Bindu Bhargavi1 Vivek Kumar Gupta3 and Girish Radhakrishnan1
1National Institute of Animal Biotechnology Hyderabad 500049 India2Ella Foundation Genome Valley Turkapally Shameerpet Hyderabad 500078 India3Central Institute for Research on Goats Makhdoom Mathura 281122 India
Correspondence should be addressed to Girish Radhakrishnan girishniaborgin
Received 11 April 2016 Revised 26 May 2016 Accepted 29 May 2016
Academic Editor Shen Liang Chen
Copyright copy 2016 Sarwar Azam et al This is an open access article distributed under the Creative Commons Attribution Licensewhich permits unrestricted use distribution and reproduction in any medium provided the original work is properly cited
Brucellosis is themost frequent zoonotic diseaseworldwide with over 500000 newhuman infections every yearBrucellamelitensisthe most virulent species in humans primarily affects goats and the zoonotic transmission occurs by ingestion of unpasteurizedmilk products or through direct contact with fetal tissues Brucellosis is endemic in India but no information is available onpopulation structure and genetic diversity of Brucella spp in India We performed multilocus sequence typing of four Bmelitensisstrains isolated from naturally infected goats from India For more detailed genetic characterization we carried out whole genomesequencing and comparative genome analysis of one of the Bmelitensis isolates Bm IND1 Genome analysis identified 141 uniqueSNPs 78 VNTRs 51 Indels and 2 putative prophage integrations in the Bm IND1 genome Our data may help to develop improvedepidemiological typing tools and efficient preventive strategies to control brucellosis
1 Introduction
Brucellosis is a worldwide zoonotic disease that accounts forhuge loses to the livestock sector and poses a serious threat topublic health The disease is caused by bacteria of the genusBrucella a member of the 120572-2 Proteobacteria [1] Brucellaeare Gram-negative facultative intracellular bacteria that caninfect a wide range of domestic and wild animals as well ashumans Six classical species were initially recognized withinthe genus Brucella namely B abortus Bmelitensis B suis Bovis B canis and B neotomae [2] Brucella invades and repli-cates in professional phagocytic cells such as macrophagesand dendritic cells as well as nonprofessional phagocytes suchas trophoblasts [3ndash5] Brucella mechanisms and virulencefactors that mediate invasion and intracellular persistencehave been poorly characterized
Brucellosis remains endemic and is a reemerging diseasein many regions of the world including Latin AmericaMiddle East Africa Central Asia and the Mediterraneanbasin that affects human health and animal productivity
The disease leads to a significant detrimental effect on localeconomies resulting in the perpetuation of poverty [6]Brucellosis is a serious veterinary and public health problemin India and the disease is reported in cattle buffalo sheepgoats pigs camel dogs and humans [7] Brucellosis is anendemic disease in India and the country experienced asharply increasing rate of human brucellosis in recent yearsand the species of main concern is Bmelitensis
B melitensis the most virulent species in humans pri-marily affects goats and the zoonotic transmission occurs byingestion of unpasteurized milk products or through directcontact with fetal tissues Genetic diversity and populationstructure of Brucella spp remain unknown in India Multi-locus sequence typing (MLST) has been considered as therobust tool for dissecting genetic diversity and populationstructure within the bacterial species The established MLSTschema for Brucella spp employed nine highly distinctgenomic loci [8] However MLST resolution is limited andoften fails to differentiate closely related strains With theadvent of next generation sequencing whole genome offers
Hindawi Publishing CorporationInternational Journal of GenomicsVolume 2016 Article ID 3034756 13 pageshttpdxdoiorg10115520163034756
2 International Journal of Genomics
new opportunities to analyze the genetic diversity amongthe groups [9] Genome sequencing and analysis of Brucellaspp from diverse hosts and geographical regions have beenreported [9] Until now 61 genomes of Bmelitensis have beensequenced and made available in the GenBank B melitensiscontains more than 3000 genes that are distributed overtwo circular chromosomes These sequenced species serve asvast resources for comparative genomics and understandingthe evolutionary history Comparative genomics will provideinsights into the virulence mechanisms of the Brucella sppsuch as novel genomic islands and integration of prophagesand SNPs that regulate the expression of certain genes oraffect the function of important virulence-associated pro-teins One of the previous studies demonstrated conservationof genes and genomic islands across the different Brucellaspp [10] In a recent study the evolutionary relationship of Bmelitensis on the basis of whole genome SNPs revealed spatialclustering of Bmelitensis isolates into five genotypes [11] Allthe Asian isolates of B melitensis clustered into genotype IIwhereas the isolates from Europe and America clustered intogenotypes IV and V respectively
We performed genotyping by MLST on B melitensisstrains isolated from naturally infected goats To performdetailed genetic characterization and comparative genomeanalysis we performed whole genome sequencing of one ofthe strains B melitensis IND1 using the Illumina platformThe analysis revealed the extent of genetic variation of Bmelitensis IND1 in comparison to B melitensis isolates fromother geographical locationsData generated fromour studiesmay help to develop new diagnostic assays based on stablemarkers such as SNPs (Single Nucleotide Polymorphisms)and Variable Number of Tandem Repeats (VNTRs) formolecular epidemiological studies Identification of SNPsIndels and novel phage integration sites will provide insightsinto the virulence mechanisms of this stealthy pathogenwhich could ultimately lead to the development of noveltherapeutic and preventive strategies to control brucellosis
2 Materials and Methods
21 Isolation of B melitensis For isolation of Brucella mate-rials from four different sources are listed in the Supplemen-tary Table 1 (in Supplementary Material available online athttpdxdoiorg10115520163034756) Samples were inoc-ulated on sterile plates of Brucella selective agar containingHemin and Vitamin K
1media (Hi Media India) and incu-
bated at 37∘C for 48 hours The plates were observed at every24 hours for the development of growth After obtainingthe growth the colonies suspected for Brucella on the basisof cultural characteristics were selected and streaked againon plates containing Brucella selective agar with Hemin andVitamin K
1and incubated at 37∘C for 2 days to obtain the
pure culture
22 Biotyping of Brucella Isolates Cultures showing typicalBrucella characteristics were subjected to biotyping tech-niques such as H
2S production growth in the presence
of thionin and basic fuchsin (10ndash40120583gmL) dye incorpo-rated into tryptic soy agar at different concentrations andCO2requirement immediately after the primary isolation
as described [12] Lead acetate strips were used to identifythe production of H
2S during growth and the growth was
evaluated on media containing streptomycin (25 120583gmL)to discriminate the isolates from vaccine strain Rev1 asdescribed [13]
Genomic DNA of all five strains was isolated using theWizard Genomic DNA Purification Kit (Promega USA)Isolated DNA was used for polymerase chain reactions toamplify 16S rRNA and the Omp 31 gene for the confirmatoryidentification of Brucella melitensis using the Taq PCRmastermix kit (Qiagen) 16S rRNA is specific to the genus Brucellawhile Omp 31 is a species-specific gene to Brucella melitensis[14ndash16]
23 MLST Analysis of B melitensis Isolates For MLSTanalysis 4396 nucleotide sequences spanning nine genomicfragments from Brucella were selected as described [8] Ofthe nine loci seven belong to classical housekeeping genesone locus derived from the outer membrane protein 25gene and one from an intergenic region Genomic DNA wasisolated from Bm IND isolates using the Wizard GenomicDNA Purification Kit (Promega) Genomic fragments wereamplified by PCR using the following cycling parameters94∘C for 2min 35 cycles of 94∘C for 30 sec 53∘C for 30 secand 72∘C for 1min and 72∘C for 5min Primers used forMLST analysis are listed in the Additional file 13 An aliquotof the PCR amplicons was analyzed by 1 agarose gel andphotographed Remaining PCR products were purified andsubjected to Sanger sequencing using the forward and reverseprimers that were used for PCR amplifications Editing ofthe sequences and generation of contigs from forward andreverse sequences was performed using Lasergene 8 software(DNA Star USA) To perform the phylogenetic analysisnine genomic fragments mentioned above were fetched fromrepresentative Brucella species and the loci were amplifiedin silico using the MLST primers All the sequences wereconcatenated to identify the allelic profile with the helpof Brucellabase [17] and the concatenated sequences weresubjected to multiple sequence alignment using MAFFTversion 7123b [18] Phylogenetic analyses were performedwith RAxML version 812 [19] using GTRGAMMAmodel ofevolutionThe phylogram was visualized using Dendroscopeversion 3 [20]
24 Genome Sequencing Assembly and Annotations Thecomplete genome of Brucella melitensis IND1 (Bm IND1)was sequenced using Illumina technology The sequenceddata have been deposited at DDBJEMBLGenBank underthe accession number JMKL00000000 [21] Paired end datagenerated were filtered for low quality reads using in-housescript After preprocessing high quality data were used tomake a scaffold level assembly using the SOAPdenovo version201 assembler [22] Scaffolds were further mapped withraw reads using bowtie2 version 224 [23] and coverage ateach base was calculated using SAMtools version 0119 [24]
International Journal of Genomics 3
Graphs for coverage analysis were plotted using GNUplotversion 46 (httpwwwgnuplotinfo)
Completely sequenced genomes ofBmelitensis such as Bmelitensis 16M B melitensis M28 B melitensis ATCC23457andBmelitensisNIwere initially considered as candidates forthe template to construct the chromosomal assembly of BmIND1 The raw data of Bm IND1 were aligned onto all thegenomes using bowtie2 and the SNPs were identified usingSAMtools [24] BCFtools and VCFtools (httpvcftoolssourceforgenet) Highly confident SNPs were filtered outusing scripts from ISMU pipeline [25] based on the criteriathat the raw read depth is greater than 10 and there isno reference base in the alignment Finally B melitensisATCC 23457 that showed the minimum number of SNPswith Bm IND1 was used as the template for chromosomelevel assembly Abacas version 131 [26] was used to assem-ble Bm IND1 scaffolds into two chromosomes Bm IND1chromosomal level assembly was further manually curatedusing the BLAST output of Bm IND1 scaffolds with the Bmelitensis ATCC 23457 genome We compared the syntenicrelationships between Bm IND1 B melitensis ATCC 23457and B melitensis 16M using Mauve version 231 [27] Thestructural and functional annotations of Bm IND1 genomewere carried out by RAST server [28]
25 Whole Genome Phylogeny Genomes of B melitensisisolates were downloaded from GenBank (ftpftpncbinihgovgenomesgenbankbacteriaBrucella melitensis) Firstwe assessed the completeness of the assembly and annota-tions of each sequenced genome and filtered out the incom-plete ones To assess the core genome and single copyorthologs Orthomcl v14 [29] was used with default param-eters We used 2319 single copy orthologs to construct amaximum-likelihood tree following the approach of Wattamet al [10] MAFFT version 7123b [18] was used to alignsequences from each gene family independently All thealignments were further processed and concatenated usingGblocks version 091b [30] RAxML version 8121 [19]was used to generate a tree for all the dataset using thePROTGAMMALG model of evolution The tree was visual-ized using Dendroscope version 3 [20]
26 Detection of Prophages The genome was searched forprophage sequences and phage attachment site using PHAST(phage search tool) available at httpphastwishartlabcom[31]
27 Identification of Variable Number of Tandem Repeats(VNTRs) Tandem repeats in each chromosomes of Bm IND1were identified using Tandem repeat finder [32] A precom-piled Tandem repeat finder version 407b was downloadedfromhttptandembuedutrftrfdownloadhtml and runonLinux (64-bit) platform using the parameter of a minimumalignment score of 80
28 Identification of SNPs We downloaded the genomesequences of Bmelitensis 16M BmelitensisM28 Bmeliten-sis ATCC 23457 B melitensis M5-90 B melitensis NI
B melitensis Ether and B abortus 2308 for SNP analysisWe considered only the completely assembled genomes foranalysis of SNPs We established a pipeline for finding SNPsbetween two reference sequences using Nucmer and show-snps program from the Mummer3 package [33] Show-snpsprovide SNPs derived only from uniquely aligned regionsSNPs were extracted from each strain against Bm IND1 andthe data were further annotated using SnpEff [34] to predictSNP effects in the genome
29 Indels Analysis To find insertions and deletions in thecoding region VCF files generated against B melitensisATCC 23457 using Bm IND1 reads for template genomeselection were annotated with SnpEff Indels in the codingregions and their corresponding functions were extractedfrom Bmelitensis ATCC 23457 using in-house Perl script
3 Results and Discussion
31 Isolation and Genotyping of B melitensis IND Strains Weisolated four strains of B melitensis from naturally infectedgoat followed by MLST analysis to understand the geneticdiversity among the B melitensis IND strains To performMLST analysis we amplified nine loci that included sevenhousekeeping genes one locus from the outer membraneprotein 25 (omp25) and one locus from an intergenicregion (Supplementary Figure 1) The inclusion of loci fromomp25 and intergenic region was reported to have providedmore discriminatory power in the phylogenetic analysis [8]Subsequently we compared the allelic profiles of the fourB melitensis IND isolates with each other and with otherreported Brucella species All the B melitensis IND isolatesdisplayed identical sequences with an allelic profile of 3-2-3-2-1-5-3-8-2 which belong to Sequence Typing- (ST-)8 The phylogram was rooted with B microti and in thephylogram all the B melitensis isolates clustered into onelineage (Figure 1) Bm IND strains grouped with other Asianstrains that is B melitensis M28 B melitensis M5-90 andBmelitensisNI whereas Bmelitensis Ether and Bmelitensis16M branched separatelyThis was anticipated as all the Asianstrains belong to ST-8 andBmelitensisEther andBmelitensis16M falls into ST-9 and ST-7 respectively In fact the supportvalue is very low for the branches of Asian isolates in thephylogram owing to the same allelic profile (ST-8) of MLSTloci that were considered for the phylogenetic analysis Asexpected B suis and B ovis clustered into different cladesin the phylogram (Figure 1) The analysis indicates that Bmelitensis with ST-8 is prevalent in Asia
32 Whole Genome Sequencing Assembly and Annotationof B melitensis Strain Bm IND1 We performed the wholegenome sequencing of Bm IND1 to analyze the geneticdivergence and genomic features in detail The raw datagenerated using the Illumina sequencing platform wereassembled using SOAPdenovoThis provided 102 contigs thatwere further assembled into 29 scaffolds (Table 1) Mappingreads onto them further validated these scaffolds On averageeach scaffold base was covered more than 100 times (100x)
4 International Journal of Genomics
B pinnipedialis B294B ovis ATCC 25840
B suis 1330B canis ATCC 23365
B abortus A13334
B abortus 9-941 B melitensis Ether
B melitensis NI
B melitensis 16M
B melitensis ATCC 23457 B melitensis M5-90 B melitensis M28
B melitensis IND3
B melitensis IND4 B melitensis IND1 B melitensis IND2
B microti CCM 4915
20
30
100
100
100
0001
60
100
0
0
0
0
0
10
100
Figure 1 Phylogenetic tree based on MLST analysis Seven housekeeping genes and two loci from outer membrane protein 25 and anintergenic region respectively were used for MLST analysis Bootstrap percentages retrieved in 100 replications are shown at the nodes
Table 1 Genome assembly statistics for Bm IND1 genome
All scaffoldsNumber of scaffolds 29N50 (scaffolds) 298927 bpLongest scaffold (pseudo molecule) 609256 bpSmallest scaffold 599 bpNumber of contigs 102N50 (contigs) 64911 bpLongest contig 151060 bpSmallest contig 384 bpLength of Chromosome I 2128440 bpLength of Chromosome II 1185949 bp
however some scaffolds like scaffolds 9 14 18 20 and 22 haveshown high depth of coverage (Supplementary Figure 2)This is likely due to the presence of duplicated or repeatregions of the genome
To generate chromosome level assembly the scaffoldswere assigned to two Brucella chromosomes with properorder and orientation Generally a reference genome of aclosely related species is used as the template to align andorder the scaffolds To achieve this we considered genomesof B melitensis 16M reported from USA B melitensis NIfrom Mongolia B melitensis Ether from Italy B melitensisATCC 23457 from India and the B melitensis M28 fromChina All these genomes are completely sequenced and wellannotated and the genome assembly of few has previouslybeen used as reference genomes for other strains [35 36]We aligned the raw data of Bm IND1 on the genomes ofabove B melitensis strains using bowtie2 and identified theSNPs (Supplementary Table 2) Since B melitensis ATCC
23457 displayed the least number of SNPs which indicatesminimum genetic divergence we selected the genome of thisstrain as the reference genome for chromosomal assembly ofBm IND1 B melitensis IND1 and B melitensis ATCC 23457were isolated from India however they belong to differentbiovars
Bm IND1 scaffolds were assembled into two chromo-somes using B melitensis ATCC 23457 as the template Totalnumber of scaffolds assigned on Chromosomes I and II are23 and 6 respectively Subsequently assembly was manuallycurated with a focus on those scaffolds that showed higherphysical coverage to fix the duplications We observed theduplications of scaffolds 20 22 27 28 and 29 on Chro-mosome I and scaffold 26 on Chromosome II with respectto the B melitensis ATCC 23457 genome These manuallyplaced scaffolds were in concordance with observed physicalcoverage However no duplication in other scaffolds wasobserved especially scaffolds 9 and 14 that showed highcoverage (Supplementary Figure 2(b)) We assume that thehigher coverage in these scaffolds may be due to internalrepeats or sequencing bias Next we aligned the genomes ofB melitensis ATCC 23457 B melitensis 16M and Bm IND1and observed for macrolevel synteny and large genomic rear-rangements In fact theywere highly syntenicwith each otherexcept for one segment of Bmelitensis 16M on ChromosomeII which was in reverse orientation in B melitensis ATCC23457 and Bm IND1 (Figure 2(a)) All general features of thegenome are summarized in Figures 2(b) and 2(c)
We annotated the genome of Bmelitensis Bm IND1 usingRapid Annotations with Subsystems Technology (RAST) toobtain the coding and noncoding genes [28] A total of55 tRNA 12 rRNA and 3191 protein coding genes with anaverage CDS length of 874 bp were annotated (Table 2) Inaddition RAST annotates the genomic structures and assigns
Figure 2 (a) Alignment of Bmelitensis 16M BmelitensisATCC 23457 and Bmelitensis IND1 genome Mauve alignment shows the syntenyregions between the three strains Bm IND1 and B melitensis ATCC 23457 aligned well with each other however a segment (olive colorblock) on Chromosome II of Bm IND1 is in reverse orientation in B melitensis 16M ((b) and (c)) Circular representation of B melitensisIND1 Chromosome I (b) and Chromosome II (c) Chromosomal coordinates are indicated on outer most circle Circles are represented fromouter to inner as circle 1 CDS on the positive strand (green for annotated red for hypothetical) circle 2 CDS on negative strand (blue forannotated red for hypothetical) circle 3 RNA genes (orange for tRNA and purple for rRNA) circle 4 VNTRS (turquoise) circle 5 GCcontent (olive for positive and purple for negative) circle 6 GC skew (olive for positive and purple for negative) Red blocks above circle 1represent phage integration site in Chromosome I
Figure 3 Distribution of subsystem category for B melitensis IND1 Bm IND1 genome sequence was annotated using Rapid AnnotationSystem Technology server Features of each subsystem and their coverage are summarized in the pie chart
their functions on the basis of presence of subsystems in thegenome This makes functional annotation of genes moreaccurate than simply assigning the functions on the basis ofsequence similarity of known genes Functions of 2562 geneswere assignedwhile 629 geneswere annotated as hypothetical(Table 2) A total of 1649 genes were assigned for differentsubsystems where maximum number (405) was assignedfor metabolism of amino acids followed by carbohydratemetabolism (331) (Figure 3)
321 Whole Genome Phylogeny Determining the phyloge-netic relationship in a bacterial population is essential tounderstand the population structure evolutionary historyand host relationship and to develop diagnostic assays formolecular epidemiological studies [9] To perform com-parative phylogenomics we downloaded all the currentlyavailable Bmelitensis genomes (59 genomes) from GenBankand considered B abortus 2308 as the outgroup speciesWe evaluated the completeness of assembly and annotationof the genomes by assessing the number of orthologousgenes which are highly conserved among the strains Any Bmelitensis strain showing less number of orthologous genesthan the number of orthologous genes present in B abortuswith respect to B melitensis were ignored for the down-stream phylogenomic studies This facilitates more accuratecore genome estimation and identification of single copy
orthologs present in the species which improves resolutionof the phylogenetic tree Therefore we ignored the genomesof 12 Bmelitensis strains and considered the entire repertoireof coding genes of 48 B melitensis strains including BmIND1 for the analysis (Supplementary Table 3) A total of151361 genes of B melitensis strains were clustered in 3800gene families of which 25124 gene families present in all the48 strains However 2461 genes only showed exact singlecopy orthology in each strain that could be considered asthe core genome of B melitensis clade The core genomeincludes 73ndash82of genes from each of theBmelitensis strainWattam et al [10] reported that 2285 core genes are presentin the Brucella genus by analyzing the genomes of 40 Brucellaspecies Conceivably the core genome of B melitensis cladewas higher than the total number of core genes presentacross the Brucella genus We used B abortus 2308 as theoutgroup for whole genome phylogeny that increased thetotal cluster of genes to 3829 After including the B abortusstrain the total number of single copy orthologs decreasedto 2319 genes In the whole genome phylogram Bm IND1clusteredwith other Asian isolates ofBmelitensis as observedin the MLST analysis (Figure 4) Bm IND1 grouped with Bmelitensis NI which was originated from Mongolia and boththe strains belong to biovar 3 (Figure 4) The phylogeneticrelationship established here is in agreement with the earlierreports [10 11] Tan et al [11] performed a comparative wholegenome SNP analysis of B melitensis strains from around
Figure 4 Phylogenetic tree showing relationship between B melitensis IND1 and other B melitensis strains Maximum likelihood tree of 49whole genome sequenced B melitensis strains inferred from concatenated partitioned alignment of 2319 core genes using RAxML Supportvalues of branches are calculated from 100 bootstrap replicates and the branch length is proportional to the number of substitutions per siteB abortus 2308 has been used as outgroup species
Table 3 Prophage regions detected in B melitensis isolate
the world and reported clustering of B melitensis isolatesinto five genotypes In agreement with this observationour phylogenomic studies also revealed the clustering of Bmelitensis isolates into five groups where Group 1 formedthe earliest diverging clade Group II represents most of theAsian isolates of B melitensis including Bm IND1 Parallelto Group II another lineage evolved which further branchedinto groups III IV and V Group III represents isolates fromAfrica and groups IV and V constitute isolates from Europeand America respectively
322 Prophages Prophages that are integrated into thegenome of bacteria can contributemany biological propertiesto their bacterial hosts such as virulence biosynthesis andsecretion of toxins genomic divergence and evolution [37]We analyzed the genome of Bm IND1 for prophages usingPHAST that was designed to identify and annotate prophagesequences in bacterial genomes [31] The analysis identified 2putative prophage integrations in Chromosome I of Bm IND1
(Table 3 and Figure 5) Region 1 is composed of a fragmentof 137 kb size that encoded 18 genes out of which 14 geneswere phage specific and 4 genes were bacteria specific RegionII is composed of 226 kb with 14 genes where 8 genes werephage specific and remaining 6 genes belonged to BrucellaNotably region 1 is considered as intact prophage upstreamof QseB locus and the RAST server could identify the genesin this region Region II is predicted as incomplete prophagebut flanked with attachment sites Region I is present in theChinese isolate of B melitensis M28 also We identified twoputative phage integrations in Chromosome I of Bmelitensis16M genome which did not show any similarity to that ofBm IND1 or B melitensis M28 The analyses clearly indicatethat the prophage integration events contribute to the geneticdiversity of Bmelitensis
323 VNTRs VNTRs play an important role in evolutiongene regulation genome structure antigenic variation andvirulence [38ndash40] Mutations in VNTRs produce a wide
8 International Journal of Genomics
Region 1
Region 2
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
new opportunities to analyze the genetic diversity amongthe groups [9] Genome sequencing and analysis of Brucellaspp from diverse hosts and geographical regions have beenreported [9] Until now 61 genomes of Bmelitensis have beensequenced and made available in the GenBank B melitensiscontains more than 3000 genes that are distributed overtwo circular chromosomes These sequenced species serve asvast resources for comparative genomics and understandingthe evolutionary history Comparative genomics will provideinsights into the virulence mechanisms of the Brucella sppsuch as novel genomic islands and integration of prophagesand SNPs that regulate the expression of certain genes oraffect the function of important virulence-associated pro-teins One of the previous studies demonstrated conservationof genes and genomic islands across the different Brucellaspp [10] In a recent study the evolutionary relationship of Bmelitensis on the basis of whole genome SNPs revealed spatialclustering of Bmelitensis isolates into five genotypes [11] Allthe Asian isolates of B melitensis clustered into genotype IIwhereas the isolates from Europe and America clustered intogenotypes IV and V respectively
We performed genotyping by MLST on B melitensisstrains isolated from naturally infected goats To performdetailed genetic characterization and comparative genomeanalysis we performed whole genome sequencing of one ofthe strains B melitensis IND1 using the Illumina platformThe analysis revealed the extent of genetic variation of Bmelitensis IND1 in comparison to B melitensis isolates fromother geographical locationsData generated fromour studiesmay help to develop new diagnostic assays based on stablemarkers such as SNPs (Single Nucleotide Polymorphisms)and Variable Number of Tandem Repeats (VNTRs) formolecular epidemiological studies Identification of SNPsIndels and novel phage integration sites will provide insightsinto the virulence mechanisms of this stealthy pathogenwhich could ultimately lead to the development of noveltherapeutic and preventive strategies to control brucellosis
2 Materials and Methods
21 Isolation of B melitensis For isolation of Brucella mate-rials from four different sources are listed in the Supplemen-tary Table 1 (in Supplementary Material available online athttpdxdoiorg10115520163034756) Samples were inoc-ulated on sterile plates of Brucella selective agar containingHemin and Vitamin K
1media (Hi Media India) and incu-
bated at 37∘C for 48 hours The plates were observed at every24 hours for the development of growth After obtainingthe growth the colonies suspected for Brucella on the basisof cultural characteristics were selected and streaked againon plates containing Brucella selective agar with Hemin andVitamin K
1and incubated at 37∘C for 2 days to obtain the
pure culture
22 Biotyping of Brucella Isolates Cultures showing typicalBrucella characteristics were subjected to biotyping tech-niques such as H
2S production growth in the presence
of thionin and basic fuchsin (10ndash40120583gmL) dye incorpo-rated into tryptic soy agar at different concentrations andCO2requirement immediately after the primary isolation
as described [12] Lead acetate strips were used to identifythe production of H
2S during growth and the growth was
evaluated on media containing streptomycin (25 120583gmL)to discriminate the isolates from vaccine strain Rev1 asdescribed [13]
Genomic DNA of all five strains was isolated using theWizard Genomic DNA Purification Kit (Promega USA)Isolated DNA was used for polymerase chain reactions toamplify 16S rRNA and the Omp 31 gene for the confirmatoryidentification of Brucella melitensis using the Taq PCRmastermix kit (Qiagen) 16S rRNA is specific to the genus Brucellawhile Omp 31 is a species-specific gene to Brucella melitensis[14ndash16]
23 MLST Analysis of B melitensis Isolates For MLSTanalysis 4396 nucleotide sequences spanning nine genomicfragments from Brucella were selected as described [8] Ofthe nine loci seven belong to classical housekeeping genesone locus derived from the outer membrane protein 25gene and one from an intergenic region Genomic DNA wasisolated from Bm IND isolates using the Wizard GenomicDNA Purification Kit (Promega) Genomic fragments wereamplified by PCR using the following cycling parameters94∘C for 2min 35 cycles of 94∘C for 30 sec 53∘C for 30 secand 72∘C for 1min and 72∘C for 5min Primers used forMLST analysis are listed in the Additional file 13 An aliquotof the PCR amplicons was analyzed by 1 agarose gel andphotographed Remaining PCR products were purified andsubjected to Sanger sequencing using the forward and reverseprimers that were used for PCR amplifications Editing ofthe sequences and generation of contigs from forward andreverse sequences was performed using Lasergene 8 software(DNA Star USA) To perform the phylogenetic analysisnine genomic fragments mentioned above were fetched fromrepresentative Brucella species and the loci were amplifiedin silico using the MLST primers All the sequences wereconcatenated to identify the allelic profile with the helpof Brucellabase [17] and the concatenated sequences weresubjected to multiple sequence alignment using MAFFTversion 7123b [18] Phylogenetic analyses were performedwith RAxML version 812 [19] using GTRGAMMAmodel ofevolutionThe phylogram was visualized using Dendroscopeversion 3 [20]
24 Genome Sequencing Assembly and Annotations Thecomplete genome of Brucella melitensis IND1 (Bm IND1)was sequenced using Illumina technology The sequenceddata have been deposited at DDBJEMBLGenBank underthe accession number JMKL00000000 [21] Paired end datagenerated were filtered for low quality reads using in-housescript After preprocessing high quality data were used tomake a scaffold level assembly using the SOAPdenovo version201 assembler [22] Scaffolds were further mapped withraw reads using bowtie2 version 224 [23] and coverage ateach base was calculated using SAMtools version 0119 [24]
International Journal of Genomics 3
Graphs for coverage analysis were plotted using GNUplotversion 46 (httpwwwgnuplotinfo)
Completely sequenced genomes ofBmelitensis such as Bmelitensis 16M B melitensis M28 B melitensis ATCC23457andBmelitensisNIwere initially considered as candidates forthe template to construct the chromosomal assembly of BmIND1 The raw data of Bm IND1 were aligned onto all thegenomes using bowtie2 and the SNPs were identified usingSAMtools [24] BCFtools and VCFtools (httpvcftoolssourceforgenet) Highly confident SNPs were filtered outusing scripts from ISMU pipeline [25] based on the criteriathat the raw read depth is greater than 10 and there isno reference base in the alignment Finally B melitensisATCC 23457 that showed the minimum number of SNPswith Bm IND1 was used as the template for chromosomelevel assembly Abacas version 131 [26] was used to assem-ble Bm IND1 scaffolds into two chromosomes Bm IND1chromosomal level assembly was further manually curatedusing the BLAST output of Bm IND1 scaffolds with the Bmelitensis ATCC 23457 genome We compared the syntenicrelationships between Bm IND1 B melitensis ATCC 23457and B melitensis 16M using Mauve version 231 [27] Thestructural and functional annotations of Bm IND1 genomewere carried out by RAST server [28]
25 Whole Genome Phylogeny Genomes of B melitensisisolates were downloaded from GenBank (ftpftpncbinihgovgenomesgenbankbacteriaBrucella melitensis) Firstwe assessed the completeness of the assembly and annota-tions of each sequenced genome and filtered out the incom-plete ones To assess the core genome and single copyorthologs Orthomcl v14 [29] was used with default param-eters We used 2319 single copy orthologs to construct amaximum-likelihood tree following the approach of Wattamet al [10] MAFFT version 7123b [18] was used to alignsequences from each gene family independently All thealignments were further processed and concatenated usingGblocks version 091b [30] RAxML version 8121 [19]was used to generate a tree for all the dataset using thePROTGAMMALG model of evolution The tree was visual-ized using Dendroscope version 3 [20]
26 Detection of Prophages The genome was searched forprophage sequences and phage attachment site using PHAST(phage search tool) available at httpphastwishartlabcom[31]
27 Identification of Variable Number of Tandem Repeats(VNTRs) Tandem repeats in each chromosomes of Bm IND1were identified using Tandem repeat finder [32] A precom-piled Tandem repeat finder version 407b was downloadedfromhttptandembuedutrftrfdownloadhtml and runonLinux (64-bit) platform using the parameter of a minimumalignment score of 80
28 Identification of SNPs We downloaded the genomesequences of Bmelitensis 16M BmelitensisM28 Bmeliten-sis ATCC 23457 B melitensis M5-90 B melitensis NI
B melitensis Ether and B abortus 2308 for SNP analysisWe considered only the completely assembled genomes foranalysis of SNPs We established a pipeline for finding SNPsbetween two reference sequences using Nucmer and show-snps program from the Mummer3 package [33] Show-snpsprovide SNPs derived only from uniquely aligned regionsSNPs were extracted from each strain against Bm IND1 andthe data were further annotated using SnpEff [34] to predictSNP effects in the genome
29 Indels Analysis To find insertions and deletions in thecoding region VCF files generated against B melitensisATCC 23457 using Bm IND1 reads for template genomeselection were annotated with SnpEff Indels in the codingregions and their corresponding functions were extractedfrom Bmelitensis ATCC 23457 using in-house Perl script
3 Results and Discussion
31 Isolation and Genotyping of B melitensis IND Strains Weisolated four strains of B melitensis from naturally infectedgoat followed by MLST analysis to understand the geneticdiversity among the B melitensis IND strains To performMLST analysis we amplified nine loci that included sevenhousekeeping genes one locus from the outer membraneprotein 25 (omp25) and one locus from an intergenicregion (Supplementary Figure 1) The inclusion of loci fromomp25 and intergenic region was reported to have providedmore discriminatory power in the phylogenetic analysis [8]Subsequently we compared the allelic profiles of the fourB melitensis IND isolates with each other and with otherreported Brucella species All the B melitensis IND isolatesdisplayed identical sequences with an allelic profile of 3-2-3-2-1-5-3-8-2 which belong to Sequence Typing- (ST-)8 The phylogram was rooted with B microti and in thephylogram all the B melitensis isolates clustered into onelineage (Figure 1) Bm IND strains grouped with other Asianstrains that is B melitensis M28 B melitensis M5-90 andBmelitensisNI whereas Bmelitensis Ether and Bmelitensis16M branched separatelyThis was anticipated as all the Asianstrains belong to ST-8 andBmelitensisEther andBmelitensis16M falls into ST-9 and ST-7 respectively In fact the supportvalue is very low for the branches of Asian isolates in thephylogram owing to the same allelic profile (ST-8) of MLSTloci that were considered for the phylogenetic analysis Asexpected B suis and B ovis clustered into different cladesin the phylogram (Figure 1) The analysis indicates that Bmelitensis with ST-8 is prevalent in Asia
32 Whole Genome Sequencing Assembly and Annotationof B melitensis Strain Bm IND1 We performed the wholegenome sequencing of Bm IND1 to analyze the geneticdivergence and genomic features in detail The raw datagenerated using the Illumina sequencing platform wereassembled using SOAPdenovoThis provided 102 contigs thatwere further assembled into 29 scaffolds (Table 1) Mappingreads onto them further validated these scaffolds On averageeach scaffold base was covered more than 100 times (100x)
4 International Journal of Genomics
B pinnipedialis B294B ovis ATCC 25840
B suis 1330B canis ATCC 23365
B abortus A13334
B abortus 9-941 B melitensis Ether
B melitensis NI
B melitensis 16M
B melitensis ATCC 23457 B melitensis M5-90 B melitensis M28
B melitensis IND3
B melitensis IND4 B melitensis IND1 B melitensis IND2
B microti CCM 4915
20
30
100
100
100
0001
60
100
0
0
0
0
0
10
100
Figure 1 Phylogenetic tree based on MLST analysis Seven housekeeping genes and two loci from outer membrane protein 25 and anintergenic region respectively were used for MLST analysis Bootstrap percentages retrieved in 100 replications are shown at the nodes
Table 1 Genome assembly statistics for Bm IND1 genome
All scaffoldsNumber of scaffolds 29N50 (scaffolds) 298927 bpLongest scaffold (pseudo molecule) 609256 bpSmallest scaffold 599 bpNumber of contigs 102N50 (contigs) 64911 bpLongest contig 151060 bpSmallest contig 384 bpLength of Chromosome I 2128440 bpLength of Chromosome II 1185949 bp
however some scaffolds like scaffolds 9 14 18 20 and 22 haveshown high depth of coverage (Supplementary Figure 2)This is likely due to the presence of duplicated or repeatregions of the genome
To generate chromosome level assembly the scaffoldswere assigned to two Brucella chromosomes with properorder and orientation Generally a reference genome of aclosely related species is used as the template to align andorder the scaffolds To achieve this we considered genomesof B melitensis 16M reported from USA B melitensis NIfrom Mongolia B melitensis Ether from Italy B melitensisATCC 23457 from India and the B melitensis M28 fromChina All these genomes are completely sequenced and wellannotated and the genome assembly of few has previouslybeen used as reference genomes for other strains [35 36]We aligned the raw data of Bm IND1 on the genomes ofabove B melitensis strains using bowtie2 and identified theSNPs (Supplementary Table 2) Since B melitensis ATCC
23457 displayed the least number of SNPs which indicatesminimum genetic divergence we selected the genome of thisstrain as the reference genome for chromosomal assembly ofBm IND1 B melitensis IND1 and B melitensis ATCC 23457were isolated from India however they belong to differentbiovars
Bm IND1 scaffolds were assembled into two chromo-somes using B melitensis ATCC 23457 as the template Totalnumber of scaffolds assigned on Chromosomes I and II are23 and 6 respectively Subsequently assembly was manuallycurated with a focus on those scaffolds that showed higherphysical coverage to fix the duplications We observed theduplications of scaffolds 20 22 27 28 and 29 on Chro-mosome I and scaffold 26 on Chromosome II with respectto the B melitensis ATCC 23457 genome These manuallyplaced scaffolds were in concordance with observed physicalcoverage However no duplication in other scaffolds wasobserved especially scaffolds 9 and 14 that showed highcoverage (Supplementary Figure 2(b)) We assume that thehigher coverage in these scaffolds may be due to internalrepeats or sequencing bias Next we aligned the genomes ofB melitensis ATCC 23457 B melitensis 16M and Bm IND1and observed for macrolevel synteny and large genomic rear-rangements In fact theywere highly syntenicwith each otherexcept for one segment of Bmelitensis 16M on ChromosomeII which was in reverse orientation in B melitensis ATCC23457 and Bm IND1 (Figure 2(a)) All general features of thegenome are summarized in Figures 2(b) and 2(c)
We annotated the genome of Bmelitensis Bm IND1 usingRapid Annotations with Subsystems Technology (RAST) toobtain the coding and noncoding genes [28] A total of55 tRNA 12 rRNA and 3191 protein coding genes with anaverage CDS length of 874 bp were annotated (Table 2) Inaddition RAST annotates the genomic structures and assigns
Figure 2 (a) Alignment of Bmelitensis 16M BmelitensisATCC 23457 and Bmelitensis IND1 genome Mauve alignment shows the syntenyregions between the three strains Bm IND1 and B melitensis ATCC 23457 aligned well with each other however a segment (olive colorblock) on Chromosome II of Bm IND1 is in reverse orientation in B melitensis 16M ((b) and (c)) Circular representation of B melitensisIND1 Chromosome I (b) and Chromosome II (c) Chromosomal coordinates are indicated on outer most circle Circles are represented fromouter to inner as circle 1 CDS on the positive strand (green for annotated red for hypothetical) circle 2 CDS on negative strand (blue forannotated red for hypothetical) circle 3 RNA genes (orange for tRNA and purple for rRNA) circle 4 VNTRS (turquoise) circle 5 GCcontent (olive for positive and purple for negative) circle 6 GC skew (olive for positive and purple for negative) Red blocks above circle 1represent phage integration site in Chromosome I
Figure 3 Distribution of subsystem category for B melitensis IND1 Bm IND1 genome sequence was annotated using Rapid AnnotationSystem Technology server Features of each subsystem and their coverage are summarized in the pie chart
their functions on the basis of presence of subsystems in thegenome This makes functional annotation of genes moreaccurate than simply assigning the functions on the basis ofsequence similarity of known genes Functions of 2562 geneswere assignedwhile 629 geneswere annotated as hypothetical(Table 2) A total of 1649 genes were assigned for differentsubsystems where maximum number (405) was assignedfor metabolism of amino acids followed by carbohydratemetabolism (331) (Figure 3)
321 Whole Genome Phylogeny Determining the phyloge-netic relationship in a bacterial population is essential tounderstand the population structure evolutionary historyand host relationship and to develop diagnostic assays formolecular epidemiological studies [9] To perform com-parative phylogenomics we downloaded all the currentlyavailable Bmelitensis genomes (59 genomes) from GenBankand considered B abortus 2308 as the outgroup speciesWe evaluated the completeness of assembly and annotationof the genomes by assessing the number of orthologousgenes which are highly conserved among the strains Any Bmelitensis strain showing less number of orthologous genesthan the number of orthologous genes present in B abortuswith respect to B melitensis were ignored for the down-stream phylogenomic studies This facilitates more accuratecore genome estimation and identification of single copy
orthologs present in the species which improves resolutionof the phylogenetic tree Therefore we ignored the genomesof 12 Bmelitensis strains and considered the entire repertoireof coding genes of 48 B melitensis strains including BmIND1 for the analysis (Supplementary Table 3) A total of151361 genes of B melitensis strains were clustered in 3800gene families of which 25124 gene families present in all the48 strains However 2461 genes only showed exact singlecopy orthology in each strain that could be considered asthe core genome of B melitensis clade The core genomeincludes 73ndash82of genes from each of theBmelitensis strainWattam et al [10] reported that 2285 core genes are presentin the Brucella genus by analyzing the genomes of 40 Brucellaspecies Conceivably the core genome of B melitensis cladewas higher than the total number of core genes presentacross the Brucella genus We used B abortus 2308 as theoutgroup for whole genome phylogeny that increased thetotal cluster of genes to 3829 After including the B abortusstrain the total number of single copy orthologs decreasedto 2319 genes In the whole genome phylogram Bm IND1clusteredwith other Asian isolates ofBmelitensis as observedin the MLST analysis (Figure 4) Bm IND1 grouped with Bmelitensis NI which was originated from Mongolia and boththe strains belong to biovar 3 (Figure 4) The phylogeneticrelationship established here is in agreement with the earlierreports [10 11] Tan et al [11] performed a comparative wholegenome SNP analysis of B melitensis strains from around
Figure 4 Phylogenetic tree showing relationship between B melitensis IND1 and other B melitensis strains Maximum likelihood tree of 49whole genome sequenced B melitensis strains inferred from concatenated partitioned alignment of 2319 core genes using RAxML Supportvalues of branches are calculated from 100 bootstrap replicates and the branch length is proportional to the number of substitutions per siteB abortus 2308 has been used as outgroup species
Table 3 Prophage regions detected in B melitensis isolate
the world and reported clustering of B melitensis isolatesinto five genotypes In agreement with this observationour phylogenomic studies also revealed the clustering of Bmelitensis isolates into five groups where Group 1 formedthe earliest diverging clade Group II represents most of theAsian isolates of B melitensis including Bm IND1 Parallelto Group II another lineage evolved which further branchedinto groups III IV and V Group III represents isolates fromAfrica and groups IV and V constitute isolates from Europeand America respectively
322 Prophages Prophages that are integrated into thegenome of bacteria can contributemany biological propertiesto their bacterial hosts such as virulence biosynthesis andsecretion of toxins genomic divergence and evolution [37]We analyzed the genome of Bm IND1 for prophages usingPHAST that was designed to identify and annotate prophagesequences in bacterial genomes [31] The analysis identified 2putative prophage integrations in Chromosome I of Bm IND1
(Table 3 and Figure 5) Region 1 is composed of a fragmentof 137 kb size that encoded 18 genes out of which 14 geneswere phage specific and 4 genes were bacteria specific RegionII is composed of 226 kb with 14 genes where 8 genes werephage specific and remaining 6 genes belonged to BrucellaNotably region 1 is considered as intact prophage upstreamof QseB locus and the RAST server could identify the genesin this region Region II is predicted as incomplete prophagebut flanked with attachment sites Region I is present in theChinese isolate of B melitensis M28 also We identified twoputative phage integrations in Chromosome I of Bmelitensis16M genome which did not show any similarity to that ofBm IND1 or B melitensis M28 The analyses clearly indicatethat the prophage integration events contribute to the geneticdiversity of Bmelitensis
323 VNTRs VNTRs play an important role in evolutiongene regulation genome structure antigenic variation andvirulence [38ndash40] Mutations in VNTRs produce a wide
8 International Journal of Genomics
Region 1
Region 2
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Graphs for coverage analysis were plotted using GNUplotversion 46 (httpwwwgnuplotinfo)
Completely sequenced genomes ofBmelitensis such as Bmelitensis 16M B melitensis M28 B melitensis ATCC23457andBmelitensisNIwere initially considered as candidates forthe template to construct the chromosomal assembly of BmIND1 The raw data of Bm IND1 were aligned onto all thegenomes using bowtie2 and the SNPs were identified usingSAMtools [24] BCFtools and VCFtools (httpvcftoolssourceforgenet) Highly confident SNPs were filtered outusing scripts from ISMU pipeline [25] based on the criteriathat the raw read depth is greater than 10 and there isno reference base in the alignment Finally B melitensisATCC 23457 that showed the minimum number of SNPswith Bm IND1 was used as the template for chromosomelevel assembly Abacas version 131 [26] was used to assem-ble Bm IND1 scaffolds into two chromosomes Bm IND1chromosomal level assembly was further manually curatedusing the BLAST output of Bm IND1 scaffolds with the Bmelitensis ATCC 23457 genome We compared the syntenicrelationships between Bm IND1 B melitensis ATCC 23457and B melitensis 16M using Mauve version 231 [27] Thestructural and functional annotations of Bm IND1 genomewere carried out by RAST server [28]
25 Whole Genome Phylogeny Genomes of B melitensisisolates were downloaded from GenBank (ftpftpncbinihgovgenomesgenbankbacteriaBrucella melitensis) Firstwe assessed the completeness of the assembly and annota-tions of each sequenced genome and filtered out the incom-plete ones To assess the core genome and single copyorthologs Orthomcl v14 [29] was used with default param-eters We used 2319 single copy orthologs to construct amaximum-likelihood tree following the approach of Wattamet al [10] MAFFT version 7123b [18] was used to alignsequences from each gene family independently All thealignments were further processed and concatenated usingGblocks version 091b [30] RAxML version 8121 [19]was used to generate a tree for all the dataset using thePROTGAMMALG model of evolution The tree was visual-ized using Dendroscope version 3 [20]
26 Detection of Prophages The genome was searched forprophage sequences and phage attachment site using PHAST(phage search tool) available at httpphastwishartlabcom[31]
27 Identification of Variable Number of Tandem Repeats(VNTRs) Tandem repeats in each chromosomes of Bm IND1were identified using Tandem repeat finder [32] A precom-piled Tandem repeat finder version 407b was downloadedfromhttptandembuedutrftrfdownloadhtml and runonLinux (64-bit) platform using the parameter of a minimumalignment score of 80
28 Identification of SNPs We downloaded the genomesequences of Bmelitensis 16M BmelitensisM28 Bmeliten-sis ATCC 23457 B melitensis M5-90 B melitensis NI
B melitensis Ether and B abortus 2308 for SNP analysisWe considered only the completely assembled genomes foranalysis of SNPs We established a pipeline for finding SNPsbetween two reference sequences using Nucmer and show-snps program from the Mummer3 package [33] Show-snpsprovide SNPs derived only from uniquely aligned regionsSNPs were extracted from each strain against Bm IND1 andthe data were further annotated using SnpEff [34] to predictSNP effects in the genome
29 Indels Analysis To find insertions and deletions in thecoding region VCF files generated against B melitensisATCC 23457 using Bm IND1 reads for template genomeselection were annotated with SnpEff Indels in the codingregions and their corresponding functions were extractedfrom Bmelitensis ATCC 23457 using in-house Perl script
3 Results and Discussion
31 Isolation and Genotyping of B melitensis IND Strains Weisolated four strains of B melitensis from naturally infectedgoat followed by MLST analysis to understand the geneticdiversity among the B melitensis IND strains To performMLST analysis we amplified nine loci that included sevenhousekeeping genes one locus from the outer membraneprotein 25 (omp25) and one locus from an intergenicregion (Supplementary Figure 1) The inclusion of loci fromomp25 and intergenic region was reported to have providedmore discriminatory power in the phylogenetic analysis [8]Subsequently we compared the allelic profiles of the fourB melitensis IND isolates with each other and with otherreported Brucella species All the B melitensis IND isolatesdisplayed identical sequences with an allelic profile of 3-2-3-2-1-5-3-8-2 which belong to Sequence Typing- (ST-)8 The phylogram was rooted with B microti and in thephylogram all the B melitensis isolates clustered into onelineage (Figure 1) Bm IND strains grouped with other Asianstrains that is B melitensis M28 B melitensis M5-90 andBmelitensisNI whereas Bmelitensis Ether and Bmelitensis16M branched separatelyThis was anticipated as all the Asianstrains belong to ST-8 andBmelitensisEther andBmelitensis16M falls into ST-9 and ST-7 respectively In fact the supportvalue is very low for the branches of Asian isolates in thephylogram owing to the same allelic profile (ST-8) of MLSTloci that were considered for the phylogenetic analysis Asexpected B suis and B ovis clustered into different cladesin the phylogram (Figure 1) The analysis indicates that Bmelitensis with ST-8 is prevalent in Asia
32 Whole Genome Sequencing Assembly and Annotationof B melitensis Strain Bm IND1 We performed the wholegenome sequencing of Bm IND1 to analyze the geneticdivergence and genomic features in detail The raw datagenerated using the Illumina sequencing platform wereassembled using SOAPdenovoThis provided 102 contigs thatwere further assembled into 29 scaffolds (Table 1) Mappingreads onto them further validated these scaffolds On averageeach scaffold base was covered more than 100 times (100x)
4 International Journal of Genomics
B pinnipedialis B294B ovis ATCC 25840
B suis 1330B canis ATCC 23365
B abortus A13334
B abortus 9-941 B melitensis Ether
B melitensis NI
B melitensis 16M
B melitensis ATCC 23457 B melitensis M5-90 B melitensis M28
B melitensis IND3
B melitensis IND4 B melitensis IND1 B melitensis IND2
B microti CCM 4915
20
30
100
100
100
0001
60
100
0
0
0
0
0
10
100
Figure 1 Phylogenetic tree based on MLST analysis Seven housekeeping genes and two loci from outer membrane protein 25 and anintergenic region respectively were used for MLST analysis Bootstrap percentages retrieved in 100 replications are shown at the nodes
Table 1 Genome assembly statistics for Bm IND1 genome
All scaffoldsNumber of scaffolds 29N50 (scaffolds) 298927 bpLongest scaffold (pseudo molecule) 609256 bpSmallest scaffold 599 bpNumber of contigs 102N50 (contigs) 64911 bpLongest contig 151060 bpSmallest contig 384 bpLength of Chromosome I 2128440 bpLength of Chromosome II 1185949 bp
however some scaffolds like scaffolds 9 14 18 20 and 22 haveshown high depth of coverage (Supplementary Figure 2)This is likely due to the presence of duplicated or repeatregions of the genome
To generate chromosome level assembly the scaffoldswere assigned to two Brucella chromosomes with properorder and orientation Generally a reference genome of aclosely related species is used as the template to align andorder the scaffolds To achieve this we considered genomesof B melitensis 16M reported from USA B melitensis NIfrom Mongolia B melitensis Ether from Italy B melitensisATCC 23457 from India and the B melitensis M28 fromChina All these genomes are completely sequenced and wellannotated and the genome assembly of few has previouslybeen used as reference genomes for other strains [35 36]We aligned the raw data of Bm IND1 on the genomes ofabove B melitensis strains using bowtie2 and identified theSNPs (Supplementary Table 2) Since B melitensis ATCC
23457 displayed the least number of SNPs which indicatesminimum genetic divergence we selected the genome of thisstrain as the reference genome for chromosomal assembly ofBm IND1 B melitensis IND1 and B melitensis ATCC 23457were isolated from India however they belong to differentbiovars
Bm IND1 scaffolds were assembled into two chromo-somes using B melitensis ATCC 23457 as the template Totalnumber of scaffolds assigned on Chromosomes I and II are23 and 6 respectively Subsequently assembly was manuallycurated with a focus on those scaffolds that showed higherphysical coverage to fix the duplications We observed theduplications of scaffolds 20 22 27 28 and 29 on Chro-mosome I and scaffold 26 on Chromosome II with respectto the B melitensis ATCC 23457 genome These manuallyplaced scaffolds were in concordance with observed physicalcoverage However no duplication in other scaffolds wasobserved especially scaffolds 9 and 14 that showed highcoverage (Supplementary Figure 2(b)) We assume that thehigher coverage in these scaffolds may be due to internalrepeats or sequencing bias Next we aligned the genomes ofB melitensis ATCC 23457 B melitensis 16M and Bm IND1and observed for macrolevel synteny and large genomic rear-rangements In fact theywere highly syntenicwith each otherexcept for one segment of Bmelitensis 16M on ChromosomeII which was in reverse orientation in B melitensis ATCC23457 and Bm IND1 (Figure 2(a)) All general features of thegenome are summarized in Figures 2(b) and 2(c)
We annotated the genome of Bmelitensis Bm IND1 usingRapid Annotations with Subsystems Technology (RAST) toobtain the coding and noncoding genes [28] A total of55 tRNA 12 rRNA and 3191 protein coding genes with anaverage CDS length of 874 bp were annotated (Table 2) Inaddition RAST annotates the genomic structures and assigns
Figure 2 (a) Alignment of Bmelitensis 16M BmelitensisATCC 23457 and Bmelitensis IND1 genome Mauve alignment shows the syntenyregions between the three strains Bm IND1 and B melitensis ATCC 23457 aligned well with each other however a segment (olive colorblock) on Chromosome II of Bm IND1 is in reverse orientation in B melitensis 16M ((b) and (c)) Circular representation of B melitensisIND1 Chromosome I (b) and Chromosome II (c) Chromosomal coordinates are indicated on outer most circle Circles are represented fromouter to inner as circle 1 CDS on the positive strand (green for annotated red for hypothetical) circle 2 CDS on negative strand (blue forannotated red for hypothetical) circle 3 RNA genes (orange for tRNA and purple for rRNA) circle 4 VNTRS (turquoise) circle 5 GCcontent (olive for positive and purple for negative) circle 6 GC skew (olive for positive and purple for negative) Red blocks above circle 1represent phage integration site in Chromosome I
Figure 3 Distribution of subsystem category for B melitensis IND1 Bm IND1 genome sequence was annotated using Rapid AnnotationSystem Technology server Features of each subsystem and their coverage are summarized in the pie chart
their functions on the basis of presence of subsystems in thegenome This makes functional annotation of genes moreaccurate than simply assigning the functions on the basis ofsequence similarity of known genes Functions of 2562 geneswere assignedwhile 629 geneswere annotated as hypothetical(Table 2) A total of 1649 genes were assigned for differentsubsystems where maximum number (405) was assignedfor metabolism of amino acids followed by carbohydratemetabolism (331) (Figure 3)
321 Whole Genome Phylogeny Determining the phyloge-netic relationship in a bacterial population is essential tounderstand the population structure evolutionary historyand host relationship and to develop diagnostic assays formolecular epidemiological studies [9] To perform com-parative phylogenomics we downloaded all the currentlyavailable Bmelitensis genomes (59 genomes) from GenBankand considered B abortus 2308 as the outgroup speciesWe evaluated the completeness of assembly and annotationof the genomes by assessing the number of orthologousgenes which are highly conserved among the strains Any Bmelitensis strain showing less number of orthologous genesthan the number of orthologous genes present in B abortuswith respect to B melitensis were ignored for the down-stream phylogenomic studies This facilitates more accuratecore genome estimation and identification of single copy
orthologs present in the species which improves resolutionof the phylogenetic tree Therefore we ignored the genomesof 12 Bmelitensis strains and considered the entire repertoireof coding genes of 48 B melitensis strains including BmIND1 for the analysis (Supplementary Table 3) A total of151361 genes of B melitensis strains were clustered in 3800gene families of which 25124 gene families present in all the48 strains However 2461 genes only showed exact singlecopy orthology in each strain that could be considered asthe core genome of B melitensis clade The core genomeincludes 73ndash82of genes from each of theBmelitensis strainWattam et al [10] reported that 2285 core genes are presentin the Brucella genus by analyzing the genomes of 40 Brucellaspecies Conceivably the core genome of B melitensis cladewas higher than the total number of core genes presentacross the Brucella genus We used B abortus 2308 as theoutgroup for whole genome phylogeny that increased thetotal cluster of genes to 3829 After including the B abortusstrain the total number of single copy orthologs decreasedto 2319 genes In the whole genome phylogram Bm IND1clusteredwith other Asian isolates ofBmelitensis as observedin the MLST analysis (Figure 4) Bm IND1 grouped with Bmelitensis NI which was originated from Mongolia and boththe strains belong to biovar 3 (Figure 4) The phylogeneticrelationship established here is in agreement with the earlierreports [10 11] Tan et al [11] performed a comparative wholegenome SNP analysis of B melitensis strains from around
Figure 4 Phylogenetic tree showing relationship between B melitensis IND1 and other B melitensis strains Maximum likelihood tree of 49whole genome sequenced B melitensis strains inferred from concatenated partitioned alignment of 2319 core genes using RAxML Supportvalues of branches are calculated from 100 bootstrap replicates and the branch length is proportional to the number of substitutions per siteB abortus 2308 has been used as outgroup species
Table 3 Prophage regions detected in B melitensis isolate
the world and reported clustering of B melitensis isolatesinto five genotypes In agreement with this observationour phylogenomic studies also revealed the clustering of Bmelitensis isolates into five groups where Group 1 formedthe earliest diverging clade Group II represents most of theAsian isolates of B melitensis including Bm IND1 Parallelto Group II another lineage evolved which further branchedinto groups III IV and V Group III represents isolates fromAfrica and groups IV and V constitute isolates from Europeand America respectively
322 Prophages Prophages that are integrated into thegenome of bacteria can contributemany biological propertiesto their bacterial hosts such as virulence biosynthesis andsecretion of toxins genomic divergence and evolution [37]We analyzed the genome of Bm IND1 for prophages usingPHAST that was designed to identify and annotate prophagesequences in bacterial genomes [31] The analysis identified 2putative prophage integrations in Chromosome I of Bm IND1
(Table 3 and Figure 5) Region 1 is composed of a fragmentof 137 kb size that encoded 18 genes out of which 14 geneswere phage specific and 4 genes were bacteria specific RegionII is composed of 226 kb with 14 genes where 8 genes werephage specific and remaining 6 genes belonged to BrucellaNotably region 1 is considered as intact prophage upstreamof QseB locus and the RAST server could identify the genesin this region Region II is predicted as incomplete prophagebut flanked with attachment sites Region I is present in theChinese isolate of B melitensis M28 also We identified twoputative phage integrations in Chromosome I of Bmelitensis16M genome which did not show any similarity to that ofBm IND1 or B melitensis M28 The analyses clearly indicatethat the prophage integration events contribute to the geneticdiversity of Bmelitensis
323 VNTRs VNTRs play an important role in evolutiongene regulation genome structure antigenic variation andvirulence [38ndash40] Mutations in VNTRs produce a wide
8 International Journal of Genomics
Region 1
Region 2
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
B melitensis ATCC 23457 B melitensis M5-90 B melitensis M28
B melitensis IND3
B melitensis IND4 B melitensis IND1 B melitensis IND2
B microti CCM 4915
20
30
100
100
100
0001
60
100
0
0
0
0
0
10
100
Figure 1 Phylogenetic tree based on MLST analysis Seven housekeeping genes and two loci from outer membrane protein 25 and anintergenic region respectively were used for MLST analysis Bootstrap percentages retrieved in 100 replications are shown at the nodes
Table 1 Genome assembly statistics for Bm IND1 genome
All scaffoldsNumber of scaffolds 29N50 (scaffolds) 298927 bpLongest scaffold (pseudo molecule) 609256 bpSmallest scaffold 599 bpNumber of contigs 102N50 (contigs) 64911 bpLongest contig 151060 bpSmallest contig 384 bpLength of Chromosome I 2128440 bpLength of Chromosome II 1185949 bp
however some scaffolds like scaffolds 9 14 18 20 and 22 haveshown high depth of coverage (Supplementary Figure 2)This is likely due to the presence of duplicated or repeatregions of the genome
To generate chromosome level assembly the scaffoldswere assigned to two Brucella chromosomes with properorder and orientation Generally a reference genome of aclosely related species is used as the template to align andorder the scaffolds To achieve this we considered genomesof B melitensis 16M reported from USA B melitensis NIfrom Mongolia B melitensis Ether from Italy B melitensisATCC 23457 from India and the B melitensis M28 fromChina All these genomes are completely sequenced and wellannotated and the genome assembly of few has previouslybeen used as reference genomes for other strains [35 36]We aligned the raw data of Bm IND1 on the genomes ofabove B melitensis strains using bowtie2 and identified theSNPs (Supplementary Table 2) Since B melitensis ATCC
23457 displayed the least number of SNPs which indicatesminimum genetic divergence we selected the genome of thisstrain as the reference genome for chromosomal assembly ofBm IND1 B melitensis IND1 and B melitensis ATCC 23457were isolated from India however they belong to differentbiovars
Bm IND1 scaffolds were assembled into two chromo-somes using B melitensis ATCC 23457 as the template Totalnumber of scaffolds assigned on Chromosomes I and II are23 and 6 respectively Subsequently assembly was manuallycurated with a focus on those scaffolds that showed higherphysical coverage to fix the duplications We observed theduplications of scaffolds 20 22 27 28 and 29 on Chro-mosome I and scaffold 26 on Chromosome II with respectto the B melitensis ATCC 23457 genome These manuallyplaced scaffolds were in concordance with observed physicalcoverage However no duplication in other scaffolds wasobserved especially scaffolds 9 and 14 that showed highcoverage (Supplementary Figure 2(b)) We assume that thehigher coverage in these scaffolds may be due to internalrepeats or sequencing bias Next we aligned the genomes ofB melitensis ATCC 23457 B melitensis 16M and Bm IND1and observed for macrolevel synteny and large genomic rear-rangements In fact theywere highly syntenicwith each otherexcept for one segment of Bmelitensis 16M on ChromosomeII which was in reverse orientation in B melitensis ATCC23457 and Bm IND1 (Figure 2(a)) All general features of thegenome are summarized in Figures 2(b) and 2(c)
We annotated the genome of Bmelitensis Bm IND1 usingRapid Annotations with Subsystems Technology (RAST) toobtain the coding and noncoding genes [28] A total of55 tRNA 12 rRNA and 3191 protein coding genes with anaverage CDS length of 874 bp were annotated (Table 2) Inaddition RAST annotates the genomic structures and assigns
Figure 2 (a) Alignment of Bmelitensis 16M BmelitensisATCC 23457 and Bmelitensis IND1 genome Mauve alignment shows the syntenyregions between the three strains Bm IND1 and B melitensis ATCC 23457 aligned well with each other however a segment (olive colorblock) on Chromosome II of Bm IND1 is in reverse orientation in B melitensis 16M ((b) and (c)) Circular representation of B melitensisIND1 Chromosome I (b) and Chromosome II (c) Chromosomal coordinates are indicated on outer most circle Circles are represented fromouter to inner as circle 1 CDS on the positive strand (green for annotated red for hypothetical) circle 2 CDS on negative strand (blue forannotated red for hypothetical) circle 3 RNA genes (orange for tRNA and purple for rRNA) circle 4 VNTRS (turquoise) circle 5 GCcontent (olive for positive and purple for negative) circle 6 GC skew (olive for positive and purple for negative) Red blocks above circle 1represent phage integration site in Chromosome I
Figure 3 Distribution of subsystem category for B melitensis IND1 Bm IND1 genome sequence was annotated using Rapid AnnotationSystem Technology server Features of each subsystem and their coverage are summarized in the pie chart
their functions on the basis of presence of subsystems in thegenome This makes functional annotation of genes moreaccurate than simply assigning the functions on the basis ofsequence similarity of known genes Functions of 2562 geneswere assignedwhile 629 geneswere annotated as hypothetical(Table 2) A total of 1649 genes were assigned for differentsubsystems where maximum number (405) was assignedfor metabolism of amino acids followed by carbohydratemetabolism (331) (Figure 3)
321 Whole Genome Phylogeny Determining the phyloge-netic relationship in a bacterial population is essential tounderstand the population structure evolutionary historyand host relationship and to develop diagnostic assays formolecular epidemiological studies [9] To perform com-parative phylogenomics we downloaded all the currentlyavailable Bmelitensis genomes (59 genomes) from GenBankand considered B abortus 2308 as the outgroup speciesWe evaluated the completeness of assembly and annotationof the genomes by assessing the number of orthologousgenes which are highly conserved among the strains Any Bmelitensis strain showing less number of orthologous genesthan the number of orthologous genes present in B abortuswith respect to B melitensis were ignored for the down-stream phylogenomic studies This facilitates more accuratecore genome estimation and identification of single copy
orthologs present in the species which improves resolutionof the phylogenetic tree Therefore we ignored the genomesof 12 Bmelitensis strains and considered the entire repertoireof coding genes of 48 B melitensis strains including BmIND1 for the analysis (Supplementary Table 3) A total of151361 genes of B melitensis strains were clustered in 3800gene families of which 25124 gene families present in all the48 strains However 2461 genes only showed exact singlecopy orthology in each strain that could be considered asthe core genome of B melitensis clade The core genomeincludes 73ndash82of genes from each of theBmelitensis strainWattam et al [10] reported that 2285 core genes are presentin the Brucella genus by analyzing the genomes of 40 Brucellaspecies Conceivably the core genome of B melitensis cladewas higher than the total number of core genes presentacross the Brucella genus We used B abortus 2308 as theoutgroup for whole genome phylogeny that increased thetotal cluster of genes to 3829 After including the B abortusstrain the total number of single copy orthologs decreasedto 2319 genes In the whole genome phylogram Bm IND1clusteredwith other Asian isolates ofBmelitensis as observedin the MLST analysis (Figure 4) Bm IND1 grouped with Bmelitensis NI which was originated from Mongolia and boththe strains belong to biovar 3 (Figure 4) The phylogeneticrelationship established here is in agreement with the earlierreports [10 11] Tan et al [11] performed a comparative wholegenome SNP analysis of B melitensis strains from around
Figure 4 Phylogenetic tree showing relationship between B melitensis IND1 and other B melitensis strains Maximum likelihood tree of 49whole genome sequenced B melitensis strains inferred from concatenated partitioned alignment of 2319 core genes using RAxML Supportvalues of branches are calculated from 100 bootstrap replicates and the branch length is proportional to the number of substitutions per siteB abortus 2308 has been used as outgroup species
Table 3 Prophage regions detected in B melitensis isolate
the world and reported clustering of B melitensis isolatesinto five genotypes In agreement with this observationour phylogenomic studies also revealed the clustering of Bmelitensis isolates into five groups where Group 1 formedthe earliest diverging clade Group II represents most of theAsian isolates of B melitensis including Bm IND1 Parallelto Group II another lineage evolved which further branchedinto groups III IV and V Group III represents isolates fromAfrica and groups IV and V constitute isolates from Europeand America respectively
322 Prophages Prophages that are integrated into thegenome of bacteria can contributemany biological propertiesto their bacterial hosts such as virulence biosynthesis andsecretion of toxins genomic divergence and evolution [37]We analyzed the genome of Bm IND1 for prophages usingPHAST that was designed to identify and annotate prophagesequences in bacterial genomes [31] The analysis identified 2putative prophage integrations in Chromosome I of Bm IND1
(Table 3 and Figure 5) Region 1 is composed of a fragmentof 137 kb size that encoded 18 genes out of which 14 geneswere phage specific and 4 genes were bacteria specific RegionII is composed of 226 kb with 14 genes where 8 genes werephage specific and remaining 6 genes belonged to BrucellaNotably region 1 is considered as intact prophage upstreamof QseB locus and the RAST server could identify the genesin this region Region II is predicted as incomplete prophagebut flanked with attachment sites Region I is present in theChinese isolate of B melitensis M28 also We identified twoputative phage integrations in Chromosome I of Bmelitensis16M genome which did not show any similarity to that ofBm IND1 or B melitensis M28 The analyses clearly indicatethat the prophage integration events contribute to the geneticdiversity of Bmelitensis
323 VNTRs VNTRs play an important role in evolutiongene regulation genome structure antigenic variation andvirulence [38ndash40] Mutations in VNTRs produce a wide
8 International Journal of Genomics
Region 1
Region 2
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Figure 2 (a) Alignment of Bmelitensis 16M BmelitensisATCC 23457 and Bmelitensis IND1 genome Mauve alignment shows the syntenyregions between the three strains Bm IND1 and B melitensis ATCC 23457 aligned well with each other however a segment (olive colorblock) on Chromosome II of Bm IND1 is in reverse orientation in B melitensis 16M ((b) and (c)) Circular representation of B melitensisIND1 Chromosome I (b) and Chromosome II (c) Chromosomal coordinates are indicated on outer most circle Circles are represented fromouter to inner as circle 1 CDS on the positive strand (green for annotated red for hypothetical) circle 2 CDS on negative strand (blue forannotated red for hypothetical) circle 3 RNA genes (orange for tRNA and purple for rRNA) circle 4 VNTRS (turquoise) circle 5 GCcontent (olive for positive and purple for negative) circle 6 GC skew (olive for positive and purple for negative) Red blocks above circle 1represent phage integration site in Chromosome I
Figure 3 Distribution of subsystem category for B melitensis IND1 Bm IND1 genome sequence was annotated using Rapid AnnotationSystem Technology server Features of each subsystem and their coverage are summarized in the pie chart
their functions on the basis of presence of subsystems in thegenome This makes functional annotation of genes moreaccurate than simply assigning the functions on the basis ofsequence similarity of known genes Functions of 2562 geneswere assignedwhile 629 geneswere annotated as hypothetical(Table 2) A total of 1649 genes were assigned for differentsubsystems where maximum number (405) was assignedfor metabolism of amino acids followed by carbohydratemetabolism (331) (Figure 3)
321 Whole Genome Phylogeny Determining the phyloge-netic relationship in a bacterial population is essential tounderstand the population structure evolutionary historyand host relationship and to develop diagnostic assays formolecular epidemiological studies [9] To perform com-parative phylogenomics we downloaded all the currentlyavailable Bmelitensis genomes (59 genomes) from GenBankand considered B abortus 2308 as the outgroup speciesWe evaluated the completeness of assembly and annotationof the genomes by assessing the number of orthologousgenes which are highly conserved among the strains Any Bmelitensis strain showing less number of orthologous genesthan the number of orthologous genes present in B abortuswith respect to B melitensis were ignored for the down-stream phylogenomic studies This facilitates more accuratecore genome estimation and identification of single copy
orthologs present in the species which improves resolutionof the phylogenetic tree Therefore we ignored the genomesof 12 Bmelitensis strains and considered the entire repertoireof coding genes of 48 B melitensis strains including BmIND1 for the analysis (Supplementary Table 3) A total of151361 genes of B melitensis strains were clustered in 3800gene families of which 25124 gene families present in all the48 strains However 2461 genes only showed exact singlecopy orthology in each strain that could be considered asthe core genome of B melitensis clade The core genomeincludes 73ndash82of genes from each of theBmelitensis strainWattam et al [10] reported that 2285 core genes are presentin the Brucella genus by analyzing the genomes of 40 Brucellaspecies Conceivably the core genome of B melitensis cladewas higher than the total number of core genes presentacross the Brucella genus We used B abortus 2308 as theoutgroup for whole genome phylogeny that increased thetotal cluster of genes to 3829 After including the B abortusstrain the total number of single copy orthologs decreasedto 2319 genes In the whole genome phylogram Bm IND1clusteredwith other Asian isolates ofBmelitensis as observedin the MLST analysis (Figure 4) Bm IND1 grouped with Bmelitensis NI which was originated from Mongolia and boththe strains belong to biovar 3 (Figure 4) The phylogeneticrelationship established here is in agreement with the earlierreports [10 11] Tan et al [11] performed a comparative wholegenome SNP analysis of B melitensis strains from around
Figure 4 Phylogenetic tree showing relationship between B melitensis IND1 and other B melitensis strains Maximum likelihood tree of 49whole genome sequenced B melitensis strains inferred from concatenated partitioned alignment of 2319 core genes using RAxML Supportvalues of branches are calculated from 100 bootstrap replicates and the branch length is proportional to the number of substitutions per siteB abortus 2308 has been used as outgroup species
Table 3 Prophage regions detected in B melitensis isolate
the world and reported clustering of B melitensis isolatesinto five genotypes In agreement with this observationour phylogenomic studies also revealed the clustering of Bmelitensis isolates into five groups where Group 1 formedthe earliest diverging clade Group II represents most of theAsian isolates of B melitensis including Bm IND1 Parallelto Group II another lineage evolved which further branchedinto groups III IV and V Group III represents isolates fromAfrica and groups IV and V constitute isolates from Europeand America respectively
322 Prophages Prophages that are integrated into thegenome of bacteria can contributemany biological propertiesto their bacterial hosts such as virulence biosynthesis andsecretion of toxins genomic divergence and evolution [37]We analyzed the genome of Bm IND1 for prophages usingPHAST that was designed to identify and annotate prophagesequences in bacterial genomes [31] The analysis identified 2putative prophage integrations in Chromosome I of Bm IND1
(Table 3 and Figure 5) Region 1 is composed of a fragmentof 137 kb size that encoded 18 genes out of which 14 geneswere phage specific and 4 genes were bacteria specific RegionII is composed of 226 kb with 14 genes where 8 genes werephage specific and remaining 6 genes belonged to BrucellaNotably region 1 is considered as intact prophage upstreamof QseB locus and the RAST server could identify the genesin this region Region II is predicted as incomplete prophagebut flanked with attachment sites Region I is present in theChinese isolate of B melitensis M28 also We identified twoputative phage integrations in Chromosome I of Bmelitensis16M genome which did not show any similarity to that ofBm IND1 or B melitensis M28 The analyses clearly indicatethat the prophage integration events contribute to the geneticdiversity of Bmelitensis
323 VNTRs VNTRs play an important role in evolutiongene regulation genome structure antigenic variation andvirulence [38ndash40] Mutations in VNTRs produce a wide
8 International Journal of Genomics
Region 1
Region 2
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Figure 3 Distribution of subsystem category for B melitensis IND1 Bm IND1 genome sequence was annotated using Rapid AnnotationSystem Technology server Features of each subsystem and their coverage are summarized in the pie chart
their functions on the basis of presence of subsystems in thegenome This makes functional annotation of genes moreaccurate than simply assigning the functions on the basis ofsequence similarity of known genes Functions of 2562 geneswere assignedwhile 629 geneswere annotated as hypothetical(Table 2) A total of 1649 genes were assigned for differentsubsystems where maximum number (405) was assignedfor metabolism of amino acids followed by carbohydratemetabolism (331) (Figure 3)
321 Whole Genome Phylogeny Determining the phyloge-netic relationship in a bacterial population is essential tounderstand the population structure evolutionary historyand host relationship and to develop diagnostic assays formolecular epidemiological studies [9] To perform com-parative phylogenomics we downloaded all the currentlyavailable Bmelitensis genomes (59 genomes) from GenBankand considered B abortus 2308 as the outgroup speciesWe evaluated the completeness of assembly and annotationof the genomes by assessing the number of orthologousgenes which are highly conserved among the strains Any Bmelitensis strain showing less number of orthologous genesthan the number of orthologous genes present in B abortuswith respect to B melitensis were ignored for the down-stream phylogenomic studies This facilitates more accuratecore genome estimation and identification of single copy
orthologs present in the species which improves resolutionof the phylogenetic tree Therefore we ignored the genomesof 12 Bmelitensis strains and considered the entire repertoireof coding genes of 48 B melitensis strains including BmIND1 for the analysis (Supplementary Table 3) A total of151361 genes of B melitensis strains were clustered in 3800gene families of which 25124 gene families present in all the48 strains However 2461 genes only showed exact singlecopy orthology in each strain that could be considered asthe core genome of B melitensis clade The core genomeincludes 73ndash82of genes from each of theBmelitensis strainWattam et al [10] reported that 2285 core genes are presentin the Brucella genus by analyzing the genomes of 40 Brucellaspecies Conceivably the core genome of B melitensis cladewas higher than the total number of core genes presentacross the Brucella genus We used B abortus 2308 as theoutgroup for whole genome phylogeny that increased thetotal cluster of genes to 3829 After including the B abortusstrain the total number of single copy orthologs decreasedto 2319 genes In the whole genome phylogram Bm IND1clusteredwith other Asian isolates ofBmelitensis as observedin the MLST analysis (Figure 4) Bm IND1 grouped with Bmelitensis NI which was originated from Mongolia and boththe strains belong to biovar 3 (Figure 4) The phylogeneticrelationship established here is in agreement with the earlierreports [10 11] Tan et al [11] performed a comparative wholegenome SNP analysis of B melitensis strains from around
Figure 4 Phylogenetic tree showing relationship between B melitensis IND1 and other B melitensis strains Maximum likelihood tree of 49whole genome sequenced B melitensis strains inferred from concatenated partitioned alignment of 2319 core genes using RAxML Supportvalues of branches are calculated from 100 bootstrap replicates and the branch length is proportional to the number of substitutions per siteB abortus 2308 has been used as outgroup species
Table 3 Prophage regions detected in B melitensis isolate
the world and reported clustering of B melitensis isolatesinto five genotypes In agreement with this observationour phylogenomic studies also revealed the clustering of Bmelitensis isolates into five groups where Group 1 formedthe earliest diverging clade Group II represents most of theAsian isolates of B melitensis including Bm IND1 Parallelto Group II another lineage evolved which further branchedinto groups III IV and V Group III represents isolates fromAfrica and groups IV and V constitute isolates from Europeand America respectively
322 Prophages Prophages that are integrated into thegenome of bacteria can contributemany biological propertiesto their bacterial hosts such as virulence biosynthesis andsecretion of toxins genomic divergence and evolution [37]We analyzed the genome of Bm IND1 for prophages usingPHAST that was designed to identify and annotate prophagesequences in bacterial genomes [31] The analysis identified 2putative prophage integrations in Chromosome I of Bm IND1
(Table 3 and Figure 5) Region 1 is composed of a fragmentof 137 kb size that encoded 18 genes out of which 14 geneswere phage specific and 4 genes were bacteria specific RegionII is composed of 226 kb with 14 genes where 8 genes werephage specific and remaining 6 genes belonged to BrucellaNotably region 1 is considered as intact prophage upstreamof QseB locus and the RAST server could identify the genesin this region Region II is predicted as incomplete prophagebut flanked with attachment sites Region I is present in theChinese isolate of B melitensis M28 also We identified twoputative phage integrations in Chromosome I of Bmelitensis16M genome which did not show any similarity to that ofBm IND1 or B melitensis M28 The analyses clearly indicatethat the prophage integration events contribute to the geneticdiversity of Bmelitensis
323 VNTRs VNTRs play an important role in evolutiongene regulation genome structure antigenic variation andvirulence [38ndash40] Mutations in VNTRs produce a wide
8 International Journal of Genomics
Region 1
Region 2
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Figure 4 Phylogenetic tree showing relationship between B melitensis IND1 and other B melitensis strains Maximum likelihood tree of 49whole genome sequenced B melitensis strains inferred from concatenated partitioned alignment of 2319 core genes using RAxML Supportvalues of branches are calculated from 100 bootstrap replicates and the branch length is proportional to the number of substitutions per siteB abortus 2308 has been used as outgroup species
Table 3 Prophage regions detected in B melitensis isolate
the world and reported clustering of B melitensis isolatesinto five genotypes In agreement with this observationour phylogenomic studies also revealed the clustering of Bmelitensis isolates into five groups where Group 1 formedthe earliest diverging clade Group II represents most of theAsian isolates of B melitensis including Bm IND1 Parallelto Group II another lineage evolved which further branchedinto groups III IV and V Group III represents isolates fromAfrica and groups IV and V constitute isolates from Europeand America respectively
322 Prophages Prophages that are integrated into thegenome of bacteria can contributemany biological propertiesto their bacterial hosts such as virulence biosynthesis andsecretion of toxins genomic divergence and evolution [37]We analyzed the genome of Bm IND1 for prophages usingPHAST that was designed to identify and annotate prophagesequences in bacterial genomes [31] The analysis identified 2putative prophage integrations in Chromosome I of Bm IND1
(Table 3 and Figure 5) Region 1 is composed of a fragmentof 137 kb size that encoded 18 genes out of which 14 geneswere phage specific and 4 genes were bacteria specific RegionII is composed of 226 kb with 14 genes where 8 genes werephage specific and remaining 6 genes belonged to BrucellaNotably region 1 is considered as intact prophage upstreamof QseB locus and the RAST server could identify the genesin this region Region II is predicted as incomplete prophagebut flanked with attachment sites Region I is present in theChinese isolate of B melitensis M28 also We identified twoputative phage integrations in Chromosome I of Bmelitensis16M genome which did not show any similarity to that ofBm IND1 or B melitensis M28 The analyses clearly indicatethat the prophage integration events contribute to the geneticdiversity of Bmelitensis
323 VNTRs VNTRs play an important role in evolutiongene regulation genome structure antigenic variation andvirulence [38ndash40] Mutations in VNTRs produce a wide
8 International Journal of Genomics
Region 1
Region 2
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
1 Terminase 6 Tail protein2 Portal protein7 Phage-like protein
3 Hypothetical protein8 Attachment site4 Head protein9 tRNA
5 Non-phage-like protein
1 2
3 3 3
3 3
3 333 444
5 5 55
5
6
6 6
7 7 7
77
77777
8 9
526311
1903640
1133
6870
(bp)
(bp)0
1374
2266
540051
1926303
Figure 5 Genomic organization of two putative phage like regions Scales are described below the chromosomal region and legends aredescribed at the bottom
Table 4 SNPs detected in other B melitensis isolates and B abortus 2308 with respect to Bm IND1
Species Synonymous SNPs Nonsynonymous SNPs Total number of SNPsB melitensis 16M 603 1462 2561B melitensisM28 56 142 281B melitensisM5-90 55 141 308B melitensis NI 78 180 351B melitensis ATCC 23457 51 124 252B melitensis ether 649 1551 2726B abortus 2308 1458 3454 6049
range of allelic diversity and VNTRs are considered as apowerful technique in molecular typing of bacterial species[41ndash43] We have analysed the genome of Bm IND1 andidentified 78 VNTRs with DNA motif size ranges from 8to 30 bps and the copy number ranges from 19 to 104(Supplementary Table 4) The data generated in our analysiscould be used for developing rapid diagnostic assays for high-resolution molecular epidemiological and clinical studies
324 SNPs SNPs serve as a powerful tool to describethe phylogenetic framework of a species [44] SNPs datawill help to develop novel high-resolution molecular typingtechniques for inter- and intraspecies discrimination ofpathogenic microorganisms We compared the genome ofBm IND1 with seven other B melitensis strains that is Bmelitensis 16M B melitensisM28 B melitensis ATCC 23457BmelitensisM5-90 Bmelitensis NI Bmelitensis Ether andB abortus 2308 for SNPs (Table 4 and Figure 6) The highestnumber of SNPs was detected with B abortus as it belongedto a different species (Table 4) Four B melitensis strains thatare originated from Asia namely B melitensis M28 bv1 Bmelitensis M5-90 bv1 B melitensis ATCC 23457 bv2 and Bmelitensis NI bv3 exhibited fewer SNPs ranging from 252
to 351 indicating their close genetic relatedness irrespectiveof their biovars This observation was in agreement withthe reported SNP-based phylogenetic analysis by Tan et al[11] However the SNPs observed with different strains inAdditional file 4 were less than the SNPs detected from NGSraw reads for template genome selection This is because thepolymorphisms extracted in these cases were derived fromuniquely aligned regions between two genome sequencesMost of the identified SNPs were in the coding regions ofthe genomes that may be attributed to the high proportionof coding regions in bacteria
While analysing the distribution of SNP locus amongall Asian strains 142 SNPs were shared by four strainsnamely B melitensis M28 B melitensis ATCC 23457 Bmelitensis M5-90 and B melitensis NI (Figure 7) Out of142 SNPs 141 are shared by all the 7 strains included in thepolymorphism analysis (Table 5)Therefore these 141 uniqueloci in Bm IND1 could be employed for genotyping andother molecular epidemiological studies B abortus 2308 Bmelitensis Ether and B melitensis 16M shared the maximumnumber of SNPs (952) against Bm IND1 These 952 lociare conserved in all Asian strains which may indicate thatAsian strains evolved from a common ancestor and theseloci mutated before its differentiation This finding is in
International Journal of Genomics 9
Chromosome I
210000
420000
630000
840000
1050000
1260000
1470000
1680000
1890000
2100000 0
2128440bp
(a)
Chromosome II
0
1100000
990000
880000
770000
660000550000
440000
330000
220000
110000
1185949bp
(b)
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Figure 6 Circular representation of identified Single Nucleotide Polymorphisms in Bmelitensis IND1 Chromosome I (a) and ChromosomeII (b) Chromosomal coordinates are indicated on outer most circle From outer to inner circles represented as circle 1 CDS (black) circle 2SNPs against B abortus 2308 (purple) circle 3 SNPs against B melitensis Ether (blue) circle 4 SNPs against B melitensis 16M (pink) circle5 SNPs against Bmelitensis NI (green) circle 6 SNPs against BmelitensisM5-90 (Orange) circle 7 SNPs against BmelitensisM 28
4
5
105
209
142
130
31
0
0
00
0
0
0
0
B melit
ensis
M28
B melitensis M5-90 B melitensis NIB melitensis ATCC 23457
Figure 7 The Venn diagram of SNPs detected in different Asianstrains against Bmelitensis IND1
agreement with whole genome phylogenetic analysis whereB melitensis M28 B melitensis M5-90 B melitensis NI andBm IND1 clustered together as a separate clade (Figure 4)However 4864 SNP loci are highly specific toB abortus 2308which are not shared by any of the six B melitensis strainsThese loci with interspecific polymorphisms can differentiatethese two species in clinical and epidemiological studiesAlso identified SNPs that are unique to each B melitensisstrain could be employed for in-depthmolecular analysis anddevelopment of novel molecular typing tools
We also categorized the genes containing SNPs basedon their functions assigned by RAST server (Supplemen-tary Figure 3) The subsystem category which has shownthe highest proportion of genes containing SNPs is nitro-gen metabolism followed by phosphorous carbohydrate
and amino acid metabolism Our findings are in agreementwith differential utilization of carbohydrates and amino acidsby closely related Brucella species and biovars A biotypingsystem has recently been developed to discriminate Brucellaspecies and biovars based on their differential metabolicactivity [45]
325 Indels Indels refers to deletions or insertions ofnucleotides in one genome with respect to another whichcould be employed as sequence signatures to characterizeevolution of diverged organisms [46] Indels can have adrastic effect on a gene leading to frameshift truncationsor extensions of an encoded protein We have identified 51Indels in the Bm IND1 genome with respect to B melitensisATCC 23457 of which 25 are located in the coding regions(Table 6) One noted INDEL is in the glycerol-3-phosphatedehydrogenase gene (glpD) of Bm IND1 Insertion of aguanosine residue in the 31015840 end of glpD resulted in two aminoacid mismatch followed by deletion of 69 amino acids fromthe C-terminus of this protein We verified this Indel by PCRamplification and sequencing of corresponding regions fromthe glpD gene of B melitensis IND strains including BmIND1 Insertion of G was present in the glpD gene of all thefour B melitensis IND strains with respect to ATCC23457and 16M (Figure 8) It has been reported that B melitensis16M deficient in glpD was attenuated in human and mousemacrophages [47 48] However our preliminary infectionstudies did not indicate attenuation of Bm IND1 in mouseperipheral blood mononuclear cells Our future studies willfocus on in vitro and in vivo infections studies using BmIND1 to analyse its virulence properties with the objectiveof developing novel live attenuated vaccines for livestockbrucellosis
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Figure 8 Sequence alignment of 3 terminus regions of glpD gene (a) Insertion of ldquoGrdquo in the glpD gene (position 1286) of Bm IND strainsin comparison to Bmelitensis 16M (16M) and Bmelitensis ATCC23457 (Bm ATCC) (b) Deletion of 69 nucleotides from the C-terminus ofglpD of Bm IND1 as a result of the nucleotide insertion
Table 5 Distribution of shared SNPs among 7 different strains of Bmelitensis identified against Bm IND1
Names of species sharing the SNPs in canonicalmanner
Numberof SNPs
Bm 16M BmM28 BmM5-90 Bm NI B abortusBm ATCC-23457 Bm Ether 141
Bm 16M BmM28 BmM5-90 B abortus BmATCC-23457 Bm Ether 1
Bm 16M BmM28 BmM5-90 Bm NI BmATCC-23457 Bm Ether 1
Bm 16M B abortus Bm ATCC-23457 Bm Ether 2Bm 16M B abortus Bm Ether 952Bm 16M B abortus Bm NI 1Bm 16M BmM28 BmM5-90 1Bm 16M B abortus 16Bm 16M Bm ATCC-23457 1Bm 16M Bm Ether 16Bm 16M BmM28 2B abortus Bm Ether 66B abortus Bm NI 1Bm Ether Bm NI 2BmM28 BmM5-90 128Bm 16M 1426B abortus 4864Bm ATCC-23457 102Bm Ether 1540BmM5-90 31BmM28 2Bm NI 205Bm denoted as Brucella melitensis
4 Conclusions
In conclusion genomic characterization and comparativegenome analysis of Bm IND1 revealed genetic structure ofB melitensis from India as well as from other geograph-ical locations Comparative genome analysis identified thesources of genetic variation in the form of SNPs VNTRsprophages and Indels These genetic markers could beemployed for developing high-resolution epidemiologicaltyping tools to understand the structure of Brucella popula-tion and for outbreak analysis Information on prophage inte-gration events and Indels in the virulence-associated geneswill provide important leads for the further experimentalcharacterization of virulence properties of Bm IND1 Thismay ultimately lead to the development of efficient therapeu-tic and preventive strategies to control animal and humanbrucellosis
Abbreviations
DIVA Differentiating infected from vaccinated animalsMLST Multilocus sequence typingSNP Single Nucleotide PolymorphismIndels Insertions and deletionsVNTRs Variable Number of Tandem Repeats
Disclosure
Present address for Sashi Bhushan Rao is as follows BiosafetySupport Unit Department of Biotechnology New Delhi110001 India Present address for Vivek Kumar Gupta is asfollows Centre for Animal Disease Research and DiagnosisIzatnagar 243122 India
Competing Interests
The authors declare that they have no competing interests
International Journal of Genomics 11
Table6IN
DEL
sidentified
inBm
IND1w
ithrespecttotheB
melitensisAT
CC23457in
thec
odingregion
Sno
GeneID
ATCC
23457
BmIN
D1
Genen
ame
1BM
EARS
00885
GTT
TCGCG
GAAG
CTTT
CGCG
GAA
GTT
TCGCG
GAA
Methion
ines
ynthase
2BM
EARS
00935
CGGG
CGGGG
Glycerol-3
-pho
sphate
dehydrogenase
3BM
EARS
00980
GCC
GCC
CABC
transporterp
ermease
4BM
EARS
01770
ATTT
TTT
ATTT
TTFA
D-binding
molybdo
pterin
dehydrogenase
5BM
EARS
02325
ATAT
ATAT
TAT
4-Hydroxy-3-m
ethylbut-2-
enyldiph
osph
ate
redu
ctase
6BM
EARS
03470
TGGGGGG
TGGGGGGG
Hypothetic
alprotein
7BM
EARS
04970
GAAAAAAA
GAAAAAAAA
Mechano
sensitive
ion
channelprotein
MscS
8BM
EARS
06240
ACCA
GAAG
CCCG
GCC
AGAAG
CCCG
GC
ACCA
GAAG
CCCG
GC
Cob
alttranspo
rter
9BM
EARS
07030
GCC
GAspartyl-tRN
Aam
idotransferase
subu
nitB
10BM
EARS
07320
GCG
CGGCG
GCT
CTCG
CGGCG
GCT
CTGCG
CGGCG
GCT
CTMultid
rugABC
transporter
ATP-bind
ingprotein
11BM
EARS
07865
AGCG
GGTA
AAC
GGGCG
GGTA
AAC
GGGC
AGCG
GGTA
AACG
GGCG
GGTA
AACG
GGCG
GGTA
AACG
GGC
Sensor
histidinek
inase
12BM
EARS
08810
TCC
TCTransporter
13BM
EARS
10105
GT
GAlanine
acetyltransfe
rase
14BM
EARS
10390
TGCG
TCTG
CGTC
ACCT
TTCA
TGGCG
TCHypothetic
alprotein
15BM
EARS
10570
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
TGGTG
GCG
GGGGTG
GCG
GGGGTG
GC
Type
IVsecretionprotein
VirB10
16BM
EARS
1200
0GCG
CGTT
CATC
GCC
GCG
GCG
CGTT
CATC
GCC
GCG
TTCA
TCGCC
GCG
Branched-chain
aminoacid
ABC
transporter
ATP-bind
ingprotein
17BM
EARS
13145
TGGGGG
TGGGGGG
Hypothetic
alprotein
18BM
EARS
13295
TTA
Aminotransferase
19BM
EARS
13570
TCCC
CCCC
CCC
TCCC
CCCC
CCPy
ridine
nucle
otide-disulfide
oxidoreductase
20BM
EARS
13580
AGGGGGGGGG
AGGGGGGGGGG
N-fo
rmylglutam
ate
amidoh
ydrolase
21BM
EARS
13730
CTTT
TTTT
CTTT
TTTT
THypothetic
alprotein
22BM
EARS
14825
GAAAAC
GCC
AAAA
GAAA
ABC
transporterp
ermease
23BM
EARS
14895
TCCC
CCCC
CCTC
CCCC
CCCC
CCC
Amidase
24BM
EARS
15830
CAAA
CAAAA
Aminoacid
ABC
transporter
substrate-bind
ingprotein
12 International Journal of Genomics
Authorsrsquo Contributions
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
Girish Radhakrishnan conceived the idea and designed thestudy Girish Radhakrishnan Sarwar Azam Sashi BhushanRao and Vivek Kumar Gupta worked out the methodologySashi Bhushan Rao Padmaja Jakka and Bindu Bhargaviperformed wet lab experiments and Girish Radhakrishnananalyzed the data Sarwar Azam and Veera NarasimhaRaoanalyzed the data for assembly annotations and comparativegenomics Girish Radhakrishnan and Sarwar Azamwrote thepaper
Acknowledgments
This work was supported by funding from the Departmentof Biotechnology Ministry of Science and Technology Gov-ernment of India through the National Institute of AnimalBiotechnology Hyderabad India
References
[1] G Pappas N Akritidis M Bosilkovski and E Tsianos ldquoBru-cellosisrdquoThe New England Journal of Medicine vol 352 no 22pp 2325ndash2367 2005
[2] S J Cutler A M Whatmore and N J Commander ldquoBruce-llosismdashnew aspects of an old diseaserdquo Journal of AppliedMicrobiology vol 98 no 6 pp 1270ndash1281 2005
[3] T D Anderson and N F Cheville ldquoUltrastructural morpho-metric analysis of Brucella abortus-infected trophoblasts inexperimental placentitis bacterial replication occurs in roughendoplasmic reticulumrdquoTheAmerican Journal of Pathology vol124 no 2 pp 226ndash237 1986
[4] J Celli C De Chastellier D-M Franchini J Pizarro-CerdaE Moreno and J-P Gorvel ldquoBrucella evades macrophagekilling via VirB-dependent sustained interactions with theendoplasmic reticulumrdquo Journal of Experimental Medicine vol198 no 4 pp 545ndash556 2003
[5] J Ko and G A Splitter ldquoMolecular host-pathogen interactionin brucellosis current understanding and future approaches tovaccine development for mice and humansrdquo Clinical Microbiol-ogy Reviews vol 16 no 1 pp 65ndash78 2003
[6] G Pappas P Papadimitriou N Akritidis L Christou and E VTsianos ldquoThe new globalmap of human brucellosisrdquoTheLancetInfectious Diseases vol 6 no 2 pp 91ndash99 2006
[7] A Kumar ldquoBrucellosis need of public health intervention inrural Indiardquo ContributionsMacedonian Academy of Sciencesand Arts Section of Biological and Medical Sciences vol 31 no1 pp 219ndash231 2010
[8] A M Whatmore L L Perrett and A P MacMillan ldquoChar-acterisation of the genetic diversity of Brucella by multilocussequencingrdquo BMCMicrobiology vol 7 article 34 2007
[9] D OrsquoCallaghan and AMWhatmore ldquoBrucella genomics as weenter the multi-genome erardquo Briefings in Functional Genomicsvol 10 no 6 Article ID elr026 pp 334ndash341 2011
[10] A R Wattam J T Foster S P Mane et al ldquoComparativephylogenomics and evolution of the brucellae reveal a path tovirulencerdquo Journal of Bacteriology vol 196 no 5 pp 920ndash9302014
[11] K-K Tan Y-C Tan L-Y Chang et al ldquoFull genome SNP-basedphylogenetic analysis reveals the origin and global spread ofBrucella melitensisrdquo BMC Genomics vol 16 article 93 2015
[12] I F Huddleson ldquoDifferentiation of the species of the genusBrucellardquo American Journal of Public Health and the NationsHealth vol 21 no 5 pp 491ndash498 1931
[13] S Erdelling and A Sen ldquoIsolation and biotyping of Brucellaspecies from aborted sheep fetusesrdquo Pendik Veterinary Micro-biology vol 31 no 2 pp 31ndash42 2000
[14] L Herman and H De Ridder ldquoIdentification of Brucellaspp by using the polymerase chain reactionrdquo Applied andEnvironmental Microbiology vol 58 no 6 pp 2099ndash2101 1992
[15] C Romero C Gamazo M Pardo and I Lopez-Goni ldquoSpecificdetection of Brucella DNA by PCRrdquo Journal of Clinical Micro-biology vol 33 no 3 pp 615ndash617 1995
[16] A Singh V K Gupta A Kumar V K Singh and S Nayakwadildquo16S rRNA and omp31 gene based molecular characterizationof field strains of B melitensis from aborted foetus of goats inIndiardquoThe ScientificWorld Journal vol 2013 Article ID 1603767 pages 2013
[17] J Sankarasubramanian U S Vishnu L A Khader J SridharP Gunasekaran and J Rajendhran ldquoBrucellaBase genomeinformation resourcerdquo Infection Genetics and Evolution vol 43pp 38ndash42 2016
[18] K Katoh andDM Standley ldquoMAFFTmultiple sequence align-ment software version 7 improvements in performance andusabilityrdquo Molecular Biology and Evolution vol 30 no 4 pp772ndash780 2013
[19] A Stamatakis ldquoRAxML version 8 a tool for phylogeneticanalysis and post-analysis of large phylogeniesrdquo Bioinformaticsvol 30 no 9 pp 1312ndash1313 2014
[20] D H Huson D C Richter C Rausch T Dezulian M Franzand R Rupp ldquoDendroscope an interactive viewer for largephylogenetic treesrdquo BMC Bioinformatics vol 8 article 4602007
[21] S B Rao V K Gupta M Kumar et al ldquoDraft genome sequenceof the field isolate Brucella melitensis strain Bm IND1 fromIndiardquoGenome Announcements vol 2 no 3 Article ID e00497-14 2014
[22] R Luo B Liu Y Xie et al ldquoSOAPdenovo2 an empiricallyimproved memory-efficient short-read de novo assemblerrdquoGigaScience vol 1 no 1 article 18 2012
[23] B Langmead and S L Salzberg ldquoFast gapped-read alignmentwith Bowtie 2rdquo Nature Methods vol 9 no 4 pp 357ndash359 2012
[24] H Li B Handsaker A Wysoker et al ldquoThe sequence align-mentmap format and SAMtoolsrdquoBioinformatics vol 25 no 16pp 2078ndash2079 2009
[25] S Azam A Rathore T M Shah et al ldquoAn Integrated SNPMining and Utilization (ISMU) pipeline for next generationsequencing datardquo PLoS ONE vol 9 no 7 article e101754 2014
[26] S Assefa T M Keane T D Otto C Newbold and MBerriman ldquoABACAS algorithm-based automatic contiguationof assembled sequencesrdquo Bioinformatics vol 25 no 15 pp1968ndash1969 2009
[27] A C E Darling B Mau F R Blattner and N T PernaldquoMauve multiple alignment of conserved genomic sequencewith rearrangementsrdquoGenome Research vol 14 no 7 pp 1394ndash1403 2004
[28] R K Aziz D Bartels A Best et al ldquoThe RAST Server rapidannotations using subsystems technologyrdquo BMCGenomics vol9 article 75 2008
[29] L Li C J Stoeckert Jr and D S Roos ldquoOrthoMCL identi-fication of ortholog groups for eukaryotic genomesrdquo GenomeResearch vol 13 no 9 pp 2178ndash2189 2003
International Journal of Genomics 13
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014
[30] G Talavera and J Castresana ldquoImprovement of phylogeniesafter removing divergent and ambiguously aligned blocks fromprotein sequence alignmentsrdquo Systematic Biology vol 56 no 4pp 564ndash577 2007
[31] Y Zhou Y Liang K H Lynch J J Dennis and D S WishartldquoPHAST a fast phage search toolrdquo Nucleic Acids Research vol39 no 2 pp W347ndashW352 2011
[32] G Benson ldquoTandem repeats finder a program to analyze DNAsequencesrdquo Nucleic Acids Research vol 27 no 2 pp 573ndash5801999
[33] S Kurtz A Phillippy A L Delcher et al ldquoVersatile and opensoftware for comparing large genomesrdquo Genome Biology vol 5article R12 2004
[34] P Cingolani A Platts L L Wang et al ldquoA program forannotating and predicting the effects of single nucleotidepolymorphisms SnpEff SNPs in the genome of Drosophilamelanogaster strain w 1118 iso-2 iso-3rdquo Fly vol 6 no 2 pp80ndash92 2012
[35] F Wang S Hu Y Gao Z Qiao W Liu and Z Bu ldquoCompletegenome sequences of Brucella melitensis strains M28 and M5-90 with different virulence backgroundsrdquo Journal of Bacteriol-ogy vol 193 no 11 pp 2904ndash2905 2011
[36] V G Delvecchio V Kapatral R J Redkar et al ldquoThe genomesequence of the facultative intracellular pathogen Brucellamelitensisrdquo Proceedings of the National Academy of Sciences ofthe United States of America vol 99 no 1 pp 443ndash448 2002
[37] A M Varani C B Monteiro-Vitorello H I Nakaya andM-AVan Sluys ldquoThe role of prophage in plant-pathogenic bacteriardquoAnnual Review of Phytopathology vol 51 pp 429ndash451 2013
[38] A Van Belkum S Scherer L Van Alphen and H VerbrughldquoShort-sequence DNA repeats in prokaryotic genomesrdquoMicro-biology and Molecular Biology Reviews vol 62 no 2 pp 275ndash293 1998
[39] A van Belkum S Scherer W van Leeuwen D Willemse Lvan Alphen and H Verbrugh ldquoVariable number of tandemrepeats in clinical strains of Haemophilus influenzaerdquo Infectionand Immunity vol 65 no 12 pp 5017ndash5027 1997
[40] J C Tan A Tan L Checkley C M Honsa and M T FerdigldquoVariable numbers of tandem repeats inPlasmodium falciparumgenesrdquo Journal of Molecular Evolution vol 71 no 4 pp 268ndash278 2010
[41] S P Yazdankhah andB-A Lindstedt ldquoVariable number tandemrepeat typing of bacteriardquo Methods in Molecular Biology vol396 pp 395ndash405 2007
[42] C-S Chiou HWatanabe Y-WWang et al ldquoUtility of multilo-cus variable-number tandem-repeat analysis as amolecular toolfor phylogenetic analysis of Shigella sonneirdquo Journal of ClinicalMicrobiology vol 47 no 4 pp 1149ndash1154 2009
[43] P KeimMN Van Ert T Pearson A J Vogler L Y Huynh andDMWagner ldquoAnthraxmolecular epidemiology and forensicsusing the appropriate marker for different evolutionary scalesrdquoInfection Genetics and Evolution vol 4 no 3 pp 205ndash213 2004
[44] P H Brito and S V Edwards ldquoMultilocus phylogeography andphylogenetics using sequence-based markersrdquo Genetica vol135 no 3 pp 439ndash455 2009
[45] S Al Dahouk H C Scholz H Tomaso et al ldquoDifferentialphenotyping of Brucella species using a newly developed semi-automatedmetabolic systemrdquo BMCMicrobiology vol 10 article269 2010
[46] O K Kamneva D A Liberles and N L Ward ldquoGenome-wide influence of indel substitutions on evolution of bacteria
of the PVC superphylum revealed using a novel computationalmethodrdquo Genome Biology and Evolution vol 2 pp 870ndash8862010
[47] Q Wu J Pei C Turse and T A Ficht ldquoMariner mutagenesisof Brucella melitensis reveals genes with previously uncharac-terized roles in virulence and survivalrdquo BMCMicrobiology vol6 article 102 2006
[48] A Zuniga-Ripa T Barbier R Conde-Alvarez et al ldquoBrucellaabortus depends on pyruvate phosphate dikinase and malicenzyme but not on fbp and glpX fructose-16-bisphosphatasesfor full virulence in laboratory modelsrdquo Journal of Bacteriologyvol 196 no 16 pp 3045ndash3057 2014





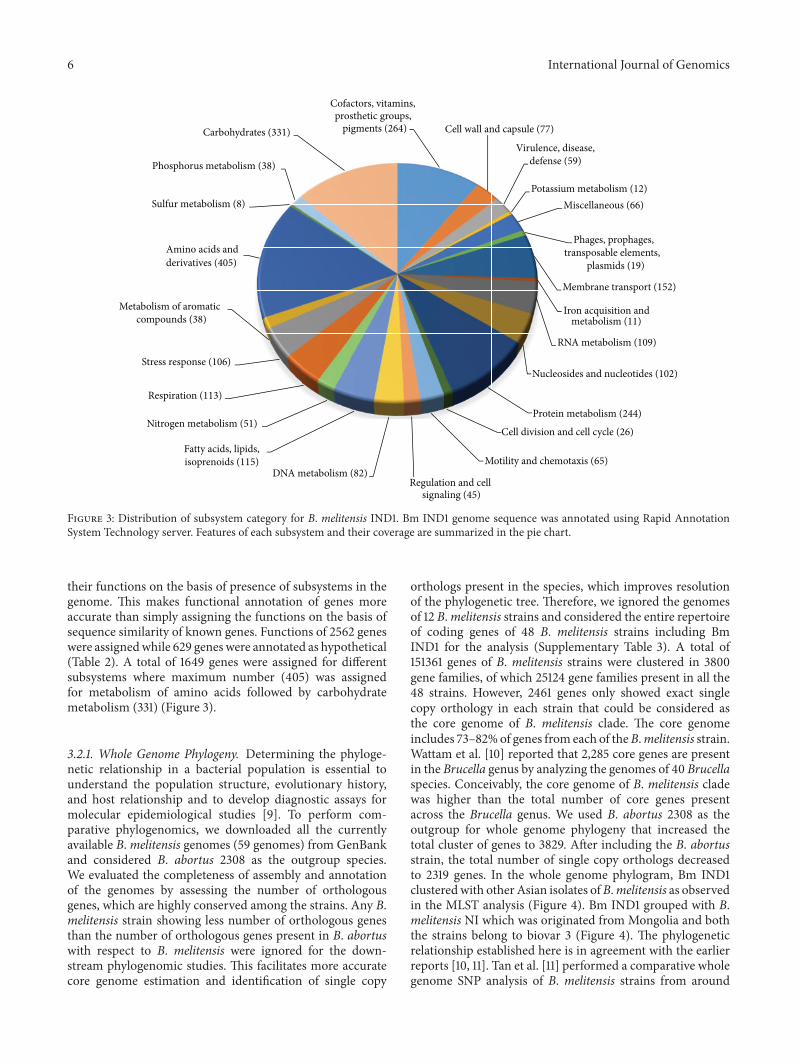




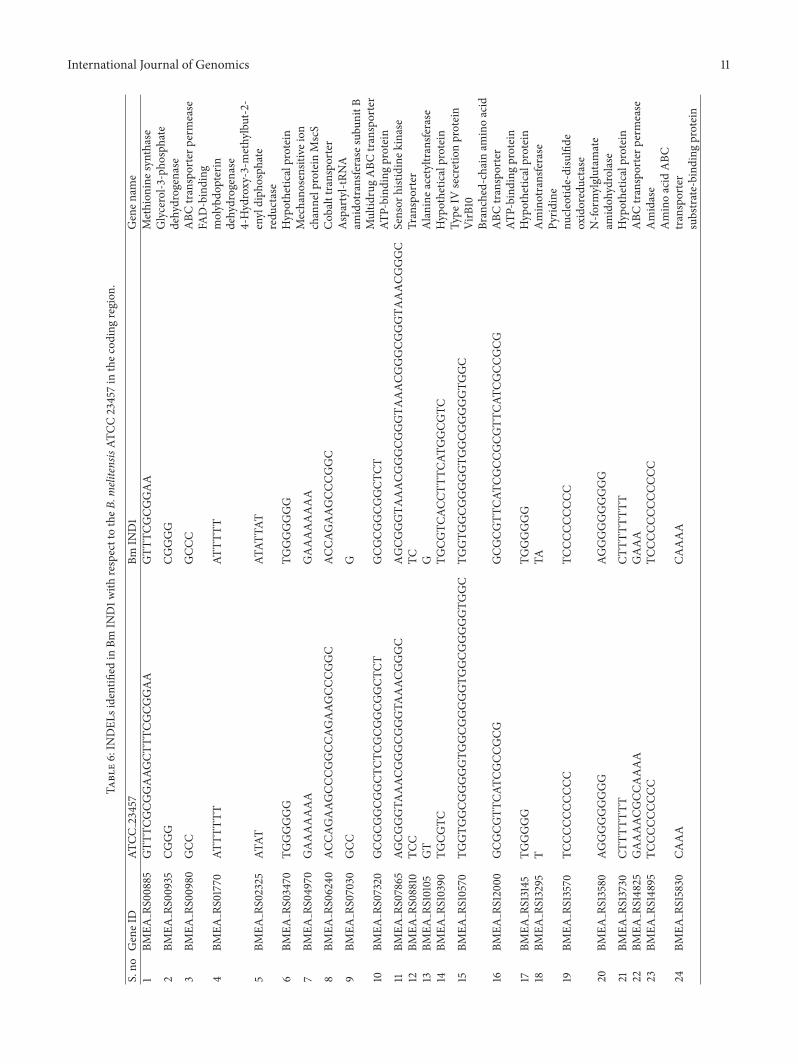













![RESEARCH ARTICLE Open Access Epidemiological features … · B. melitensis, B. abortus, B. suis and B. canis often cause abortion and infertility in these natural hosts [4,8-11].](https://static.documents.pub/doc/80x56/5ca9d6b588c9931f068d2547/research-article-open-access-epidemiological-features-b-melitensis-b-abortus.jpg)






