Revista Peruana de Pediatría Publicación Oficial de la Sociedad Peruana de Pediatría La Revista Peruana de Pediatría, publicación oficial de difusión científica de la Sociedad Peruana de Pediatría desde 1942, tiene como objetivo la publicación de la producción científica en el contexto pediátrico en todas las áreas, especialmente en trabajos de investigación con el fin de contribuir a mejorar la calidad de atención del paciente pediátrico en el país. Además propicia el intercambio de información con entidades similares en el país y en el extranjero, a fin de promover el avance y la aplicación de la investigación en salud. Los artículos son arbitrados por pares. Está prohibida la reproducción total o parcial de los artículos publicados en esta revista, solo se hará mediante autorización del Consejo Editorial. El contenido de cada artículo es de responsabilidad exclusiva del autor o autores y no compromete la opinión de la revista. Rev. peru. pediatr. 67 (1) 2014 ISSN 1993-6826 FUNDADOR Dr. Carlos Krumdieck Evin DIRECTOR HONORARIO Dr. Manuel Ytahashi Córdova DIRECTOR Dr. Rafael Gustin García Médico Pediatra del Grupo Pediátrico. COMITÉ EDITORIAL Dr. Luis Huicho Oriundo Médico Pediatra del Instituto Nacional de Salud del Niño. Magister y Doctor en Medicina. Profesor de Pediatría de la Universidad Nacional Mayor de San Marcos y Universidad Peruana Cayetano Heredia. Dra. Theresa Ochoa Woodell Médico Infectólogo Pediatra. Profesor Auxiliar de Pediatría de la Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Profesor Asociado de Epidemiología en la Escuela de Salud Pública de la Universidad de Texas Houston. Dr. Miguel Chávez Pastor Médico Endocrinólogo Pediatra y Genetista del Instituto Nacional de Salud del Niño. Profesor Asociado del Departamento Académico de Ciencias Morfológicas de la Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Dra. Virginia Garycochea Cannon Médico Pediatra del Instituto Nacional de Salud del Niño. Profesor Auxilar de Pediatría de la Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Dr. Carlos Álamo Solís Médico Pediatra del Instituto Nacional de Salud del Niño. Maestría en Medicina. Profesor de Pediatría en la Universidad de San Martín de Porres. Jefe de la Unidad de Diseño y Elaboración de Proyectos de Investigación del Instituto Nacional de Salud del Niño. Dr. Raúl Rojas Galarza Médico Pediatra del Instituto Nacional de Salud del Niño. Magister en Salud Pública con Mención en Gestión Hospitalaria. Auditor Médico. Diplomatura en Salud Ocupacional y Seguridad en el Trabajo. Docente del Diplomado de Auditoría Médica Basada en la Evidencia (Universidad Peruana de Ciencias Aplicadas). Revisor Cochrane. Miembro del Critical Appraisal Skills Programme (CASP) - Perú. Dr. Daniel Torres Vela Médico Pediatra del Instituto Nacional de Salud del Niño. Maestría en Salud Pública. Secretario del Comité de Bioética Clínico del Instituto Nacional de Salud del Niño. Dr. Carlos Gonzáles Saravia Médico Pediatra del Instituto nacional de Salud del Niño. Magister en Nutrición Clínica. Miembro del Comité de Evaluación, Monitoreo y Asesoría de Proyectos de Investigación y Asesor en Valoración y Elaboración de Guías de Práctica Clínica del Instituto Nacional de Salud del Niño.
Transcript
1
Revista Peruana de PediatríaPublicación Oficial de la Sociedad Peruana de Pediatría
La Revista Peruana de Pediatría, publicación oficial de difusión científica de la Sociedad Peruana de Pediatría desde 1942, tiene como objetivo la publicación de la producción científica en el contexto pediátrico en todas las áreas, especialmente en trabajos de investigación con el fin de contribuir a mejorar la calidad de atención del paciente pediátrico en el país. Además propicia el intercambio de información con entidades similares en el país y en el extranjero, a fin de promover el avance y la aplicación de la investigación en salud. Los artículos son arbitrados por pares. Está prohibida la reproducción total o parcial de los artículos publicados en esta revista, solo se hará mediante autorización del Consejo Editorial. El contenido de cada artículo es de responsabilidad exclusiva del autor o autores y no compromete la opinión de la revista.
Rev. peru. pediatr. 67 (1) 2014
ISSN 1993-6826
FUNDADOR Dr. Carlos Krumdieck Evin
DIRECTOR HONORARIO Dr. Manuel Ytahashi Córdova
DIRECTOR Dr. Rafael Gustin García Médico Pediatra del Grupo Pediátrico. COMITÉ EDITORIAL Dr. Luis Huicho Oriundo Médico Pediatra del Instituto Nacional de Salud del Niño. Magister y
Doctor en Medicina. Profesor de Pediatría de la Universidad Nacional Mayor de San Marcos y Universidad Peruana Cayetano Heredia.
Dra. Theresa Ochoa Woodell
Médico Infectólogo Pediatra. Profesor Auxiliar de Pediatría de la Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Profesor Asociado de Epidemiología en la Escuela de Salud Pública de la Universidad de Texas Houston.
Dr. Miguel Chávez PastorMédico Endocrinólogo Pediatra y Genetista del Instituto Nacional de Salud del Niño. Profesor Asociado del Departamento Académico de Ciencias Morfológicas de la Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia.
Dra. Virginia Garycochea CannonMédico Pediatra del Instituto Nacional de Salud del Niño. Profesor Auxilar de Pediatría de la Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia.
Dr. Carlos Álamo SolísMédico Pediatra del Instituto Nacional de Salud del Niño. Maestría en Medicina. Profesor de Pediatría en la Universidad de San Martín de Porres. Jefe de la Unidad de Diseño y Elaboración de Proyectos de Investigación del Instituto Nacional de Salud del Niño.
Dr. Raúl Rojas GalarzaMédico Pediatra del Instituto Nacional de Salud del Niño. Magister en Salud Pública con Mención en Gestión Hospitalaria. Auditor Médico. Diplomatura en Salud Ocupacional y Seguridad en el Trabajo. Docente del Diplomado de Auditoría Médica Basada en la Evidencia (Universidad Peruana de Ciencias Aplicadas). Revisor Cochrane. Miembro del Critical Appraisal Skills Programme (CASP) - Perú.
Dr. Daniel Torres VelaMédico Pediatra del Instituto Nacional de Salud del Niño. Maestría en Salud Pública. Secretario del Comité de Bioética Clínico del Instituto Nacional de Salud del Niño.
Dr. Carlos Gonzáles SaraviaMédico Pediatra del Instituto nacional de Salud del Niño. Magister en Nutrición Clínica. Miembro del Comité de Evaluación, Monitoreo y Asesoría de Proyectos de Investigación y Asesor en Valoración y Elaboración de Guías de Práctica Clínica del Instituto Nacional de Salud del Niño.
2
COMITÉ HONORARIO Dr. Melitón Arce Rodríguez Dr. Alejandro Castro Franco Dr. Carlos Manuel Díaz Rodríguez Dra. Rosa Elvira Jiménez La Rosa Dra. Rebeca Kuniyoshi Kiyan Dr. Eduardo Balbín Luna Victoria Dr. Roberto Rivero Quiroz Dra. Lidia Hernández Barba
COMITÉ CONSULTIVO Dra. Nelly Valenzuela Espejo Médico Pediatra Nefróloga. Magíster en Salud Pública Universidad Nacional Federico Villarreal. Dr. José Tantaleán Da Fieno Médico Pediatra Intensivista del Instituto Nacional de Salud del Niño (INSN). Magíster en Medicina Universidad Nacional Federico Villarreal. Dra. Elsa Chea Woo Médico Pediatra. Magíster en Medicina Universidad Peruana Cayetano Heredia. Dra. Angela Castillo Vilela Médico Pediatra del Hospital Nacional Guillermo Almenara Irigoyen.
Magíster en Medicina.
Dr. Juan Francisco Rivera Medina Médico Pediatra Gastroenterólogo del Instituto Nacional de Salud del
Niño. Magister en Nutrición Infantil. Dirección Los Geranios 151, Lince. Lima 14, Perú. Teléfono: 51-1-4411570 Telefax: 51-1-4226397
Hecho el Depósito Legal en la Biblioteca Nacional del Perú Nº 2007-01985
3
SOCIEDAD PERUANA DE PEDIATRÍACARGOS DIRECTIVOS
JUNTA DIRECTIVA
Presidente Dr. José Alberto Tantaleán Da FienoVicepresidenta Dra. Sarah María Vega SanchezSecretario General Dr. Abel Hernán Salinas RivasSecretario de Actas Dr. Oscar Alfredo Aller FernandiniTesorero Dr. José Luis Wong Mayuri Secretaria de Acción Científica Dra. Claudia María Ugarte TaboadaPro Secretarios de Acción Científica Dra. María Esther Castillo Díaz Dr. Mario Humberto Encinas AranaSecretario de Publicaciones y Biblioteca Dr. Rafael Gustin GarcíaVocales de Ética y Calificación Profesional Dra. Angela Castillo Vilela Dra. Carmen Manuela Risco Denegri Dra. Giovanna Alida Punis ReyesPast-Presidenta Dra. Carmen Rosa Maldonado FaverónSecretario de Filiales Dr. Ildauro Aguirre Sosa
COORDINADORES DE CAPÍTULOS
Capítulo de Alergia e Inmunología Dr. José Gereda SolariCapítulo de Cardiología Dra. María Lapoint MontesCapítulo de Endocrinología Dra. Cecilia Bonilla SuárezCapítulo de Gastroenterología, Hepatología y Nutrición Dra. Janetliz Cucho JuradoCapítulo de Infectología Dra. Olguita Del Aguila Del AguilaCapítulo de Medicina del Adolescente Dra. Rosario Del Solar PonceCapítulo de Neumología Dra. Lucrecia Monsante CarrilloCapítulo de Neonatología Dra. Carmen Dávila AliagaCapítulo de Neuropediatría Dra. Inés Caro KahnCapítulo de Pediatría Ambulatoria Dra. Emma Obeso LedesmaCapítulo de Oncología Dra. Clara Pérez SamitierCapítulo de Terapia Intensiva Dra. Rosa Arana SunoharaCapítulo de Pediatría Social Dra. María Del Carmen Calle Dávila
FILIALES
Arequipa Dra. Maritza Ramos MedinaAyacucho Dr. Juan Rondinelli ZagaCajamarca Dr. Rubén Alvarado RevoredoChiclayo Dra. Carmen Gutiérrez GutiérrezChimbote Dra. Carmen Ormaeche MacassiCusco Dr. Jimmy Borja CastroHuancayo Dr. Fernando Gonzáles VivasIca Dr. Otto Chuy CastroIquitos Dr. Jhonny Flores ChávezJuliaca Dra. Licelly Aguilar ZuñigaPiura Dr. Ricardo Olea ZapataTacna Dra. Lourdes Lombardi BacigalupoTarapoto Dra. Mirtha Huayanca NavarroTrujillo Dra. Herminia Lázaro RodriguezUcayali Dr. Óscar Mendoza Sevilla
4
Revista Peruana de PediatríaPublicación Oficial de la Sociedad Peruana de Pediatría
Rev. peru. pediatr. 67 (1) 2014ISSN 1993-6826
CONTENIDO
EDITORIAL La Educación Médica en el Perú: ¿Quién es el responsable de los futuros pediatras en el país?Alejandro Bermúdez García.
TRABAJOS ORIGINALES Estudio Clínico Epidemiológico de la Fibrosis Quística en el Instituto Nacional de Salud del Niño, Lima 2002-2011. Daniel Torres Vela, Hernán Del Castillo Barrientos, Carol Flores Fernández, Amparo Zavaleta Pesantes.
Edad de Diagnóstico Clínico del Hipotiroideo Congénito: Veinte Años Después. Natalia Tovar Gutiérrez, Alex Rojas Hernández, Fiorella Torres Matta, Rubén Susaya Campos, Carlos Del Águila Villar, Juan Manuel Falen Boggio.
Raquitismo Renal Hipofosfatémico Experiencia en 10 Años. Graciela Sakihara Asato, Héctor Choqque Tacca, Alfredo Mendoza Bedriñaña, Victoria López Córdova, Gisela Pimentel Klose, Mario Encinas Arana, Adriana Ancieta Zerda.
CASO CLINÍCOSíndrome Moebius en el Hospital Apoyo Iquitos. Posible Causa por Exposición a Misoprostol. Reporte de un Caso. Hermann Silva Delgado, Javier Villaverde Montoya. TEMA DE REVISIÓNTelemedicina, ya está acá, ¿nosotros dónde estamos?Javier Ferreyros Kuppers.
ÉTICA¿Existe un módulo moral?Miguel Chávez.
REGLAMENTO DE PUBLICACIÓN
Pág.
6
9
17
22
32
38
44
48
5
Revista Peruana de PediatríaPublicación Oficial de la Sociedad Peruana de Pediatría
Rev. peru. pediatr. 67 (1) 2014ISSN 1993-6826
CONTENT
6
9
17
22
32
38
44
48
Pág.
EDITORIAL Medical Education in Peru. Who is responsible for future pediatricians in the country?Alejandro Bermúdez García.
ORIGINAL ARTICLESClinical Epidemiology Study of Cystic Fibrosis in the National Institute of Child Health, 2002-2011 Lima. Daniel Torres Vela, Hernán Del Castillo Barrientos, Carol Flores Fernández, Amparo Zavaleta Pesantes.
Age of Clinical Diagnosis of Congenital Hypothyroid: Twenty Years Later. Natalia Tovar Gutiérrez, Alex Rojas Hernández, Fiorella Torres Matta, Rubén Susaya Campos, Carlos Del Águila Villar, Juan Manuel Falen Boggio.
Hypophosphatemic Renal Rickets: Experience in 10 Years. Graciela Sakihara Asato, Héctor Choqque Tacca, Alfredo Mendoza Bedriñaña, Victoria López Córdova, Gisela Pimentel Klose, Mario Encinas Arana, Adriana Ancieta Zerda. CASE REPORTMoebius Syndrome in the Hospital Apoyo Iquitos. Possible Cause by Exposure to Misoprostol. Report of one Case. Hermann Silva Delgado, Javier Villaverde Montoya. REVIEW TOPICTelemedicine, is already here, where are we?Javier Ferreyros Kuppers.
ETHICSIs there a moral module?Miguel Chávez.
PUBLICATION GUIDELINES
6 Editorial
La Educación Médica en el Perú: ¿Quién es el responsable de los futuros pediatras en el país?
Hace algunos días escuchaba el comentario de unos colegas en mi hospital, ellos decían:¿Cómo es posible que exista tanta diferencia entre estos 2 residentes de 3er año y pensar que ambos tendrán el título de nuestra especialidad en un mes?
Ese comentario ya lo había escuchado muchas veces en casi todos los servicios de mi hospital y en otros muchos hospitales, así que decidí preguntar a mis colegas sobre su percepción de la formación de especialistas en el Perú. Muchos me respondieron intentando darme diferentes explicaciones, algunos incluso siendo profesores en prestigiosas universidades del medio, me daban respuestas contradictorias, por lo que llegué a concluir que la gran mayoría de médicos ingresan a una especialidad en la universidad que pueden y al hospital que alcancen de acuerdo al puntaje obtenido en la evaluación y no necesariamente al más adecuado para aprender. Observando este panorama, realmente creo que es necesaria una reflexión para generar “algo” que permita en el futuro encontrar especialistas competentes, con estándares formativos similares que se necesitan con tanta urgencia en nuestro país.
La primera queja contra nuestros residentes es: ¡Estos médicos no leen nada! Este comentario, desde mi punto de vista tiene un sesgo. He visto a más de uno cuando surge una discusión clínica, meter la mano al bolsillo, sacar su teléfono inteligente y buscar, aunque sea en google, alguna información que permita aclarar sus dudas. El concepto de obtener información ha cambiado mucho, el que tiene la información, no es ahora el que pasó madrugadas leyendo el “Tratado de Nelson” en edición inglesa de hace más de 10 años traducida al español. Yo considero que el residente tiene mucho más información hoy, entre las búsquedas que realiza en su teléfono al día podría leer mucho más que muchos asistentes con gran experiencia, el problema surge en relación al acceso, validación y motivación para realizar las búsquedas.
He utilizado con mucho cuidado el término acceso a la información porque es muy diferente al término aprender.
Aprender es un cambio de conducta frente a una situación producto de alguna intervención. Aprender implica 3 elementos básicos: El cognitivo: Es fundamental manejar información. El psicomotor: El quehacer de cada día va cambiando tu conducta. El emocional: Que va a motivarte a cambiar la conducta de manera efectiva.
Este proceso de aprender puede ocurrir de manera circunstancial, sin ningún tipo de estructuracióny es el responsable de que tengamos muchos especialistas muy buenos en diferentes hospitales. Recuerdo alguno de mis maestros decir:“La especialidad se aprende en la emergencia” y sigue siendo muy válido en la actualidad.
El hecho de poder participar en muchas situaciones, vivir muchos errores, sufrir sus consecuenticas, ver manejos exitosos, explorarlos y tener el propósito de corregir nuestra conducta frente a situaciones similares futuras hace que ocurra el aprendizaje. Pero debemos recalcar algo muy importante, aprendemos a partir de errores y en nuestras especialidades esto significa quizá poner en riesgo a otros seres humanos, situación que es prohibitiva éticamente y cada vez tiene mayor implicancia legal. Reflejo de esto es la cantidad de restricciones que aparecen en el extranjero y van llegando a nuestro país, situación que dificulta más el acceso al campo clínico de quienes están en formación.
7
Este problema actualmente es posible de enfrentar y de aquí surge el segundo componente del aprendizaje, el psicomotor. Este es responsabilidad de las instituciones formadoras como son las universidades. El proceso desestructurado de la formación (no es suficiente un librito que indique lo que creemos debe “aprender” un especialista para egresar, sino estructurar herramientas para provocar el cambio de conducta de forma progresiva y oportuna). No podemos permitir que una universidad deje a un alumno de post grado en un hospital sin tutores ni profesores responsables que guíen el aprendizaje de los futuros profesionales y tampoco pedir residentes, solo con el propósito de tener “mano de obra” en nuestros servicios (recuerdo el caso de servicios que tienen más residentes que médicos asistentes). Es aquí donde entran las metodologías actuales de educación médica, el aprendizaje basado en problemas, los escenarios de simulación, clínica, la formación basada en competencias, los programas y equipos de entrenamiento, etc. Incluso si el acceso a tecnologías es difícil por el costo, con solo el hecho de tener nuevos enfoques como por ejemplo la rotación de los futuros especialistas en centros de atención primaria y no solamente en servicios de alta especialización, permitirá ampliar aún más el panorama de salud en el país. Ver que en este nivel de atención los programas de prevención son muy importantes y quien mejor que un especialista en formación para diseñar nuevos enfoques que ayuden a solucionar los problemas de miles de pacientes, disminuir las complicaciones que llegan a centros de mediana y alta complejidad, a la larga no todos trabajarán en servicios de alta especialización, por el contrario, desarrollarán su especialidad en este campo.
Por último, en base a nuestro juramento como médicos, el tercer implicado en la formación de futuros especialistas, somos nosotros mismos. Por más que no tengamos ninguna afiliación universitaria, somos modelos en los cuales se inspiran los futuros especialistas para su formación. Nuestros actos, nuestra conducta, nuestra forma de enfrentar las situaciones diarias y el trato a nuestros pacientes influyen de manera importante en los resultados de formación. Este es el tercer componente del aprendizaje, el emocional, vital como los otros para aprender.
En conclusión somos 3 los actores que participamos en la educación de los médicos futuros: el propio aprendiz, la universidad y los actuales especialistas. No debemos evadir nuestra responsabilidad, por el contrario debemos asumirla y obligar a los otros actores a hacerlo. Es la única manera como podremos, al exhalar nuestro último aliento de vida, estar orgullosos de haber vestido un mandil blanco y haber trascendido en la vida de nuestros hermanos de profesión como un verdadero médico.
Alejandro Bermúdez GarcíaEspecialista en Cirugía Ortopédica y Traumatología – Hospital de Emergencias Pediátricas
Profesor de Medicina – Universidad Peruana de Ciencias Aplicadas (UPC)Maestro en Docencia Universitaria – Universidad Andrés Bello (Chile)
8
9Rev. Perú. pediatr. 67 (1) 2014
Estudio Clínico Epidemiológico de la Fibrosis Quística en el Instituto Nacional de Salud del Niño, Lima 2002-2011. Clinical Epidemiology Study of Cystic Fibrosis in the National Institute of Child Health, 2002-2011 Lima. Daniel Torres Vela1, Hernán Del Castillo Barrientos2, Carol Flores Fernández3, Amparo Zavaleta Pesantes3.
RESUMENIntroducción: La Fibrosis Quística (FQ) es una enfermedad autosómica recesiva que se manifiesta principalmente por su compromiso a nivel pulmonar y digestivo. En Perú, la prevalencia de FQ es desconocida, así como las características clínicas y epidemiológicas de la enfermedad. Objetivos: Determinar las características clínicas y epidemiológicas de la FQ en el Instituto Nacional de Salud del Niño en el periodo comprendido entre los años 2008 y 2011. Materiales y Métodos: Se realizó una investigación clínica epidemiológica con tipo de diseño observacional, retrospectivo y descriptivo. Se seleccionaron casos de pacientes del Instituto Nacional de Salud del Niño que en sus historias clínicas consignaban el diagnóstico de la enfermedad, en el periodo comprendido entre enero del 2002 y diciembre del 2011. Resultados: Se consignaron 37 casos que cumplieron los criterios de inclusión según el consenso de la Fundación Americana de FQ. Las edades de diagnóstico fluctuaron desde un mes hasta 16 años, siendo el 51,4% mujeres. Las características clínicas principales fueron predominantemente respiratorias y digestivas, se encontró que el 94,6% de los pacientes presentó síntomas respiratorios agudos o crónicos y el 54,1% esteatorrea. Conclusiones: En todos los pacientes se encontró enfermedad sinopulmomar crónica, de los cuales el 75,7% presentó tos crónica productiva. Las anomalías gastrointestinales se presentaron en el 81,1% de los pacientes, encontrándose falla en el crecimiento en todos ellos. La complicación más frecuente fue la neumonía en un 59,5%. El agente patógeno más frecuente en las vías respiratorias fue Staphylococcus aureus en el 51,4%, seguido de Pseudomonas aeruginosa en el 48,6%. Palabras claves: Fibrosis Quística, clínica, epidemiología, diagnóstico.
SUMMARY Introduction:Cystic Fibrosis (CF) is an autosomal recessive disease that occurs primarily for their commitment in the lungs and digestive system. In Peru, the prevalence of CF is unknown as well as the clinical and epidemiological characteristics of the disease. Objective:To determine the clinical and epidemiological characteristics of CF at National Institute of Child Health from 2008 to 2011. Methods: We conducted a clinical epidemiological research which design is observational, retrospective and descriptive. Patient cases were selected from the National Institute of Child Health with diagnosis of the disease in their medical records, between January 2002 and December 2011. Results: We recorded 37 cases that met the criteria for inclusion in the consensus of the American
Foundation for CF. The ages ranged diagnostic from one month to 16 years, 51.4% being women. The main clinical features were predominantly respiratory and digestive, it was found out that 94.6% of patients had acute or chronic respiratory symptoms and 54.1% steatorrhea. Conclusion: In all patients sinopulmomar disease was chronic, of which 75.7% had chronic productive cough. Gastrointestinal abnormalities occurred in 81.1% of patients, being growth a failure in all of them. The most common complication was pneumonia by 59.5%. The most common pathogen in the respiratory system was Staphylococcus aureus in 51.4%, followed by Pseudomonas aeruginosa in 48.6%. Keywords: Cystic Fibrosis, clinical, epidemiology, diagnosis.
1 Médico Pediatra del Instituto Nacional de Salud del Niño. Lima - Perú. Correo electrónico: [email protected] Médico Neumólogo del Instituto Nacional de Salud del Niño. Lima - Perú. 3 Químico Farmacéutico. Laboratorio de Biología Molecular. Facultad de Farmacia y Bioquímica. Universidad Nacional Mayor de San Marcos. Lima - Perú. Recibido: 26 de febrero del 2014 Aceptado: 27 de marzo del 2014
TRABAJOS ORIGINALES
10
INTRODUCCIÓNLa Fibrosis Quística (FQ) es una enfermedad autosómica recesiva causada por mutaciones en el gen CFTR codifica a una proteína de 1480 aminoácidos, llamada proteína reguladora de la conductancia transmembrana de la FQ (CFTR) (1) (2) (3). La alteración de esta proteína conduce a un transporte anormal de electrolitos en las células epiteliales de todos los órganos y sistemas, lo que condiciona la disfunción de diversas glándulas (4) (5) (6).
La principales manifestaciones clínicas se dan a nivel de las vías respiratorias, gastrointestinales y las glándulas sudoríparas de la piel (7) (8), siendo las más importantes: infecciones respiratorias recurrentes causadas por Staphylococcus aureus, Pseudomonas aeruginosa, Haemofilus influencia y Burkholderia cepacia, síndrome de mala absorción intestinal, íleo meconial al nacer, deficiencia en el crecimiento, cirrosis u otras formas de disfunción hepática o insuficiencia pancreática y alteración del contenido de sodio y cloro en el sudor (1) (4) (9). La FQ es la principal causa de enfermedad pulmonar grave en la infancia y la responsable de la mayor parte de las insuficiencias pancreáticas endo y exocrinas en la primera etapa de la vida. Adicionalmente, se puede presentar disminución del crecimiento y cirrosis u otras formas de disfunción hepática. Es por ello que si bien la FQ es primariamente reconocida por la morbilidad pulmonar asociada a ella, las manifestaciones más tempranas que se notan en la mayoría de los pacientes están relacionadas con alteraciones gastrointestinales y nutricionales (10).
Actualmente los criterios para el diagnóstico incluyen, junto a la presencia de las características clínicas, dos o más determinaciones de concentraciones de cloro en sudor superior a 60 mmol/l, o la detección de dos mutaciones reconocidas de FQ; sin embargo puede existir una variante fenotípica atípica en donde se encuentra concentraciones de cloro en sudor borderline de 40 a 60 mmol/L o normal menor a 40 mmol/L (1) (11).
Debido a las diferencias en la composición étnica latinoamericana, hay menor incidencia de FQ en la población de Centro y Sudamérica que en otros caucásicos, lo cual es un indicador del diferente comportamiento de la enfermedad de acuerdo a la población en que se presenta (12) (13)
(14). En América Latina se han descrito estudios de incidencia de FQ entre sus poblaciones en Cuba, México, Costa Rica, Argentina, Brasil y
Colombia (15). En el Perú, en la última década Monsante y colaboradores reportaron 19 casos de FQ en el Hospital Edgardo Rebagliati Martins en el periodo 2000-2007, de los cuales 6 fallecieron (17). Así mismo, Castilla y colaboradores realizaron un estudio epidemiológico de las infecciones en pacientes con FQ en el hospital antes mencionado (16) (17). Durante el 2011 el Perú, según el Instituto Nacional de Estadística e Informática tendrá una población de 29. 797. 694 hab. La tasa bruta de natalidad será de 19. 4 x 1000 hab. Tomando la incidencia 1 en 9000 de los recién nacidos vivos hijos de hispanos en los Estados Unidos, se puede inferir que en el país se tendría unos 64 pacientes por año con Fibrosis Quística.
MATERIALES Y MÉTODOSSe realizó una investigación clínica epidemiológica con tipo de diseño observacional, retrospectivo, descriptivo, en los niños atendidos en la Consulta Externa y Hospitalización, en el Instituto de Salud del Niño en el periodo 2002-2012. Se seleccionó a los pacientes que reúnan los criterios de inclusión: bacteriológico, mediante la baciloscopía y el cultivo de los especímenes obtenidos con relación a los órganos o sistemas comprometidos, que nos confirma el diagnóstico etiológico o de certeza, clínico, epidemiológico, inmunológico, radiológico, e histopatológico, que son criterios de presunción, debiendo presentarse por lo menos tres de estos criterios para apoyar el diagnóstico presuntivo de FQ en el niño. Se consideró los criterios de exclusión a los casos que no reúnan por lo menos tres criterios presuntivos, las fichas sanitarias incompletas e historias clínicas eliminadas del archivo central de la Institución. En total se incluyeron a 155 pacientes del Instituto Nacional de Salud del Niño (INSN) que en sus historias clínicas tenían consignado el diagnóstico de FQ de enero del 2002 a diciembre del 2011. Se analizó la información de cada paciente de la Unidad de FQ, del registro del test de sudor del Laboratorio de Bioquímica y de la base de datos proporcionada por la Unidad de Estadística del INSN.
Se utilizó una ficha de recolección de datos, en la cual se consignaron los datos clínicos generales (sexo, edad , procedencia , fecha de diagnóstico, antecedentes familiares y antecedentes patológicos), características fenotípicas (enfermedad sino-pulmonar crónica, anormalidades gastrointestinales, pérdida de sodio y alcalosis metabólica crónica, azoospermia), examen físico (peso y talla), exámenes auxiliares
Estudio Clínico Epidemiológico de la Fibrosis Quística en el Instituto Nacional de Salud del Niño, Lima 2002-2011.
11
(test del sudor, análisis de mutaciones, test de Sudán en heces y cultivos), hospitalizaciones, consultas, estado nutricional, complicaciones y tratamiento. Para el diagnóstico se tuvo en cuenta los criterios del consenso americano de FQ (18), que incluye una o más características de los rasgos fenotípicos y aumento de la concentración de cloro en la prueba de sudor por iontoforesis con pilocarpina en dos o más ocasiones, o en su defecto identificar dos mutaciones para FQ, historia de FQ en un familiar, tamizaje neonatal positivo para FQ o transporte iónico epitelial nasal anormal.
RESULTADOSCaracterísticas de los pacientesSe obtuvo una muestra de 155 registros con diagnóstico de FQ desde enero del 2002 hasta diciembre del 2011 en el INSN, se excluyeron 118 historias clínicas por no cumplir con los criterios de diagnóstico establecidos. De las cuales 20 contaban con una sola prueba de sudor positiva y cuadro clínico sugerente, 1 con valores límite de la prueba de sudor más cuadro clínico compatible pero sin estudio de análisis de mutaciones de FQ por reacción en cadena de la polimerasa (PCR). No se encontraron 10 historias clínicas en los archivos del INSN. Finalmente, el presente estudio se realizó con 37 historias clínicas que cumplieron los criterios antes mencionados. La distribución de los pacientes por edad al momento del diagnóstico se muestra en la figura 1.
Figura 1. Distribución de los pacientes con fibrosis quística del Instituto Nacional de Salud del Niño. Lima del 2002 al 2011. Las edades de diagnóstico fluctuaron desde un mes hasta 16 años con 5 meses. En este periodo de estudio se presentaron 51,4% (N=19) casos de pacientes mujeres y 48,6% (N= 18) de hombres. Se encontró que el 100% (N = 37) de los pacientes fueron mestizos, pacientes nacidos en
0%
10%
20%
30%
40%
50%
60%
0 a 1 6 a 10 11 a 14 15 a 17
56,8
Porc
enta
je
Edad (años)
- - -- -
13,5
2,75,4
2 a 5
21,6
Las características fenotípicas de los pacientes se detallan en la tabla 2. La enfermedad sinopulmonar crónica se presentó en un 100% (N=37), mientras que las anomalías gastrointestinales en un 81,1% (N = 30). El 48,6% (N=18) presentó síndromes de pérdida de Na+ y alcalosis metabólica crónica. El diagnóstico fue realizado en un 91,9% (N=34) por presentar uno o más rasgos fenotípicos y por contar con el incremento de la concentración de cloro en el test de sudor por iontoforesis con pilocarpina en dos o más ocasiones; en tanto sólo en un 8,1% (N=3) de los casos se realizó el diagnóstico por características fenotípicas más el estudio de análisis de mutaciones de FQ por PCR, los resultados obtenidos indican que los 3 pacientes son heterocigotos para la mutación ∆F508.
el país e hijos de padres peruanos. Con respecto al lugar de nacimiento el 48,6% nacieron en Lima, mientras que el 51,4% fueron de provincias, donde el mayor número procede de Piura con 22,2% (N=4), Lambayeque e Ica con 16,7% (N= 3), Amazonas y Cajamarca con 11,1% (N= 2) y Ancash, Huancayo, Apurímac y Junín con 5,6 % (N=1).
DiagnósticoLas principales características clínicas de los pacientes con FQ fueron síntomas respiratorios agudos o crónicos y falla en el crecimiento, representando el 94, 6% (N= 35) y 81,1% (N= 30) de pacientes respectivamente. Además, el 54,1% (N=20) de los pacientes presentó esteatorrea (tabla 1).
Tabla 1. Características clínicas de los pacientes con
fibrosis quística del Instituto Nacional de Salud del Niño. Lima del 2002 al 2011.
Daniel Torres Vela, Hernán Del Castillo Barrientos, Carol Flores Fernández, Amparo Zavaleta Pesantes.
12
ComplicacionesLa complicación más frecuente fue la neumonía, la cual se presentó en el 59,5% (N=22) de los pacientes. Se presentaron otras complicaciones como bronquiectasias en el 48,6% (N=18), reflujo gastroesofágico (RGE) en el 29,7% (N=11), atelectasias e insuficiencia pancreática en el 8,1% (N= 3) en ambos casos y tuberculosis, hepatomegalia e ictericia en el 5,4% (N= 2). Se encontró eosinofilia en el 32,4% (N= 12). El número de hospitalizaciones en promedio fue de 3 con una estancia hospitalaria de 16,9 días en promedio, con un total de 13,5 consultas por paciente (figura 2).
TratamientoEn la tabla 3 se presenta el tratamiento prescrito a los pacientes con FQ, los fármacos más empleados fueron los antibióticos en el 97,3 % (N=36). En muchas historias clínicas no se encontraron la frecuencia de administración y duración del tratamiento.
Tabla 3. Tratamiento recibido por los pacientes con fibrosis quística del Instituto Nacional de Salud
del Niño. Lima del 2002 al 2011.
TRATAMIENTOPacientes
N (%)
Antibióticos 36 97,3
Agonistas adrenérgicos 24 64,9
Corticoides 23 62,2
Lipasas 18 48,6
Inhibidores de los receptores H2 8 21,6
Vitaminas 7 18,9
Procinéticos 6 16,2
Antagonistas colinérgicos 6 16,2
Inhibidores de la bomba de
protones3 8,1
Diuréticos 3 8,1
Hierro 1 2,7
Tabla 2. Características fenotípicas de los pacientes con fibrosis quística del Instituto Nacional de Salud
del Niño. Lima del 2002 al 2011.
Figura 2. Complicaciones más frecuentes en pacientes con fibrosis quística del Instituto Nacional
de Salud del Niño. Lima del 2002 al 2011.
Microbiología Los agentes patógenos más frecuentemente encontrados en las vías respiratorias fueron Staphylococcus sp. y Pseudomonas aeruginosa en el 51,4% (N=19) y 48,6% (N= 18) de los pacientes respectivamente. Además, se encontraron agentes patógenos como Stretococcus sp y Cándida albicans en el 18,9% (N=7) y 13,5% (N=5) respectivamente. En la figura 3 se presenta los patógenos más frecuentes encontrados en los pacientes con FQ.
Figura 3. Microorganismos más frecuentes en pacientes con fibrosis quística del Instituto Nacional
de Salud del Niño. Lima del 2002 a 2011.
Estudio Clínico Epidemiológico de la Fibrosis Quística en el Instituto Nacional de Salud del Niño, Lima 2002-2011.
13
DISCUSIÓN En los últimos años se están notificando algunos casos de FQ en nuestro medio, sin embargo no hay datos sobre la incidencia real, dado que no existe un registro y seguimiento adecuado de los casos. Las políticas de salud peruanas no permiten hacer un estudio sistemático de nuevos casos de FQ, a diferencia de otros países como Chile, que cuenta con un Programa Nacional de Fibrosis Quística a cargo del Ministerio de Salud. En la población atendida en el INSN del 2002 al 2011, la edad promedio fue de 2 años 10 meses, similar a Chile y Costa Rica (13) (15).
La prevalencia de la enfermedad en función al sexo ha variado gradualmente en nuestro país en los últimos años. En 1994, Aguirre reportó que el 67% de los pacientes eran mujeres (5), en el 2002 Torres (1) y en el 2008 Castilla reportaron que el 61,5 y 65% respectivamente eran varones (6), mientras
que en este estudio hay un mayor porcentaje de mujeres diagnosticadas, las cuales representan el 51,4% de los casos. La prevalencia de la enfermedad es independiente del sexo ya que la FQ no está asociada a un cromosoma sexual. Deficiencias en el registro y seguimiento de los pacientes no permitieron determinar la sobrevida. Se encontró que el lugar de nacimiento del mayor porcentaje de pacientes (51,4%) fue en provincias, a diferencia de los hallazgos de Torres en el 2002, quién reportó que el 69,2% nació en Lima.
DiagnósticoLas características clínicas se manifiestan predominantemente a nivel respiratorio y digestivo, siendo la enfermedad respiratoria la responsable de la mayor morbimortalidad. El 94,6% de los pacientes presentó síntomas respiratorios agudos o crónicos como característica clínica inicial. En el 100 % se encontró enfermedad sinopulmonar crónica como característica fenotípica, dentro de la cual el 75,7% de ellos presentó tos crónica productiva y el 24,3% anomalías como atelectasias y bronquiectasias. Según se reporta la enfermedad pulmonar es la principal causa de morbilidad y es responsable del 95% de la mortalidad. En el pulmón, las vías aéreas son el sitio inicial de compromiso, con aclaramiento mucociliar anormal, se dan infecciones a repetición y bronquiectasias que llevan a una disfunción pulmonar progresiva y falla respiratoria (15) (19). Las anomalías gastrointestinales suelen presentarse en un menor porcentaje. La esteatorrea producida por deficiencia de enzimas pancreáticas por daños a nivel del páncreas y se manifiesta con deposiciones abundantes, fétidas y con características grasas, para algunos autores se presenta en el 85 a 90% de los casos de los pacientes con FQ (11) (20), en este estudio se presentó en el 54,1% de los casos y el diagnóstico se realizó por el test de Sudán en heces mayor a 1+. La enfermedad hepatobiliar se presentó en un 10,8 % de los pacientes. En el hígado, la alteración de las secreciones en los conductos biliares conlleva a cirrosis biliar focal en algunos pacientes (15). El íleo meconial se encontró en el 10,8%; según se describe en la literatura, se presenta en el 15 a 20% de los niños con FQ y es causado por la deshidratación del contenido intestinal (19) (21). El prolapso rectal es reportado en cerca del 20% de los pacientes menores de 5 años no tratados, es un signo de mal nutrición. Sin embargo, en nuestro estudio sólo se encontró en el 5,4% de los pacientes, a pesar que el 94,3% presentó
Servicios más frecuentesLos servicios a los cuales acudieron más frecuentemente los pacientes con FQ fueron a neumología en el 78,4% (N=29) de los casos y gastroenterología en el 67,6% (N=25). El número de consultas fue de 192 (38,5%) para neumología y de 115 (23%) para gastroenterología. Todos los servicios a los cuales acudieron los pacientes con FQ, así como el número de consultas por servicio se detalla en la tabla 4.
Tabla 4. Número de consultas y servicios más visitados por los pacientes con fibrosis quística
del Instituto Nacional de Salud del Niño. Lima del 2002 al 2011.
Daniel Torres Vela, Hernán Del Castillo Barrientos, Carol Flores Fernández, Amparo Zavaleta Pesantes.
14
algún grado de desnutrición. La desnutrición que sufren los pacientes con FQ es el resultado de una malabsorción de nutrientes, aumento del gasto energético, infecciones pulmonares recurrentes y una inadecuada ingesta calórica (19). La incidencia de pólipos nasales varía del 6 al 56%, en nuestro estudio se presentó en un 2,7% de los pacientes (1). Las principales anormalidades en los pacientes con FQ están dadas por concentraciones anormales de iones inorgánicos en secreciones glandulares (7). El 48,6% presentó electrolitos alterados, síndromes de pérdida de sodio y alcalosis metabólicas. El signo más consistente de la FQ es la concentración elevada de cloro, sodio y potasio en la secreción de las glándulas sudoríparas. La pérdida excesiva de agua y electrolitos puede ocasionar alcalosis hiperclorémica e hiponatremia (21).
En total fueron tres pacientes a los que se les realizó el estudio de mutaciones para FQ por PCR, se detectó la mutación Delta F508 heterocigota con otra mutación no identificada en estos pacientes.
ComplicacionesLos resultados evidencian que las complicaciones son predominantes a nivel respiratorio, lo cual concuerda con lo descrito, que menciona que las pulmonares son la principal causa de morbilidad y muerte prematura. La complicación más frecuente fue la neumonía, presentándose en el 59,5% de los pacientes, seguido de bronquiectasias en el 48,6%. Otra complicación a nivel respiratorio fue la atelectasia en 8,1% y tuberculosis en 5,4%. Se sabe que el compromiso respiratorio está presente en el nacimiento con inflamación de la vía aérea, este se ve agravado por el espesamiento de las secreciones bronquiales, las cuales son el lugar ideal para el crecimiento de gérmenes no habituales que inducen una respuesta inflamatoria, principalmente en base a polimorfonucleares, los cuales liberan enzimas proteolíticas y radicales libres que destruyen el parénquima pulmonar, llevando a la pérdida de la estructura elástica, con lo cual se producen bronquiectasias y fibrosis, que llevan a la insuficiencia respiratoria y la muerte. El compromiso pulmonar progresivo deriva en insuficiencia con hipoxemia e hipercapnia con corpulmonar en etapa terminal (22). Otras manifestaciones relevantes son las que comprometen el tracto gastrointestinal y glándulas anexas, dentro de ellas se encontró que las más frecuentes fueron el reflujo gastroesofágico (RGE) y la insuficiencia pancreática con un 29,7 y 8,1 %
respectivamente. Torres en el 2002 reportó al RGE como la principal complicación en pacientes con FQ con un 30,8%. La bibliografía menciona que el RGE se presenta en alrededor del 30 % de los casos (1). En la segunda década de la vida, la afección en la función endocrina del páncreas puede desarrollar diabetes insulino-dependiente, sin embargo en nuestro estudio no se encontró ningún caso. Otras complicaciones gastrointestinales son las que se presentan a nivel hepático, dentro de ellas tenemos hepatomegalia, ictericia y síndrome colestásico, las cuales se presentaron en el 5,4 %. La literatura refiere que la afección hepática se ha encontrado en un 25% de las autopsias de los pacientes con FQ (21).
MicrobiologíaEl agente infectante inicial predominante aislado de cultivos de las vías respiratorias fue Staphylococcus sp, el cual se presentó en el 51, 4 % de los pacientes, siendo Staphylococcus aureus el más frecuente, seguido por Pseudomonas aeruginosa en el 48,6%. Se observa un incremento respecto a lo reportado por Castilla en el 2008 en su estudio realizado sobre “Epidemiología de las infecciones respiratorias en pacientes con FQ en el Hospital Nacional Edgardo Rebagliati Martins” (6), en donde reportó que el agente infectante inicial fue Pseudomonas aeruginosa en el 35% de los pacientes, seguida de Staphylococcus aureus en el 25%. Sin embargo Torres reportó en un estudio realizado en el Instituto Nacional de Salud del Niño en el 2002 que el 81,3% de los casos presentó Pseudomonas aeruginosa y el 54,8 % Staphylococcus aureus(1). En este estudio 32,4% presentó ambos agentes. S.aureus es usualmente el primer patógeno aislado de la vía aérea de los pacientes con FQ, con una tasa de infección que disminuye con la edad, mientras que por su parte la tasa de P. aeruginosa se incrementa. La presencia de S. aureus en el tracto respiratorio deteriora la vía aérea y a través de este invasor primario, la P. aeruginosa ve facilitada su entrada (13) (22). Psedomonas aeruginosa produce la infección pulmonar crónica más severa y se asocia a un deterioro progresivo de la función pulmonar, causante de la mayor mortalidad en pacientes con FQ, su aparición por primera vez en las secreciones bronquiales es indicación de antibioticoterapia agresiva aunque no exista ningún indicador clínico de exacerbación. En cuanto a la colonización crónica por Pseudomonas aeruginosa, algunos autores refieren que se presenta en el 60 a 80 % de los casos (6) (17) (22).
Estudio Clínico Epidemiológico de la Fibrosis Quística en el Instituto Nacional de Salud del Niño, Lima 2002-2011.
15
Además se encontró la presencia de otros agentes infecciosos como Streptococcus sp, Candida albicans y Klebsiella sp en un menor porcentaje de pacientes. Nuevos organismos, como la Stenotrophomonas maltophilia, han sido descritos, aunque aparentemente sin un impacto claro en la función pulmonar o en la sobrevida
(13), en nuestro estudio solo se encontró que un 2,7% de los pacientes la presentó. Al 18,9% de los pacientes no se le encontró la presencia de ningún microorganismo, esto puede deberse a que no se le tomó ningún cultivo de esputo de control.
Tratamiento Debido a que las infecciones respiratorias son una de las causas más importantes de la morbilidad y mortalidad en pacientes con FQ afectando su pronóstico y calidad de vida, los antibióticos constituyen el grupo de fármacos más empleados (6). Es importante iniciar el tratamiento precoz y agresivo de la infección bacteriana en los pacientes con FQ. El 97,3% de los pacientes recibió tratamiento antibiótico, aunque sólo un 48,6% presentó colonización bacteriana. En la gran mayoría de los casos el tratamiento puede comenzar por vía oral, y si no hay una respuesta adecuada, utilizar la hospitalización y la vía parenteral (23). Muchos de los agentes infecciosos desarrollan mecanismo de resistencia antibiótica o forman biopelículas que los protegen de la actividad de éstos como es el caso de la Pseudomonas aeruginosa (6).
Otro grupo de fármacos empleados fueron los agonistas adrenérgicos B2 en un 64,9%, estos fármacos administraron solos o asociados a antagonistas colinérgico para tratar el Síndrome de Obstrucción Bronquial Agudo (SOBA) e hiperreactividad bronquial presentes en los pacientes con FQ.Los agonistas B2 y los anticolinérgicos tienen efecto directo en la relajación de la musculatura lisa produciendo broncodilatación y aumentando la frecuencia del barrido ciliar (23).Los agonistas colinérgicos se administraron a un 16,2% de los pacientes. Un 4% de los pacientes recibió agonistas B2 asociado a antagonistas colinérgicos.
Además se emplearon corticoides tanto de acción local como de acción sistémica en un 62,2% de los pacientes. Los corticoides se emplean para tratar procesos inflamatorios causados por infecciones bacterianas y para tratar hiperreactividad bronquial. Un 4% de los pacientes recibió
agonistas B2 asociado a antagonistas colinérgicos y corticoides. El 29,7 % de los pacientes recibió fisioterapia respiratoria como parte dela terapia respiratoria.
En el páncreas la obstrucción de los conductos lleva a atrofia y a la necesidad de remplazo enzimático pancreático en aproximadamente el 85,0% de los pacientes. Al 48,6 % de los pacientes se le suministró lipasa pancreática. Para tratar el RGE se indicaron procinéticos en un 16,2% de los pacientes. Para reducir la acidez gástrica en estos pacientes se indicaron inhibidores de los receptores H2 e inhibidores de la bomba de protones en un 21,6 y 8,1% respectivamente (22). Los pacientes portadores de FQ presentan carencias vitamínicas, como consecuencia del arrastre de las mismas por esteatorrea o por menor ingesta, debido a anorexia, condición siempre presente. Las más afectadas son las vitaminas liposolubles A, D, E, K (23), es por ello que a los pacientes con FQ se les debe administrar suplementos vitamínicos y minerales. El 18,9% de los pacientes recibió suplemento vitamínico, mientras que el 2,7% recibió suplemento de hierro. Un 8.1% de los pacientes recibió tratamiento con diuréticos para casos de edema.
CONCLUSIONESEn los casos estudiados, todos los pacientes presentaron enfermedad sinopulmomar crónica, de los cuales el 75,7% presentó tos crónica productiva. Las anomalías gastrointestinales se presentaron en el 81,1% de los pacientes, encontrándose falla en el crecimiento en todos ellos. La complicación más frecuente fue la neumonía en un 59,5%. El agente patógeno más frecuente en las vías respiratorias fue Staphylococcus aureus en el 51,4%, seguido de Pseudomonas aeruginosa en el 48,6%. El 97,3% de los pacientes recibió tratamiento antibiótico, aunque sólo un 48,6% presentó colonización bacteriana. En el páncreas la obstrucción de los conductos lleva a atrofia y a la necesidad de remplazo enzimático pancreático en aproximadamente el 85% de los pacientes. Al 48,6 % de los pacientes se le suministró lipasa pancreática.
RECOMENDACIONESEs necesario impulsar la Unidad Clínica de Fibrosis Quística del Instituto de Salud del Niño y desarrollar un programa nacional para uniformizar y divulgar los criterios de diagnóstico y tratamiento de la FQ. Esto permitirá un adecuado registro y seguimiento de los pacientes con FQ,
Daniel Torres Vela, Hernán Del Castillo Barrientos, Carol Flores Fernández, Amparo Zavaleta Pesantes.
16
REFERENCIAS BIBLIOGRÁFICAS1. Torres D. Estudio clínico epidemiológico de la
fibrosis quística en el instituto de salud del niño, Lima 1991-2001 [tesis de especialidad] . Lima: Universidad Nacional Mayor de San Marcos; 2002.
2. Vega L. CFTR: Más que un canal de cloro. Pediátrica 2010; 5(1):10-14.
3. Silva C. Mutaciones más frecuentes en el gen CFTR de pacientes diagnosticados con fibrosis quística del Instituto Especializado de Salud del Niño [Tesis para optar el título profesional de Químico Farmacéutico] . Universidad Nacional Mayor de San Marcos; Lima, 2008.
4. Lay – Son G, Repetto G. Genética y fibrosis quística: Desde el gen CFTR a los factores modificadores. Pediátrica 2010; 5(1):4-9.
5. Aguirre I, Chiarella P, Hernández H, Chaparro E, Accinelli R, Zegarra O. Fibrosis quística: Reporte de casos en un hospital de Lima, Perú. Rev Méd Hered 1994; 5 (4): 204-8.
6. Vásquez C, Aristizábal R, Daza W. Fibrosis quística en Colombia. Pediátrica 2010; 5(1):44-50.
7. Repetto G, Poggi H, Harris P, Navarro H, Sánchez I, Guiraldes E, Rérez M, Boza M, Hunter B, Wevar M, Mediavilla M, Foradori A. Identification of cystic fibrosis transmembrane regulator (CFTR) mutations in Chilean patients with cystic fibrosis. Rev Méd Chile 2001; 129(8):841-847.
8. De Abreu e Silva F, Caldeira F. Fibrosis quística en Brasil. Pediátrica 2010; 5(1):39-41.
9. Largo I. Fibrosis quística. Rev. Ped. Elec. [en línea] 2009; 6(1):1-18
10. Milla C. Nutrición en fibrosis quística. Pediátrica 2010; 5(1):23-28
11. Rosenstein BJ, Zeitlin PL. Cystic fibrosis. The lancet 1998;351:277-82
12. Vega L, Sánchez I. Aspectos básicos de la fibrosis quística. Pediátrica 2010; 5(1):96-101.
13. Guzmán K, Del Campo E, Nápoles N, Toledano Y, Coello D. Main clinical-epidemiological characteristics of patients withcystic fibrosis in Santiago de Cuba province. MEDISAN [online] 2011; 15(2):152-161.
14. Murillo C, Gutiérrez J. Phenotypic expression in patients with cystic fibrosis. Actapediátr. Costarric [online] 2009; 21(1): 26-32.
16. Castilla G. Epidemiología de infecciones respiratorias en pacientes con Fibrosis Quística en el Hospital Nacional Edgardo Rebagliati Martins. Rev Perú Pediatr 2008; 61: 82- 8.
17. Aguirre I. Fibrosis Quística en el Perú. Pediátrica 2010;5(1):52
18. Rosenstein B, Cutting G. The diagnosis of cystic fibrosis: A consensus statement. J Pediatr 1998; 132: 589-95.
19. Vega L, Guiraldes E, Sanchez I. Cystic fibrosis: facing patient transition from the pediatrician to the internist. Rev. Méd. Chile 2006; 134(3): 365-371.
20. Solís A, Gutiérrez J. Fibrosis quística. Acta méd. Costarric [online] 2003; 45(1):42-48.
21. Orozco L, Chávez M, Saldaña Y, Velázquez R, Carnevale A, Gonzales A, et al. Fibrosis quística: La frontera del conocimiento molecular y sus aplicaciones clínicas. Revista de investigación clínica 2006; 58(2):139-152.
22. Largo I. Fibrosis quística. Rev. Ped. Elec. [en línea] 2009:6(1).
23. Sánchez I, Pérez A, Boza M, Lezana V, Vila M, Repetto G, et al. Consenso nacional de fibrosis quística. Rev. Chil. Pediatr. [Online] 2001; 72(4):356-380.
para poder satisfacer las necesidades sociales, económicas, emocionales y problemas médicos de los pacientes. Se requiere de un esfuerzo
multidisciplinario para el trabajo en equipo, que redunde en beneficio de los pacientes.
Estudio Clínico Epidemiológico de la Fibrosis Quística en el Instituto Nacional de Salud del Niño, Lima 2002-2011.
17
Edad de Diagnóstico Clínico del Hipotiroideo Congénito: Veinte Años Después. Age of Clinical Diagnosis of Congenital Hypothyroid: Twenty Years Later. Natalia Tovar Gutiérrez2, Alex Rojas Hernández2, Fiorella Torres Matta2, Rubén Susaya Campos2, Carlos Del Águila Villar1 y Juan Manuel Falen Boggio1, 2.
RESUMEN Objetivo: El hipotiroidismo congénito es una de las principales causas de retardo mental prevenibles, por esta razón su diagnóstico debe ser temprano y el tratamiento oportuno. El objetivo del presente trabajo es el de determinar la edad actual de diagnóstico del hipotiroideo congénito mediante criterios clínicos. Materiales y Métodos: Se trata de un estudio de tipo descriptivo, longitudinal y retrospectivo. Se analizaron 247 historias clínicas, de las cuales sólo 37 reunieron los criterios de inclusión. Los datos fueron consignados en una ficha confeccionada para este propósito, siguiendo los criterios de diagnóstico clínico de Blanco López et al. , los datos de antropometría fueron analizados con el programa EPI-INFO 2002. El análisis de los datos recogidos fue realizado con el programa estadístico Statistical Package for the Social Science v15.0 (SPSS Inc. Chicago III). Resultados: Se halló una mayor incidencia en el sexo femenino (78%) que en el sexo masculino (22%), con una relación de 3. 62/1. La edad promedio al momento del diagnóstico fue de 5.93 ± 5.28 meses para el total de la muestra. Las características clínicas al momento del diagnóstico fueron: facies abotagada (73%), piel seca (70.3%), fontanelas amplias (70.3%), estreñimiento (64.9%), hipotonía (64.9%) e ictericia (64.9%). Así mismo, se evidenció un marcado retardo del crecimiento. Posterior al diagnóstico y con la administración de L-tiroxina, el patrón de crecimiento muestra encarrilamiento del crecimiento en lo que debería ser su patrón de crecimiento y mejora del peso. Conclusiones: La edad promedio de diagnóstico del hipotiroideo congénito fue de 5.93 ± 5.28 meses, que contrasta notablemente con la edad de diagnóstico encontrada en el reporte anterior (1992) y fue más frecuente en el sexo femenino. El entrenamiento de los residentes de Pediatría en la detección clínica precoz de esta entidad ha permitido disminuir la edad de diagnóstico de esta entidad, no siendo aún óptima, por lo que debe ampliarse el Programa de Tamizaje de Hipotiroidismo Congénito a nivel nacional.
1 Servicio de Endocrinología del Instituto Nacional de Salud del Niño. Lima – Perú. 2 Facultad de Medicina Humana de la Universidad Particular San Martín de Porres. Correo electrónico Dr. Carlos Del Águila Villar: [email protected] Trabajo presentado al XXI Congreso Peruano de Pediatría, Iquitos – 2010. Recibido: 20 de Diciembre del 2013. Aceptado: 24 de Febrero del 2014.
SUMMARY Objective: Congenital hypothyroidism (CH) is the most common preventable cause of mental retardation, for this reason it is important the early identification of infants with CH and prompt institution of treatment. The purpose of this work is determine the current age of diagnosis of the congenital hypothyroid by clinical criteria. Methods: We performed a retrospective, descriptive, and longitudinal study. We review 247 Medical records and only 37 met the inclusion criteria and were analyzed. The data were entered on a form drawn up for this purpose, following the diagnosis by Blanco-Lopez et al. , anthropometry data were analyzed with EPI-INFO 2002 program. The analysis of the collected data was performed with statistical program Statistical Package for the Social Science (SPSS Inc. Chicago III) v15. 0. Results: An increased incidence in females (78) than in males (22), was found with a ratio of 3.621. The average
age at the time of diagnosis was 5.93 ± 5.28 month to the total sample. The clinical characteristics at the time of diagnosis were: bloated facies (73%), dry skin (70. 3%), large fontanelles (70.3%), constipation (64.9%), hypotonia (64.9%) and jaundice (64.9%). Likewise, evidenced a marked growth retardation. Subsequent to diagnosis and administration of l-thyroxine, the growth pattern shows tracking of growth in what should be its pattern of growth and improvement of the weigh. Conclusions: The average age of diagnosis of congenital hypothyroidism was 5.93 ± 5.28 months, which contrasts sharply with the age at diagnosis found by a previous report (1992), with a higher frequency in females. The training of Pediatric fellows at the early clinical detection of this entity has allowed to decrease the age of diagnosis of this entity, being not yet optimal, so congenital hypothyroidism screening program be extended to national level.Keywords: Congenital hypothyroidism, diagnosis, age.
INTRODUCCIÓNEl hipotiroidismo congénito es una de las principales causas de retardo mental prevenibles, para lo que se requiere detección temprana y terapia oportuna (1).
El hipotiroidismo primario tiene una incidencia de 1/3000-4000 recién nacidos (RN) en los países desarrollados, se presenta con mayor frecuencia en sexo femenino que en el masculino (2, 3). El hipotiroidismo secundario y terciario es poco frecuente, con una incidencia 1/50000-100000 RN. En nuestro país, un estudio piloto mostró una incidencia de 1/1254 recién nacidos (4). Mediante el Programa de Tamizaje Neonatal que se realiza a los recién nacidos en el Instituto Nacional Materno Perinatal (INMP), se reportó entre octubre del 2003 y agosto del 2005 una incidencia de 1/2939 recién nacidos vivos (5). Se ha reportado que la incidencia es mayor en zonas bocio endémico y el impacto del hipotiroidismo congénito en las comunidades con deficiencia de yodo radica en la posibilidad de daño cerebral no específico con reducción de la función intelectual a nivel poblacional(6). Debe señalarse que si esta entidad no es identificada al nacimiento, los signos y síntomas se van acentuando conforme transcurre el tiempo y el retardo mental que se produce (7).
Cabe destacar que en el momento del nacimiento los signos y síntomas del hipotiroidismo neonatal son variables y su frecuencia e intensidad dependen de la severidad, tipo y duración de la falla tiroidea por lo que no todos son diagnosticados clínicamente. El diagnóstico tardío se traduce en retardo pondo-estatural, alteraciones del desarrollo psicomotor y retardo mental(1, 2, 7), por lo que el tamizaje constituye el método ideal de diagnóstico temprano(3). Con la finalidad de detectar precozmente a los pacientes con hipotiroidismo congénito Letarte et al.(8) ideó un sistema de puntuación de los signos y síntomas, los cuales fueron adaptados por Blanco-López et al.(9). En 1992, Del Águila et al.(10), utilizando la puntuación desarrollada por Blanco-López encontraron que la edad de diagnóstico clínico del hipotiroideo congénito que asiste a la consulta endocrinológica en el Instituto Nacional de Salud del Niño era de 17,24 ± 23,8 meses de edad en los varones y de 17,45 ± 22,5 en las mujeres, y sólo un 21,7% fueron diagnosticados antes de los 3 meses; las manifestaciones clínicas que se presentaron con mayor frecuencia fueron estreñimiento, hipoactividad, piel seca y macroglosia, así como un marcado retardo del crecimiento.
Por tal motivo, en la formación del pediatra, en el Instituto Nacional de Salud del Niño, se insistía en la detección precoz de los pacientes que asistían a la consulta, el aplicar los criterios de Blanco-López en aquellos pacientes con sospecha clínica de hipotiroidismo congénito.
El objetivo del presente trabajo es el de determinar cuál es la edad actual de diagnóstico clínico de los pacientes con hipotiroidismo clínico y cuáles son las manifestaciones clínicas principales del hipotiroideo congénito al momento de su diagnóstico. Así mismo, se desea conocer el momento de la recuperación del crecimiento del niño hipotiroideo congénito durante los cinco primeros años de vida.
MATERIALES Y MÉTODOSEl presente es un estudio de tipo descriptivo, longitudinal y retrospectivo, para lo cual se revisó el archivo de datos de las historias clínicas de pacientes con diagnóstico de hipotiroidismo congénito recolectadas del Servicio de Endocrinología del Instituto de Salud del Niño en el periodo comprendido entre los años 1995 hasta el 2005. Se revisaron un total de 247 historias clínicas, de las cuales sólo 37 cumplían con los criterios de inclusión y contar, por lo menos, con cinco años de control en el Servicio de Endocrinología del Instituto Nacional de Salud del Niño. Los datos fueron recolectados en una Ficha de Datos, elaborada para este estudio y que incluía los criterios diagnóstico de Blanco López et al.(7). Se siguió la misma metodología utilizada en el trabajo de Del Águila et al. (Tabla1).
Tabla 1. Puntaje por evaluación clínica para sospecha diagnóstica de hipotiroidismo.
Edad de Diagnóstico Clínico del Hipotiroideo Congénito: Veinte Años Después.
19
El puntaje Z para los indicadores de talla/edad, peso/edad y peso/talla en los cinco primeros controles fue calculado con el programa EPI-INFO 2002, el cual luego fue promediado para determinar el patrón de crecimiento del hipotiroideo congénito.
El análisis de los datos recogidos fue realizado con el programa estadístico Statistical Package for the Social Science v15.0 (SPSS Inc. Chicago III).
RESULTADOSEn la tabla 2 se muestran la edad de diagnóstico en ambos géneros así como el peso y talla de nacimiento y en la figura 1 la distribución por sexos.
Tabla 2. Edad de diagnóstico y antropometría al nacimiento de pacientes con hipotiroidismo
congénito.
Femenino (n= 29) Masculino (n = 8)
M
± D. .E
4.4 ± 4.673.33 ± 0.5349 ± 1.9
± D. E.M
3.12 ± 7.53.55 ± 0.5850 ± 1.85
Edad de diagnóstico(meses)
Peso (kg)
la (cm)Tal
SEXO
78%
22%
FEMENINO (n=29)
MASCULINO (n=8)
En la figura 2 se muestra la frecuencia de signos y síntomas presentes en el momento del diagnóstico, siguiendo los criterios de Blanco López et al. (7),
Se halló una mayor incidencia en el sexo femenino (78%) que en el sexo masculino (22%), con una relación de 3.62/1 (Figura1). No se encontraron diferencias estadísticamente significativas entre los pesos y las tallas de ambos sexos al momento del nacimiento (Tabla2).
Figura 1. Distribución por sexos de los pacientes estudiados.
La edad promedio del contingente estudiado al momento del diagnóstico fue de 5.93 ± 5.28. En el sexo femenino la edad promedio de diagnóstico fue de 4.4 ± 4.67 meses, mientras que para el sexo masculino fue de 3.12 ± 7.5 meses.
Figura 2. Frecuencia de signos y síntomas clínicos en 37 pacientes con hipotiroidismo
congénito.
Natalia Tovar Gutiérrez, Alex Rojas Hernández, Fiorella Torres Matta, Rubén Susaya Campos, Carlos Del Águila Villar yJuan Manuel Falen Boggio.
Figura 3. Puntaje Z para T/E, P/E, y P/T al nacimiento y al momento del diagnóstico.
En la figura 3 se muestra el puntaje Z del peso, talla y peso/talla de los pacientes hipotiroideos al nacer y al momento del diagnóstico; el hipotiroidismo compromete estos parámetros, en especial el P/E.
El patrón de crecimiento del hipotiroideo congénito, evaluado en los cinco primeros controles, demuestra que el peso para la talla se mantiene adecuado dentro de los límites normales altos. Mientras que los valores de talla para la edad y peso para la edad muestran un patrón
20
Figura 4. Patrón de crecimiento del hipotiroideo congénito según puntaje Z para T/E, P/E y P/T.
diagnóstico clínico fue de 5,93 meses que fue mucho menor que la edad promedio encontrado por Del Águila et al. (11)
en 1992, que fue de 17,4 meses. Sin embargo, esta notable disminución en la edad de diagnóstico clínico es aún tardía desde que Raiti et al (12) han señalado que el diagnóstico y tratamiento antes de los 3 meses de edad mejora el pronóstico de los pacientes.
En relación al sexo, en el presente estudio se encontró una mayor incidencia en el sexo femenino, al igual que lo señalado en estudios anteriores (10, 11, 12)
. No se encontraron diferencias estadísticamente significativas entre los pesos y las tallas de ambos sexos al momento del nacimiento. Además, a diferencia del mencionado estudio, las características predominantes al momento del diagnóstico del hipotiroideo congénito, han variado según lo obtenido en nuestro estudio, desde que corresponden a los signos y síntomas tempranos.
Los datos de P/E y T/E en el momento del diagnóstico se encontraron fuertemente disminuidos, como se muestra en la figura 3, correspondiendo a retardo pondo-estatural; sin embargo, el peso para la talla se encontraba dentro de rango normal.
La instauración del tratamiento con L-tiroxina produce catch-up growth de la talla y del peso como se muestra en la figura 4, produciéndose un fenómeno de encarrilamiento tanto del peso y de la talla en lo que será su patrón de crecimiento y mejorando el pronóstico de estos pacientes (13, 14), sin embargo, en algunos casos se suele ver un cierto retardo en el desarrollo, lo cual parece estar relacionado con el momento de inicio de la enfermedad y del tipo de lesión en el eje tiroideo (2, 15, 16).
El método de Blanco-López ofrece gran sensibilidad y especificidad antes de los tres meses de edad (9), lo que hace de él un método práctico para el diagnóstico temprano de hipotiroidismo congénito. En todo paciente sospechoso mediante este método, debe realizarse un estudio de TSH y T4 Libre para la confirmación del diagnóstico.
En tanto no se establezca el Programa Nacional de Tamizaje para Hipotiroidismo Congénito, creemos que el entrenamiento de los futuros pediatras es fundamental para la detección temprana de esta patología y así evitar secuelas irreversibles.
-2.1 -2.0 -1.6 -1.1 -1.2
-0.8 -0.6 -0.4 -0.1 0.1
0.9 0.9 1.0 0.8 0.9
1° Control 2° Control 3° Control 4° Control 5° Control
PATRÓN DE CRECIMIENTO DEL HIPOTIROIDEO CONGÉNITO
z sco
re
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
2.5
Talla / EdadPeso / EdadPeso / Talla
DISCUSIÓN El hipotiroidismo congénito sigue siendo una de las primeras causas de retardo mental prevenible en el país. Por tal motivo, desde comienzos del presente siglo tanto el Ministerio de Salud como EsSalud (Seguridad Social), han iniciado el tamizaje para hipotiroidismo congénito de los recién nacidos en algunos centros hospitalarios de ambas instituciones. Sin embargo, este procedimiento no cubre a todos los recién nacidos a nivel del país. Por ello es importante que el futuro pediatra sepa identificar los signos tempranos presentes en el paciente hipotiroideo congénito a fin que puedan beneficiarse de un diagnóstico y tratamiento tempranos y evitar así las complicaciones de la enfermedad.
El diagnóstico clínico del hipotiroidismo congénito en el recién nacido se efectúa tardíamente debido a que los signos y síntomas clínicos se van instaurando en forma progresiva (10), por ello se ha tratado de confeccionar nomogramas con los signos y síntomas más frecuentes y de presentación temprana para detectar estos pacientes. Así, uno de los primeros en idear este tipo de instrumento fue Letarte et al (8)
, el cual fue desarrollado y mejorado por Blanco et al. (9), quien señaló que un puntaje ≥ 4 constituía diagnóstico de hipotiroidismo congénito.
Cuando se evaluó el puntaje para sospecha diagnóstica de hipotiroidismo congénito utilizando los criterios de Blanco-López se pudo constatar que en todos los casos estudiados el puntaje obtenido fue mayor o igual a 4. De otro lado, la edad de
ascendente a lo largo del periodo de control luego del diagnóstico y tratamiento (Figura 4).
Edad de Diagnóstico Clínico del Hipotiroideo Congénito: Veinte Años Después.
21
CONCLUSIONESA manera de conclusión podemos señalar que la edad de diagnóstico clínico del hipotiroidismo congénito en el Instituto Nacional de Salud del Niño desde 1992 ha disminuido en casi un año; de 17,4 a 5,93 ± 5,28 meses. Las manifestaciones clínicas más frecuentes son: facies abotagada, piel seca, fontanelas amplias, estreñimiento, hipotonía e ictericia. En el momento del diagnóstico, además de los signos y síntomas antes señalados, existe
retardo pondo-estatural, siendo el peso para la talla es adecuado.
RECOMENDACIONESEl tamizaje de hipotiroidismo congénito debe implementarse a nivel nacional a fin que los hipotiroideos congénitos sean detectados y tratados oportunamente y de esta manera evitar el retardo mental que tiene un alto costo social.
Natalia Tovar Gutiérrez, Alex Rojas Hernández, Fiorella Torres Matta, Rubén Susaya Campos, Carlos Del Águila Villar yJuan Manuel Falen Boggio.
2. Korbage de Araújo MC, Baptista Nunes da Silva MH, de Albuquerque Diniz EM et al. A tireóide no feto e no recém-nacido: peculiaridades funcionais e principais doenças tireoidianas. Pediatria (Sao Paulo) 2003; 25: 51 – 60
3. American Academic of Pediatrics Section on Endocrinology. New born screening for congenital hypothyroidism: recommended guidelines. Pediatrics 1993; 91: 1203 – 09
4. Rojas MI, Garmendia F, Solís-Rosas C et al. Detección precoz de Hipotiroidismo Congénito. Encuesta Preliminar. Diagnóstico 1984; 14:125-9.
5. Dávila Aliaga CR. Tamizaje neonatal: diagnóstico precoz para evitar discapacidad neurológica en niños. Boletín “MINSALUD” 2005; 9 (5 octubre)
6. Ermans A-M, Burdoux P, Delange F. Congenital hypothyroidism in endemic goiter. En: Copngenital Hypothyroidism, JH Dussault y P Walker, Eds. Marcel Dekker, Inc. New York. Basel, 1983, pág. 317 – 29
7. Mäempää J. Congenital hypothyroidism. Aetiological and clinical aspects. Arch Dis Child 1972; 47: 914 – 23
8. Letarte J, Dussault JH, Guyda H. Investigación clínica y por laboratorio de los lactantes hipotiroideos detectados precozmente. En: Collu R, Ducharme JR, Guyda H (ed): Endocrinología Pediátrica. Barcelona: A. Garrido Juan 1983; 435-36
REFERENCIAS BIBLIOGRÁFICAS
9. Blanco López A. , Amarilla T, Dorantes-Alvarez L. , Evaluación clínica y de laboratorio en pacientes con Hipotiroidismo congénito. Bol Med Hosp Infant Mex 1986, 43(4): 228-32
15. Simic N, Asztalos EV, Rovet J. Impact of neonatal thyroid hormone insufficiency and medical morbidity on infant neurodevelopment and attention following preterm birth. Thyroid 2009; 19: 395 – 401
16. Ramírez Y, Marchena H. Características neuropsicológicas del niño preescolar con hipotiroidismo congénito en la provincia de Cienfuegos. Rev Cubana Neuropsico l 2009; 4: 36 – 43 .
22
Raquitismo Renal Hipofosfatémico: Experiencia en 10 Años. Hypophosphatemic Renal Rickets: Experience in 10 Years. Graciela Sakihara Asato1, Héctor Choqque Tacca1, Alfredo Mendoza Bedriñaña1, Victoria López Córdova1, Gisela Pimentel Klose1, Mario Encinas Arana1, Adriana Ancieta Zerda1.
RESUMEN Objetivos: Los objetivos del estudio son los siguientes: analizar la frecuencia de raquitismo renal hipofosfatémico ligado al cromosoma X (XLH) en el lapso de 10 años, determinar hallazgos clínicos, de laboratorio y radiológicos, analizar el Z score en desviación standard (DS) en talla (HSDS) al diagnóstico y en el seguimiento y analizar eficacia de la terapia en el crecimiento longitudinal en relación a la edad de inicio del tratamiento. Materiales y métodos: El siguiente es un estudio retrospectivo, observacional y longitudinal de pacientes con raquitismo renal hipofosfatémico ligado al cromosoma X vistos en el Servicio de Nefrología del Instituto Nacional de Salud del Niño (INSN) en Lima, durante los años 2002-2011. Para el análisis estadístico se usó el programa Microsoft Office Excel 2007, tabla de NCHS 2005 y WHO Anthro 2009 para el cálculo de Z score talla. Resultados: De 123 pacientes con diagnóstico de raquitismo en la consulta ambulatoria del INSN, basados en criterios clínicos, de laboratorio y radiológicos, 22 (18%) correspondieron al diagnóstico de XLH. De ellos, el 82% fueron niñas y 73% pre-escolares. La edad promedio al inicio de la enfermedad fue 1.8 años y al diagnóstico (1raconsulta) 4.3 años. Cuatro (18%) proceden de provincias y el 55% tenía antecedente familiar. Hubo deformidad en miembros inferiores y retardo estatural en más del 70%, alteración en la marcha en el 50% y en un porcentaje menor ensanchamiento epifisiario, rosario raquítico y disminución de la fuerza muscular. Todos presentaron hipofosfatemia y fosfatasa alcalina aumentada. En 10 casos se realizó estudio de paratohormona (PTH), que fue normal y vitamina D activa, la que estuvo disminuida. En la orina se observó hiperfosfaturia con calciuria normal. En 19 pacientes se realizó porcentaje de reabsorción de fosfato, el que estuvo disminuido. Para el análisis de HSDS se agruparon en 3 series: a) Zscore> -1. 88: seis pacientes, b)Zscore< -1. 88 en menores de 6 años: siete pacientes, c)Zscore < -1. 88 en mayores de 6 años: tres pacientes. En el grupo(a) hubo mayor compromiso en la talla final en aquellos con terapia irregular. En el grupo (b) en tres de 7 pacientes el Z score final se deterioró profundamente en aquellos con tratamiento irregular. En el grupo (c), a pesar del inicio tardío del tratamiento, hay ligera mejoría en la talla final, ya que todos cumplieron la terapia regularmente, aunque no alcanzaron el crecimiento normal. El 73% recibió sales de fosfato, 12 pacientes tomaron calcitriol y 27% recibió diuréticos. Hubo rarefacción ósea en el 100% de los casos evaluados radiológicamente, edad ósea disminuida en el 64% y nefrocalcinosis en 5 casos. Conclusiones: El XLH es una causa importante de retardo del crecimiento, la forma esporádica puede presentarse en un porcentaje importante, la nefrocalcinosis es una complicación que se observa con relativa frecuencia, hay mayor incidencia en el sexo femenino por el tipod de herencia ligado a X.Palabras Claves: Raquitismo hipofosfatémico ligado al cromosoma X (XLH), factor de crecimiento fibroblástico 23(FGF23), Z score DS de talla (HSDS).
SUMMARY Objetives: Analyze frequency of X- linked renal hypophosphatemic rickets (XLH) in the period of 10 years, determine clinical, laboratory and radiological finding, analyze the height Z score DS (HSDS) at diagnostic and follow up and analyze the efficacy of therapy in the longitudinal growth in relation to age and at the starting of the treatment. Methods:This is a retrospective, observational and longitudinal study in patient with X- linked renal hypophosphatemic rickets, which were seen, in the Nephrology Service of the Instituto
Nacional de Salud del Niño (INSN) in Lima, during the years 2002-2011. We used the Microsoft Office Excel 2007 program, NCHS 2007 table and WHO Anthro 2009 for the calculation of Z score size. Results: In 123 outpatients in the INSN with diagnosis of rickets, 22 (18%) were XLH based on clinical, laboratory and radiologicical criteria, of those, 82% were girls and 73% pre-school. The average age at onset was 1.8 years and at diagnosis (1st query) was 4.3 years. Four (18%) came from provinces and 55% had a familiar history. There were lower limb deformity, statural
1 Servicio de Nefrología del Instituto Nacional de Salud del Niño. Lima – Perú Correo electrónico Dra. Graciela Sakihara Asato: [email protected] Recibido: 29 de Abril del 2013. Aceptado: 6 de Enero del 2014
Rev. Perú. pediatr. 67 (1) 2014
23
siendo su trasporte estrechamente regulado. El fosfato dietético, la paratohormona, el 1-25 dihidroxicolecaciferol (1-25(OH)2D3) y el factor de crecimiento fibroblástico 23 (FGF23), uno de los péptidos de la fosfatonina, son los principales reguladores de la reabsorción de fosfato en el riñón (2, 3)
. El fosfato es un componente clave de muchos caminos metabólicos, como desarrollo esquelético, mineralización ósea, composición de la membrana, estructura del nucleótido, mantenimiento de pH plasmático, señalamiento celular, metabolismo energético y regulación de la síntesis de proteínas(4)
, por lo que el mantenimiento de fosfatointra y extraceluar es importante. Avances recientes en técnicas genéticas y modelos animales han identificado muchos desórdenes genéticos de la homeostasis del fosfato. Las mutaciones en CoNaPi2a y CoNaPi2c, la desregulación hormonal de PTH, FGF23 y Klotho (cofactor esencial del receptor 1 FGF23) son los responsables primarios en la mayoría de los desordenes genéticos del trasporte de fosfato(2, 5, 6)
.
El FGF23 es una proteína sintetizada por osteocitos y osteoblastos, que actúa como un factor fosfatúrico, inhibidor de la 1-25(OH)2 D3 y regulador de la síntesis y secreción de PTH. Inhibe la actividad de 1α hidroxilasa y estimula la actividad de 24 hidroxilasa, llevando a niveles disminuidos de la 1-25(OH)2 D3
(3, 7).
El FGF23 media sus acciones interactuando con su receptor 1(FGFR1), siendo el receptor primario en la regulación del CoNaPi2a y 2c (8)
. Existen varios receptores FGF23 presentes en muchos tejidos, pero las acciones directas del FGF23 están limitadas sólo al riñón y la glándula paratiroidea. Esta especificidad es provista por una proteína transmenbrana Klotho(1,
3, 4), un cofactor esencial para la interacción de FGF23
con sus receptores (2, 9), como se puede observar en
la siguiente figura 1.
delay by more than 70%, gait disturbance 50% and a lower percentage of epiphyseal enlargement, raquitic rosary and diminished of muscle strength. All of them had hypophosphatemia and high level of alkaline phosphatase. In 10 cases it was performed dosage of parathyroid hormone, which was normal, but active vitamin D was diminished. Hyperphosphaturia was observed in urine, with calciuriawas normal. In 19 patients we calculated phosphate tubular reabsorption percentage, which was diminished. HSDS analysis results can be grouped in 3 series: a) Z Score>-1.88 in six patients, b) Z Score< -1.88 under 6years in 7patients, c) Z Score < -1.88 over 6 years sin three patients. In group(a) there was a greater commitment to final height in those with irregular therapy. In group (b) 3 from 7 patients, the final Z score was profoundly impaired in those with irregular therapy. In group(c) despite its late start of treatment, there is aslight improvement in final height, since all therapy was met regularly, but did not reach the normal growth. 73% of the patients received phosphate salts, 12 patients took calcitriol and 27% diuretics. There was bone rarefaction in 100% of the X-ray performed cases; diminished bone age was seen in 64% and nephrocalcinosis in 5 cases. Conclusions: The XLH is a major cause of growth retardation, there may be the sporadically form in a significant percentage, nephrocalcinosis is a common complication and there is a higher incidence in women because of X-linked inheritance. Keywords: X- linked hypophosphatemic ricket(HLH), fibroblastic growth factor 23 (FGF23), height score S, (HSDS).
INTRODUCCIÓN La regulación de la homeostasis del fosfato es fundamental para muchos procesos biológicos y, tanto la hipofosfatemia como la hiperfosfatemia, pueden tener consecuencias clínicas adversas. Sólo un porcentaje pequeño(1%) del fosfato corporal total está presente en el fluido extracelular (medición rutinaria en laboratorio), no reflejando éste el fosfato corporal total(1, 2)
.
El fosfato es absorbido del tracto gastrointestinal, vía la ruta transcelular mediada por el cotrasportador sodio fosfato 2b (CoNaPi2b) y através del camino para celular. Aproximadamente el 85% del fosfato filtrado es reabsorbido en el riñón, predominantemente en el túbulo proximal por los cotrasportadores sodio fosfato 2a y 2c (CoNaPi2a, CoNaPi2c), presentes en la membrana en borde de cepillo del túbulo proximal,
Heparin
Heparin
FGF23
FGF Receptor
FGF Receptor
1a-hydroxylase expression
1,25(OH)2 Vitamin D3
klotho
klothoFGF23
P P
P P
NaPi. 2a
Lumen
3Na+
2Na+
HPO42.
HPO42.
NaPi. 2c
Figura 1: Mecanismo de acción FGF23 con receptor FGF, Klotho y heparina estabilizando el complejo (2)
La hipofosfatemia es una condición frecuente y la forma severa puede inducir hemólisis, rabdomiolisis, trombocitopenia, insuficiencia respiratoria, disfunción cardiaca y compromiso neurológico (3)
, por lo que su corrección rápida es necesaria para evitar complicaciones severas. La presencia de cantidades adecuadas de fosfato es crucial para el proceso de apoptosis de los condrocitos maduros en la placa de crecimiento. Sin la presencia suficiente de este mineral, los condrocitos no irán a la apoptosis y la cadena de eventos fisiológicos normal que incluye invasión de vasos sanguíneos y la generación de nuevo hueso será bloqueado. La regulación aberrante de este proceso de desarrollo condiciona desorden de la placa de crecimiento, produciendo acúmulo y engrosamiento de ella, resultando en raquitismo y crecimiento retardado(10, 11). En el tejido óseo restante la hipofosfatemia resultará en osteomalacia, debido a la formación insuficiente de hidroxiapatita(10, 11).
Dentro de las causas de raquitismo renal, la hipofosfatémica ligada a X (XLH), es la forma heredada más frecuente, representando el 80% de los casos, teniendo una prevalencia de 1/20000 niños. Existen otras formas de raquitismo renal, como se muestra en la siguiente clasificación (12, 13, 14).
En el esquema se muestra la clasificación de las alteraciones de la regulación del fosfato en las diferentes formas clínicas de raquitismo hipofosfatémico en relación a la alteración fenotípica y circulación de fosfatoninas(15).El raquitismo renal hipofosfatémico ligado al cromosoma X (XLH) se manifiesta en los primeros
CLASIFICACIÓN DE RAQUITISMO RENAL
1. - Por trastorno en el trasporte de fosfatos: a) Hipofosfatemia familiar:
1. Ligada al cromosoma X2. Autosómica dominante3. Autosómica recesiva4. Raquitismo hipofosfatémico hereditario con
hipercalciuria. b) Esporádico c) Raquitismo oncogénico. 2. - Por raquitismo seudocarencial:
1. Tipo I: por trastorno de la 1-hidroxilasa2. Tipo II: por trastorno de receptores de la
ALTERACIONES DE LA HOMEOSTASIS DEL FOSFATO ASOCIADA CON ALTERACIONESDE LA PRODUCCIÓN Y CIRCULACIÓN DE FOSFATINAS (15)
ALTERACIONES CLÍNICAS
Osteomalacia inducidapor tumor
Raquitismo hipofosfatémico ligado al x
Raquitismo hipofosfatémicoautosómico dominante
Mutación en el gen que codi�ca la endopeptidasaPHEX que produce un in-dremento de la concentrade FGF23, sFRP-4 y MEPE
Mutación en el gen FGF23que induce la formación de
Mutación en gen OMP-1 asociado con de FGF23
Mutaciones en los genes GALNT3,
Concentracionesde FGF23 y FGF7
fGF23, concentraciones FGF23 yklotho (niveles de FGF23)
una forma mutante de FGF23resistente a proteolisis i
similiar
similiar
Raquitismo hipofosfatémicoautosómico recesivo
Calcinosis tumoral Hiperfosfatemia, fosfaturia, calcitriol N, calci�caciones ectópicas
insu�ciencia renal
similiar
Hipofosfatemia, hiperfosfaturia, calcitriol,osteomalacia, defecto de mineralización
Exceso producción defosfatoninas ( FGF23,sFRP.4,MEPE,FGF7)
FENOTIPO CLÍNICO FISIOPATOLOGÍA
Hiperfosfatemia, hipofosfaturia, decalcitriol
2 años de la vida, cursando con hipofosfatemia, fosfaturia, niveles bajos de 1-25(OH)2 D3, FGF23 incrementado, PTH normal, retardo estatural progresivo, arqueamiento de miembros inferiores y aparición de raquitismo en la infancia (defecto de formación delosteoide debido a carencia de fosfato en el hueso) (12-15).
Raquitismo Renal Hipofosfatémico: Experiencia en 10 Años.
GENÉTICA DEL PADECIMIENTO DOMINANTE LIGADO AL CROMOSOMA X
Los padres afectados trasmiten la enfermedad a todas sus hijas a través del cromosoma X. Las madres afectadas trasmiten la enfermedad al 50% de su descendencia independientemente del sexo.
El gen responsable es el PHEX (gen regulador del fosfato con homología a endopeptidasa en el cromosoma X). Codifica una metaloproteasa dependiente del zinc que es hallada en la membrana de la superficie celular de los osteoblastos, osteocitos, y odontoblastos. Han sido descritas mutaciones múltiples en el gen PHEX, la mayoría de los cuales resultan en una pérdida funcional de la proteína PHEX, que se encuentra localizada en el brazo corto del cromosoma X, en la región Xp22. 1.
Ocasiona una reabsorción deficitaria de fosfato en el túbulo proximal y una disminución en la producción del calcitriol, debido a que el gen mutado impide la degradación del FGF23 circulante y su incremento altera el CoNaPi2a y 2c en pacientes con XLH (2, 15, 16, 17)
.
El tratamiento es suplementar sales de fosfato y vitamina D activa. Puede usarse además diuréticostiazidas y ahorradores de potasio.
Debemos tener en cuenta que la nefrocalcinosis e hiperparatiroidismo son complicaciones secundarias de la terapia. El uso de calcimiméticos en los casos de hiperparatiroidismo primario o secundario puede ser beneficioso. Tales opciones terapéuticas no han sido usadas en estos pacientes, pero se reportan resultados prometedores (3, 18, 19)
.
En el futuro, los anticuerpos específicos probablemente serán el enfoque más fisiológico. A la fecha, aunque los resultados son prometedores en modelos animales, ningún reporte ha sido publicado en humanos(20)
.
La corrección quirúrgica de las deformidades óseas en miembros inferiores se deben realizar en la adultez temprana,cuando el cartílago de crecimiento se ha consolidado(13, 14, 15)
.
Los objetivos del estudio son los siguientes: analizar la frecuencia de XLH en el lapso de 10 años (2002-2011), determinar los hallazgos
clínicos, de laboratorio y radiológicos, analizar el score de desviación standard en talla (HSDS) al diagnóstico y seguimiento y analizar la eficacia de la terapia en el crecimiento longitudinal con relación a la edad de inicio del tratamiento.
MATERIALES Y MÉTODOSEl siguiente es un estudio retrospectivo, observacional y longitudinal basado en los datos clínicos obtenidos de la consulta ambulatoria del Servicio de Nefrología del Instituto Nacional de Salud del Niño (INSN) durante los años 2002-2011.
Se revisaron las historias clínicas de los pacientes con diagnóstico de raquitismo, los que fueron proporcionadas por la Oficina de Estadística, vistos ambulatoriamente en los consultorios de genética, traumatología, endocrinología y nefrología. De 123 paciente con diagnóstico de raquitismo, 22 (18%) correspondieron al diagnóstico de XLH, basados en a) criterios clínicos, que incluyen retardo de crecimiento, arqueamiento de miembros inferiores, alteración en la marcha, disminución en la fuerza muscular y ensanchamiento epifisiario; b) criterio de laboratorio, que incluye hipofosfatemia, normocalcemia, fosfatasa alcalina elevada, PTH normal o elevada, disminución de 1-25 (OH)2 D3, hiperfosfaturia, normocalciuria y porcentaje de reabsorción tubular de fósforo (%RTP) disminuido; c) criterio radiológico,que considera signos radiológicos de raquitismo.
Se analizaron parámetros demográficos, clínicos, de laboratorio, radiológicos, terapéuticos y complicaciones en los 22 pacientes.
En el análisis estadístico se utilizó el programa Microsoft Office Excel 2007. El procesamiento de datos se realizó por estadística descriptiva standard (tabulación computarizada, elaboración de tablas, gráficas para las variables estudiadas). Para el cálculo de Z score talla (HSDS), se aplicó el programa en Excel 2005 de la NCHS y WHO Anthro 2009.
RESULTADOSDe 123 pacientes con diagnóstico de raquitismo, 22 (18%) correspondieron al diagnóstico de XLH, según los criterios señalados previamente.
El 82% fueron de sexo femenino. La edad promedio al inicio de la enfermedad fue 1.8 años, (DS 0.88), y al diagnóstico (1ra consulta) 4.3 años, (DS 2.7).
26
Tabla 1: Sexo, Edad, seguimiento y Procedencia de los 22 Pacientes con Raqui�smo Renal Hipofosfatémico
Ligado al Cromosoma X.
Paciente Sexo Edad * inicio
Edad * diagnós�co Seguimiento* Procedencia
A f 1.6 2.7 1.3 Lima B f 1.5 2.5 3 Lima C f 1.5 6.8 4.3 Lima D f 4 10 1 Lima E f 1.6 2.5 6.3 Lima F f 1.7 2.8 13.8 Lima G f 1 2.1 9.4 Lima H f 2 2.4 15.3 Lima I f 2.1 7.1 3.7 Lima J f 1 1.8 5.4 Lima K f 1.6 2.1 9.1 Lima L f 1.2 2.1 3.8 Lima
M m 1.5 2.6 1.3 Chimbote N f 4.2 5.8 3.6 Lima O m 0.6 1.9 1.7 Lima P f 1.5 5 0.5 Chimbote Q f 1.7 3.6 Lima R f 1.3 2.7 Lima S m 1.5 11 Lima T m 3 6 Lima U f 2 6.6 Huancavelica V f 2 4.3 Cajamarca
MMII: Miembros inferiores , OM: osteo muscular, FM: fuerza muscular.
77% 73%
50%32%
27%18%
5%
6
1
Gráfico 1: Manifestaciones Clínicas de los Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X.
Dieciséis (73%) fueron pre-escolares y de ellos, 5 menores a 2 años. Seis (27%) fueron mayores a 6 años. De los 22 pacientes, 18% proceden de provincias (tabla 1). Hubo antecedente familiar en 12 casos (55%).
Las manifestaciones clínicas se señalan en el gráfico 1. Hubo deformidad en miembros inferiores y retardo pondoestatural en más del 70% de los casos, dificultad en la marcha en el 50% y en un porcentaje menor se observó ensanchamiento epifisiario, rosario raquítico, dolor osteo-muscular y disminución de fuerza muscular.
En el análisis de laboratorio, el calcio, creatinina, pH y bicarbonato séricos fueron normales al diagnóstico y en el último control. El fósforo sérico fue en promedio 2.5 (DS 0.31) al momento del diagnóstico y 2.7(DS 0.45) al seguimiento, así como la fosfatasa alcalina en promedio 1126 (DS 690) y 1119 (DS 323)respectivamente. En diez casos se realizó estudio de PTH siendo en promedio 65.8 (DS 16.4) y se determinó1-25(OH)2 D3 con un valor promedio de 19.8(DS 12.4)en las evaluaciones iniciales(tabla 2).
Tabla 2: Laboratorio Sérico en Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X.
Todos tuvieron fosfaturia y el calcio en orina fue normal. En 19 pacientes se determinó %RTP, siendo en promedio 69%, (DS 16) (tabla 3). En el análisis de orina no hubo proteinuria ni glucosuria y en 2 casos se evidenció hemoglobina y leucocituria
leve (no mostrados en la tabla 3).
Tabla 3: Laboratorio en Orina en Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X.
De los 3 casos que no se realizaron %RTP, dos de ellos tuvieron estudio de PTH normal y 1-25(OH)2 D3 disminuida y el tercer paciente tuvo fósforo sérico en 2.8 mg/dl y fosfatasa alcalina 969 U/L.
En los estudios radiológicos, se observó rarefacción ósea en 18 pacientes, que corresponden al 100% de los casos determinados y se evidenció edad ósea retrasada en el 64% de los casos. Hubo nefrocalcinosis en 5 pacientes (28%) y urolitiasis en 1 (5. 5%) de 18 ecografías renales realizadas (tabla 4).
Tabla 1: Sexo, edad, seguimiento y procedencia de los 22 Pacientes con Raquitismo Renal
Hipofosfatémico Ligado al Cromosoma X.
Raquitismo Renal Hipofosfatémico: Experiencia en 10 Años.
27
Tabla 4: Estudios de Imágenes en Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X.
Para el análisis del score de desviación standard en talla (HSDS) se agruparon en 3 categorías: a) pacientes con Z score >-1.88, seis casos, siendo la mayoría menores de 3 años, a la edad del diagnóstico, excepto un paciente de 5.8 años al momento de la primera consulta, pero quien procedía de España, estando ya en tratamiento; b) siete pacientes con Z score < -1.88 menores de 6 años; c) tres pacientes con Z score < -1.88 mayores de 6 años (tablas 5 y 6).
Tabla 5: Z Score en Talla en Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X. Grupo A
En el grupo A se puede observar, que hubo mayor afectación en la medición final, en aquellos casos que siguieron tratamiento irregular. En el grupo B, en 3 de siete pacientes, su Z score final tuvo un marcado deterioro, en correlación con terapia irregular, versus aquellos que si recibieron tratamiento regular, en los que hubo mejoría o se mantuvieron con igual Z score. En el grupo C por el contrario, observamos que a pesar del inicio tardío del tratamiento, hubo ligera mejoría en su talla final con relación a la inicial, ya que todos cumplieron con el tratamiento (tablas 5 y 6).
Tabla 6: Z Score en Talla en Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X. Grupos B y C.
Seis pacientes, a pesar de haber tenido una sola consulta, fueron incluidos en el estudio, ya que reunieron criterios clínicos, de laboratorio y signos radiológicos de raquitismo. Cuatro de ellos tuvieron Z score muy deteriorado (tabla 7).
Tabla 7: Z Score en Talla en Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X.
En el gráfico 2 se muestra el score de desviación standard en talla (HSDS) de los 4 niños y en el gráfico 3 el HSDS de las 18 niñas a través del tiempo de seguimiento en la consulta, haciendo la salvedad de que 2 varones y 4 mujeres tuvieron una sóla consulta como podemos observar en ambas gráficas.
Gráfico2: Z Score T/E. Niños con Raquitismo Renal Hipofosfatémico Ligado al Cromosoma X.
Gráfico 3: Z Score T/E. Niñas con Raquitismo Renal Hipofosfatémico Ligado al Cromosoma X.
El tiempo promedio de seguimiento ambulatorio fue de 5.2 años (DS 4. 15) (tabla 1). Dieciséis pacientes (73%) recibieron tratamiento con sales de fosfato, siendo regular la terapia en 10 de ellos. Doce pacientes (54%) recibieron calcitriol (1-25(OH)2 D3), siendo en 5 su uso constante. Seis pacientes (27%) recibieron hidroclorotiazida-amiloride, siendo su consumo regular en 3 de ellos (gráfico 4).
Gráfico 4: Tratamiento recibido por los Pacientes con Raquitismo Renal Hipofosfatémico Ligado al
Cromosoma X.
DISCUSIÓN El siguiente estudio es un estudio retrospectivo, observacional y longitudinal de pacientes con diagnóstico de Raquitismo Renal Hipofosfatémico Ligado al Cromosoma X (XLH) atendidos en la consulta ambulatoria del Servicio de Nefrología del Instituto Nacional de Salud del Niño durante un periodo de 10 años (2002-2011), con la finalidad de analizar los 4 objetivos señalados previamente.
Los 22 casos reportados correspondieron al diagnóstico de XLH, cuya variedad clínica es la más frecuente, siendo su inicio generalmente antes de los 2 años.
Se observó que hay retraso en acudir a la primera consulta, pues el diagnóstico se realizó
0
2
4
6
8
10
12
14
16
18
Fósforo Vitamina D Modure�c
N
p
a
c
i
e
n
t
e
s
Si
No
54.5
45.5
en promedio a los 4. 3 años, habiendo iniciado la enfermedad a la edad promedio de 1. 8 años. La edad al diagnóstico, en 3/4 de los casos, fue en la etapa pre-escolar y de ellos sólo cuatro (18%) fueron menores a 2 años y 6 acudieron tardíamente a la consulta. Esto significa que sólo 1/5 de los casos acuden a la edad probable de inicio de la enfermedad.
No hubo diferencia en la edad de diagnóstico entre los pacientes que vienen de provincias en comparación con los que viven en Lima. Hay el mismo grado de desconocimiento en ambas poblaciones. Hubo un predominio en el sexo femenino por ser un padecimiento dominante ligado al cromosoma X (1, 14, 21)
.
Doce pacientes (55%) tuvieron antecedentes familiares, los otros 10 pacientes probablemente fueron formas esporádicas. Se reporta que el XLH es causado por mutaciones inactivantes en el PHEX en el 87% y 72% de los casos familiares y esporádicos respectivamente (1, 22). Otros autores refieren que el análisis genómico del PHEX detecta más de 3/4 de las mutaciones en los pacientes con XLH familiar y 1/3 en los casos de mutaciones esporádicas (23, 24). Los hallazgos clínicos, los de laboratorio y radiológicos, son similares a los reportados en otros estudios (1, 12, 14, 15).
El análisis de score de desviación standard en talla (HSDS) fue mejor en aquellos que recibieron tratamiento en forma regular, independientemente del grupo etario, tal como se ve en la tabla 6, aunque no alcanzaron el crecimiento normal. En el grupo A (tabla 5), se puede observar que en los últimos controles hubo deterioro del Z score en 5 de ellos (Z S >-2) excepto el paciente procedente de España, quien tuvo un Z score final de -1. 81, pero que empeoró en relación a su valor inicial, que fue -0. 4, debido, en todos ellos, al tratamiento irregular que tuvieron en el periodo de seguimiento.
Una observación a tener en cuenta es, que el fósforo sérico usualmente no se normaliza con el suplemento de sales de fósforo, pero se observa mejoría en el crecimiento. La fosfatasa alcalina se incrementa para tratar de proveer más fosfato a las células óseas, una vez que cantidades adecuadas de fósforo son suplementadas, la actividad enzimática disminuye, por tal motivo la fosfatasa alcalina es un buen indicador de la homeostasis
Raquitismo Renal Hipofosfatémico: Experiencia en 10 Años.
29
del fósforo dentro del tejido óseo (1). En relación
al uso de vitamina D activa, se reporta un efecto directo corrigiendo la deformación ósea (1, 25, 26)
.
El tratamiento es con sales de fósforo, pero la mejoría de la mineralización ósea es menos efectiva que en el raquitismo hipofosfatémico autosómico dominante en que el tratamiento con sales de fósforo es más eficaz, pero su presentación clínica es sumamente rara, siendo su aparición mayormente en la adolescencia o en la adultez(13)
.
El tratamiento recibido por nuestros pacientes fue en base a sales de fosfato (50-100 mg/kg de fósforo elemental), calcitriol 15-60 ng/kg, hidroclorotiazida 1-2. 5 mg/kg y amiloride 0. 3-0. 5 mg/kg (1, 2, 3, 5)
. En cinco casos (28%) hubo nefrocalcinosis, complicación frecuente de la terapia con calcitriol, aunque en nuestro reporte se encuentra por debajo de lo reportado en otras series(1, 27). La patogénesis de la nefrocalcinosis es pobremente entendida, los factores de riesgo incluyen episodios de hipercalciuria e hipercalcemia, en adición a la cristalización de oxalato de calcio, secundario a la hiperoxaluria entérica(1, 3, 10).
Estudios recientes sugieren que dosis menores de fosfatos y calcitriol pueden ser administrados con la introducción de calcimiméticos como terapia adyuvante y evitar así complicaciones como la nefrocalcinosis(1, 28).
La hipofosfatemia crónica en niños se debe fundamentalmente a desórdenes genéticos, tanto de trasportadores de fósforo primarios como desregulación hormonal en la homeostasis del fósforo, esto implica que debemos realizar estudios hormonales y genéticos en aquellos pacientes que tienen criterios diagnósticos de raquitismo hipofosfatémico y diferenciar a que forma clínica pertenecen y de esa manera indicar la terapia más adecuada(1, 2)
.
CONCLUSIONESEl Raquitismo Renal Hipofosfatémico Ligado al Cromosoma X es una causa importante de retardo en el crecimiento en la edad pediátrica. Hay mayor incidencia en el sexo femenino por el tipo de herencia dominante ligado al cromosoma X. La forma esporádica puede observarse en un porcentaje importante. La nefrocalcinosis es una complicación que se debe tener presente, no así la nefrolitiasis, cuya presencia es excepcional. Nuestros hallazgos enfatizan la importancia del diagnóstico temprano, que nos permita dar tratamiento antes de que el crecimiento se vea más comprometido. No hubo influencia del género en el crecimiento. El crecimiento progresivo insuficiente se pudo evitar con el inicio precoz del tratamiento.
1. Quinlan C, Guegan C, et al. Growth in PHEX-associated X-linked hypophosphatemic rickets: the importance of early treatment. Ped. Nephrol. 2012; 27: 581-588.
2. Gattineni J, Baumi M. Genetic disosrders of phosphate regulation. Ped Nephrol. 2012; 27:1477-1487.
3. Bacchetta J, Salusky I. Evaluation of hypophosphatemia: Lesson from patients with genetic disorders. AJKD. 2012; 59:152-159.
4. Bugg NC, Jones JA. Hypophosphatemia. Pathophysiology, effects and management on the intensive care unit. Anesthesia. 1998; 53:895-902.
5. Gattineni J, Baum M. Regulation of phosphate transport by fibroblastic growth factor 23 (FGF23) implications for disorders of phosphate metabolism. Ped Nephrol. 2010; 25:591-601.
6. Li H, Martin AC et al. Compound delection of FGF 23 and FGFR4, Partially rescues the Hyp Mouse phenotype. Am J Physiol Endocrinol Metab. 2010; 300:E08-517.
7. Yamasaki Y, Tamada T et al. Anti-FGF23 neutralizing antibiodies show the physiological role and structural features of FGF23. J Bone Miner Res. 2008; 23:1509-1518.
8. Gattineni J, Bates C et al. FGF23 decreases renal NaPi-2a and NaPi2c expression and indices hypophosphatemia in vivo predominantly via FGF receptor1. Am J Physsiol RenalPhysiol. 2009; 297:F282-F291.
10. Goretti M, Penido M, Alon U. Phosphate homeostasis and its role in bone health. Ped Nephrol. 2012; 27:2039-2048.
11. Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab. 2009; 27:392-401.
12. Gordillo G. Raquitismo hipofosfatemico. Nefrología Pediátrica. Editorial Elsevier España SA 2da Ed. 2003; 301-307.
13. Negri AL, Negrotti T. Distintas formas de presentación clínica del raquitismo hipofosfatémico autosómico dominante por mutación del FGF23 en una familia. Medicina. 2004; 64:103-106.
14. Quintana López G. Raquitismo hipofosfatémico ligado a X (XLH). Rev Colombiana Reumat. 2008; 15:117-122.
15. Luis Yanes Ma. L. Raquitismo hipofosfatémico familiar. Bol Pediatr (España). 2010; 50:56-61.
17. Amir S, Ahzadeh N et al. Hereditary disorder of renal phosphate wasting. Nature Rev Nephrol. 2010; 6:657-665.
18. RaederH, Shaw N et al. A case of X-linked hypophosphatemic rickets: complications and the therapeutic use of cinacalcet. Eur J Endocrinol. 2008; 159 suppls. :5101-5105.
19. Alon US, Levy Olomucky R et al. Calcimimetics as an adyuvant treatment for familiar hypophosphatemic rickets. Clin J Am Soc Nephrol. 2008; 3:658-664.
20. Aono Y, Yamazaki Y et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Rev. 2009; 24:1879-1888.
21. Yuan B, Takaiwa M et al. Aberrant PHEX function in osteoblast and osteocytes alone underlies murine X-linked hypophosphatemia. J Clin Invest. 2008; 118:723-734.
22. Gauchet C, Walrant-Debray O et al. PHEX analysis in 118 pedigrees reveals mew genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125:401-411.
23. Holm JA, Nelson AE et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001; 86:3889-3899.
24. Christie P26T, Harding B et al. X-linked hypophosphatemia attributable to pseudoexons of the PHEX gene. J Clin Endocrinol Metab. 2001; 86:3040-3044.
25. Boskey A, Frank A et al. he PHEX transgenic corrects mineralization defects in 9 month-old hypophosphatemic mice. Calcif. tissue Int. 2009; 84:126-137.
26. Ecarot B, Glurieus FH et al. Effect of 1-25 dihydroxyvitamim D3 treatment on bone formation by transplanted cells from normal and X-linked hypophospphatemic mice. J Bone Miner Res. 1995; 10:424-431.
27. Kooh SW, Binet A et al. Nephrocalcinosis in X-linked hypophosphatemic rickets. Its relationship to treatment, kiney function and growth. Clin Invest Med. 1994; 17:123-130.
28. Alon US, Levy-Olomucki R et al. Calcimimetics as an adjuvant treatment for familiar hypophosphatemic rickets. Clin J Am Soc Nephrol. 2008; 3:658-664.
Raquitismo Renal Hipofosfatémico: Experiencia en 10 Años.
31
¦>UL£¥>>>O>>>NSMNQMPNOQ>>>OPXSVXQN>LªL
32
RESUMEN Describimos una recién nacida afectada con Síndrome Moebius, cuya madre usó Misoprostol a las 9 semanas de la gestación con fines abortivos, ingiriendo dos tabletas vía bucal (400ug) y aplicándose vaginalmente tres tabletas (600ug) sin provocar el aborto. Se describe el perfil clínico de un paciente y su evolución hasta el alta. Además se comparan las diferencias clínicas presentadas en nuestro paciente con los casos descritos en la literatura. Se postulan varias hipótesis respecto a la etiopatogenia del Síndrome y su probable asociación al uso de Misoprostol (Citotec®) con fines abortivos en el primer trimestre de la gestación. El Misoprostol ha sido descrito como responsable de disrupción vascular, secundaria a sus efectos uterotónicos y alteración del flujo sanguíneo fetal, que deriva en hipoxia y hemorragia. Palabras claves: Misoprostol, Síndrome Moebius.
Síndrome Moebius en el Hospital Apoyo Iquitos. Posible Causa por Exposición a Misoprostol. Reporte de un Caso. Moebius Syndrome in the Hospital Apoyo Iquitos. Possible Cause by Exposure to Misoprostol. Report of one Case. Hermann Silva Delgado1, Javier Villaverde Montoya2.
CASO CLÍNICO
Rev. Perú. pediatr. 67 (1) 2014
SUMMARY
We describe a newborn patient affected with Moebius Syndrome whose mother used Misoprostol for abortive purpose at the ninth week of pregnancy, by taking two tablets (400ug) through the oral tract and applying three tablets (600ug) in the vagina without cause miscarriage. It is describe the clinical profile of a patient as well as his evolution up to the discharge day. Also, clinical findings in our patient are compared with cases described in literature. Several hypotheses are postulated concerning the Syndrome’s ethiopathogenesis and its probable association with the use of Misoprostol (Citotec®) during the first trimester of pregnancy with abortive purpose. Misoprostol has been described as responsible of vascular disruption, secondary to its uterus tonic effects with alteration of fetal blood flow which causes hypoxia and hemorrhage. Keywords: Misoprostol, Moebius Syndrome.
INTRODUCCIÓN
El Síndrome Moebius (MIM 157900) antes llamado Secuencia de Moebius, es una anomalía congénita infrecuente, asociada a parálisis de los pares VI y VII, generalmente bilateral, lo que conlleva a un cierre incompleto de los párpados, estrabismo convergente, ausencia de movimientos de los músculos de la expresión facial y escurrimiento permanente de saliva. Fue descrita por Von
Graefe en 1880 e identificada como una entidad nosológica con características clínicas clásicas por el neurólogo Paul Julius Moebius en 1888, asignándose el epónimo correspondiente. En la actualidad, al síndrome se le vincula el compromiso de otros pares craneales, principalmente el V, IX, X y XII, causando trastornos de la deglución, reflujo gastroesofágico, hipoglosia, dificultades para succionar, regurgitación gástrica y dificultades para el vaciamiento gástrico; se han descrito casos con compromiso de hasta siete pares craneanos y otros que afectan sólo el VIII y XI con paresia facial izquierda, parálisis unilateral del VI y VIII par, con estenosis severa del conducto auditivo interno, compatible con una aplasia o hipoplasia del nervio vestíbulococlear, además de la presencia de deformidades del tórax, escoliosis, hipoplasia pectoral y de extremidades como el pie equino varo, según lo reportado por Lora et. al.1. Villafranca refiere además, agenesia de músculos pectorales y glándulas mamarias (Síndrome Poland MIM 1738000), asociada a luxación congénita de cadera2. Villagra-Sanjurjo describe un caso con agenesia de músculo pectoral derecho, braquisindactilia en extremidad superior derecha e hipoplasia de segundo dedo de mano izquierda 3. Entre las anomalías orofaciales se observa microtia, micrognatia, paladar hendido, paladar ojival, microftalmia y finalmente retardo mental 4, 5, 23. Roig Quilis, propone una denominación de carácter
1.Médico Pediatra. Servicio de neonatología del hospital Apoyo Iquitos. Facultad de Medicina Universidad Nacional Peruana. Correo electrónico: [email protected]édico Pediatra. Servicio de Pediatría del Hospital Apoyo Iquitos. Ricibido: 1 de Agosto del 2013. Aceptado: 30 de Enero del 2104
33
anatómico funcional al síndrome descrito, como disgenesia del tronco encefálico6. Cammarata-Scalisi afirma que el cuadro sólo puede explicarse si se entiende como un síndrome de “mal desarrollo” del rombencéfalo7.
No está claro por completo la etiología y patogénesis del Síndrome, sin embargo se han propuesto dos causas: una genética y otra isquémica. La causa genética de este Síndrome se encuentra a nivel del cromosoma 13q12. 2-3 y se hereda con un patrón autosómico dominante8, pero también se han reportado casos con herencia autosómico recesiva o ligado al cromosoma X6, o de aparición esporádica y otras debidas a anomalías cromosómicas10. Además la teratogenicidad de ciertos medicamentos o tóxicos durante la gestación, tales como alcohol, cocaína, talidomida, ergotamina y el misoprostol son propuestos como un factor importante 8, 9. Con respecto a la segunda causa, la hipótesis más aceptada es la disrupción morfogénica de las arterias basilar o vertebral en la 17ava semana de gestación, que llevaría a la involución de los núcleos de los pares craneales VI y VII, y a la aparición de calcificaciones en el piso del cuarto ventrículo5. El resultado de esta serie de sucesos es lo que se conoce como disgenesia del tallo cerebrall5.
El Misoprostol medicamento sintético, análogo a la prostlaglandina E1, genera un efecto vasoconstrictor, que resulta en isquemia transitoria fetal, afectando principalmente los núcleos de los nervios craneanos abducens y facial, como uno de los factores que podría influir en la presentación de Síndrome Moebius. La FDA reconoce al Misoprostol como útil en el tratamiento de la úlcera péptica y en la prevención de lesiones gastrointestinales inducidas por antiinflamatorios no esteroideos. Lo clasifica en la categoría X, según el riesgo de inducción de toxicidad sobre la gestación. Sin embargo también es aceptada como droga útero-tónica, útil en la maduración del cuello uterino y de uso en obstetricia. Las contracciones uterinas intensas y prolongadas que causa, generarían una restricción del crecimiento y del movimiento fetal. Este medicamento es utilizado sin las restricciones debidas (receta médica), por la facilidad de ser adquirido en el mercado nacional y en otros países de Latinoamérica como Paraguay y Brasil y en algunos países europeos incluso. En algunos países su uso está legalizado para el aborto. El mecanismo de acción teratogénico del Misoprostol estaría asociado a su efecto de
disrupción vascular, secundaria a sus efectos úterotónicos, con alteración del flujo sanguíneo fetal, que deriva en hipoxia y hemorragia11, 12.
Si la exposición tóxica del Misoprostol ocurre entre las cinco y medio a siete y medio semanas de gestación, contadas a partir del último día de la menstruación (FUM), se considera el período de mayor sensibilidad para que ocurran las anomalías congénitas de mayor severidad a nivel de las extremidades. Para la afectación del sistema nervioso central, el período de mayor sensibilidad se extiende desde la quinta semana hasta la decimoctava semana posterior a la FUM. Si la exposición ocurre entre las siete y medio y nueve y medio semanas posteriores a la FUM, dicho periodo es considerado de menor sensibilidad y se asocia a anomalías congénitas menos severas, como defectos de los dedos de las manos, equinovaro o anomalías cardiacas 24.
En el presente estudio, documentamos el caso de un neonato de sexo femenino que muestra las características clínicas del Síndrome Moebius, en quien podría haber influido la ingesta materna de Misoprostol - en el primer trimestre del embarazo- con fines abortivos. La niña nació con parálisis facial y alteración en miembros inferiores.
DESCRIPCIÓN DEL PACIENTE
El Servicio de Neonatología del Hospital Apoyo Iquitos, el 6 de mayo del 2013, recibe una recién nacida, con un peso de 2,820g (percentil 50-90), talla de 45,5cms (percentil10-50) y PC de 31.5cms (percentil 10-50), con APGAR 8 y 9 al minuto y cinco minutos respectivamente y de 36 semanas de gestación por test de Capurro. La madre de 30 años en su tercera gestación, con historia familiar de un hijo de 4 años portador de cardiopatía congénita cianótica tipo Tetralogía de Fallot y un aborto, es natural y procedente del distrito de Iquitos. El padre de 43 años, natural de Iquitos, no es consanguíneo y ambos con nivel de instrucción secundaria completa. La madre es ama de casa y el padre se dedica al comercio ambulatorio. No refirieron antecedentes patológicos familiares (Figura 1: Genograma). La gestación no planificada fue rechazada por los padres; la madre decide abortar a las 9 semanas de gestación e ingiere Misoprostol dos tabletas y se aplica tres tabletas intravaginalmente. El embarazo prosiguió sin mayores complicaciones, reportando infección urinaria en el segundo trimestre y flujo vaginal durante toda la gestación.
Hermann Silva Delgado , Javier Villaverde Montoya.
34
La gestante tuvo seis controles prenatales en el centro de salud de Iquitos, donde se le practican dos ecografías reportadas como normales.
Figura 1. Genograma: Mostrando la historia familiar y el propositus con Síndrome Moebius.
Examen Físico: llama la atención el llanto sordo y monótono, con la boca entreabierta, se observa parálisis facial bilateral y ausencia de mímica facial (Foto 1), sin embargo, puede cerrar parcialmente los ojos cuando duerme. Al nacimiento muestra dificultad para deglutir y se alimenta por sonda orogástrica, también se observa sialorrea moderada. Sus ojos abiertos no muestran expresividad emocional (Foto 2). Inicialmente es alimentado por sonda orogástrica (Foto 3) y recién a los 14 días muestra reflejo de succión y posteriormente se alimenta por succión directa. El examen de tórax y pulmones fueron normales. Desde el nacimiento muestra baja saturación de oxígeno, entre 70 a 86% y que no mejora con la administración de oxígeno. El estudio ecocardiográfico reportó leve derrame pericárdico, atresia tricúspide con persistencia del ductus arterioso. En abdomen no se palpó visceromegalia. Las extremidades superiores normales y las inferiores mostraban pie equino varo bilateral (Foto 4). La ecografía transfontanelar reveló ventrículos laterales asimétricos y con leve dilatación. A partir de los 14 días de edad se alimenta por succión directa y se le coloca primeros yesos de corrección del pie equino varo y sale de alta a los 16 días de nacida.
Foto 2: Mirada fija y rostro de “careta” inexpresivo.
Foto 3: A los 8 días de edad. Recibe leche materna por sonda orogástrica.
Foto 4: Pie equino varo bilateral.
Síndrome Moebius en el Hospital Apoyo Iquitos. Posible Causa por Exposición a Misoprostol. Reporte de un Caso.
35Hermann Silva Delgado , Javier Villaverde Montoya.
Las fotografías de la paciente se muestran con autorización de la madre.
DISCUSIÓNNuestra paciente presentaba parálisis facial, lo que se evidenciaba cuando lloraba, entreabría la boca y emitía un llanto sordo y monótono, que más parecía un ronquido. Cuando dormía ocluía parcialmente los párpados. La paciente, al nacimiento presentó dificultad para deglutir, requiriendo ser alimentada por sonda orogástrica, razón por la cual se sospechó Síndrome Moebius. Este síndrome suele asociarse a alteraciones cráneo-buco-faciales como paladar ojival, xerostomía13, mordida abierta, cierre inadecuado de la boca por hipoplasia mandibular14, implantación baja de orejas15 y micrognatia1, esta última característica presente en la paciente (Foto 5).
La paciente presentó baja saturación de oxígeno desde el nacimiento y el estudio ecocardiográfico reveló leve derrame pericárdico, atresia tricúspide con persistencia del ductus arterioso a diferencia de lo encontrado por otros autores. Cammarata-Scalisi, describe el tipo de alteraciones cardiacas que se asocian al Síndrome Moebius como son la conexión anómala venosa pulmonar total, defectos septales ventriculares, asociados o no a dextrocardia y transposición de grandes vasos7.
Las anomalías de las extremidades son frecuentes en el Síndrome Moebius. Perez Aytes describe que los pies zambos es la malformación esquelética más frecuentemente asociada al Síndrome Moebius (casi un 25% lo tienen) y pueden causar trastornos en la marcha10. Nosotros encontramos pie equino en nuestra paciente (Foto 4). No encontramos, sindactilia, amputación congénita y presencia de dedos rudimentarios, tanto en miembros inferiores como en superiores5,16 como en otros estudios.
Debemos plantear el diagnóstico diferencial del Síndrome Moebius, con la Distrofia
Foto 5: Micrognatia.
Muscular Miotónica Congénita o enfermedad de Steiner (DMM), la cual se asocia a hipotonía generalizada en el recién nacido, quien no muestra movilidad espontánea; además, presenta insuficiencia respiratoria desde el nacimiento, requiriendo asistencia ventilatoria, signos clínicos no presentes en nuestra paciente; también se describe debilidad de los músculos faciales, nuestra paciente presentaba parálisis facial central y si bien inicialmente no succionaba, pasados los 14 días, se alimentaba por succión directa, lo que no sucede en la DMM, en quienes persiste la dificultad en la alimentación por varios meses. La mortalidad de la DMM congénita es de 16%21. Otra patología necesaria en el diagnóstico diferencial es el Síndrome de Carey Fineman Ziter (MIM 254940), en el cual se describe secuencia de Moebius asociada a secuencia de Robin y miopatía. Presentan dificultad respiratoria al nacimiento, requiriendo asistencia ventilatoria. Se describe además períodos de apnea recurrente, características no presentes en nuestra paciente. Presentan además, hipotonía generalizada e hipoplasia muscular 22.
Se sostiene que la ingestión de Misoprostol durante la gestación se ha asociado al Síndrome Moebius y a malformaciones en las extremidades, esto debido a disrupción vascular transitoria de las arterias subclavias o vertebrales o sus ramas, que limitan el aporte sanguíneo embrionario en la fase temprana, como el mecanismo de acción teratógena7, 17.
Vargas y colaboradores reportó un estudio de casos y controles, multicéntrico, llevado a cabo en Brasil, donde compara la frecuencia del uso de Misoprostol en gestantes, cuyos productos presentaron defectos de disrupción vascular y un grupo control en el que se diagnosticaron otros tipos de defectos. La exposición prenatal se identificó en 34,4% de niños con diagnóstico de disrupción vascular, respecto de sólo 4,3% en el grupo control, resultados estadísticamente significativos19.
Goldberg y colaboradores en una revisión del tema sobre Misoprostol y gestación, reportaron un estudio colaborativo de malformaciones congénitas en Latinoamérica, con un incremento de las anomalías congénitas de las extremidades, defectos de los miembros por bridas congénitas, artrogriposis, hidrocefalia, holoprosencefalia y extrofia de la vejiga, pero no Síndrome Moebius
36
en gestantes expuestas a Misoprostol in utero20.
En el caso de nuestro paciente habría que considerar lo siguiente, tenemos que tener en cuenta que a las 9 semanas de gestación empieza el período fetal, donde la teratogenicidad es menor que en el período embrionario (momento de la ingesta del fármaco). Si uno de los mecanismos del Misoprostol es causar disrupción del flujo sanguíneo con la subsiguiente dificultad de crecimiento de órganos en el feto y considerando que los mayores daños ocurren entre la quinta y sétima (período embrionario), la posibilidad que haya sido afectado el paciente por el fármaco no es directa o poco probable (ver estudio citado de Golberg). La evidencia nos lleva a pesar que el Síndrome Moebius se produjo más temprano, es decir tuvo un origen genético, en vez de teratogénico. Lo que parece es que en un Síndrome Moebius ya establecido, el misoprostol pudo haber causado daños adicionales por falta de irrigación a los tejidos (puesto que en relación
a lo manifestado en la introducción, los daños son menores después de la séptima semana), por lo tanto, no se puede concluir que haya sido debido a la acción del Misoprostol como causa primaria.
CONCLUSIONESEl presente caso es una neonata nacida en Iquitos, que cumple con los criterios clínicos de Síndrome o Secuencia de Moebius, si bien es cierto, hay una asociación entre el Síndrome de Moebius y la ingesta de Misoprostol, no podemos concluir en nuestra paciente, que haya sido la causa, debido a que la ingesta del fármaco fue a partir de la novena semana. Recomendamos tener presente que la prescripción o el uso indebido de éste fármaco durante el primer trimestre de embarazo es riesgoso.
Síndrome Moebius en el Hospital Apoyo Iquitos. Posible Causa por Exposición a Misoprostol. Reporte de un Caso.
37
1. José Gabriel Lora F. y Jean Pierre Certain Yepes. Síndrome de Mobius con compromiso del VIII Par Craneal: a propósito de un caso. Acta de Otorrinolaringología &Cirugía de Cabeza y Cuello. 2009; 37(1):48-51
2. Jorge Villafranca A, Paulo Castillo D, Mario Garcés S, Eduardo Villalón F, Edith Grez L, Alejandra Díaz G. Síndrome de Moebius. Rev. Chilena de Cirugía. 2003; 55 (1):75-80.
3. Tania Villagra-Sanjurjo, Dra. Iris Violeta Vizzuett-López. : Síndrome Möbius-Poland: Presentación de un caso. Rev Mex Oftalmol; Septiembre-Octubre 2009; 83(5):314-317.
4. VIII Congreso Nacional e Internacional de Odontopediatría: Estela Novoa, Catalina Martínez, Claudia Espinoza, Jimena Montenegro, José Hassi. Manifestaciones orales del Síndrome de Mobius asociado a Poland: descripción de un caso clínico. Hospital Dr. Luis Calco Mackenna. Universidad de Chile. Revista de la Sociedad Chilena de Odontopediatría 2010; 25(2):27.
5. Ricardo Poveda J. : Secuencia de Moebius en anestesiología. Rev Chil Anest, 2012; 41: 140-143.
6. M Roig Quilis. Disgenesia tronco encefálica: los síndromes de Möbius, Cogan y Pierre Robin en revisión. An Pediatr (Barc) 2005; 62(4): 346-351
7. Francisco Cammarata-Scalisi. : Espectro clínico y etiológico del síndrome de Möbius. Arch Argent Pediatr 2007; 105(5):444-446.
8. Govaert P, Vanhaesebrouck P, De Praeter C, et al. Moebius sequence and prenatal brainstem ischemia. Pediatrics 1989; 84: 570-573.
9. Cecilia Fernández-Ponce, Enio Hernández, Carlos Silvera-Redondo, Beatriz Jiménez, Eder Quintero, Álvaro Idrovo, Manneh Ray. : Síndrome de Moebius: Genopatía vs efecto teratogénico. Salud Uninorte. Barranquilla (Col) 2006; 22 (2): 182-187.
10. A Pérez Aytés. : Síndrome de Moebius. Protoc diagn ter pediatr. 2010;1:80-84.
11. V. Etayo Etayo, I. Garralda Torres, M. E. Yoldi Petri, T. Durá Travé. : Síndrome de Moebius: diagnóstico neonatal. Acta Pediatr Esp. 2008; 66(10): 513-516.
12. Pastuszak AL, Schuler L, Speck-Martins CE, Coelho KE, Cordello SM, Vargas F, Brunoni D, Schwarz IV, Larrandaburu M, Safattle H, Meloni VF, Koren G. Use of misoprostol during pregnancy and Moebius syndrome in infants. N Engl J Med. 1998; 338:1881-1885.
13. Catalina Martínez, Claudia Espinoza, Jimena Montenegro, José Hassi. Manifestaciones orales del síndrome de Mobius asociado a Poland: descripción de un caso clínico. Rev. Soc. Chil. Odontopediatría. 2010; Vol. 25(2): 27.
14. Cándida Aleyda Carrillo Hernández, Héctor Flores Romo Chávez. : Síndrome de Moebius. Revista de Especialidades Médico-Quirúrgicas 2010;15(4):261-265.
15. Beatriz Milet L, Ximena Alegría P. : Síndrome de Moebius. Rev. Obstet. Ginecol. - Hosp. Santiago Oriente Dr. Luis Tisné Brousse. 2006; 1 (3): 215-216.
16. Andrea Osorio O., Juan Guillermo Rodríguez A. , Oscar Pizarro R., Axel Paredes V., Susana Aguilera P. Embriopatías asociadas al uso de misoprostol. Rev Chil Obstet Ginecol 2007;72 (5):346-351.
17. Vieira Ventura B., Tiller Miller M., Danda1 D., Carta A., Teixeira Brandt C., Oliveira Ventura L., Profile of ocular and systemic characteristics in Möbius sequence patients from Brazil and Italy. Arq Bras Oftalmol. 2012;75(3):202-6.
18. MINSA. Hospital Apoyo Iquitos. Reporte Estadístico año 2011.
19. Vargas FR, Schuler-Faccini L, Brunoni D, et al. Prenatal exposure to misoprostol and vascular disruption defects: a case-control study. Am J Med Genet 2000; 95:302-306.
20. Goldberg A., L., Greenberg M., Arney P. :M isoprostol and pregnancy. N Engl J Med, 2001; 344(1):38-47.
21. Aviña Fierro J. A., Hernández Aviña D.A. : Síndrome de Steinert neonatal: distrofia miotónica tipo 1 congénita. Arch Pediatr Urug 2009; 80(1): 33-36.
22. Tanchi P.P.D., Edmonds L.K., Guan Koh T., H., H. : Carey-Fineman-Ziter syndrome: a spectrum of presentations. Case Rep. Perinat. Med. 2012; 1(1-2): 99–102.
23. Figueiredo MC, Faustino-Silva DD, Bez AS, Rincón DPC.: Manifestaciones orales del síndrome de Mobius asociado a Poland: descripción de un caso clínico. Avances en Odontoestomatología 2009; 25 (4):179-183.
24. Sitruk-Ware R., Davey A., Sakiz E.: Fetal malformation and failed medical termination of pregnancy. The LANCET 1998; 352: 323.
REFERENCIAS BIBLIOGRÁFICAS
Hermann Silva Delgado , Javier Villaverde Montoya.
38
Telemedicine, is already here, where are we?Javier Ferreyros Kuppers1
1 Médico Pediatra. Director de Pediatras Asociados. Correo electrónico: [email protected] Recibido: 10 de Marzo del 2014 Aceptado: 28 de Marzo del 2014
hablar con imagen y audio con los doctores que requiriésemos. Muy parecido a lo que actualmente hacemos desde nuestra casa, entonces lo podíamos hacer transmitiendo imágenes desde un oftalmoscopio, otoscopio, sonidos desde un estetoscopio y las imágenes mostradas directamente a la cámara. De ésta manera solucionábamos un problema tan común en nuestro país, la distancia, la falta de especialistas cerca, el costo de un viaje para encontrar atención de primera calidad. No quiero imaginarme lo que costó en ese entonces aquél programa. Hoy, sorprendentemente, aún no aprovechamos una tecnología que, superando por lejos a la de entonces, está a nuestra disposición.
La Asociación Americana de Telemedicina la define como el intercambio de información médica de un sitio a otro, a través de comunicación electrónica, para la salud y educación del paciente o profesional, con el propósito de mejorar el cuidado de los pacientes.
Hace diez años la Academia Americana de Pediatría publicaba en el Pediatrics2 que en el Steering Committee on Clinical Information Technology, abarcarían el uso de tecnología de comunicaciones electrónicas para proveer y apoyar cuidados de salud a infantes, niños, adolescentes y adultos jóvenes cuando la distancia separa al profesional del paciente, padres o un profesional a cargo. Específicamente excluyeron la comunicación telefónica entre profesionales o entre profesionales y pacientes y además también el uso de ésta tecnología para la educación de profesionales, tema al que nosotros sí quisiéramos referirnos por tratarse de un medio
RESUMENLa Telemedicina es una herramienta moderna que ya se encuentra entre nosotros. Hacemos una revisión sobre algunas aplicaciones actuales y lo que se está haciendo en el Perú. Se analiza si los pediatras, familias y compañías de seguro estamos preparados para estos cambios. Palabras claves: Telemedicina, niños.
SUMMARYTelemedicine is a modern tool already among us. We make a review of some of its current applications and what is being done in Peru. We analyze if pediatricians, families and insurance companies are ready for the changes. Keywords: Telemedicine, children.
Ya en 19061 Einthoven era capaz de leer electrocardiogramas realizados a distancia. Ahora, más de 100 años después, los pacientes nos envían fotos de sus niños pidiéndonos consejo por algún exantema que le ha brotado a su hijo o que opinemos sobre una radiografía que le acaban de hacer a su niño en Máncora. No hay que ser muy imaginativo para predecir que en poco tiempo nuestros pacientes nos estarán haciendo consultas por Skype, Facetime, Whatsapp o algún nuevo software por el estilo.
Todo esto nos genera una serie de preguntas, ¿será bueno para la medicina? ¿Será bueno para nuestros pacientes? ¿Podremos ofrecer un mejor servicio a nuestros pacientes? ¿Cómo se nos compensará económicamente por ello? Mientras que sigamos atónitos esperando respuestas, la Telemedicina seguirá avanzando y si no reaccionamos, nos pasará por encima.
Hace veinte años, trabajando en un Hospital rural en las montañas de West Virginia, a dos horas y media del centro hospitalario con las subespecialidades pediátricas necesarias y que en época de nieve se hacía inaccesible, el gobierno de los EEUU puso a nuestra disposición un sistema por el cual nos comunicábamos con el Hospital y podíamos
Rev. Perú. pediatr. 67 (1) 2014
Telemedicina, ya está acá, ¿nosotros dónde estamos?
TEMA DE REVISIÓN
39
poco usado aún entre nosotros, así como también sobre el “e-health” o uso de internet para proveer cuidados de salud sin videoconferencia.
Si bien el espectro que la Telemedicina puede abarcar en pediatría es enorme, algunas áreas la han explotado más que otras. El más evidente a discutir acá y más espectacular es la posibilidad de intercambiar en vivo por videoconferencia, aspectos de cuidados de la salud entre una institución considerada de alto nivel y otra que por problemas de distancia o económicos, de infraestructura o de carencia de profesionales, pueden entablar.
Es evidente que como profesionales, en algunas ocasiones sentimos la frustración de no poder proveer de los cuidados que quisiéramos a uno de nuestros pacientes. Sea por falta de infraestructura o de conocimientos o experiencia, siempre hemos sabido consultar a alguien que sabe más que nosotros. Esta práctica se hace mucho más evidente en lugares alejados como contaba en mi experiencia trabajando en un hospital rural. Las consultas pueden variar desde temas de cómo mejorar la gestión en la atención médica hasta solicitar recomendación en el tratamiento de un paciente que por las condiciones del caso no puede ser trasladado y deben proporcionar cuidados médicos para los cuales no están preparados. Puede ser que en un hospital distante están encontrando infecciones que sospechan pudieran ser intrahospitalarias y solicitan recomendaciones de qué hacer para identificar la fuente y evitar futuros casos o el paciente en insuficiencia cardiaca que requiere la interpretación de una radiografía y la terapéutica a instalar.
Veo la necesidad de poner en práctica esta herramienta desde provincias alejadas de nuestro país con especialistas en nuestras instituciones de alto nivel o entre éstas instituciones e instituciones más especializadas que las nuestras en el extranjero.
Para realizar esto se requiere de una preparación en ambos polos de la comunicación y debemos actuar ya para que estas condiciones se den. En provincias deberán tener no solo la mínima tecnología, una computadora con capacidad de video y sonido comunicada a internet, la posibilidad de tener imágenes digitales, un escáner y el software de comunicación necesario, sino el personal dispuesto a hacer un resumen del caso clínico con la información relevante para poder utilizar el recurso del tiempo de manera apropiada.
En el otro extremo de la comunicación están las instituciones que deben estar preparadas para
recibir la consulta, debe haber principalmente la voluntad de querer hacerlo, el personal disponible, el horario disponible, el espacio con la computadora, cámara y audio apropiados. La dedicación a analizar la información recibida con anticipación a la comunicación, cuando la situación lo permita, para no perder tiempo durante la comunicación. La capacidad de recibir imágenes, audio, videos, documentos escaneados, etc. Analizarlos, discutirlos, prepararlos y luego, una vez comunicados transmitirlos de una manera amigable, sin ánimos de reproche o tono de fiscalización sino de ayudar en el cuidado de la salud del paciente.
La consulta puede surgir de un pediatra que necesita una opinión experta o de una familia insatisfecha con los resultados de los cuidados que el niño viene recibiendo, así las cosas, es fácil imaginar las complicaciones que la Telemedicina también puede ocasionar y a los cuales nos referiremos también.
La Academia Americana de Pediatría en el 2003 incluía en sus políticas de atención pediátrica la Telemedicina para mejorar el acceso y la calidad de la atención. 4
Una de las incógnitas que hay que resolver es el impacto socio económico que la Telemedicina traerá a la atención de la salud. Si bien, hay algunos estudios que demuestran el abaratamiento de costos, disminución de hospitalizaciones, menor tiempo de hospitalización, diminución de mortalidad y mejoría en la calidad de vida, no hay suficientes estudios para afirmar que así ocurrirá en todo intento de utilizarla5, 6.
Conceptos relacionados a la Telemedicina:
Debemos ir acostumbrándonos a los términos que iremos poco a poco familiarizándonos, estos son:
Telesalud: Que comprende conceptos de servicios y atención sanitaria, gestión, educación, informática médica como historia clínica electrónica7.
Teleducación: Puede incluir educación a profesionales o a pacientes. Como ejemplos en nuestro medio podemos mencionar la Teleconferencia anual del Miami Children’s Hospital donde accedemos en vivo a conferencias dictadas en Miami y las recogemos en Piura, Trujillo, Arequipa y Lima desde hace ya varios años o la Discusión de casos clínicos realizada entre un grupo de profesionales en Lima que remitieron los casos a los subespecialistas en el Miami Children’s Hospital y pudieron discutir los casos en vivo en abril de éste año.
Javier Ferreyros Kuppers.
40
Teleconsulta: Actividad que motiva este comentario, la posibilidad de que un profesional o un paciente o su familia puedan comunicarse con otro especialista a quien se consulta para obtener mayor información encontrándose a distancia. Estas pueden ser:
En tiempo real (Sincrónicas): cuando el paciente está presente y transmite sus síntomas al profesional en vivo.
En tiempo diferido (store & forward) (Asincrónicas): cuando la información es almacenada, transmitida para su revisión y comentario posterior.
Telepráctica: Las diferentes áreas de aplicación.
Teleradiología: Una de las más antiguas (30 años) por su estandarización y rara necesidad de requerir la presencia del paciente en el momento de la lectura.
Telepatología: El hecho de acceder a imágenes obtenidas en el microscopio y tampoco requerir la presencia del paciente.
Telesalud en sistemas penitenciarios: De más está resaltar lo práctico que puede resultar para evitar la movilización de reclusos muchas veces para atenciones que no requieren hospitalización.
Teleoftalmología: Muy utilizada actualmente al poder compartir imágenes ya sea desde una lámpara de hendidura con video, microscopio quirúrgico o imágenes retinianas ya sea en vivo a través del tele oftalmoscopio o con fotografías digitales.
Telecardiología: Ya que existe el tele estetoscopio y la posibilidad de transmitir ruidos cardiacos y sus ritmos mediante electrocardiogramas así como ecocardiografías.
Teledermatología: Una de las aplicaciones más usadas globalmente por ser básicamente ectoscópica.
Telepsiquiatría: También viene usándose por la carencia de profesionales en lugares alejados y no tener que examinarse al paciente sino mantener una comunicación dialogante.
TeleUCI: Por la posibilidad del monitoreo remoto.
Telecirugía: con la aplicación de la cirugía robótica con el operador a distancia del paciente intervenido, conocida desde su reporte en la revista Nature en el 2001 de un paciente en Estrasburgo operado de colecistectomía laparoscópica desde Nueva York.
Teleinvestigación: campo del que escucharemos cada vez más, pues facilitaría la recopilación de información a distancia o la agrupación de información de investigadores a distancia.
Otro beneficio con el que nos veremos involucrados en poco tiempo es la Historia Clínica Electrónica la cual ya ha sido regulada como obligatoria desde este año para poder participar del Medicare y Medicaid.
Revistas y Asociación:
Contamos ya con dos revistas que se encargan de éste tema y La American Telemedicine Association (www.americantelemed.org) que existe desde 1993.
¿Qué se está haciendo en el Perú?La Asociación Peruana de Telesalud y Telemedicina (APTT), filial de la Asociación Iberoamericana de Telesalud y Telemedicina se fundó en Mayo del 2013 y ya viene organizando el I Congreso Iberoamericano de Telesalud y Telemedicina y VII Reunión Regional de ATALACC (American Telemedicine Association, Latin American & Caribbean Chapter) el 16 - 18 de Octubre, 2014 en Lima.
El Ministerio de Salud promulgó:
Resolución Ministerial 365-2008/MINSA Norma Técnica de Salud en Tele salud
Decreto Supremo N° 028-2005-MTC Plan Nacional de Tele salud
Resolución Ministerial 009-2003-MTC Creación de la Comisión Nacional de Telesanidad
El Director General de la Oficina de Estadística de Informática del Ministerio de Salud de Perú, Walter Curioso, compartió las experiencias y los avances en Telesalud de Perú durante su participación en el 9° Congreso Regional de Información y Comunicación
Telemedicina, ya está acá, ¿nosotros dónde estamos?
41Javier Ferreyros Kuppers.
a cargo del paciente, si fuera posible, para escuchar al especialista del Miami Children’s Hospital durante 30 minutos resolviendo las preguntas realizadas y las que surjan durante la consulta. De ésta manera estamos ahorrando significativamente los gastos de viaje, hospedaje, tiempo y lo que es más importante, la opinión de un especialista altamente calificado con la experiencia de una institución reconocida internacionalmente.
en Salud (CRICS9).
EsSalud, desde Junio del 2013 viene trabajando creando el CENATE (Centro Nacional de Telemedicina) ubicado en la Av. Arenales como el primer centro referente de telemedicina en el Perú brindando servicio a provincias. Aquí realizan programas de Teleconsulta y Teleradiología. Para la Teleconsulta cuentan con dos médicos internistas, un dermatólogo y un oftalmólogo que prestan atención a distancia al Hospital de Pucallpa (Red Ucayali), Hospital El Buen Samaritano (Red Amazonas), Posta médica de Huamachuco (Red La Libertad) y Posta médica de Tocache (Red Huánuco) y para la Teleradiología cuentan con cuatro radiólogos que leen imágenes médicas de 23 centros asistenciales en todo el país coordinado por la Dra. Bernardette Isabel Cotrina.
EsSalud también cuenta con La Red Cardiológica de Telemedicina del Instituto Nacional del Corazón (INCOR).
El Colegio Médico del Perú realizó una clase magistral en cirugía en donde se apreció por Internet de alta velocidad en el auditorio del Colegio Médico del Perú.
Así mismo, existe La Red Andina de Vigilancia Epidemiológica (RAVE) del Organismo Andino de la Salud (OAS) conformada porBolivia, Colombia, Chile, Ecuador, Perú y Venezuela.
El Enlace Hispano Americano de Salud (EHAS - Perú), brinda servicios de comunicación y acceso a información de salud al personal que trabaja en zonas rurales del país utilizando tecnologías de bajo costo para mejorar las condiciones de salud en localidades remotas.
Pediatras Asociados, nuestro centro pediátrico en Miraflores firmó un convenio en el 2013 con el Miami Children’s Hospital para llevar a cabo el programa de Telemedicina o Telehealth y realizar teleconsultas de segunda opinión con unas 30 especialidades pediátricas de ésta institución. El pediatra o la familia interesada en Lima solicita a través de Pediatras Asociados la consulta completando un paquete de información con el resumen de la historia clínica, resultados de laboratorio, imágenes pertinentes como fotos, radiografías, tomografías, resonancias, videos o lo que contribuya a ayudar al especialista consultado a responder a las preguntas específicas que le quieran hacer. Luego de ser revisado el caso por el especialista solicitado, se programa la Teleconsulta con la familia solicitante y el profesional
El programa de Telemedicina del Miami Children’s Hospital busca extenderse en el futuro a formar parte de las actividades cotidianas de hospitales con herramientas para realizar la Teleconsulta desde la cama del enfermo o trasladarse incluso como parte de la visita con herramientas móviles.
Como otra muestra de e-Salud hemos editado en conjunto con el Dr. Enrique Massa un Manual de Terapéutica Pediátrica que aprovechará las telecomunicaciones para actualizarse de manera regular y así mantener a los suscritos al día, ya sea con nuevas guías de manejo terapéutico o con nuevos productos que ingresan a la farmacopea local.
Así mismo Pediatras Asociados viene solucionando consultas de cualquier familia interesada a través de su página su página web o en Facebook.
42
¿Estamos preparados?
Luego de revisar todos estos aspectos tan interesantes por novedosos, nos plantean un reto, pues para hacer uso de ellos debemos estar preparados, de allí que surja la pregunta.
¿Están los pacientes preparados?
En muchas encuestas se ha evidenciado la conformidad de los pacientes luego de haber hecho uso de la Telemedicina, en nuestro caso, de las 6 teleconsultas que hemos realizado, las familias han quedado satisfechas en todos los casos. Sin embargo aún hay temas delicados a analizar. Cuando la familia es la que solicita la teleconsulta generalmente es porque no está satisfecha con los cuidados que su niño viene recibiendo, lo que plantea una decisión que no necesariamente incluye a su médico tratante de hacer la teleconsulta. ¿Están las familias preparadas para entender que el resultado de la teleconsulta puede no coincidir con los cuidados que su niño viene recibiendo?¿Podría prestarse esto a problemas de tipo legal?. En la práctica vemos que rápidamente las familias hacen uso de la tecnología y se sienten cómodos realizando la consulta.
Con respecto a los médicos, la resistencia al cambio ha sido evidenciada en la aplicación de las historias médicas electrónicas. Hay múltiples experiencias descritas y casi siempre ha tenido que sufrirse diversos niveles de resistencia pero habría que admitir que en la mayoría de casos los resultados superan al vía crucis de la adaptación. Con respecto a las teleconsultas aún me queda mucho por descubrir pero tengo motivos para sospechar la resistencia que existe a solicitar ayuda. De los 6 casos realizados hasta ahora han sido las familias las que han solicitado las teleconsultas y no el médico tratante.
Si bien la referencia de pacientes a especialistas es práctica común, veo con sospecha que el enfrentarse a pediatras especialistas en el extranjero plantea un nivel de ansiedad superior, tal vez temor a ser corregidos delante de la familia, es por esto también que resulta fundamental el ánimo con que deben ser contestadas las teleconsultas, la voz, la entonación, las pausas, la empatía, todas las habilidades que hacen a uno un buen comunicador. Más evidente es la incomodidad del profesional de tener que trasladarse a otro local para realizar la teleconsulta por el tiempo que esto aqueja. El tiempo y la experiencia nos responderán estas preguntas.
En muchos centros, la persona encargada de la coordinación de las teleconsultas es personal para-médico bien capacitado en tecnología.
¿Están las compañías de seguros preparadas?
Me he reunido en repetidas ocasiones con dos de las compañías de seguros más importantes en el país para analizar éste tema con el obvio interés de que éstas lleguen a pagar por los servicios prestados a través de la telemedicina. Si bien ya hay compañías aseguradoras que están utilizando la telemedicina para abaratar costos en otros países8, aún no conocemos de ningún intento para hacerlo así en nuestro medio. El tema que más parece preocuparles es el uso indiscriminado del sistema, pretendiendo consultar a un especialista en el extranjero por temas que fácilmente puedan ser solucionados localmente, lo que hace obvio algún tipo de triaje, ya sea con copagos o a través de una solicitud generada por el médico tratante o por un equipo auditor de la misma compañía de seguros. Por otro lado ya están viendo las ventajas al poder evitar el carrusel de terceras, cuartas y quintas opiniones en casos complejos y el evidente ahorro en tener que pagar por consultas llevadas a cabo en el extranjero. No dudamos que en un futuro próximo veremos a las compañías de seguro como actores principales de este sistema de telesalud.
Complicaciones:
El ilustrador Al Murphy nos deleita con algunas de ellas.
Telemedicina, ya está acá, ¿nosotros dónde estamos?
43
REFERENCIAS BIBLIOGRÁFICAS
1. Einthoven W. Le telecardiogramme. Arch Int de Physiol1906;4:132–64 (translated into English, Am Heart J 1957;53:602–15).
2. Spooner A, Gotlieb E. Telemedicine: Pediatric Applications. Pediatrics 2004;113:e639–43.
3. Linkous JD. Toward A Rapidly Evolving Definition of Telemedicine. Washington, DC: American Telemedicine Association; 2001.
4. American Academy of Pediatrics. Scope of Practice Issues in the Delivery of Pediatric Health Care. Committee on Pediatric Workforce. Pediatrics 2003;111;426-35.
5. Strehle E, Shabde N. One hundred years of telemedicine: does this new technology have a place in paediatrics?. Arch Dis Child 2006;91:956-59.
6. Jennett P, Affleck Hall L, Hailey D, Ohinmaa A, Anderson C, Thomas R, Young B, Lorenzetti D, Scott R. The socio-economic impact of telehealth: a systematic review. J Telemed Telecare 2003;9:311-20.
7. Giselle Ricur. Manual de salud electrónica para directivos de servicios y sistemas de salud de la Sociedad Española de Informática de la Salud. Telemedicina: generalidades y áreas de aplicación clínicas. Organización de las Naciones Unidas. 2012, 169-94.
8. Kharif O. Telemedicine: Doctor Visits Via Video Calls. Bloomberg Businessweek Technology. 2014. http://www.businessweek.com/articles/2014-02-27/health-insurers-add-telemedicine-services-to-cut-costs.
Javier Ferreyros Kuppers.
44
RESUMEN Muchas investigaciones (1980-2012) afirman que los humanos estamos equipados de lo que se llama un normative moral sense que resulta de una red neuronal que incluye diferentes regiones del cerebro con su principal centro representado por la Corteza Pre-frontal Ventro Medial (VMPFC) especialmente del lado derecho. El juicio moral y la conducta requieren, de hecho, de la integración de diferentes procesos como: la decodificación de señales por órganos sensitivos (tálamo), la activación de emociones básicas (lóbulo temporal antero medial, tronco cerebral y núcleos de los centros visceromotores), la conciencia de la relevancia e importancia de los estímulos (VMPFC y la corteza orbito frontal) y la implementación de control de potenciales formas relacionadas de conducta. De acuerdo a este modelo una alteración en cualquiera de los centros corticales o subcoiticales podría constituir la base para los cambios en la conducta social. Entonces el concepto que ha surgido en muchos estudios, es que puede existir un módulo moral o un sentido moral innato el cual puede ser la base de las emociones, sentimientos y conductas con el fin de promover la cohesión del grupo y cooperación. Como argumento de la existencia de este módulo innato, hay evidencias en base a observaciones clínicas y de radio imágenes que muestran que la patología se debe a alteraciones de esta "red moral". Hay que tener en cuenta que los procesos cognitivos y emocionales juegan un papel competitivo en la aparición del juicio moral. Palabras Clave: Sentido moral innato, juicio moral.
Uno de los retos más apasionantes de la investigación neurocientífica actual está representado por el descubrimiento de posibles sustratos de base neuronal de la moralidad. El supuesto es que en los seres humanos podría existir un sentido moral innato o módulo moral. Mi punto de vista es que si lo hay, y por ello sustentaré las
1 Pediatra con especialidad en Endocrinología y Genética. Profesor Asociado de la Facultad de Medicina Alberto Hurtado Universidad Peruana Cayetano Heredia. Asistente del Servicio de Genética del INSN.
Correo electrónico: [email protected] Recibido: 15 de Enero del 2014 Aceptado: 10 de Febrero del 2014
SUMMARY Many studies (1980-2012) researchs suggest that humans are equipped with what is called a normative moral sense that results from a neural network which includes different regions of the brain with its principal place represented by the Pre-frontal Cortex Medial Ventro (VMPFC) especially the side right. The moral judgment and behavior required, in fact, the integration of different processes such as decoding of signals by sensory organs (thalamus), activation of basic emotions (anterior medial temporal lobe, and brainstem nuclei visceromotor centers), awareness of the relevance and importance of the stimuli (VMPFC and orbito frontal cortex) and the implementation of control related potential forms
conduct. According to this model a change in any of the cortical or sub-corticals centers could form the basis for changes in social behavior. So the concept that has emerged in many studies is that there may be a moral module or an innate moral sense which can be the basis of emotions, feelings and behaviors in order to promote group cohesion and cooperation. As an argument for the existence of this innate module, evidence based on clinical observations and radio images show that the disease is due to alterations of this "moral network. "Keep in mind that the cognitive and emotional processes play a competitive role in the development of moral judgment. Keywords: Normative moral sense, moral judgment.
evidencias. Diferentes estudios sugieren que existe una moral innata a diferencia de lo que plantean por ejemplo Jesse J. Prinz o Patricia Churchland (plataforma neuronal de conducta y el apego como plataforma neurológica de la moralidad) y para quien el marco animal social determina los tipos de moral.
Rev. Perú. pediatr. 67 (1) 2014
¿Existe un módulo moral?
ÉTICA
Is there a moral module?Miguel Chávez1
45
Hay muchos estudios (1980-2012) afirman que los humanos estamos equipados de lo que se llama un normative moral sense que resulta de una red neuronal que incluye diferentes regiones del cerebro con su principal centro representado por la Corteza Pre-frontal Ventro Medial (VMPFC) especialmente del lado derecho1. El juicio moral y la conducta requieren, de hecho, de la integración de diferentes procesos como: la decodificación de señales por órganos sensitivos (tálamo), la activación de emociones básicas (lóbulo temporal antero medial, tronco cerebral y núcleos de los centros visceromotores), la conciencia de la relevancia e importancia de los estímulos (VMPFC y la corteza orbito frontal) y la implementación de control de potenciales formas relacionadas de conducta2. De acuerdo a este modelo una alteración en cualquiera de los centros corticales o sub-coiticales podría constituir la base para los cambios en la conducta social.
El VMPFC esta estrechamente conectado al sistema límbico y medias reacciones automáticas que son evidentes cuando uno tiene que enfrentarse a violaciones morales3. Las lesiones cerebrales que afectan el VMPFC derecho parecen mitigar emociones morales y respuestas a dilemas relativos tanto a perjudicar a los demás y el sentido de equidad y justicia. Los pacientes con estas
lesiones pueden mostrar diferentes alteraciones en la función emocional, los cuales incluyen apatía afectiva, reducción de la empatía y dificultad en el control de ira y frustración. La Corteza orbito frontal y ventro lateral (OFC/VL) puede controlar las emociones aversivas inherentes a la esfera social e inhibe respuestas inmediatas que vienen de la amígdala y suprimen el comportamiento impulsivo y la desinhibición 4, 5. La amígdala media respuestas al miedo/ansiedad, disgusto y los estímulos sociales negativos y modula la comprensión de los límites morales y sociales, como también ciertas expresiones faciales, principalmente negativas 6. La falta de reactividad a algunos estímulos estresantes registrados en algunos psicópatas se ha atribuido a las lesiones en la amígdala. Lesiones bilaterales más amplias que involucran la corteza parietal anterior adyacente subyacen en el Síndrome de Klüver-Bucy caracterizado por hipersexualidad y una exagerada tendencia hacia la exploración oral y táctil de los objetos, agnosia visual, bulimia, ausencia de miedo, gran agresividad, déficit de memoria, epilepsia y dificultad para reconocer personas y objetos7. Veamos como estaría diseñada “la red moral”:
Unos pocos estudios han propuesto que esta red moral pudiera ser dejada de lado por los procesos
Miguel Chávez.
46
racionales mediados por la Corteza pre-frontal dorso lateral (DLPFC) llevando a utilitarismo, es decir, actuando en función del mayor bien para el mayor número de personas. El resultado es la creación de una tensión psicológica positiva (¿empatía?) lo cual permite entender el estado mental de los otros (teoría de la mente)9. A menos que sea inhibido activamente, la iniciación de este proceso ocurre automáticamente a través de las neuronas espejo y los resultados en la empatía, las emociones y la conducta moral10
. Estos nuevos hallazgos. Aunque todavía preliminares, tienen una serie de implicaciones. Por ejemplo: cuando un individuo presenta un comportamiento inusual por primera vez o un cambio completo de personalidad en comparación a un previo modelo de conducta, la posibilidad de un trastorno neurológico subyacente debe tomarse en cuenta 11. Han y col. 12 muestra en su estudio el sustrato neural del proceso de decisiones morales entre norteamericanos y coreanos. Los coreanos mostraron una mayor actividad en el putamen derecho relacionado con los procesos socio-intuitivos y la circunvolución frontal superior derecha con los procesos de control cognitivo bajo una condición personal moral y en el sulcus post central con el cálculo mental en un contexto familiar bajo una condición moral impersonal. Los norteamericanos mostraron un significativo mayor grado de actividad bilateral en la corteza cingulada anterior (ACC) asociada con la resolución de conflictos bajo condición moral-personal y en la circunvolución frontal medial derecha (MFG) asociada con un simple cognitive branching en un contexto no familiar bajo una condición de moral impersonal cuando se aplico un umbral más indulgente que los coreanos. Estos hallazgos sugieren que las interacciones entre la cultura, y el desarrollo juegan un papel importante.
Entonces el concepto que ha surgido en muchos estudios, es que puede existir un módulo moral o un sentido moral innato el cual puede ser la base de las emociones, sentimientos y conductas con el fin de promover la cohesión del grupo y cooperación.
Como argumento de la existencia de éste módulo innato, hay evidencias en base a observaciones clínicas y de radio imágenes que muestran que la patología se debe a alteraciones de esta "red moral". Hay que tener en cuenta que los procesos cognitivos y emocionales juegan un papel competitivo en la aparición del juicio moral 13, 14. De hecho los juicios utilitarios principalmente regulados por la cognición requieren emociones para ser motivados y lo opuesto es verdad para los juicios no utilitarios.
Existe todavía una cuestión abierta en lo referente a las influencias ambientales y en particular del attachment, y las interrelaciones personales en la modulación de la organización del sentido moral 15.
Sin ánimo de ser cientista, creo que las evidencias estructurales cerebrales trabajan en red la cual se está interrelacionada dinámicamente, constituyendo ese módulo moral innato, que no es un lugar específico sino una serie de comunicaciones neuronales que genética y epigenéticamente han ido reguladas y han aparecido paulatinamente en el desarrollo del sistema nervioso humano, si bien es cierto las influencias ambientales y de otro tipo juegan un papel importante en nuestro juicio moral, ello va a depender de la integridad de la "red moral", y no sólo del marco social como decía Patricia Churchland 16.
Estoy de acuerdo con Marc Hauser, en el sentido de que "la idea de que estamos dotados de un instinto moral, una facultad de la mente humana que guía inconscientemente nuestros juicios acerca de lo correcto y lo incorrecto, estableciendo un abanico de sistemas morales susceptibles de aprendizaje, cada uno de ellos con conjunto de marcas exclusivas o compartidas…17".
Cabe resaltar el aporte de Hauser con su aporte del módulo moral y el análisis de Patricia Churchland en el sentido que la plataforma moral con sustento neurológico de la vinculación emocional que fue modificada por presiones evolutivas y los valores culturales que han desembocado en diversos estilos de conducta moral que están arraigados en la conducta habitual de los humanos, sobre todo, en el apego y lo natural del deseo de cooperación con los otros (nuestro entorno familiar y la sociedad) y su cuidado, es una muestra de la "química moral" de nuestro cerebro. Para ello también tendríamos que mencionar a la oxitocina que disminuye el estrés y permite que desarrollemos la confianza entre unos y otros, necesaria para producir vínculos de familiaridad y moralidad social; lo cual a mi parecer no excluye el innatismo si no lo refuerza y no lo excluye.
Existe un módulo moral?
?
47Miguel Chávez.
REFERENCIAS BIBLIOGRÁFICAS
1. Marazziti D, Baroni S, Land, Ceresoli D, Dell’Osso L. The neurobiology of moral sense: facts or hypotheses? Annals of General Psychiatry 2013, 12:6
2. Moll J, de Oliveira-Souza R, Eslinger PJ, Bramati IE, Mourao-Miranda J, Andreiuolo PA, Pessoa L: The neural correlates of moral sensitivity: a functional magnetic resonance imaging investigation of basic and moral emotions. J Neurosci 2002, 22(7):2730–2736.
3. Greene JD, Nystrom LE, Engell AD, Darley JM, Cohen JD: The neural bases of cognitive conflict and control in moral judgment. Neuron 2004, 44:389–400.
4. Baxter MG, Parker A, Lindner CC, Izquierdo AD, Murray EA: Control of response selection by reinforcer value requires interaction of amygdala and orbital prefrontal cortex. J Neurosci 2000, 20(11):4311–4319.
5. Blair RJ: The roles of orbital frontal cortex in the modulation of antisocial behavior. Brain Cogn 2004, 55(1):198–208.
6. Adolphs R, Tranel D, Damasio AR: The human amygdala in social judgment. Nature 1998, 393(6684):470–474.
7. Devinsky J, Sacks O, Devinsky O: Kluver-Bucy syndrome, hypersexuality, and the law. Neurocase 2010, 16(2):140–145.
8. Moll J, de Oliveira-Souza R: Moral judgments, emotions and the utilitarian brain. Trends Cogn Sci 2007, 11(8):319–321.
9. Moll J, de Oliveira-Souza R, Garrido GJ, Bramati IE, Caparelli-Daquer EM, Paiva ML, Zahn R, Grafman J: The self as a moral agent: linking the neural bases of social agency and moral sensitivity. Soc Neurosci 2007, 2(3–4):336–352.
10. Calarge C, Andreasen NC, O’Leary DS: Visualizing how one brain understands another: a PET study of theory of mind. Am J Psychiatry 2003, 160(11):1954–1964
11. Shallice T: ‘Theory of mind’ and the prefrontal cortex. Brain 2001, 124(Pt 2):247–248.
12. Han H, Glover GH, Jeong C: Cultural influences on the neural correlate of moral decision making processes. 2013, 18(259C):215-228.
13. Damasio AR, Tranel D, Damasio H: Individuals with sociopathic behavior caused by frontal damage fail to respond autonomically to social stimuli. Behav Brain Res 1990, 41(2):81–94.
14. Greene JD, Nystrom LE, Engell AD, Darley JM, Cohen JD: The neural bases of cognitive conflict and control in moral judgment. Neuron 2004, 44:389–400. 4.
15. Kahneman D: Experiences of collaborative research. Am Psychol 2003, 58(9):723–730.
16. Churchland P. El cerebro moral. Barcelona: Paidós; 2012. pp 35
17. Hauser, M. La mente moral. Barcelona : Paidós ; 2008 pp 492
48
Reglamento de publicación 1. DE LA REVISTALa Revista Peruana de Pediatría es la publicación oficial de la Sociedad Peruana de Pediatría, destinado a divulgar y propagar los conocimientos sobre Medicina Preventiva y Asistencial entre los médicos pediatras y profesionales interesados. Dicha acción la ejerce a través de la publicación de trabajos científicos de sus asociados y profesionales médicos no asociados, nacionales e internacionales, que por su calidad y rigor científico merezcan su publicación para ser difundidos dentro y fuera del país.
2. DE LAS SECCIONES DE LA REVISTA La Revista Peruana de Pediatría cuenta con
las siguientes secciones:
2. 1. EDITORIAL Destinada a aspectos doctrinarios de la
Sociedad Peruana de Pediatría o a temas que por su trascendencia y actualidad requieren su publicación.
2. 2. TRABAJOS ORIGINALES Comprende la sección de trabajos originales e
inéditos de las diversas áreas de la Pediatría o ciencias afines. No excederá de 15 páginas. Para considerar su publicación deben reunir los siguientes requisitos:
El título debe estar en español e inglés.
• RESUMEN: En el que se expondrá en forma precisa la esencia del trabajo y deberá incluir los siguientes subtítulos: Objetivo, Material y métodos, Resultados y Conclusiones, y tendrá su respectiva traducción al inglés, la cual no deberá exceder las 200 palabras. Al final se escribirán las palabras clave en número no mayor de cinco (http://www. nlm. nih. gov).
• INTRODUCCIÓN: Que incluye la exposición de motivos del trabajo y una breve referencia de la literatura, que debe ser clara, explícita y concisa.
• MATERIAL Y MÉTODOS: Se describen las características del material empleado en el trabajo y la metodología usada en el estudio en forma concreta y precisa.
• RESULTADOS: Deberán ser de carácter objetivo, con el análisis estadístico en los casos pertinentes, sin interpretación personal y serán acompañados de las tablas y/o figuras
respectivas (figuras, radiografías, fotografías).
• DISCUSIÓN: Comprenderá la interpretación de los resultados comparándolos con los trabajos realizados por otros autores y las conclusiones que se derivan de ello cuando sea apropiado.
• AGRADECIMIENTOS (si viene al caso). • REFERENCIAS BIBLIOGRÁFICAS: Se harán
de acuerdo a las Normas de Vancouver y serán presentadas en el orden como aparecen en el texto. Todas las referencias deberán estar citadas ya sea en el texto o en las ilustraciones con un número arábigo entre paréntesis, en superíndice. Estas llamadas de cita precedidas de un espacio, se colocan antes del punto, coma u otro signo de puntuación. El máximo de referencias bibliográficas permitidas es de 40.
2. 3. TEMAS DE REVISIÓN Son trabajos de actualizaciones sobre temas
de la especialidad, en particular de trabajos originales o académicos o médico–quirúrgicos, no excederá de 12 páginas. Deberá acompañarse de un resumen con su respectiva traducción en inglés y palabras clave no más de 200 palabras.
2. 4. CASOS O REPORTES CLÍNICOS Se considera para su publicación únicamente
aquellos casos que signifiquen interés diagnóstico, anomalía de evolución, rareza de observación, evidente interés por la respuesta terapéutica. Deberá acompañarse de un resumen con su respectiva traducción en inglés y palabras clave. No excederá de 6 páginas.
Se tomarán en cuenta los siguientes puntos:a) Introducciónb) Reporte del caso clínicoc) Comentarioc) Bibliografía
2. 5. SALUD MENTAL Se publicarán artículos sobre algún aspecto
de salud mental del niño y el adolescente. No excederá de 4 páginas. Deberá acompañarse de un resumen con su respectiva traducción en inglés y palabras clave.
2. 6. ÉTICA Se considera en esta sección algún tema de
interés sobre ética médica. No excederá de 3 páginas.
49
2. 7. CULTURAL Se consideran para esta sección artículos de
interés en el área cultural. No excederá de 3 páginas.
3. NORMAS PARA SU PUBLICACIÓN Se refiere a los aspectos generales de los
trabajos.
3. 1. Los trabajos se reciben en el local de la Sociedad Peruana de Pediatría: Calle Los Geranios Nº 151, Urb. Jardín, Lince, Lima 14 – Perú; de lunes a viernes de 10 a. m. a 5 p. m. Teléfono Nº 4226397, correo electrónico: [email protected] Al entregar el trabajo recibirá una constancia de recepción.
3. 2. El artículo debe estar acompañado por una Carta de Presentación dirigida al presidente del Comité de Publicaciones solicitando la evaluación de dicho artículo para su publicación y debe certificarse mediante una Declaración Jurada que no ha sido publicado en otra revista y estar firmada por el autor y coautores.
3. 3. La Revista Peruana de Pediatría acepta la solicitud de publicación de contribuciones nacionales y extranjeras de la especialidad siempre que se ajusten a los requerimientos del presente Reglamento.
3. 4. La Revista Peruana de Pediatría se reserva el derecho de aceptar los trabajos que se presenten y de solicitar las modificaciones que considere necesarias para poder cumplir con las exigencias de la publicación.
3. 5. La publicación de trabajos en la Revista Peruana de Pediatría, en sus diferentes secciones, no obliga necesariamente a solidarizarse con las opiniones vertidas por él o los autores.
3. 6. Los trabajos presentados para su publicación en la revista son evaluados por árbitros externos.
3. 7. El trabajo deberá enviarse en original impreso y en forma electrónica debidamente grabado en un disquete o CD en el programa Word para Windows 97 o posterior, letra Arial, tamaño de fuente 10 puntos, espacio sencillo.
3. 8. El artículo debe usar el Sistema Internacional de Medidas.
3. 9. Las ilustraciones (tablas, figuras, fotografías y otros documentos similares) no excederán de 10 y deben estar insertadas dentro del artículo en Word, con el título correspondiente y en el orden de aparición, con letra tipo oración. Ejemplo: Tabla 1. Índices de salud.
3. 9. 1. Las tablas, gráficas, radiografías, fotografías, etc. , deben tener un título breve y claro, escrito en letra arial y serán numeradas según el orden de llamada que aparece en el texto. Precisar número de fuente en negrita. Ejemplo:
Tabla 1. Letalidad de acuerdo al grado de hemorragia intraventricular.
3. 9. 2. Las tablas y las ilustraciones serán considerados para el entendimiento del texto y no deberán ser reiterativos entre sí o con relación al texto (no trace líneas horizontales ni verticales en el interior de los cuadros).
3. 9. 3. Las figuras, radiografías, fotografías, etc. deben tener leyenda.
3. 9. 4. Las publicaciones de reproducciones a color deben consultarse con el director responsable.
3. 10. En la primera página del trabajo debe anotarse. Título: breve, que represente el contenido del
artículo, en español y en inglés. Nombre y los dos apellidos del autor y coautores. Grado académico y afiliación institucional. Nombre del Departamento y/o institución en que se
realizó el trabajo. Dirección del autor, adonde se le dirigirá la
correspondencia, así como teléfono y dirección de correo electrónico.
3. 11. Los autores que hayan presentado artículos recibirán una carta de la Secretaría de Comité de Publicación y Biblioteca, señalando la “aceptación” o “no aceptación” de dichos trabajos en un plazo máximo de 60 días. Los originales de los trabajos no aceptados serán remitidos con la carta de “no aceptación”.
3. 12. Una vez aceptado y publicado el artículo, los derechos pertenecen a la Revista Peruana de Pediatría, y serán reproducidos por terceros sólo con la autorización correspondiente.



















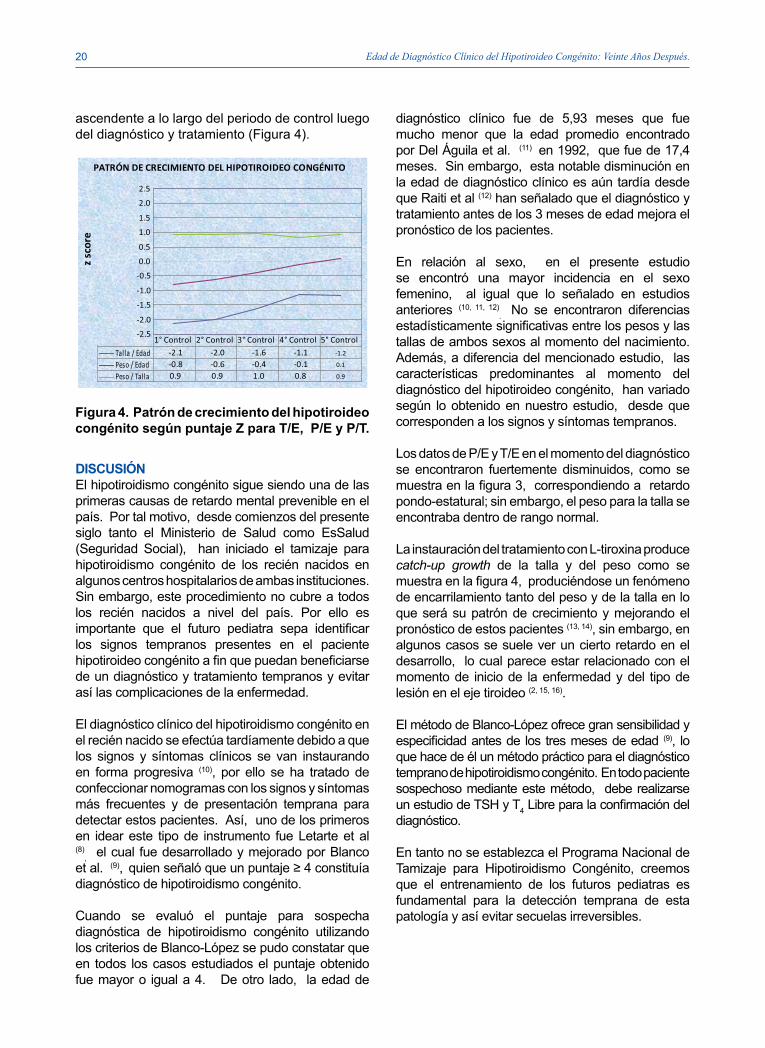



































![Power. MIAMI. Reforma Constitucional. Veronica Roman Quiroz[1]](https://static.documents.pub/doc/80x56/56d6bf311a28ab301695423f/power-miami-reforma-constitucional-veronica-roman-quiroz1.jpg)









