Page 1
University of Tennessee Health Science Center University of Tennessee Health Science Center
UTHSC Digital Commons UTHSC Digital Commons
Theses and Dissertations (ETD) College of Graduate Health Sciences
12-2013
Determinants of Upper Genital Tract Complications in a Determinants of Upper Genital Tract Complications in a
Chlamydial Urogenital Mouse Model Chlamydial Urogenital Mouse Model
Enitra N. Jones University of Tennessee Health Science Center
Follow this and additional works at: https://dc.uthsc.edu/dissertations
Part of the Medical Sciences Commons
Recommended Citation Recommended Citation Jones, Enitra N. , "Determinants of Upper Genital Tract Complications in a Chlamydial Urogenital Mouse Model" (2013). Theses and Dissertations (ETD). Paper 347. http://dx.doi.org/10.21007/etd.cghs.2013.0156.
This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected] .
Page 2
Determinants of Upper Genital Tract Complications in a Chlamydial Urogenital Determinants of Upper Genital Tract Complications in a Chlamydial Urogenital Mouse Model Mouse Model
Abstract Abstract Genital Chlamydia trachomatis infection is a major public health concern. Chlamydia is the most commonly reported infection in the United States and the most common bacterial sexually transmitted infection worldwide. Unrecognized infection endangers female reproductive health by serious complications such as Pelvic Inflammatory Disease, ectopic pregnancy, and involuntary infertility. Widespread Chlamydia control programs were implemented more than two decades ago to improve women's reproductive health but, despite initial success, the number of chlamydial infections reported have increased.
One of the hypotheses put forth to explain increased chlamydial reporting suggests that a longterm caveat of control initiatives is interference with the development of natural occurring immunity as a result of mass screening and rapid treatment. It is proposed that human cohorts are more susceptible to subsequent chlamydiae infection and their increased susceptibility drive the current increase in sexually transmitted chlamydiae case notifications.
In these studies we describe a comprehensive approach to assessing the role of early antichlamydial intervention, an integral component of control initiatives, on the subsequent development and severity of upper genital tract sequelae in a murine model of recurrent chlamydiae urogenital infection. The development of an in vivo model of urogenital Chlamydia trachomatis infection is central to defining the risk of developing long term reproductive complications, delineating potential biomarkers for chlamydial-induced genital tract disease, interrogating host factors that may contribute to the development of adverse complications, and anti-chlamydial vaccine development.
Document Type Document Type Dissertation
Degree Name Degree Name Doctor of Philosophy (PhD)
Program Program Biomedical Sciences
Research Advisor Research Advisor Gerald I. Byrne, Ph.D.
Keywords Keywords Chlamydia, Disease, Genital Tract, Mouse, Sequelae
Subject Categories Subject Categories Medical Sciences | Medicine and Health Sciences
Comments Comments One year embargo expired December 2014
This dissertation is available at UTHSC Digital Commons: https://dc.uthsc.edu/dissertations/347
Page 3
Determinants of Upper Genital Tract Complications in a Chlamydial Urogenital
Mouse Model
A Dissertation
Presented for
The Graduate Studies Council
The University of Tennessee
Health Science Center
In Partial Fulfillment
Of the Requirements for the Degree
Doctor of Philosophy
From The University of Tennessee
By
Enitra N. Jones
December 2013
Page 4
ii
Copyright © 2013 by Enitra N. Jones.
All rights reserved.
Page 5
iii
DEDICATION
This work is lovingly dedicated to my mother, Evelyn T. Jones. You believed in
me when I was filled with self-doubt. You encouraged me when others said I could not
accomplish the goal. Your faith never wavered and you fervently prayed. For this and
more I am eternally grateful.
Page 6
iv
ACKNOWLEDGEMENTS
It takes a village . . . .
–African Proverb
First, I would like to thank my research advisor, Dr. Gerald I. Byrne, for the
opportunity to carry out the enclosed research in his laboratory. His passion for science
and enthusiasm for learning have been both inspiring and motivational throughout my
doctoral journey. Dr. Byrne has also allowed me the freedom to integrate my interest in
public health with my academic pursuits and for that I am grateful.
The guidance offered by each member of my graduate committee was essential. I
would like to thank Drs. Robert Belland, B. Keith English, Elizabeth Fitzpatrick, and P.
David Rogers for providing scientific insight throughout this process and for being
genuinely invested in my success while matriculating at the University of Tennessee
Health Science Center and beyond. Whether it was help with a laboratory technique, a
recommendation letter, or an encouraging word, each member was more than willing to
lend a helping hand. Thank you for sharing your expertise and offering thoughtful
critiques on my work.
I would also like to acknowledge past and present members of the Byrne
Laboratory. My first teachers in the laboratory were Drs. O. Sadia Mahdi and Isao
Miyairi who patiently taught me the “ins and outs” of cell culture and chlamydial growth.
I am thankful for Vijaya Onguri’s contributions to early vaccination studies and her
ongoing friendship. The technical help throughout the years provided by Dr. Jan Peters
and Jonathan Laxton, now members of the RBL staff, is greatly appreciated. The
histological studies discussed in this dissertation would not have been possible without
Xeofei Wang. Conversations with Dr. Yin Su concerning animal studies were invaluable.
I also want to acknowledge the RBL staff and the many summer students that have been
active participants in laboratory discussions throughout the years. Thank you all!
My time at the University of Tennessee Health Science Center has been enriched
by several members of the faculty/staff and student body. I was truly blessed to have
made the acquaintance of my “UT Mom”, Mrs. Ruby McNeal. Your unwavering support
and encouragement will never be forgotten and is appreciated beyond words. Thank you
to Mrs. Devonia Cage, Mrs. Carolyn Fields, and Mrs. Evelyn Lewis for your prayer and
encouragement. The guidance and mentorship offered by Executive Vice Chancellor and
Chief Operations Officer, Dr. Kennard Brown, was invaluable and greatly appreciated. I
also want to thank the Microbiology, Immunology, and Biochemistry Department, the
members of the Black Graduate Student Association, and many of my fellow graduate
students for their help throughout the years.
I would like to thank my immediate and extended family for their love and
support. To my parents, Ezra and Evelyn Jones, who prayed without ceasing, dried my
tears when things seemed too difficult to continue, and continue to love me
Page 7
v
unconditionally. I love you to life forever. To my grandmother, Annie Sue Gibbs, thank
you for reminding me that God doesn’t always “move the mountains”, sometimes He
gives us the strength to climb them! To my aunts, uncles, siblings, cousins, friends, and
sorority sisters- thank you for the comedic relief and unwavering support. To my best
friends (outside of my parents), Tron Foster, Jason Hughes and Luviska Nicholas, thank
you for nourishing my spirit and believing in my dreams. Sincere thanks to two of my
ongoing mentors and friends, Drs. Oswald D’Auvergne and Ivory Toldson. Last but
certainly not least, I thank the Lord for granting me the serenity to accept the things I can
not change, the courage to change the things I can, and the wisdom to know the
difference.
Page 8
vi
ABSTRACT
Genital Chlamydia trachomatis infection is a major public health concern.
Chlamydia is the most commonly reported infection in the United States and the most
common bacterial sexually transmitted infection worldwide. Unrecognized infection
endangers female reproductive health by serious complications such as Pelvic
Inflammatory Disease, ectopic pregnancy, and involuntary infertility. Widespread
Chlamydia control programs were implemented more than two decades ago to improve
women’s reproductive health but, despite initial success, the number of chlamydial
infections reported have increased.
One of the hypotheses put forth to explain increased chlamydial reporting
suggests that a long-term caveat of control initiatives is interference with the
development of natural occurring immunity as a result of mass screening and rapid
treatment. It is proposed that human cohorts are more susceptible to subsequent
chlamydiae infection and their increased susceptibility drive the current increase in
sexually transmitted chlamydiae case notifications.
In these studies we describe a comprehensive approach to assessing the role of
early anti-chlamydial intervention, an integral component of control initiatives, on the
subsequent development and severity of upper genital tract sequelae in a murine model of
recurrent chlamydiae urogenital infection. The development of an in vivo model of
urogenital Chlamydia trachomatis infection is central to defining the risk of developing
long term reproductive complications, delineating potential biomarkers for chlamydial-
induced genital tract disease, interrogating host factors that may contribute to the
development of adverse complications, and anti-chlamydial vaccine development.
Page 9
vii
TABLE OF CONTENTS
CHAPTER 1. CHLAMYDIAE .........................................................................................1
Historical Perspective ....................................................................................................1
Taxonomy ......................................................................................................................1 Developmental Cycle .....................................................................................................2 Chlamydiae Clinical Significance ..................................................................................4
Chlamydia pneumoniae .............................................................................................4 Chlamydia psittaci .....................................................................................................4
Chlamydia trachomatis ..............................................................................................6 Ocular Chlamydia trachomatis ..................................................................................6
Genital Chlamydia trachomatis .................................................................................6 Mouse Model of Genital Infection .................................................................................7
Murine Genital Tract Pathology ................................................................................9 Murine Model Limitations .........................................................................................9
Immunological Response to Genital Infection ............................................................11 Intracellular Immune Response Overview...................................................................11
Chlamydia Innate Response .....................................................................................11 Chlamydia Adaptive Response ................................................................................12 Vaccination ..............................................................................................................13
Control Measures .........................................................................................................14 Sexually Transmitted Chlamydia Epidemiology .....................................................15
Hypotheses for Rebounding Chlamydia Rates ........................................................16
Arrested Immunity Hypothesis ................................................................................18
Dissertation Rationale ..................................................................................................19
CHAPTER 2. ARRESTED IMMUNITY: IMPACT OF EARLY TREATMENT
ON UPPER GENITAL TRACT SEQUELAE ..............................................................20
Introduction ..................................................................................................................20
Materials and Methods .................................................................................................21 Mice .........................................................................................................................21 Chlamydia Strain and Titration ................................................................................21 Mouse Infection .......................................................................................................21 Antibiotic Treatment ................................................................................................21
Bacterial Shedding ...................................................................................................21
Fluorescence-Linked Immunosorbent Assay (FLISA) Antibody Analysis .............22
Pathology .................................................................................................................22 Organ-Total Body Weight Ratio Analysis ...............................................................22 Statistical Analysis ...................................................................................................23
Results ..........................................................................................................................23 Kinetics of Genital Tract Infection in Antibiotic-Treated Mice ..............................23
Immune Arrest and Pathology Following Primary Infection of Antibiotic-
Treated Mice ............................................................................................................23 Disease Severity Following Reinfection in Antibiotic-Treated Mice .....................26
Page 10
viii
IL-4 Is Detectable Late Following Reinfection and Positively Correlates with
Disease Severity in Antibiotic-Treated Mice ...........................................................31 Chlamydia and Cellular Infiltrates Following Reinfection in Antibiotic-Treated
Mice ..........................................................................................................................31
Discussion ....................................................................................................................36
CHAPTER 3. CHLAMYDIAL GENITAL TRACT SEQUELAE IS AGE AND
STRAIN-DEPENDENT ..................................................................................................42
Introduction ..................................................................................................................42 Materials and Methods .................................................................................................42
Mice .........................................................................................................................42 Chlamydia Strain .....................................................................................................43
Chlamydia muridarum Infection .............................................................................43 Pathology Assessment .............................................................................................43
Results ..........................................................................................................................44 C57BL/6J Exhibit Age-Dependent Upper Genital Tract Sequelae Post-Primary
Intravaginal Infection ...............................................................................................44 Secondary Intravaginal Infection Exacerbates Chlamydial-Induced Upper
Genital Tract Complications ....................................................................................44 C57BL/6 Mice Are More Susceptible to Chlamydial-Induced Upper Genital
Tract Complications When Compared to DBA/2J Mice .........................................44
Discussion ....................................................................................................................47
CHAPTER 4. IMMUNIZATION WITH C. MURIDARUM OUTER
MEMBRANE COMPLEX FAILS TO PROTECT AGAINST UPPER
GENITAL TRACT COMPLICATIONS IN A MURINE MODEL OF
GENITAL INFECTION .................................................................................................50
Introduction ..................................................................................................................50
Materials and Methods (Systemic Lethal Model) ........................................................51 Mice .........................................................................................................................51
Chlamydiae ..............................................................................................................51 COMC Extraction ....................................................................................................51 Immunization and Infection .....................................................................................52
Results (Systemic Lethal Model) .................................................................................52 Materials and Methods (Urogenital Model) ................................................................55
Mice .........................................................................................................................55
Chlamydiae ..............................................................................................................55
COMC Extraction ....................................................................................................55 Immunization and Infection .....................................................................................55
Results (Urogenital Model) ..........................................................................................56 Materials and Methods (Alum Study)..........................................................................56
Mice .........................................................................................................................56
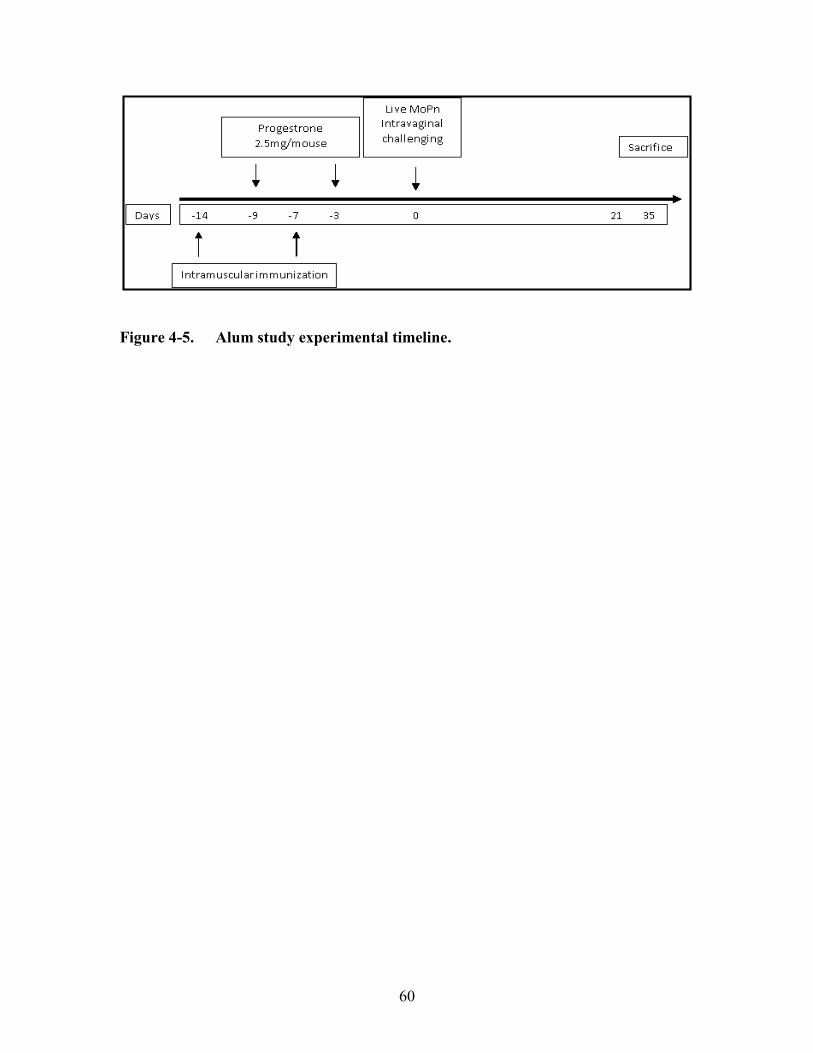
Chlamydiae ..............................................................................................................56 Immunization and Infection .....................................................................................59 Pathology Assessment .............................................................................................59
Page 11
ix
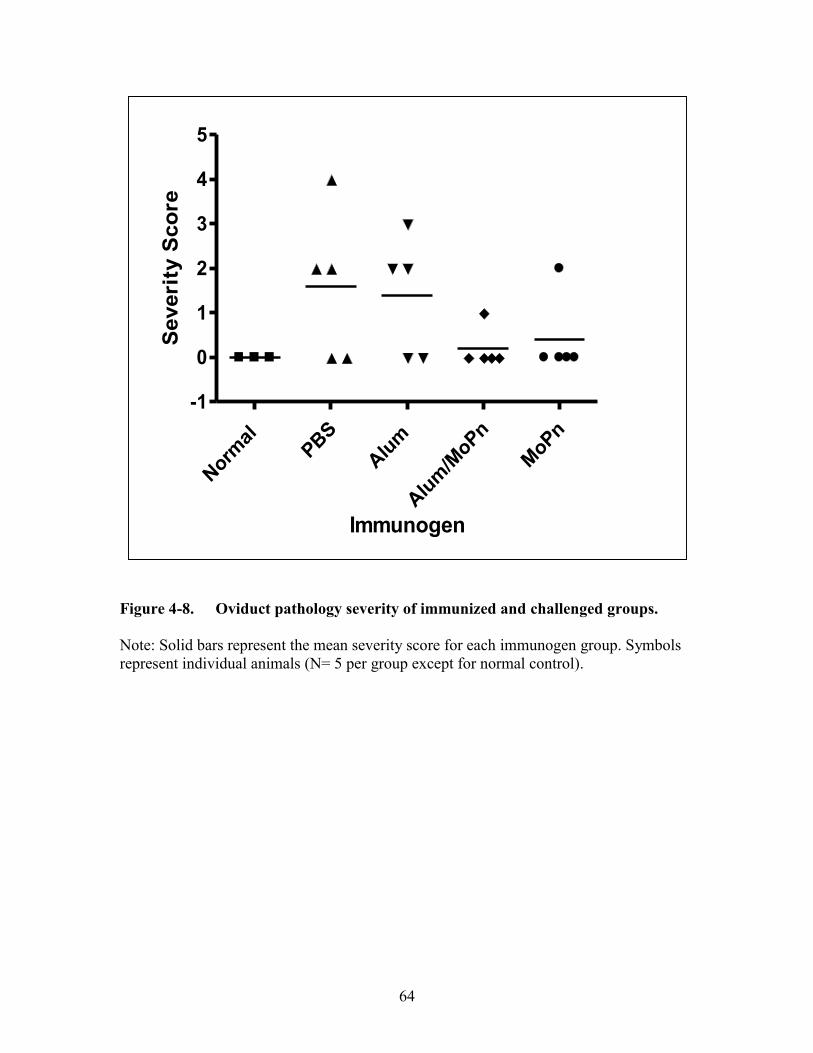
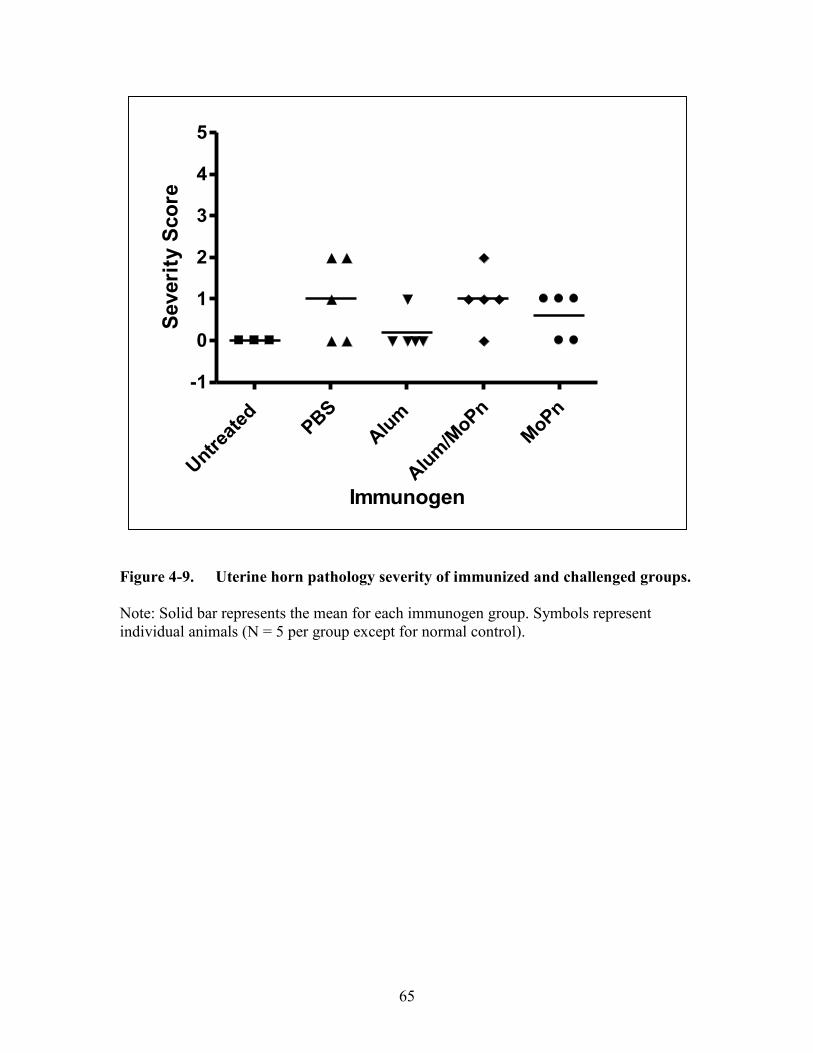
Results (Alum Study) ...................................................................................................59
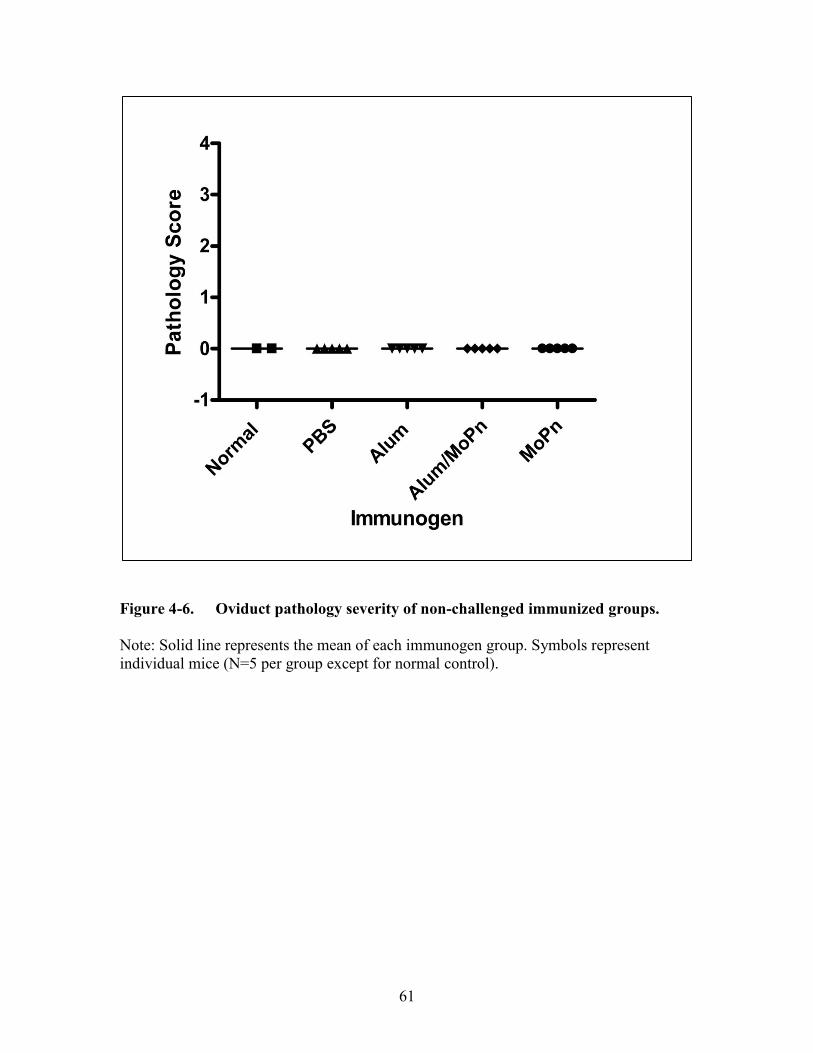
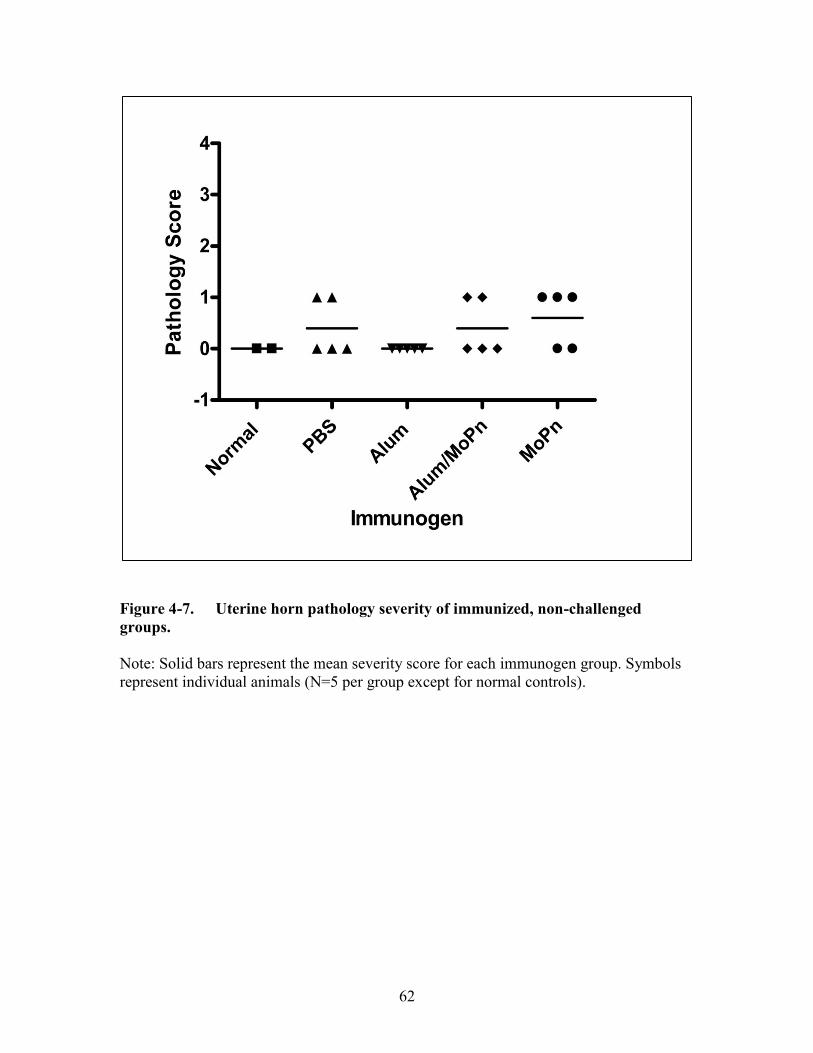
Immunogens without Intravaginal Chlamydia muridarum Challenge Do Not
Induce Severe Upper Genital Tract Sequelae Formation .........................................59 Alum Alone Does Not Negate the Development of Severe Upper Genital Tract
Complications ...........................................................................................................63 Discussion ....................................................................................................................63
CHAPTER 5. CONCLUSIONS ......................................................................................68
Testing the IL-4 Hypothesis.........................................................................................68 Identifying Genetic Link to Age-Dependent Disease Severity....................................68
Implications for Vaccine Development .......................................................................69
LIST OF REFERENCES ............................................................................................... 70
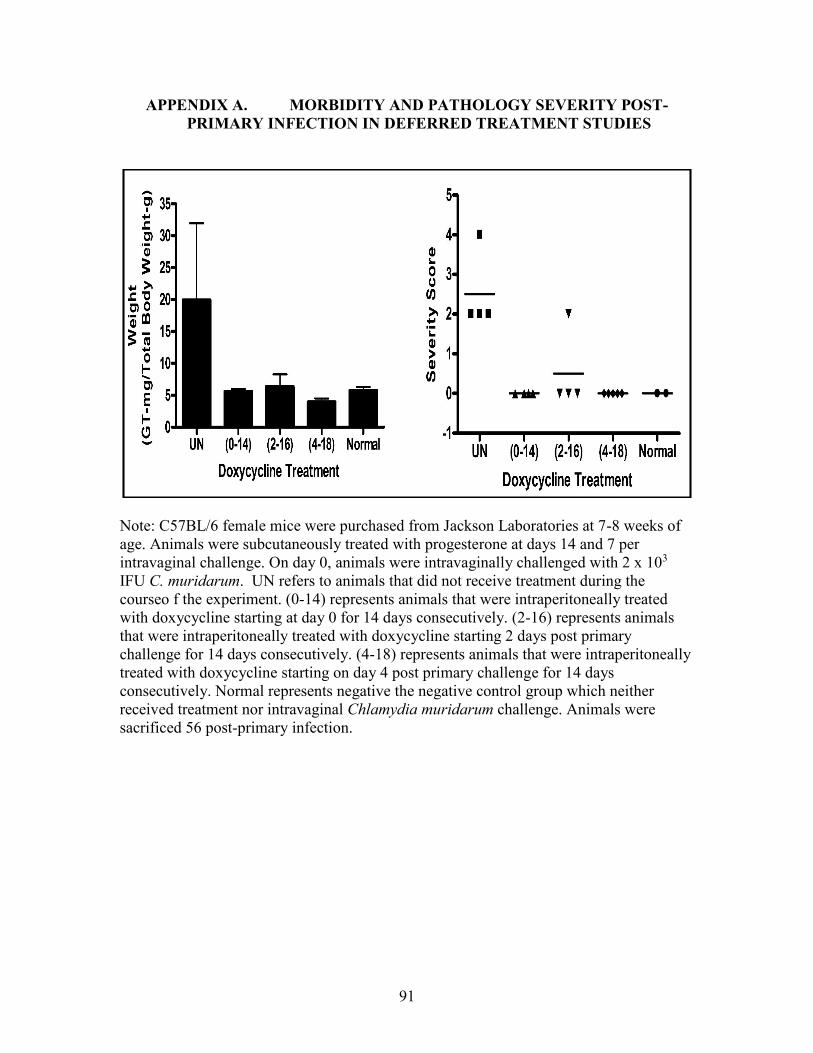
APPENDIX A. MORBIDITY AND PATHOLOGY SEVERITY POST-
PRIMARY INFECTION IN DEFERRED TREATMENT STUDIES .......................91
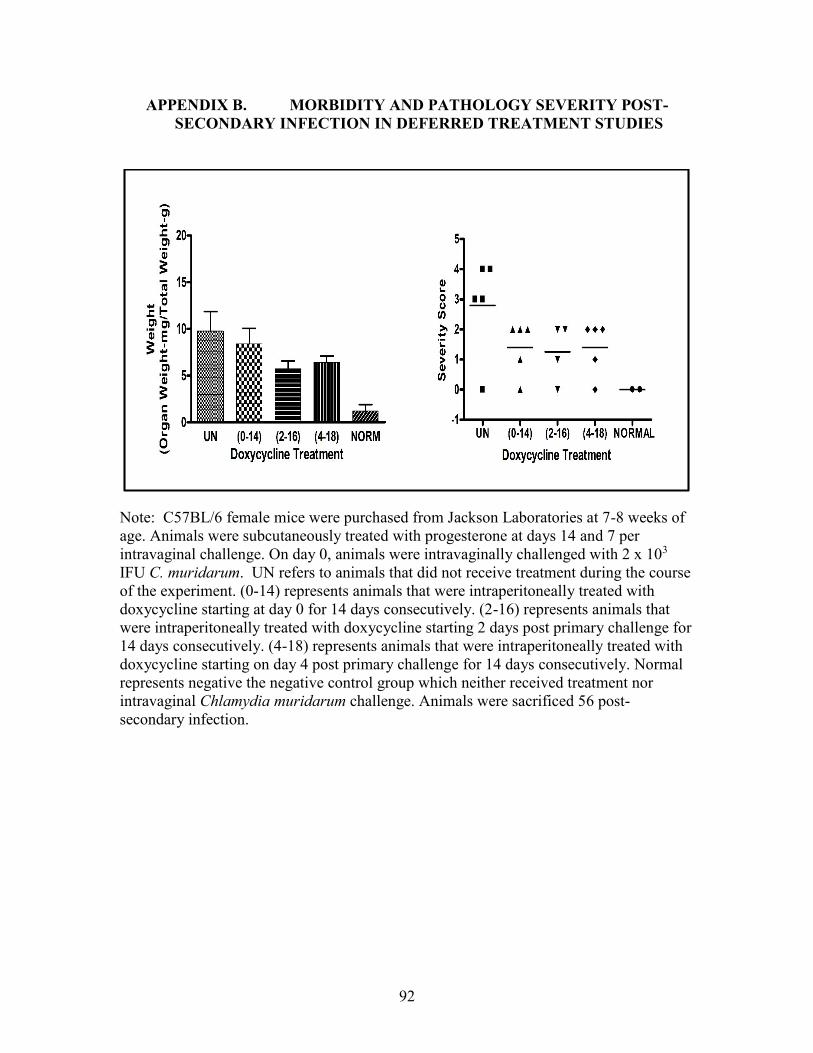
APPENDIX B. MORBIDITY AND PATHOLOGY SEVERITY POST-
SECONDARY INFECTION IN DEFERRED TREATMENT STUDIES .................92
APPENDIX C. TH1-RELATED CYOKINES POST RECURRENT
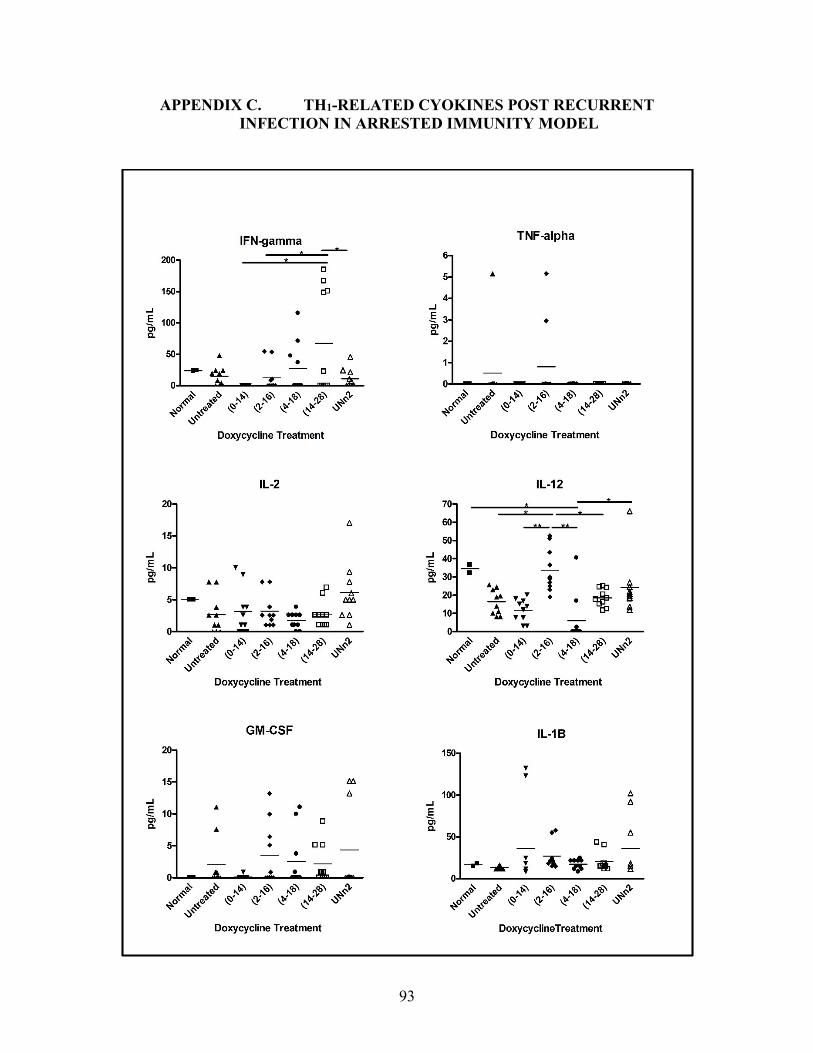
INFECTION IN ARRESTED IMMUNITY MODEL..................................................93
APPENDIX D. TH2-RELATED CYTOKINES POST RECURRENT
INFECTION IN ARRESTED IMMUNITY MODEL..................................................94
VITA................................................................................................................................. 95
Page 12
x
LIST OF TABLES
Table 1-1. Chlamydiae that cause human disease. ...........................................................5
Table 1-2. Increased chlamydial rate hypotheses. ..........................................................17
Page 13
xi
LIST OF FIGURES
Figure 1-1. Chlamydiae developmental cycle. ..................................................................3
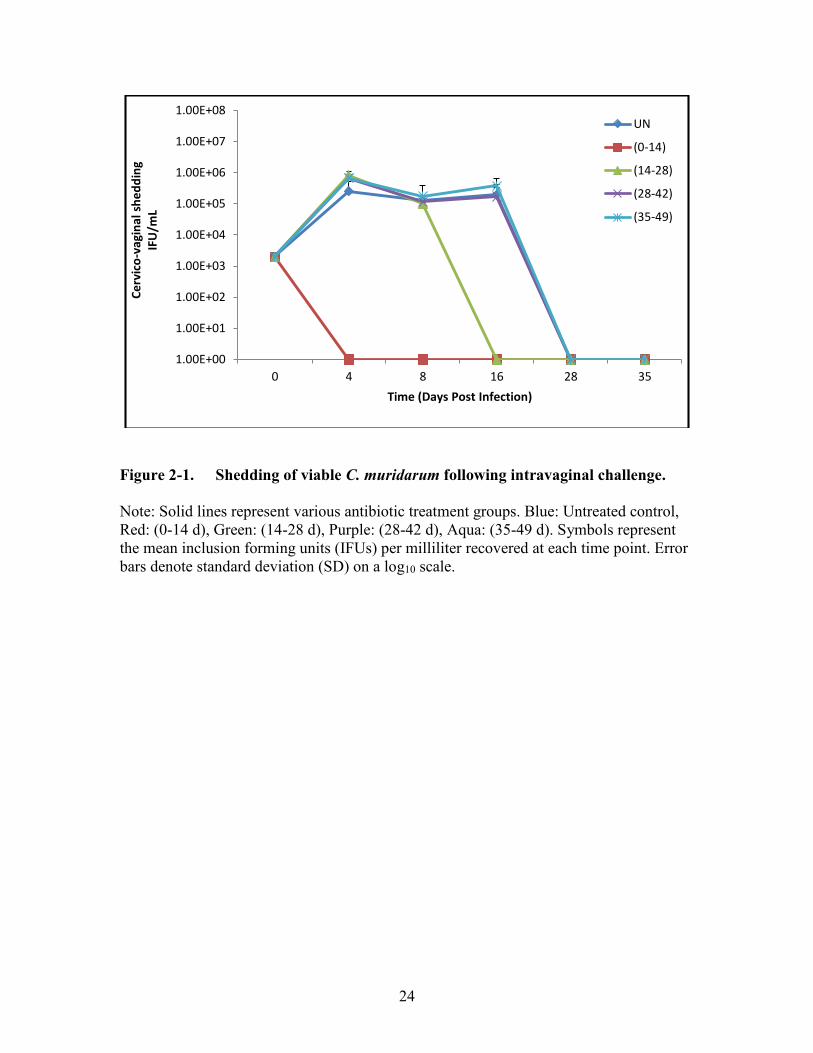
Figure 2-1. Shedding of viable C. muridarum following intravaginal challenge. ...........24
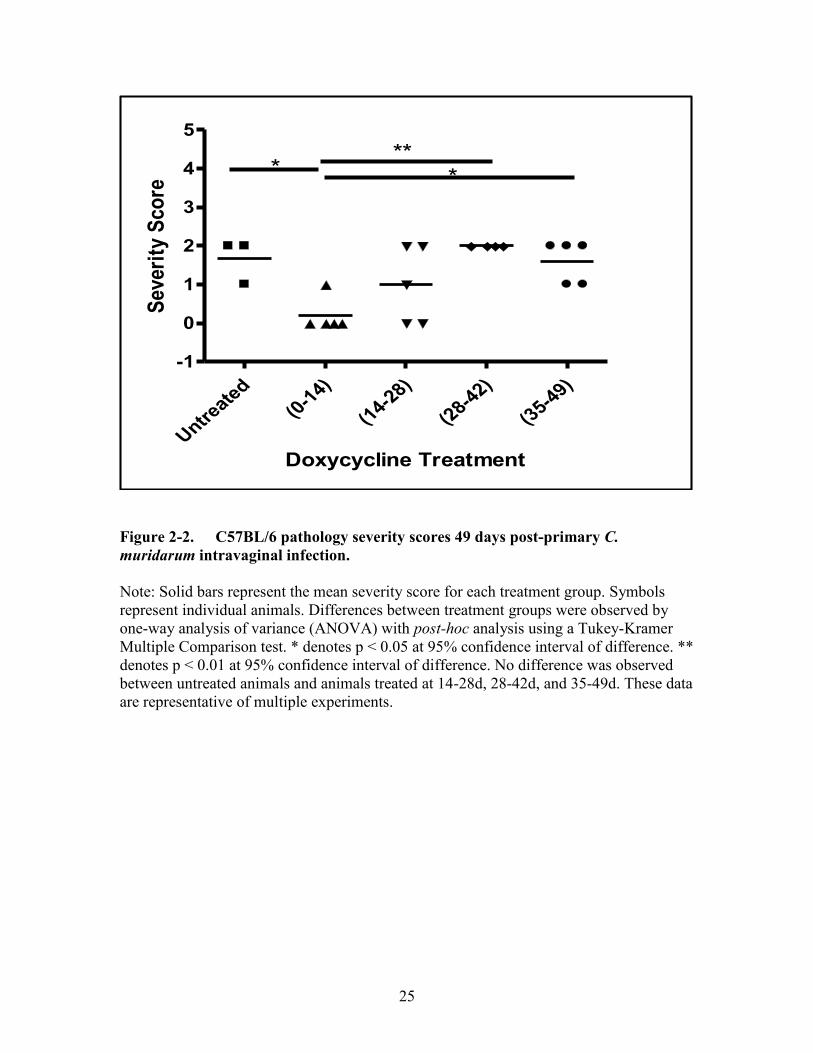
Figure 2-2. C57BL/6 pathology severity scores 49 days post-primary C. muridarum
intravaginal infection. ..................................................................................25
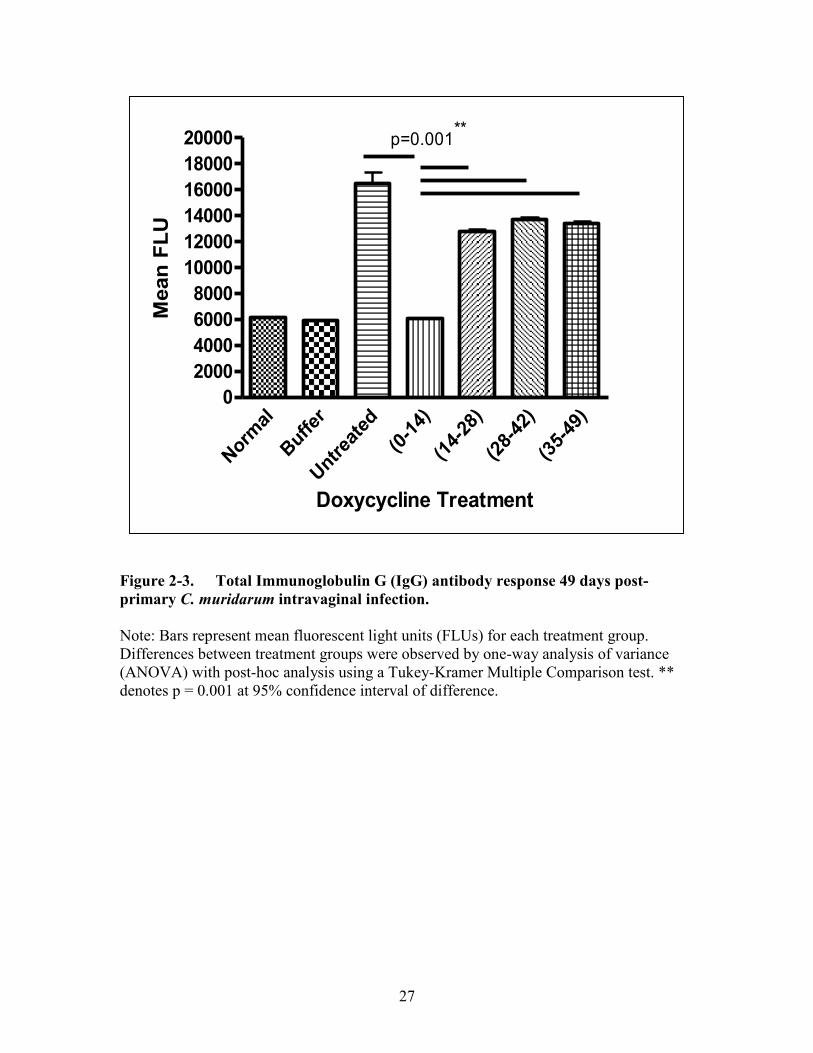
Figure 2-3. Total Immunoglobulin G (IgG) antibody response 49 days post-primary
C. muridarum intravaginal infection. ...........................................................27
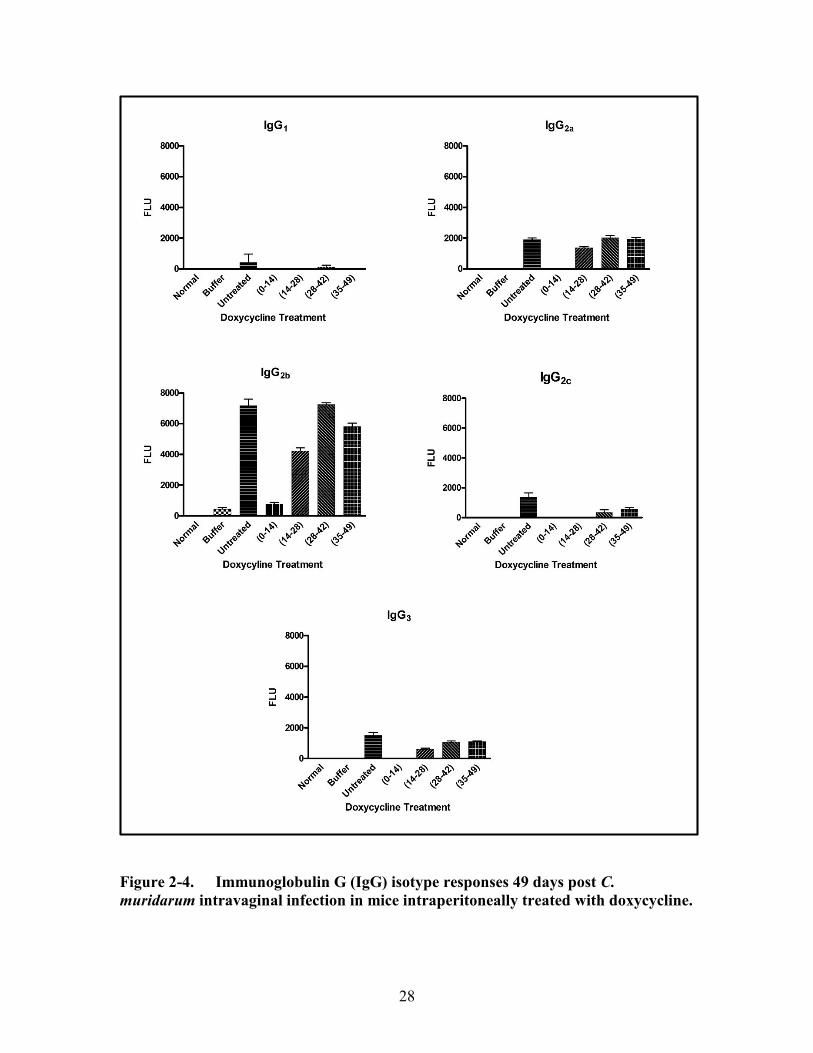
Figure 2-4. Immunoglobulin G (IgG) isotype responses 49 days post C. muridarum
intravaginal infection in mice intraperitoneally treated with doxycycline...28
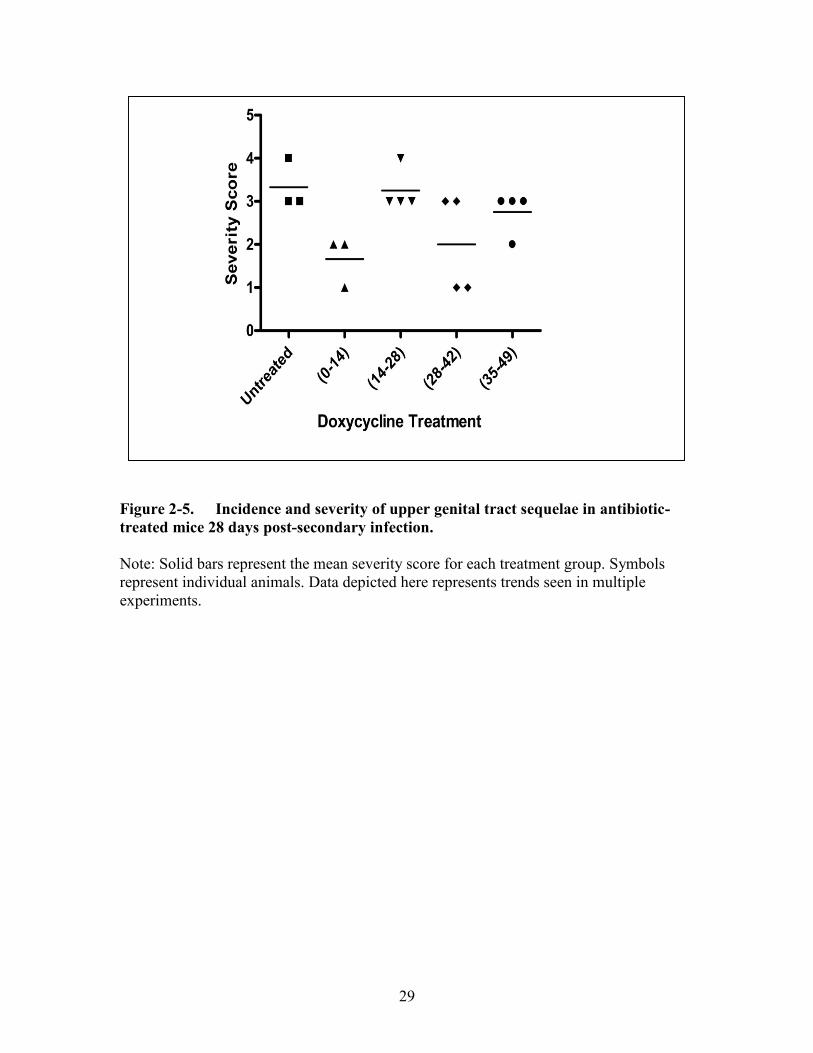
Figure 2-5. Incidence and severity of upper genital tract sequelae in antibiotic-
treated mice 28 days post-secondary infection. ...........................................29
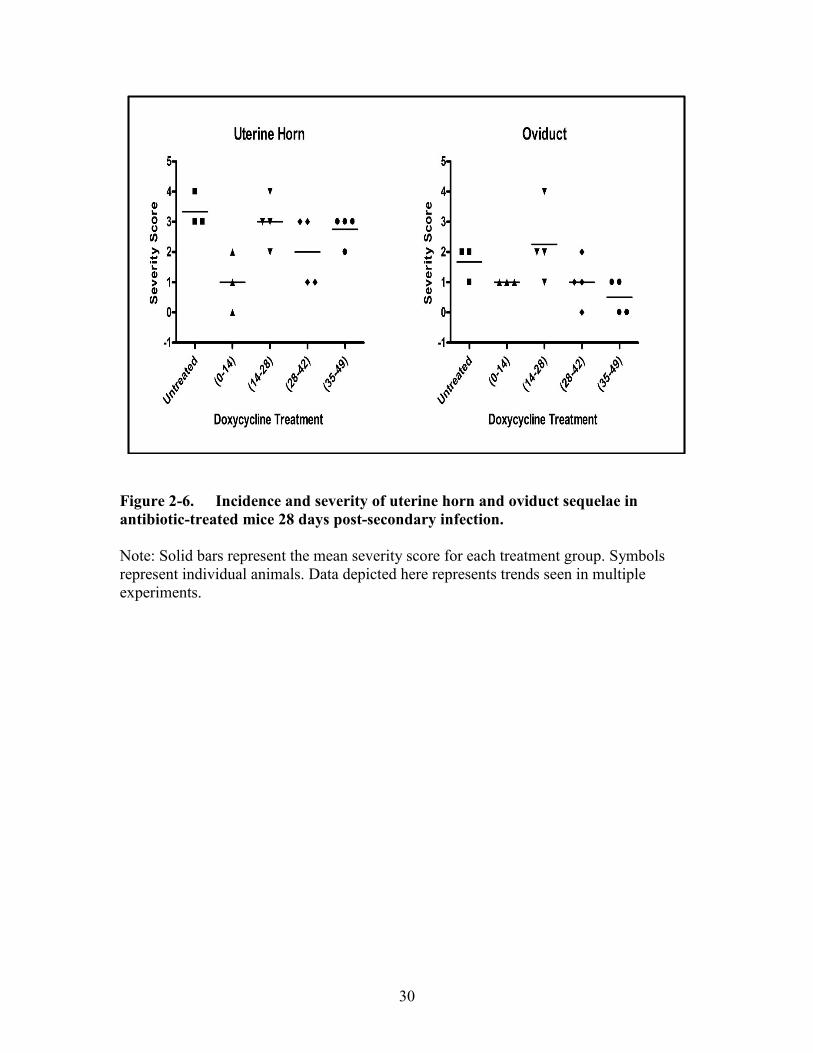
Figure 2-6. Incidence and severity of uterine horn and oviduct sequelae in antibiotic-
treated mice 28 days post-secondary infection. ...........................................30
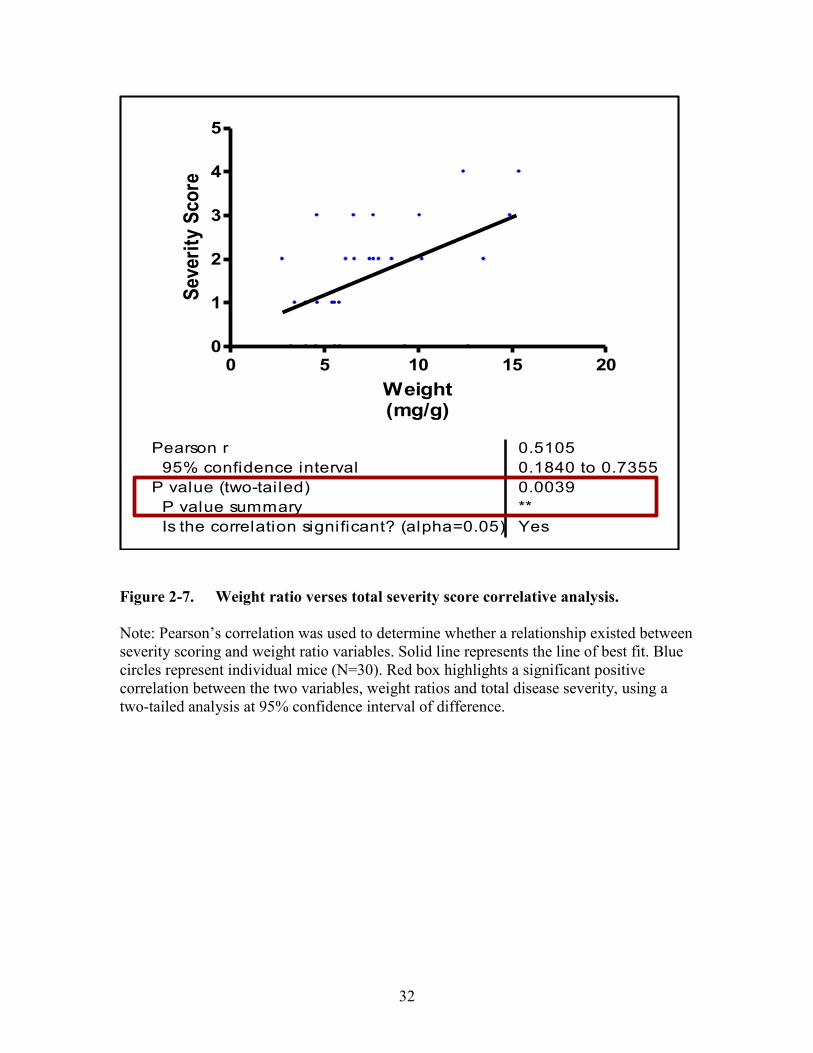
Figure 2-7. Weight ratio verses total severity score correlative analysis. .......................32
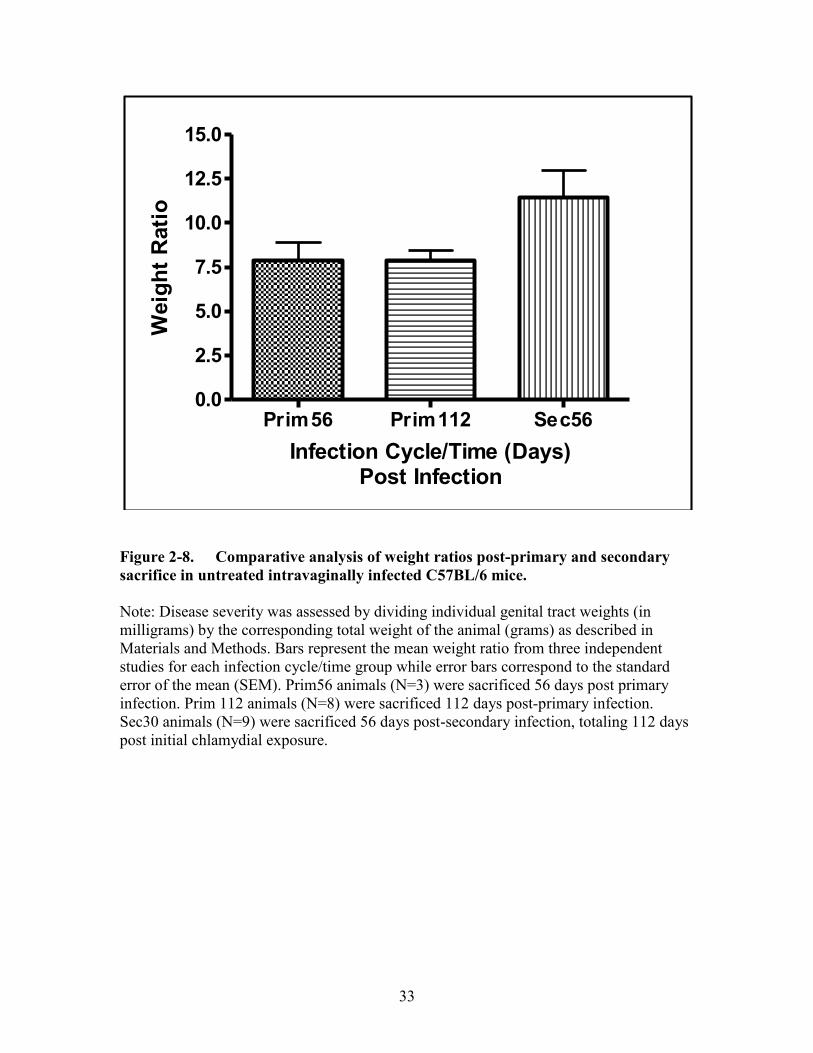
Figure 2-8. Comparative analysis of weight ratios post-primary and secondary
sacrifice in untreated intravaginally infected C57BL/6 mice. .....................33
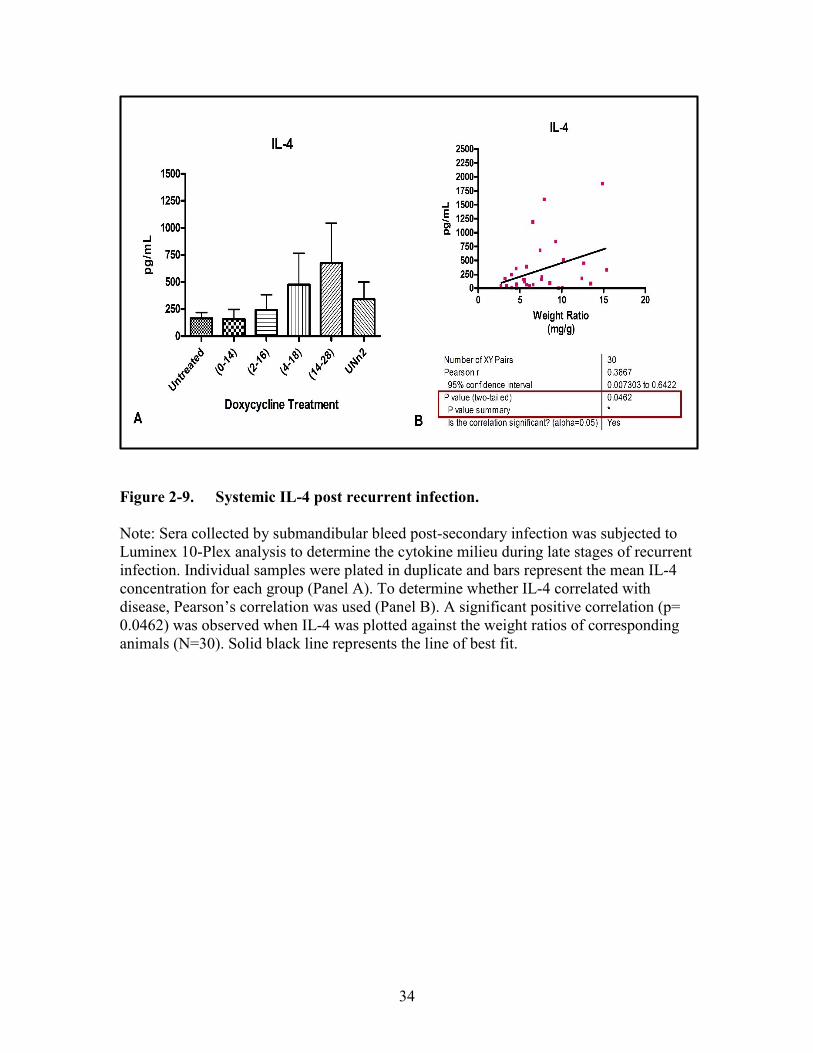
Figure 2-9. Systemic IL-4 post recurrent infection. ........................................................34
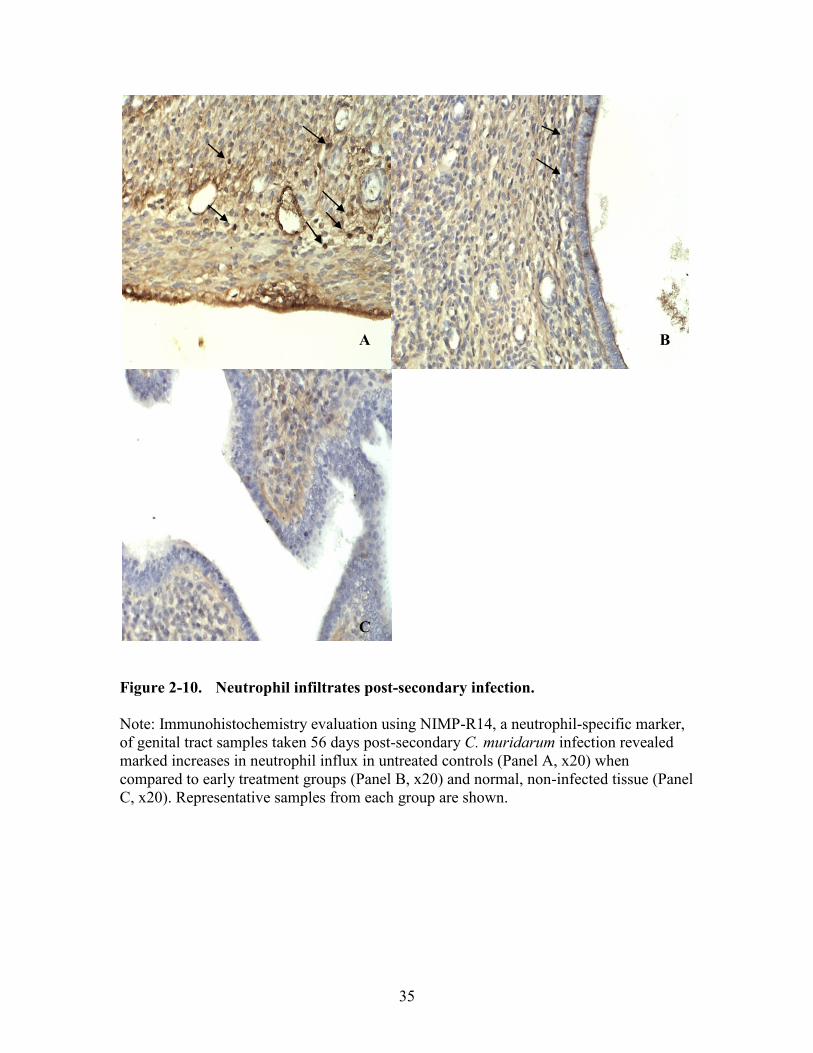
Figure 2-10. Neutrophil infiltrates post-secondary infection. ...........................................35
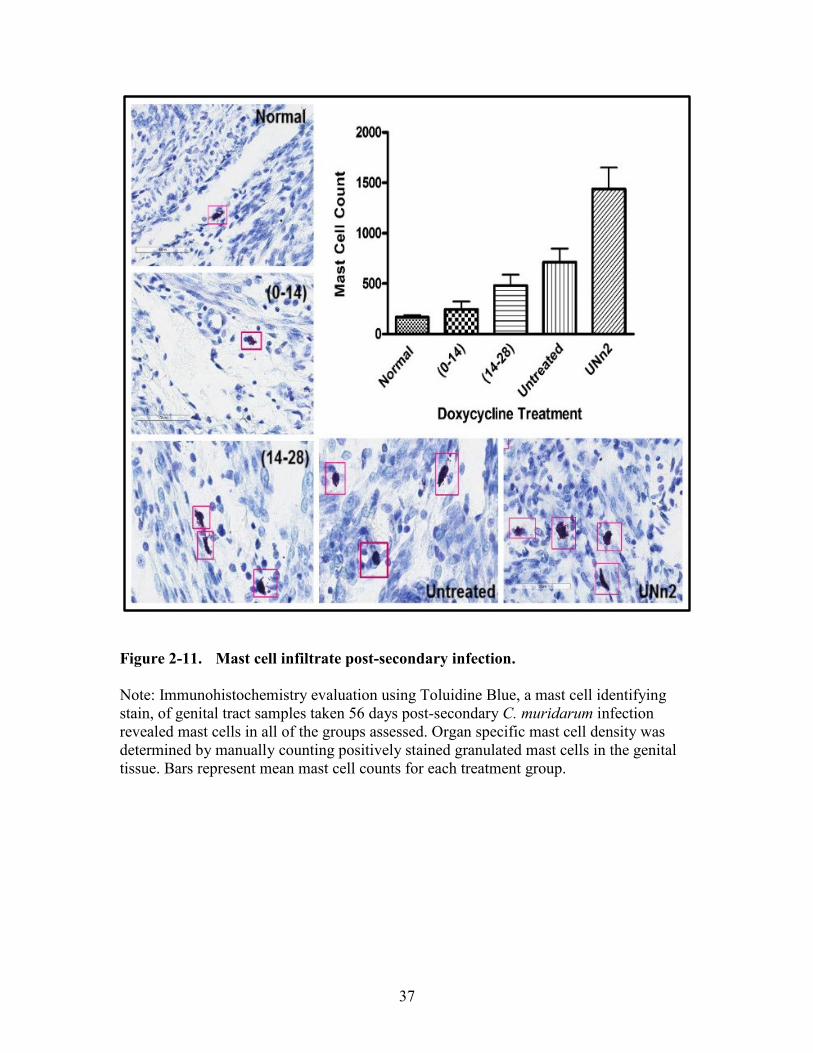
Figure 2-11. Mast cell infiltrate post-secondary infection.................................................37
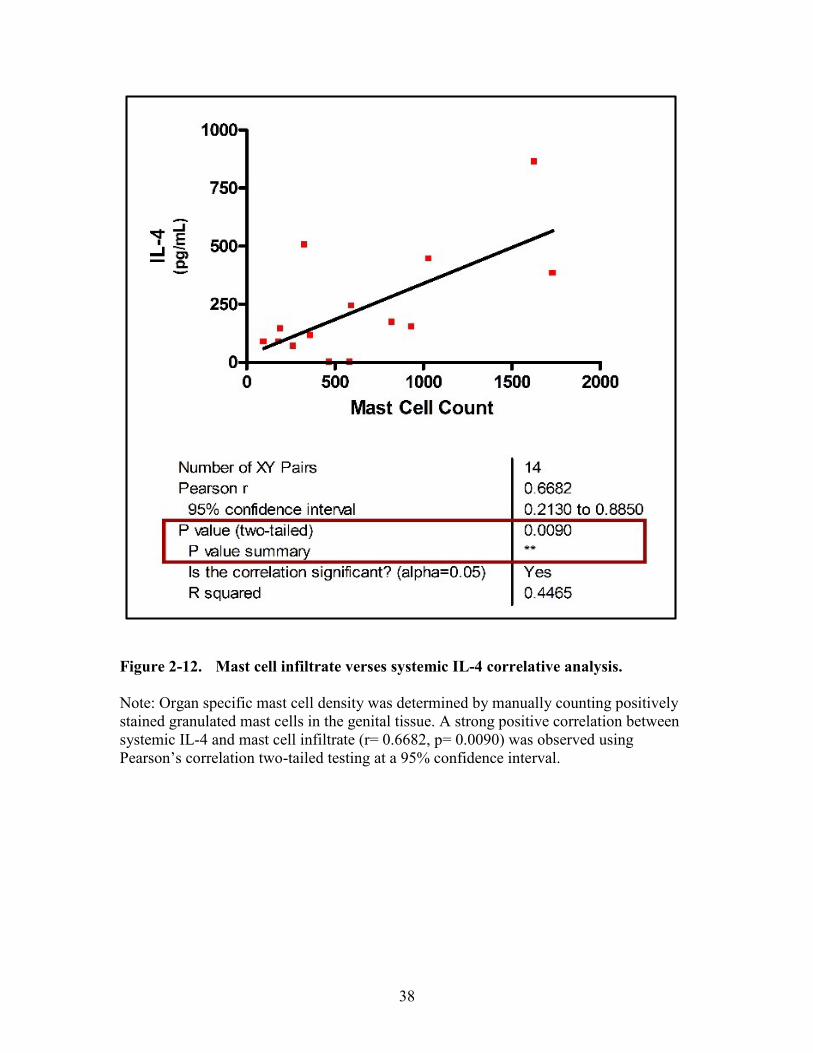
Figure 2-12. Mast cell infiltrate verses systemic IL-4 correlative analysis. ......................38
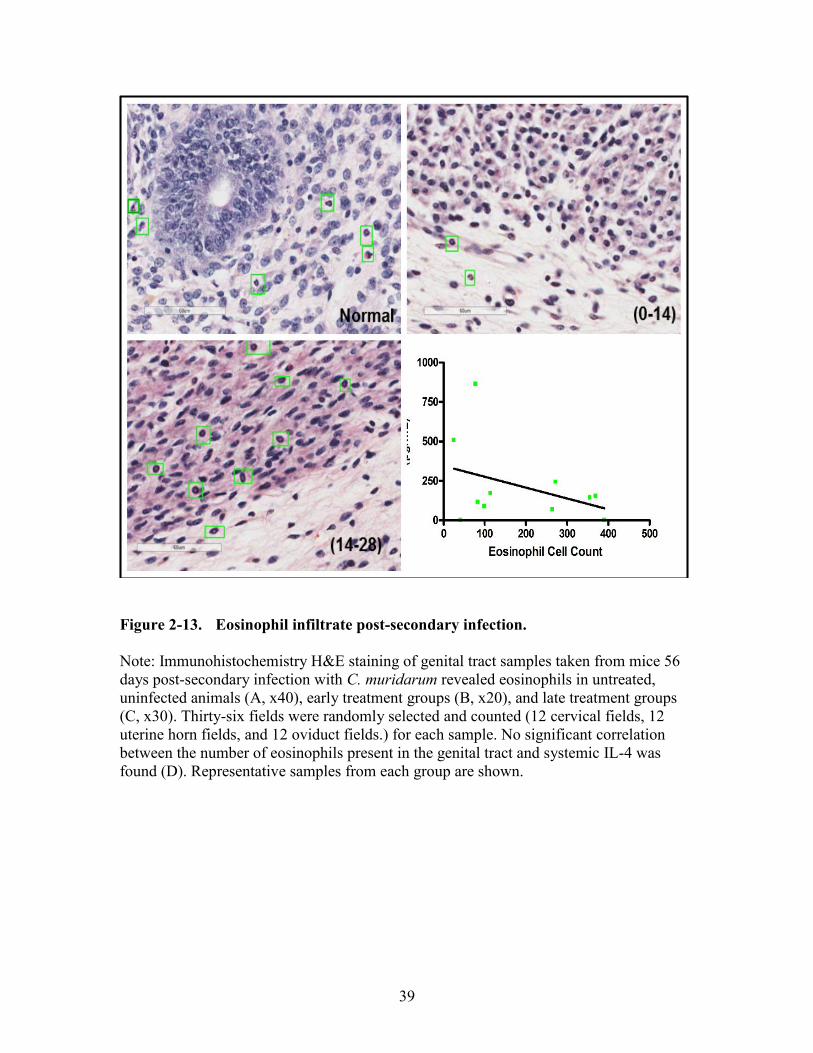
Figure 2-13. Eosinophil infiltrate post-secondary infection. .............................................39
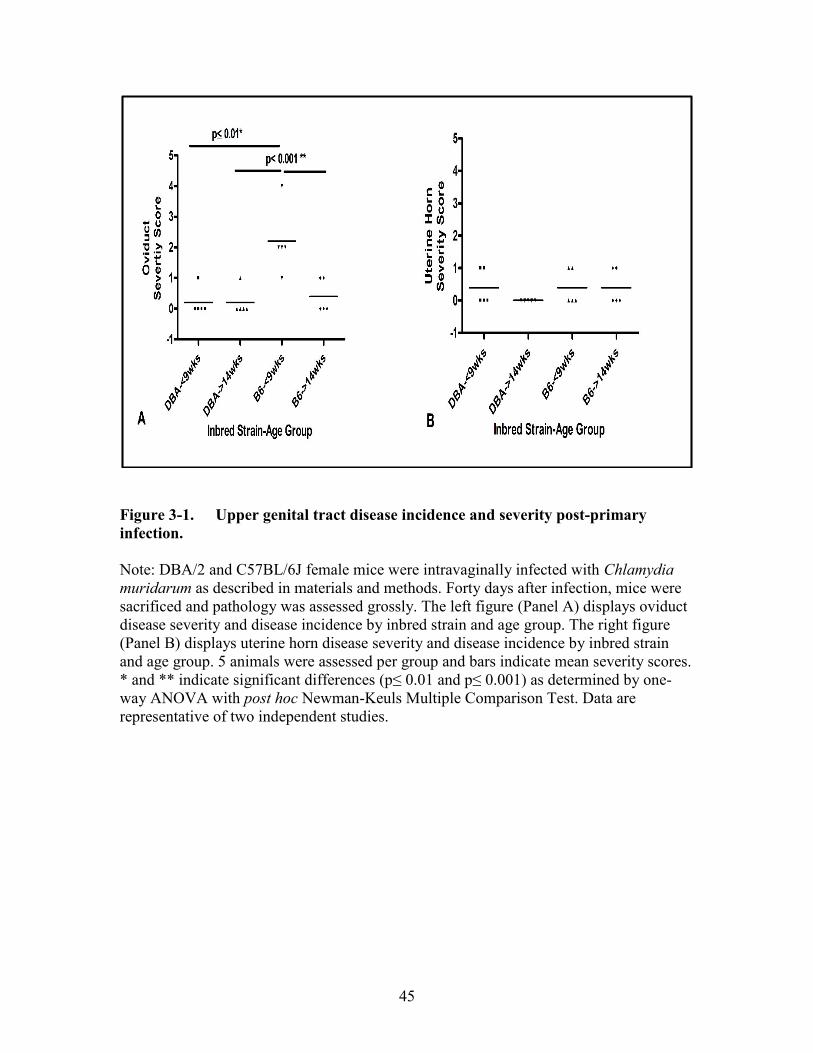
Figure 3-1. Upper genital tract disease incidence and severity post-primary
infection........................................................................................................45
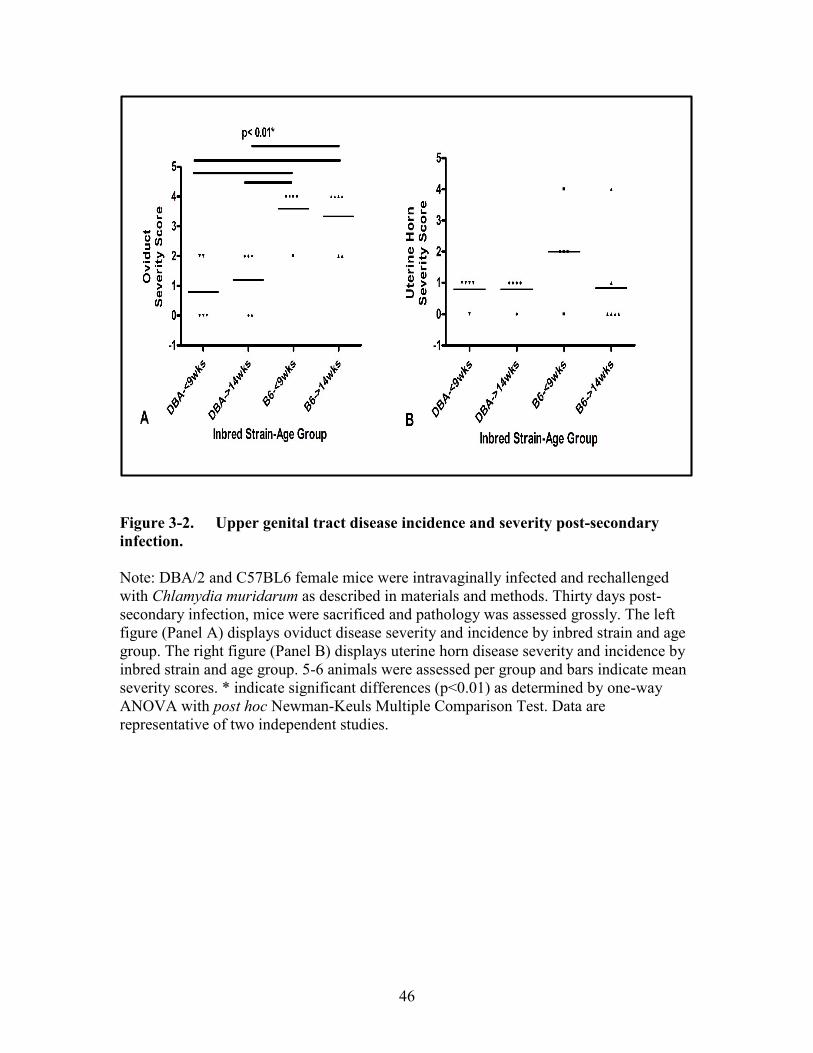
Figure 3-2. Upper genital tract disease incidence and severity post-secondary
infection........................................................................................................46
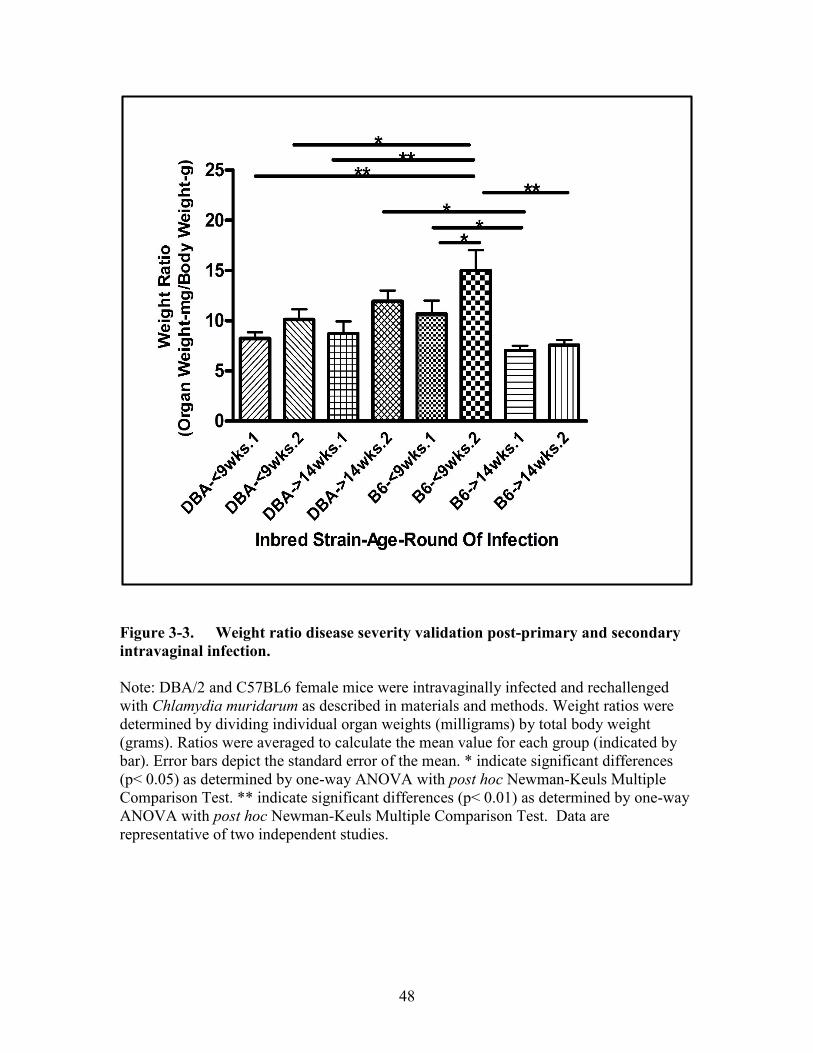
Figure 3-3. Weight ratio disease severity validation post-primary and secondary
intravaginal infection. ..................................................................................48
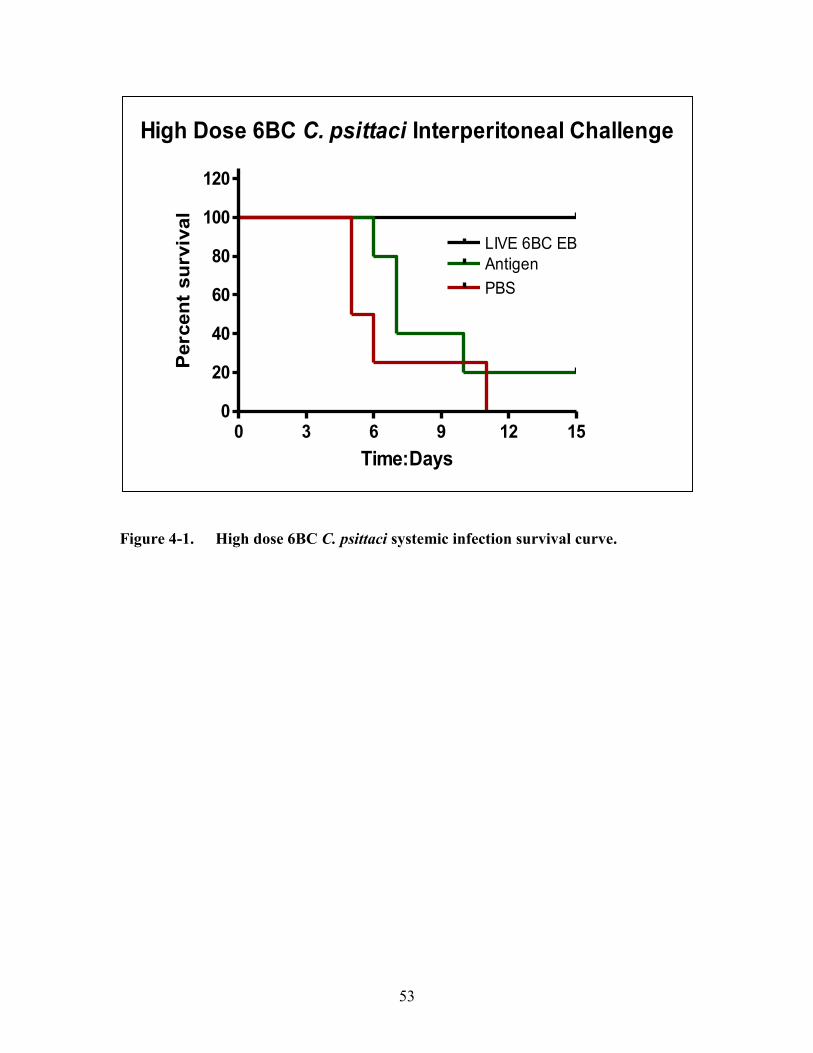
Figure 4-1. High dose 6BC C. psittaci systemic infection survival curve. .....................53
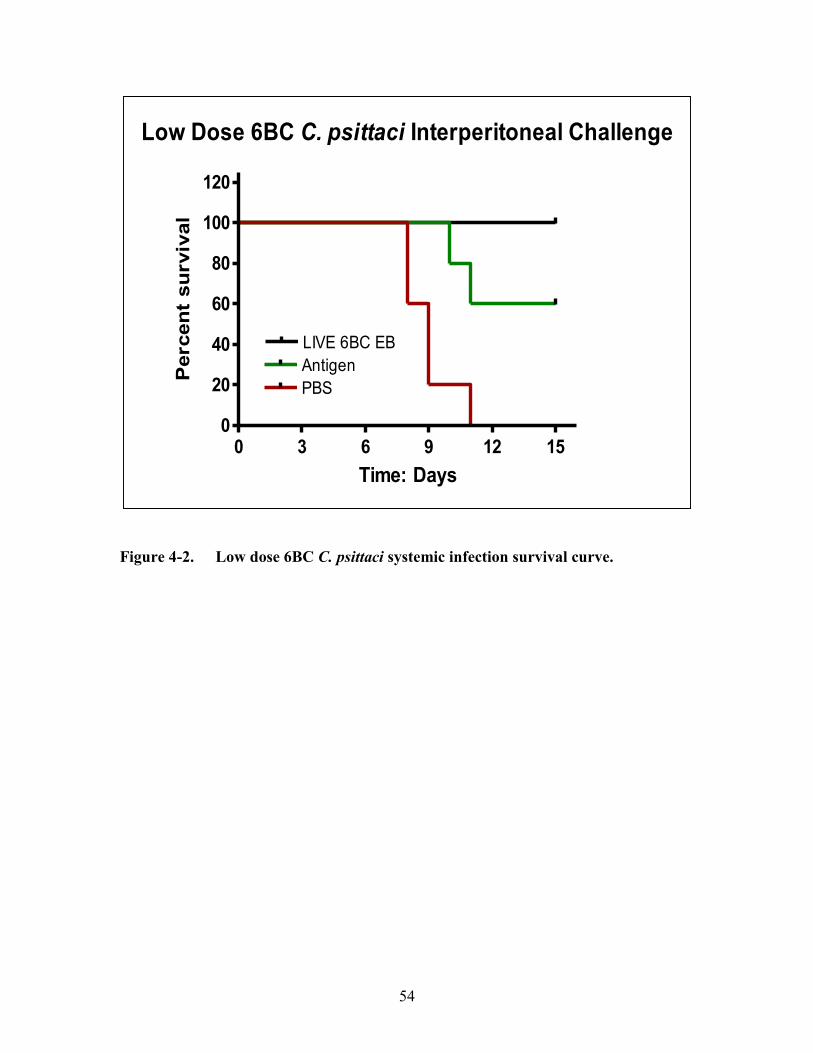
Figure 4-2. Low dose 6BC C. psittaci systemic infection survival curve. ......................54
Page 14
xii
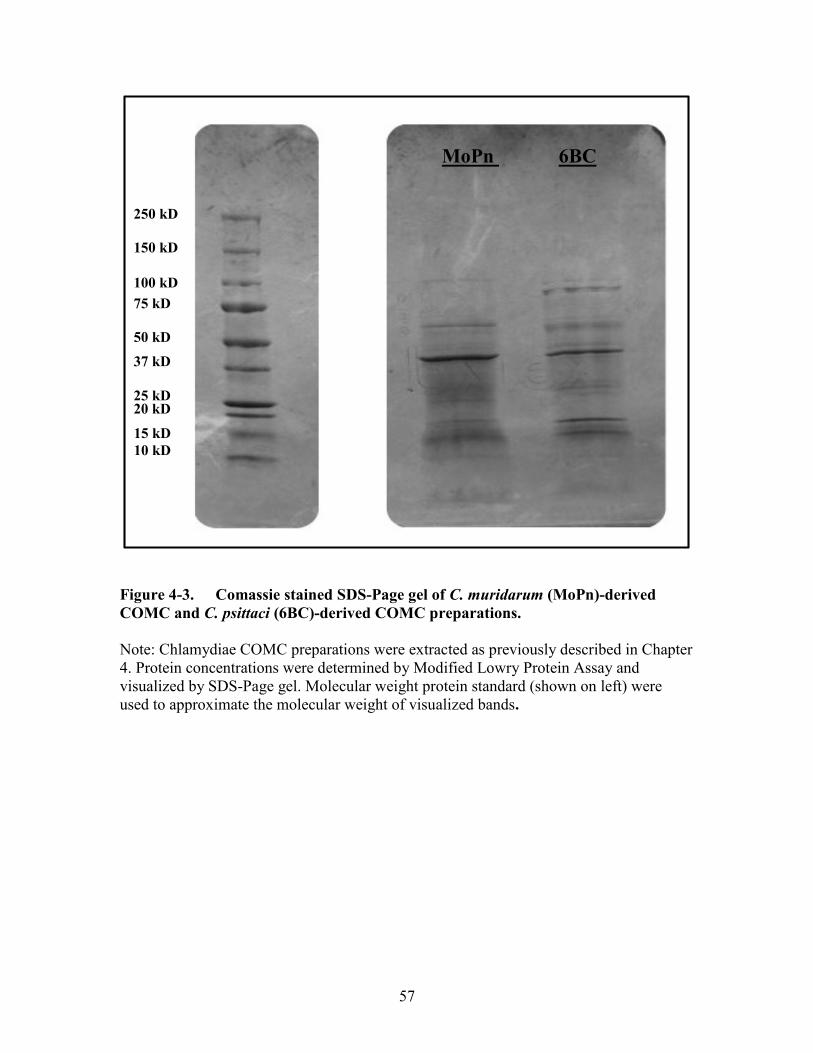
Figure 4-3. Comassie stained SDS-Page gel of C. muridarum (MoPn)-derived
COMC and C. psittaci (6BC)-derived COMC preparations. .......................57
Figure 4-4. C. muridarum-derived COMC immunization study gross pathology. .........58
Figure 4-5. Alum study experimental timeline. ...............................................................60
Figure 4-6. Oviduct pathology severity of non-challenged immunized groups. .............61
Figure 4-7. Uterine horn pathology severity of immunized, non-challenged groups. .....62
Figure 4-8. Oviduct pathology severity of immunized and challenged groups. ..............64
Figure 4-9. Uterine horn pathology severity of immunized and challenged groups. ......65
Page 15
xiii
LIST OF ABBREVIATIONS
APC Antigen-presenting cells
CDC Centers for Disease Control and Prevention
Cpn Chlamydia pneumoniae
Ct Chlamydia trachomatis
DC Dendritic cell
EB Elementary bodies
ELISA Enzyme-linked immunosorbent assay
FLISA Fluorescent-linked immunosorbent assay
GM-CSF Granulocyte-macrophage coloy-stimulating factor
H&E Hematoxylin and eosin stain
HPV Human papillomavirus
HSP Heat shock protein
IACUC Institutional Animal Care and Use Committee
IFN Interferon
IFU Inclusion-forming units
IgG Immunoglobulin G
IL Interleukin
LD50 Lethal dose, 50%
LGV Lymphogranuloma venereum
LPS Lipopolysaccharide
MHC Major histocompatibility complex
MOMP Major outer membrane protein
MoPn Chlamydia muridarum
NAAT Nucleic acid amplification testing
PAMP Pathogen-associated molecular pattern
PID Pelvic inflammatory disease
PMN Polymorphonuclear leukocyte
PRR Pattern recognition receptor
RB Reticulate bodies
RPM Rotations per minute
SD Standard deviation
STD Sexually transmitted disease
STI Sexually transmitted infection
TH0 Naive T Cells
TH1 T Helper 1 Cells
TH2 T Helper 2 Cells
TNF Tumor necrosis factor
UGT Upper genital tract
WHO World Health Organization
Page 16
1
CHAPTER 1. CHLAMYDIAE
Historical Perspective
Tales as old as time . . . .
-Howard Ashman and Alan Menken
Descriptions of chlamydial disease date back to ancient Chinese and Egyptian
texts detailing trachoma, a blinding chlamydial ocular disease. In 2002 Dimitrakov
reviewed the expedition in which Ludwig Halberstaedter and Stanislaus von Prowazek
discovered the causative agent of trachoma [1]. In 1907, while on the island of Java,
Halberstaedter and von Prowazek pioneered early chlamydial etiology studies by
inoculating orangutans with material obtained from trachoma patients. They were able to
demonstrate the infectious nature of the pathogen by describing intracellular vacuoles in
Giemsa-stained epithelial cells derived from conjunctival scrapings of the infected
animals but incorrectly characterized the agent as protozoan.
In 1910, Linder reported finding the same type of intracytoplasmic inclusions in
the eyes of neonates and linked their neonatal conjunctivitis to intrapartum exposure in
women with unrecognized and untreated infection. Linder went on to speculate that the
infection was transmitted sexually after identifying inclusion bodies in the mother’s
cervical scrapings, urethral cells from the fathers, and in individuals with non-
gonnococcal uretheritis [2] [3] [4].
In the 1930’s Samuel Bedson and colleagues characterized the developmental
cycle of psittacosis particles, which at the time were thought to be viruses given their
dependence on eukaryotic cells for replication. In 1957, fifty years after the Java
excursion, C. trachomatis was successfully isolated and cultured in yolk sacs by Feifan
T’ang et al [5] [6]. It was not until the mid 1960’s that chlamydiae would be defined as
prokaryotic bacteria that possessed a non-infectious intracellular replication phase [7]. In
1969, Gordon et al described culturing chlamydiae in irradiated McCoy cells, a less time
consuming technique alternative to isolating chlamydiae in yolk sac that would
revolutionize diagnostic procedures for chlamydial infection [8].
Taxonomy
What is in a name?
–William Shakespeare
Chlamydiae are prokaryotic obligate intracellular bacteria that belong to the order
Chlamydiales. Under the Chlamydiale umbrella are the families Chlamydiaceae,
Parachlamydiaceae, Waddliaceae, and Simkaniaceae [9]. In 1999, it was recommended
that the Chlamydiaceae family be subdivided into two genera, Chlamydia and
Chlamydophila, based on the phylogenetic analysis of the 16s and 23s rRNA [10] but this
recommendation was riddled with inconsistencies and controversial amongst those in the
Page 17
2
chlamydial field. As reviewed by Stephens and colleagues [11], the overwhelming
majority of publications (81% in the year 2006), continued to use the single genus name
Chlamydia, despite the implementation of the two genera system. To that end, nine
species are recognized in the Chlamydia genus: Chlamydia abortus, Chlamydia caviae,
Chlamydia felis, Chlamydia muridarum, Chlamydia pecorum, Chlamydia pneumoniae,
Chlamydia psittaci, Chlamydia suis, and Chlamydia trachomatis. Historically,
chlamydiae serovars were based on conventional immunoepitiope analysis by
monoclonal antibody directed against the major outer membrane protein (MOMP) [12].
Today, chlamydial classification includes genovar sequence data of ompA, the gene that
encodes MOMP [13] [14]. The ompA-based classifying system, when compared to the
conventional sera-based analysis, more accurately reflects strain virulence and genotypic
diversity on the population level [15].
Developmental Cycle
In the circle, the circle of life.
–Sir Timothy Miles Bendon Rice
Originally described in the 1930’s by Samuel Bedson et al [16], the unique
biphasic developmental cycle of chlamydiae has been extensively reviewed [17] [18] [19]
[20] [21] [22]. Two morphologically different forms characterize the chlamydial
developmental cycle, the elementary body (EB) and the reticulate body (RB). The
elementary body is the infectious form of the organism. Although small in size, about 0.2
to 0.4 microns in diameter, EBs are resistant to extracellular conditions and are able to
attach and enter susceptible host cells. Once endocytosed into the host cell, EBs
differentiate into RBs inside a membrane bound vacuole called an inclusion. Reticulate
bodies are the metabolically active and non-infectious intracellular forms of chlamydiae
which multiply via binary fusion within the inclusion. After repeated cycles of cell
division, RBs undergo a second differentiation stage resulting in infectious chlamydial
EBs. Depending on the Chlamydia species, infectious progeny exit the initially infected
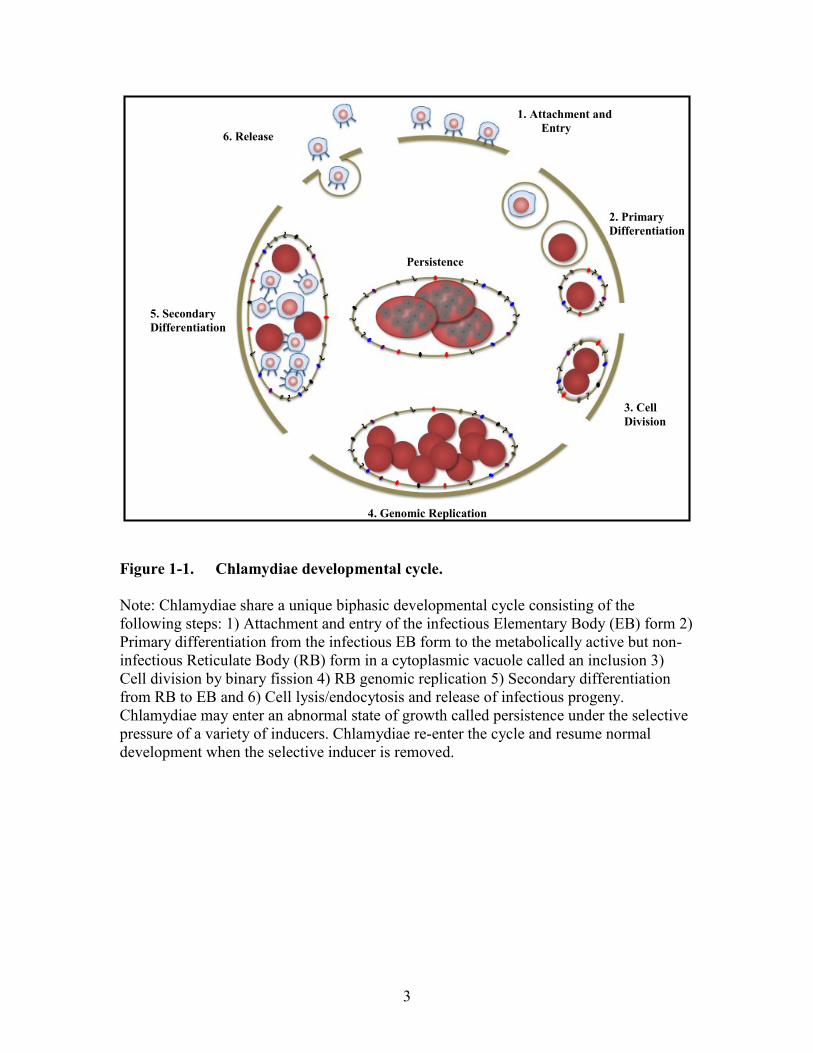
cell between 30-72 hours post infection and infect neighboring host cells (Figure 1-1).
For millions of years Chlamydia species have infected eukaryotic cells and many
researchers believe that they have ensured their survival by deviating from their normal
biphasic developmental cycle. Persistence, a reversible interruption in the productive
intracellular chlamydial growth cycle by environmental factors, has been reviewed many
times over [23] [24] [25] [26] [27] and is characterized by large aberrant non-replicating
RBs which are unable to alternate between EB and RB morphological forms. In vitro
inducers of this abnormal growth state include physiological changes in the host cell,
gamma interferon treatment [28] [29] [30] [31] [32], beta lactam administration [33] [34]
[35], nutrient restriction [36] [37] [38] [39] [40], and concurrent herpes infection [41]
[42] [43]. By definition, restoration of normal chlamydiae development occurs when the
environmental stressor that induced the persistent state is removed. While the likelihood
of persistence being involved in chlamydiae-induced pathogenesis has been documented
in a variety of culture-based systems, particularly as it relates to antibiotic treatment and
Page 18
3
Figure 1-1. Chlamydiae developmental cycle.
Note: Chlamydiae share a unique biphasic developmental cycle consisting of the
following steps: 1) Attachment and entry of the infectious Elementary Body (EB) form 2)
Primary differentiation from the infectious EB form to the metabolically active but non-
infectious Reticulate Body (RB) form in a cytoplasmic vacuole called an inclusion 3)
Cell division by binary fission 4) RB genomic replication 5) Secondary differentiation
from RB to EB and 6) Cell lysis/endocytosis and release of infectious progeny.
Chlamydiae may enter an abnormal state of growth called persistence under the selective
pressure of a variety of inducers. Chlamydiae re-enter the cycle and resume normal
development when the selective inducer is removed.
1. Attachment and
Entry
2. Primary
Differentiation
3. Cell
Division
4. Genomic Replication
5. Secondary
Differentiation
6. Release
Persistence
Page 19
4
host immunological pressures, conclusive evidence of human chlamydial persistence
remains to be demonstrated.
Chlamydiae Clinical Significance
Chlamydial infection is of global importance given its broad host tropism, the
scope of its geographic distribution, and the risk of developing debilitating sequelae. Of
the nine Chlamydia species, three are known to cause a diverse spectrum of diseases in
human populations: Chlamydia pneumoniae, Chlamydia psittaci, and Chlamydia
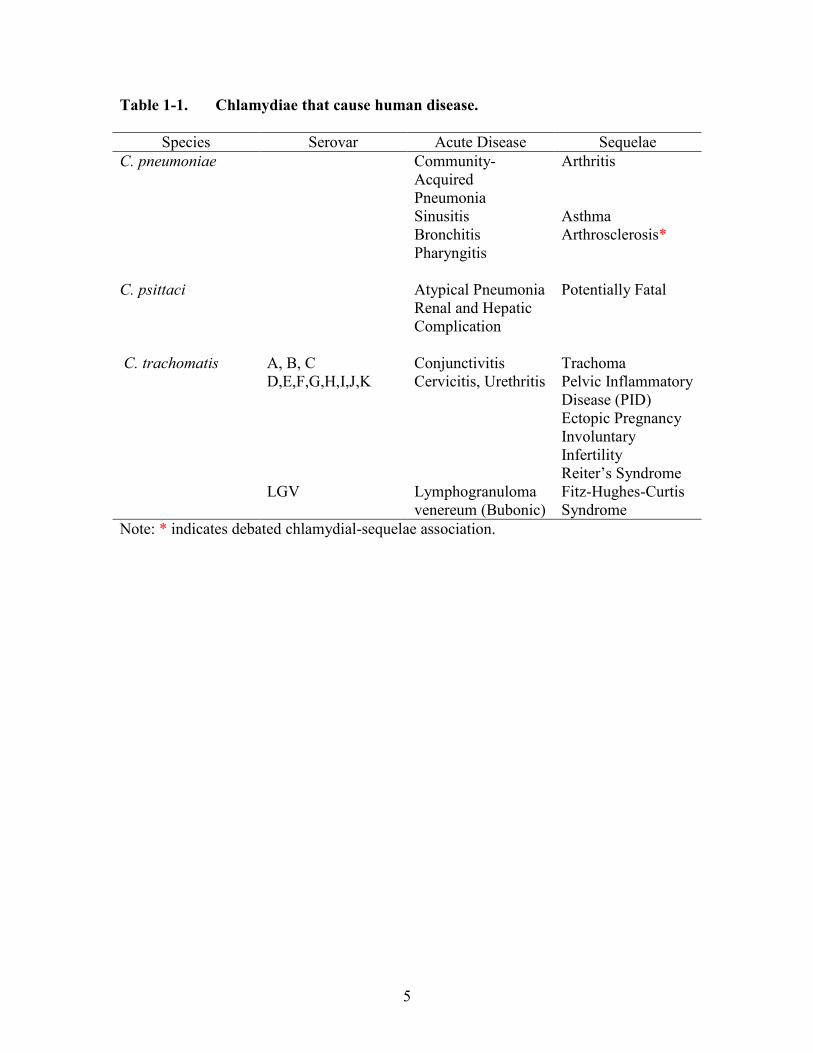
trachomatis (Table 1-1).
Chlamydia pneumoniae
So you’re telling me that you can catch Chlamydia by just breathing? There goes the
neighborhood.
–Corey Dannard
Chlamydia pneumoniae was introduced as a novel member of the genus
Chlamydia by Grayston et al in 1989 and is commonly associated with upper respiratory
infections. C. pneumoniae causes approximately ten percent of community-acquired
pneumonia and five percent of pharyngitis, bronchitis, and sinusitis [44]. In addition,
strong associations exist between C. pneumoniae infection and atherosclerosis, a chronic
cardiovascular disease whose complications lead to half of the adult deaths in the United
States and other parts of the western world (Reviewed by Belland et al [45]). It is
important to note that the association between Chlamydia pneumoniae and
atherosclerosis is a point of debate given the failure to improve clinical outcomes with the
administration of antichlamydial antibiotics in large scale clinical trials including patients
with cardiovascular disease [46] [47] [48].
Chlamydia psittaci
You do know, of course, that zoonotic doesn’t mean the viruses came from the zoo.
–Law and Order: Criminal Intent
Chlamydia psittaci, a zoonotic pathogen whose natural reservoir is avian, is
recognized by the Centers for Disease Control and Prevention as a category B select
agent due to the ease of respiratory dissemination and associated morbidity and mortality
rates [49] [50]. As reviewed by Harkinezhad and colleagues, human C. psittaci infection
is called psittacosis and is acquired by inhalation or, to a lesser extent, ingestion of bird
excretions [51]. After inhalation, the organism infects the respiratory epithelium and
remains latent for up to three weeks void of clinical symptoms. Following the incubation
period those infected experience flu-like symptoms such as headaches, chills, fever,
cough, and in rare instances, neurological and cardiac-related complications. In extreme,
untreated cases the infection can be fatal.
Page 20
5
Table 1-1. Chlamydiae that cause human disease.
Species Serovar Acute Disease Sequelae
C. pneumoniae Community-
Acquired
Pneumonia
Arthritis
Sinusitis Asthma
Bronchitis Arthrosclerosis*
Pharyngitis
C. psittaci Atypical Pneumonia Potentially Fatal
Renal and Hepatic
Complication
C. trachomatis A, B, C Conjunctivitis Trachoma
D,E,F,G,H,I,J,K Cervicitis, Urethritis Pelvic Inflammatory
Disease (PID)
Ectopic Pregnancy
Involuntary
Infertility
Reiter’s Syndrome
LGV Lymphogranuloma
venereum (Bubonic)
Fitz-Hughes-Curtis
Syndrome
Note: * indicates debated chlamydial-sequelae association.
Page 21
6
Chlamydia trachomatis
Chlamydia trachomatis is composed of four biovars, biological strains, based on
the target cells they infect and whole genome sequencing [52] [53] [54]. As implied by
the names, the ocular trachoma lineage commonly infects eye mucosa while urogenital
linages most commonly infect the genital epithelia. Lymphogranuloma venereum (LGV)
is a disseminating biovar that thrives within the lymphatic niche. Trachoma biovars are
subdivided into fifteen serovars based on ompA antigenic variation encoding the major
outer membrane protein (MOMP). Loosely, serovars A, B, Ba, and C serve as the
causative agent of Trachoma, the leading cause of infectious blindness throughout the
world. Serovars D-K are commonly associated with sexually transmitted infection. While
L1, L2, L3 are the three main LGV serovars, the newly identified L2b serotype has
emerged as the causative agent of the current European and North America epidemic
[54].
Ocular Chlamydia trachomatis
The earliest documented historical accounts of chlamydial infection detail
trachoma, a contagious disease of the conjunctiva, the outside covering of the eye, and
cornea caused by Chlamydia trachomatis. While trachoma rarely occurs in western
societies, ocular serovars of C. trachomatis are epidemic in parts of Africa, Asia, South
America, Australia, and the Middle East making it the world’s leading cause of
preventable infectious blindness.
Active infection, a self-limiting inflammation of the conjunctiva, is primarily seen
in children and is transmitted by direct contact with mucosal secretions, poor sanitation,
inanimate objects such as towels and clothing that have infected secretions on them, and
natural vectors such as flies (Reviewed by [55] [56]). Recurrent infection, which is
common in developing countries, leads to the development of scarred tissue and,
eventually, trichiasis, inversion of the eyelid. Inwardly turned eyelashes cause physical
damage to the cornea. This results in the blindness seen primarily in endemic adult
populations. The World Health Organization endeavors to eliminate trachoma by the year
2020 with the implementation of the S.A.F.E. campaign which includes surgery,
antibiotics, facial cleanliness, and environmental improvements [57] [58].
Genital Chlamydia trachomatis
Sexually transmitted diseases are hidden epidemics of tremendous health and
economic consequence in the United States…the scope, impact and consequences of
STDs are under recognized by the public and health care professionals.
–Institute of Medicine, 1997
Sexually transmitted Chlamydia trachomatis has a significant impact on human
health given its adverse effects on reproduction. The World Health Organization
Page 22
7
estimates that ninety million cases of chlamydial infection occur worldwide each year
and an estimated four million cases are reported annually in the United States. Many
believe these estimates are much lower than the actual incidence due to the fact that
infection is largely asymptomatic. While individuals who are infected may not experience
symptoms, it is important to note that they are still at risk of developing long term
sequelae.
In men, C. trachomatis primarily infects the urethra making it the most common
cause of non-gonococcal urethritis. In some instances, the infection spreads from the
urethra to the epididymis resulting in epididymitis, a condition primarily associated with
sexually active males under the age of thirty-five. Infection may also result in Reiter’s
Syndrome in male and female populations. Whether or not Chlamydia infection plays a
direct role in male infertility is still controversial despite the fact that chlamydial DNA
can be recovered from a substantial number of male partners in infertile couples [59] [60]
and has been recovered attached to spermatozoa from the peritoneal fluid of women with
salpingitis [61].
Genital serovars of chlamydiae are of particular importance to women due to the
irreversible reproductive sequelae that may result post infection. Infection of the cervix
can ascend causing endometritis, inflammation of the endometrium, and salpingitis,
inflammation of the fallopian tubes. Untreated C. trachomatis infection has been linked
to chronic complications such as Pelvic Inflammatory Disease (PID), involuntary
infertility and ectopic pregnancy. Moreover, chlamydial infection increases the risk of
contracting human immunodeficiency virus (HIV) and has been implicated in the
development of human papilloma virus (HPV)-induced cervical neoplasia [62] [63].
Although rare, C. trachomatis-induced salpingitis spreading beyond the upper genital
tract into the peritoneum has been documented. The resulting peritonitis and perihepatitis
is called Fitz-Hugh-Curtis Syndrome and may be accompanied by upper quadrant
abdominal pain and the development of adhesions that resemble the strings of a violin
[64] [65].
Mouse Model of Genital Infection
Life is hard for insects. And don’t think mice are having any fun either.
–Woody Allen
Humans are not the only hosts in which chlamydiae can establish an infection and
cause disease. Guinea pigs, turkeys, sheep, and higher order primates have been used to
study chlamydiae-associated disease but an extensive amount of data has been extracted
from mouse models. Mice infected with either C. muridarum or human biovars of C.
trachomatis are most frequently used as models of chlamydial genital infection due to the
ease of reproducibility afforded by inbred strains, commercial ability of reagents, and
genetically engineered animals that allow for immunological interrogation [66].
While nonhuman primate [67] [68] [69] and guinea pig [70] [71] models of
infection were established, Barron and colleagues [72] developed a novel system in
Page 23
8
which researchers intravaginally infected mice with Chlamydia muridarum, previously
known as C. trachomatis mouse pneumonitis strain or MoPn, in 1981. In the Barron
study, chlamydial inclusions were identified by examining Giemsa stained vaginal smear
preparations and chlamydiae-specific immunofluorescent cervical scrapings and
epithelial tissue. These findings were significant because they identified a convenient
model using a natural mouse pathogen that induced pathologies remarkable similar to
those observed in humans infected with C. trachomatis serovar D [73].
Mouse models of chlamydial infection have been used to evaluate the role a
host’s genetic background may play in chlamydiae infection resistance and Chlamydia-
associated outcomes. De la Maza and colleagues intravaginally infected mice with
varying H-2 complexes to determine its effect on chlamydial-related infertility [74]. The
H-2 complex defines the major histocompatibility complex (MHC) in mice and is
homologous to HLA in humans. Six weeks after challenge BALB/c, C57BL/6, and C3H
mice were mated with male breeders and the embryos were counted. Seventy-five
percent (N=20) of C57BL/6 mice became pregnant and had a mean of 4.5 embryos per
mouse. Forty percent (N=20) of BALB/c mice became pregnant and had a mean score of
1.5 embryos per mouse. Thirty percent (N=20) of C3H animals became pregnant and had
a mean score of 1.7 embryos per mouse. From these studies the researchers concluded
that the genetic makeup of the host modulates the degree of chlamydial-induced
infertility. In 1997, Darville et al expanded the study by comparing C3H/HeN mice with
C57BL/6 mice using varying strains of Chlamydia [75]. When intravaginally infected
with C. trachomatis, serovar E or C. muridarum, C3H mice had an increased incidence of
hydrosalpinx, increased chlamydial shedding, and prolonged infection course when
compared to C57BL/6 animals. This suggested that genetic factors played a role in
chlamydial resistance and that the murine model could be used to understand the
mechanisms responsible for resistance variability in the human population.
Advances in human genetics, such as the human genome project, have
revolutionized our understanding of the host’s role in human health and disease by
allowing for inter- and intra-species genetic comparisons [76] [77]. Indeed, whole
genome association studies would allow researchers to interrogate genes and their
associated disease phenotypes relatively quickly when compared to the previous method
of “knocking out” genes in in vivo models and looking for changes in the initially
observed response. As one would expect, identifying conserved genetic sequences across
diverse species would require vast amounts of genetic information. This requirement
grants the mouse model a significant advantage over other animal models given the fact
that genetic sequences and many gene function relationships have been identified and are
readily available using informatics tools like the Mouse Genome Database
(http://www.informatics.jax.org/).
Although chlamydial comparative studies are still in their infancy, high-
throughput genomic analyses have the potential to transform how we currently identify
and therapeutically treat those infected. As proof of principle, Miyairi and colleagues
recently sought to predict outcomes of systemic chlamydial infection using recombinant
inbred mice (BXD) and computational modeling [78]. Infection of parental strains,
Page 24
9
C57BL/6 and DBA/2J, an extensive panel of B (C57BL/6) x D (DBA/2J) mice, in
conjunction with gene mapping and computational Bayesian network modeling were
used to define underlying pathways contributing to variations in disease severity. The
researchers validated predictions that Ctrq3 or polymorphisms in immunological relevant
GTPases conferred resistance in B6 dominant genetic backgrounds, whereas,
susceptibility was heightened in D2 dominant backgrounds as a function of neutrophilic
influx modulation. While there are no homologs of interferon-inducible p47 GTPases in
humans, Miyairi’s findings implicate neutrophils as a tentative therapeutic target, validate
computational chlamydiae-related modeling as a way of predicting disease outcomes, and
highlight recombinant inbred strains as a way of elucidating previously unknown host-
derived pathways contributing to disease.
Murine Genital Tract Pathology
C. muridarum, although originally isolated from the murine respiratory tract [79]
[80], closely mimicked human sexually transmitted chlamydiae disease when used to
infect the genital tracts of mice. In 1983, Swenson and colleagues reported Chlamydia-
induced genital pathology mirrored that seen in human populations [81]. In these
experiments mice were inoculated with C. muridarum in the ovarian bursa, a thin
membrane that encapsulates the ovary and separates it from the interperitoneal cavity.
Hydrosalpinx, blockage of the fallopian tube(s) with serous fluid, was observed in mice
between 25-30 days post infection. Salpingitis and hydrosalpinx formation have been
linked to involuntary infertility in human female populations by irreversible scarring in
the reproductive system and similar outcomes were demonstrated in the model. Although
the natural route of infection was not used in this study, it validated that the mouse model
could be used to explore mechanisms associated with the development and severity of
chlamydiae-induced upper tract complications. Since, several studies, including those
outlined in this body of work, have demonstrated vaginal inoculation of the mouse results
in reproducible upper genital tract pathology.
Murine Model Limitations
Although the murine model has been advantageous, there are some caveats. For
instance, there is great variability in chlamydiae strains, innoculum doses used, and the
inbred strain used. The efficiency of infection using human strains of C. trachomatis is
significantly lessened when compared to the strain isolated from the murine respiratory
tract, C. muridarum [82]. As a result, investigators commonly use higher doses of
Chlamydia trachomatis to establish murine infections. Inoculating doses have also been a
point of debate in the chlamydial field. In 2004, Maxion and colleagues evaluated
differences in BALB/c cell infiltration and pathology formation as a result of innoculum
doses ranging from 104 to 107 inclusion-forming units (IFUs). They found that dose
variation altered immune cell representation in the genital tract noting increases in PMN
and DC infiltrates in the lower genital tract as chlamydial dose increased [83]. Carey et al
investigated the effects of inoculum dose on pathology development in BALB/c female
Page 25
10
mice [84]. They concluded higher doses of Chlamydia muridarum lead to greater oviduct
infection.
Lastly, and perhaps the most relevant argument, is that C. trachomatis is
transmitted by oral, vaginal, and anal sexual contact with an infected individual. The
likelihood of large amounts of infectious organisms being transmitted by the routes
mentioned is low. In support of this argument, the infectious chlamydial load in humans
is low with a median IFU of 72 from male-derived urethral swabs and 450 IFU from
cervical swabs taken from women [85]. Collectively, these studies underscore the need to
standardize infection parameters in chlamydial models across the board to reflect the
likely transmission inoculums seen in human scenarios of infection.
Another caveat of the mouse model is the five day estrous cycle. The frequency of
epithelial uterine sloughing proved to be a problem in establishing chlamydial infection
in mice because the target population (epithelial cells) was turning over prior to the
completion of the chlamydial developmental cycle. Tuffery et al performed experiments
that showed the female genital tract epithelium could be stabilized by subcutaneously
injecting progesterone one to two weeks prior to intravaginal infection (Tuffrey, and
Taylor-Robinson, 1981). Hormone treatment stabilized murine menses and enhanced
chlamydiae genital infection by preventing the normal renewal of genital epithelium. As
a result, the mouse model can be used to study the natural course of infection and
chlamydiae-induced pathology with increased reproducibility. Currently, the use of
progesterone in animal models is debated because sex hormones have been shown to
affect susceptibility to a number of sexually transmitted infections in human and animal
studies [86] [87] [88].
Another concern is the availability of a persistent murine model. In human
female populations, persistent infections defined by chronic asymptomatic chlamydial
genital tract infections, may give way to the development of reproductive complications
such as PID, chronic abdominal pain, and tubal infertility. As mentioned previously, in
vitro persistent states have been induced by environmental stresses such as the interferon
gamma inducible tryptophan decyclizing enzyme 2, 3-indoleamine dioxygenase, iron
depletion, and by treatment with penicillin. In efforts to reproduce persistence in vivo,
Ramsey et al used iNOS knockout mice [89]. When NOS2-/- mice were infected with C.
muridarum, they exhibited higher rates of upper genital tract sequelae but culture-based
resolution was comparable to that observed in wild-type mice. In 1997, Cotter et al
intravaginally infected wild-type immunocompetent mice and were able to reactivate
chlamydiae shedding after initial clearance, evidenced by the inability to recover viable
organisms from the vaginal vault, with the immunosuppressive drug cyclophosphamide
[90]. While these studies illustrate persistence in the mouse, the mouse model may not
be a useful tool for exploring mechanisms of persistent infection because it difficult to get
reactivation. Further investigation for a suitable animal model is warranted given the lack
of conclusive evidence for persistent infection in the human population.
Page 26
11
Immunological Response to Genital Infection
Before discussing the host immune response to sexually transmitted chlamydial
infection, it is important to note key distinctions of the female genital mucosa when
compared to other mucosal sites of the body. The reproductive environment is unique in
that it must exhibit a certain level of tolerance for commensal flora that colonize the
lower genital tract and withstand the presence of an immunologically foreign fetus in the
uterus during pregnancy, all the while maintaining its ability to mount a response to
pathogenic organisms. In addition, the genital environment differs from other mucosal
sites like the proximal intestinal system in that many of its effector functions, including
immunological properties, are hormonally regulated [91]. These distinctions and the lack
of local concentrations of lymphoid tissue such as the gut-associated lymphoid tissue
(GALT) component Peyer’s Patches in the intestine or bronchular-associated lymphoid
tissue (BALT) in the lung contribute to the complexity of chlamydiae infection in the
genital tract.
Intracellular Immune Response Overview
After “self verses non-self” discrimination, the host orchestrates appropriate
immunological responses based on the niche in which pathogens thrive [92]. Bacteria
such as Mycobacterium tuberculosis[93] [94], species associated with the genus
Rickettsia [95] [96], chlamydial species [82] [97] [98] [99] [100] [101], and viruses like
HIV [102] [103] and influenza [104] are hallmark intracellular pathogens that require
specialized approaches to achieve clearance. Antigen-presenting cells (APCs) and T-cells
are crucial to intracellular pathogen elimination. For instance, pattern recognition
receptors (PRRs) associated with APCs like dendritic cells or macrophages recognize
pathogen-associated molecular patterns (PAMPs) which, in turn, initiate antimicrobial
compounds such as interferon-gamma, tumor necrosis factor-alpha, and interleukin-two
[105]. These cytokines assist in the activation of other APCs and push naïve T-cells
(Th0) toward an appropriate TH1 pathogen-specific linage. This cascade culminates in B-
cell activation and the development of plasma cells, antibody-producing B-cells that
ready the host for subsequent encounters.
Chlamydia Innate Response
Induction of the innate immune response is central to mounting an effective attack
against chlmaydial genital pathogens. The process begins with the recognition of
pathogen associated molecular patterns (PAMPS) by Pattern Recognition Receptors
(PRRs) of Antigen Presenting Cells (APCs) or host cell membranes. Various chlamydiae
components or the entire organism may serve as ligands for toll-like receptors (TLRs), a
membrane bound family of PRRs. For example, chlamydiae-derived lipopeptide was
shown to stimulate TLRs 2, 1, and 6 in macrophages [106]. Although less stimulatory
than E. coli lipopolysaccride (LPS), chlamydial LPS may serve as a ligand for TLR4
[107] and to a lesser extent TLR2 [108]. Moreover, whole organism was used to
determine differences in chlamydiae-induced oviduct pathology after discriminatory
Page 27
12
stimulation of TLRs 2 and 4 [109]. Stimulation of TLRs give way to activation of the
NF-K B pathway which, in turn, results in production of pro-inflammatory cytokines and
chemokines.
Almost immediately after infection, a cascade of proinflammatory cytokines are
secreted by the target epithelium. This was first reported by Rasmussen and colleagues in
1997 [110]. Using the in vitro HeLa 229 epithelial cell line, these studies showed that
interleukin-8, growth-related oncogene- alpha (GRO-alpha), neutrophil-derived
granulocyte-macrophage colony stimulating factor (GM-CSF), interleukin-6, and
interleukin-1alpha were secreted following infection. Experiments using mouse derived
oviduct epithelial cells performed by Johnson et al showed that proinflammatory
cytokines and chemokines like tumor necrosis factor alpha, GM-CSF, interleukin-6, MIP-
2, KC, MCP-1 and MCP-5 were produced following C. muridarum infection [111]. The
secretion of these effector molecules lead to the massive influx of innate immune cells.
Darville and colleagues, by flow cytometry, and Morrison and Morrison, by in situ
immmunohistochemistry, characterized the primary infiltrate and found that monocytes,
natural killer cells, and neutrophils were prevalent in these cell populations [75] [112].
The newly recruited cells expanded the repertoire of effector molecules being produced
and, depending on the cytokines and chemokines released, recruited pathogen-specific
lymphocytes to the site of infection.
Chlamydia Adaptive Response
Effective host responses require the induction of, both, the innate and acquired
arms of the immune system. Indeed, resident and recruited innate immune cells such as
neutrophils and professional antigen presenting cells such as macrophages and dendritic
cells begin producing tumor necrosis factor-alpha, interleukin-12, and interferon-gamma.
This stimulates naïve T cells toward CD4 T-helper 1 lineages which are necessary for
chlamydial clearance in the genital tract. Studies in interferon gamma [113] and
interferon gamma receptor knockout mice [114] illustrate a prominent role for the
cytokine in the resolution of primary chlamydial infection. Rank and colleagues first
demonstrated an important role for CD4 T cells in the genital tract by vaginally infecting
athymic nude mice [115]. Nude animals had an active infection evidenced by chlamydial
shedding for more than 200 days post infection, whereas animals with intact T cell
populations resolved infection within 21 days. Landers et al reinforced this finding in
1991 when they used CD4 antigen specific anti-L2T4 antibodies to deplete CD4 in mice
vaginally infected with C. muridarum [116]. These depletion studies resulted in
increased vaginally shedding of Chlamydia and an increased number of organisms
recovered from the oviduct. Su et al later transferred CD4 and CD8-enriched spleen cells
from immune mice to naïve mice and found that adoptive transfer of CD4 lymphocytes
conferred immunity in C. muridarum genital tract infection [117]. Kelly and colleagues
published work illustrating that CD4+ cells were abundantly recruited to the genital tract
following intravaginal infection due to interaction between CD4+ cell home receptor
alpha-4-beta-7 and adhension molecules, ICAM-1, VCAM-1, and MadCAM-1 [118]
[119]. Most recently, Gondek and colleagues reported that CD4+ T cells were necessary
Page 28
13
and sufficient to clear genital tract infection [120]. Using transcervical inoculation, a
method that directly infects the uterine lining of mice, the researchers demonstrated C.
trachomatis, LGV and C. muridarum infected mice are protected from infection and
reinfection when treated with pathogen-specific CD4+ lymphocytes. Furthermore, when
animals were treated with anti-CD4 antibody, Chlamydia 16S DNA levels where
comparable to those observed in naïve mice. Collectively, these studies demonstrate the
importance of T helper 1 type cytokines and CD4 T cells in the resolution and protection
of chlamydial-induced genital infection.
The humoral response has also been implicated in chlamydial immunity. Plasma
cells, commonly known as antibody producing B cells, are thought to play a leading role
in protecting against reinfection, while playing a secondary role to CD4-mediated
resolution of primary infection [99]. One mechanism by which B cells may control
subsequent infection is by antibody driven neutralization, which is plausible given the
biphasic developmental cycle of chlamydiae. Data from Peeling et al using serum from
guinea pigs to neutralize C. trachomatis with UM-4, a strain specific monoclonal
antibody in an in vitro neutralization assay support this theory [121]. Furthermore,
Morrison and colleagues showed that B-cell deficient mice, when depleted of CD4+ T
cells during secondary infection, where unable to clear infection [98] [99]. Although the
experiments by Morrison et al did not rule out the feasibility of direct neutralization, they
implicated a more collaborative mechanism between CD4 lymphocytes and plasma cells,
perhaps by the enhancement of antigen presentation during recurrent infection episodes.
Although the exact mechanism remains unclear, antibody, particularly IgG given it is the
predominant antibody produced in the genital mucosa, is thought to play a collaborative
role in chlamydial immunity.
Vaccination
The prevention of disease today is one of the most important factors in the line of human
endeavor.
–Charles H. Mayo, M.D.
The immune response is the host’s natural defense mechanism to foreign antigens
but the system can be manipulated to respond to foreign antigens quicker and more
efficiently than if it were the first encounter. The previously mentioned form of
manipulation serves as the basis for vaccination or, what some refer to as immunization.
Although current anti-chlamydial therapies effectively clear the organism from
the lower genital tract, they do not address the potential consequences of reinfection.
Batteiger and colleagues reported that almost one third of previously infected individuals
are reinfected with the same serovar due to sexual interaction with the same sexual
partner [122], potentially increasing the likelihood of irreversible tissue damage. One
way of addressing reoccurring infection is through the development of an effective and,
most importantly, safe vaccine.
Page 29
14
Perhaps the most well-known C. trachomatis vaccination studies were the
trachoma human trials performed in the 1960’s [123] [124]. During these trials children
were intramuscularly vaccinated with formalin-fixed whole organisms and traced for
three years. Unfortunately, a portion of those immunized developed severe disease upon
exposure to chlamydiae while others developed partial, serovar-specific immunity when
compared to unvaccinated controls. Since then, vaccination studies have been exclusively
performed in animal models and frequently exploit components of chlamydiae for
vaccination.
First purified by Caldwell and colleagues [125], the major outer membrane
protein (MOMP) has been the focal point of chlamydiae subunit vaccination studies for
the past thirty years. Recently reviewed by Farris and Morrison, native or recombinant
protein, DNA, plasmid, and outer membrane complexed MOMP have been the most
frequently studied MOMP-derived antigens [126]. At best, these studies have only
elicited partial immunity by either failing to reduce bacterial burden, failing to protect
animals from reinfection, and/or failing to protect against sequelae formation.
Interestingly, recombinant vault nanoparticle delivery of MOMP immunogens has
renewed hope in MOMP’s potential as an effective vaccine candidate. Intranasal
immunization with rMOMP nanoparticles results in a T-helper 1 driven immune response
and significantly reduces chlamydial vaginal shedding when compared to animals
immunized with live C. muridarum [127].
Additional antigens have been identified based, mainly, on human seroactivity.
Some of these include chlamydial protease-like activity factor (CPAF), other outer
membrane proteins such as OmcB and Pmps, porin protein B (PorB), and the type III
secretion protein, Tarp. Of the previously mentioned chlamydial components, CPAF was
thought to have the most promise. Intranasal immunization with recombinant CPAF
(rCPAF) plus interleukin-12 reduced bacterial shedding and sequelae formation in
BALB/c intravaginally challenged with C. muridarum [128]. In addition, rCPAF and
Cpg-1826 was used to intranasally immunize BALB/c mice prior to multiple rounds of C.
muridarum intravaginal challenge [129]. rCPAF plus CpG vaccination resulted in a
significantly greater number of pregnancies when compared to mock immunized controls
indicating protection post primary and secondary exposures to genital chlamydial
infection. In 2012, Chen et al called into question the entire body of work done using
CPAF and its proposed substrates after reporting CPAF lost its ability to cleave or
degrade 11 of the 16 previously reported host proteins when a CPAF- specific inhibitor
was used prior to cell lysis [130]. These results suggest that the proteolysis activity
observed in all previous CPAF studies was likely due to the way in which the
preparations were prepared and discredit, at least for now, the idea that CPAF protease
activity is a virulence factor important in chlamydial pathogenesis.
Control Measures
Chlamydia trachomatis infection is the most commonly reported sexually
transmitted disease in the United States and a significant threat to public health world-
wide. Chlamydia control programs were implemented to alleviate the public health
Page 30
15
burden of chlamydial infection by improving detection mechanisms, shortening the
duration of infection, and tracking source-associated sexual networks [131]. These
programs ushered in an initial lag phase denoted by a decline in chlamydial cases.
However, over the last two decades, case notifications of Chlamydia-associated infection
have steadily increased and, in many countries, exceed rates recorded prior to the
implementation of intervention strategies [132] [133] [134]. Norway, Finland, Sweden,
and Canada have documented similar trends with an initial decline following the
introduction of Chlamydia control programs [132] [135] [136] [137]. In recent years,
these countries have witnessed chlamydial rates exceeding those recorded prior to the
establishment of surveillance systems. Given the magnitude and scope of current
epidemiological trends, researchers and policy makers alike are interested in pinpointing
the causal agent(s) for increased chlamydial case notifications.
Sexually Transmitted Chlamydia Epidemiology
The insidious nature of Chlamydia trachomatis infection has made it the world’s
most common cause of curable sexually transmitted disease. In 2010, over a million cases
(1,307,893) of chlamydial genital tract infection were reported to the Centers for Disease
Control and Prevention from fifty states and the District of Columbia [63]. This was the
largest number of cases reported to the CDC for any reportable condition with a case rate
of 426.0 per 100,000 people, representing a 5.1% increase in reported cases over the
previous year. Although these rates are staggering, the actual number is more than likely
higher than that reported seeing as many cases remain undiagnosed.
Gender disparities have consistently been reported among individuals infected
with sexually transmitted C. trachomatis. According to the National Sexually Transmitted
Disease Surveillance Report 2010, women bear a heavier Chlamydia burden than men
with reported rates two and half times more than their male counterparts despite the fact
that Quinn and colleagues reported a nearly identical frequency of transmission among
494 male and female sexually transmitted disease clinic patients [138]. This discrepancy
may be attributed to the fact that women are more likely than men to be screened for
chlamydial infection [139]. However, the switch from urethral swabbing to urine-based
non-invasive nucleic acid amplification testing as the main detection method may result
in more men opting to get tested [140] [141]. This is supported, at least in part, by the
36% increase in chlamydiae case notification since the year 2006. A 19.5% increase was
observed in the female population between 2006 and 2010, indicating the increase in
male reporting was not solely because of an increase in population-based chlamydiae
prevalence [63].
Age has been implicated as a risk factor for urogenital chlamydial infection [142]
[143] [144] [145] [146] [147] [148]. The highest notification rates correspond to sexually
active female adolescents between the ages of 15-19. The second highest rate among
females is young adults between the ages of 20-24. Age-specific rates among males are
higher among 20-24 year olds with 1,187 cases per 100,000 people reported in the year
2010. These numbers are particularly troubling considering chronic chlamydial
Page 31
16
complications have been linked to infertility in both genders and the age groups greatest
affected represent peak reproductive years [63].
Significant ethnic and racial disparities exist among reported sexually transmitted
infections, specifically Chlamydia cases. For instance, African-Americans only represent
14% of the national demographic, but they account for approximately half of all reported
syphilis and chlamydiae cases and 75% of all reported gonorrhea infections [63]. With an
incidence rate 1,167.5 per 100,000 persons, African-American chlamydial rates are eight
times higher than white Americans. American Indian/Alaska Natives chlamydial rates
are 4.3 times higher than white Americans while Hispanic American chlamydial rates are
2.7 times greater than those reported among Caucasian-Americans. While it would be
easy for researchers and epidemiologists to dismiss these health inequalities based on the
idea that these communities are more susceptible to infection and/or are more likely to
participate in risky behaviors, the reasons are more complex [149]. While individual
behaviors can influence health, surveillance trends are, in large part, influenced by
cultural, economical, environmental, educational, and social factors [150] [151] [152].
For instance, in a study conducted by Kaplan and colleagues in 77 different Chicago
communities, sexually transmitted infections were higher in neighborhoods that had high
poverty rates, high unemployment rates, and a low percentage of high school graduates
[153]. In a cross-sectional analysis of over 12,000 young adults, Nguyen et al concluded
that geographical location and economic status were factors associated with chlamydial
reporting with study participants living in the southern region and no functional income
being more likely to report chlamydiae infection [154]. While the studies citied highlight
findings seen in relatively small participant groups, they are representative of health
determinants influencing national surveillance profiles.
Hypotheses for Rebounding Chlamydia Rates
Seven hypotheses were put forth to explain this phenomena and were published
by Rekart and Brunham [133] and are adapted in Table 1-2. Hypotheses one through
four relate to the use of nucleic acid amplification testing for detecting genital infection
with chlamydiae. Hypothesis one speaks to an increased probability of “false positive”
tests because of a loss of specificity in detection methods. Watson and colleagues
performed a meta-analysis including the following diagnostic methods for urogenital
chlamydia: nucleic acid amplification testing (NAAT), gene probe, enzyme immunoassay
(EIA), direct immunofluorescence (DFA), and cell culture [141]. The study concluded
that NAAT testing was the best option for massive screening programs because the
results were comparable to those observed using the “gold standard”, cell culture, and
urine samples did not constitute a gender bias. Men and women would be willing to
undergo screening using this non-invasive technique.
Hypotheses two through four argue increased sensitivity, improved screening
amongst high-risk populations, and greater screening among the male population are the
basis for surveillance data trends. In 2006, Burckhardt et al assessed the impact of
changing from culture-based chlamydial detection to nucleic acid amplification testing in
Page 32
17
Table 1-2. Increased chlamydial rate hypotheses.
Hypothesis
Number
Hypothesis Description
H1 False positive increase due to lower specificity of NAAT methods
compared to culture methods
H2 Increase in case detection due to increased sensitivity of NAAT testing
compared to non-NAAT testing
H3 Higher testing rates among men due to non-invasive NAT urine-based
testing
H4 Targeted screening of high-risk populations and NAAT self-collected
sampling among female population(s)
H5 Development of antibiotic resistance
H6 Increase in unsafe sexual behaviors
H7 Chlamydial arrested immunity
Page 33
18
a genitourinary medicine clinic cohort [155]. Although an initial increase in positive
testing occurred in the two and half years immediately following the implementation of
NAAT testing, positive test percentages returned to those observed prior to the detection
method change. The researchers concluded that the upturn of chlamydial incidence was
not exclusively due to the improvement of detection methods.
Hypothesis five suggests antimicrobial resistance is responsible for the
rebounding rates. Indeed, antibiotic resistance is a serious issue, particularly for
individuals infected with syphilis and/or gonorrhea [156] [157] [133] [158], two major
sexually transmitted infections. Although tetracycline resistance has been documented in
chlamydiae serovars that infect livestock [159], resistance among human isolates are
rarely observed.
Hypothesis six implicates the safe sex practices, or lack thereof, as a potential
cause. Undoubtedly, the behavioral choices made by individuals influence population-
based surveillance data. Perhaps the ideal example is that of HIV/AIDS. After the initial
recognition of HIV in the early 1980’s, huge awareness campaigns were implemented
that led to a decline in STI transmission across the board [160] [161]. After effective
treatment was introduced and the general population no longer viewed HIV/AIDS as a
“death sentence”, STI incidence rates rebounded. This demonstrates that effect of human
behavior on modulating infection cases and confirms the likelihood of current trends
being a result of several factors.
The final hypothesis is the arrested immunity hypothesis. Although it is important
to remember that none of the previously mentioned postulates are likely to be mutually
exclusive, the arrested immunity hypothesis is the cornerstone of my dissertation research
and the primary theme hereafter.
Arrested Immunity Hypothesis
The arrested immunity hypothesis asserts that prompt treatment of genital tract
infection is coupled with a reduction in naturally acquired immunity, resulting in
increased incidence and prevalence of chlamydial infection [132]. Interference with the
development of host immunity to chlamydiae genital infection has been shown using the
urogenital murine model by the Caldwell research group [162]. Su and colleagues treated
C57BL/10 female mice with doxycycline at different timepoints postinfection. No IgG
antibody was detected in sera from animals that received treatment at day zero, the onset
of infection. This demonstrated the effects of early treatment on the development of anti-
chlamydial immune responses.
Brunham et al applied the Cox proportional hazards model to evaluate the effects
of antibiotic treatment on the development of population-based immunity [163]. The Cox
model is a statistical survival model developed by Sir David R. Cox that relates the time
between an “event” occurring to one or more covariates that make also occur within that
time frame. The Brunham model is hinged on findings by Molano and colleagues [164]
suggesting immunity in human populations may take years to develop. These findings,
Page 34
19
when considered with the early treatment component of control programs, suggest that
human hosts don’t develop an adequate immunological response to chlamydiae due to the
truncated exposure time with chlamydial antigen(s) in the genital tract. Using the Cox
model, Brunham’s groups concluded that early treatment renders the general population
more susceptible to reinfection.
Dissertation Rationale
Despite the implementation of wide-spread control initiatives, sexually
transmitted Chlamydia rates continue to rise and are coupled with a decline in
chlamydiae-associated chronic complications such as PID, ectopic pregnancy, and
involuntary infertility [131]. Several rebounding hypotheses have been put forth to
explain current surveillance trends and, optimally, humans studies would provide the
most insight as to why intensive screening and treatment strategies are, seemingly,
ineffective. For obvious reasons, these studies are impractical given the ethical concerns
surrounding trials undertaken with the potential of chlamydial-induced upper genital tract
complications. Taking into account current therapeutic intervention, immune arrest and
infection history, we used the chlamydiae urogenital mouse model to determine the
effects of antibiotic treatment on duration of infection, immunological parameters and,
most importantly, chlamydial-induced upper tract complications in primary and
reoccurring cycles of infection. Moreover, we documented the effects of host factors,
such as age and genetic make-up, and immunization on the development of severe
chlamydial-induced genital tract disease.
Page 35
20
CHAPTER 2. ARRESTED IMMUNITY: IMPACT OF EARLY TREATMENT
ON UPPER GENITAL TRACT SEQUELAE
Introduction
Sexually transmitted Chlamydia trachomatis is the most commonly reported
notifiable infection in the United States [147]. Although up to 75% of women and 50% of
men with lower tract chlamydial infections are asymptomatic [165] [166] [167], the
potential to develop genital tract sequelae remains unchanged when compared to
individuals that experience symptoms such as postcoital bleeding, fever, abdominal pain,
and mucopurulent vaginal discharge [168]. This is of particular concern given the
reported rate of chlamydial infection among women is over two and half times greater
than the reported rates for men [147] and C. trachomatis infection can ascend into the
female upper genital tract causing long term reproductive complications such as ectopic
pregnancy, involuntary infertility, miscarriage, and increased risk of coinfection and
transmission of other pathogenic agents [169] [170] [171] [172]. As a result, in the
absence of a protective chlamydial vaccine, Chlamydia prevention efforts mainly focus
on increasing the rate of screening and treating infected individuals [134] [170] [173].
While current antibiotic regimens are highly effective in clearing the organism
during uncomplicated lower genital tract infection [174] [175], it has been reported that
doxycycline treatment prevents the development of host immune responses in a murine
model of chlamydial genital infection [162]. Furthermore, Brunham and Rekart
hypothesized that populations were susceptible to reinfection due to a shortened duration
of C. trachomatis encounters which may limit natural immunity [132] [133]. Indeed,
repeated chlamydial exposure has been identified as a risk factor for worse disease in
animal and human studies when compared to reference groups that have only been
exposed once or are considered low-risk sexually transmitted disease populations [176]
[177] [178]. Among a high- risk population of sex workers, reoccurring C. trachomatis
infection was associated with Pelvic Inflammatory Disease [179]. Similarly, in a
retrospective study of 11,000 Wisconsin women, ≥3 positive chlamydial tests increased
the chances of Pelvic Inflammatory Disease diagnosis by a factor of six [177]. Although
these studies indicate an increased risk for severe disease following multiple positive
Chlamydia tests, they fail to distinguish between prolonged exposure and reinfection, or
the role antibiotic intervention played, if any, in the development of disease. This is
further complicated by the initial decline in reported chlamydial infection rates following
the introduction of screening and treatment control programs which have since rebounded
to levels comparable or exceeding those observed prior to the implementation of
intervention strategies.
In this study, we sought to determine if data derived from population surveillance,
human studies, and in vivo experiments could be recapitulated using the urogenital mouse
model as a framework to determine how treatment at various time-points throughout the
course of an active primary infection affects disease outcomes during repeated infection
cycles.
Page 36
21
Materials and Methods
Mice
7 to 8 week old C57BL/6J mice were purchased from the Jackson Laboratory
(Bar Harbor, ME) and maintained at our facility, which is fully accredited by the
Association for Accreditation and Assessment of Laboratory Animal Care International.
The Animal Care and Use Committee at the University of Tennessee Health Science
Center (Memphis, TN) approved all animal studies.
Chlamydia Strain and Titration
The Chlamydia muridarum Weiss strain was propagated in HeLa 229 cells and
stored at -80oC. Chlamydial titer was evaluated by infecting HeLa 229 cells and counting
Inclusion Forming Units (IFUs) by fluorescence microscopy as previously described
[113].
Mouse Infection
Mice were subcutaneously treated with 2.5 mg of medroxyprogesterone acetate
(Sicor or Depo-Provera, Pfizer, New York, NY) in 0.1 mL of PBS at fourteen and seven
days prior to vaginal infection. Mice were infected by vaginal inoculation with 2 x 103
IFU of C. muridarum in 10 uL of sucrose phosphate glutamic acid (SPG) buffer. For
rechallenge experiments, mice were treated with progesterone and vaginally challenged,
as previously described, fifty-six days post primary infection unless otherwise specified.
Antibiotic Treatment
Mice were treated daily with 0.3 mg of doxycycline hyclate (APP
Pharmaceuticals, Schaumburg, Il) diluted in distilled, endotoxin-free water (HyClone,
Logan, Ut) by interperitoneal injection during the primary infection course only. All
treatment groups received antibiotic for fourteen days consecutively except for
uninfected, negative control mice treated with progesterone. Treatment groups included
treatment from 0-14 d, 14-28 d, 28-42 d, 35-49 d, and Untreated-(UN).
Bacterial Shedding
Vaginal swabs were taken on interval days throughout infection and collected in
2-mL microcentrifuge tubes that contained 0.5 mL of SPG (with three 4mm diameter
round glass beads) and stored at -80oC. Swab samples were thawed and vortexed.
Infectivity was quantified by inoculating Hela 229 monolayers seeded in 48-well plates
with swab supernatant fluid. After 44 h incubation at 37oC in an atmosphere of 5% CO2,
Page 37
22
plates were washed with PBS-Azide and methanol fixed. Chlamydial inclusions were
visualized and counted by fluorescence microscopy.
Fluorescence-Linked Immunosorbent Assay (FLISA) Antibody Analysis
For total immunoglobulin G (IgG) and IgG isotype studies, HeLa 229 cells were
seeded in 48-well plates (CoStar, Corning, NY) at a cell number of 105 cells/well
overnight. HeLa 229 monolayers were infected with C. muridarum at a multiplicity of
infection (MOI) of two for 2 hours with consistent rocking at 37oC. The innoculum was
immediately aspirated and plates were incubated in Dulbecco’s Modified Eagle Medium
(DMEM) (Life Technologies, Carlsbad, California) complete containing 2ug/mL of
cycloheximide for 36 hours at 37oC in an atmosphere of 5% CO2. Following incubation,
plates were washed 3 times with PBS-Azide and fixed with 500 uL of methanol per well
for 10 minutes. After fixation, plates were washed 3 times with PBS-Azide and incubated
at 37oC with a 1:500 dilution of murine sera, pooled based on antibiotic treatment group,
in PBS/0.1% grade IV ovalbumin/0.05% Tween 20. After incubation, plates were washed
3 times and incubated with FitC-conjugated rat anti-mouse IgG, IgG1, IgG2a, IgG2b
(Southern Biotech, Birmingham, AL), FitC-conjugated goat anti-mouse IgG2c, or Alexa
Fluor ® 488 -conjugated goat anti-mouse IgG3 (Invitrogen by Life Technologies,
Carlsbad, California) for thirty minutes. Plates were washed 3 times with PBS-Azide and
fluorescence intensity was immediately read as fluorescent light units (FLUs) at
sensitivity 70 using a Biotex Synergy 2 Microplate Reader. Results represent the mean of
triplicate values for each pooled sample group while the solid horizontal bar denotes
background intensity as measured by normal sera from uninfected untreated animals.
Pathology
Immediately following sacrifice, individual mice were grossly examined for
genital tract pathology abnormalities and photographs were taken. The uterine horns and
oviducts were collectively and separately scored based on dilation and visual
inflammation (reddening and blockage) of the aseptically removed urogenital tissue.
Scoring for dilation of uterine horn and oviduct tissues was as follows: 0, normal/no
significant dilation; 1, mild dilation of a single cross-section; 2, one to three dilated cross-
sections; 3, more than three dilated cross-sections; 4, confluent dilation. Excised tissues
were methanol fixed and paraffin-embedded. Longitudinal sections were later stained
with one of the following: hematoxylin and eosin (H&E), NIMP-R14 (neutrophil
identifying marker), CD68 (macrophage identifying marker), CD4 (T-cell identifying
marker), and Toluidine Blue (mast cell identifying stain).
Organ-Total Body Weight Ratio Analysis
Mice were individually weighed to obtain the total body weight in grams.
Immediately following sacrifice, the genital tract was aseptically removed and weighed in
Page 38
23
milligrams. Organ-total body weights were expressed as a function of organ weight
divided by total body weight to generate weight ratios. Weight ratios for each animal
were averaged by corresponding antibiotic treatment group.
Statistical Analysis
Statistical significance of differences between treatment groups was analyzed
with GraphPad Prsm 4 software (La Jolla, CA) using one-way analysis of variance
(ANOVA) with post hoc Tukey testing comparing all possible pairs of means unless
otherwise noted. The strength of linear correlations was determined using Pearson
product moment correlation coefficient unless otherwise noted. All analyses were
performed using two-tailed testing at a 95% confidence interval of difference.
Results
Kinetics of Genital Tract Infection in Antibiotic-Treated Mice
To evaluate the effect of antibiotic treatment on the duration of chlamydial
infection, cervicovaginal swabs were collected throughout the primary course of
infection. Progesterone treated mice were vaginally infected with 2 x 103 IFU
chlamydiae, treated with doxycycline and swabbed as described in Materials and
Methods. Mice infected but not treated displayed kinetics of an active infection shedding
3-5 log10 of viable organisms within the first 3 weeks (Figure 2-1). These untreated
animals were culture negative, indicating a natural resolution of lower genital tract
infection, by approximately week four. We were unable to recover infectious organisms
at day four after inoculation in early treated animals, the earliest time point analyzed.
This suggests that antibiotic treatment prevented intravaginal chlamydial colonization,
negating the establishment of an active urogenital tract infection. Mice that started
treatment at an intermediate time point (14-28 d) exhibited a rapid reduction in
recoverable organisms after the onset of treatment, whereas, groups treated late during
the infection cycle (treatment initiation at day 28 or 35) paralleled untreated clearance
rates.
Immune Arrest and Pathology Following Primary Infection of Antibiotic-Treated
Mice
Antibiotic treatment has been shown to prevent the onset of host immune
responses and the development of upper tract pathology post primary infection [162]
[180]. To assess whether an immunological response had occurred we measured systemic
chlamydial specific IgG antibody titers post primary infection by FLISA in addition to
scoring the severity of disease. As hypothesized, pathology following primary challenge
was significantly reduced in early treatment groups when compared to later treatment
groups or untreated controls (Figure 2-2). Animals that began treatment at 14 days post
Page 39
24
Figure 2-1. Shedding of viable C. muridarum following intravaginal challenge.
Note: Solid lines represent various antibiotic treatment groups. Blue: Untreated control,
Red: (0-14 d), Green: (14-28 d), Purple: (28-42 d), Aqua: (35-49 d). Symbols represent
the mean inclusion forming units (IFUs) per milliliter recovered at each time point. Error
bars denote standard deviation (SD) on a log10 scale.
1.00E+00
1.00E+01
1.00E+02
1.00E+03
1.00E+04
1.00E+05
1.00E+06
1.00E+07
1.00E+08
0 4 8 16 28 35
Ce
rvic
o-v
agin
al s
he
dd
ing
IFU
/mL
Time (Days Post Infection)
UN
(0-14)
(14-28)
(28-42)
(35-49)
Page 40
25
Figure 2-2. C57BL/6 pathology severity scores 49 days post-primary C.
muridarum intravaginal infection.
Note: Solid bars represent the mean severity score for each treatment group. Symbols
represent individual animals. Differences between treatment groups were observed by
one-way analysis of variance (ANOVA) with post-hoc analysis using a Tukey-Kramer
Multiple Comparison test. * denotes p < 0.05 at 95% confidence interval of difference. **
denotes p < 0.01 at 95% confidence interval of difference. No difference was observed
between untreated animals and animals treated at 14-28d, 28-42d, and 35-49d. These data
are representative of multiple experiments.
Untr
eated
(0-1
4)
(14-
28)
(28-
42)
(35-
49)
-1
0
1
2
3
4
5
***
*
Doxycycline Treatment
Sev
erit
y S
core
Page 41
26
primary infection displayed reduced disease scores (mean of 1, mild disease) when
compared to moderately diseased untreated animals and late treatment groups. In
addition, the severity of upper genital tract complications followed the same trend
established with the development of chlamydiae-specific immune responses (Figure 2-3).
In treatment groups where Chlamydia- specific antibody titers were observed as
determined by FLISA analysis, chlamydiae-related genital pathology was more severe.
Total anti-chlamydial IgG antibody titer was undetectable in early treated animals and
was significantly different in comparison to untreated animals and animals treated at 14-
28d, 28-42d, and 35-49d. Anti-chlamydial IgG levels resembling those of untreated
controls suggested a role for immune-mediated upper tract pathology in the development
of adverse disease outcomes.
Given the importance of anti-chlamydial Th1-dominant adaptive responses in the
resolution of chlamydiae genital infection [181] [182] [183] [184], we evaluated the
effects of antibiotic intervention on isotype switching as a marker for Th1 verses Th2
biased responses in a FLISA assay with C. muridarum-infected HeLa 229 cells as
antigen. IgG isotypes were undetectable following early treatment but data for untreated
and late treated animals strongly suggested a Th1 bias, evidenced by IgG2 dominance,
regardless of the onset of antibiotic intervention (Figure 2-4). Collectively, these data
implicate a bias for Th1 immunity irrespective of severe disease development or the
initiation of anti-chlamydial drug intervention.
Disease Severity Following Reinfection in Antibiotic-Treated Mice
Repeated infections are common in women and early treatment and have been
postulated to increase reinfection susceptibility on the population level [163] [185] [186].
Therefore, it was important to assess whether immune arrest during primary infection
contributed to disease severity post-secondary infection in our model. As a result, mice
were rechallenged 56 days after primary vaginal encounter to assess disease severity
following repeated infection. The animal groups are reflective of the onset of antibiotic
treatment during primary infection as animals did not receive treatment during the
secondary infection course. Gross examination of early treatment samples showed
significant differences in the magnitude and localization of Chlamydia-induced
pathology. Data presented in (Figure 2-5) show that reinfected animals treated at day
zero during primary infection had a mean disease severity score of 1.6, indicating mild to
no disease. Conversely, animals treated at days fourteen, twenty-eight, and thirty-five
displayed moderate to severe disease, with mean severity scores ranging from 2-3.1. To
ascertain differences in the localization of disease, we separately scored uterine horn
(Figure 2-6A) and oviduct (Figure 2-6B) pathology. Early treatment resulted in
significantly less endometrial disease when compared to untreated controls or animals
treated at fourteen, twenty-eight, and thirty-five days post primary infection. Gross
evaluation of the oviduct pathology revealed mild disease irrespective of the onset of
treatment.
In contrast to human isolates, C. muridarum may result in pathology and
Page 42
27
Figure 2-3. Total Immunoglobulin G (IgG) antibody response 49 days post-
primary C. muridarum intravaginal infection.
Note: Bars represent mean fluorescent light units (FLUs) for each treatment group.
Differences between treatment groups were observed by one-way analysis of variance
(ANOVA) with post-hoc analysis using a Tukey-Kramer Multiple Comparison test. **
denotes p = 0.001 at 95% confidence interval of difference.
Nor
mal
Buf
fer
Unt
reat
ed
(0-1
4)
(14-
28)
(28-
42)
(35-
49)
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000 p=0.001**
Doxycycline Treatment
Mean
FL
U
Page 43
28
Figure 2-4. Immunoglobulin G (IgG) isotype responses 49 days post C.
muridarum intravaginal infection in mice intraperitoneally treated with doxycycline.
Page 44
29
Figure 2-5. Incidence and severity of upper genital tract sequelae in antibiotic-
treated mice 28 days post-secondary infection.
Note: Solid bars represent the mean severity score for each treatment group. Symbols
represent individual animals. Data depicted here represents trends seen in multiple
experiments.
Page 45
30
Figure 2-6. Incidence and severity of uterine horn and oviduct sequelae in
antibiotic-treated mice 28 days post-secondary infection.
Note: Solid bars represent the mean severity score for each treatment group. Symbols
represent individual animals. Data depicted here represents trends seen in multiple
experiments.
Page 46
31
infertility after a single infection in mice [187]. Consequently, we questioned whether the
the increase in disease severity following secondary infection was a result of primary
disease worsening over time or if repeated infection compounded sequelae severity. To
determine whether secondary challenge resulted in more severe disease when compared
to disease post primary infection, we compared weight ratios as a measure of objective
disease severity for three independent untreated groups as weight positively correlated
with disease severity score (Figure 2-7). As depicted in (Figure 2-8), untreated C.
muridarum challenged animals were sacrificed fifty-six days post primary infection
(Prim56) and were compared to untreated C.muridarum challenged mice sacrificed one
hundred and twelve days post primary infection (Prim112). Prim56 and Prim112 mg/g
ratio means remained stable despite a fifty-six day gap between sacrifice dates. Albeit not
statistically significant, untreated C. muridarum challenged animals sacrificed fifty-six
days post-secondary infection (Sec56), displayed an increase in mg/g ratio when
compared to animals that only received primary infection. These data suggest severe
disease is compounded with repeated chlamydial exposure as opposed to residual primary
disease progressively worsening over time.
IL-4 Is Detectable Late Following Reinfection and Positively Correlates with Disease
Severity in Antibiotic-Treated Mice
Little is known about the cytokine milieu in the late stages of recurrent
chlamydial infection. To address the question of which, if any, cytokines were detectable
systemically during reinfection cycles, IFN-gamma, TNF-alpha, GM-CSF, IL-1B, IL-2,
IL-4, IL-6, IL-8, IL-10, and IL-12 were simultaneously analyzed from individual sera
samples collected from mice sacrificed 56 days post-secondary infection. Our results
implicate a relatively short-lived TH1 response as suggested by a dominant IgG2 antibody
response (Figure 2-2) and unremarkable detection of IFN-gamma, TNF-alpha, GM-CSF,
IL-1B, IL-2, and IL-12 post recurrent infection (See appendices). Interestingly, IL-4, a
potent inducer of TH2 immunity and a key defensive player against extracellular
pathogens, was detected in the circulation and positively correlated with an objective
measure of disease severity, weight ratios (Figure 2-9). Moreover, IL-4 detection in the
observed sera analytes gradually increased with the deferred treatment of primary
infection suggesting a TH2-independent role for IL-4 in this model.
Chlamydia and Cellular Infiltrates Following Reinfection in Antibiotic-Treated Mice
Genital tract sections were stained with H&E, NIMP-R14, CD68, CD4, or
Toluidine to identify abundant cell types present in the genital tract after recurrent
infection, including potential populations of known IL-4 secreting cells. Late treated
tissues stained with neutrophil-specific marker, NIMP-R14, revealed dense
polymorphonuclear populations when compared to the residential populations observed
in uninfected, untreated controls. Tissues excised from early treated animals
(Figure 2-10B) displayed far fewer neutrophils than late treated groups (Figure 2-10A).
Levels of T-cells expressing CD4 and macrophages expressing CD68 were comparable to
Page 47
32
Figure 2-7. Weight ratio verses total severity score correlative analysis.
Note: Pearson’s correlation was used to determine whether a relationship existed between
severity scoring and weight ratio variables. Solid line represents the line of best fit. Blue
circles represent individual mice (N=30). Red box highlights a significant positive
correlation between the two variables, weight ratios and total disease severity, using a
two-tailed analysis at 95% confidence interval of difference.
0 5 10 15 200
1
2
3
4
5
Pearson r
95% confidence interval
P value (two-tailed)
P value summary
Is the correlation significant? (alpha=0.05)
0.5105
0.1840 to 0.7355
0.0039
**
Yes
Weight(mg/g)
Severi
ty S
co
re
Page 48
33
Figure 2-8. Comparative analysis of weight ratios post-primary and secondary
sacrifice in untreated intravaginally infected C57BL/6 mice.
Note: Disease severity was assessed by dividing individual genital tract weights (in
milligrams) by the corresponding total weight of the animal (grams) as described in
Materials and Methods. Bars represent the mean weight ratio from three independent
studies for each infection cycle/time group while error bars correspond to the standard
error of the mean (SEM). Prim56 animals (N=3) were sacrificed 56 days post primary
infection. Prim 112 animals (N=8) were sacrificed 112 days post-primary infection.
Sec30 animals (N=9) were sacrificed 56 days post-secondary infection, totaling 112 days
post initial chlamydial exposure.
Prim56 Prim112 Sec560.0
2.5
5.0
7.5
10.0
12.5
15.0
Infection Cycle/Time (Days)Post Infection
Weig
ht
Rati
o
Page 49
34
Figure 2-9. Systemic IL-4 post recurrent infection.
Note: Sera collected by submandibular bleed post-secondary infection was subjected to
Luminex 10-Plex analysis to determine the cytokine milieu during late stages of recurrent
infection. Individual samples were plated in duplicate and bars represent the mean IL-4
concentration for each group (Panel A). To determine whether IL-4 correlated with
disease, Pearson’s correlation was used (Panel B). A significant positive correlation (p=
0.0462) was observed when IL-4 was plotted against the weight ratios of corresponding
animals (N=30). Solid black line represents the line of best fit.
Page 50
35
Figure 2-10. Neutrophil infiltrates post-secondary infection.
Note: Immunohistochemistry evaluation using NIMP-R14, a neutrophil-specific marker,
of genital tract samples taken 56 days post-secondary C. muridarum infection revealed
marked increases in neutrophil influx in untreated controls (Panel A, x20) when
compared to early treatment groups (Panel B, x20) and normal, non-infected tissue (Panel
C, x20). Representative samples from each group are shown.
A B
C
Page 51
36
levels observed in untreated, uninfected controls (data not shown) assessed at the same
time-point.
As previously mentioned, we detected unexpected concentrations of systemic IL-
4 (Figure 2-9) despite seeing the antibody surrogate marker for TH1-related cytokines,
IgG2 (Figure 2-4). As a result we sought to histologically identify known IL-4 producing
cell populations. Using Toluidine Blue stain we identified a treatment dependent increase
in genital tract mast cell populations, the number of mast cells increased the longer
treatment was deferred (Figure 2-11). Furthermore, there was a significant correlation
(p= 0.0090) between the severity of disease as assessed by weight ratios and mast cell
density (Figure 2-12). We also identified eosinophil infiltrates in H&E stained genital
tissue preparations but no statistically significant association was observed (Figure 2-13).
Discussion
In this study, the main objective was to integrate various Chlamydia observations
into a single, comprehensive mouse model of chlamydial genital infection. The
reproductive health of women is of particular concern with current CDC Chlamydia
surveillance data indicating increases in reported chlamydial infections, perhaps, in part,
as a result of rapid antibiotic intervention. In contrast, rising infection rates are coupled
with reduced complication rates in screened populations [131] [134] [188] despite
evidence of adverse complications being linked to multiple or prolonged chlamydial
exposures in human epidemiological studies [176] [177] [179]. Collectively, these
observations underscore the importance of determining the impact of antibiotic
intervention on the development of severe upper tract disease following repeated
infection.
In this C. muridarum model of genital infection, we showed that mice
intravaginally infected and treated early during primary infection displayed rapid
bacterial clearance kinetics in the lower genital tract, significantly reduced anti-
chlamydial IgG titer, and virtually no upper genital tract disease. These results verify and
validate a previous antibiotic study in C57BL/10 mice that concluded early antibiotic
treatment reduced the development of natural immunity [162]. Our study expands on this
model by extending observation on disease severity to post-secondary infection and by
defining differences in disease localization. Mice treated early during primary infection
displayed mild disease severity scores (mean score of 1) in both uterine horn dilation and
hydrosalpinx formation. Conversely, untreated infected mice and mice treated later
during primary infection displayed moderate to severe disease severity scores in the
uterine horn (mean range of 2 to 3.3). However, in the oviduct, significant differences
were not observed between treatment groups suggesting either chlamydiae-related
responses differ between these two tissues or severe endometrial disease limits the
likelihood of oviduct disease. Interestingly, and perhaps not logically, mice that received
treatment midway through the natural course of an active primary infection (14-28d)
displayed elevated disease severity scores in both the uterine horn and the oviduct, albeit
not significantly different from other treatment groups. So far, we have not studied this
phenomenon extensively but eradication of the organism soon after the initiation of
Page 52
37
Figure 2-11. Mast cell infiltrate post-secondary infection.
Note: Immunohistochemistry evaluation using Toluidine Blue, a mast cell identifying
stain, of genital tract samples taken 56 days post-secondary C. muridarum infection
revealed mast cells in all of the groups assessed. Organ specific mast cell density was
determined by manually counting positively stained granulated mast cells in the genital
tissue. Bars represent mean mast cell counts for each treatment group.
Page 53
38
Figure 2-12. Mast cell infiltrate verses systemic IL-4 correlative analysis.
Note: Organ specific mast cell density was determined by manually counting positively
stained granulated mast cells in the genital tissue. A strong positive correlation between
systemic IL-4 and mast cell infiltrate (r= 0.6682, p= 0.0090) was observed using
Pearson’s correlation two-tailed testing at a 95% confidence interval.
Page 54
39
Figure 2-13. Eosinophil infiltrate post-secondary infection.
Note: Immunohistochemistry H&E staining of genital tract samples taken from mice 56
days post-secondary infection with C. muridarum revealed eosinophils in untreated,
uninfected animals (A, x40), early treatment groups (B, x20), and late treatment groups
(C, x30). Thirty-six fields were randomly selected and counted (12 cervical fields, 12
uterine horn fields, and 12 oviduct fields.) for each sample. No significant correlation
between the number of eosinophils present in the genital tract and systemic IL-4 was
found (D). Representative samples from each group are shown.
Page 55
40
antibiotic treatment and detectable humoral antibody responses fail to explain the
observation. Future characterization of the model, including but not limited to the cellular
immune response, is of interest and should be investigated to elucidate the underlying
immune mechanism involved.
Histological analyses of various cellular populations suggest an influx of
polymorphonuclear cells during late stages of recurrent urogenital infection. Using
NIMP-R14, a neutrophil –specific marker, we observed an increase in neutrophils among
late treated animals when compared to uninfected controls and early treated groups. This
follows previous observations by our group [189] [190] and others [191] [192] [193] that
cellular infiltrates composed of neutrophils correlate with the development of more
severe disease. Data from our studies illustrate the sustained presence of neutrophils
during late stages of recurrent infection (fifty six days post-secondary infection) and
suggest that disease severity may be a function of sustained innate reactivity rather than
the TH1-biased response that was observed in all animals, irrespective of disease severity.
This expands the current paradigm of neutrophil influx as an early marker of infection
into a potential mediator of disease outcomes and implies that modulation of neutrophil
influx may improve chlamydiae-related disease severity and may be investigated as a
potential way to improve reproductive health.
Perhaps the most interesting finding in this investigation was the systemic
detection of TH2-linked immune regulator, IL-4 (Figure 2-9), with greater detection of
IL-4 corresponding with late treatment groups displaying the most severe disease. While
our data support the current dogma of TH1-type responses playing a role in the resolution
of infection as evidenced by a detectable IgG2-biased chlamydial-specific antibody
response (Figure 2-4), they also suggest an alternative TH2 independent role for IL-4
production during recurrent chlamydiae infection. Our data illustrate the development of
chlamydial-induced genital tract pathology, with the severity of disease increasing the
longer you defer antichlamydial treatment and with multiple rounds of chlamydial
exposure. Following secondary exposure, a scenario in which we report exacerbated
genital tissue damage, large amounts of IL-4 were detectable and gradually increased as
the magnitude of disease increased. Our data compliments observations made by Holland
et al of increased TH2-related cytokine production in patients with more severe disease
[194]. Miquel et al reported that women with a history of Chlamydia trachomatis
infection secrete cytokines consistent with TH2 immunity, specifically significant levels
of IL-4, in response to ex vivo stimulation [195]. Collectively, these observations lead us
to hypothesis a role for IL-4 in tissue repair. Recent reports have linked IL-4 to tissue
repair and wound healing by alternatively activating macrophages and stimulating the
expansion of IL-4 producing cells [196] [197] [198] [199] [200]. In line with these
reports, we were able to detect both eosinophils and mast cells, known IL-4 producing
cells, in our model. Furthermore, it is important to keep in mind that we detected a TH1
response, at least early on during infection, as evidenced by an IgG2 biased antibody
response. This suggests that the production of IL-4 occurs later in the immunological
cascade (following the production and secretion of chlamydial-specific antibody), which
decreases the likelihood of IL-4 initiating the genital tract tissue damage observed (the
Page 56
41
idea of IL-4 being an injury promoter) and points to the possibility of IL-4 playing a role
in “injury response.”
While we present data in support of the IL-4-driven injury response hypothesis,
further study is necessary for hypothesis validation and mechanism delineation. For
instance, we proposed the transient production of TH1-related cytokines led to the
detection of IgG2 isotypes during early infection cycles. Time course studies following
the onset and dissipation of TH1-linked cytokines and IL-4 are key to verifying that
proposal. While we know eosinophils and mast cells are capable of producing IL-4 and
their presence in the genital tract implicates them as IL-4 sources, it would be useful to
know if they are actually responsible for producing the detected IL-4. This can be
accomplished by microarray analysis of IL-4 gene expression and subsequent ELISA
assay for IL-4 production ex vivo. Additional experiments may also include looking at
the upregulation of genes known to particulate in repair signaling pathways in genital
tract-derived tissue and dispelling the possibility of IL-4 participating in the initial stages
of disease development using IL-4-/- knockout mice.
Rapid antibiotic treatment is a key component of current Chlamydia control
initiatives that may influence infection rates and, more importantly, disease outcome
trends on the population level, but assessing the impact of antibiotic intervention on
severe disease development is difficult to address due to ethical concerns associated with
delaying or withholding treatment in the human population. To this end, we interrogated
the murine model for its potential to serve as a translational bridge between population-
based epidemiology and previous animal and human studies. Our results reaffirm the
benefit of rapid antibiotic intervention in Chlamydia control efforts and suggest an added
benefit during repeated infection even in the absence of antibiotic treatment. Although
early-treated mice developed some upper tract disease after re-challenge, it was
substantially less severe than the disease observed in groups where treatment was
delayed. This is of particular interest since previously screened and treated women
commonly re-enter susceptible sexual networks, acquire a secondary infection, and fail to
seek treatment due to the asymptomatic nature of Chlamydia trachomatis infection. Most
importantly, we provide support for IL-4 dependent tissue damage response following the
induction of chlamydial-induced genital tract complications. We conclude that the
integrated murine model is useful for studying the efficacy of antibiotic intervention at
varying time-points with respect to disease severity but requires further characterization
to elucidate potential mechanisms by which chlamydial pathogenesis occurs and is
regulated, particularly as it pertains to the induction of IL-4. The dichotomy between cell-
mediated chlamydiae-related immune responses may not be as straightforward as we
previously thought.
Page 57
42
CHAPTER 3. CHLAMYDIAL GENITAL TRACT SEQUELAE IS AGE AND
STRAIN-DEPENDENT
Introduction
Sexually transmitted Chlamydia trachomatis is the causative agent of the most
commonly reportable disease in the United States [147] and a small percentage of
infected women develop reproductive and gynecologic complications such as chronic
pelvic pain [170, 201-204], pelvic inflammatory disease (PID)[170] [201] [205] [206],
involuntary infertility [170] [172] [201] [203] [206], and ectopic pregnancy [170] [172]
[176] [201] [204] [206]. While many factors contribute to individual disease variability to
chlamydiae [74] [75] [100] [207], the effects of biological determinants such as genetic
predisposition and host age at the time of infection [208] [209] are largely unknown.
Identification of host genetic factors is central to improving the quality of current disease
animal models and understanding chlamydial-induced disease heterogeneity. Moreover,
murine genetic studies may implicate novel pathway targets for individualized
therapeutic intervention in humans.
Inbred mouse models have been extensively used to study host contributions to
chlamydiae infection susceptibility [74] [75] [210] [211] [212] [213] [214] [215].
Cytokine induction and expression, H2 haplotypes, chlamydial species, and Th-1-
mediated responses have all been identified as factors that contribute to varying murine
susceptibility. More recently, unbiased genome-wide approaches have been used to
identify p47 GTPase genes encoded on chromosome 11 that confer resistance in mice to
systemic models of C. trachomatis and C. psittaci infection, respectively [190] [216]
[217]. Since, Miyairi et al have combined the high throughput forward genetic approach
of BxD recombinant inbred strains with Bayesian network modeling to identify host
pathways that modulate susceptibility to systemic chlamydial infection [78].
In the present investigation, we considered the age of the animal at time of
infection and the genetic composition of the host when identifying differences in upper
genital tract disease severity. We conclude that age and host genetic make-up are both
important determinants of disease.
Materials and Methods
Mice
Seven week old C57BL/6J (H-2b) and DBA/2J (H-2d) female mice (referred to as
≤9wk animals) were commissioned from The Jackson Laboratory (Bar Harbor, ME,
USA). Fourteen to 32-week old C57BL/6J and DBA/2J female mice (referred to as
>14wks) were bred and maintained at our facility, which is fully accredited by the
Association for Accreditation and Assessment of Laboratory Animal Care International.
Page 58
43
The Animal Care and Use Committee at The University of Tennessee Health Science
Center (Memphis, TN) approved all animal studies.
Chlamydia Strain
The Chlamydia muridarum (Weiss) strain was propagated in HeLa 229 cells and
elementary bodies (EBs) were purified from infected cells by 30% density gradient
centrifugation in Renograffin (E.R. Squibb and Sons, Princeton, NJ). Chlamydial titer
was evaluated by infecting HeLa 229 cells and enumerating by indirect fluorescence
microscopy as previously described. Stocks were stored at -80oC until use.
Chlamydia muridarum Infection
DBA/2J, C57BL/6J, and BXD mice were pretreated with progesterone to
synchronize the estrous cycle and stabilize the target genital epithelium. Pretreatment
consisted of two subcutaneous injections of 2.5 mg medroxyprogesterone acetate
injectable suspension (Sicor, Irvine, CA) in 0.1 mL phosphate buffered saline (PBS),
given at 10 and 3 days prior to infection. Mice were inoculated with 5000 inclusion-
forming units (IFUs) contained in 5 uL sucrose phosphate-buffered glutamic acid (SPG)
via the vaginal vault with a filtered micropipettor on day 0. Five animals from each group
(DBA/2J-≤9wks, DBA/2J->14wks, C57BL/6J-≤9wks, and C57BL/6-2J->14wks) were
sacrificed 40-days post infection. For rechallenge studies, five animals from each group
were progesterone treated at day 30 and intravaginally challenged with 5000 IFU at day
40. All rechallenged animals were euthanized 30 days post reinfection, seventy days post-
primary infection.
Pathology Assessment
Individual mice were weighed to obtain total body weight in grams. In situ
macroscopic examinations were performed for evidence of upper genital tract
abnormalities immediately following sacrifice. Genital tissues were then aseptically
removed, photographed, and weighed to obtain organ weight in micrograms. Severity
scores were determined for the uterine horn and oviducts separately: 0, no significant
dilatation; 1, mild dilatation of a single cross-section; 2, one to three dilated cross-
sections; 3, greater than three dilated cross-sections; 4, confluent pronounced dilation.
Scores assigned to individual mice were averaged to obtain the mean severity score for
each group of animals. Weight ratios (organ weight-milligrams/ total body weight-grams)
were used as an objective validation tool for upper genital tract severity scoring.
Page 59
44
Results
C57BL/6J Exhibit Age-Dependent Upper Genital Tract Sequelae Post-Primary
Intravaginal Infection
DBA/2 and C57BL/6 female animals of varying ages were intravaginally infected
with C. muridarum as described in materials and methods. Individual mice were scored
according to the localization of disease 40 days post infection. Nine out of 20 animals
(45%) assessed developed hydrosalpinx with B6-≤ 9wk animals displaying significantly
more oviduct disease (p< 0.001) when compared to all other animal groups (Figure
3-1A). Twenty percent of DBA/2, regardless of age, displayed mild oviduct dilatation
indicated by a mean severity score of 0.23. Conversely, discordant oviduct severity
scores were observed among C57BL/6J-≤ 9wk and C57BL/6J->14wk groups. A
moderate oviduct severity score of 2.2 was observed in B6-≤ 9wk animals while B6-
>14wk animals displayed mild oviduct involvement with a mean score of 0.4. No
significant differences were observed in endometrial dilatation among groups,
irrespective of inbred strain or age (Figure 3-1B).
Secondary Intravaginal Infection Exacerbates Chlamydial-Induced Upper Genital
Tract Complications
We hypothesized that recurrent chlamydial exposures would result in worse
disease when compared to animals that received primary infection alone. To test this
hypothesis, DBA and B6 female mice were intravaginally infected at day 0 and
rechallenged at day 40 as described in materials and methods. Thirty days after
rechallenge, genital tissues were aseptically harvested and assessed for oviduct and
uterine horn dilatation. As we hypothesized, secondary disease exacerbated negative
disease outcomes in both strains and age groups (Figure 3-2) when compared to the
corresponding groups that only received a primary infection (Figure 3-1). Sixteen out of
21 animals (76%) examined after secondary infection developed oviduct pathology as
opposed to 45% of primarily infected animals. Fourteen out of 21 animals (66.6%)
examined after secondary infection developed uterine horn pathology as opposed to 30%
of primarily infected animals.
C57BL/6 Mice Are More Susceptible to Chlamydial-Induced Upper Genital Tract
Complications When Compared to DBA/2J Mice
DBA and B6 female mice were intravaginally infected at day 0 and rechallenged
at day 40 as described in materials and methods. Thirty days after rechallenge, genital
tissues were aseptically harvested and assessed for oviduct and uterine horn dilatation.
Oviduct disease observed in C57BL/6-≤ 9wk animals was significantly worse (mean
severity score 3.6) than that observed in DBA/2-≤ 9wk and DBA/2->14wk animals with
mean scores of 0.8 and 1.2, respectively. Additively, C57BL/6->14wk animals displayed
significantly more severe oviduct disease than that seen in DBA/2-≤ 9wk or DBA/2
Page 60
45
Figure 3-1. Upper genital tract disease incidence and severity post-primary
infection.
Note: DBA/2 and C57BL/6J female mice were intravaginally infected with Chlamydia
muridarum as described in materials and methods. Forty days after infection, mice were
sacrificed and pathology was assessed grossly. The left figure (Panel A) displays oviduct
disease severity and disease incidence by inbred strain and age group. The right figure
(Panel B) displays uterine horn disease severity and disease incidence by inbred strain
and age group. 5 animals were assessed per group and bars indicate mean severity scores.
* and ** indicate significant differences (p≤ 0.01 and p≤ 0.001) as determined by one-
way ANOVA with post hoc Newman-Keuls Multiple Comparison Test. Data are
representative of two independent studies.
Page 61
46
Figure 3-2. Upper genital tract disease incidence and severity post-secondary
infection.
Note: DBA/2 and C57BL6 female mice were intravaginally infected and rechallenged
with Chlamydia muridarum as described in materials and methods. Thirty days post-
secondary infection, mice were sacrificed and pathology was assessed grossly. The left
figure (Panel A) displays oviduct disease severity and incidence by inbred strain and age
group. The right figure (Panel B) displays uterine horn disease severity and incidence by
inbred strain and age group. 5-6 animals were assessed per group and bars indicate mean
severity scores. * indicate significant differences (p<0.01) as determined by one-way
ANOVA with post hoc Newman-Keuls Multiple Comparison Test. Data are
representative of two independent studies.
Page 62
47
->14wk animals. Although uterine horn severity assessments did not result in statistically
significant differences, B6-≤ 9wk animals consistently displayed moderate disease
severity as indicated by a mean score of 2 while B6->14wk endometrial involvement was
comparable to that observed in DBA/2 animals.
To remove any unintentional disease bias imposed by using the quantitative
severity scoring system, we assessed disease severity as a function of organ weight in
milligrams divided by total body weight in grams (Figure 3-3). We have previously
observed a direct correlation between severe disease and increased weight ratios given
genital abnormalities that involve dilatation of murine oviduct and uterine horn tissues
increase the ratio numerator value-organ weight in milligrams (Figure 2-7). Weight
ratios compliment severity score data in that secondary infection results in worse disease
when compared to animals of the same strain and age group. Moreover, C57BL/6-≤ 9wk
animals are most susceptible to chlamydial-induced upper genital tract complications
evidenced by significant differences observed in recurrent infection oviduct severity
scores and objective weight ratio analysis.
Discussion
Individual differences in disease susceptibility as a result of infectious agents
exist in the population. For example only a small subset of women with uncomplicated
chlamydial genital infection go on to develop severe upper genital tract
complications[170]. Furthermore, adolescent females are more likely to contract
Chlamydia than any reported age group [185] [218] [219]. In the United States alone, the
reported 2011 rates of sexually transmitted chlamydial infection for women between the
ages of 15-19 and 20-24 were 3416.5 and 3722.5 per 100,000 population, respectively
[147]. The reported rate for adolescents between the ages of 15-19 yrs is over five times
the national rate of 648.9 per 100,000 population and almost six times the national rate
for young women between the ages of 20-24 yrs. These rates are particularly alarming
considering the rate for women in their mid-forties to early fifties is only 35.8 per
100,000 population [147]. Taken together, current epidemiological trends highlight the
need for chlamydial models to account for biological factors such as host age when
assessing incidence and severity of chlamydiae-induced reproductive complications.
In this study, chlamydial-induced disease outcomes were largely dependent on the
mouse age and strain used to model urogenital infection. C57BL/6-≤ 9wk mice were
more susceptible to oviduct pathology when compared to DBA/2 mice as evidenced by
mean severity score post primary (Figure 3-1A) and secondary infection cycles (Figure
3-2A). In addition to extra-strain disease variation, intra-strain disease variation among
B6 was shown to be age-dependent in the oviduct post primary infection and, albeit not
statistically significant, in the oviduct and uterine horn post-secondary infection. In
addition to the four-tiered semi-quantitative severity scoring system, these results were
objectively validated with weight ratio analysis (Figure 3-3). BL/6-≤ 9wk animals
display significantly more disease as evidenced by weight ratios when compared to
Page 63
48
Figure 3-3. Weight ratio disease severity validation post-primary and secondary
intravaginal infection.
Note: DBA/2 and C57BL6 female mice were intravaginally infected and rechallenged
with Chlamydia muridarum as described in materials and methods. Weight ratios were
determined by dividing individual organ weights (milligrams) by total body weight
(grams). Ratios were averaged to calculate the mean value for each group (indicated by
bar). Error bars depict the standard error of the mean. * indicate significant differences
(p< 0.05) as determined by one-way ANOVA with post hoc Newman-Keuls Multiple
Comparison Test. ** indicate significant differences (p< 0.01) as determined by one-way
ANOVA with post hoc Newman-Keuls Multiple Comparison Test. Data are
representative of two independent studies.
Page 64
49
> 14wk animals with the same genetic background. Interestingly, disease severity as a
function of weight observed in DBA/2 animals remained consistent irrespective of age or
disease localization.
While inter-strain susceptibility variations have been appreciated for some time
[74] [75] [210] [220], our studies suggest C57BL/6 animals are susceptible to
chlamydial-induced genital tract complications when compared to DBA/2 mice as
determined by semi-quantitative severity scoring (Figures 3-1 and 3-2) and weight ratios
(Figures 3-3). Kaltenboeck et al also found that C57BL/6 animals were susceptible to
chlamydial-induced disease using a respiratory murine model of infection [221]. In their
studies C57BL/6 mice developed severe pneumonia while BALB/C mice were
completely void of symptoms following intranasal priming with C. psittaci and
subsequent challenge. Interestingly, our results and those seen in the Kaltenboeck model
conflict with genital studies proposing a resistant phenotype for C57BL/6 when
compared to C3H and BALB inbred strains, both of which are commonly referred to as
susceptible strains given their increased chlamydial shedding and longer courses of
infection when compared to C57BL/6 mice. Although we have not delineated a
mechanism for the results seen in our study, immunological validation is warranted and
currently underway. The discrepancies observed between our results and those published
by others may also be attributed, at least in part, to differences in the strains of Chlamydia
used, inoculum dosage, murine strains, multiple rounds of infection, and variation in what
is defined as resistance or susceptibility (I. E. reduced shedding, shorter infection course,
infertility, eradication of the infectious agent in the lower and upper genital tract, or the
development of upper genital tract sequelae).
As many researchers developing infectious disease models, we are interested in
identifying human correlates of phenotypic differences seen in our model. Studies are
currently underway using B (C57BL/6) x D (DBA/2J) recombinant inbred mouse strains
to identify genetic factors that may modulate disease susceptibility in an age-dependent
manner. By interrogating the GeneNetwork database using a forward genomic approach,
we hope to identify genes associated with the modulation of oviduct and uterine horn
disease severity in an age and strain dependent manner. More importantly, we are
interested in identifying human correlates for the potential murine-derived phenotypic-
genetic links observed in our model.
While studies aimed at delineating causal mechanisms for the age and strain
dependence reported here are currently underway, these initial observations have
immediate implications for the use of mouse models in understanding chlamydial-
induced disease. Furthermore, these data suggest that the host genetic contribution to
genital pathogenesis may be more complex than previously appreciated.
Page 65
50
CHAPTER 4. IMMUNIZATION WITH C. MURIDARUM OUTER MEMBRANE
COMPLEX FAILS TO PROTECT AGAINST UPPER GENITAL TRACT
COMPLICATIONS IN A MURINE MODEL OF GENITAL INFECTION
Introduction
Chlamydia trachomatis is the causative agent of the most commonly reported
sexually transmitted disease in the United States, Chlamydia, and a public health threat
worldwide [56] [63] [147] [222]. Despite the overwhelming global presence of
chlamydial infection, many cases are undiagnosed due to asymptomatic infection and
may serve as a reservoir for infection [175] [223]. Regardless of whether Chlamydia
trachomatis infection is symptomatic or asymptomatic, women with genital infections
may develop long-term sequelae like pelvic inflammatory disease (PID), ectopic
pregnancy, involuntary infertility, and miscarriage [169] [170]. Antibiotic therapy is
currently effective in clearing the organism from the lower genital tract but the
emergence of antibiotic resistance, although rare, is a viable concern [224] [225] [226]
227]. Currently, rapid antibiotic intervention is a key component of Chlamydia control
programs [173] [175] and may serve as the selective pressure necessary for the evolution
of antibiotic resistant human strains. Taken together, the high rate of asymptomatic
infection and the potential for developing antibiotic resistance despite the current
availability of effective antimicrobial therapies underscore the need for a safe and
efficacious vaccine.
Trachoma vaccine trials using whole organisms, both inactivated and live, were
performed decades ago and are perhaps the most notable vaccination studies to date.
Unfortunately, a subset of individuals who were vaccinated and reexposed to Chlamydia
experienced significantly more severe disease when compared to non-vaccinated cohorts
[124] [228] [229]. While no human trials have been untaken since the trachoma study,
researchers have continued the quest to identify safe chlamydial antigens that elicit
immunological memory- the basis of all vaccines.
A number of Chlamydia trachomatis- associated antigenic proteins have been
described, particularly proteins associated with the cell surface. Many studies have
focused on developing a subunit-based vaccine using Chlamydia trachomatis major outer
membrane protein (MOMP) as the antigen [230] [231]. MOMP has been an attractive
candidate because it constitutes an approximated 60% of the total outer membrane mass
and is immune-assessable due to its location on the chlamydial surface [21] [125].
Although MOMP seemed promising, its ability to induce protection has been sketchy at
best, displaying varying protection in small animal models and higher order primates
[232] [233] [234] [235] [236] [237] [238] [239] [240] [241]. There may be several
reasons for the variation seen in the previously mentioned studies including but not
limited to the animal model and strain(s) used, immunization site, challenge route,
definition of protection (I.E. reduction in shedding, fertility, cytokine production,
antibody titer, or sequelae formation) , and antigen preparation. Indeed, one of the main
lessons taken from previous MOMP studies is that conformational integrity of the
chlamydial membrane may increase protection following chlamydiae challenge. As a
Page 66
51
result, we elected to use detergent-extracted chlamydial outer membrane complexes
(COMCs) as described by Caldwell et al [125] in the preliminary studies outlined later in
this Chapter. Sarkosyl-Insoluble COMC was found to be structurally intact and
maintained the shape observed in protein-rich infectious chlamydial particles, elementary
bodies (EBs).
In the pages to follow, I outline experiments evaluating the efficacy of COMC as
a potential antigen in both systemic and genital models of chlamydiae infection. As a
disclaimer, these studies are in their infancy at best and should not be interpreted as
complete studies. Several gaps exist in the experiments outlined hereafter but these data
do provide interesting tangents that may serve as the basis for future experimental
direction. To that end, our initial results in the systemic model showed promise by
negating the effects of lethal interperitoneal chlamydial doses. Conclusions originating
from studies in the urogenital murine model were more muddled when compared to the
lethal model. Despite the inconclusive nature of the genital tract model results, we
examined the potential benefits of alum, an adjuvant known to generate a TH2 cell-
mediated response, in the prevention of severe genital sequelae.
Materials and Methods (Systemic Lethal Model)
Mice
Female DBA/2J mice were purchased from Jackson Laboratories at 7-8 weeks of
age. They were housed in the University of Tennessee Health Science Center animal
facility and animal protocols were performed in compliance with institutional IACUC
and federal mandates.
Chlamydiae
Chlamydia psittaci 6BC was propagated in the murine fibroblast cell line L929
(ATCC; Manassas, VA) and stored at -80o C. Titers were determined by infecting L929
cells plated at a density of 2 x 105 cells/well in 24- well plates (Corning Costar
Corporation, Cambridge, MA), with tenfold dilutions of thawed stock. Infected
monolayers were incubated for 48 hours, methanol fixed, and chlamydial inclusions were
enumerated using fluorescent staining with FITC-conjugated anti-chlamydial
lipopolysaccride antibody (Fitzgerald Industries Internation Incorporated, Concord, MA).
COMC Extraction
Chlamydia psittaci 6BC COMC was purified as described by Caldwell et al [125].
In brief, C. psittaci 6BC stock was thawed at room temperature and suspended in 5mL of
pH 8 phosphate buffered saline (PBS) containing 2% dipolar ionic Zwittergonic detergent
and 1.5 mm EDTA. A dry ice bath was used to precipitate proteins and the resulting
Page 67
52
suspension was sonicated, in series, 3 times with 1 minute iced incubation periods
between each round of sonication. The cell suspension was incubated for 3 hours at 37o
C and ultracentrifuged at 32, 700 rotations per minute (RPM) for 1hour.
Immunization and Infection
DBA/2J mice were intramuscularly mock immunized with 100mL of sterile
Dulbecco’s phosphate buffered saline (PBS), viable whole elementary bodies (EBs) of
6BC (5 x 102 IFU in 100mL of sterile PBS), or 6BC-derived COMC (40mg total) in
100mL of PBS, respectively. Mice received a total of two immunizations at days -14 and
-7. On day 0, mice were intraperitoneally challenged with a low dose (1 x 103 IFU,
equivalent to 1000 LD50) or a high dose (1 x 105 IFU, equivalent to 100000 LD50) and
monitored for survival. Surviving animals were sacrificed 15 days post infection
irrespective of immunization regimen.
Results (Systemic Lethal Model)
Our laboratory was instrumental in developing a systemic interperitoneal model
of chlamydial disease using C. psittaci 6BC, a zoonotic to which mice are highly
susceptible. Using the systemic model Byrne and colleagues were able to elucidate basic
concepts concerning interferon-gamma and Chlamydia psittaci clearance [242] [243]
[244]. Unpublished data from our laboratory showed that doses as low as 1 EB can cause
mice to succumb to infection within 10 days of initial C. psittaci challenge. These data
provided the basis for using the systemic model of chlamydiae infection to elucidate
COMC immunogenicity. One hundred percent of mice immunized with viable 6BC
Chlamydia psittaci elementary bodies (EBs) survived challenge and recovered from
infection irrespective of the challenge dose. Conversely, all mock PBS immunized mice
succumbed to infection within 11 days of interperitoneal challenge. 20% of COMC
antigen immunized mice survived an inordinately high dose challenge and went on to
recover from infection (Figure 4-1). Moreover, 60% of mice immunized with 6BC
Chlamydia psittaci -derived antigen survived and recovered from lethal challenge
(Figure 4-2).
Despite the rigor associated with the systemic chlamydial intraperitoneal murine
model, COMC preparations were capable of eliciting partial protection against lethal
challenge. Both immunizing doses, 102 and 105 IFU, supersede the LD50 and highlight the
sensitivity of DBA/2J inbred mice to 6BC Chlamydia psittaci intraperitoneal infection as
previously illustrated by the Byrne laboratory. Collectively, these data suggested COMC-
induced partial protection with homologous challenge and justified transitioning these
studies into the urogenital model which would display more subtle read-outs.
Page 68
53
Figure 4-1. High dose 6BC C. psittaci systemic infection survival curve.
High Dose 6BC C. psittaci Interperitoneal Challenge
0 3 6 9 12 150
20
40
60
80
100
120
LIVE 6BC EB
Antigen
PBS
Time:Days
Perc
en
t su
rviv
al
Page 69
54
Figure 4-2. Low dose 6BC C. psittaci systemic infection survival curve.
Low Dose 6BC C. psittaci Interperitoneal Challenge
0 3 6 9 12 150
20
40
60
80
100
120
LIVE 6BC EB
Antigen
PBS
Time: Days
Percen
t su
rviv
al
Page 70
55
Materials and Methods (Urogenital Model)
Mice
Female DBA/2J mice were purchased from Jackson Laboratories at 7-8 weeks of
age. They were housed in the University of Tennessee Health Science Center animal
facility and animal protocols were performed in compliance with institutional IACUC
and federal mandates.
Chlamydiae
Chlamydia muridarum (Weiss) was propagated in the human epithelial cell line
HeLa 229 (ATCC; Manassas, VA) and stored at -80o C. Titers were determined by
infecting HeLa 229 cells seeded at a density of 2 x 105 cells/well in 24- well plates
(Corning Costar Corporation, Cambridge, MA), with tenfold dilutions of thawed stock.
Infected monolayers were incubated for 48 hours, methanol fixed, and chlamydial
inclusions were enumerated using fluorescent staining with FITC-conjugated anti-
chlamydial lipopolysaccride antibody (Fitzgerald Industries Internation Incorporated,
Concord, MA).
COMC Extraction
Chlamydia muridarum COMC was purified as described by Caldwell et al [125].
In brief, C. muridarum stock was thawed at room temperature and suspended in 5mL of
pH 8 phosphate buffered saline (PBS) containing 2% dipolar ionic Zwittergonic detergent
and 1.5 mm EDTA. A dry ice bath was used to precipitate proteins and the resulting
suspension was sonicated, in series, 3 times with 1 minute iced incubation periods
between each round of sonication. The cell suspension was incubated for 3 hours at 37o
C and ultracentrifuged at 32, 700 rotations per minute (RPM) for 1hour.
Immunization and Infection
DBA/2J mice were intramuscularly immunized with 100mL of sterile Dulbecco’s
phosphate buffered saline (PBS), 100mL of sterile Dulbecco’s phosphate buffered saline
(PBS) and 25ug of alum, live C. muridarum elementary bodies (5 x 102 IFU in 100mL of
sterile PBS), 25ug of Chlamydia muridarum-derived COMC in 100mL of PBS, or 25ug
of Chlamydia muridarum-derived COMC in 100mL of PBS and 25ug of alum. Mice
received a total of two immunizations at days -14 and -7. One week prior to challenge,
the estrous cycle was synchronized by subcutaneous injection of 2.5mg of
medroxyprogesterone acetate (Sicor, CA). On day 0, mice were intravaginally challenged
with 2 x 103 IFU, a non-lethal dose of C. muridarum.
Page 71
56
Results (Urogenital Model)
C. muridarum and C. psittaci-derived COMC preparations were compared by
commassie-stained SDS-Page gel and displayed comparable banding patterns
(Figure 4-3). As expected, animals intramuscularly immunized with viable whole C.
muridarum elementary bodies and intravaginally challenged with 5 x 103 IFU displayed
the most severe disease when compared to other immunization groups as evidenced by
dilation of the uterine horns and reddening of the genital tract (Figure 4-4A). Mock
immunized animals (Figure 4-4B) also displayed fluid accumulation in the uterine horn,
although to a lesser extent when compared to the live immunized group. COMC
immunized animals displayed less severe disease when compared to live immunized
groups but still displayed mild to moderate dilation and reddening (Figure 4-4D). While
animals immunized with COMC-alum displayed mild reddening, dilation was not
observed (Figure 4-4E). Lastly but most intriguingly, animals immunized with the PBS-
alum combination displayed no dilation or substantial reddening (Figure 4-4C).
While we hypothesized that alum would augment protection elicited by COMC
antigen immunization despite its polarity toward a Th2-type response, we were perplexed
by the observation seen in PBS-alum immunized groups. Unexpectedly, the alum
adjuvant seemed to negate the development of severe genital tract sequelae induced by
exposure to C. muridarum intravaginal infection. The study described below followed as
an attempt to assess whether alum alone was capable of negating the development of
upper genital tract sequelae following C. muridarum intravaginal infection.
Materials and Methods (Alum Study)
Mice
Female C57BL/6J mice were purchased from Jackson Laboratories at 7-8 weeks
of age. They were housed in the University of Tennessee Health Science Center animal
facility and animal protocols were performed in compliance with institutional IACUC
and federal mandates.
Chlamydiae
Chlamydia muridarum (Weiss) was propagated in the human epithelial cell line
HeLa 229 (ATCC; Manassas, VA) and stored at -80o C. Titers were determined by
infecting HeLa 229 cells seeded at a density of 2 x 105 cells/well in 24- well plates
(Corning Costar Corporation, Cambridge, MA), with tenfold dilutions of thawed stock.
Infected monolayers were incubated for 48 hours, methanol fixed, and chlamydial
inclusions were enumerated using fluorescent staining with FITC-conjugated anti-
chlamydial lipopolysaccride antibody (Fitzgerald Industries Internation Incorporated,
Concord, MA).
Page 72
57
Figure 4-3. Comassie stained SDS-Page gel of C. muridarum (MoPn)-derived
COMC and C. psittaci (6BC)-derived COMC preparations.
Note: Chlamydiae COMC preparations were extracted as previously described in Chapter
4. Protein concentrations were determined by Modified Lowry Protein Assay and
visualized by SDS-Page gel. Molecular weight protein standard (shown on left) were
used to approximate the molecular weight of visualized bands.
MoPn 6BC
250 kD
150 kD
100 kD
75 kD
50 kD
37 kD
25 kD 20 kD
15 kD
10 kD
Page 73
58
Figure 4-4. C. muridarum-derived COMC immunization study gross pathology.
Note: Representative photos of genital gross pathology post homologous C. muridarum immunization and challenge. Photo (A) is
representative of live immunizations. Photo (B) is representative of mock immunized animals. Photo (C) is representative of mock-
alum immunized animals. Photo (D) is representative of COMC immunized animals and photo (E) represents animal cohorts
immunized with COMC-alum. Black arrows point out selected areas of dilatation, reddening, and hydros0alpinx formation.
Page 74
59
Immunization and Infection
C57BL/6J mice were intramuscularly immunized with 100mL of sterile
Dulbecco’s phosphate buffered saline, 100mL of sterile Dulbecco’s phosphate buffered
saline and 25ug of alum (Thermo Scientific), live C. muridarum elementary bodies (5 x
102 IFU in 100mL of sterile PBS), or a mixture of live C. muridarum (5 x 102 IFU in
100mL of sterile PBS) and 25ug of alum. Mice received two immunizations one week
apart and were treated subcutaneously with 2.5mg of medroxyprogesterone acetate
(Sicor, CA) at days -9 and -3 to synchronize the esterous cycle and enhance mouse
susceptibility to chlamydial infection. On day 0, mice were intravaginally challenged
with 2 x 103 IFU Chlamydia muridarum in 10uL of PBS. Animals that were immunized
and challenged were sacrificed 35 days post infection. Immunized animals that did not
receive an infection were sacrificed 21 days after day 0.
Pathology Assessment
Before removing the genital tissue, an in situ gross examination was performed
for evidence of chlamydial-induced abnormalities such as uterine horn dilation and
hydrosalpinx formation. Genital tracts were aseptically removed, photographed,
subjected to macroscopic inspection, and assigned a severity score on the day of sacrifice.
Results (Alum Study)
Immunogens without Intravaginal Chlamydia muridarum Challenge Do Not Induce
Severe Upper Genital Tract Sequelae Formation
Female C57BL/6J mice were intramuscularly immunized with alum adjuvant,
viable C. muridarum and alum adjuvant, viable C. muridarum, or mock immunized with
sterile PBS according to the timeline displayed in (Figure 4-5). Genital tracts from each
animal were aseptically removed, weighed, photographed, and grossly assessed for upper
genital tract sequelae formation three weeks after day 0. Two non-immunized C57BL/6
mice, of the same age and gender, were used as a reference. As hypothesized,
hydrosalpinx formation was not observed in any of the immunization groups (Figure
4-6). Negligible increases in dilation of the uterine horns were observed in mock, C.
muridarum and alum adjuvant, and C. muridarum alone immunized animals. Although
these increases were minimal, they were documented by granting the lowest severity
score possible (Figure 4-7). Overall, genital tract tissues, regardless of immunization
group were comparable to that observed in the two non-immunized, non-challenged
reference samples.
Page 75
60
Figure 4-5. Alum study experimental timeline.
Page 76
61
Figure 4-6. Oviduct pathology severity of non-challenged immunized groups.
Note: Solid line represents the mean of each immunogen group. Symbols represent
individual mice (N=5 per group except for normal control).
Page 77
62
Figure 4-7. Uterine horn pathology severity of immunized, non-challenged
groups.
Note: Solid bars represent the mean severity score for each immunogen group. Symbols
represent individual animals (N=5 per group except for normal controls).
Page 78
63
Alum Alone Does Not Negate the Development of Severe Upper Genital Tract
Complications
Female C57BL/6J mice were intramuscularly immunized with alum adjuvant,
viable C. muridarum and alum adjuvant, viable viable C. muridarum, or mock
immunized with sterile PBS according to the timeline displayed in (Figure 4-5). All
immunization groups were intravaginally challenged with 2 x 103 IFU of Chlamydia
muridarum. Genital tracts from each animal were aseptically removed, weighed,
photographed, and grossly assessed for upper genital tract sequelae formation and
severity 35 days post infection. Three female C57BL/6 mice which were not immunized
or challenged were used as a negative reference.
Unlike their non-challenged counterparts (Figure 4-6), all immunization groups
displayed some level of oviduct disease (Figure 4-8). 40% of mock immunized animals
displayed unilateral hydrosalphinx formation while 20% displayed bilateral oviduct
involvement. 20% of animals immunized with C. muridarum or C. muridarum and alum
adjuvant exhibited unilateral oviduct disease. Interestingly, 20% of animals immunized
with alum adjuvant alone displayed bilateral hydrosalpinx while unilateral oviduct
pathology was observed in 40% of the same immunization group.
Uterine horn dilation was seen in all groups immunized (Figure 4-9). 60% of
animals immunized with C. muridarum alone displayed mild dilation with a mean
severity score of approximately 1. Eighty percent of animals immunized with C.
muridarum and alum adjuvant were found to have dilated uterine horns with 60% of
those being mildly dilated and 20% displaying moderate dilation. Only 20% of alum
adjuvant immunized animals displayed uterine involvement, whereas, 60% of mock
immunized animals experienced uterine horn dilation.
Discussion
Although C. trachomatis lower genital tract infections may resolve without
adverse reproductive complications [164] [245] [246], a subset of those infected may
develop upper genital tract sequelae. Presently, Chlamydia control initiatives focus on
identifying and treating individuals before, at least in humans, irreversible tissue damage
occurs. This approach is complicated by the fact that most human chlamydial infections
are often asymptomatic. As a result, the most effective way of controlling chlamydiae
infections would be to develop a vaccine. Although the studies outlined in this Chapter
fail to provide definitive answers pertaining to protection against chlamydial infection,
they do highlight the need for ongoing vaccine development initiatives to consider the
pathogenesis approach given the overarching goal of any Chlamydia trachomatis
vaccination effort would be to preserve reproductive health by preventing the
development of severe disease manifestations.
In the systemic model of chlamydial infection we showed that a detergent-
extracted preparation of C. psittaci-derived COMC protected mice from lethal
intraperitoneal challenge. To our knowledge, this is the first study of its kind and the
Page 79
64
Figure 4-8. Oviduct pathology severity of immunized and challenged groups.
Note: Solid bars represent the mean severity score for each immunogen group. Symbols
represent individual animals (N= 5 per group except for normal control).
Norm
al
PBS
Alu
m
Alu
m/M
oPn
MoP
n
-1
0
1
2
3
4
5
Immunogen
Severi
ty S
co
re
Page 80
65
Figure 4-9. Uterine horn pathology severity of immunized and challenged groups.
Note: Solid bar represents the mean for each immunogen group. Symbols represent
individual animals (N = 5 per group except for normal control).
Untr
eate
d
PBS
Alu
m
Alu
m/M
oPn
MoPn
-1
0
1
2
3
4
5
Immunogen
Severi
ty S
co
re
Page 81
66
results inspired us to examine the efficacy of COMC in the C. muridarum urogenital
model given the fact that mice intravaginally infected with Chlamydia muridarum
develop genital sequelae similar to that observed in human populations. Seeing as the
development of genital morbidities such as hydrosalpinx and endometritis are more
subtle than the mortality (survival vs death) end point observed in the systemic model, we
opted to co-administer alum adjuvant in subsequent genital tract experiments as a way of
enhancing the responses observed with homologous COMC antigen. Additionally,
aluminum hydroxide, aluminum phosphate, and aluminum sulfate are the only vaccine
adjuvant formations licensed in the United States and are currently being used in vaccines
directed against the intracellular pathogens, hepatitis B virus (HBV) and human
papilloma virus (HPV) [247].
In our studies, COMC-alum immunized groups (Figure 4-1E), as hypothesized,
fared better than mock immunized groups (Figure 4-1B) and live immunized groups
(Figure 4-1A) displaying no increases in uterine horn or hydrosalpinx formation. As we
hypothesized, DBA/2J mice immunized with live C. muridarum and challenged with C.
muridarum displayed the most severe pathology with both endometrial dilation and
oviduct inflammation (Figure 4-1A). These findings complement our studies and the
studies of others that suggest chlamydial infection recurrence increases the risk for
reproductive sequelae [177] [179] [248].
By far the most interesting observation was the fact that alum adjuvant and PBS
immunized animals displayed no signs of chlamydial-induced pathology or reddening
which is indicative of inflammation. While Th2-driven responses such as those prompted
by the alum adjuvant have been shown to promote immunopathology in chronic diseases
such as schistosomiasis [249] [250], autoimmune diseases, and allergies, more recent
studies have implicated TH2-linked responses to tissue repair and wound healing. As a
result, we hypothesized that a Th2-type response may play a protective role in upper
genital tract disease formation.
To experimentally address the effects of alum adjuvant on chlamydial-induced
genital tract pathology we immunized female C57BL/6J mice with alum adjuvant, alum
and whole organism C. muridarum, or intact C. muridarum organisms alone. Based on
the observations seen in the COMC urogenital studies, we hypothesized that alum only
severity scores would be on par with severity scores associated with non-immunized,
non-challenges animals (negative controls). Interestingly, animals immunized with alum
adjuvant alone displayed severity scores comparable to those observed in alum/C.
muridarum and C. muridarum alone immunization groups (Figure 4-9). While the alum
alone observation was unexpected, we were encouraged by the fact that all immunization
groups exhibited less severe disease, as indicated by mean severity score, than the mock-
immunized PBS group which complemented the results seen in the preliminary COMC
urogenital studies (Figure 4-4 and Figure 4-9).
There are at least two, not mutually exclusive, explanations for the discrepant
results seen in the alum adjuvant alone group. One is the mouse inbred strain we used in
the follow-up studies. We substituted the DBA/2J mouse strain originally used in the
Page 82
67
COMC studies for the C57BL/6J inbred strain because B6 animals used in ongoing,
unrelated studies in our laboratory routinely exhibit more severe genital tract disease
when compared to DBA/2J animals. We hypothesized that by using the ‘more severe’
B6 background we would stratify the results seen between groups, making the potential
benefits of alum immunization easier to identify. We know that the enhancement effects
of alum are less pronounced than that of other adjuvants like Freud’s complete adjuvant
(FCA) and are optimal when used in conjunction with an immunogenic compound.
Assuming the upper genital tract sequelae observed in our model is mediated, at least in
part, by the host immune response, it is possible that the B6 TH1-bias was too much for
alum adjuvant to overcome which manifested as mild disease. Secondly, but not less
important, is the chlamydial preparation used in the study. Chlamydiae-derived outer
membrane complex preparations were used as immunogen in the preliminary studies
while whole viable organisms were used in the alum efficacy studies. This is important in
that the alum alone immunized group scores (Figure 4-9) are indistinguishable from
Chlamydia muridarum-alum and Chlamydia muridarum alone group scores. In using
concentrated COMC preparations, we directed the host response against known
immunogenic proteins associated with the chlamydial membrane. Perhaps statistical
differences were not observed amongst immunization groups, including alum alone,
because we diversified the anti-chlamydial antibody pool by using whole organisms over
purified COMC preparations. While it is conceivable that either or both of these
explanations contributed to differences seen in our alum efficacy experiments, further
study is needed to draw definitive conclusions.
While data presented in this Chapter would, seemingly, negate the hypothesis put
forth in Chapter 2 relating to the potential benefits of having TH2-related IL-4 present late
during recurrent infection that is not the case. Our hypothesis hinges on the idea that IL-4
is detectable late during recurrent infection cycles as a way for chlamydial hosts to repair
severe tissue damage. In our studies, alum adjuvant was administered concurrently with
chlamydial immunogen, prior to the development of severe upper genital tract
complications. It is logically to hypothesize that a portion of time in which the host
perceives ‘self-damage’ is necessary for the gearing up of potentially protective TH2-
related responses. As stated previously, this is only a hypothesis albeit formed from data
derived from ongoing chlamydial genital studies in our laboratory. The continuation and
translational relevance of the studies reported in this Chapter are currently being
investigated in our laboratory and are supported, at least in part, by the recent report
published by Rodolfo et al suggesting an evolutionary genital tract bias for Th2 responses
as a mechanism to prevent chlamydial-induced genital tract complications in humans
[195].
Page 83
68
CHAPTER 5. CONCLUSIONS
The reproductive health of women is of particular concern with current CDC
Chlamydia surveillance data indicating increases in reported chlamydial infections,
perhaps, in part, as a result of rapid antibiotic intervention. To this end, we interrogated
the murine model for its potential to serve as a translational bridge between population-
based epidemiology and previous animal and human studies. Our studies reaffirm the
benefit of rapid antibiotic intervention in Chlamydia control efforts and suggest an added
benefit during repeated infection even in the absence of antibiotic treatment. Moreover,
our results suggest a TH2-independent role for IL-4 in genital tract tissue repair.
Testing the IL-4 Hypothesis
IL-4 as a mediator of TH2-driven immunity is well established but the role IL-4
may play in genital tract tissue repair is largely unknown. In our studies we show
elevated levels of systemic IL-4 late during recurrent chlamydial genital infection and
cellular infiltrates that are known to produce IL-4 in the genital tract. We also show a
correlation between systemic IL-4 and disease severity. To assess the role of IL-4 in the
arrested immunity model, longitudinal studies are necessary. In these studies we would
measure the onset, duration, and resolution of TH1 and TH2-linked cytokines and disease
progression. This is important given the fact that tissue repair mechanisms rely on the
presence of tissue damage. If IL-4 is indeed a driver of in vivo tissue repair, at least in
this model, it is plausible that the production and secretion of the cytokine would begin
with or after the development of chlamydial-induced pathology. Future studies should
also measure IL-4 transcription levels in implicated cell types (mast cells and
eosinophils) to confirm that they are the primary source of IL-4. These studies would be
followed by IL-4 knockout studies that examine the effect of IL-4 on collateral tissue
damage during multiple rounds of chlamydiae infection.
Identifying Genetic Link to Age-Dependent Disease Severity
Studies are currently underway using B (C57BL/6) x D (DBA/2J) recombinant
inbred mouse strains to identify genetic factors that may modulate disease susceptibility
in an age-dependent manner. By interrogating the GeneNetwork database using a forward
genomic approach, we hope to identify genes associated with the modulation of oviduct
and uterine horn disease severity in an age and strain dependent manner. If disease
severity is associated with specific genetic loci, human cell culture studies focused on
RNA silencing will ensue. These studies will be followed with in vivo studies using
knockout mice specific for the identified gene(s).
Page 84
69
Implications for Vaccine Development
It is generally accepted that an effective anti-chlamydial vaccine would need to
elicit a robust TH1-type CD4+ T cell response but our studies suggest that prophylactic
control of chlamydiae may be more complex than originally appreciated. If the goal is to
clear the pathogen from the site of infection and minimize the likelihood of developing
adverse reproductive upper genital tract complications, eliciting TH1-linked immune
responses may not be enough. It may be beneficial for the host to mount a mixed
immunological response (I.E. TH1/ TH2). Our work highlights the need for additional
studies aimed at addressing the role of mixed cell-mediated responses in chlamydial
infection and immunopathological genital tract damage.
Page 85
70
LIST OF REFERENCES
1. Dimitrakov J. Chlamydia- A Journey From Embryonated Egg To LCR Available
at: http://www.prostatitis.org/chlamydia.html.
2. Lindner K. Zur aetiologie der gonokokkenfrien urethritis. Wiener klinische
Wochenschrift 1910; 23:283-4.
3. Lindner K. Gonoblennorrhoe, einschlussblennorrhoe und trachoma Albrecht Von
Graefes Arch Ophthalmol 1911; 78:380.
4. Csonka GW. Non-Gonococcal Urethritis. The British journal of venereal diseases
1965; 41:1-8.
5. Tang FF, Chang HL, Huang YT, Wang KC. Studies on the etiology of trachoma
with special reference to isolation of the virus in chick embryo. Chinese medical
journal 1957; 75:429-47.
6. Tang FF, Huang YT, Chang HL, Wong KC. Isolation of trachoma virus in chick
embryo. Journal of hygiene, epidemiology, microbiology, and immunology 1957;
1:109-20.
7. Moulder JW. The relation of the psittacosis group (Chlamydiae) to bacteria and
viruses. Annual review of microbiology 1966; 20:107-30.
8. Gordon FB, Harper IA, Quan AL, Treharne JD, Dwyer RS, Garland JA. Detection
of Chlamydia (Bedsonia) in certain infections of man. I. Laboratory procedures:
comparison of yolk sac and cell culture for detection and isolation. The Journal of
infectious diseases 1969; 120:451-62.
9. Bush RM, Everett KD. Molecular evolution of the Chlamydiaceae. International
journal of systematic and evolutionary microbiology 2001; 51:203-20.
10. Everett KD, Bush RM, Andersen AA. Emended description of the order
Chlamydiales, proposal of Parachlamydiaceae fam. nov. and Simkaniaceae fam.
nov., each containing one monotypic genus, revised taxonomy of the family
Chlamydiaceae, including a new genus and five new species, and standards for
the identification of organisms. International journal of systematic bacteriology
1999; 49 Pt 2:415-40.
11. Stephens RS, Myers G, Eppinger M, Bavoil PM. Divergence without difference:
phylogenetics and taxonomy of Chlamydia resolved. FEMS immunology and
medical microbiology 2009; 55:115-9.
12. Wang SP, Grayston JT. Immunologic relationship between genital TRIC,
lymphogranuloma venereum, and related organisms in a new microtiter indirect
immunofluorescence test. American journal of ophthalmology 1970; 70:367-74.
Page 86
71
13. Stephens RS, Sanchez-Pescador R, Wagar EA, Inouye C, Urdea MS. Diversity of
Chlamydia trachomatis major outer membrane protein genes. Journal of
bacteriology 1987; 169:3879-85.
14. Baehr W, Zhang YX, Joseph T, et al. Mapping antigenic domains expressed by
Chlamydia trachomatis major outer membrane protein genes. Proceedings of the
National Academy of Sciences of the United States of America 1988; 85:4000-4.
15. Millman K, Black CM, Johnson RE, et al. Population-based genetic and
evolutionary analysis of Chlamydia trachomatis urogenital strain variation in the
United States. Journal of bacteriology 2004; 186:2457-65.
16. Bedson SP BJ. A morphological study of psittacosis virus, with the decription of a
developmental cycle. British journal of experimental pathology 1932; 13:461-6.
17. Stirling P, Richmond S. The developmental cycle of Chlamydia trachomatis in
McCoy cells treated with cytochalasin B. Journal of general microbiology 1977;
100:31-42.
18. Ward ME. Chlamydial classification, development and structure. British medical
bulletin 1983; 39:109-15.
19. Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiological
reviews 1991; 55:143-90.
20. Hackstadt T, Fischer ER, Scidmore MA, Rockey DD, Heinzen RA. Origins and
functions of the chlamydial inclusion. Trends in microbiology 1997; 5:288-93.
21. Hatch TP. Developmental Biology In: Stephens RS, ed. Chlamydia: Intracellular
Biology, Pathogenesis, and Immunity. Washington, D.C.: American Society for
Microbiology 1999:29-67.
22. Abdelrahman YM, Belland RJ. The chlamydial developmental cycle. FEMS
microbiology reviews 2005; 29:949-59.
23. Beatty WL, Morrison RP, Byrne GI. Persistent chlamydiae: from cell culture to a
paradigm for chlamydial pathogenesis. Microbiological reviews 1994; 58:686-99.
24. Hogan RJ, Mathews SA, Mukhopadhyay S, Summersgill JT, Timms P.
Chlamydial persistence: beyond the biphasic paradigm. Infect Immun 2004;
72:1843-55.
25. Wyrick PB. Chlamydia trachomatis persistence in vitro: an overview. The Journal
of infectious diseases 2010; 201 Suppl 2:S88-95.
26. Byrne GI BW. Chlamydial Persistence Redux In: Tan M BP, ed. Intracellular
Pathogens I: Chlamydiales. Washignton, D.C.: ASM Press, 2012.
Page 87
72
27. Schoborg RV. Chlamydia persistence- a tool to dissect Chlamydia-host
interactions. Microbes and infection / Institut Pasteur 2011; 13:649-62.
28. Byrne GI, Lehmann LK, Kirschbaum JG, Borden EC, Lee CM, Brown RR.
Induction of tryptophan degradation in vitro and in vivo: a gamma-interferon-
stimulated activity. Journal of interferon research 1986; 6:389-96.
29. Byrne GI, Lehmann LK, Landry GJ. Induction of tryptophan catabolism is the
mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia
psittaci replication in T24 cells. Infect Immun 1986; 53:347-51.
30. Beatty WL, Byrne GI, Morrison RP. Morphologic and antigenic characterization
of interferon gamma-mediated persistent Chlamydia trachomatis infection in
vitro. Proceedings of the National Academy of Sciences of the United States of
America 1993; 90:3998-4002.
31. Belland RJ, Nelson DE, Virok D, et al. Transcriptome analysis of chlamydial
growth during IFN-gamma-mediated persistence and reactivation. Proceedings of
the National Academy of Sciences of the United States of America 2003;
100:15971-6.
32. Kokab A, Jennings R, Eley A, Pacey AA, Cross NA. Analysis of modulated gene
expression in a model of Interferon-gamma-induced persistence of Chlamydia
trachomatis in HEp-2 cells. Microbial pathogenesis 2010; 49:217-25.
33. Galasso GJ, Manire GP. Effect of antiserum and antibiotics on persistent infection
of HeLa cells with meningopneumonitis virus. J Immunol 1961; 86:382-5.
34. Matsumoto A, Manire GP. Electron microscopic observations on the effects of
penicillin on the morphology of Chlamydia psittaci. Journal of bacteriology 1970;
101:278-85.
35. Skilton RJ, Cutcliffen LT, Barlow D, et al. Penicillin induced persistence in
Chlamydia trachomatis: high quality time lapse video analysis of the
developmental cycle. PloS one 2009; 4:e7723.
36. Moulder JW. Inhibition of onset of overt multiplication of Chlamydia psittaci in
persistently infected mouse fibroblasts (L cells). Infect Immun 1983; 39:898-907.
37. Coles AM, Reynolds DJ, Harper A, Devitt A, Pearce JH. Low-nutrient induction
of abnormal chlamydial development: a novel component of chlamydial
pathogenesis? FEMS microbiology letters 1993; 106:193-200.
38. Ouellette SP, Hatch TP, AbdelRahman YM, Rose LA, Belland RJ, Byrne GI.
Global transcriptional upregulation in the absence of increased translation in
Chlamydia during IFNgamma-mediated host cell tryptophan starvation.
Molecular microbiology 2006; 62:1387-401.
Page 88
73
39. Ibana JA, Belland RJ, Zea AH, et al. Inhibition of indoleamine 2,3-dioxygenase
activity by levo-1-methyl tryptophan blocks gamma interferon-induced
Chlamydia trachomatis persistence in human epithelial cells. Infect Immun 2011;
79:4425-37.
40. Thompson CC, Carabeo RA. An optimal method of iron starvation of the obligate
intracellular pathogen, Chlamydia trachomatis. Frontiers in microbiology 2011;
2:20.
41. Deka S, Vanover J, Dessus-Babus S, et al. Chlamydia trachomatis enters a viable
but non-cultivable (persistent) state within herpes simplex virus type 2 (HSV-2)
co-infected host cells. Cellular microbiology 2006; 8:149-62.
42. Deka S, Vanover J, Sun J, Kintner J, Whittimore J, Schoborg RV. An early event
in the herpes simplex virus type-2 replication cycle is sufficient to induce
Chlamydia trachomatis persistence. Cellular microbiology 2007; 9:725-37.
43. Prusty BK, Bohme L, Bergmann B, et al. Imbalanced oxidative stress causes
chlamydial persistence during non-productive human herpes virus co-infection.
PloS one 2012; 7:e47427.
44. Kuo CC, Jackson LA, Campbell LA, Grayston JT. Chlamydia pneumoniae
(TWAR). Clinical microbiology reviews 1995; 8:451-61.
45. Belland RJ, Ouellette SP, Gieffers J, Byrne GI. Chlamydia pneumoniae and
atherosclerosis. Cellular microbiology 2004; 6:117-27.
46. Andraws R, Berger JS, Brown DL. Effects of antibiotic therapy on outcomes of
patients with coronary artery disease: a meta-analysis of randomized controlled
trials. JAMA: the journal of the American Medical Association 2005; 293:2641-7.
47. Higgins JP. Chlamydia pneumoniae and coronary artery disease: the antibiotic
trials. Mayo Clinic proceedings Mayo Clinic 2003; 78:321-32.
48. Higgins JP, Higgins JA, Higgins PM, Ahuja S, Higgins DL. Chlamydia
pneumoniae and coronary artery disease: legitimized linkages? Expert review of
cardiovascular therapy 2003; 1:367-84.
49. Biosafety in microbiological and biomedical laboratories (BMBL). 5th ed.
Atlanta, GA: Centers for Disease Control and Prevention 2007 Chosewood L.C.
WDE, editors, ed.
50. Grinblat-Huse V, Drabek EF, Creasy HH, et al. Genome sequences of the
zoonotic pathogens Chlamydia psittaci 6BC and Cal10. Journal of bacteriology
2011; 193:4039-40.
Page 89
74
51. Harkinezhad T, Geens T, Vanrompay D. Chlamydophila psittaci infections in
birds: a review with emphasis on zoonotic consequences. Veterinary
microbiology 2009; 135:68-77.
52. Alexandra Nunes PJN, Maria J Borrego, João P Gomes. Chlamydia trachomatis
diversity viewed as a tissue-specific coevolutionary arms race. Genome Biology
2008; 9.
53. Borges V NA, Ferreira R, Borrego MJ, Gomes JP. Directional Evolution of
Chlamydia trachomatis towards Niche-Specific Adaptation. Journal of
bacteriology 2012; 194 6143-53.
54. Harris SR CI, Seth-Smith HMB, Solomon AW, Cutcliffe LT, Marsh P, Skilton
RJ, Holland MJ, Mabey D, Peeling RW, Lewis DA, Spratt BG, Unemo M,
Persson K, Bjartling C, Brunham R, de Vries HJC, Morré SA, Speksnijder A,
Bébéar CM, Clerc M, de Barbeyrac B, Parkhill J, Thomson NR. Whole-genome
analysis of diverse Chlamydia trachomatis strains identifies phylogenetic
relationships masked by current clinical typing. Nature Genetics 2012; 44:413-9.
55. Frick KD, Hanson CL, Jacobson GA. Global burden of trachoma and economics
of the disease. The American journal of tropical medicine and hygiene 2003;
69:1-10.
56. Burton MJ, Mabey DC. The global burden of trachoma: a review. PLoS neglected
tropical diseases 2009; 3:e460.
57. Schachter J, Dawson CR. Elimination of blinding trachoma. Current opinion in
infectious diseases 2002; 15:491-5.
58. Mathew AA, Turner A, Taylor HR. Strategies to control trachoma. Drugs 2009;
69:953-70.
59. Gdoura R, Daoudi F, Bouzid F, et al. Detection of Chlamydia trachomatis in
semen and urethral specimens from male members of infertile couples in Tunisia.
The European journal of contraception & reproductive health care: the official
journal of the European Society of Contraception 2001; 6:14-20.
60. Gdoura R, Keskes-Ammar L, Bouzid F, Eb F, Hammami A, Orfila J. Chlamydia
trachomatis and male infertility in Tunisia. The European journal of contraception
& reproductive health care: the official journal of the European Society of
Contraception 2001; 6:102-7.
61. Friberg J, Confino E, Suarez M, Gleicher N. Chlamydia trachomatis attached to
spermatozoa recovered from the peritoneal cavity of patients with salpingitis. The
Journal of reproductive medicine 1987; 32:120-2.
Page 90
75
62. Anttila T, Saikku P, Koskela P, et al. Serotypes of Chlamydia trachomatis and
risk for development of cervical squamous cell carcinoma. JAMA: the journal of
the American Medical Association 2001; 285:47-51.
63. CDC. Sexually Transmitted Disease Surveillance 2010: Centers for Disease
Control and Prevention 2011.
64. Vieira JT. Acute gonococcic peritonitis in the right upper quadrant in women;
Fitzhugh syndrome. Revista brasileira de medicina 1950; 7:287-9.
65. Paavonen J, Saikku P, von Knorring J, Aho K, Wang SP. Association of infection
with Chlamydia trachomatis with Fitz-Hugh-Curtis syndrome. The Journal of
infectious diseases 1981; 144:176.
66. Miyairi I, Ramsey KH, Patton DL. Duration of untreated chlamydial genital
infection and factors associated with clearance: review of animal studies. The
Journal of infectious diseases 2010; 201 Suppl 2:S96-103.
67. Jacobs NF, Jr., Arum ES, Kraus SJ. Experimental infection of the chimpanzee
urethra and pharynx with Chlamydia trachomatis. Sexually transmitted diseases
1978; 5:132-6.
68. Moller BR, Mardh PA. Experimental epididymitis and urethritis in grivet
monkeys provoked by Chlamydia trachomatis. Fertility and sterility 1980;
34:275-9.
69. Moller BR, Mardh PA. Experimental salpingitis in grivet monkeys by Chlamydia
trachomatis: Modes of spread of infection to the Fallopian tubes. Acta
pathologica et microbiologica Scandinavica Section B, Microbiology 1980;
88:107-14.
70. Mount DT, Bigazzi PE, Barron AL. Infection of genital tract and transmission of
ocular infection to newborns by the agent of guinea pig inclusion conjunctivitis.
Infect Immun 1972; 5:921-6.
71. Mount DT, Bigazzi PE, Barron AL. Experimental genital infection of male guinea
pigs with the agent of guinea pig inclusion conjunctivitis and transmission to
females. Infect Immun 1973; 8:925-30.
72. Barron AL WH, Rank RG, Soloff BL, Moses EB. A new animal model for the
study of Chlamydia trachomatis genital infections: Infection of mice with the
agent of mouse pneumonitis. Journal of Infectious Diseases 1981; 141:63-6.
73. Read TD, Brunham RC, Shen C, et al. Genome sequences of Chlamydia
trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic acids research
2000; 28:1397-406.
Page 91
76
74. de la Maza LM, Pal S, Khamesipour A, Peterson EM. Intravaginal inoculation of
mice with the Chlamydia trachomatis mouse pneumonitis biovar results in
infertility. Infect Immun 1994; 62:2094-7.
75. Darville T, Andrews CW, Jr., Laffoon KK, Shymasani W, Kishen LR, Rank RG.
Mouse strain-dependent variation in the course and outcome of chlamydial genital
tract infection is associated with differences in host response. Infect Immun 1997;
65:3065-73.
76. Chakravarti A. Single nucleotide polymorphisms: to a future of genetic medicine.
Nature 2001; 409:822-3.
77. Peltonen L MV. Dissecting Human Disease in the Postgenomic Era. Science
2001; 291:1224-9.
78. Miyairi I, Ziebarth J, Laxton JD, et al. Host genetics and Chlamydia disease:
prediction and validation of disease severity mechanisms. PloS one 2012;
7:e33781.
79. Nigg C. An unidentified virus which produces pneumonia and systemic infection
in mice. Science 1942; 95:49-50.
80. Nigg C, Eaton MD. Isolation from normal mice of a pneumotropic virus which
forms elementary bodies. The Journal of experimental medicine 1944; 79:497-
510.
81. Swenson CE, Donegan E, Schachter J. Chlamydia trachomatis-induced salpingitis
in mice. The Journal of infectious diseases 1983; 148:1101-7.
82. Perry LL, Su H, Feilzer K, et al. Differential sensitivity of distinct Chlamydia
trachomatis isolates to IFN-gamma-mediated inhibition. J Immunol 1999;
162:3541-8.
83. Maxion HK, Liu W, Chang MH, Kelly KA. The infecting dose of Chlamydia
muridarum modulates the innate immune response and ascending infection. Infect
Immun 2004; 72:6330-40.
84. Carey AJ, Cunningham KA, Hafner LM, Timms P, Beagley KW. Effects of
inoculating dose on the kinetics of Chlamydia muridarum genital infection in
female mice. Immunology and cell biology 2009; 87:337-43.
85. Eckert LO SR, Hawes SE, Stamm WE. Quantitative Chlamydia trachomatis
cultures: correlation of chlamydial inclusion-forming units with serovar, age, sex,
and race. The Journal of infectious diseases 2000; 182:540-4.
86. Brabin L. Interactions of the female hormonal environment, susceptibility to viral
infections, and disease progression. AIDS patient care and STDs 2002; 16:211-
21.
Page 92
77
87. Baker DA, Plotkin SA. Enhancement of vaginal infection in mice by herpes
simplex virus type II with progesterone. Proc Soc Exp Biol Med 1978; 158:131-4.
88. Kaushic C, Murdin AD, Underdown BJ, Wira CR. Chlamydia trachomatis
infection in the female reproductive tract of the rat: influence of progesterone on
infectivity and immune response. Infect Immun 1998; 66:893-8.
89. Ramsey KH, Sigar IM, Rana SV, Gupta J, Holland SM, Byrne GI. Role for
inducible nitric oxide synthase in protection from chronic Chlamydia trachomatis
urogenital disease in mice and its regulation by oxygen free radicals. Infect
Immun 2001; 69:7374-9.
90. Cotter TW, Miranpuri GS, Ramsey KH, Poulsen CE, Byrne GI. Reactivation of
chlamydial genital tract infection in mice. Infect Immun 1997; 65:2067-73.
91. Wira CR, Fahey JV, Sentman CL, Pioli PA, Shen L. Innate and adaptive
immunity in female genital tract: cellular responses and interactions.
Immunological reviews 2005; 206:306-35.
92. Janeway CA. The immune system evolved to discriminate infectious nonself from
noninfectious self. Immunol Today 1992; 13:11-6.
93. Harding CV BW. Regulation of antigen presentation by Mycobacterium
tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol 2010; 8:296-307.
94. Khan N GU, Pahari S, Agrewala JN. Manipulation of costimulatory molecules by
intracellular pathogens: Veni, Vidi, Vici!!. PLoS pathogens 2012; 8.
95. Jordan JM WM, Olano J, Walker DH. The Absence of Toll-Like Receptor 4
Signaling in C3H/HeJ Mice Predisposes Them to Overwhelming Rickettsial
Infection and Decreased Protective Th1 Responses. Infection and Immunity
2008; 76:3717-37224.
96. Mansueto P VG, Di Lorenzo G, Arcoleo F, Mansueto S, Cillari E. Immunology of
human rickettsial diseases. J Biol Regul Homeost Agents 2008; 22:131-9.
97. Perry LL FK, Caldwell HD. Immunity to Chlamydia trachomatis is mediated by
T helper 1 cells through IFN-gamma dependent and independent pathways. J
Immunol 1997; 158:3344-52.
98. Morrison SG, Su H, Caldwell HD, Morrison RP. Immunity to murine Chlamydia
trachomatis genital tract reinfection involves B cells and CD4(+) T cells but not
CD8(+) T cells. Infect Immun 2000; 68:6979-87.
99. Morrison SG, Morrison RP. A predominant role for antibody in acquired
immunity to chlamydial genital tract reinfection. J Immunol 2005; 175:7536-42.
Page 93
78
100. Yang X, HayGlass KT, Brunham RC. Genetically determined differences in IL-
10 and IFN-gamma responses correlate with clearance of Chlamydia trachomatis
mouse pneumonitis infection. J Immunol 1996; 156:4338-44.
101. Del Río L BA, Sánchez J, Garcés B, Caro MR, Gallego MC, et al. Chlamydophila
abortus (Chlamydia psittaci serotype 1) clearance is associated with the early
recruitment of neutrophils and CD8+ T cell in a mouse model. J Comp Pathol
2000; 132:171-81.
102. Bhardwaj N HF, Moir S. The immune response to HIV Nature Reviews
Immunology, 2012.
103. Chinen J SW. Molecular virology and immunology of HIV infection. J Allergy
Clin Immunol 2002; 110:189-98.
104. Sun J BT. Role of T cell immunity in recovery from influenza virus infection.
Curr Opin Virol 2013.
105. Jr Janeway CA MR. Innate Immune Recognition. Annu Rev Immunol 2002;
20:197-216.
106. Bas S, Neff L, Vuillet M, et al. The proinflammatory cytokine response to
Chlamydia trachomatis elementary bodies in human macrophages is partly
mediated by a lipoprotein, the macrophage infectivity potentiator, through
TLR2/TLR1/TLR6 and CD14. J Immunol 2008; 180:1158-68.
107. Prebeck S, Brade H, Kirschning CJ, et al. The gram-negative bacterium
Chlamydia trachomatis L2 stimulates tumor necrosis factor secretion by innate
immune cells independently of its endotoxin. Microbes and infection / Institut
Pasteur 2003; 5:463-70.
108. Erridge C, Pridmore A, Eley A, Stewart J, Poxton IR. Lipopolysaccharides of
Bacteroides fragilis, Chlamydia trachomatis and Pseudomonas aeruginosa signal
via toll-like receptor 2. J Med Microbiol 2004; 53:735-40.
109. Darville T, O'Neill JM, Andrews CW, Jr., Nagarajan UM, Stahl L, Ojcius DM.
Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of
oviduct pathology in chlamydial genital tract infection. J Immunol 2003;
171:6187-97.
110. Rasmussen SJ, Eckmann L, Quayle AJ, et al. Secretion of proinflammatory
cytokines by epithelial cells in response to Chlamydia infection suggests a central
role for epithelial cells in chlamydial pathogenesis. The Journal of clinical
investigation 1997; 99:77-87.
111. Johnson RM. Murine oviduct epithelial cell cytokine responses to Chlamydia
muridarum infection include interleukin-12-p70 secretion. Infect Immun 2004;
72:3951-60.
Page 94
79
112. Morrison SG, Morrison RP. In situ analysis of the evolution of the primary
immune response in murine Chlamydia trachomatis genital tract infection. Infect
Immun 2000; 68:2870-9.
113. Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. Dissemination of
Chlamydia trachomatis chronic genital tract infection in gamma interferon gene
knockout mice. Infect Immun 1997; 65:2145-52.
114. Ito JI, Lyons JM. Role of gamma interferon in controlling murine chlamydial
genital tract infection. Infect Immun 1999; 67:5518-21.
115. Rank RG SL, Barron AL. Chronic chlamydial genital infection in congenitally
athymic nude mice. Infection and Immunity 1985; 48:847-9.
116. Landers DV EK, Sung M, Schachter J. Role of L3T4-bearing t-cell populations in
experimental murine chlamydial salpingitis. Infection and Immunity 1991;
59:3774-7.
117. Su H CH. CD4+ T cells play a significant role in adoptive immunity to
Chlamydia trachomatis infection of the mouse genital tract. Infection and
Immunity 1995; 63:3302-8.
118. Kelly KA, Rank RG. Identification of homing receptors that mediate the
recruitment of CD4 T cells to the genital tract following intravaginal infection
with Chlamydia trachomatis. Infection and Immunity 1997; 65:5198-208.
119. Hawkins RA, Rank RG, Kelly KA. Expression of mucosal homing receptor
alpha4beta7 is associated with enhanced migration to the Chlamydia-infected
murine genital mucosa in vivo. Infection and Immunity 2000; 68:5587-94.
120. Gondek DC, Olive AJ, Stary G, Starnbach MN. CD4+ T Cells are necessary and
sufficient to confer protection against Chlamydia trachomatis infection in the
murine upper genital tract. J Immunol 2012.
121. Peeling R, Maclean IW, Brunham RC. In vitro neutralization of Chlamydia
trachomatis with monoclonal antibody to an epitope on the major outer
membrane protein. Infection and Immunity 1984; 46:484-8.
122. Batteiger BE, Fraiz J, Newhall WJ, Katz BP, Jones RB. Association of recurrent
chlamydial infection with gonorrhea. The Journal of infectious diseases 1989;
159:661-9.
123. Bell SD, Jr., Nichols RL, Haddad NA. The immunology of the trachoma agent
with a preliminary report on field trials vaccine. Investigative ophthalmology
1963; 2:471-81.
Page 95
80
124. Grayston JT, Wang SP, Yang YF, Woolridge RL. The effect of trachoma virus
vaccine on the course of experimental trachoma infection in blind human
volunteers. The Journal of experimental medicine 1962; 115:1009-22.
125. Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization
of the major outer membrane protein of Chlamydia trachomatis. Infection and
Immunity 1981; 31:1161-76.
126. Farris CM, Morrison RP. Vaccination against Chlamydia genital infection
utilizing the murine C. muridarum model. Infection and Immunity 2011; 79:986-
96.
127. Champion CI, Kickhoefer VA, Liu G, et al. A vault nanoparticle vaccine induces
protective mucosal immunity. PloS one 2009; 4:e5409.
128. Murthy AK, Chambers JP, Meier PA, Zhong G, Arulanandam BP. Intranasal
vaccination with a secreted chlamydial protein enhances resolution of genital
Chlamydia muridarum infection, protects against oviduct pathology, and is highly
dependent upon endogenous gamma interferon production. Infection and
Immunity 2007; 75:666-76.
129. Murthy AK, Li W, Guentzel MN, Zhong G, Arulanandam BP. Vaccination with
the defined chlamydial secreted protein CPAF induces robust protection against
female infertility following repeated genital chlamydial challenge. Vaccine 2011;
29:2519-22.
130. Chen AL JK, Lee JK, Sutterlin C, Tan M. CPAF: A chlamydial protease in search
of an authentic substrate. PLoS pathogens 2012; 8:e1002842.
131. Brunham RC, Rekart ML. Considerations on Chlamydia trachomatis disease
expression. FEMS immunology and medical microbiology 2009; 55:162-6.
132. Brunham RC, Rekart ML. The arrested immunity hypothesis and the
epidemiology of chlamydia control. Sexually transmitted diseases 2008; 35:53-4.
133. Rekart ML, Brunham RC. Epidemiology of chlamydial infection: are we losing
ground? Sexually transmitted infections 2008; 84:87-91.
134. Gottlieb SL, Brunham RC, Byrne GI, Martin DH, Xu F, Berman SM.
Introduction: The natural history and immunobiology of Chlamydia trachomatis
genital infection and implications for Chlamydia control. The Journal of
infectious diseases 2010; 201 Suppl 2:S85-7.
135. Valdic CM MM, Bowden FJ, Fairley CK, Kaldor JM. The prevalence of genital
Chlamydia trachomatis in Australia 1997-2004: a systematic review. Sex Health
2005; 2:169-83.
Page 96
81
136. Chen MY DB. Genital Chlamydia trachomatis infection in Australia:
epidemiology and clincial implications. Sex Health 2004; 1:189-96.
137. Lyytikainen E, Kaasila M, Koskela P, et al. Chlamydia trachomatis
seroprevalence atlas of Finland 1983-2003. Sexually transmitted infections 2008;
84:19-22.
138. Quinn TC, Gaydos C, Shepherd M, et al. Epidemiologic and microbiologic
correlates of Chlamydia trachomatis infection in sexual partnerships. JAMA: the
journal of the American Medical Association 1996; 276:1737-42.
139. Mishori R ME, WinklerPrins V. Chlamydia trachomatis infections: screening,
diagnosis, and management. American family physician 2012; 86:1127-32.
140. Gaydos CA TM, Dalesio N, Wood BJ, Quinn TC. Comparison of three nucleic
acid amplification tests for detection of Chlamydia trachomatis in urine
specimens. Journal of clinical microbiology 2004; 42:3041-5.
141. Watson EJ, Templeton A, Russell I, et al. The accuracy and efficacy of screening
tests for Chlamydia trachomatis: a systematic review. J Med Microbiol 2002;
51:1021-31.
142. Nsuami MJ, Nsa M, Brennan C, Cammarata CL, Martin DH, Taylor SN.
Chlamydia Positivity in New Orleans Public High Schools, 1996-2005:
Implications for Clinical and Public Health Practices. Academic pediatrics 2013.
143. Low N, Forster M, Taylor SN, Nsuami MJ. Repeat Chlamydia screening among
adolescents: cohort study in a school-based programme in New Orleans. Sexually
transmitted infections 2013; 89:20-4.
144. Torrone EA, Geisler WM, Gift TL, Weinstock HS. Chlamydia trachomatis
infection among women 26 to 39 years of age in the United States, 1999 to 2010.
Sexually transmitted diseases 2013; 40:335-7.
145. Beharry MS. Adolescent sexuality. Pediatric annuals 2013; 42:64.
146. Beharry MS, Shafii T, Burstein GR. Diagnosis and treatment of chlamydia,
gonorrhea, and trichomonas in adolescents. Pediatric annals 2013; 42:26-33.
147. Sexually Transmitted Disease Surveillance Report. Atlanta Centers for Disease
Control and Prevention (CDC), 2012.
148. Geisler WM, Tang J, Wang C, Wilson CM, Kaslow RA. Epidemiological and
genetic correlates of incident Chlamydia trachomatis infection in North American
adolescents. The Journal of infectious diseases 2004; 190:1723-9.
Page 97
82
149. Sznitman S, Stanton BF, Vanable PA, et al. Long term effects of community-
based STI screening and mass media HIV prevention messages on sexual risk
behaviors of African American adolescents. AIDS and behavior 2011; 15:1755-
63.
150. Dean HD, Fenton KA. Addressing social determinants of health in the prevention
and control of HIV/AIDS, viral hepatitis, sexually transmitted infections, and
tuberculosis. Public Health Rep 2010; 125 Suppl 4:1-5.
151. Hoffman S, Beckford Jarrett ST, Kelvin EA, et al. HIV and sexually transmitted
infection risk behaviors and beliefs among Black West Indian immigrants and
US-born Blacks. American journal of public health 2008; 98:2042-50.
152. Hogben M, Leichliter JS. Social determinants and sexually transmitted disease
disparities. Sexually transmitted diseases 2008; 35:S13-8.
153. Kaplan MS, Crespo CJ, Huguet N, Marks G. Ethnic/racial homogeneity and
sexually transmitted disease: a study of 77 Chicago community areas. Sexually
transmitted diseases 2009; 36:108-11.
154. Nguyen TQ, Ford CA, Kaufman JS, Leone PA, Suchindran C, Miller WC.
Infrequent chlamydial testing among young adults: financial and regional
differences. Sexually transmitted diseases 2008; 35:725-30.
155. Burckhardt F, Warner P, Young H. What is the impact of change in diagnostic
test method on surveillance data trends in Chlamydia trachomatis infection?
Sexually transmitted infections 2006; 82:24-30.
156. Stephenson J. Rise in drug-resistant gonorrhea cases: spurs new treatment advice
for gay, bisexual men. JAMA: the journal of the American Medical Association
2004; 291:2420.
157. Ohnishi M, Saika T, Hoshina S, et al. Ceftriaxone-resistant Neisseria
gonorrhoeae, Japan. Emerging infectious diseases 2011; 17:148-9.
158. Stamm LV. Global challenge of antibiotic-resistant Treponema pallidum.
Antimicrobial agents and chemotherapy 2010; 54:583-9.
159. Sandoz KM, Rockey DD. Antibiotic resistance in Chlamydiae. Future
microbiology 2010; 5:1427-42.
160. Nicoll A, Hughes G, Donnelly M, et al. Assessing the impact of national anti-HIV
sexual health campaigns: trends in the transmission of HIV and other sexually
transmitted infections in England. Sexually transmitted infections 2001; 77:242-7.
161. Pisani E, Lazzari S, Walker N, Schwartlander B. HIV surveillance: a global
perspective. J Acquir Immune Defic Syndr 2003; 32 Suppl 1:S3-11.
Page 98
83
162. Su H, Morrison R, Messer R, Whitmire W, Hughes S, Caldwell HD. The effect of
doxycycline treatment on the development of protective immunity in a murine
model of chlamydial genital infection. The Journal of infectious diseases 1999;
180:1252-8.
163. Brunham RC, Pourbohloul B, Mak S, White R, Rekart ML. The unexpected
impact of a Chlamydia trachomatis infection control program on susceptibility to
reinfection. The Journal of infectious diseases 2005; 192:1836-44.
164. Molano M, Meijer CJ, Weiderpass E, et al. The natural course of Chlamydia
trachomatis infection in asymptomatic Colombian women: a 5-year follow-up
study. The Journal of infectious diseases 2005; 191:907-16.
165. Gonzales GF, Munoz G, Sanchez R, et al. Update on the impact of Chlamydia
trachomatis infection on male fertility. Andrologia 2004; 36:1-23.
166. Stamm WE. Chlamydia trachomatis infections: progress and problems. The
Journal of infectious diseases 1999; 179 Suppl 2:S380-3.
167. Schautteet K, Stuyven E, Cox E, Vanrompay D. Validation of the Chlamydia
trachomatis genital challenge pig model for testing recombinant protein vaccines.
J Med Microbiol 2011; 60:117-27.
168. Patton DL, Moore DE, Spadoni LR, Soules MR, Halbert SA, Wang SP. A
comparison of the fallopian tube's response to overt and silent salpingitis.
Obstetrics and gynecology 1989; 73:622-30.
169. Baud D, Goy G, Jaton K, et al. Role of Chlamydia trachomatis in miscarriage.
Emerging infectious diseases 2011; 17:1630-5.
170. Haggerty CL, Gottlieb SL, Taylor BD, Low N, Xu F, Ness RB. Risk of sequelae
after Chlamydia trachomatis genital infection in women. The Journal of
infectious diseases 2010; 201 Suppl 2:S134-55.
171. Fleming DT, Wasserheit JN. From epidemiological synergy to public health
policy and practice: the contribution of other sexually transmitted diseases to
sexual transmission of HIV infection. Sexually transmitted infections 1999; 75:3-
17.
172. Westrom L, Joesoef R, Reynolds G, Hagdu A, Thompson SE. Pelvic
inflammatory disease and fertility. A cohort study of 1,844 women with
laparoscopically verified disease and 657 control women with normal
laparoscopic results. Sexually transmitted diseases 1992; 19:185-92.
173. Gottlieb SL, Berman SM, Low N. Screening and treatment to prevent sequelae in
women with Chlamydia trachomatis genital infection: how much do we know?
The Journal of infectious diseases 2010; 201 Suppl 2:S156-67.
Page 99
84
174. Lau CY, Qureshi AK. Azithromycin versus doxycycline for genital chlamydial
infections: a meta-analysis of randomized clinical trials. Sexually transmitted
diseases 2002; 29:497-502.
175. Miller KE. Diagnosis and treatment of Chlamydia trachomatis infection.
American family physician 2006; 73:1411-6.
176. Bakken IJ, Skjeldestad FE, Lydersen S, Nordbo SA. Births and ectopic
pregnancies in a large cohort of women tested for Chlamydia trachomatis.
Sexually transmitted diseases 2007; 34:739-43.
177. Hillis SD, Owens LM, Marchbanks PA, Amsterdam LF, Mac Kenzie WR.
Recurrent chlamydial infections increase the risks of hospitalization for ectopic
pregnancy and pelvic inflammatory disease. American journal of obstetrics and
gynecology 1997; 176:103-7.
178. Patton DL, Kuo CC. Histopathology of Chlamydia trachomatis salpingitis after
primary and repeated reinfections in the monkey subcutaneous pocket model.
Journal of reproduction and fertility 1989; 85:647-56.
179. Kimani J, Maclean IW, Bwayo JJ, et al. Risk factors for Chlamydia trachomatis
pelvic inflammatory disease among sex workers in Nairobi, Kenya. The Journal
of infectious diseases 1996; 173:1437-44.
180. Swenson CE, Sung ML, Schachter J. The effect of tetracycline treatment on
chlamydial salpingitis and subsequent fertility in the mouse. Sexually transmitted
diseases 1986; 13:40-4.
181. Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary
protective role for major histocompatibility complex class II-restricted responses
in Chlamydia trachomatis genital tract infection. Infect Immun 1995; 63:4661-8.
182. Rank RG, Whittum-Hudson JA. Protective immunity to chlamydial genital
infection: evidence from animal studies. The Journal of infectious diseases 2010;
201 Suppl 2:S168-77.
183. Cain TK RR. Local Th1-like responses are induced by intravaginal infection of
mice with the mouse pneumonitis biovar of Chlamydia trachomatis. Infection and
Immunity 1995; 63:1784-9.
184. Morrison RP, Caldwell HD. Immunity to murine chlamydial genital infection.
Infection and Immunity 2002; 70:2741-51.
185. Batteiger BE, Tu W, Ofner S, et al. Repeated Chlamydia trachomatis genital
infections in adolescent women. The Journal of infectious diseases 2010; 201:42-
51.
Page 100
85
186. Whittington WL, Kent C, Kissinger P, et al. Determinants of persistent and
recurrent Chlamydia trachomatis infection in young women: results of a
multicenter cohort study. Sexually transmitted diseases 2001; 28:117-23.
187. Lyons JM, Ito JI, Jr., Pena AS, Morre SA. Differences in growth characteristics
and elementary body associated cytotoxicity between Chlamydia trachomatis
oculogenital serovars D and H and Chlamydia muridarum. Journal of clinical
pathology 2005; 58:397-401.
188. Gottlieb SL, Martin DH, Xu F, Byrne GI, Brunham RC. Summary: The natural
history and immunobiology of Chlamydia trachomatis genital infection and
implications for Chlamydia control. The Journal of infectious diseases 2010; 201
Suppl 2:S190-204.
189. Miyairi I, Laxton JD, Wang X, et al. Chlamydia psittaci genetic variants differ in
virulence by modulation of host immunity. The Journal of infectious diseases
2011; 204:654-63.
190. Miyairi I, Tatireddigari VR, Mahdi OS, et al. The p47 GTPases Iigp2 and Irgb10
regulate innate immunity and inflammation to murine Chlamydia psittaci
infection. J Immunol 2007; 179:1814-24.
191. Frazer LC, O'Connell CM, Andrews CW, Jr., Zurenski MA, Darville T. Enhanced
neutrophil longevity and recruitment contribute to the severity of oviduct
pathology during Chlamydia muridarum infection. Infect Immun 2011; 79:4029-
41.
192. Darville T, Hiltke TJ. Pathogenesis of genital tract disease due to Chlamydia
trachomatis. The Journal of infectious diseases 2010; 201 Suppl 2:S114-25.
193. Ramsey KH, Sigar IM, Schripsema JH, Shaba N, Cohoon KP. Expression of
matrix metalloproteinases subsequent to urogenital Chlamydia muridarum
infection of mice. Infect Immun 2005; 73:6962-73.
194. Holland MJ, Bailey RL, Conway DJ, et al. T helper type-1 (Th1)/Th2 profiles of
peripheral blood mononuclear cells (PBMC); responses to antigens of Chlamydia
trachomatis in subjects with severe trachomatous scarring. Clinical and
experimental immunology 1996; 105:429-35.
195. Vicetti Miguel RD, Harvey SA, LaFramboise WA, Reighard SD, Matthews DB,
Cherpes TL. Human female genital tract infection by the obligate intracellular
bacterium Chlamydia trachomatis elicits robust Type 2 immunity. PloS one 2013;
8:e58565.
196. Mann CJ, Perdiguero E, Kharraz Y, et al. Aberrant repair and fibrosis
development in skeletal muscle. Skeletal muscle 2011; 1:21.
Page 101
86
197. Serrano AL, Mann CJ, Vidal B, Ardite E, Perdiguero E, Munoz-Canoves P.
Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair
and disease. Current topics in developmental biology 2011; 96:167-201.
198. Van Dyken SJ, Locksley RM. Interleukin-4 and interleukin-13-mediated
alternatively activated macrophages: roles in homeostasis and disease. Annual
review of immunology 2013; 31:317-43.
199. Molofsky AB, Nussbaum JC, Liang HE, et al. Innate lymphoid type 2 cells
sustain visceral adipose tissue eosinophils and alternatively activated
macrophages. The Journal of experimental medicine 2013; 210:535-49.
200. Pulendran B, Artis D. New paradigms in type 2 immunity. Science 2012;
337:431-5.
201. Safrin S, Schachter J, Dahrouge D, Sweet RL. Long-term sequelae of acute pelvic
inflammatory disease: A retrospective cohort study. American journal of
obstetrics and gynecology 1992; 166:1300-5.
202. Lepine LA, Hillis SD, Marchbanks PA, Joesoef MR, Peterson HB, Westrom L.
Severity of pelvic inflammatory disease as a predictor of the probability of live
birth. American journal of obstetrics and gynecology 1998; 178:977-81.
203. Stacey CM, Munday PE, Taylor-Robinson D, et al. A longitudinal study of pelvic
inflammatory disease. British journal of obstetrics and gynaecology 1992; 99:994-
9.
204. Buchan H, Vessey M, Goldacre M, Fairweather J. Morbidity following pelvic
inflammatory disease. British journal of obstetrics and gynaecology 1993;
100:558-62.
205. McCormack WM. Pelvic inflammatory disease. The New England journal of
medicine 1994; 330:115-9.
206. Brunham RC, Binns B, Guijon F, et al. Etiology and outcome of acute pelvic
inflammatory disease. The Journal of infectious diseases 1988; 158:510-7.
207. Pal S, Peterson EM, de la Maza LM. Susceptibility of mice to vaginal infection
with Chlamydia trachomatis mouse pneumonitis is dependent on the age of the
animal. Infection and Immunity 2001; 69:5203-6.
208. Bortz WM. Biological basis of determinants of health. American journal of public
health 2005; 95:389-92.
209. U.S. Department of Health and Human Services. Office of Disease Prevention
and Health Promotion. Healthy People 2020 Available at:
http://www.healthypeople.gov/2020/about/DOHAbout.aspx#biology. Accessed
June 26 2013.
Page 102
87
210. Tuffrey M, Alexander F, Woods C, Taylor-Robinson D. Genetic susceptibility to
chlamydial salpingitis and subsequent infertility in mice. Journal of reproduction
and fertility 1992; 95:31-8.
211. Qiu H, Yang J, Bai H, et al. Less inhibition of interferon-gamma to organism
growth in host cells may contribute to the high susceptibility of C3H mice to
Chlamydia trachomatis lung infection. Immunology 2004; 111:453-61.
212. Qiu H, Wang S, Yang J, et al. Resistance to chlamydial lung infection is
dependent on major histocompatibility complex as well as non-major
histocompatibility complex determinants. Immunology 2005; 116:499-506.
213. Darville T, Andrews CW, Jr., Sikes JD, Fraley PL, Braswell L, Rank RG. Mouse
strain-dependent chemokine regulation of the genital tract T helper cell type 1
immune response. Infection and Immunity 2001; 69:7419-24.
214. Darville T, Andrews CW, Jr., Sikes JD, Fraley PL, Rank RG. Early local cytokine
profiles in strains of mice with different outcomes from chlamydial genital tract
infection. Infection and Immunity 2001; 69:3556-61.
215. Buzoni-Gatel D, Bernard F, Pla M, Rodolakis A, Lantier F. Role of H-2 and non-
H-2-related genes in mouse susceptibility to Chlamydia psittaci. Microbial
pathogenesis 1994; 16:229-33.
216. Bernstein-Hanley I, Balsara ZR, Ulmer W, Coers J, Starnbach MN, Dietrich WF.
Genetic analysis of susceptibility to Chlamydia trachomatis in mouse. Genes and
immunity 2006; 7:122-9.
217. Bernstein-Hanley I, Coers J, Balsara ZR, Taylor GA, Starnbach MN, Dietrich
WF. The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis
susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proceedings of
the National Academy of Sciences of the United States of America 2006;
103:14092-7.
218. Oh MK, Cloud GA, Fleenor M, Sturdevant MS, Nesmith JD, Feinstein RA. Risk
for gonococcal and chlamydial cervicitis in adolescent females: incidence and
recurrence in a prospective cohort study. The Journal of adolescent health :
official publication of the Society for Adolescent Medicine 1996; 18:270-5.
219. Scott Lamontagne D, Baster K, Emmett L, et al. Incidence and reinfection rates of
genital chlamydial infection among women aged 16-24 years attending general
practice, family planning and genitourinary medicine clinics in England: a
prospective cohort study by the Chlamydia Recall Study Advisory Group.
Sexually transmitted infections 2007; 83:292-303.
220. Tuffrey M GF, Quinn R, Taylor-Robinson D. Infertility in mice infected genitally
with a human strain of Chlamydia trachomatis. Journal of Reproduction &
Fertility 1986; 78:251-60.
Page 103
88
221. Huang J, DeGraves FJ, Lenz SD, et al. The quantity of nitric oxide released by
macrophages regulates Chlamydia-induced disease. Proceedings of the National
Academy of Sciences of the United States of America 2002; 99:3914-9.
222. Miller WC, Ford CA, Morris M, et al. Prevalence of chlamydial and gonococcal
infections among young adults in the United States. JAMA: the journal of the
American Medical Association 2004; 291:2229-36.
223. LaMontagne DS, Fine DN, Marrazzo JM. Chlamydia trachomatis infection in
asymptomatic men. American journal of preventive medicine 2003; 24:36-42.
224. Jones RB, Van der Pol B, Martin DH, Shepard MK. Partial characterization of
Chlamydia trachomatis isolates resistant to multiple antibiotics. The Journal of
infectious diseases 1990; 162:1309-15.
225. Lefevre JC, Lepargneur JP. Comparative in vitro susceptibility of a tetracycline-
resistant Chlamydia trachomatis strain isolated in Toulouse (France). Sexually
transmitted diseases 1998; 25:350-2.
226. Somani J, Bhullar VB, Workowski KA, Farshy CE, Black CM. Multiple drug-
resistant Chlamydia trachomatis associated with clinical treatment failure. The
Journal of infectious diseases 2000; 181:1421-7.
227. Lenart J, Andersen AA, Rockey DD. Growth and development of tetracycline-
resistant Chlamydia suis. Antimicrobial agents and chemotherapy 2001; 45:2198-
203.
228. Rockey DD, Wang J, Lei L, Zhong G. Chlamydia vaccine candidates and tools
for chlamydial antigen discovery. Expert review of vaccines 2009; 8:1365-77.
229. Grayston JT, Wang SP, Yeh LJ, Kuo CC. Importance of reinfection in the
pathogenesis of trachoma. Reviews of infectious diseases 1985; 7:717-25.
230. Igietseme JU, Black CM, Caldwell HD. Chlamydia vaccines: strategies and
status. BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene
therapy 2002; 16:19-35.
231. de la Maza LM, Peterson EM. Vaccines for Chlamydia trachomatis infections.
Curr Opin Investig Drugs 2002; 3:980-6.
232. Campos M, Pal S, O'Brien TP, Taylor HR, Prendergast RA, Whittum-Hudson JA.
A chlamydial major outer membrane protein extract as a trachoma vaccine
candidate. Investigative ophthalmology & visual science 1995; 36:1477-91.
233. Taylor HR, Whittum-Hudson J, Schachter J, Caldwell HD, Prendergast RA. Oral
immunization with chlamydial major outer membrane protein (MOMP).
Investigative ophthalmology & visual science 1988; 29:1847-53.
Page 104
89
234. Pal S, Theodor I, Peterson EM, de la Maza LM. Immunization with an acellular
vaccine consisting of the outer membrane complex of Chlamydia trachomatis
induces protection against a genital challenge. Infect Immun 1997; 65:3361-9.
235. Pal S, Barnhart KM, Wei Q, Abai AM, Peterson EM, de la Maza LM.
Vaccination of mice with DNA plasmids coding for the Chlamydia trachomatis
major outer membrane protein elicits an immune response but fails to protect
against a genital challenge. Vaccine 1999; 17:459-65.
236. Zhang DJ, Yang X, Shen C, Brunham RC. Characterization of immune responses
following intramuscular DNA immunization with the MOMP gene of Chlamydia
trachomatis mouse pneumonitis strain. Immunology 1999; 96:314-21.
237. Cunningham KA, Carey AJ, Hafner L, Timms P, Beagley KW. Chlamydia
muridarum major outer membrane protein-specific antibodies inhibit in vitro
infection but enhance pathology in vivo. Am J Reprod Immunol 2011; 65:118-26.
238. Igietseme JU, Murdin A. Induction of protective immunity against Chlamydia
trachomatis genital infection by a vaccine based on major outer membrane
protein-lipophilic immune response-stimulating complexes. Infection and
Immunity 2000; 68:6798-806.
239. Cheng C, Bettahi I, Cruz-Fisher MI, et al. Induction of protective immunity by
vaccination against Chlamydia trachomatis using the major outer membrane
protein adjuvanted with CpG oligodeoxynucleotide coupled to the nontoxic B
subunit of cholera toxin. Vaccine 2009; 27:6239-46.
240. Pal S, Theodor I, Peterson EM, de la Maza LM. Immunization with the
Chlamydia trachomatis mouse pneumonitis major outer membrane protein can
elicit a protective immune response against a genital challenge. Infection and
Immunity 2001; 69:6240-7.
241. Su H, Parnell M, Caldwell HD. Protective efficacy of a parenterally administered
MOMP-derived synthetic oligopeptide vaccine in a murine model of Chlamydia
trachomatis genital tract infection: serum neutralizing IgG antibodies do not
protect against chlamydial genital tract infection. Vaccine 1995; 13:1023-32.
242. Byrne GI, Faubion CL. Lymphokine-mediated microbistatic mechanisms restrict
Chlamydia psittaci growth in macrophages. J Immunol 1982; 128:469-74.
243. Byrne GI, Faubion CL. Inhibition of Chlamydia psittaci in oxidatively active
thioglycolate-elicited macrophages: distinction between lymphokine-mediated
oxygen-dependent and oxygen-independent macrophage activation. Infection and
Immunity 1983; 40:464-71.
Page 105
90
244. Byrne GI, Murray HW. Lymphokine stimulated macrophages inhibit intracellular
Chlamydia psittaci replication by mechanisms distinct from intracellular
inhibition of Toxoplasma gondii replication. Advances in experimental medicine
and biology 1983; 162:361-4.
245. Morre SA, van den Brule AJ, Rozendaal L, et al. The natural course of
asymptomatic Chlamydia trachomatis infections: 45% clearance and no
development of clinical PID after one-year follow-up. International journal of
STD & AIDS 2002; 13 Suppl 2:12-8.
246. Sheffield JS, Andrews WW, Klebanoff MA, et al. Spontaneous resolution of
asymptomatic Chlamydia trachomatis in pregnancy. Obstetrics and gynecology
2005; 105:557-62.
247. Tritto E, Mosca F, De Gregorio E. Mechanism of action of licensed vaccine
adjuvants. Vaccine 2009; 27:3331-4.
248. Woolridge RL, Grayston JT, Chang IH, Yang CY, Cheng KH. Long-term follow-
up of the initial (1959-1960) trachoma vaccine field trial on Taiwan. American
journal of ophthalmology 1967; 63:Suppl:1650-5.
249. de Oliveira Fraga LA, Torrero MN, Tocheva AS, Mitre E, Davies SJ. Induction of
type 2 responses by schistosome worms during prepatent infection. The Journal of
infectious diseases 2010; 201:464-72.
250. Cheever AW, Lenzi JA, Lenzi HL, Andrade ZA. Experimental models of
Schistosoma mansoni infection. Memorias do Instituto Oswaldo Cruz 2002;
97:917-40.
Page 106
91
APPENDIX A. MORBIDITY AND PATHOLOGY SEVERITY POST-
PRIMARY INFECTION IN DEFERRED TREATMENT STUDIES
Note: C57BL/6 female mice were purchased from Jackson Laboratories at 7-8 weeks of
age. Animals were subcutaneously treated with progesterone at days 14 and 7 per
intravaginal challenge. On day 0, animals were intravaginally challenged with 2 x 103
IFU C. muridarum. UN refers to animals that did not receive treatment during the
courseo f the experiment. (0-14) represents animals that were intraperitoneally treated
with doxycycline starting at day 0 for 14 days consecutively. (2-16) represents animals
that were intraperitoneally treated with doxycycline starting 2 days post primary
challenge for 14 days consecutively. (4-18) represents animals that were intraperitoneally
treated with doxycycline starting on day 4 post primary challenge for 14 days
consecutively. Normal represents negative the negative control group which neither
received treatment nor intravaginal Chlamydia muridarum challenge. Animals were
sacrificed 56 post-primary infection.
Page 107
92
APPENDIX B. MORBIDITY AND PATHOLOGY SEVERITY POST-
SECONDARY INFECTION IN DEFERRED TREATMENT STUDIES
Note: C57BL/6 female mice were purchased from Jackson Laboratories at 7-8 weeks of
age. Animals were subcutaneously treated with progesterone at days 14 and 7 per
intravaginal challenge. On day 0, animals were intravaginally challenged with 2 x 103
IFU C. muridarum. UN refers to animals that did not receive treatment during the course
of the experiment. (0-14) represents animals that were intraperitoneally treated with
doxycycline starting at day 0 for 14 days consecutively. (2-16) represents animals that
were intraperitoneally treated with doxycycline starting 2 days post primary challenge for
14 days consecutively. (4-18) represents animals that were intraperitoneally treated with
doxycycline starting on day 4 post primary challenge for 14 days consecutively. Normal
represents negative the negative control group which neither received treatment nor
intravaginal Chlamydia muridarum challenge. Animals were sacrificed 56 post-
secondary infection.
Page 108
93
APPENDIX C. TH1-RELATED CYOKINES POST RECURRENT
INFECTION IN ARRESTED IMMUNITY MODEL
Page 109
94
APPENDIX D. TH2-RELATED CYTOKINES POST RECURRENT
INFECTION IN ARRESTED IMMUNITY MODEL
Page 110
95
VITA
Enitra N. Jones was born in Houston, Texas in 1983, the daughter of Ezra C.
Jones and Evelyn T. Jones. In 2001, she graduated from West Jefferson High School of
Harvey, Louisiana in the top three percent of her class. In July 2005, Enitra earned her
Bachelor of Science degree in Biology with a concentration in Microbiology from the
Honors College at Southern University and Agricultural and Mechanical College in
Baton Rouge, Louisiana (Cum Laude Latin Honors). In 2006, Enitra enrolled in the
Integrated Program of Biomedical Sciences at the University of Tennessee Health
Science Center in Memphis, Tennessee. She graduated in 2013 with a Doctorate of
Philosophy in Microbial Pathogenesis, Immunology, and Inflammation. Enitra is
currently a two-year American Public Health Laboratories (APHL)/ Centers for Disease
Control and Prevention (CDC) Emerging Infectious Diseases (EID) Postdoctoral
Research Fellow in Atlanta, Georgia.