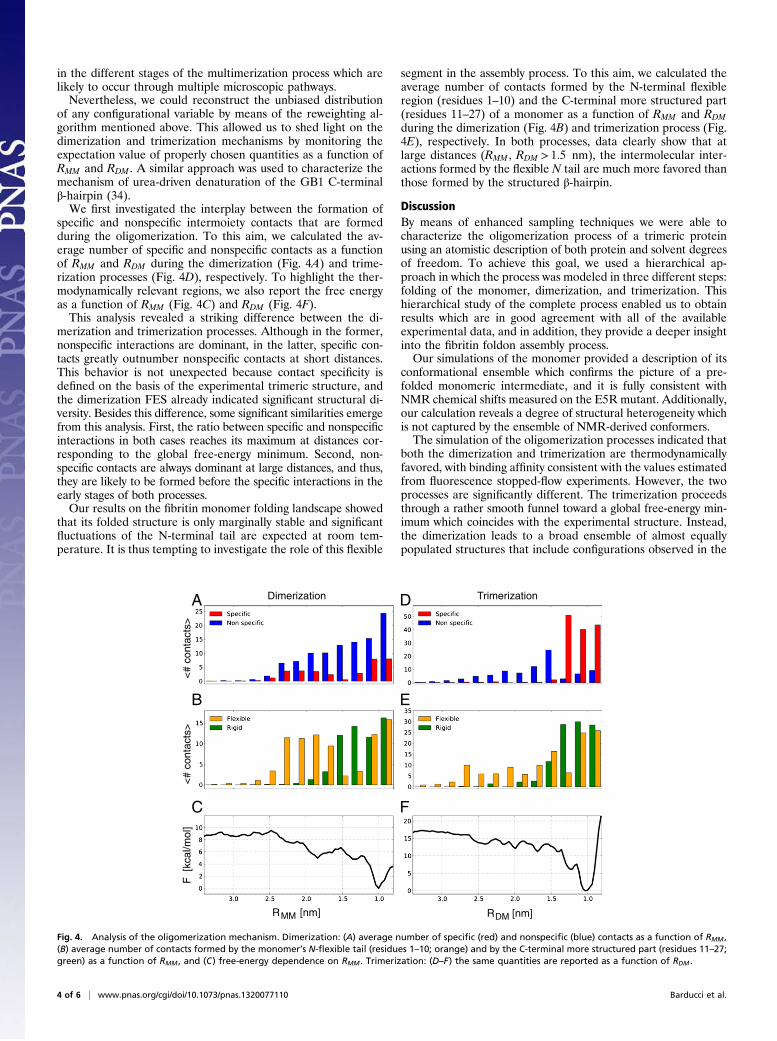
specific and nonspecific intermoiety contacts that are formedduring the oligomerization. To this aim, we calculated the av-erage number of specific and nonspecific contacts as a functionof RMM and RDM during the dimerization (Fig. 4A) and trime-rization processes (Fig. 4D), respectively. To highlight the ther-modynamically relevant regions, we also report the free energyas a function of RMM (Fig. 4C) and RDM (Fig. 4F).This analysis revealed a striking difference between the di-
merization and trimerization processes. Although in the former,nonspecific interactions are dominant, in the latter, specific con-tacts greatly outnumber nonspecific contacts at short distances.This behavior is not unexpected because contact specificity isdefined on the basis of the experimental trimeric structure, andthe dimerization FES already indicated significant structural di-versity. Besides this difference, some significant similarities emergefrom this analysis. First, the ratio between specific and nonspecificinteractions in both cases reaches its maximum at distances cor-responding to the global free-energy minimum. Second, non-specific contacts are always dominant at large distances, and thus,they are likely to be formed before the specific interactions in theearly stages of both processes.Our results on the fibritin monomer folding landscape showed
that its folded structure is only marginally stable and significantfluctuations of the N-terminal tail are expected at room tem-perature. It is thus tempting to investigate the role of this flexible
segment in the assembly process. To this aim, we calculated theaverage number of contacts formed by the N-terminal flexibleregion (residues 1–10) and the C-terminal more structured part(residues 11–27) of a monomer as a function of RMM and RDMduring the dimerization (Fig. 4B) and trimerization process (Fig.4E), respectively. In both processes, data clearly show that atlarge distances (RMM , RDM > 1:5 nm), the intermolecular inter-actions formed by the flexible N tail are much more favored thanthose formed by the structured β-hairpin.
DiscussionBy means of enhanced sampling techniques we were able tocharacterize the oligomerization process of a trimeric proteinusing an atomistic description of both protein and solvent degreesof freedom. To achieve this goal, we used a hierarchical ap-proach in which the process was modeled in three different steps:folding of the monomer, dimerization, and trimerization. Thishierarchical study of the complete process enabled us to obtainresults which are in good agreement with all of the availableexperimental data, and in addition, they provide a deeper insightinto the fibritin foldon assembly process.Our simulations of the monomer provided a description of its
conformational ensemble which confirms the picture of a pre-folded monomeric intermediate, and it is fully consistent withNMR chemical shifts measured on the E5R mutant. Additionally,our calculation reveals a degree of structural heterogeneity whichis not captured by the ensemble of NMR-derived conformers.The simulation of the oligomerization processes indicated that
both the dimerization and trimerization are thermodynamicallyfavored, with binding affinity consistent with the values estimatedfrom fluorescence stopped-flow experiments. However, the twoprocesses are significantly different. The trimerization proceedsthrough a rather smooth funnel toward a global free-energy min-imum which coincides with the experimental structure. Instead,the dimerization leads to a broad ensemble of almost equallypopulated structures that include configurations observed in the
DA
R [nm]MM R [nm]DM
B
C
E
F
<#
cont
acts
>
noitaziremirTnoitaziremiD
>stcatnoc#
<]lo
m/ lac k[F
Fig. 4. Analysis of the oligomerization mechanism. Dimerization: (A) average number of specific (red) and nonspecific (blue) contacts as a function of RMM ,(B) average number of contacts formed by the monomer’s N-flexible tail (residues 1–10; orange) and by the C-terminal more structured part (residues 11–27;green) as a function of RMM , and (C) free-energy dependence on RMM . Trimerization: (D–F) the same quantities are reported as a function of RDM .
4 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1320077110 Barducci et al.
trimeric experimental structure. The most favored dimericstructure, which has been so far elusive to experimental investi-gation, corresponds to an almost antiparallel packing of the twomonomers. This scenario suggests that the addition of a thirdmonomer operates a conformational selection of the dimer en-semble by stabilizing the conformation observed in the native trimer.Although our simulations could not directly provide dynamical
information, we were able to gain a significant insight into thefibritin assembly mechanism by analyzing how interprotomercontacts are formed during the oligomerization processes. First,we quantified the formation of nonspecific interactions, and weshowed that they play a major role in the early recognition stageof both dimerization and trimerization. This finding is fullyconsistent with the general idea that nonspecific encounter com-plexes are of key importance in accelerating protein–proteinbinding (35, 36). Transient encounter complexes have been elu-cidated by paramagnetic relaxation enhancement NMR spectros-copy (37) and studied computationally by means of coarse-grainedmodels (38, 39).We further investigated the nature of these interactions by
identifying the fragments that most likely interact in the earlystages of binding. Our analysis indicated that the monomerN-terminal tail has a crucial function in both monomer–monomerand dimer–monomer recognition. As discussed before, boththe NMR experiments and our simulations clearly show that theN-terminal tail is the most flexible and disordered region of themonomer. Thus, it is tempting to relate this finding to the “fly-casting” mechanism (40) proposed for intrinsically disorderedproteins (IDPs). According to this model, the lack of a well-defined structure in IDPs leads to an increased capture radiusand a reduction in free-energy barriers, ultimately resulting ina fast binding kinetics (40, 41). Similarly, here the presence ofresidual disorder in an almost completely folded monomer mightbe beneficial in the recognition stage of the binding process.Fibritin foldon domain shows a remarkably fast assembly ki-
netics, and it is considered an evolutionarily optimized trimeri-zation motif. In the current view, this property is a consequenceof the structural preorganization of single monomers whichprovide a scaffold for the oligomerization. Our results confirmthis picture but suggest that an interplay between thermody-namic bias toward native structure formation and residual flex-ibility could provide additional kinetic advantage. These findingsmight be extended to the interactions between other structuredproteins and thus pave the way to further investigation in thisdirection. In conclusion, our study demonstrates that the com-bination of different enhanced sampling techniques makes MDsimulations a powerful computational microscope to investigatecomplex biomolecular processes such as protein oligomerization.
Materials and MethodsGeneral Setup. All of the simulations were performed using GROMACS4 MDcode (42) and PLUMED plugin (43). The Amber99SB-ILDN (44) and TIP3P (45)models were used for protein and water molecules, respectively. Initialconformations were obtained from the trimeric NMR structure of fibritinfoldon domain (PDB code: 1RFO). All of the simulations were performedusing a rhombic dodecahedral box with periodic boundary conditions. Well-tempered MetaD (46) was used to accelerate sampling in all of the steps ofthe oligomerization process in combination with PT, as described below. ThePT replica temperatures were chosen according to the distribution proposedin ref. (47). Additional details of the MD parameters and system equilibra-tion are provided in Supporting Information, Simulation Details.
Monomer Folding. Fibritin monomer was solvated in 3,461 water moleculescorresponding to an equilibrated box size of 110.9 nm3. In the PTMetaDscheme, 80 replicas were simulated in the temperature range 275–650 Kwith a resulting exchange acceptance probability of ’0.2. The simulationwas carried out for 85 ns, corresponding to an aggregated simulation timeof 6.8 μs. The MetaD bias was applied to three CVs. The first CV was theradius of gyration of the Cα atoms of the monomer. Hb was calculated usinga sum of switching functions:
Hb ¼ ∑ij
1−�dij
r0
�n
1−�dij
r0
�m ,
where r0 was set to 0.25 nm and n and m were set to 6 and 12, respectively.The sum runs over all of the pairs of hydrogen and oxygen backbone atomswith a sequence separation equal to or larger than four residues. The samefunctional form was used to evaluate the number of native contact NM . Inthis case, r0 was set to 0.85 nm, n and m were set to 6 and 12, and the sumincluded all of the Cα–Cα atom pairs which are closer than 0.9 nm in theexperimental structure and separated by more than four residues insequence.
Dimerization and Trimerization. Both systems were solvated in 8,419 watermolecules corresponding to equilibrated box sizes of 265.0 nm3 for the di-merization and 270.5 nm3 for the trimerization. The PTMetaD-WTE schemewas applied using 16 replicas distributed in the temperature range 290–656K. First, the bias in the energy space was converged for all of the replicas in20 ns. This protocol allowed us to significantly increase the potential energyfluctuations and to obtain an average exchange acceptance probability of’0.2 for both systems. The MetaD bias was then applied to two configura-tional variables to enhance sampling: RMM and ND for the dimerization andRDM and NT for the trimerization. RMM and RDM were calculated as the dis-tance between the centers of mass of the two monomers (dimerization) andbetween the dimer and the monomer (trimerization). The total number ofspecific contacts formed by the dimer ðNDÞ and the trimer ðNT Þ was calcu-lated analogously to NM but taking into account only contacts between thetwo monomers and between the monomer and the dimer, respectively. Thelength of the PTMetaD-WTE simulation was 140 ns for the dimerization and200 ns for the trimerization. As discussed previously, structural restraintswere implemented to focus sampling in the most relevant regions of theconfigurational space. These restraints included the following: (i) in bothdimerization and trimerization, two restraints on each monomer in the rmsdspace to avoid monomer unfolding while allowing significant fluctuation ofthe N-terminal tail and (ii) in the trimerization, a restraint on the dimer in ND
space to limit its sampling to basins N and C (Fig. 3).
Analysis. The equilibrium probability distributions of all quantities other thanthe MetaD CVs were reconstructed using a reweighting algorithm (30). In thedimerization process, the monomers can actually be arranged into twosymmetric, native-like structures (AB and BA). These conformations are de-generate but characterized by distinct sets of interchain contacts (ND,AB andND,BA). Hence, a symmetrized definition of ND, i.e., ND ¼ maxðND,AB,ND,BAÞ,was used in all of our analyses. In the analysis of the dimerization and tri-merization mechanism (Fig. 4), the contacts were calculated as the totalnumber of monomer–monomer and monomer–dimer residue pairs witha Cα–Cα distance smaller than a sharp cutoff (0.75 nm).
ACKNOWLEDGMENTS. We acknowledge Prof. Thomas Kiefhaber for in-troducing us to the system and for several useful discussions. This work wassupported by a grant from the Swiss National Supercomputing Centre underProject ID s223. We acknowledge European Union Grant ERC-2009-AdG-247075 for funding.
1. Marianayagam NJ, Sunde M, Matthews JM (2004) The power of two: Protein di-
merization in biology. Trends Biochem Sci 29(11):618–625.2. Hashimoto K, Nishi H, Bryant S, Panchenko AR (2011) Caught in self-interaction:
Evolutionary and functional mechanisms of protein homooligomerization. Phys Biol
8(3):035007.3. Bosshard HR (2008) Concurrent Association and Folding of Small Oligomeric Proteins
7. Chong LT, Snow CD, Rhee YM, Pande VS (2005) Dimerization of the p53 oligomeri-zation domain: Identification of a folding nucleus by molecular dynamics simulations.J Mol Biol 345(4):869–878.
8. Piana S, Lindorff-Larsen K, Shaw DE (2013) Atomistic description of the folding ofa dimeric protein. J Phys Chem B 117(42):12935–12942.
9. Laio A, Parrinello M (2002) Escaping free-energy minima. Proc Natl Acad Sci USA99(20):12562–12566.
10. Barducci A, Bonomi M, Parrinello M (2011) Metadynamics. Wiley Interdiscip RevComput Mol Sci 1(5):826–843.
11. Hansmann U (1997) Parallel tempering algorithm for conformational studies of bi-ological molecules. Chem Phys Lett 281(1):140–150.
12. Tao Y, Strelkov SV, Mesyanzhinov VV, Rossmann MG (1997) Structure of bacterio-phage T4 fibritin: A segmented coiled coil and the role of the C-terminal domain.Structure 5(6):789–798.
13. Boudko SP, et al. (2002) Domain organization, folding and stability of bacteriophageT4 fibritin, a segmented coiled-coil protein. Eur J Biochem 269(3):833–841.
14. Miroshnikov KA, et al. (1998) Engineering trimeric fibrous proteins based on bacte-riophage T4 adhesins. Protein Eng 11(4):329–332.
15. Frank S, et al. (2001) Stabilization of short collagen-like triple helices by protein en-gineering. J Mol Biol 308(5):1081–1089.
16. Yang X, et al. (2002) Highly stable trimers formed by human immunodeficiency virustype 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fi-britin. J Virol 76(9):4634–4642.
17. Papanikolopoulou K, et al. (2004) Adenovirus fibre shaft sequences fold into thenative triple beta-spiral fold when N-terminally fused to the bacteriophage T4 fibritinfoldon trimerisation motif. J Mol Biol 342(1):219–227.
18. Güthe S, et al. (2004) Very fast folding and association of a trimerization domain frombacteriophage T4 fibritin. J Mol Biol 337(4):905–915.
19. Dima RI, Thirumalai D (2002) Exploring protein aggregation and self-propagationusing lattice models: Phase diagram and kinetics. Protein Sci 11(5):1036–1049.
20. Nguyen PH, Li MS, Stock G, Straub JE, Thirumalai D (2007) Monomer adds to pre-formed structured oligomers of Abeta-peptides by a two-stage dock-lock mechanism.Proc Natl Acad Sci USA 104(1):111–116.
21. Reddy G, Straub JE, Thirumalai D (2009) Influence of preformed Asp23-Lys28 saltbridge on the conformational fluctuations of monomers and dimers of Abeta pep-tides with implications for rates of fibril formation. J Phys Chem B 113(4):1162–1172.
22. Habazettl J, Reiner A, Kiefhaber T (2009) NMR structure of a monomeric intermediateon the evolutionarily optimized assembly pathway of a small trimerization domain.J Mol Biol 389(1):103–114.
23. Bussi G, Gervasio FL, Laio A, Parrinello M (2006) Free-energy landscape for betahairpin folding from combined parallel tempering and metadynamics. J Am Chem Soc128(41):13435–13441.
24. Meier S, Güthe S, Kiefhaber T, Grzesiek S (2004) Foldon, the natural trimerizationdomain of T4 fibritin, dissociates into a monomeric A-state form containing a stablebeta-hairpin: Atomic details of trimer dissociation and local beta-hairpin stabilityfrom residual dipolar couplings. J Mol Biol 344(4):1051–1069.
25. Neal S, Nip AM, Zhang H, Wishart DS (2003) Rapid and accurate calculation of protein1H, 13C and 15N chemical shifts. J Biomol NMR 26(3):215–240.
26. Kohlhoff KJ, Robustelli P, Cavalli A, Salvatella X, Vendruscolo M (2009) Fast and ac-curate predictions of protein NMR chemical shifts from interatomic distances. J AmChem Soc 131(39):13894–13895.
27. Camilloni C, De Simone A, Vranken WF, Vendruscolo M (2012) Determination ofsecondary structure populations in disordered states of proteins using nuclear mag-netic resonance chemical shifts. Biochemistry 51(11):2224–2231.
28. Camilloni C, Robustelli P, De Simone A, Cavalli A, Vendruscolo M (2012) Character-ization of the conformational equilibrium between the two major substates of RNaseA using NMR chemical shifts. J Am Chem Soc 134(9):3968–3971.
29. Kragelj J, Ozenne V, Blackledge M, Jensen MR (2013) Conformational propensities ofintrinsically disordered proteins from NMR chemical shifts. ChemPhysChem 14(13):3034–3045.
30. Bonomi M, Barducci A, Parrinello M (2009) Reconstructing the equilibrium Boltzmanndistribution from well-tempered metadynamics. J Comput Chem 30(11):1615–1621.
31. Barducci A, Bonomi M, Parrinello M (2010) Linking well-tempered metadynamicssimulations with experiments. Biophys J 98(9):L44–L46.
32. Bonomi M, Parrinello M (2010) Enhanced sampling in the well-tempered ensemble.Phys Rev Lett 104(19):190601.
33. Deighan M, Bonomi M, Pfaendtner J (2012) Efficient simulation of explicitly solvatedproteins in the well-tempered ensemble. J Chem Theory Comput 8(7):2189–2192.
34. Berteotti A, Barducci A, Parrinello M (2011) Effect of urea on the β-hairpin confor-mational ensemble and protein denaturation mechanism. J Am Chem Soc 133(43):17200–17206.
35. Schreiber G, Haran G, Zhou HX (2009) Fundamental aspects of protein-protein asso-ciation kinetics. Chem Rev 109(3):839–860.
36. Ubbink M (2009) The courtship of proteins: Understanding the encounter complex.FEBS Lett 583(7):1060–1066.
38. Spaar A, Dammer C, Gabdoulline RR, Wade RC, Helms V (2006) Diffusional encounterof barnase and barstar. Biophys J 90(6):1913–1924.
39. Kim YC, Tang C, Clore GM, Hummer G (2008) Replica exchange simulations oftransient encounter complexes in protein-protein association. Proc Natl Acad SciUSA 105(35):12855–12860.
40. Shoemaker BA, Portman JJ, Wolynes PG (2000) Speeding molecular recognition byusing the folding funnel: The fly-casting mechanism. Proc Natl Acad Sci USA 97(16):8868–8873.
41. Huang Y, Liu Z (2009) Kinetic advantage of intrinsically disordered proteins in coupledfolding-binding process: A critical assessment of the “fly-casting” mechanism. J MolBiol 393(5):1143–1159.
42. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: Algorithms forhighly efficient, load-balanced, and scalable molecular simulation. J Chem TheoryComput 4(3):435–447.
43. Bonomi M, et al. (2009) PLUMED: A portable plugin for free-energy calculations withmolecular dynamics. Comput Phys Commun 180(10):1961–1972.
44. Lindorff-Larsen K, et al. (2010) Improved side-chain torsion potentials for the Amberff99SB protein force field. Proteins 78(8):1950–1958.
45. Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M (1983) Comparison ofsimple potential functions for simulating liquid water. J Chem Phys 79(2):926–935.
46. Barducci A, Bussi G, Parrinello M (2008) Well-tempered metadynamics: A smoothlyconverging and tunable free-energy method. Phys Rev Lett 100(2):020603.
47. Prakash MK, Barducci A, Parrinello M (2011) Replica temperatures for uniform ex-change and efficient roundtrip times in explicit solvent parallel tempering simu-lations. J Chem Theory Comput 7(7):2025–2027.
6 of 6 | www.pnas.org/cgi/doi/10.1073/pnas.1320077110 Barducci et al.