Journal of PathologyJ Pathol 2015; 235: 342–354Published online in Wiley Online Library(wileyonlinelibrary.com) DOI: 10.1002/path.4425
INVITED REVIEW
Human Beta-papillomavirus infection and keratinocytecarcinomasKoen D Quint,1,2 Roel E Genders,1,3 Maurits NC de Koning,2 Cinzia Borgogna,4 Marisa Gariglio,4Jan Nico Bouwes Bavinck,1 John Doorbar5# and Mariet C Feltkamp6,*#
1 Department of Dermatology, Leiden University Medical Centre, The Netherlands2 DDL Diagnostic Laboratory, Rijswijk, The Netherlands3 Department of Dermatology, Roosevelt Clinic, Leiden, The Netherlands4 Virology Unit, Department of Translational Medicine, Novara Medical School, Italy5 Division of Virology, Department of Pathology, University of Cambridge, UK6 Department of Medical Microbiology, Leiden University Medical Centre, Leiden, The Netherlands
*Correspondence to: M Feltkamp, Department of Medical Microbiology, Leiden University Medical Centre, Leiden, The Netherlands.E-mail: [email protected]
Received 30 June 2014; Revised 4 August 2014; Accepted 11 August 2014
No conflicts of interest were declared.
Introduction
Human papillomaviruses (HPVs) are small (8 kb) cir-cular double-stranded DNA viruses with the abilityto infect human epithelium at different body sites.Currently more than 170 different HPV types havebeen described, which can be clustered into five genera(Alpha-, Beta-, Gamma-, Mu- and Nu-papillomavirus),based on their taxonomy [1,2] (http://pave.niaid.nih.gov/#home). Papillomaviruses are classified on thebasis of DNA heterogeneity, with most types within agenus showing< 60% L1 sequence identity with typesfrom other genera [1,3].
The infectious role of HPV in cutaneous warts (ver-rucae vulgaris as well as condylomata acuminata) wasalready appreciated in the 1960s, although the phyloge-netic multiplicity of HPV types was not at this time fullyrealized [4]. In 1977, Harald zur Hausen [5] proposed arole for HPV in the development of cervical cancer, forwhich he received a Nobel prize in 2008. During this
time, a large body of research has established a criticalrole for HPV in the development of different types ofcancer, leading to the statement of the InternationalAgency for Research on Cancer (IARC), that strongevidence of a causal role for HPV has been found in can-cers derived from the cervix, vagina, vulva, anus, penisand oropharynx and that approximately 4.8% of the newcancers occurring in 2008 worldwide were due to HPVinfections [6]. These statements refer to the carcino-genic (and potentially carcinogenic) ‘high-risk’ HPVtypes from the Alpha-genus (AlphaPV), which have aparticular tropism for mucosal epithelium, and whichgenerally do not cause cutaneous squamous cell carci-nomas outside of the anogenital area. However, a rolefor HPV in human skin carcinogenesis was suggestedeven earlier than for HPV in cervical cancer [7].
Keratinocyte carcinomas (KCs; basal and squamouscell carcinomas) are by far the most common can-cers in Caucasian populations [8]. In Australia, forexample, a country with many fair-skinned inhabitantsand high exposure to UV radiation (the most important
Figure 1. Genome and life cycle organization of the Beta-papillomaviruses. The genomes of the BetaPV HPV5 and the AlphaPV HPV16 areshown (A); the positions of the major open reading frames and the long control region are indicated. BetaPV types lack an E5 ORF and havea much longer E4 ORF than the AlphaPV types. The positions of major promoters and splice donor/acceptor sites are indicated by arrows(promoters) and circles (d, donor; a, acceptor), respectively, alongside the positions of the early (PAE) and late polyadenylation sites (PAL).Infection and life cycle, and lesion formation, by BetaPV types are shown in the right panels. To initiate infection, HPV virions must gainaccess to the epithelial basal cells and probably an epithelial stem cell (coloured purple), either through a wound or possibly through thehair follicle (B). Once the wound has been repaired and a BetaPV lesion has formed (C), it is thought that the viral genome persists as a lowcopy number episome in the basal layer, often with limited viral gene expression. Cell cycle entry (cells with red nuclei) can be driven bythe viral E6/E7 proteins, with E4 expression (green), genome ampification (dark blue) and L1 expression (yellow) occurring as the infectedcell migrates towards the epithelial surface. It is thought that viral gene expression may become deregulated in EV and immunosuppressedindividuals.
environmental risk factor for KC), approximately 3% ofthe population develops KC each year [9,10]. Solar UVBradiation can cause DNA damage in keratinocytes as theresult of a photochemical reaction in which cyclobutanepyrimidine dimers are formed in addition to reactiveoxygen species [11]. Other factors, including chemicals,Röntgen radiation, smoking and immunosuppressivemedication, can also contribute to the development ofKC. In addition, there is growing evidence in the liter-ature that HPVs belonging to the Beta-genus (BetaPV)can act as co-factors in the development of UV-inducedKC, especially in immunocompromized patients.Although ongoing research performed in patients withprimary and acquired immunodeficiencies has foundassociations between markers of BetaPV infection andKC [12,13], the role of BetaPV in skin carcinogenesisis not yet fully established. This review summarizes theavailable evidence for a role of BetaPV in the develop-ment of UV-induced KC, and attempts to consider thisin the context of the biology of these viruses.
Human papillomaviruses – BetaPV versusAlphaPV
Infection and replicationThe HPV genome, regardless of genus, can be dividedinto a long control region (LCR) that contains regulatory
elements (eg transcription factor-binding sites, enhancerelements and promoter elements) and the viral origin ofreplication (ori) [14], and a coding region that encodesthe early (E) and late (L) proteins (Figure 1A)[15,16].The early proteins (typically E1–E7) have two majorfunctions, namely the regulation of viral transcriptionand replication, and the modulation of host cell proteinfunction (Table 1). The modulation of host cell proteinfunction is necessary to create a cellular environmentoptimal for viral replication, and eventually also forvirion assembly and release. The late proteins L1 and L2are the major and minor capsid proteins, respectively,with L2 also having a role in genome packaging duringvirion assembly [17]. Viral gene expression is also con-trolled by promoter elements that lie outside of the LCR,and by the differential use of splice donor and acceptorsites that regulate the levels of different viral transcriptsas the HPV-infected keratinocyte differentiates.
To initiate an infection, HPVs need to gain accessto the basal keratinocytes, and probably an epidermalstem cell within the basal cell layer, which is nor-mally protected by differentiated stratified epithelium(Figure 1). In many cases, it is thought that this isfacilitated by an exogenous (micro)trauma or otherlocalized break in the epithelial barrier [18]. However,at particular epithelial sites, infection may be lessdependent on epithelial damage (Figure 1B). For thehigh-risk AlphaPVs, such ‘vulnerable’ sites include thecervical and anal transformation zones, where stratified
E1 ATP-dependent helicase involved in viral DNA replication. Functionally highly conserved between typesE2 Viral transcription factor, also important for viral genome segregation, the recruitment of E1 to the viral replication origin and
encapsidation. Conserved between typesE4 Abundantly expressed as an E1–E4 fusion protein during the late phase of the virus life cycle. Accumulates as cytoplasmic inclusion
granules in Beta, Gamma and Mu HPV types. Binds to cytokeratin filaments and disrupts cell structureE5 Divergent between types. In Alpha types, E5 interacts with PDGF receptors and activates mitogenic signalling pathways via
transcription factors. Interferes with HLA display of peptide antigens. E5 not present in Beta PV typesE6 E6 of Alpha HPV types binds p53. High-risk Alpha types also degrade p53 through association with E6AP, and can activate telomerase.
Beta types bind MAML1 but not E6AP and inhibit keratinocyte differentiation. All associate with the p300 transcriptionalco-activator
E7 High-risk Alpha types interact with members of the pRB family. Low-risk Alpha and Beta types specifically associate with p130 and arethought to have a lower ability to associate with pRb
Late proteinsL1 Major capsid protein. Assembles into pentomeric capsomeres, which are the primary components of the icosahedral virion shellL2 Minor capsid protein, also involved in encapsidation of viral DNA and in viral uncoating after infection
Papillomaviruses do not contain an E3 ORF. Initial designation of E3 in BPV resulted from a sequencing error that was later corrected.
and columnar epithelium abut, and the tonsillar crypts,which comprise a poorly structured epithelium [19].Eccrine sweat glands have been proposed as a siteof infection for MuPV types [20], and the frequentdetection of BetaPV on plucked hairs points to the hairfollicle as an important site of infection for BetaPV(and possibly GammaPV) types [21]. In general, HPVentry is thought to require the initial binding to heparansulphate on the surface of basal cells and the basallamina [22], which facilitates subsequent associationwith a specific secondary receptor to allow virus entryand viral genome replication.
The establishment of viral genomes as low copy num-ber episomes in infected basal cells involves the dis-connection of viral and cellular DNA replication as aresult of the expression of the HPV E1 and E2 pro-teins. These proteins bind specifically to the viral originof replication (ori) to recruit cellular replication fac-tors. After binding to specific sequence motifs in theviral genome, the E2 protein can activate or represstranscription, depending on the context of these bind-ing sites and the nature of the associated cellular fac-tors [23]. This results in an initial burst of HPV genomeamplification immediately post-infection, followed bylow-level genome maintenance, in which viral and cel-lular DNA replication are thought to coincide during Sphase [24,25]. In addition to its important roles in repli-cation and transcription, E2 also has a key function inensuring viral genome partitioning during mitosis bytethering replicated HPV episomes to the host cell chro-mosome [26].
Although the high-risk AlphaPVs have genes thatdrive cell proliferation, resulting in productive infec-tions and neoplasia, these same genes generally act dif-ferently in the BetaPVs, which typically cause onlyasymptomatic infections in the general population. Cur-rent thinking suggests that BetaPVs infect a long-lived,slow-cycling epidermal stem (or stem-like) cell, such asmay be found in the hair follicle bulge (Figure 1B). Divi-sion of these infected cells populates the surroundingbasal layer with transiently amplifying cells that containviral episomes, with completion of the BetaPV life cycle
upon differentiation. Indeed, recent work has suggestedthat infection by BetaPV types 5 and 8 can enhancethe stem-like characteristics of the infected cell [27].These clinical and molecular observations suggest fun-damental differences in the way that BetaPV types andhigh-risk AlphaPV types cause cancers (see below).
When keratinocytes exit the epithelial basal layer, theylose their ability to proliferate and embark on a termi-nal differentiation programme as they progress throughthe spinous and granular layers. A key role of the E6and E7 proteins of all papillomaviruses is to ensure thetimely re-entry of the infected cell into the cell cycle toallow genome amplification in the mid-epithelial layers.However, the potential to induce keratinocyte prolifer-ation by E6 and E7 appears to differ between the HPVgenera, and in particular between Beta- and AlphaPVs[28]. Importantly, many HPV types appear able to drivecell cycle entry to allow genome amplification in thesuprabasal cell layers, but do not typically support exten-sive cell proliferation and progression through mitosis,as seen with the high-risk AlphaPVs. Thus althoughBetaPVs are not generally associated with neoplasiaafter infection, it remains to be fully established whetherBetaPV gene expression can become deregulated in cer-tain situations (eg in association with sun exposure),with corresponding changes in lesional pathology. Theviral E4 (and E5 in some genera) proteins are expressedin the spinous and granular layers, with the E4 proteinsof cutaneous HPV types from the Beta, Gamma andMu genera assembling into cytoplasmic inclusion gran-ules that are pathognomonic of disease (see Figure 2)[29]. The E4 and E5 proteins of the high-risk AlphaPVtypes contribute to genome amplification success, withE4 having a proposed role in virus release and dis-semination. In many HPV types, E5 is a membraneprotein located in the Golgi and endoplasmic reticu-lum (ER), but is absent in HPV types from the Betagenus. During the HPV life cycle, it is thought that theelevation in E2 levels that results from late promoteractivation during genome amplification leads eventu-ally to a down-regulation of E6 and E7 expression.This down-regulation allows terminal differentiation of
Figure 2. Histological features and distribution of viral markers of infection in an intra-epidermal proliferative lesion from an EV patient. (A)Haematoxylin and eosin (H&E) staining in a biopsy section from a representative intra-epidermal proliferative lesion typically found in EV,whose clinical appearance is shown in Figure 3. The epithelium displays the unequivocal histological features associated with productiveBetaPV infection, including the presence of enlarged cells with prominent grey pallor. In these areas, co-immunostaining for the viral proteinE4 (red) and the cell proliferation marker MCM7 (green) (B) reveals intense cytoplasmic E4 staining in the large ballooning keratinocytes andincreased MCM7 expression; scale bar= 100 μm; the boxed regions are magnified in the right-hand images (scale bar= 10 μm). Expressionof the major coat protein L1 (C) occurs in a subset of E4-positive cells in the upper epithelial layers; white arrows, L1 nuclear staining. Allsections were counterstained with DAPI (blue) to visualize cell nuclei.
the keratinocytes and expression of the HPV capsid pro-teins L1 and L2, followed by the assembly of infectiousvirions [30], which are shed with the dead keratinocytesto facilitate infection of a new host.
Cell cycle deregulationDuring productive infection, HPVs express a complexpattern of spliced mRNAs, with E6 and E7 ORFsoften being described as oncogenes. In cells infectedwith high-risk AlphaPVs, E6 expression can induceimportant physiological changes through interactionswith a number of different host proteins [31–34].Importantly for the high-risk AlphaPVs, E6 expressioninduces the proteolytic degradation of the ‘gate-keeper’tumour suppressor protein p53 by recruiting p53 tothe E6AP ubiquitin ligase complex [35]. At the sametime, E2F-dependent cell cycle arrest is inhibited bythe binding of the HPVE7 protein to members of theretinoblastoma suppressor protein family (pRb, p105,p130), causing the release of E2F transcription factors.This release results in the transcription of cellular genesinvolved in DNA replication, which, in conjunction withother E6/E7 functions (eg p21 binding [36]), finallyleads to progression towards S phase [37,38].
In general, it is believed that BetaPVs disrupt the hostcell cycle and its regulatory pathways less rigorously.For instance, the capacity of E6 to induce degradation ofp53 is less efficient for BetaPVs compared to AlphaPVs.Intriguingly however, some BetaPV types show prop-erties that are comparable with the high-risk AlphaPVHPV16. Several genus Beta HPV E6 proteins inhibitat least some p53 target genes, although perhaps notby the same mechanism or to the same degree as thehigh-risk genus Alpha HPV E6 proteins [39]. BetaPV38E7, for instance, can bind and degrade pRb to a levelsimilar to that of the AlphaPV16 E7 protein [40], whilethe HPV38E6 and E7 proteins can increase the lifespanof human keratinocytes by inducing telomerase activ-ity and by deregulating the cell cycle and senescenceprogrammes [41–43]. Moreover, these proteins lead tothe accumulation of a specific phosphorylated speciesof p53 that can selectively induce δ-Np73 [44,45], anisoform of the p53-related protein p73, which inhibitsthe capacity of p53 to induce the transcription of genesinvolved in apoptosis and growth suppression [45]. Inaddition, the activation of NF-κB is thought to pro-tect BetaPV immortalized human keratinocytes againstTNFα and UV-mediated apoptosis [46].
Studies on cell cycle regulation have further shownthat the E6 proteins of BetaPV5 and -8 also inhibit thetransforming growth factor β (TGFβ) signalling pathwaythrough degradation of the SMAD3 transcription factor[47]. TGFβ-triggered pathways normally lead to the syn-thesis of CDK inhibitors (p17, p21 and p27) that play acrucial role in the cell cycle. It can be postulated that spe-cific degradation of SMAD3 could negatively regulateinhibitors of the cell cycle and favour cell cycle progres-sion from G1 to S phase, allowing ‘unscheduled’ viralDNA replication and possibly also cell transformation.
Recently, evidence has been found that the BetaPV5and -8 E6 proteins can recruit MAML1, which causesrepression of the cutaneous tumour-suppressive Notchsignalling pathway, thereby disturbing the balancebetween Notch and the oncogene RAS in favour of RAS[48–51]. Abrogation of Notch signalling also leads toup-regulation of AP1-activating Wnt5a signalling inkeratinocytes, which is a classical skin carcinogenesispathway that is likely to favour cell proliferation overdifferentiation [52]. Interestingly, we find that, whencompared to the high-risk AlphaPV types, the HPV5genome is progressively lost from rapidly dividingkeratinocytes in tissue culture (CB and JD, unpublishedresults), supporting a model in which these virusesmay predispose to the accumulation of genetic errors,but with the loss of viral episomes as the neoplasticphenotype develops [53].
DNA repair and apoptosisAs the vast majority of cutaneous squamous cellcarcinomas (SCCs) occur on sun-exposed sites, it isgenerally assumed that BetaPVs somehow cooperatewith UV radiation in transforming cutaneous epithelialkeratinocytes. This assumption is supported by epi-demiological studies that suggest a joint effect of UVradiation and BetaPV infection [54]. Unlike anogenitalSCCs caused by AlphaPV, generally no integration ofBetaPV is observed in cutaneous SCCs and its pre-cursor lesions, which again suggests that a differentmechanism of cellular transformation is employed byBetaPV.
It is known that the E6 proteins of BetaPV8, -20,-22, -38, -76, -92 and -96 can protect UV-treatedkeratinocytes from apoptosis [55–58]. In transgenicmice, the E6 and E7 proteins from BetaPV38 con-tribute to cutaneous SCC development by makingkeratinocytes more susceptible to UV radiation [53,59],and we suspect that these proteins may cooperate withUV light in the development of actinic keratosis-likedysplastic lesions. Interestingly, these mice only devel-oped (pre)cancerous lesions when exposed to UVlight, whereas wild-type mice exposed to UV lightdid not [59]. Spontaneous development of papillomasand dysplasia occurred in transgenic mice expressingthe complete or parts of the early region (E2 or E6) ofBetaPV8 and, when exposed to a single dose of UV radi-ation, squamous cell carcinomas developed [60–62].Although both transgenic mouse models confirm the
combined effect of UV radiation and BetaPV expres-sion, in this model, the HPV transgenes were controlledby a keratin promoter which resulted in constitutivelyhigh levels of epithelial E6 and E7 expression. Thus,although Beta HPV gene expression can clearly pre-dispose to the development of cancers, expressionlevels during natural infection of immunocompetentindividuals is likely to be less extensive and at lowerlevels. This very likely underlies differences in theperceived cancer risk between immunocompetent indi-viduals and those that are immunosuppressed or whohave a genetic susceptibility to infection.
In addition to their interference with cell cycle reg-ulatory pathways, as previously outlined by BouwesBavinck and Feltkamp in 2004 [63], BetaPVs mayalso confer a carcinogenic effect via their ability toinhibit normal DNA repair and apoptosis pathways inUV-damaged cells, eg by up-regulating δ-Np73, asmentioned above. Interestingly, BetaPV5 E6 inhibitsapoptosis via the degradation of Bak, a tumour suppres-sor protein that induces apoptosis [64,65]. In addition,BetaPV E6 proteins have also been shown to act indi-rectly to disrupt p53-mediated repair of UVB-damagedDNA by reducing the levels of the proteins ATR, ATMand p300 [66–69].
A
B
Understanding Disease
Figure 3. Clinical manifestation of epidermodysplasia verruci-formis. (A) Pityriasis-like scaly lesions on the back of an EV patient.(B) Hyperkeratotic verrucous plaques typical of EV on the dorsalaspect of the right hand of the same patient.
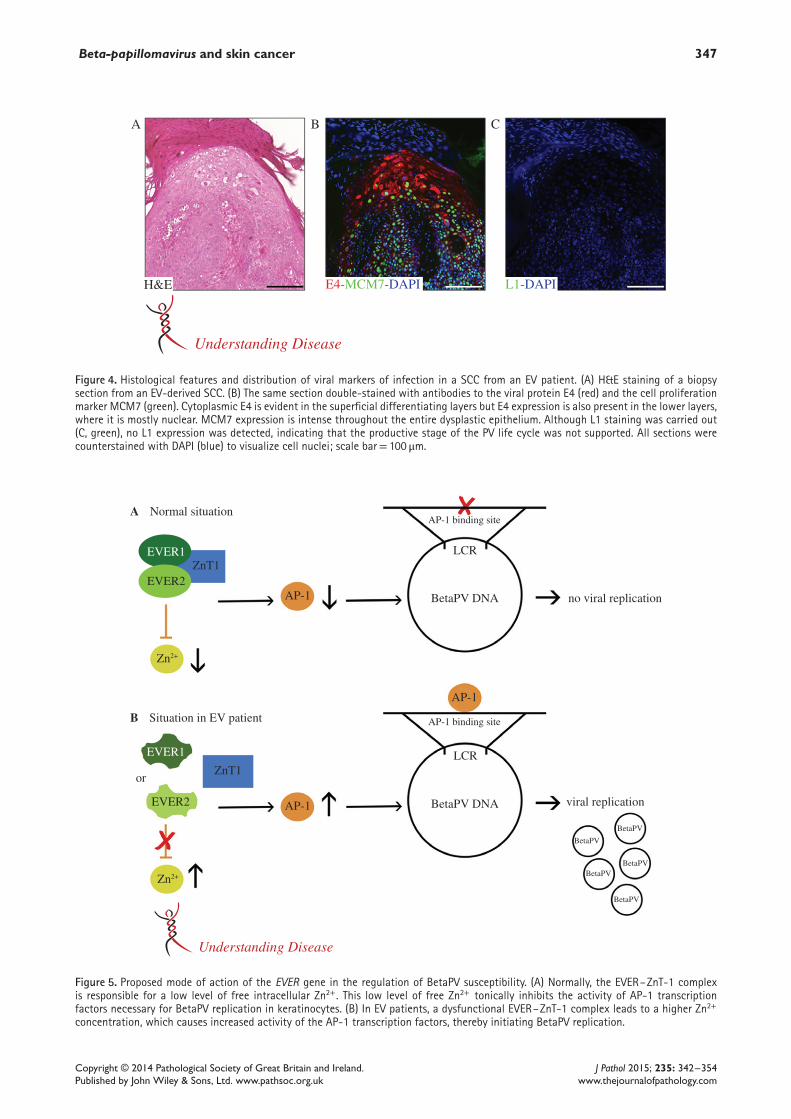
Figure 4. Histological features and distribution of viral markers of infection in a SCC from an EV patient. (A) H&E staining of a biopsysection from an EV-derived SCC. (B) The same section double-stained with antibodies to the viral protein E4 (red) and the cell proliferationmarker MCM7 (green). Cytoplasmic E4 is evident in the superficial differentiating layers but E4 expression is also present in the lower layers,where it is mostly nuclear. MCM7 expression is intense throughout the entire dysplastic epithelium. Although L1 staining was carried out(C, green), no L1 expression was detected, indicating that the productive stage of the PV life cycle was not supported. All sections werecounterstained with DAPI (blue) to visualize cell nuclei; scale bar= 100 μm.
Zn2+
Zn2+
EVER2 AP-1
AP-1
AP-1
BetaPV DNA
BetaPV DNA
Normal situation
Situation in EV patient
or
LCR
no viral replication
LCR
AP-1 binding site
AP-1 binding site
viral replication
BetaPV
BetaPVBetaPV
BetaPV
BetaPV
EVER1
ZnT1
ZnT1EVER1
EVER2
Understanding Disease
A
B
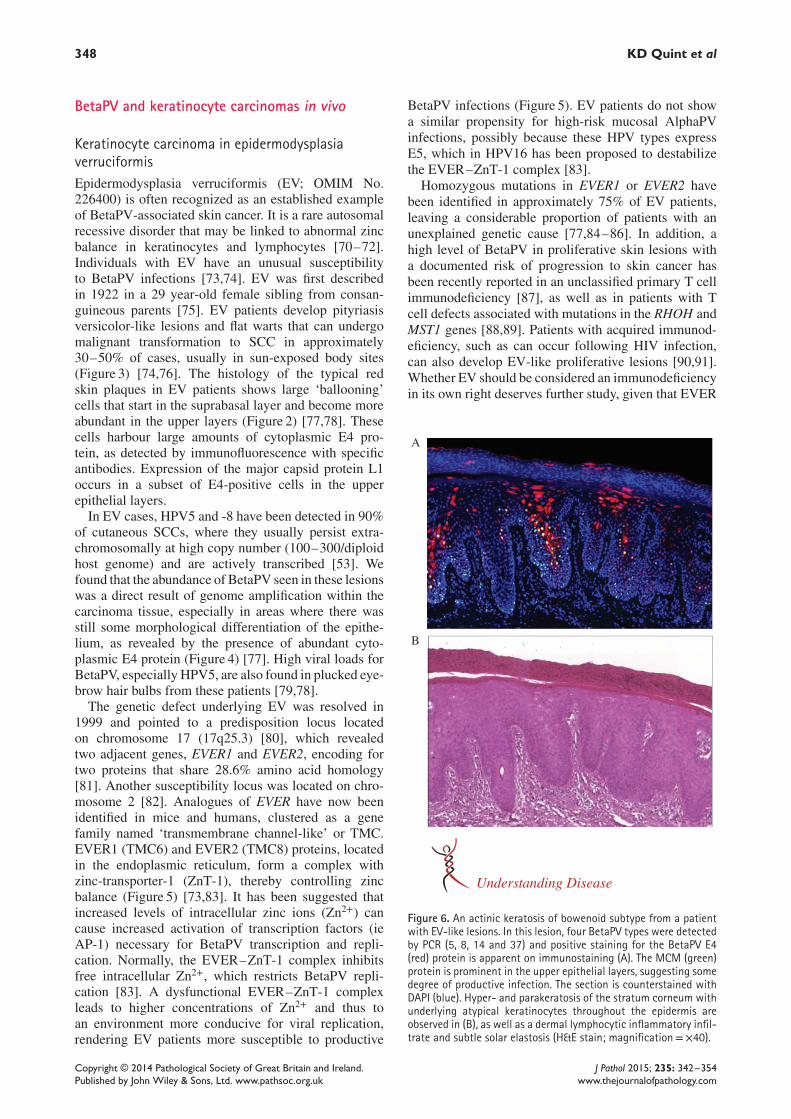
Figure 5. Proposed mode of action of the EVER gene in the regulation of BetaPV susceptibility. (A) Normally, the EVER–ZnT-1 complexis responsible for a low level of free intracellular Zn2+. This low level of free Zn2+ tonically inhibits the activity of AP-1 transcriptionfactors necessary for BetaPV replication in keratinocytes. (B) In EV patients, a dysfunctional EVER–ZnT-1 complex leads to a higher Zn2+
concentration, which causes increased activity of the AP-1 transcription factors, thereby initiating BetaPV replication.
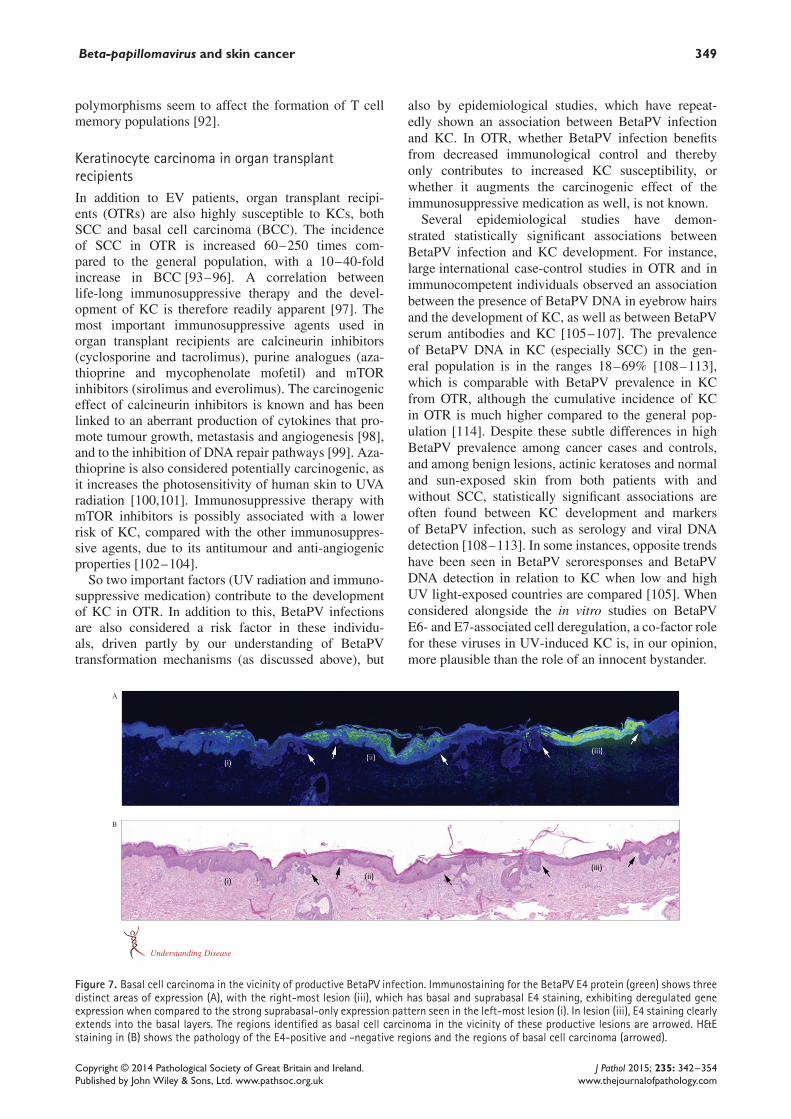
Keratinocyte carcinoma in epidermodysplasiaverruciformisEpidermodysplasia verruciformis (EV; OMIM No.226400) is often recognized as an established exampleof BetaPV-associated skin cancer. It is a rare autosomalrecessive disorder that may be linked to abnormal zincbalance in keratinocytes and lymphocytes [70–72].Individuals with EV have an unusual susceptibilityto BetaPV infections [73,74]. EV was first describedin 1922 in a 29 year-old female sibling from consan-guineous parents [75]. EV patients develop pityriasisversicolor-like lesions and flat warts that can undergomalignant transformation to SCC in approximately30–50% of cases, usually in sun-exposed body sites(Figure 3) [74,76]. The histology of the typical redskin plaques in EV patients shows large ‘ballooning’cells that start in the suprabasal layer and become moreabundant in the upper layers (Figure 2) [77,78]. Thesecells harbour large amounts of cytoplasmic E4 pro-tein, as detected by immunofluorescence with specificantibodies. Expression of the major capsid protein L1occurs in a subset of E4-positive cells in the upperepithelial layers.
In EV cases, HPV5 and -8 have been detected in 90%of cutaneous SCCs, where they usually persist extra-chromosomally at high copy number (100–300/diploidhost genome) and are actively transcribed [53]. Wefound that the abundance of BetaPV seen in these lesionswas a direct result of genome amplification within thecarcinoma tissue, especially in areas where there wasstill some morphological differentiation of the epithe-lium, as revealed by the presence of abundant cyto-plasmic E4 protein (Figure 4) [77]. High viral loads forBetaPV, especially HPV5, are also found in plucked eye-brow hair bulbs from these patients [79,78].
The genetic defect underlying EV was resolved in1999 and pointed to a predisposition locus locatedon chromosome 17 (17q25.3) [80], which revealedtwo adjacent genes, EVER1 and EVER2, encoding fortwo proteins that share 28.6% amino acid homology[81]. Another susceptibility locus was located on chro-mosome 2 [82]. Analogues of EVER have now beenidentified in mice and humans, clustered as a genefamily named ‘transmembrane channel-like’ or TMC.EVER1 (TMC6) and EVER2 (TMC8) proteins, locatedin the endoplasmic reticulum, form a complex withzinc-transporter-1 (ZnT-1), thereby controlling zincbalance (Figure 5) [73,83]. It has been suggested thatincreased levels of intracellular zinc ions (Zn2+) cancause increased activation of transcription factors (ieAP-1) necessary for BetaPV transcription and repli-cation. Normally, the EVER–ZnT-1 complex inhibitsfree intracellular Zn2+, which restricts BetaPV repli-cation [83]. A dysfunctional EVER–ZnT-1 complexleads to higher concentrations of Zn2+ and thus toan environment more conducive for viral replication,rendering EV patients more susceptible to productive
BetaPV infections (Figure 5). EV patients do not showa similar propensity for high-risk mucosal AlphaPVinfections, possibly because these HPV types expressE5, which in HPV16 has been proposed to destabilizethe EVER–ZnT-1 complex [83].
Homozygous mutations in EVER1 or EVER2 havebeen identified in approximately 75% of EV patients,leaving a considerable proportion of patients with anunexplained genetic cause [77,84–86]. In addition, ahigh level of BetaPV in proliferative skin lesions witha documented risk of progression to skin cancer hasbeen recently reported in an unclassified primary T cellimmunodeficiency [87], as well as in patients with Tcell defects associated with mutations in the RHOH andMST1 genes [88,89]. Patients with acquired immunod-eficiency, such as can occur following HIV infection,can also develop EV-like proliferative lesions [90,91].Whether EV should be considered an immunodeficiencyin its own right deserves further study, given that EVER
A
B
Understanding Disease
Figure 6. An actinic keratosis of bowenoid subtype from a patientwith EV-like lesions. In this lesion, four BetaPV types were detectedby PCR (5, 8, 14 and 37) and positive staining for the BetaPV E4(red) protein is apparent on immunostaining (A). The MCM (green)protein is prominent in the upper epithelial layers, suggesting somedegree of productive infection. The section is counterstained withDAPI (blue). Hyper- and parakeratosis of the stratum corneum withunderlying atypical keratinocytes throughout the epidermis areobserved in (B), as well as a dermal lymphocytic inflammatory infil-trate and subtle solar elastosis (H&E stain; magnification=×40).
polymorphisms seem to affect the formation of T cellmemory populations [92].
Keratinocyte carcinoma in organ transplantrecipientsIn addition to EV patients, organ transplant recipi-ents (OTRs) are also highly susceptible to KCs, bothSCC and basal cell carcinoma (BCC). The incidenceof SCC in OTR is increased 60–250 times com-pared to the general population, with a 10–40-foldincrease in BCC [93–96]. A correlation betweenlife-long immunosuppressive therapy and the devel-opment of KC is therefore readily apparent [97]. Themost important immunosuppressive agents used inorgan transplant recipients are calcineurin inhibitors(cyclosporine and tacrolimus), purine analogues (aza-thioprine and mycophenolate mofetil) and mTORinhibitors (sirolimus and everolimus). The carcinogeniceffect of calcineurin inhibitors is known and has beenlinked to an aberrant production of cytokines that pro-mote tumour growth, metastasis and angiogenesis [98],and to the inhibition of DNA repair pathways [99]. Aza-thioprine is also considered potentially carcinogenic, asit increases the photosensitivity of human skin to UVAradiation [100,101]. Immunosuppressive therapy withmTOR inhibitors is possibly associated with a lowerrisk of KC, compared with the other immunosuppres-sive agents, due to its antitumour and anti-angiogenicproperties [102–104].
So two important factors (UV radiation and immuno-suppressive medication) contribute to the developmentof KC in OTR. In addition to this, BetaPV infectionsare also considered a risk factor in these individu-als, driven partly by our understanding of BetaPVtransformation mechanisms (as discussed above), but
also by epidemiological studies, which have repeat-edly shown an association between BetaPV infectionand KC. In OTR, whether BetaPV infection benefitsfrom decreased immunological control and therebyonly contributes to increased KC susceptibility, orwhether it augments the carcinogenic effect of theimmunosuppressive medication as well, is not known.
Several epidemiological studies have demon-strated statistically significant associations betweenBetaPV infection and KC development. For instance,large international case-control studies in OTR and inimmunocompetent individuals observed an associationbetween the presence of BetaPV DNA in eyebrow hairsand the development of KC, as well as between BetaPVserum antibodies and KC [105–107]. The prevalenceof BetaPV DNA in KC (especially SCC) in the gen-eral population is in the ranges 18–69% [108–113],which is comparable with BetaPV prevalence in KCfrom OTR, although the cumulative incidence of KCin OTR is much higher compared to the general pop-ulation [114]. Despite these subtle differences in highBetaPV prevalence among cancer cases and controls,and among benign lesions, actinic keratoses and normaland sun-exposed skin from both patients with andwithout SCC, statistically significant associations areoften found between KC development and markersof BetaPV infection, such as serology and viral DNAdetection [108–113]. In some instances, opposite trendshave been seen in BetaPV seroresponses and BetaPVDNA detection in relation to KC when low and highUV light-exposed countries are compared [105]. Whenconsidered alongside the in vitro studies on BetaPVE6- and E7-associated cell deregulation, a co-factor rolefor these viruses in UV-induced KC is, in our opinion,more plausible than the role of an innocent bystander.
B
A
Understanding Disease
(i)
(i)(ii)
(iii)
(ii)(iii)
Figure 7. Basal cell carcinoma in the vicinity of productive BetaPV infection. Immunostaining for the BetaPV E4 protein (green) shows threedistinct areas of expression (A), with the right-most lesion (iii), which has basal and suprabasal E4 staining, exhibiting deregulated geneexpression when compared to the strong suprabasal-only expression pattern seen in the left-most lesion (i). In lesion (iii), E4 staining clearlyextends into the basal layers. The regions identified as basal cell carcinoma in the vicinity of these productive lesions are arrowed. H&Estaining in (B) shows the pathology of the E4-positive and -negative regions and the regions of basal cell carcinoma (arrowed).
When considering the involvement of BetaPV in thedevelopment of KC, it is important to examine wherein the carcinogenic process BetaPV may play a role.Interestingly, the BetaPV types 5 and 8, which are asso-ciated with KC in EV patients, are detected at higherrates in actinic keratoses compared to SCCs [115]. Fur-thermore, BetaPV viral load or copy number was foundto be significantly higher in actinic keratoses comparedto SCCs, suggesting some degree of productive infection
in such premalignant lesions [113]. These observationswere supported by the detection of BetaPV E4 and L1protein expression in four of 19 actinic keratoses, as wellas at the pathological borders of SCCs and BCCs froman OTR-derived skin tumour series [116] (Figure 6).Interestingly, increased expression of MCM7, a cellularreplication marker, was found to extend into the upperepithelial layers in all the E4-positive areas, a pattern
immune competent patientA
B immune suppressed patient
UV-B
UV-B
DNA-repair
DNADamage
Apoptosis
Keratinocyte
Keratinocytecancer
immune system
immune systemsuppressed
BetaPV
BetaPV
BetaPV
BetaPV
sun
sun
Understanding Disease
DNA-repair
DNADamage
Apoptosis
Figure 8. Proposed mechanism for BetaPV infection and keratinocyte carcinoma development. In immunocompetent individuals (A), BetaPVinfections are suppressed by the immune system. In this case, only low levels of BetaPV protein will be present to interact with host cellproteins involved in DNA damage repair and apoptosis, and genotoxic damage imposed by UV radiation will be managed adequately. In thepresence of immunosuppression, however (B), productive BetaPV infection occurs and sufficient amounts of BetaPV protein, particularlyE6, are expressed, inhibiting DNA damage repair mechanisms and apoptosis. As a result, genomic instability develops in the infectedkeratinocytes, which may lead ultimately to the development of keratinocye carcinoma.
of expression that typically identifies sites of produc-tive viral infection. The close association of viral geneexpression with at least some actinic keratoses is con-sistent with an early causal role for BetaPV in the devel-opment of such lesions in OTR. It is apparent, however,that not all studies have observed marked differences inDNA copy number between normal skin/benign lesionsand (pre-)cancerous skin [117,118], which leads us tosuspect that actinic keratoses may in fact be a hetero-geneous group with regard to BetaPV copy number andgene expression patterns.
Occasionally, an association between BetaPV infec-tion and BCC has been observed, and in particular this isnoticeable when HPV20 serological responses and BCCare compared [119–121]. In the vicinity of sporadicBCCs that carry a high viral BetaPV load, E4 and L1expression is sometimes observed (Figure 7). In the caseof E4, the pattern of expression suggests some degreeof life cycle deregulation, possibly as a result of accu-mulation of genetic changes in the infected basal cell oran over-expression of viral early genes (cf. high-gradedisease caused by high-risk AlphaPV types) (Figure 7).We suspect, in this case, that the different levels of E4expression in the different lesional areas may be linkedto different viral copy numbers (and consequently differ-ent levels of expression) in the immunosuppressed hostbackground. Interestingly, certain BetaPV types, such asBetaPV20, have been shown to up-regulate the p16INK4a
and Akt–PI3K pathway and thus potentially interferewith the cell cycle pathways involved in progression toBCC [122]. Overall, the evidence that HPV is involvedin BCC carcinogenesis is, however, much weaker thanthe possible involvement of these HPV types in cuta-neous SCC carcinogenesis.
Conclusion
Although the role of BetaPV in KC development is notyet fully understood, both experimental and epidemio-logical evidence suggests a carcinogenic role of BetaPVin this type of skin cancer. It is assumed that BetaPVsact as a co-factor in addition to UV-radiation early inthe carcinogenic process (Figure 8), and that they act in amanner that is different from that which has been workedout for high-risk AlphaPV. For instance, they generallydo not integrate into the human cellular DNA and donot seem to be involved in maintenance of the malig-nant phenotype of KC. Rather, BetaPV types have anestablished capability to impair DNA repair and apopto-sis following excessive UV exposure. As result of this,persistent presence and persistent expression of theseBetaPV types in the skin could over a long period oftime facilitate the accumulation of genetic errors inthe infected cell, thereby facilitating the developmentof actinic keratoses and KC, especially at sun-exposedsites, where the need for functional DNA repair mecha-nisms is paramount (Figure 8).
If we believe that BetaPV types have evolved to persistin a long-lived, slow-cycling stem cell, then it is per-haps not surprising that, unlike the high-risk AlphaPVtypes, the BetaPV episome is incapable of being reli-ably maintained in faster-cycling cancer cells. Given thisscenario, epidemiological studies are unlikely to detectBetaPV DNA in each cell of end-point cancers. As dis-cussed in this review, different BetaPV types have dif-ferent effects on cellular mechanisms, and a combina-tion of these HPV types may further increase the risk ofKC. Although conclusive demonstration of a hit-and-runmechanism of cancer development remains to be con-clusively demonstrated for any virus, the argument thatBetaPV types could work in this way is substantial.We sincerely hope that further research will resolve thisquestion, and provide further insight into the involve-ment of BetaPV in KC carcinogenesis.
Acknowledgements
We acknowledge the help of Heather Griffin (NationalInstitute for Medical Research, London, and Universityof Cambridge, UK) in developing the E4/multiple stain-ing regime, and in preparing some of the images used inthis review, including Figure 7. Thanks also to JenniferMcNiff (Yale University, USA) for the tissue section thatwas stained for E4 and MCM shown in Figure 6. Thehelp of Wai Han Yau from NIMR, London, in the prepa-ration of Figure 1, and Thomas Donker, medical photog-rapher, Leiden University Medical Centre, for producingFigures 5 and 8, is also acknowledged. JD is supportedby the UK Medical Research Council. MG is supportedby the AIRC (Associazione Italiana per la Ricerca sulCancro).
Author contributions
KQ wrote the manuscript and all other authors reviewedand improved it; JD and CB provided histological pic-tures; RG provided the clinical imaging and checked thehistology and text regarding EV; MK, CB and MG con-tributed to and checked the molecular part of the review;JB was the clinical expert; and MF and JD coordinatedand supervised the writing of the manuscript and con-tributed to the overall opinions presented in the text.
References1. Bernard HU, Burk RD, Chen Z, et al. Classification of papillo-
maviruses (PVs) based on 189 PV types and proposal of taxonomicamendments. Virology 2010; 401: 70–79.
2. Kocjan BJ, Steyer A, Sagadin M, et al. Novel human papillomavirustype 174 from a cutaneous squamous cell carcinoma. Genome
Announc 2013; 1: e00445–13.3. de Villiers EM, Fauquet C, Broker TR, et al. Classification of
86. Zavattaro E, Azzimonti B, Mondini M, et al. Identification of defec-tive Fas function and variation of the perforin gene in an epi-dermodysplasia verruciformis patient lacking EVER1 and EVER2
mutations. J Invest Dermatol 2008; 128: 732–735.87. Landini MM, Borgogna C, Peretti A, et al. Alpha- and
Beta-papillomavirus infection in a young patient with an unclassi-fied primary T-cell immunodeficiency and multiple mucosal andcutaneous lesions. J Am Acad Dermatol 2014; 71: 108–115.
88. Crequer A, Picard C, Patin E, et al. Inherited MST1 deficiencyunderlies susceptibility to EV-HPV infections. PLoS One 2012; 7:e44010.
89. Crequer A, Troeger A, Patin E, et al. Human RHOH deficiencycauses T cell defects and susceptibility to EV-HPV infections. J Clin
verruciformis due to multiple and unusual HPV infection amongvertically infected, HIV-positive adolescents in Zimbabwe. ClinInfect Dis 2012; 54: e119–123.
91. Meireles SI, de Andrade SM, de Lima CLJ, et al. Epidermodyspla-sia verruciformis in a young man with HIV since birth – case report.An Bras Dermatol 2013; 88: 190–192.
92. Crequer A, Picard C, Pedergnana V, et al. EVER2 deficiency isassociated with mild T-cell abnormalities. J Clin Immunol 2013;33: 14–21.
93. Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ trans-plantation. N Engl J Med 2003; 348: 1681–1691.
94. Hartevelt MM, Bavinck JN, Kootte AM, et al. Incidence of skin can-cer after renal transplantation in The Netherlands. Transplantation1990; 49: 506–509.
95. Moloney FJ, Comber H, O’Lorcain P, et al. A population-basedstudy of skin cancer incidence and prevalence in renal transplantrecipients. Br J Dermatol 2006; 154: 498–504.
96. Tessari G, Naldi L, Boschiero L, et al. Incidence and clinical pre-dictors of a subsequent nonmelanoma skin cancer in solid organtransplant recipients with a first non-melanoma skin cancer: a mul-ticenter cohort study. Arch Dermatol 2010; 146: 294–299.
97. Terhorst D, Drecoll U, Stockfleth E, et al. Organ transplant recipi-ents and skin cancer: assessment of risk factors with focus on sunexposure. Br J Dermatol 2009; 161(suppl 3): 85–89.
98. Guba M, Graeb C, Jauch KW, et al. Pro- and anti-cancer effectsof immunosuppressive agents used in organ transplantation. Trans-
plantation 2004; 77: 1777–1782.99. Ori Y, Herman-Edelstein M, Zingerman B, et al. Effect of immuno-
suppressive drugs on spontaneous DNA repair in human periph-eral blood mononuclear cells. Biomed Pharmacother 2012; 66:409–413.
100. O’Donovan P, Perrett CM, Zhang X, et al. Azathioprine and UVAlight generate mutagenic oxidative DNA damage. Science 2005;309: 1871–1874.
101. Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatmentphotosensitizes human skin to ultraviolet A radiation. Br J Dermatol2008; 159: 198–204.
102. Euvrard S, Morelon E, Rostaing L, et al. Sirolimus and secondaryskin-cancer prevention in kidney transplantation. N Engl J Med2012; 367: 329–339.
103. Hoogendijk-van den Akker JM, Harden PN, Hoitsma AJ, et al.
Two-year randomized controlled prospective trial converting treat-ment of stable renal transplant recipients with cutaneous invasivesquamous cell carcinomas to sirolimus. J Clin Oncol 2013; 31:1317–1323.
104. Kauffman HM, Cherikh WS, Cheng Y, et al. Maintenance immuno-suppression with target-of-rapamycin inhibitors is associated with areduced incidence of de novo malignancies. Transplantation 2005;80: 883–889.
105. Bouwes Bavinck JN, Neale RE, Abeni D, et al. Multicenter study ofthe association between betapapillomavirus infection and cutaneoussquamous cell carcinoma. Cancer Res 2010; 70: 9777–9786.
106. Karagas MR, Nelson HH, Sehr P, et al. Human papillomavirusinfection and incidence of squamous cell and basal cell carcinomasof the skin. J Natl Cancer Inst 2006; 98: 389–395.
107. Proby CM, Harwood CA, Neale RE, et al. A case-control studyof betapapillomavirus infection and cutaneous squamous cell car-cinoma in organ transplant recipients. Am J Transpl 2011; 11:1498–1508.
108. Asgari MM, Kiviat NB, Critchlow CW, et al. Detection of humanpapillomavirus DNA in cutaneous squamous cell carcinoma amongimmunocompetent individuals. J Invest Dermatol 2008; 128:1409–1417.
109. Forslund O, Ly H, Reid C, et al. A broad spectrum of humanpapillomavirus types is present in the skin of Australian patientswith non-melanoma skin cancers and solar keratosis. Br J Dermatol2003; 149: 64–73.
110. Forslund O, Iftner T, Andersson K, et al. Cutaneous human papillo-maviruses found in sun-exposed skin: Beta-papillomavirus species2 predominates in squamous cell carcinoma. J Infect Dis 2007; 196:876–883.
111. Meyer T, Arndt R, Christophers E, et al. Importance of humanpapillomaviruses for the development of skin cancer. Cancer Detect
Prev 2001; 25: 533–547.112. Patel AS, Karagas MR, Perry AE, et al. Exposure profiles and
human papillomavirus infection in skin cancer: an analysis of 25genus Beta types in a population-based study. J Invest Dermatol2008; 128: 2888–2893.
113. Weissenborn SJ, Nindl I, Purdie K, et al. Human papillomavirusDNA loads in actinic keratoses exceed those in non-melanoma skincancers. J Invest Dermatol 2005; 125: 93–97.
114. Mackintosh LJ, de Koning MN, Quint WG, et al. Presence of betahuman papillomaviruses in non-melanoma skin cancer from organtransplant recipients and immunocompetent patients in the west ofScotland. Br J Dermatol 2009; 161: 56–62.
115. Pfister H, Fuchs PG, Majewski S, et al. High prevalence of epider-modysplasia verruciformis-associated human papillomavirus DNAin actinic keratoses of the immunocompetent population. Arch Der-
matol Res 2003; 295: 273–279.116. Borgogna C, Lanfredini S, Peretti A, et al. Improved detection
reveals active beta-papillomavirus infection in skin lesions fromkidney transplant recipients. Mod Pathol 2014; 27: 1101–1115.
117. Harwood CA, Surentheran T, McGregor JM, et al. Human papil-lomavirus infection and non-melanoma skin cancer in immunosup-pressed and immunocompetent individuals. J Med Virol 2000; 61:289–297.
118. Shamanin V, Glover M, Rausch C, et al. Specific types of humanpapillomavirus found in benign proliferations and carcinomas ofthe skin in immunosuppressed patients. Cancer Res 1994; 54:4610–4613.
119. Bzhalava D, Guan P, Franceschi S, et al. A systematic review of theprevalence of mucosal and cutaneous human papillomavirus types.Virology 2013; 445: 224–231.
120. Feltkamp MC, Broer R, di Summa FM, et al. Seroreactivity to epi-dermodysplasia verruciformis-related human papillomavirus typesis associated with non-melanoma skin cancer. Cancer Res 2003; 63:2695–2700.
121. Iannacone MR, Gheit T, Waterboer T, et al. Case-control study ofcutaneous human papillomavirus infection in basal cell carcinomaof the skin. J Invest Dermatol 2013; 133: 1512–1520.
122. Paolini F, Carbone A, Benevolo M, et al. Human papillomaviruses,p16INK4a and Akt expression in basal cell carcinoma. J Exp ClinCancer Res 2011; 30: 108.